DOI:
10.1039/D3RA05858A
(Paper)
RSC Adv., 2023,
13, 32424-32432
Development of a chromium oxide loaded mesoporous silica as an efficient catalyst for carbon dioxide-free production of ethylene oxide
Received
28th August 2023
, Accepted 25th October 2023
First published on 3rd November 2023
Abstract
Ethylene oxide (EO) is a significant raw material used in many commodities for consumers, particularly ethoxylates, polymers, and certain other glycol derivatives. We synthesized a catalyst by incorporation of chromium oxide into a mesoporous silica material (Cr/MSM) via the hydrothermal method, an effective catalyst for partial ethylene oxidation for producing carbon dioxide (CO2) free EO. Subsequently, XRD, BET, XPS, and TEM were used to analyse the structural characteristics of the Cr/MSM catalyst. The catalytic performance of the synthesized catalyst was assessed in the liquid-phase epoxidation (LPE) of ethylene, utilizing peracetic acid (PAA) as an oxidant. This approach not only circumvented the generation of CO2 but also mitigated the risk of metal leaching. Confirmation of the successful production of EO was achieved through GC chromatography, where the presence of a peak with a retention time (RT) of 8.91 minutes served as conclusive evidence. We systematically explored a range of reaction parameters, including temperature, catalyst concentration, the molar ratio of ethylene to PAA, and solvent effect. This comprehensive investigation aimed to fine-tune the reaction conditions, ultimately improving ethylene conversion and enhancing the selectivity of the catalyst for EO production. This approach can effectively resolve the issues of greenhouse gas emissions and metal leaching that had been associated with previously reported catalysts.
Introduction
In the field of epoxidation, numerous materials have been investigated to evaluate their capabilities as effective catalysts for the production of polyethylene, polyvinyl chloride, and ethylene oxide (EO). In particular, EO is the third-largest ethylene derivative that is produced on a million–ton scale worldwide annually for various applications.1 It is the main building block of antifreeze, surfactants, polymers, and different glycols for sterilization and fumigation purposes.2 The conventional approach for EO production involves using oxygen as the oxidizing agent at elevated temperatures ranging from 392 to 500 °F and pressures between 10 and 30 bar, typically in the presence of a catalyst based on silver.3,4 Nevertheless, this method exhibits low selectivity in the epoxidation reaction, around 85%. This results in the substantial generation of carbon dioxide (CO2) as a by-product, positioning it as the second-largest emitter of CO2 among all chemical processes.3,4 Scientists have tried to replace the CO2-emitting gas-phase epoxidation (GPE) with a CO2-free liquid-phase epoxidation (LPE).4 In the LPE process, a methyltrioxorhenium (MTO)-based homogeneous catalyst and hydrogen peroxide (H2O2), as an oxidant, are employed, resulting in a highly efficient catalytic performance with nearly 100% selectivity for producing EO.5 However, this method employing MTO presents two drawbacks: (1) the catalyst deactivates due to the accumulation of water (a by-product), and (2) the use of expensive Re metal, which is also prone to supply instability. Therefore, exploring more cost-effective catalytic materials with high H2O2 utilization efficiency, as alternatives to Re, could enhance process viability and economic feasibility.6 As a consequence, there is a need for more research efforts to explore alternative heterogeneous epoxidation catalysts.7–9 In this pursuit, prior investigations have focused on catalysts composed of transition metals in higher oxidation states, which are supported on mesoporous silica materials (MSM), particularly MCM-41 and SBA-15.10–14 It was noted that the alkene substrate underwent catalysis through a redox reaction, enabling the transfer of oxygen from the oxidant to the alkene double bond.15–19 Among the various metal options for effective catalysts, niobium (Nb)-based catalysts have demonstrated good catalytic activity in the epoxidation of cyclic alkenes, propene, and various chemical substrates like carvone and limonene. These catalysts exhibit excellent thermal stability and resistance to both metal leaching and hydrolysis, making them well-suited for the LPE process.20–28 In this regard, the utilization of Nb/MSM has been reported, employing Nb/MCM-41 and MCM-48, in the oxidation of cyclohexene. However, it was found that efficient selectivity in epoxidation was achieved only with a low incorporation of Nb metal into the MCM-48 support.29–34 Yan et al. conducted research on MSM catalysts based on tungsten (W) and niobium (Nb) for the epoxidation of ethylene, utilizing hydrogen peroxide (H2O2) as the oxidizing agent.35 It was revealed that both W-KIT-6 and Nb-KIT-6 metal catalysts exhibited significant efficiency in EO production under standard conditions. Nevertheless, a notable limitation observed with W-KIT-6 and Nb-KIT-6-based catalysts was significant metal leaching and the decomposition of H2O2.36 In line with the research conducted by Yan et al., and other similar research efforts, we have successfully developed a highly efficient Nb/MSM-based catalyst for EO production in our previous study.9 In our method, we employed peracetic acid (PAA) as an oxidant instead of H2O2,36 which yielded not only excellent efficiency but also a remarkable advantage of CO2-free EO production.
In the present research work, we have prepared Cr/MSM catalyst for an EO production method with zero greenhouse gas emissions. This catalyst was synthesized using a hydrothermal method, where CrCl3 was employed to introduce Cr metal into an ordered cubic large-pore mesoporous silicate (SiO2) matrix, generated from tetraethyl orthosilicate (TEOS). PAA was selected as the oxidant due to its reaction with ethylene gas, resulting in the CO2-free production of EO, accompanied by the formation of by-products, including acetic acid (AA), ethylene glycol (EG), and its derivatives. These by-products hold notable significance and find diverse applications across various fields. The heteroatom structure of prepared Cr/MSM was verified through XRD analysis, while BET analysis was used to examine its porosity. Likewise, the morphology of the catalyst was assessed using TEM. The Cr/MSM catalyst was then employed in the production of EO through a liquid-phase epoxidation (LPE) process. In this process, ethylene gas was pressurized (at 50 bars) and then dissolved in a liquid reaction medium containing PAA. This LPE process was applied to synthesis both EO and acetic acid, as verified through gas chromatography (GC) analysis. To maximize conversion of ethylene and catalyst's selectivity towards EO production, we optimized the reaction conditions such as temperature, ethylene to oxidant molar ratio, catalyst dose, and solvent effect to achieve the best possible results. The GC analysis affirmed an 81.8% conversion rate of ethylene to EO, with a corresponding selectivity of 88.4%. In this whole process, we successfully produced EO and valuable by-products, all while achieving a zero CO2 emission, underscoring the environment-friendliness of this EO production method.
Experimental
Chemicals
Tetraethyl orthosilicate (TEOS) (Sigma-Aldrich, 98% Germany), chromium III chloride (Sigma-Aldrich, Germany), pluronic P123 (Sigma-Aldrich, Germany), n-butanol (Sigma-Aldrich, Germany), conc. sulphuric acid (Merck, Germany), peracetic acid (Sigma Aldrich, 32%, Germany). All reagents and compounds were of analytical grade and used as received during the synthesis process.
Synthesis of catalyst (Cr/MSM)
The catalyst synthesis involved a typical hydrothermal method.37 Initially, 6 g of pluronic P123 was dissolved in 218 mL of distilled water. To this solution, 7.92 mL of 35% conc. HCl was gradually added with constant stirring at 35 °C. After the complete dissolution of pluronic P123, 7.41 mL of n-butanol solvent was added while stirring at 35 °C over a period of one hour. Then, 1.539 g of CrCl3 and 14.849 mL of TEOS were added dropwise to the mixture under the same conditions. The resulting mixture was stirred at 35 °C for 24 hours and then was heated at 100 °C for another 24 hours in a closed flask, which resulted in the formation of precipitates in the reaction mixture. Afterward, these precipitates were filtered out and left to dry overnight at 100 °C. The resulting crude product was washed with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethanol and HCl. Finally, the obtained solid material was calcinated at 550 °C. To evaluate the properties, a separate preparation of the catalyst support (MSM) was carried out using the identical synthesis procedure as Cr/MSM, excluding the addition of CrCl3 to the reaction mixture along with TEOS.
:1 mixture of ethanol and HCl. Finally, the obtained solid material was calcinated at 550 °C. To evaluate the properties, a separate preparation of the catalyst support (MSM) was carried out using the identical synthesis procedure as Cr/MSM, excluding the addition of CrCl3 to the reaction mixture along with TEOS.
Ethylene oxide production
The prepared catalyst was used for the LPE of ethylene to produce EO. During this process, PAA, acetonitrile, and the catalyst were added in the specified quantities into the reactor. The purified ethylene gas was supplied into this reactor from a separate cylinder at a pressure of 50 bar. A weighing balance was used to monitor the feed quantity of ethylene gas. An oil bath equipped with a digital heater, cooler, and variable speed pump, with a temperature range of −20 to 200 °C, was employed to control the temperature of the reactor. Simultaneously, the pressure within the reactor was regulated by a pressure controller located on the gas line tail.
The reaction was conducted at various temperatures (40, 50, and 60 °C). Likewise, the reaction time was varied from 1 to 6 hours in an effort to determine the optimal reaction conditions. In all these experiments, the pressure was maintained constantly at 50 bar. Following each experiment, a specific amount of the sample was obtained and analysed by gas chromatography. As a reference, these experiments were also conducted using MSM (catalyst support) to confirm its inertness. Furthermore, the inertness of acetonitrile was experimentally confirmed through blank runs. The simplified reaction is shown in Scheme 1.
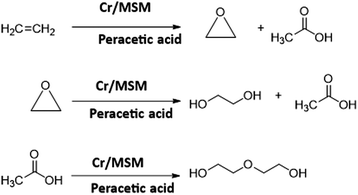 |
| | Scheme 1 Synthesis of EO and its derivatives via LPE. | |
The equations provided were used to calculate the quantity of EO produced and the EO selectivity.
| |
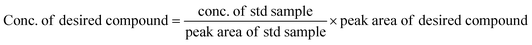 | (1) |
| |
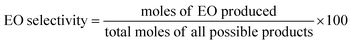 | (2) |
Similarly, a reported method was employed to calculate the utilization (UPAA) and conversion (XPAA) efficiencies of PAA.38
| |
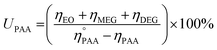 | (3) |
| |
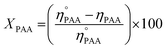 | (4) |
ηEO,
ηMEG, and
ηDEG = moles of EO, mono ethylene glycol (MEG), and diethylene glycol (DEG), respectively.
and
ηPAA = initial and final moles of PAA.
Sample characterization
The powder X-ray diffractometer (PXRD) pattern was used to confirm the successful preparation of the mesoporous silica material MSM (catalyst support) and the Cr/MSM catalyst. Monochromatic Cu Kα radiation (λ = 0.154 nm) with an output voltage of 200 kV and a current of 50 mA was used, utilizing the advanced Miniflex Benchtop power X-ray diffractometer (Rigaku, Japan). The mean size of crystallites, with a size error of 0.5 nm, was determined from the XRD data using the Scherrer's equation.39,40 In the Brunauer–Emmett–Teller (BET) analysis, the Micro-metrics Gemini VII 2390t Automated Gas Sorption instrument was used to analyse the surface of the MSM and Cr/MSM via the N2 adsorption–desorption method at 77 K, with a pressure range of 0.05–0.30 bars. The identification of both the adsorption and desorption branches of the isotherms provided valuable data for evaluating the surface properties and pore diameter of the samples. The Barrett–Joyner–Halenda (BJH) approximation was employed to determine the pore size. The samples were heated for 4 hours at 150 K in a vacuum before analysis.41 Higher-resolution TEM pictures were obtained using a TEM microscope (JEOL 2010F) operating at a high voltage of 200 kV. To prepare the samples, a suspension of the sample was diluted with methanol and subsequently drop-dried onto holey carbon-coated copper TEM grids. The chemical composition of the sample was analyzed using X-ray photoelectron spectrometer (XPS) with an Al–K X-ray source operating at 1486.6 eV and a hemispherical energy analyzer (Specs EA 10 Plus) operating in vacuum better than 10−7 Pa. The core-level binding energies of photoelectrons emitted from the surface were determined and calibrated by fixing the C (1s) core level at 284.5 eV.
Results and discussion
XRD analysis
The XRD analysis was used to confirm the successful synthesis of MSM and Cr/MSM, the XRD diffractograms of the synthesized MSM and Cr/MSM are shown in Fig. 1(a) and (b), respectively. The XRD diffractogram of MSM displays a single broad peak at 2θ = 1.7° confirming the amorphous nature of this material. Whereas in the diffractogram of Cr/MSM, a broad peak is observed at 2θ = 23.32° which can be attributed to the presence of short double bonds in Cr2O3 or Cr2O5, which might resemble Cr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.42 As reported, the calcined sample primarily comprised Cr(V) or Cr(VI) species, signifying a shift from a lower-valent state.
O bonds.42 As reported, the calcined sample primarily comprised Cr(V) or Cr(VI) species, signifying a shift from a lower-valent state.
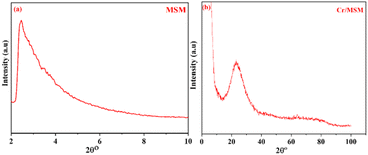 |
| | Fig. 1 XRD pattern of (a) MSM and (b) Cr/MSM. | |
Additionally, the initially prepared sample appeared pale green but changed to yellow after calcination. This colour shift may indicate the transformation of trivalent chromium ions in octahedral geometry to higher-valence chromium ions, such as chromate and/or polychromate ions, adopting a tetrahedral geometry.42 As reported, the calcined sample primarily comprised Cr(V) or Cr(VI) species, signifying a shift from a lower-valent state.40 This infers that the Cr ions are evenly distributed throughout the inner and outer channels of the MSM support, and it also implies that the amorphous structure of the MSM support was not disrupted by loading the metal ions. These findings are in good agreement with previously published work.43
BET analysis
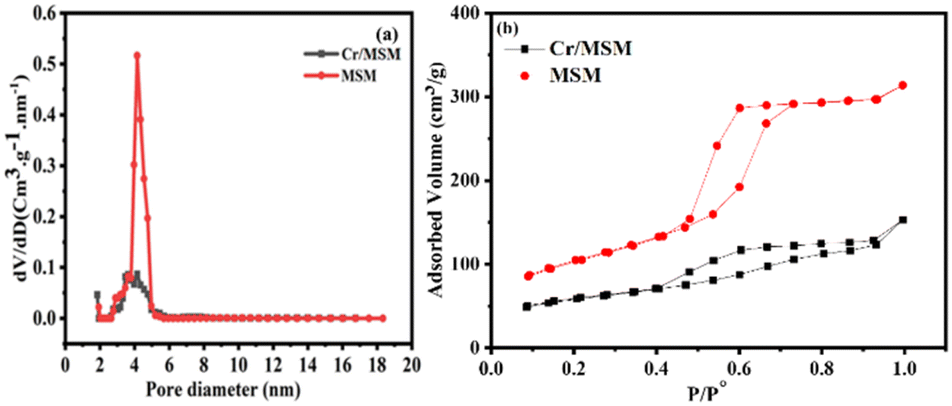
The BET technique was employed to study the surface properties of the Cr/MSM catalyst and its support at 77 K. Fig. 2 illustrates that both the catalyst and the support display type IV isotherms in accordance with IUPAC nomenclature. These isotherms are characteristic of well-ordered silica materials, as evidenced by the observed hysteresis loops. The MSM support exhibits a surface area of 270.475 m2 g−1 and a pore volume of 0.434 cm3 g−1. However, after incorporating Cr metal into the MSM support, the surface area significantly decreased from 270.475 to 153.616 m2 g−1. Furthermore, the pore volume decreased from 0.434 to 0.185 cm3 g−1 due to partial blockage of the pores, as shown in (Fig. 2(a)). The Barrett–Joyner–Halenda (BJH) method was utilized to determine the pore dimensions of both the Cr/MSM and MSM samples.37,44 Also, to assess the uniformity of pore sizes, capillary condensation analyses were performed on both the Cr/MSM catalyst and its support, utilizing the adsorption–desorption curve. As illustrated in Fig. 2(b), the detection of the sharp inflection in the adsorption isotherm, occurring at a relative pressure (p/p0) between 0.5 and 0.7, is primarily attributed to capillary condensation of nitrogen within the pores. The degree of pore size uniformity is assessed by considering the height and sharpness of the inflection. The measurements revealed a mean pore diameter of 4.532 nm for Cr/MSM and 4.152 nm for the support. These findings align with previously reported literature.45,46 Table 1 presents various textural parameters obtained from the BET analysis, including surface area (SBET), cumulative pore volume (VBJH), and mesoporous diameter (DBJH).
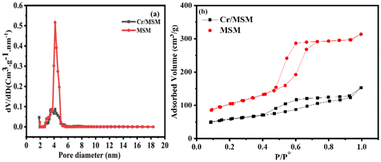 |
| | Fig. 2 (a) Pore size distribution (b) N2 adsorption–desorption isotherm. | |
Table 1 BET results of support (MSM) and catalyst Cr/MSM
| Entry |
Material |
(SBET) (m2 g−1) |
(VBJH) (mL g−1) |
(DBJH) (nm) |
| 1 |
MSM support |
270.475 m2 g−1 |
0.434 cm3 g−1 |
4.152 nm |
| 2 |
Cr/MSM |
153.616 m2 g−1 |
0.185 cm3 g−1 |
4.532 nm |
TEM
The structure and porous nature of the Cr/MSM catalyst was also examined using TEM images at resolutions of 50 nm and 100 nm, as given in Fig. 3. These TEM micrographs confirm the mesoporous nature of the calcined Cr/MSM catalyst. The images reveal the uniform distribution of Cr2O3 nanoparticles with sizes ranging from 20 to 30 nm throughout the MSM. This indicates that the morphology of the silica material remains unchanged after loading Cr2O3 nanoparticles into the MSM support.47 Furthermore, both XRD and BET analyses agreed well with this fact.
 |
| | Fig. 3 TEM images of Cr/MSM catalyst. | |
XPS
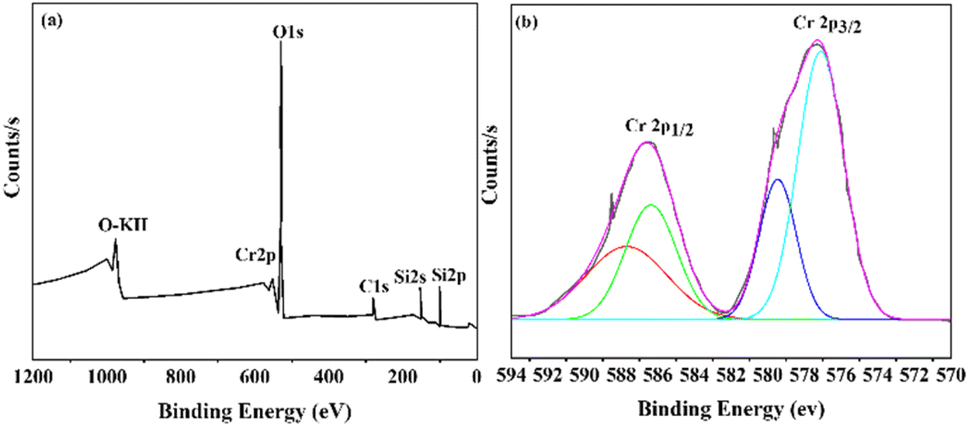
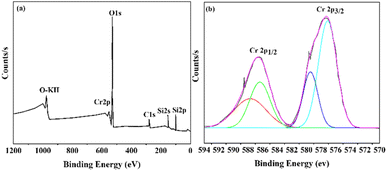
X-ray photoelectron spectroscopy (XPS) was employed to examine the surface chemistry and presence of Cr species in the synthesized Cr/MSM catalyst. The XPS survey spectrum presented in Fig. 4(a) reveals the presence of elements such as Chromium (Cr), Oxygen (O), Silicon (Si), and Carbon (C) on the surface of the Cr/MSM catalyst.
 |
| | Fig. 4 (a) XPS analysis of Cr/MSM (b) Cr 2p XPS spectrum of Cr/MSM. | |
In Fig. 4(b), the finely scanned XPS spectra of the Cr 2p orbital for the prepared catalyst unveil two prominent peaks at approximately 577 eV and 579 eV, which can be attributed to Cr3+ and Cr5+, respectively, based on prior research.48–50 To further analyze the XPS peaks corresponding to Cr 2p3/2 and Cr 2p1/2, we performed deconvolution using a Gaussian–Lorentzian–Voigt fitting method.51 This process resulted in the appearance of two additional peaks. In line with established literature, the peaks observed at approximately 579 eV and 589 eV are typically attributed to Cr5+ ions, while those at 577 eV and 587 eV are assigned to Cr3+,52,53 These findings confirm the presence of both Cr2O3 and Cr2O5 species on the MSM support.
The catalytic process
Ethylene epoxidation using Cr/MSM and MSM. The epoxidation of ethylene was carried out using the Cr/MSM catalyst in the presence of PAA. In order to confirm that the MSM does not exhibit any intrinsic catalytic activity in the epoxidation process, we conducted a control experiment. This control experiment involved the epoxidation of ethylene using PAA and pure MSM, without the addition of the Cr/MSM catalyst54 (entry 2 in Table 2). Following a typical procedure, EO was produced by heating a reaction mixture consisting of 3.29 mmol of ethylene, 5.6 mmol of oxidant peracetic acid (PAA), 0.3 g of Cr/MSM catalyst, and 20 mL of acetonitrile at 40 °C for 6 hours. Gas chromatography was employed to analyse the successful production of EO, and the chromatograms obtained are depicted in Fig. 5. The GC chromatogram in Fig. 5(a) shows the presence of an EO peak with a retention time (RT) of 8.91 minutes, indicating the successful epoxidation of ethylene. Additionally, the chromatogram displays peaks corresponding to useful by-products of the LPE method, including acetic acid (AA), monoethylene glycol (MEG), and diethylene glycol (DEG), at RT values of 23.91, 26.52, and 31.02 minutes, respectively. The GC analysis further confirms the environmental friendliness of the EO production procedure, as no peak is observed at a retention time of 3.5 minutes (Fig. 5(a)), indicating the absence of CO2 production. In contrast, in the control experiment using only the MSM support instead of Cr/MSM for EO formation, neither EO nor any by-products were formed, as exhibited in Fig. 5(b). Similarly, the Cr/MSM catalyst exhibited a good selectivity of 93.7% for EO formation with a mild ethylene conversion of 61% (Fig. 5(c)).
Table 2 Catalytic performances of Cr/MSM and MSM in ethylene epoxidation
| Entry |
Catalyst |
TOF |
XPAA (%) (±3%) |
UPAA (%) (±3%) |
| Blank run was conducted under identical conditions to the original experiment, except that ethylene was not added, while Cr/MSM and PAA were present. TOF (turnover frequency) is the no. moles of CH3COOOH converted per Cr atom per unit of time.35 |
| 1 |
Cr/MSM |
189 |
13.05 |
18.0 |
| 2 |
MSM |
0 |
0 |
0 |
| 3 |
Cr/MSMa |
0 |
12.5 |
0 |
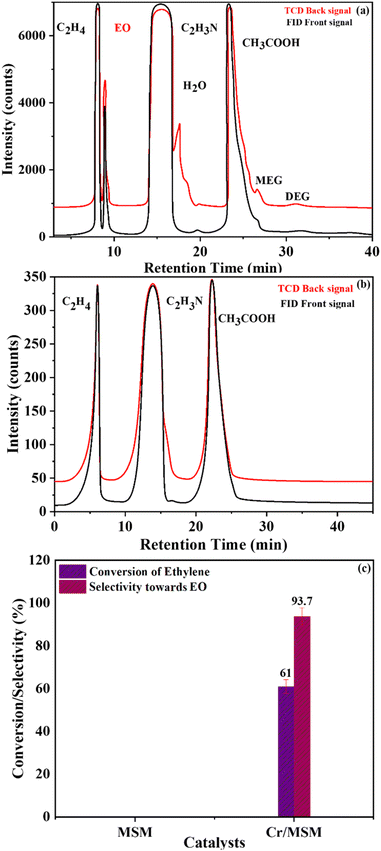 |
| | Fig. 5 GC chromatogram of (a) Cr/MSM (b) MSM support and (c) conversion of ethylene and selectivity of Cr/MSM to EO production. [Reaction condition: ethylene = 3.29 mmol, PAA (32%) = 5.6 mmol, temperature = (50 °C), dose of catalyst (10 wt%) = 0.3 g, solvent = C2H3N (20 mL), time = 6 h.] | |
Furthermore, Table 2 provides data obtained from the ethylene epoxidation in the presence of a catalyst, support, and a blank run. The data clearly shows that using Cr/MSM catalyst, 13.05% conversion efficiency (XPAA) and 18.0% utilization efficiency (UPAA) of PAA were obtained. No EO was produced when MSM was employed instead of Cr/MSM, and there was no PAA conversion (entry 2 in Table 2). However, the conversion of PAA of 12.5% was observed in the blank run (entry 3 in Table 2), where the experiment involved Cr/MSM and PAA, without the addition of ethylene. As reported by Yan et al.35 a metal peroxo complex is formed by the reaction of PAA with Cr/MSM, this complex is subsequently utilized in the oxidation of alkenic substrate to the corresponding epoxide. Such complex formation on MCM-41 material grafted with tungsten has been supported by prior experimental evidences.55 In the blank run (entry 3 in Table 2), as the ethylene was not present, this metal peroxo complex might have been decomposed during the course of the reaction. Moreover, the Turnover frequency (TOF) of Cr/MSM was also calculated using a reported method.56 As given in Table 2, the TOF was measured to be 189, using PAA and Cr/MSM.
According to a study by Wu et al., where H2O2 was used as an oxidant and Nb/MSM as a catalyst, the reported TOF value was 89.56 Therefore, it can be concluded that PAA has greater potential as an oxidant compared to H2O2 for EO formation.
In pursuit of optimizing the catalyst's selectivity for EO production and enhancing the conversion of ethylene to EO, we conducted LPE experiments, systematically varying reaction parameters such as temperature, catalyst dosage, ethylene to PAA molar ratio, and employing various solvents.
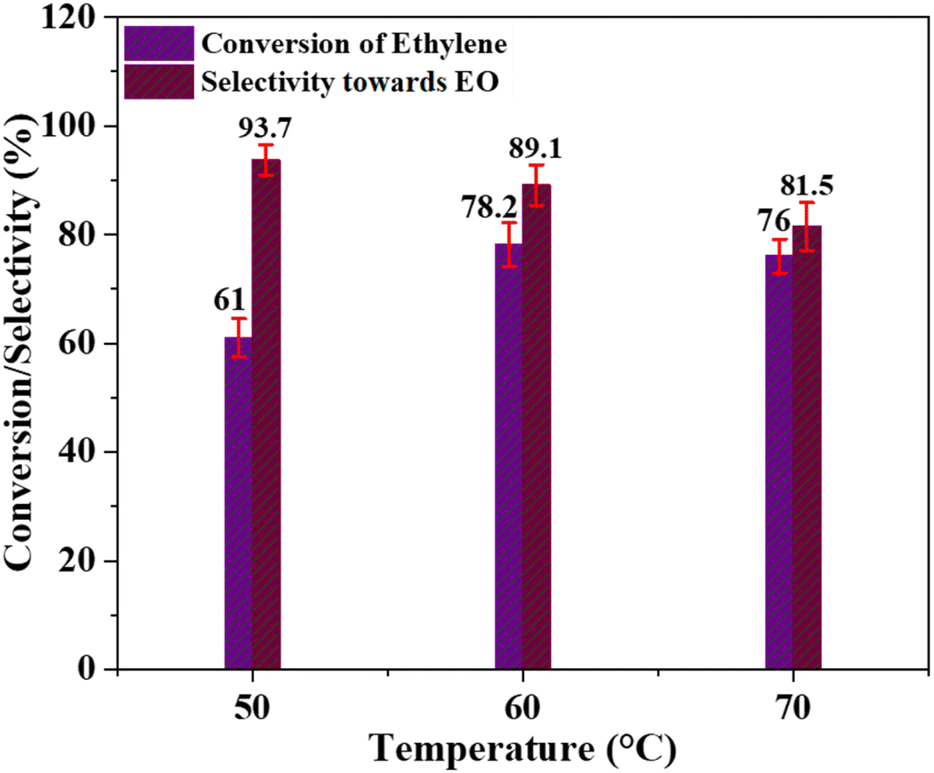
Effect of temperature. The effect of temperature on the conversion of ethylene and the selectivity of the catalyst for ethylene oxide production was examined at three different temperatures: 50 °C, 60 °C, and 70 °C. The remaining reaction parameters, such as the molar ratio of ethylene to PAA and the catalyst quantity, were kept constant. Acetonitrile was used as the solvent, and the reaction duration was set at 6 hours. The corresponding results are depicted in Fig. 6. At 50 °C, the ethylene conversion was 61% with a catalyst selectivity of 93.7%. Increasing the temperature to 60 °C improved the ethylene conversion to 78.2%, but the selectivity decreased to 89.1%. Further increase in the temperature to 70 °C resulted in an ethylene conversion of 76% and a selectivity of 81.5%. The synthesis of acetic acid (AA) and glycol derivatives as by-products likely contributes to the lower selectivity of the catalyst towards ethylene oxide production at high temperatures. The reaction mechanism of this process is widely recognized to involve the production of ethylene oxide intermediate, and at elevated temperatures, the stability of the C–C bond surpasses that of the C–O bond.57 Thus, the higher ethylene conversion and lower catalyst selectivity towards EO formation might be caused by the rapid breakdown of PAA at high temperatures.58 Therefore, it can be deduced that 60 °C is the optimal temperature for achieving the best possible conversion of ethylene and selectivity of the Cr/MSM catalyst for EO formation.
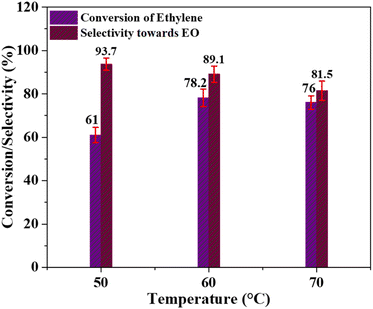 |
| | Fig. 6 Effect of temperature; [reaction conditions: ethylene = 3.29 mmol, PAA (32%) = 5.6 mmol, time = 6 h, catalyst dose = 0.3 g, solvent = acetonitrile (20 mL)]. | |
Effect of catalyst amount. The amount of catalyst has a significant effect on both the conversion of ethylene and the catalyst's selectivity for EO formation. This effect was evaluated by conducting a series of LPE reactions by varying the catalyst amounts from 0.1 to 0.5 g. All these experiments were carried out for 6 hours using a temperature of 60 °C, ethylene to oxidant (PAA) molar ratio of 1:2, and acetonitrile as an organic solvent.As shown in Fig. 7, increasing the catalyst quantity resulted in a slight improvement in selectivity towards EO production, along with a significantly higher conversion of ethylene, which can be attributed to the increase in the active catalyst species. As illustrated in Fig. 7, the ethylene conversion notably increased from 58.3 to 78.2% when the catalyst dose was increased from 0.1 to 0.3 g. Further increasing the catalyst amount only marginally improved the ethylene conversion efficiency. Likewise, increasing the catalyst amount from 0.1 to 0.5 g only led to a slight improvement in selectivity, from 84.2 to 89.7%. Consequently, under the given reaction conditions, a catalyst amount of 0.3 g can be considered optimal.
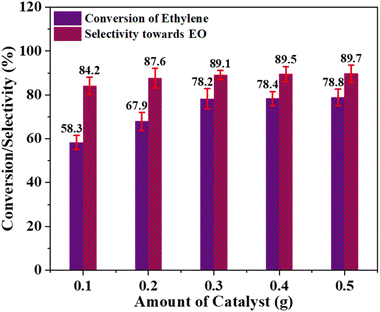 |
| | Fig. 7 Effect of catalyst quantity; [reaction conditions: ethylene = 3.29 mmol, oxidant (32%) = 5.6 mmol, time = 6 h, solvent = acetonitrile (20 mL), temperature = 60 °C]. | |
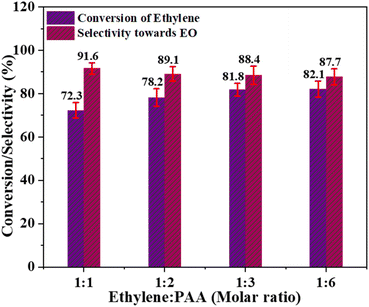
Effect of ethylene-to-oxidant molar ratio. To assess the effect of the molar ratio of ethylene to oxidant on ethylene conversion and catalyst's selectivity for EO production, the ethylene epoxidation reaction was conducted using optimal temperature conditions (60 °C), a catalyst amount of 0.3 g, and the acetonitrile as an organic solvent. Different ethylene to oxidant molar ratios, such as 1:1, 1:2, 1:3, and 1:6, were used in these experiments. Fig. 8 clearly shows that the conversion of ethylene increased continuously from 72.3% at ethylene to PAA molar ratio of 1:1 and peaked at 82.1% when the molar ratio was 1:6. However, the catalyst's selectivity for EO production exhibited a different pattern, continuously decreasing with increasing PAA levels. This may be attributed to the significant decay of the oxidant (PAA) at elevated doses, which remains in the reaction mixture instead of binding to catalytic active sites. These findings suggest that employing an excess of the oxidant is not favourable for increasing catalytic activity. Hence, the optimal molar ratio of ethylene to PAA, which achieves a balance between ethylene conversion and selectivity for EO formation, is determined to be 1:3.
 |
| | Fig. 8 Effect of ethylene to PAA molar ratio; [reaction conditions: ethylene = 3.29 mmol, catalyst dose 0.3 g, time = 6 h, solvent = acetonitrile (20 mL), temperature = 60 °C]. | |
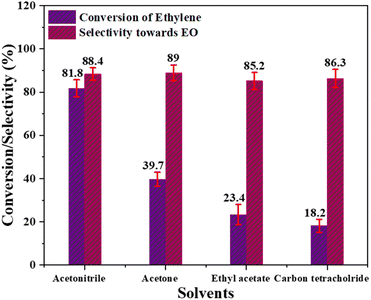
Effect of solvent. The effect of different solvents, including ethyl acetate, acetonitrile, carbon tetrachloride, and acetone, on ethylene conversion and EO production was studied under optimal reaction conditions, including temperature, molar ratio of ethylene to oxidant, and catalyst quantity. Fig. 9 clearly shows that acetonitrile solvent demonstrated the highest ethylene conversion at 81.8%. Furthermore, a decrease in solvent polarity impedes ethylene conversion, with ethylene conversion values of 39.7%, 23.4%, and 18.2% observed in acetone, ethyl acetate, and CCl4 respectively. This can be attributed to increased mass transfer and homogeneity in more polar solvents, leading to higher ethylene conversion.59 However, EO selectivity did not exhibit a clear pattern in response to changes in the solvent, indicating that it is not significantly affected by solvent variations.
 |
| | Fig. 9 Effect of different solvents; [reaction conditions: ethylene = 3.29 mmol, PAA (32%) = 5.6 mmol, temperature = 60 °C, time = 6 h, catalyst = 0.3 g, solvent = acetonitrile (20 mL)]. | |
Based on all of these experiments, it can be deduced that the for best selectivity of Cr/MSM catalyst for EO production and conversion of ethylene, the trade-off parameters are temperature (60 °C), catalyst quantity (0.3 g), and ethylene to oxidant molar ratio (1:3). The ideal solvent for EO production in LPE with Cr/MSM catalyst and PAA as an oxidant is acetonitrile. As we have reported28 that PAA may play a role in the hydrolysis of EO, potentially leading to a reduction in EO selectivity.9 Thus, epoxidation should take place at feasible lower catalyst incorporation on the support to reduce epoxide ring opening since a higher catalyst/acidity in the catalyst might lead to more breakdown of PAA and increased hydrolysis of the EO ring.38 Furthermore, it has been well reported that water produced during the process may promote brønsted acid sites by suppressing Lewis acid sites, and these brønsted acid sites may accelerate the hydrolysis of the epoxide ring.35
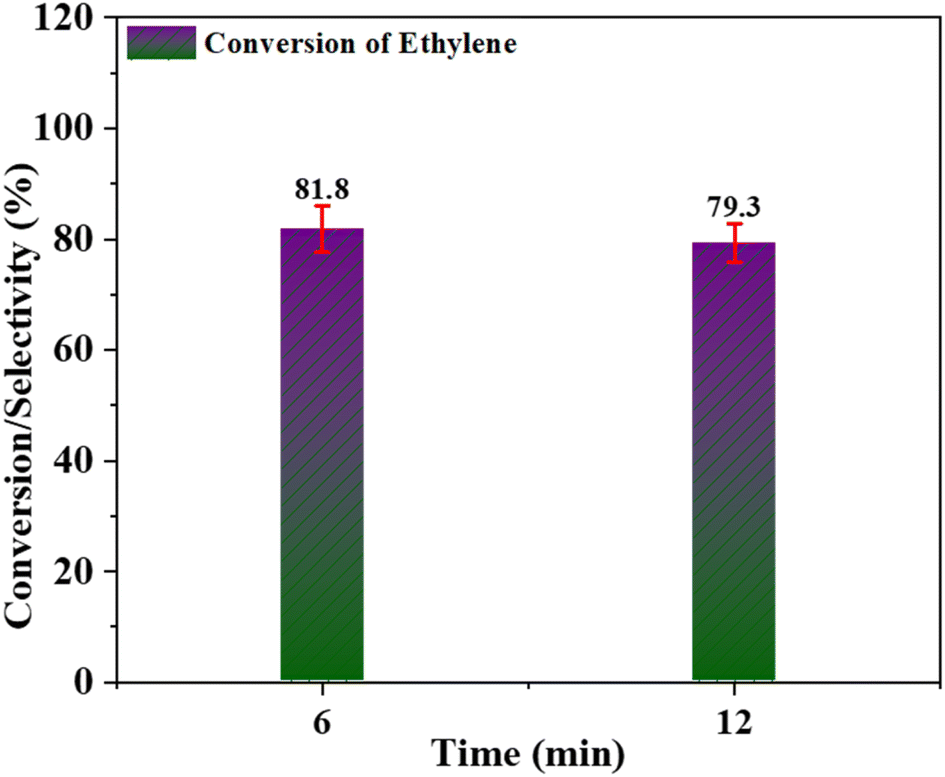
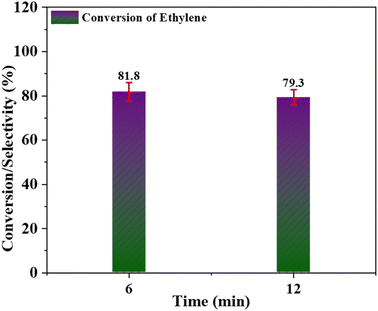
Leaching of catalyst. During the epoxidation reaction, some oligomerized metal oxide particles are formed on the surface of the catalyst in the presence of an oxidant. Such oligomerized metal oxide particles cannot catalyze the epoxidation reaction, as reported by Das et al.60 To investigate this leaching behaviour of the metal, a Cr/MSM catalyst was subjected to a leaching test. This involved separating the catalyst from the heated reaction mixture at a temperature of 60 °C after 6 hours, followed by allowing the supernatant reaction solution to continue for an additional 6 hours, resulting in a total reaction time of 12 hours.61 The ethylene conversion at 6 hours and 12 hours was analysed using the GC approach. As illustrated in Fig. 10, the study indicates that there was no further ethylene conversion. This fact clearly shows that the Cr/MSM catalyst (when used with PAA) is very significant to mitigate the problem of leaching, other than formation of some oligomerized metal oxide species.
 |
| | Fig. 10 Leaching test for Cr/MSM catalyst for ethylene conversion. | |
Additionally, Table 3 presents a comparative analysis of the catalytic properties of the catalyst prepared in the current study with those reported in previously published literature.
Table 3 A comparison of our catalyst for EO production with the previously reported catalysts is presented
| Catalyst |
Ethylene conversion |
EO selectivity |
Oxidant |
TOF (±3%) |
Ref. |
| Nb/MSM |
79.5% |
84.3% |
PAA |
163 |
9 |
| Ag/MSM |
79.3% |
82.5% |
O2 |
159 |
62 |
| W/MSM |
68.2% |
80% |
H2O2 |
153 |
35 |
| Nb/MSM |
50.1% |
58% |
H2O2 |
284 |
35 |
| Cr/MSM |
81.8% |
88.4% |
PAA |
189 |
This work |
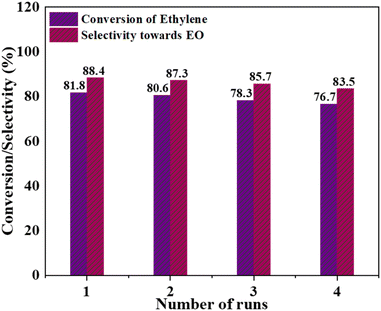
Reusability of the catalyst. An examination of the reusability of the Cr/MSM catalyst was conducted (Fig. 11). This involved separating the catalyst from the reaction mixture through filtration, followed by a thorough washing with a solvent and subsequent drying at 100 °C for 6 hours. It was observed that, under the optimized reaction conditions, both the conversion of ethylene and the selectivity towards EO exhibited a modest reduction. This reduction may be attributed to the formation of oligomerized metal oxide species, which might have been removed from the catalyst's surface during the washing process. These results confirm the reusability of the prepared catalyst.
 |
| | Fig. 11 Catalytic efficiency (%) vs. recycle times of Cr/MSM catalyst. | |
Conclusions
An eco-friendly production of ethylene oxide (EO) was accomplished using Cr/MSM catalyst via the liquid-phase epoxidation (LPE) of ethylene. This catalyst was synthesized by incorporation of chromium oxide into mesoporous silica material (MSM), and this Cr/MSM catalyst was thoroughly characterized by XRD, TEM, and BET analysis. The XRD technique was used to validate the presence of the heteroatom structure within Cr/MSM, while N2 adsorption isotherms were utilized to assess the mesoporous configuration. We assessed the catalytic activity of Cr/MSM in EO production through the liquid-phase epoxidation of ethylene, employing peracetic acid as the oxidizing agent. This process yielded an impressive 81.8% ethylene conversion with a catalyst's selectivity for EO production of 88.4%. To attain these results, we precisely fine-tuned multiple reaction parameters, including temperature (60 °C), catalyst dosage (0.3 g), the ethylene to PAA molar ratio (1:3), and reaction duration (6 hours), and acetonitrile was found to be the best organic solvent in this process. These investigations and the resulting outcomes provide strong affirmation that the synthesized Cr/MSM catalyst holds substantial promise as a catalyst for efficient and CO2-free ethylene oxide production.
Author contributions
Conceptualization, Toheed Akhter and Muhammad Maqbool; methodology, Maqbool; validation, Toheed Akhter, Sadaf Ul Hassan, Asif Mahmood, Waheed Al-Masry; formal analysis, Muhammad Maqbool; investigation, Muhammad Maqbool; resources, Toheed Akhter, Shumaila Razzaque; writing-original draft preparation, Toheed Akhter, Shumaila Razzaque; writing-review and editing, Toheed Akhter, Sadaf Ul Hassan, Asif Mahmood, Waheed Al-Masry; and; supervision, Toheed Akhter and Sadaf Ul Hassan. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the European Union Horizon 2020 research and innovations program under the Marie Sklodowska Curie grant number 847639. The authors acknowledge the funding provided by Researcher's Supporting Project Number (RSP2023R43), King Saud University, Riyadh, Saudi Arabia.
References
- V. Dal Santo, M. Guidotti, R. Psaro, L. Marchese, F. Carniato and C. Bisio, Proc. R. Soc. A, 2012, 468(2143), 1904–1926 CrossRef CAS.
- E. Alper and O. Y. Orhan, Petroleum, 2017, 3, 109–126 CrossRef.
- L. Pinaeva and A. Noskov, Pet. Chem., 2020, 60, 1191–1206 CrossRef CAS.
- S. Frankowski, A. Skrzyńska and Ł. Albrecht, Chem. Commun., 2019, 55, 11675–11678 RSC.
- S. A. Theofanidis, G. Kasun Kalhara Gunasooriya, I. Itskou, M. Tasioula and A. A. Lemonidou, ChemCatChem, 2022, 14, e202200032 CrossRef CAS.
- H. Delavar and A. Naderifar, Chem. Eng. Sci., 2022, 250, 117425 CrossRef CAS.
- P. H. Keijzer, J. E. van den Reijen, C. J. Keijzer, K. P. de Jong and P. E. de Jongh, J. Catal., 2022, 405, 534–544 CrossRef CAS.
- B. T. Egelske, W. Xiong, H. Zhou and J. R. Monnier, J. Catal., 2022, 410, 221–235 CrossRef CAS.
- M. Maqbool, T. Akhter, M. Faheem, S. Nadeem, C. H. Park and A. Mahmood, RSC Adv., 2023, 13, 1779–1786 RSC.
- M. Moliner and A. Corma, Microporous Mesoporous Mater., 2014, 189, 31–40 CrossRef CAS.
- Y. Yu, F. Li, Z. Zang, L. Xu and G. Liu, Mol. Catal., 2020, 495, 111158 CrossRef CAS.
- H. Wang, H. Cheng, F. Lai and D. Xiong, Materials, 2022, 15, 8097 CrossRef CAS PubMed.
- M. Selvaraj and P. Sinha, New J. Chem., 2010, 34, 1921–1929 RSC.
- M. Selvaraj, M. Kandaswamy, D. Park and C. Ha, Catal. Today, 2010, 158, 377–384 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485–493 CrossRef CAS.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J. M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- J. M. Thomas, Chem. Phys., 2008, 128, 182502 Search PubMed.
- Y. Huang, D. Yu, Y. Qiu, L. Chu and Y. Lin, Front. Chem., 2020, 8, 557334 Search PubMed.
- F. Zaera, Chem. Rev., 2022, 122, 8594–8757 CrossRef CAS PubMed.
- M. Ziolek, I. Sobczak, A. Lewandowska, I. Nowak, P. Decyk, M. Renn and B. Jankowska, Catal. Today, 2001, 70, 169–181 CrossRef CAS.
- F. Somma, P. Canton and G. Strukul, J. Catal., 2005, 229, 490–498 CrossRef CAS.
- D. Kryszak, K. Stawicka, V. Calvino-Casilda, R. Martin-Aranda and M. Ziolek, Appl. Catal., A, 2017, 531, 139–150 CrossRef CAS.
- J. V. Coelho, L. C. Oliveira, F. C. Moura, P. P. de Souza, C. A. Silva, K. B. Batista and M. J. da Silva, Appl. Catal., A, 2012, 419, 215–220 CrossRef.
- A. Feliczak-Guzik, A. Wawrzyńczak and I. Nowak, Microporous Mesoporous Mater., 2015, 202, 80–89 CrossRef CAS.
- H. Talukdar, S. R. Gogoi, S. Y. Sultana, R. Begum, D. Dowerah, B. Sarma and N. S. Islam, Dalton Trans., 2023, 52, 10165–10182 RSC.
- T. Mizugaki and K. Kaneda, Chem. Rec., 2019, 19, 1179–1198 CrossRef CAS PubMed.
- D. d. S. Valadares, J. O. C. de França, R. C. Fernandes, L. M. Dezaneti, S. C. Loureiro Dias and J. A. Dias, Chemistry, 2023, 5, 1138–1170 CrossRef CAS.
- N. V. Maksimchuk, V. Y. Evtushok, O. V. Zalomaeva, G. M. Maksimov, I. D. Ivanchikova, Y. A. Chesalov, I. V. Eltsov, P. A. Abramov, T. S. Glazneva and V. V. Yanshole, ACS Catal., 2021, 11, 10589–10603 CrossRef CAS.
- Y. Liu, K. Murata and M. Inaba, Chem. Lett., 2003, 32, 992–993 CrossRef CAS.
- G. Saikia, K. Ahmed, C. Rajkhowa, M. Sharma, H. Talukdar and N. S. Islam, New J. Chem., 2019, 43, 17251–17266 RSC.
- Y. Zhang, L. Qi, A. Lund, P. Lu and A. T. Bell, J. Am. Chem. Soc., 2021, 143, 8352–8366 CrossRef CAS PubMed.
- S. Pasricha, P. Gahlot, K. Mittal, D. Rai, N. Avasthi, H. Kaur and S. Rai, ChemistrySelect, 2022, 7, e202103674 CrossRef CAS.
- J. Yu, Y. Zhou, Z. Lin and R. Tong, Org. Lett., 2016, 18, 4734–4737 CrossRef CAS PubMed.
- A. Held, E. Janiszewska, J. Czerepińska and J. Kowalska-Kuś, RSC Adv., 2020, 10, 10144–10154 RSC.
- W. Yan, A. Ramanathan, M. Ghanta and B. Subramaniam, Catal. Sci. Technol., 2014, 4, 4433–4439 RSC.
- J. Zhang, Z. Xin, X. Meng, Y. Lv and M. Tao, Fuel, 2014, 116, 25–33 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. Kresge, K. Schmitt, C. Chu, D. H. Olson, E. Sheppard and S. McCullen, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- W. Yan, A. Ramanathan, P. D. Patel, S. K. Maiti, B. B. Laird, W. H. Thompson and B. Subramaniam, J. Catal., 2016, 336, 75–84 CrossRef CAS.
- S. Laha and R. Gläser, Microporous Mesoporous Mater., 2007, 99, 159–166 CrossRef CAS.
- A. Sakthivel, S. E. Dapurkar and P. Selvam, Catal. Lett., 2001, 77, 155–158 CrossRef CAS.
- T. Al-Harbi, F. Al-Hazmi and W. E. Mahmoud, Superlattices Microstruct., 2012, 52, 643–647 CrossRef CAS.
- A. Sakthivel and P. Selvam, J. Catal., 2002, 211, 134–143 CrossRef CAS.
- L. Wang, L. Wang and J. Zhang, J. Mater. Sci., 2009, 44, 6512–6518 CrossRef CAS.
- a. C. Kresge, M. Leonowicz, W. J. Roth, J. Vartuli and J. Beck, nature, 1992, 359, 710–712 CrossRef.
- G. Somorjai and R. Rioux, Catal. Today, 2005, 100, 201–215 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- V. Alfredsson and M. W. Anderson, Chem. Mater., 1996, 8, 1141–1146 CrossRef CAS.
- D. Nazimov, O. Klimov, A. Saiko, S. Trukhan, T. Glazneva, I. Prosvirin, S. Cherepanova and A. Noskov, Catal. Today, 2021, 375, 401–409 CrossRef CAS.
- B. Grzybowska, J. Słoczyński, R. Grabowski, K. Wcisło, A. Kozłowska, J. Stoch and J. Zieliński, J. Catal., 1998, 178, 687–700 CrossRef CAS.
- S. Wang, K. Murata, T. Hayakawa, S. Hamakawa and f. Suzuki, Appl. Catal., A, 2000, 196, 1–8 CrossRef CAS.
- A. S. Al-Awadi, A. M. El-Toni, J. P. Labis, A. Khan, H. Ghaithan, A. A. Al-Zahrani, A. E. Abasaeed and S. M. Al-Zahrani, Catalysts, 2021, 11, 642 CrossRef CAS.
- Y. Cheng, F. Zhang, Y. Zhang, C. Miao, W. Hua, Y. Yue and Z. Gao, Chin. J. Catal., 2015, 36, 1242–1248 CrossRef CAS.
- A. S. Al-Awadi, A. M. El-Toni, M. Alhoshan, A. Khan, J. P. Labis, A. Al-Fatesh, A. E. Abasaeed and S. M. Al-Zahrani, Ceram. Int., 2019, 45, 1125–1134 CrossRef CAS.
- S. Narayanan, J. J. Vijaya, S. Sivasanker, C. Ragupathi, T. Sankaranarayanan and L. J. Kennedy, J. Porous Mater., 2016, 23, 741–752 CrossRef CAS.
- M. Morey, J. Bryan, S. Schwarz and G. Stucky, Chem. Mater., 2000, 12, 3435–3444 CrossRef CAS.
- X. Lu, W.-J. Zhou, Y. Guan, A. Liebens and P. Wu, Catal. Sci. Technol., 2017, 7, 2624–2631 RSC.
- J. Liu, Z. Wang, P. Jian and R. Jian, J. Colloid Interface Sci., 2018, 517, 144–154 CrossRef CAS PubMed.
- U. Taralkar, P. Kalita, R. Kumar and P. Joshi, Appl. Catal., A, 2009, 358, 88–94 CrossRef CAS.
- D. R. Das and A. K. Talukdar, ChemistrySelect, 2017, 2, 8983–8989 CrossRef CAS.
- D. R. Das, P. Kalita and A. K. Talukdar, J. Porous Mater., 2020, 27, 893–903 CrossRef CAS.
- A. Boro and A. K. Talukdar, J. Porous Mater., 2019, 26, 1185–1196 CrossRef CAS.
- J. Van Den Reijen, W. Versluis, S. Kanungo, M. d'Angelo, K. de Jong and P. de Jongh, Catal. Today, 2019, 338, 31–39 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Sadaf Ul Hassanb,
Asif Mahmoodc,
Waheed Al-Masryc and
Shumaila Razzaque*d
*a,
Sadaf Ul Hassanb,
Asif Mahmoodc,
Waheed Al-Masryc and
Shumaila Razzaque*d


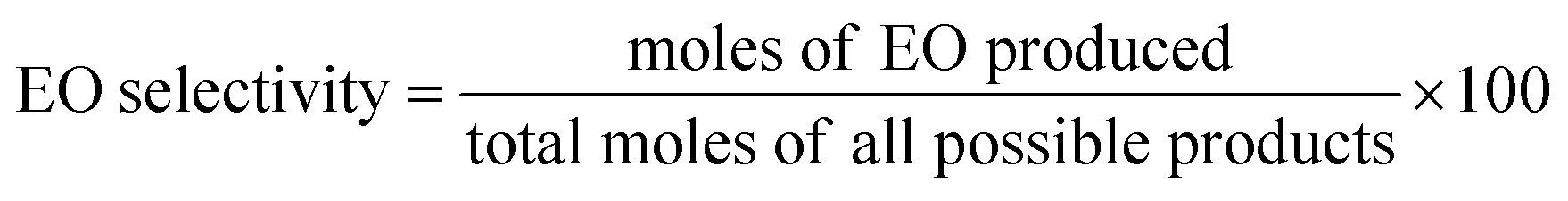

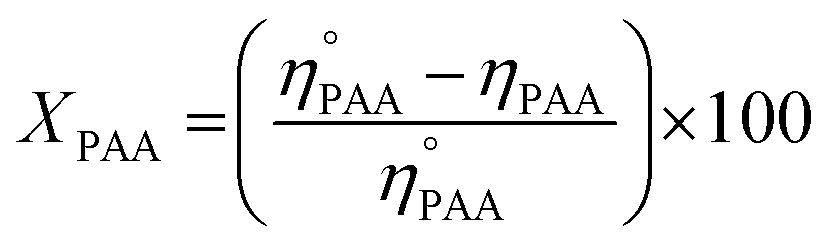
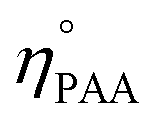 and ηPAA = initial and final moles of PAA.
and ηPAA = initial and final moles of PAA.