
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-induced polymorphism in dipodal N-donor ligands containing a biphenyl core†
Simran Chaudhary ,
Dariusz Kędziera,
Zbigniew Rafiński and
Liliana Dobrzańska*
,
Dariusz Kędziera,
Zbigniew Rafiński and
Liliana Dobrzańska*
Faculty of Chemistry, Nicolaus Copernicus University in Toruń, Gagarina 7, 87-100, Toruń, Poland. E-mail: lianger@umk.pl
First published on 18th October 2023
Abstract
Polymorph screenings for two related dipodal N-donor ligands containing a biphenyl core, namely 4,4′-bis(pyridin-4-ylmethyl)-1,1′-biphenyl (1) and 4,4′-bis(1H-imidazol-1-ylmethyl)-1,1′-biphenyl (2) were performed, and the new phases were isolated and their crystal structures analysed. Profiling included methods such as PXRD and thermal analysis. Hirshfeld surface analyses, as well as crystal lattice energy calculations provided deeper insight in the interplay of the intermolecular forces and the stability of the isolated phases. Furthermore, our studies revealed the presence of solvent-induced polymorphism, whereby the metastable phase is dominant upon crystallisation from THF (1a) and EtOH (2c). Upon heating, these phases transform into a more stable form, whereby the transformations were followed by PXRD studies (1, 2).
Introduction
Polymorphism, an intriguing phenomenon concerning the formation of crystal structures1 that can be defined as the existence of multiple crystalline forms of the same compound,2 differing by molecular conformation (conformational polymorphs),3 molecular arrangement (packing polymorphs)4 or both, has been the subject of intense research in the last few decades. The existence of multiple crystalline forms of the same composition has a big impact, especially on materials science and more specifically within the pharmaceutical industry, as it makes the design of compounds of particular build and properties very challenging.5 It almost goes without saying that it is crucial to retain the same form of a drug in order not to be surprised by sudden changes in properties caused by the appearance of another form.6 It is broadly known that various synthetic/crystallisation conditions (solvent effect,7 the level of supersaturation,8 temperature9 and pressure10) can lead to the occurrence of polymorphism. The phenomenon is related to the interplay of noncovalent intermolecular forces, for example hydrogen bonds,11 halogen bonds12 and π–π interactions,13 different combinations of which can lead to the formation of disparate crystalline phases. Taking into account the variety of crystal structures of similar lattice energy, which can be formed, it is not trivial to predict the final product of the crystallisation process. This issue is reflected by computational Crystal Structure Prediction methods (CSP) currently being developed, which for a simple organic molecule can generate hundreds of possible polymorphs.14In continuation of our studies encompassing a family of dipodal N-donor ligands,15 we would like to present the polymorphic behaviour of two related compounds (Scheme 1) namely, 4,4′-bis(pyridin-4-ylmethyl)-1,1′-biphenyl (1) and 4,4′-bis(1H-imidazol-1-ylmethyl)-1,1′-biphenyl (2). Their recrystallization screenings in different solvents allowed us to isolate a series of polymorphs.
 | ||
| Scheme 1 Representation of the presented N-donor ligands. | ||
Experimental
Reagents and materials
All commercially available chemicals and solvents were of reagent grade and were used without further purification.Synthetic procedures
Both presented ligands (Scheme 1) were synthesised earlier16 but the procedures were modified.1H NMR (CDCl3, 700 MHz) δ 8.54 (d, 4H), 7.55 (d, 4H), 7.27 (d, 4H), 7.19 (d, 4H), 4.04 (s, 4H); 13C NMR (CDCl3, 100 MHz) δ 149.9, 139.2, 138.0, 129.5, 127.3, 124.2, 40.9.
1H NMR (CDCl3, 700 MHz) δ 7.65 (s, 2H), δ 7.58 (d, 4H), δ 7.26 (d, 4H), 7.15 (t, 2H), 6.96 (t, 2H), 5.20 (s, 4H); 13C NMR (CDCl3, 100 MHz) δ 140.3, 137.4, 135.6, 129.9, 127.8, 127.6, 119.3, 50.4.
Crystallisation of different forms of 1 and 2
The compounds 1 and 2 were recrystallized from a range of solvents of different geometry and polarity, such as acetone, acetonitrile, DCM, EtOH, MeOH and THF (10 mg of compound/10 ml of solvent). Vials covered with parafilm were left to undergo slow evaporation, which allowed us to obtain good quality crystals in all vials containing 1. In the case of 2, crystals suitable for SCXRD studies could only be grown from DCM, MeOH and EtOH (poor quality). The earlier reported crystal structures of 2 (monohydrate 2H and anhydrous form 2a) were isolated as unexpected products of the metal complexation reaction by applying slow diffusion of an ethanolic solution of AgBF4 into the ligand solution dissolved in chloroform or by slow diffusion of an aqueous solution of AgNO3 into a solution of ligand dissolved in acetone, respectively.17Measurements
1H and 13C NMR spectra were recorded on Bruker Avance 700 MHz and 400 MHz instruments, respectively and referenced to residual solvent peaks (see Fig. S1 and S2†).Thermal analyses (TGA, DTA) were performed on a TA Instruments SDT 650 Analyser. All TGA experiments were performed at a heating rate of 2 °C min−1 under dry nitrogen with a flow rate of 100 ml min−1 covering the temperature range: 25–600 °C.
PXRD patterns were obtained on a Philips X'Pert X-ray diffractometer using CuKα radiation. The voltage and current were 40 kV and 30 mA, respectively. The samples were measured at the 2Θ range of 4–45° with a scan speed of 0.0089° s−1. All data were acquired at ambient temperature. The PXRD data were analysed using Powder Cell18 and Profex19 software.
Structure determination
Single-crystal X-ray diffraction data for 1a, 1b, 2b and 2c were collected on an XtaLAB Synergy-S Dualflex diffractometer equipped with monochromated CuKα radiation (λ = 1.54184 Å). The crystals were coated with Paratone-N oil and mounted on a loop. Data collection was carried out at 100(2) K to minimize solvent loss, possible structural disorder and thermal motion effects. Data frames were processed (unit cell determination, intensity data integration, correction for Lorentz and polarisation effects, and empirical absorption correction) by using the corresponding diffractometer's software package.20 The structures were solved by using direct methods with SHELXS-2018/3 (ref. 21) and refined by using full-matrix least-squares methods based on F2 by using SHELXL-2018/3.22 The programs Mercury23 and POV-Ray24 were both used to prepare molecular graphics. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were positioned geometrically with C–H = 0.95 Å (aromatic) and 0.99 Å (methylene), and refined as riding, with Uiso (H) = 1.2 Ueq (C).A summary of the data collection and structure refinement parameters are provided in Table 1. None of the crystal structures of 1 were reported previously, but as mentioned earlier, two forms of 2 (2H and 2a) were described before.17 Their unit cell parameters and basic data collection conditions are shown in Table 1. Kitaigorodskii packing indices were calculated by applying the PLATON package.25 The values shown for 2H and 2a are most likely underestimated and can not be directly compared to those of 2b and 2c, as the SCXRD data for the former two were collected at room temperature.
| Compound reference | 1a | 1b | 2H | 2a | 2b | 2c |
| Chemical formula | C24H20N2 | C24H20N2 | C20H18N4·H2O | C20H18N4 | C20H18N4 | C20H18N4 |
| Formula mass | 336.42 | 336.42 | 332.40 | 314.38 | 314.38 | 314.38 |
| a/Å | 20.7777(2) | 5.76800(5) | 4.7126(10) | 10.957(1) | 5.66180(10) | 18.2880(5) |
| b/Å | 10.58810(10) | 9.98210(10) | 15.269(3) | 9.964(1) | 14.6008(2) | 7.9130(2) |
| c/Å | 8.25020(10) | 15.60390(10) | 24.148(5) | 15.457(2) | 19.1251(3) | 22.3941(5) |
| α/° | 90 | 90 | 90 | 90 | 90 | 90 |
| β/° | 94.6050(10) | 100.4410(10) | 94.024(3) | 94.43(1) | 90.6510(10) | 90 |
| γ/° | 90 | 90 | 90 | 90 | 90 | 90 |
| Unit cell volume/Å3 | 1809.16(3) | 883.546(11) | 1733.33 | 1682.4(3) | 1580.91(4) | 3240.72(14) |
| Space group | P21/c | P21 | P21/n | P21/c | P21/n | Pbca |
| No. of formula units per unit cell, Z | 4 | 2 | 4 | 8 | ||
| Temperature/K | 100(2) | 100(2) | 293(2) | 293(2) | 100(2) | 100(2) |
| Radiation type | CuKα | CuKα | MoKα | MoKα | CuKα | CuKα |
| Absorption coefficient, μ/mm−1 | 0.556 | 0.570 | 0.633 | 0.618 | ||
| No. of reflections measured | 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 998 998 |
29193 |
18651 |
27548 |
||
| No. of independent reflections | 3774 | 3501 | 3263 | 2985 | ||
| Rint | 0.0347 | 0.0238 | 0.0290 | 0.0638 | ||
| Final R1 values (I > 2σ(I)) | 0.0373 | 0.0256 | 0.0342 | 0.0382 | ||
| Final wR(F2) values (I > 2σ(I)) | 0.0936 | 0.0674 | 0.0863 | 0.0921 | ||
| Final R1 values (all data) | 0.0440 | 0.0260 | 0.0384 | 0.0520 | ||
| Final wR(F2) values (all data) | 0.0978 | 0.0677 | 0.0892 | 0.0989 | ||
| Goodness of fit on F2 | 1.028 | 1.038 | 1.052 | 1.033 | ||
| Flack parameter | −0.13(10) | |||||
| Kitaigorodskii packing indices | 68.4 | 70.3 | 68.2 | 66.4/66.3 | 71.6 | 69.7 |
Computational methods
Results and discussion
Polymorphs of 4,4′-bis(pyridin-4-ylmethyl)-1,1′-biphenyl (1a/1b)
PXRD screening studies performed for sample 1 recrystallized from a range of solvents indicated the formation of at least two different phases. Especially the powder pattern obtained for crystalline material grown from THF stood out (Fig. S3†), even though there was no striking difference in morphology of the crystals formed in the different solvents. We isolated single-crystals from this solvent, collected SCXRD data, determined the crystal structure (1a) and generated its powder pattern, which corresponded very well with the experimentally determined trace (Fig. 1). Furthermore, a good quality single crystal was isolated from MeOH and the crystal structure was determined (1b). Powder Cell indicated the absence of 1a in the solid recrystallized from MeOH. Moreover, the phase 1b shows its dominance in all studied solids, apart from the one obtained from THF, in which its contribution is negligible, at ca. 2%. Interestingly increasing the concentration of the solute in THF (15 or 20 mg/10 ml) leads to the formation of 1b exclusively. | ||
| Fig. 1 Overlay of the simulated PXRD patterns generated from crystal structures recrystallized from THF (red, 1a) and MeOH (blue, 1b), and experimental PXRD pattern obtained for the sample after recrystallization from THF (black). | ||
The isolated polymorphs 1a and 1b crystallise in monoclinic systems of the P21/c and P21 space groups, respectively. It is worth mentioning that pairs of polymorphs crystallising in a combination of centrosymmetric and acentric space groups have previously received quite some attention, as studying these could facilitate gaining control over the formation of acentric packings.29 The molecules in these two crystalline forms adopt different conformations as shown in Fig. 2. The dihedral angles between the planes of the benzene rings are 34° and 35° for 1a and 1b, respectively, versus 57° and 20° between the planes of the pyridine rings.
 | ||
| Fig. 2 On the left: molecular structure of 1a with atomic displacement plot shown at 50% probability; the labelling refers also to 1b, on the right: overlay of 1a (red) and 1b (blue); RMSD 1.0522 Å. | ||
The molecular packing in both crystalline phases involves sets of different intermolecular forces even though their choices in the case of this compound are rather limited (Fig. S4†). As could be expected, a large contribution is coming from C–H⋯N interactions leading to the formation of 3D supramolecular assemblies. These are further supported by C–H⋯π forces (Table 2), involving methylene groups and pyridine rings as donors and benzene rings as acceptors (1a), whereas for 1b either pyridine rings act as donors and pyridine rings as acceptors or benzene and pyridine rings as donors and benzene rings as acceptors. Furthermore, weak intermolecular π–π interactions between two adjacent pyridine rings containing N24 (symmetry operator: 1 − x, 1 − y, −z) are present in 1a, with a centroid–centroid distance of 3.7092(6) Å. The most striking differences between these two crystal structures lie in the strength of the interactions formed, the involvement of different molecular units in their formation, and the presence or absence of π–π interactions.
| Compound | D–H⋯A | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) |
|---|---|---|---|---|
| a (1a) Cg1 is the centroid of benzene ring C14–C19; (1b) Cg1 is the centroid of benzene ring C8–C13, Cg2 is the centroid of pyridine ring N1–C6, symmetry codes (1a): (i) −x,1/2 + y,−1/2 − z, (ii) 1 − x,1 − y,1 − z, (iii) 1 − x,1/2 + y,1/2 − z, (iv) 1 − x,1 − y,−z, (v) x,1/2 − y,−1/2 + z, (vi) x, y, −1 + z; (1b): (i) −x + 1,y − 1/2,−z + 1, (ii) x,y,z + 1, (iii) −x,y + 1/2,−z + 2, (iv) 1 − x, 1/2 + y,2 − z, (v) 1 + x,y,−1 + z, (vi) 1 − x,−1/2 + y,2 − z, (vii) −x,−1/2 + y,2 − z. | ||||
| 1a | C5–H5⋯N1i | 2.61 | 3.553(2) | 174 |
| C13–H13⋯N1i | 2.56 | 3.485(2) | 163 | |
| C10–H10⋯N24ii | 2.86 | 3.626(2) | 139 | |
| C16–H16⋯N24iii | 2.74 | 3.617(2) | 153 | |
| C20–H20A⋯N24iv | 2.70 | 3.474(2) | 135 | |
| C25–H25⋯N24v | 2.94 | 3.790(2) | 150 | |
| C7–H7A⋯Cg1vi | 2.85 | 3.767(1) | 154 | |
| C23–H23⋯Cg1ii | 2.95 | 3.829(1) | 155 | |
| 1b | C7–H7B⋯N1i | 2.73 | 3.521(2) | 137 |
| C13–H13⋯N1i | 2.74 | 3.585(2) | 149 | |
| C22–H22⋯N1ii | 2.73 | 3.507(2) | 140 | |
| C5–H5⋯N24iii | 2.68 | 3.381(2) | 131 | |
| C16–H16⋯Cg1iv | 2.78 | 3.683(2) | 158 | |
| C2–H2⋯Cg2v | 2.90 | 3.572(2) | 129 | |
| C23–H23⋯Cg2vi | 2.89 | 3.680(2) | 141 | |
| C25–H25⋯Cg1vii | 2.95 | 3.697(2) | 136 | |
Further analyses of the intermolecular forces stabilising the crystal structures, by applying Crystal Explorer, allowed to visualise these in the form of fingerprint plots, as well as to estimate their percentage contributions, which indicate, among others, the presence of stronger C–H⋯π interactions in 1b (Fig. 3), as observed earlier.
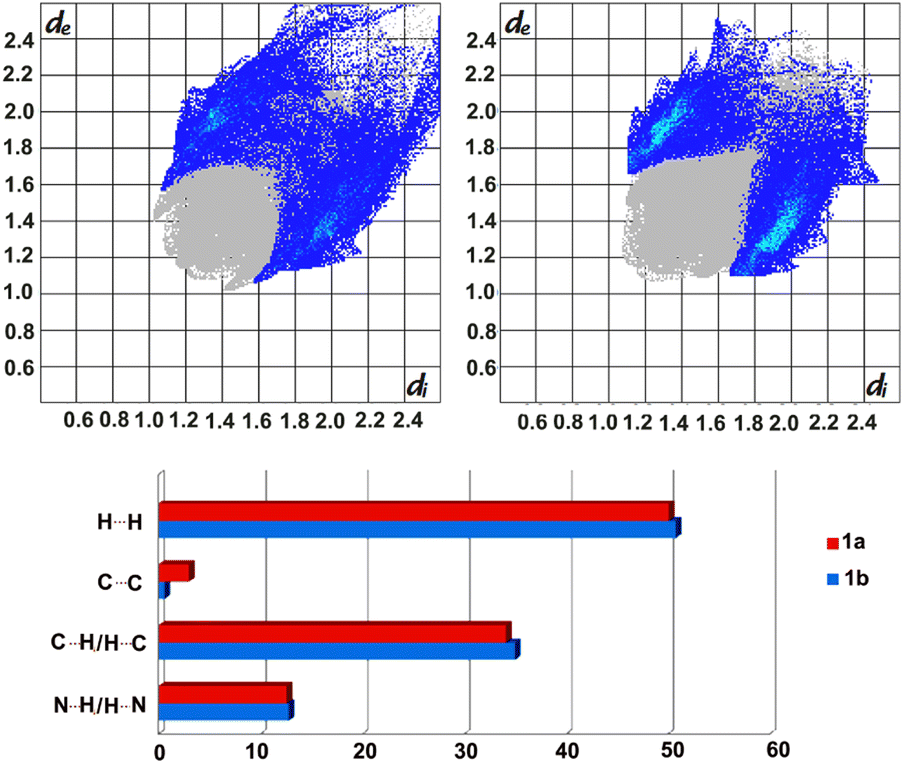 | ||
| Fig. 3 Fingerprint plots (top) for form 1a (left) and 1b (right) with the contribution of C⋯H/H⋯C contacts indicated in blue. Bottom: estimated contributions (percentages) of selected intermolecular forces stabilizing the formation of 1a and 1b. | ||
Moreover, the calculated enrichment ratios30 show once again the importance of C–H⋯N and C–H⋯π interactions in stabilising the crystal packing in 1a and 1b, with slightly higher dominance of the former in 1a (1.33 versus 1.15) and the latter in 1b (1.27 vs. 1.20).
The crystal lattice energy calculations (Table 3) indicate a lower stability of form 1a, which is in good agreement with the lower Kitaigorodskii packing index (see KI indices in Table 1). The results show once again a similar input of different forces, with the major difference in input of the dispersion term, which delivers the major contribution to stabilizing both crystalline phases.
| Energy component/form | Coulombic | Polarisation | Dispersion | Repulsion | Energy in total |
|---|---|---|---|---|---|
| 1a | −51.0 | −22.9 | −198.3 | 94.9 | −177.4 |
| 1b | −50.8 | −21.9 | −208.8 | 96.7 | −184.9 |
To analyse the system further, thermal analyses (TG/DTA) of 1a and 1b were performed (Fig. S5 and S6†), which indicated a phase transition taking place in the case of 1a at ca. 100 °C. This was confirmed by a PXRD study, as heating a sample of 1a at 110 °C for 2 min revealed that this leads to irreversible conversion to 1b.
Polymorphs of 4,4′-bis(1H-imidazol-1-ylmethyl)-1,1′-biphenyl (2a/2b/2c)
The crystal structure of 2 was reported and deposited at CSD earlier (refcode: COKCIP, 2a) as well as its corresponding mono-hydrated form (refcode: COKCOV, 2H). PXRD screening of solids grown from a range of solvents revealed the formation of at least two additional phases (Fig. 4). In DCM and THF, the form 2b was present exclusively, as shown by PXRD and by applying Powder Cell. Solvents such as MeOH, acetone and acetonitrile led to the formation of mixtures of 2H and 2b with a contribution of more than 70% of the latter (the highest contribution of 2b was noticed in acetone, at 92%). Interestingly, the powder pattern of crystalline material grown from EtOH allowed for isolation of another phase (2c, with a contribution of ca. 74%), which forms a mixture with 2H (2b was absent in this case). | ||
| Fig. 4 PXRD patterns obtained for samples of 2 after recrystallization from DCM (blue) and EtOH (red) and simulated PXRD patterns generated from crystal structures of 2H (COKCOV, pink) and 2a (COKCIP, green). | ||
Though the crystals of 2c were of poor quality, we managed to select a crystal suitable for SCXRD measurements (Fig. 5).
 | ||
| Fig. 5 Overlay of 2b (blue) and 2c (yellow) indicating the difference in position of one of the imidazole rings (RMS deviation is 0.9791 Å). | ||
2a and 2b crystallise in monoclinic systems of the P21/c and P21/n space groups respectively, whereas 2c crystallises in the space group Pbca of a higher symmetry orthorhombic system. As reported earlier, one of the imidazole rings in 2a shows positional N/C disorder (50:50), rendering a very accurate comparison of 2a with the other two phases impossible. Comparing the conformation adopted by the ligand in 2b with the orientations adopted by the two components 2a1 and 2a2 (disorder), indicated certain differences, especially in the position of one of the imidazole rings (Fig. S7,† RMS deviation of 1.1590 Å for 2a1 and RMS deviation of 0.8922 Å for 2a2).
Another orientation of the imidazole ring is present in 2c which is facilitated by the flexibility of the molecule. The torsion angles C7–C6–N1–C2 and C16–C19–N20–C21, corresponding with the labelling in Fig. 4, are as follows: 89°/102°, 87°/−99°, 92°/−148° for 2a, 2b and 2c respectively.
The dihedral angles between the planes of the benzene rings are 33°, 31°, 34° for 2a, 2b and 2c respectively, versus 10°, 11° and 61° between the corresponding planes of the imidazole rings. Like in 1, the molecular packing in 2a–2c involves weak C–H⋯N hydrogen bonds, leading to the formation of 3D supramolecular assemblies supported by C–H⋯π interactions (Table 4, Fig. S8†). π–π interactions are absent in this series.
| Compound | D–H⋯A | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) |
|---|---|---|---|---|
| a (2a1) Cg1 is the centroid of the benzene ring containing C5, Cg2 is the centroid of the imidazole ring containing N1, Cg3 is the centroid of the imidazole ring containing N2; (2a2) Cg1 is the centroid of the benzene ring containing C5, Cg2 is the centroid of the imidazole ring containing N1, Cg3 is the centroid of the imidazole ring containing N2; (2b) Cg1 is the centroid of imidazole ring N20–C24, Cg2 is the centroid of benzene ring C7–C12, Cg3 is the centroid of imidazole ring N1–C5, Cg4 is the centroid of benzene ring C13–C18; (2c) Cg1 is the centroid of imidazole ring N20–C24, Cg2 is the centroid of benzene ring C13–C18, symmetry codes (2a1): (i) 1 − x,−1/2 + y,1/2 − z, (ii) 1 − x,1/2 + y,1/2 − z, (iii) 2 − x,−1/2 + y,−1/2 − z, (iv) −x + 2,−x,−y (v) 2 − x,1/2 + y,−1/2 − z, (vi) 1 − x,−y, −z (vii) 1 − x,−1/2 + y,1/2 − z, (viii) 2 − x,1/2 + y,−1/2 − z; (2a2): (i) 1 − x,1/2 + y,1/2 − z, (ii) 1 − x,−1/2 + y,1/2 − z, (iii) 2 − x,−1/2 + y,−1/2 − z, (iv) 2 − x,−y,−z, (v) 2 − x,1/2 + y,−z − 1/2, (vi) 1 − x,−y,−z, (vii) −x,−1/2 + y,1/2 − z; (2b): (i) 1/2 − x,1/2 + y,1/2 − z, (ii) 3/2 − X,1/2 + Y,1/2 − Z, (iii) −1/2 + x,1/2 + y,1/2 − z, (iv) −x,−y,1 − z, (v) 3/2 + X,1/2 − Y,−1/2 + Z, (vi) 1 − X,−Y,1 − Z, (vii) −1 + x,y,z; (2c): (i) x,1/2 − y,1/2 + z, (ii) 3/2 − x,1 − y,1/2 + z, (iii) x,3/2 − y,−1/2 + z, (iv) 2 − x,1 − y,1 − z, (vi) 3/2 − x,1/2 + y,z, (vii) 2 − x,2 − y,1 − z. | ||||
| 2a1 | C6–H3⋯N3i | 2.94 | 3.820 | 158 |
| C10–H8⋯N3i | 2.96 | 3.798 | 150 | |
| C18–H15⋯N3ii | 2.93 | 3.792 | 155 | |
| C3–H1⋯N5iii | 2.90 | 3.752 | 153 | |
| C8–H5⋯N5iv | 2.67 | 3.524 | 154 | |
| C13–H11⋯N5v | 2.76 | 3.675 | 168 | |
| C4–H2⋯Cg1vi | 2.69 | 3.575 | 159 | |
| C10–H8⋯Cg2vii | 2.73 | 3.565 | 150 | |
| C12–H10⋯Cg3viii | 2.95 | 3.677 | 136 | |
| 2a2 | C9–H6⋯N4i | 2.95 | 3.598 | 128 |
| C10–H8⋯N4ii | 2.933 | 3.853 | 173 | |
| C3–H1⋯N5iii | 2.90 | 3.752 | 153 | |
| C8–H5⋯N5iv | 2.67 | 3.524 | 154 | |
| C13–H11⋯N5v | 2.76 | 3.675 | 169 | |
| C4–H2⋯Cg1vi | 2.69 | 3.5751 | 159 | |
| C10–H8⋯Cg2i | 2.73 | 3.5650 | 150 | |
| C12–H10⋯Cg3iii | 2.95 | 3.6773 | 136 | |
| 2b | C17–H17⋯N3i | 2.851 | 3.264(2) | 107 |
| C18–H18⋯N3ii | 2.933 | 3.681(2) | 137 | |
| C19–H19B⋯N3i | 2.733 | 3.318(2) | 118 | |
| C19–H19A⋯N3iii | 2.57 | 3.537(2) | 164 | |
| C8–H8⋯N22iv | 2.687 | 3.589(2) | 154 | |
| C6–H6B⋯N22iv | 2.833 | 3.708(2) | 159 | |
| C5–H5⋯Cg1v | 2.70 | 3.385(1) | 129 | |
| C14–H14⋯Cg2vi | 2.94 | 3.656(1) | 133 | |
| C18–H18⋯Cg3ii | 2.90 | 3.714(1) | 144 | |
| C21–H21⋯Cg4vii | 2.90 | 3.835(1) | 167 | |
| 2c | C19–H19B⋯N3i | 2.702 | 3.572(2) | 147 |
| C21–H21⋯N3ii | 2.852 | 3.629(2) | 140 | |
| C2–H2⋯N22iii | 2.825 | 3.577(2) | 137 | |
| C6–H6A⋯N22iii | 2.494 | 3.399(2) | 152 | |
| C6–H6B⋯N22iv | 2.809 | 3.677(2) | 147 | |
| C12–H12⋯N22iv | 2.960 | 3.642(2) | 130 | |
| C12–H12⋯Cg1iv | 2.78 | 3.551(1) | 139 | |
| C19–H19A⋯Cg2vi | 2.83 | 3.811(1) | 169 | |
| C23–H23⋯Cg2vii | 2.82 | 3.761(1) | 173 | |
It is worth mentioning that estimating the contributions of the different intermolecular contacts in the two separated forms of 2a, namely 2a1 and 2a2 contributing equally to molecular disorder, by applying Crystal Explorer, indicated the interplay between H⋯N (16:17.9%) and H⋯H (52.6:51.4%) forces. The results were further averaged and compared with 2b and 2c as presented in Fig. 6, indicating the largest contribution of hydrogen bonds in the case of 2b which, as shown below, is the most energetically favoured phase.
 | ||
| Fig. 6 Estimated contributions (percentages) of selected intermolecular contacts to the Hirshfeld surface in 2a (blue), 2b (purple) and 2c (green). | ||
The crystal lattice energy calculation (Table 5) indicated the higher stability of form 2b over 2c, which is in good agreement with the higher Kitaigorodskii packing index of 2b (see KI indices in Table 1). It also pointed out a lower stability of phase 2a, the presence of which was not observed in any of the studied solids, and which was previously isolated in solid form after silver salt complexation. However the results for this particular phase are not very accurate, as the data set was collected at room temperature and additionally the molecule shows disorder. The lowest input of coulombic/polarisation factors observed for 2a could be the consequence of a lower input of C–H⋯π interactions, as shown on the histogram presented in Fig. 6. The results once again reveal that the dispersion term delivers the major contribution to stabilizing these three crystal phases.
| Energy component/Form | Coulombic | Polarisation | Dispersion | Repulsion | Energy in total |
|---|---|---|---|---|---|
| a As this is an average of 2a disordered components, the values are (very) rough estimates. | |||||
| 2a1 | −43.3 | −23.6 | −172.1 | 69.3 | −169.7 |
| 2a2 | −56.5 | −27.1 | −174.2 | 82.5 | −175.2 |
| 2a(av)a | −49.9 | −25.35 | −173.15 | 75.9 | −172.5 |
| 2b | −72.7 | −31.1 | −202.8 | 103.9 | −202.7 |
| 2c | −74.8 | −29.6 | −196.1 | 100.2 | −200.3 |
To analyse the system further, thermal analyses (TG/DTA) of the solids obtained from DCM and EtOH were performed. These indicated a phase transition taking place at ca. 140 °C in the case of solid grown from EtOH (Fig. S9†). A PXRD study revealed that after heating this sample at 150 °C for 3 min, the monohydrate is completely converted to 2b, whereas the 2c phase is only partially converted. Upon extended heating at this temperature or after time (3 days in air), 2c is completely converted to 2b. Interestingly, comparing the molecular packings formed by monohydrated 2H and 2b indicates the presence of the same main packing features, which could facilitate the dehydration/hydration process. Furthermore, the results of thermal analyses pointed out much higher thermal stability of the imidazole based compounds (2) (ca. 30 °C) over the pyridine analogues (1).
Conclusions
Polymorph screenings performed in a series of solvents of different geometry and polarity, such as acetone, acetonitrile, DCM, EtOH, MeOH and THF on two compounds (1 and 2) containing a biphenyl core allowed us to reveal two new phases for each.The ability to form polymorphs is, among others, the result of the conformational flexibility of these molecules, containing aromatic rings which can rotate freely. In both cases one or more solvents could be identified, leading exclusively to the formation of the energetically more stable phase, such as MeOH in the case of 1 and DCM and THF in the case of 2. Moreover, we could also pinpoint solvents in which the metastable form was predominantly present, namely THF and EtOH, respectively, and follow the irreversible transformations of the isolated metastable forms to the stable arrangement upon heating. The presented observations show that, even in the case of similarly built compounds with a composition limiting the formation of intermolecular interactions through lack of strong hydrogen bond donors, the solvent effect on the crystallisation process can tremendously differ. Furthermore, not only the transformation of a metastable to a stable phase of different molecular packing was observed, but also the dehydration of monohydrate 2, transforming to the energetically favoured phase 2b of similar packing. Studies on related systems, as well as investigations of the solvent effect on the nucleation/crystallisation process supported by computational methods, are ongoing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
LD and SC would like to thank the National Science Centre – Poland for grant no. 2014/14/E/ST5/00611 and the University Centre of Excellence “Astrophysics and Astrochemistry”. LD, ZR and SC would also like to thank the programme Excellence Initiative – Research University for funding the research group of Crystal Engineering and Advanced Solid-State Characterisation.References
- A. J. Cruz-Cabeza, N. Feeder and R. J. Davey, Commun. Chem., 2020, 3, 142 CrossRef CAS PubMed
.
-
(a) J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, New York, 2002 Search PubMed
-
(a) A. Nangia, Acc. Chem. Res., 2008, 41, 595–604 CrossRef CAS PubMed
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC
-
(a) M. T. Ruggiero, J. Axel Zeitler and T. M. Korter, Phys. Chem. Chem. Phys., 2017, 19, 28502–28506 RSC
- R. Hilfiker and M. V. Raumer, Polymorphism in the Pharmaceuticals Industry, Wiley-VCH Verlag GmbH & Co. KGaA, 2018, pp. 1–30 Search PubMed
-
(a) P. Shi, S. Xu, S. Du, S. Rohani, S. Liu, W. Tang, L. Jia, J. Wang and J. Gong, Cryst. Growth Des., 2018, 18, 5947–5956 CrossRef CAS
-
(a) J. Ouyang, J. Chen, I. Rosbottom, W. Chen, M. Guo and J. Y. Y. Heng, CrystEngComm, 2021, 23, 813–823 RSC
-
(a) Y. Liu, L. Jia, S. Wu, S. Xu, X. Zhang, S. Jiang and J. Gong, CrystEngComm, 2019, 21, 2790–2798 RSC
-
(a) F. Safari, A. Olejniczak and A. Katrusiak, Cryst. Growth Des., 2019, 19, 5629–5635 CrossRef CAS
-
(a) T. Aizawa, K. Aratsu, S. Datta, T. Mashimo, T. Seki, T. Kajitani, F. Silly and S. Yagai, Chem. Commun., 2020, 56, 4280–4283 RSC
-
(a) P. Pang, Y. Wang, X. Miao, B. Li and W. Deng, New J. Chem., 2021, 45, 6811 RSC
-
(a) Z. Xie, L. Liu, B. Yang, G. Yang, L. Ye, M. Li and Y. Ma, Cryst. Growth Des., 2005, 5, 1959–1964 CrossRef CAS
-
(a) R. M. Bhardwaj, J. A. McMahon, J. Nyman, L. S. Price, S. Konar, I. D. H. Oswald, C. R. Pulham, S. L. Price and S. M. Reutzel-Edens, J. Am. Chem. Soc., 2019, 141, 13887–13897 CrossRef CAS PubMed
-
(a) S. Chaudhary, D. Kędziera and L. Dobrzańska, Polyhedron, 2022, 224, 115989 CrossRef CAS
-
(a) M. Fujita, M. Aoyagi, F. Ibukuro, K. Ogura and K. Yamaguchi, J. Am. Chem. Soc., 1998, 120, 611–612 CrossRef CAS
- L. Carlucci, G. Ciani, S. Maggini and D. M. Proserpio, CrystEngComm, 2008, 10, 1191–1203 RSC
- W. Kraus and G. Nolze, J. Appl. Crystallogr., 1996, 29, 301–303 CrossRef CAS
- N. Döeblin and R. Kleeberg, J. Appl. Crystallogr., 2015, 48, 1573–1580 CrossRef PubMed
- Rigaku Oxford Diffraction, CrysAlisPro Software System, version 1.171.38.41, Rigaku Corporation, Oxford, UK, 2015 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van De Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS
- http://www.povray.org/.
- A. L. Spek, Platon, Utrecht University, Utrecht, The Netherlands, 1980–2021 Search PubMed
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed
-
(a) A. Gavezzotti, J. Phys. Chem., 2003, B107, 2344–2353 CrossRef
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed
- L. N. Kuleshova, D. W. M. Hofmann and M. Y. Antipin, Crystallogr. Rep., 2005, 50, 167–176 CrossRef CAS
- C. Jelsch, K. Ejsmont and L. Huder, IUCrJ, 2014, 1, 119–128 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of 1 and 2, powder patterns obtained for 1 upon recrystallisation from a range of solvents, packing diagrams of 1a, 1b, 2b and 2c, thermograms of 1a, 1b and 2c. CCDC 2253373–2253376. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra05713e |
| This journal is © The Royal Society of Chemistry 2023 |