DOI:
10.1039/D3RA05637F
(Paper)
RSC Adv., 2023,
13, 29141-29151
Cytotoxicity, anti-diabeticity, and phytocomposition investigation of Vietnamese Euphorbia tithymaloides Linn. (Euphorbiaceae)†
Received
18th August 2023
, Accepted 12th September 2023
First published on 4th October 2023
Abstract
In this study, the aerial parts of mature Vietnamese Euphorbia tithymaloides plants were put through cytotoxic, anti-diabetic, and phytocompositional evaluations. Specifically, four extracts (petroleum ether (PE), ethyl acetate (EA), methanol (Me), and aqueous (W)) were prepared by maceration at room temperature. All extracts, together with some isolated compounds, were investigated for cytotoxicity against some human normal and cancer cell lines (fibroblasts, HeLa, NCI-H460, HepG2, MCF-7, and Jurkat) using the standardized modified sulforhodamine B (SRB) assay. Additionally, the anti-diabetic activity of extracts and compounds was evaluated via their α-glucosidase inhibitory capacity. The obtained results indicated that Vietnamese E. tithymaloides extracts exhibited moderate cytotoxic activity, among which the PE extract possessed the highest values, on the NCI-H460 cell line. Second, the aqueous extract was revealed to possess very high α-glucosidase inhibitory activity (IC50 = 113.75 ± 14.02 μg ml−1). From the PE extract, three new jatrophane diterpenoids (named tithymal A, tithymal B, and tithymal C) and two known ones were isolated and structurally elucidated using NMR and MS spectroscopies. Noticeably, tithymal A exhibited significantly high inhibitory activity against α-glucosidase (IC50 = 10.71 ± 0.52 μg ml−1). These observations have significantly highlighted the medicinal potential of Vietnamese E. tithymaloides and expanded its scientific fascination.
Introduction
Euphorbia is one of the most noticeable genera within the Euphorbiaceae family, thanks to its more than 2000 beneficial members.1–4 Among the top groups of tolerant Euphorbia species are latex-bearing Euphorbias (E. tirucalli, E. grantii, E. resinifera, and so on).5–7 In addition to their very wide range of phytoconstituents, these plants have diverse bioactivities, ranging from cytotoxic, antimicrobial, and antioxidant, to anti-diabetic, anti-inflammatory, and anti-cancer activities.8,9 In Vietnam, although species of the Euphorbia genus are not as diverse as those of others, such as Aporosa or Croton, most of them are exploited as medicinal plants. E. tirucalli, E. thymifolia, E. antiquorum, and E. cyathophora are among the most common members.10–13
The Devil's backbone, Euphorbia tithymaloides, is among the important medicinal plants in Vietnam, especially for the treatment of dermatological and oral diseases. This is a vertical succulent shrub that grows up to 2.4 m in height, with distinctive green zigzag stems, hence its common name. Opposite along the stems are sessile, glabrescent, acuminate, smooth-edged, simple angiosperm leaves, which are 35–75 mm in length. Each flower is supported by a peduncle in a dichotomous cyme, with bifid egg-shaped floral leaves and bright red, irregularly acuminate, slipper-resembling, involucral bracts. Seed pods are approximately 7.5 mm long and 9 mm wide, and ovoid in shape with truncated ends.14,15 In Vietnam, E. tithymaloides thrives in mountain sides, on sandy, microelement (B, Cu, Fe, Mo, and Zn)-rich terrains. It is also planted in familial medicinal gardens for favourable usages.10–12
Regarding the phytocomposition, E. tithymaloides is noticeable for its multiform jatrophane diterpenoids and coumarins, with more than ten compounds having been isolated and elucidated in each group.16–19 Besides, the isolation of other compounds (steroids, flavonoids, organic acids, and esters, for instance) has been reported.20,21 Bioactivities of this plant have also been demonstrated to be varied, including cytotoxic, antioxidant, antimicrobial, and wound-healing activities.1,15,21 From this study, the scientific knowledge of phytocomposition and bioactivities of E. tithymaloides will be significantly expanded, assuring its potential for medicinal use.
Experimental
Materials and methods
All extracts were prepared from the dried ground powder of mature plant specimens by maceration at room temperature. They were subsequently investigated for cytotoxicity against human normal and cancer cell lines, using the previously described modified SRB assay.22,23 Besides, the α-glucosidase inhibitory activity of these extracts was evaluated, based on the procedure described by Luo-sheng et al.24 Isolated compounds obtained in appropriate quantities were also processed in these experiments. For cytotoxicity, fibroblasts were chosen as the representative of normal cells, while HeLa (cervical cancer), HepG2 (liver cancer), Jurkat (blood cancer), MCF-7 (breast cancer), and NCI-H460 (lung cancer) (American Type Culture Collection (Virginia, United States)) were the targeted cancer cell lines. The α-glucosidase inhibitory activity was evaluated via the capacity of releasing p-nitrophenol from a p-nitrophenyl-α-D-glucopyranoside precursor. For phytocompositional investigation, column and thin-layer chromatographies (TLC) were applied, and the obtained compounds were structurally elucidated using NMR and MS spectroscopies. For TLC, silica gel 60 F254 aluminum sheets (MilliporeSigma, Darmstadt, Germany) were used, while silica gel 60 (pore size range of 230–400 mesh) (MilliporeSigma, Darmstadt, Germany) was used for column chromatography. Sulfuric acid, ethanol (Xilong Scientific, Guangdong, China), and vanillin (HIMEDIA, Mumbai, India) were used for the preparation of TLC reagents. Other chemicals were of analytical grade and used without further purification. For structure elucidation, NMR spectra were recorded using a Bruker Avance NEO (Bruker, Massachusetts, United States) spectrometer, while a SCIEX X500B QTOF (SCIEX, Toronto, Canada) spectrometer was used for recording MS spectra.
Procedures
Preparation of plant materials and extracts. Fresh aerial parts of mature Vietnamese E. tithymaloides plants were collected, processed, and extracted following the procedure described in our previous publication.25 The obtained extracts were abbreviated as Eth-PE, Eth-EA, Eth-Me, and Eth-W, for petroleum ether, ethyl acetate, methanol, and aqueous extracts respectively.
Cytotoxicity investigation. Cells were cultured in 96-well plates (BioPointe Scientific, California, United States) at 37 °C in a 5% CO2 atmosphere, at a density of 104 cells per well for HeLa, HepG2, MCF-7, and fibroblasts, 7.5 × 103 for NCI-H460; and 5 × 104 for Jurkat, in media of E'MEM (Sigma-Aldrich, Missouri, United States) for MCF-7, NCI-H460, HeLa, HepG2, and fibroblasts, and RPMI 1640 (Sigma-Aldrich, Missouri, United States) for Jurkat. The culture media were supplemented with L-glutamine (2 mM), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (20 mM), amphotericin B (0.025 μg ml−1), penicillin G (100 UI ml−1), streptomycin (100 μg ml−1), and 10% (v/v) fetal bovine serum (FBS) (Sigma-Aldrich, Missouri, United States). After 24 h of incubation, the solutions of extracts or compounds, in 100% dimethyl sulfoxide (DMSO) (MilliporeSigma, Darmstadt, Germany), at assessed concentrations, were added to each well, followed by another 48 h of incubation. Treated cells were fixed with cold 50% (w/v) trichloroacetic acid (MilliporeSigma, Darmstadt, Germany) solution for 1–3 h, washed, and stained with 0.2% (w/v) SRB (Sigma-Aldrich, Missouri, United States) for 20 min. After five washes with 1% acetic acid (MilliporeSigma, Darmstadt, Germany), the protein-bound dye was solubilized in a 10 mM Tris base solution (Sigma-Aldrich, Missouri, United States), and read for optical density values using a Synergy HT 96-well micro-titer plate reader (BioTek Instruments, Vermont, United States) at wavelengths of 492 and 620 nm. Camptothecin (Sigma-Aldrich, Missouri, United States) and neat DMSO were used as the positive and negative controls, respectively. The assay was conducted one time for the cytotoxicity preliminary screening, and in triplicate for detailed investigation. The optical density (OD) values at 492 nm (OD492) and 620 nm (OD620) of each reading were recorded. The cytotoxicity percentage (% I) of extracts or compounds was determined for each extract using the following calculations:| | |
OD492 (or OD620) of sample/control = OD492/620 TN/C − OD492/620 blank
| (1) |
| | |
ODTN/C = OD492 TN/C − OD620 TN/C
| (2) |
| |
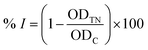 | (3) |
where ODTN and ODC are respectively the optical density of samples (with cells) and control, calculated using formulae (1) and (2). ODblank is the optical density of blank samples (without cells). The results are represented as means ± standard errors (SE) (n ≥ 3). The Kruskal–Wallis test, followed by Dunn's test, was applied for testing the cytotoxicity on cancer and normal cells (GraphPad Prism software). A p-value of <0.05 was accepted as statistically significantly different. The principal steps of cytotoxic activity evaluation for extracts and compounds of E. tithymaloides are shown in Fig. 1.
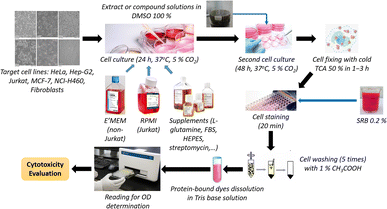 |
| | Fig. 1 Principal steps of the applied modified SRB assay. | |
α-Glucosidase inhibitory activity evaluation
First, 120 μl of each sample and 20 μl of 1 U ml−1 α-glucosidase (Sigma-Aldrich, Missouri, United States) were added to each well of a 96-well plate and incubated at 37 °C within 15 minutes. Then, 20 μl of 5 mM p-nitrophenyl-α-D-glucopyranoside (MilliporeSigma, Darmstadt, Germany) was subsequently added to each well, followed by another 15 minute incubation at 37 °C. After that, 80 μl of 0.2 M Na2CO3 (MilliporeSigma, Darmstadt, Germany) was added to halt the reaction, and the optical density of the solution was measured at 405 nm (OD405). Acarbose (Sigma-Aldrich, Missouri, United States) was used as the positive control. The α-glucosidase inhibitory percentage (% I) was calculated as follows:
where ODt and ODc are respectively the optical density of samples and control (after deducting the optical density of the blank sample without α-glucosidase); the IC50 value was determined by measuring the optical density of the sample at different concentrations. The Kruskal–Wallis test, followed by Dunn's test, was applied for the α-glucosidase inhibitory capacity evaluation (GraphPad Prism software), with R2 > 0.9. The principal steps of α-glucosidase inhibitory determination for extracts and compounds of E. tithymaloides are shown in Fig. 2.
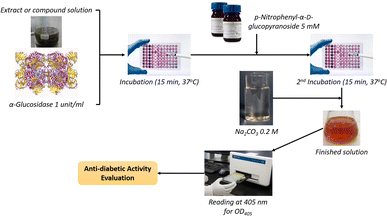 |
| | Fig. 2 Principal steps of the applied procedure for α-glucosidase inhibitory activity evaluation. | |
Phytocomposition investigation. The column chromatographic process started with the mobile phase from 100% (v/v) n-hexane to 100% (v/v) ethyl acetate, yielding nine fractions (labeled as P1 to P9, respectively). Fraction P5, giving obvious marks after TLC analysis, was chosen for further column chromatography with the mobile phase from n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate 9:1 (v/v) to 100% (v/v) ethyl acetate, obtaining 22 sub-fractions (labeled as P5.1 to P5.22, respectively). Precipitation was observed in sub-fractions P5.5, P5.6, and P5.7, leading to the decision to further isolate, purify, and recrystallize it in acetone to obtain 1.3 g of a white crystal, labeled as P1. The remaining solution of sub-fraction P5.5 was proceeded to further column chromatography (the mobile phase from 100% (v/v) n-hexane to 100% (v/v) ethyl acetate) and purification to yield 2.8 mg of a white powder (P2) and 2.7 mg of another white powder (P3). The process continued for subfractions P5.10 and P5.11 (the mobile phase from 100% (v/v) n-hexane to 100% (v/v) ethyl acetate), yielding 7 fractions for P5.10 and 8 fractions for P5.11. Respective further purification of fractions P5.10.4 and P5.11.4 resulted in 5.0 mg of a white powder (P4) and 14.0 mg of another white powder (P5). The complete chromatography procedure is summarized in Fig. 3.
:ethyl acetate 9:1 (v/v) to 100% (v/v) ethyl acetate, obtaining 22 sub-fractions (labeled as P5.1 to P5.22, respectively). Precipitation was observed in sub-fractions P5.5, P5.6, and P5.7, leading to the decision to further isolate, purify, and recrystallize it in acetone to obtain 1.3 g of a white crystal, labeled as P1. The remaining solution of sub-fraction P5.5 was proceeded to further column chromatography (the mobile phase from 100% (v/v) n-hexane to 100% (v/v) ethyl acetate) and purification to yield 2.8 mg of a white powder (P2) and 2.7 mg of another white powder (P3). The process continued for subfractions P5.10 and P5.11 (the mobile phase from 100% (v/v) n-hexane to 100% (v/v) ethyl acetate), yielding 7 fractions for P5.10 and 8 fractions for P5.11. Respective further purification of fractions P5.10.4 and P5.11.4 resulted in 5.0 mg of a white powder (P4) and 14.0 mg of another white powder (P5). The complete chromatography procedure is summarized in Fig. 3.
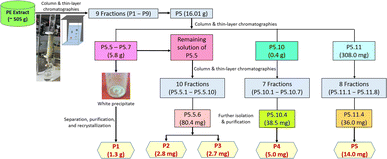 |
| | Fig. 3 Compositional investigation for the PE extract. | |
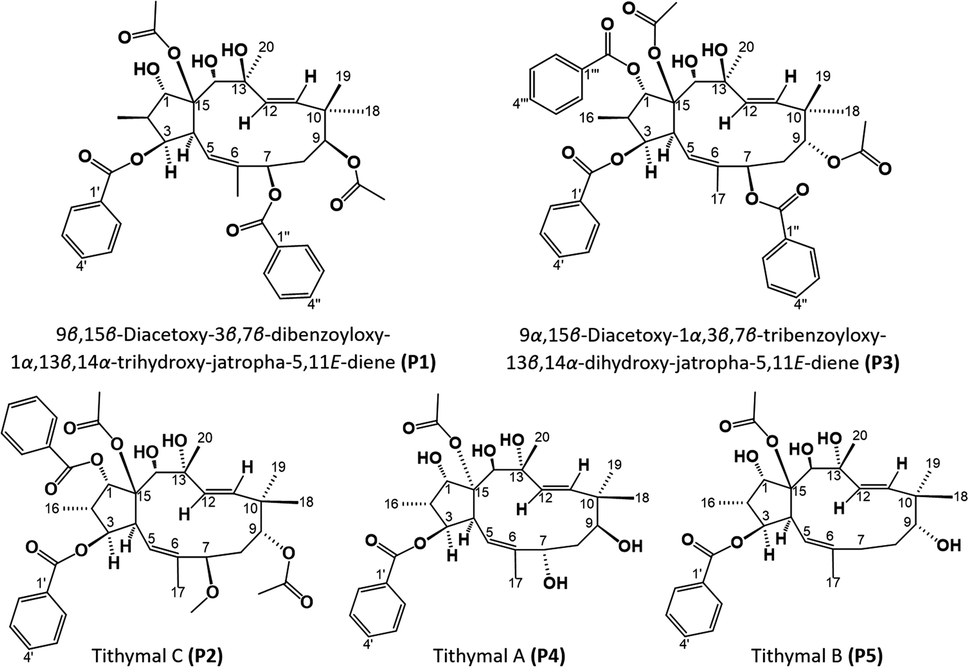
9β,15β-Diacetoxy-3β,7β-dibenzoyloxy-1α,13β,14α-trihydroxy-jatropha-5,11E-diene (P1). White crystal; soluble in acetone; TLC characters: positive reaction with 25% H2SO4 reagent (as an almost black mark), Rf = 0.73 (n-hexane:ethyl acetate 7:3, v/v). MS: [M + Na]+ pseudomolecular ion peak at m/z 701.2924 (calculated for C38H46O11, 1.4 mass of difference).
9α,15β-Diacetoxy-1α,3β-dibenzoyloxy-13α,14α-dihydroxy-7β-methoxyjatropha-5,11E-diene (tithymal C) (P2). White powder; soluble in chloroform; TLC characters: positive reaction with 25% H2SO4 reagent (as a dark brown mark), Rf = 0.31 (dichloromethane:ethyl acetate = 95:5, v/v). MS: [M + H2O]+ pseudomolecular ion peak at m/z 710.3540 (calculated for C39H48O11, 0.02 mass of difference).
9α,15β-Diacetoxy-1α,3β,7β-tribenzoyloxy-13β,14α-dihydroxy-jatropha-5,11E-diene (P3). White powder; soluble in acetone; TLC characters: positive reaction with 25% H2SO4 reagent (as an almost black mark), Rf = 0.50 (dichloromethane:ethyl acetate = 8:2, v/v). MS: [M + Na]+ pseudomolecular ion peak at m/z 805.3247 (calculated for C45H50O12, 4.7 mass of difference).
15α-Acetoxy-3β-benzoyloxy-1α,7α,9β,13α,14β-pentahydroxy-jatropha-5,11E-diene (tithymal A) (P4). White powder; soluble in chloroform; TLC characters: positive reaction with 25% H2SO4 reagent (as a dark brown mark), Rf = 0.35 (dichloromethane:acetone = 8:2, v/v). MS: [M + NH4]+ pseudomolecular ion peak at m/z 550.3009 (calculated for C29H40O9, 0.7 mass of difference).
Results and discussion
Cytotoxicity against human normal and cancer cell lines
All isolated compounds from the PE extract of E. tithymaloides were elucidated as jatrophane diterpenoids, which have only recently been studied, and their potential in bioactivity aspect attracted the attraction of scientific community.26–28 In this work, P1 and P4 were priorly chosen for the cytotoxicity and α-glucosidase inhibitory evaluations. Table 1 presents the cytotoxic activity of all extracts, P1, and P4 against human normal and cancer cell lines.
Table 1 Cytotoxicity of extracts and compounds of E. tithymaloides ( : extract;
: extract;  : compound)
: compound)
| Test concentration of all extracts and compounds was 100 μg ml−1. Abbreviation for human fibroblasts. |
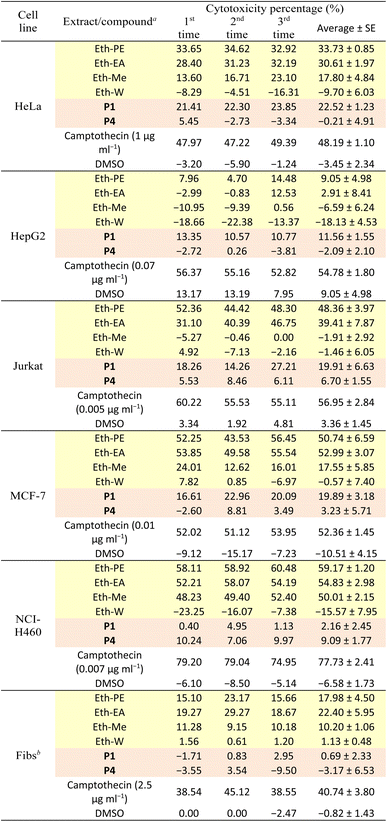 |
It was observed in Table 1 that both extracts and two selected compounds of Euphorbia tithymaloides exhibited low cytotoxic activity against fibroblasts, indicating their potential safety for human administration. Regarding cancer cell lines, only MCF-7 and NCI-H460 were affected by Euphorbia tithymaloides' extracts, with the higher cytotoxic activity on NCI-H460. In addition, only the petroleum ether and ethyl acetate extracts expressed noticeable activity against cancer cells, among which higher values belong to the petroleum ether one. For compounds of P1 and P4, none of them showed significant cytotoxic activity against cancer cell lines. Except for NCI-H460, P1 possessed higher cytotoxic values than those of P4. It could be inferred from the obtained results that Vietnamese Euphorbia tithymaloides possessed moderate cytotoxicity against human breast and lung cancer cell lines. The composition of extracts, therefore, was essential to be investigated to take deeper steps in studying their bioactivities.
α-Glucosidase inhibitory activity
Table 2 presents the α-glucosidase inhibitory activity of all extracts, compounds P1 and P4 of E. tithymaloides. It was obvious from this table that the aqueous extract of E. tithymaloides exhibited the strongest activity. For isolated compounds, both P1 and P4 showed very high inhibition of α-glucosidase, with P1 being as strong as acarbose, and P4 being significantly stronger. The obtained results contributed considerably to the anti-diabetic activity of E. tithymaloides, as well as the bioactivities of different jatrophane-type diterpenoids.
Table 2 α-Glucosidase inhibitory activity of extracts and compounds of E. tithymaloides ( : extract;
: extract;  : compound)
: compound)
Structure elucidation for isolated compounds from the PE extract
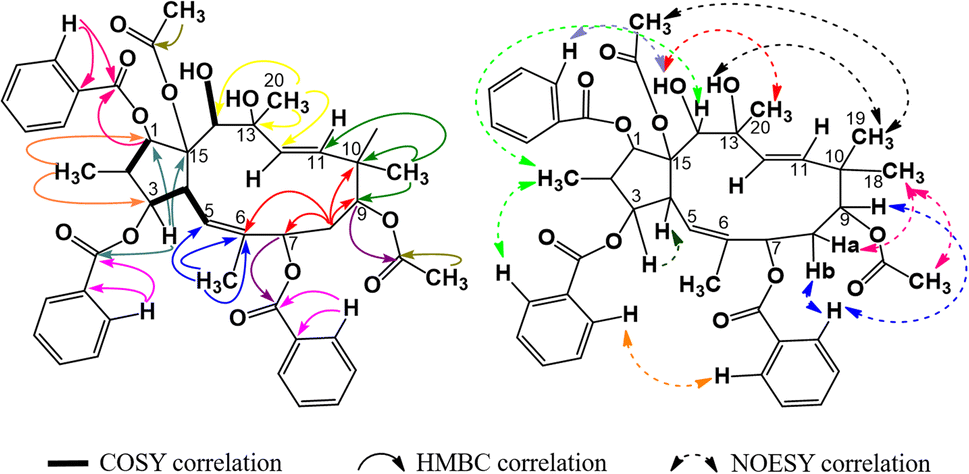
The 1H-NMR spectra of compound P1 indicated that it possessed a jatrophane skeleton, with signals of three olefin protons at δH (ppm) 5.89 (1H, d, 9.0, H-5); 5.63 (1H, d, 15.6, H-11); 5.45 (1H, d, 15.6, H-12); fifteen protons of five methyl groups at δH (ppm) 0.91 (3H, d, 6.6, CH3-16); 1.85 (3H, s, CH3-17); 1.00 (3H, s, CH3-18); 0.96 (3H, s, CH3-19); 1.30 (3H, s, CH3-20); six protons of two acetoxy groups at δH (ppm) 1.67 (3H, s, AcO-9); 2.41 (3H, s, AcO-15); aromatic protons of two benzoyloxy groups at δH (ppm) 7.72 (2H, dd, 7.2, 1.2, H-2′ and H-6′); 7.19 (2H, dd, 8.2, 7.2, H-3′ and H-5′); 7.38 (1H, ddd, 8.2, 7.2, 1.2, H-4′); 7.59 (2H, dd, 7.2, 1.2, H-2′′ and H-6′′); 7.05 (2H, dd, 8.1, 7.2, H-3′′ and H-5′′); 7.34 (1H, ddd, 8.1, 7.2, 1.2, H-4′′). P1 was revealed from those signals to be possibly an acetylbenzoyloxymethylated jatrophadiene. Such considerations on the structure of P1 were reinforced by the 13C- and DEPT-NMR spectra, with signals of seven C–OR groups at δC (ppm) 87.7 (C-1); 78.6 (C-3); 75.1 (C-7); 74.7 (C-9); 75.2 (C-13); 73.3 (C-14); 92.1 (C-15); five methyl carbons at δC (ppm) 12.1 (C-16); 16.7 (C-17); 23.5 (C-18); 21.0 (C-19); 31.5 (C-20); carbons of two acetoxy groups at δC (ppm) 32.8 and 173.6 (AcO-9); 39.3 and 170.3 (AcO-15); carbons of two benzoyloxy groups at δC (ppm) 165.6 (BzO-3); 165.3 (BzO-7); four olefin carbons at δC (ppm) 120.8 (C-5); 134.6 (C-6); 132.4 (C-11); 132.0 (C-12). From the HSQC spectra of P1, a signal of each proton of its jatrophane skeleton was defined. Third, the COSY spectra of P1 showed correlations between H-2/H-1, OH-1/H-1, H-4/H-3 and H-5, OH-14/H-14; while correlations between H-16/C-1 and C-3; H-17/C-5, C-6, and C-7; H-18 and H-19/C-9, C-10, and C-11; H-20/C-12, C-13, and C-14 were observed in the HMBC spectra, indicating that P1 was methylated at the 2, 6, 10, and 20 positions in its structure. In addition, the correlations between H-3, H-7 and benzoyloxy carbons allowed for the determination of benzoyloxylated positions for P1. Similar elucidations were made for P1's acetoxy groups (Fig. 4). Finally, the NOESY spectra of P1 resulted in its stereochemical structure determination (Fig. 9). Based on the spectral similarities to previous publication16 (Table 3), compound P1 was revealed to be 9β,15β-diacetoxy-3β,7β-dibenzoyloxy-1α,13β,14α-trihydroxy-jatropha-5,11E-diene.
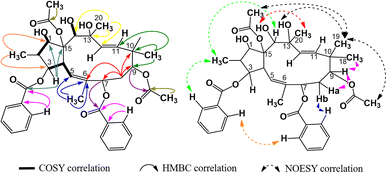 |
| | Fig. 4 Major COSY, HMBC, and NOESY correlations in P1. | |
Table 3 Spectral data comparison for P1 and 9β,15β-diacetoxy-3β,7β-dibenzoyloxy-1α,13β,14α-trihydroxy-jatropha-5,11E-diene
| Position |
P1 (acetone-d6) |
9β,15β-Diacetoxy-3β,7β-dibenzoyloxy-1α,13β,14α-trihydroxy-jatropha-5,11E-diene (CDCl3)16 |
| 1H-NMR |
13C-NMR |
1H-NMR |
13C-NMR |
| 1 |
4.25 (1H, dd, 15.0, 2.4) |
87.7 |
4.12 (1H, dd, 17.0, 2.5) |
86.7 |
| 2 |
2.32 (1H, m) |
44.3 |
2.30 (1H, m) |
42.3 |
| 3 |
5.40 (1H, s) |
78.6 |
5.99 (1H, s) |
78.0 |
| 4 |
4.23 (1H, dd, 9.0, 4.8) |
42.0 |
4.42 (1H, dd, 10.0, 5.5) |
42.8 |
| 5 |
5.89 (1H, d, 9.0) |
120.8 |
5.74 (1H, d, 9.5) |
118.2 |
| 6 |
— |
134.6 |
— |
134.4 |
| 7 |
5.25 (1H, d, 9.0) |
75.1 |
5.51 (1H, d, 8.5) |
75.0 |
| 8 |
2.18 (1H, m) |
33.1 |
1.87 (1H, m) |
34.8 |
| 2.06 (1H, m) |
1.99 (1H, s) |
| 9 |
5.24 (1H, t, 6.6) |
74.7 |
4.78 (1H, t, 6.5) |
74.0 |
| 10 |
— |
40.3 |
— |
39.2 |
| 11 |
5.63 (1H, d, 15.6) |
132.4 |
5.49 (1H, s) |
132.4 |
| 12 |
5.45 (1H, d, 15.6) |
132.0 |
5.53 (1H, m) |
129.4 |
| 13 |
— |
75.2 |
— |
75.8 |
| 14 |
4.41 (1H, m) |
73.3 |
4.09 (1H, t, 10.5) |
72.1 |
| 15 |
— |
92.1 |
— |
91.4 |
| 16 |
0.91 (3H, d, 6.6) |
12.1 |
0.84 (3H, s) |
11.7 |
| 17 |
1.85 (3H, s) |
16.7 |
1.75 (3H, s) |
16.5 |
| 18 |
1.00 (3H, s) |
23.5 |
1.01 (3H, s) |
23.0 |
| 19 |
0.96 (3H, s) |
21.0 |
0.98 (3H, s) |
20.7 |
| 20 |
1.30 (3H, s) |
31.5 |
1.22 (3H, s) |
31.6 |
| AcO-9 |
1.67 (3H, s) |
20.9 |
2.04 (3H, s) |
21.3 |
| 173.6 |
171.1 |
| AcO-15 |
2.41 (3H, s) |
22.4 |
2.33 (3H, s) |
22.3 |
| 170.3 |
172.9 |
| BzO-3 |
— |
165.6 |
— |
165.1 |
| 1′ |
— |
130.6 |
— |
130.1 |
| 2′,6′ |
7.72 (2H, dd, 7.2, 1.2) |
129.1 |
8.10 (1H, s) |
128.6 |
| 8.12 (1H, s) |
| 3′,5′ |
7.19 (2H, dd, 8.2, 7.2) |
129.7 |
7.53 (2H, m) |
129.0 |
| 4′ |
7.38 (1H, ddd, 8.2, 7.2, 1.2) |
133.5 |
7.61 (1H, m) |
133.3 |
| BzO-7 |
— |
165.3 |
— |
166.2 |
| 1′′ |
— |
130.6 |
— |
130.0 |
| 2′′,6′′ |
7.59 (2H, dd, 7.2, 1.2) |
128.7 |
7.43 (2H, d, 8.0) |
128.7 |
| 3′′,5′′ |
7.05 (2H, dd, 8.1, 7.2) |
130.0 |
7.92 (1H, d, 8.0) |
129.7 |
| 7.94 (1H, s) |
| 4′′ |
7.34 (1H, ddd, 8.1, 7.2, 1.2) |
133.1 |
7.53 (1H, m) |
133.0 |
The spectral data of compound P2 indicated that it was also an acetylbenzoyloxymethylated jatrophadiene similar to P1, with signals of three olefin protons and carbons at δH (ppm) 5.78 (1H, d, 9.9, H-5), 5.46 (1H, d, 15.6, H-11), 5.27 (1H, d, 15.6, H-12); δC (ppm) 118.5 (C-5); 133.9 (C-6); 132.4 (C-11); 129.3 (C-12); protons and carbons of five methyl groups at δH (ppm) 1.04 (3H, d, 6.8, CH3-16), 1.70 (3H, s, CH3-17), 0.93 (3H, s, CH3-18), 0.94 (3H, s, CH3-19), 1.40 (3H, s, CH3-20); δC (ppm) 11.5 (C-16); 16.6 (C-17); 20.7 (C-18); 22.8 (C-19); 31.5 (C-20); protons and carbons of two acetoxy groups at δH (ppm) 2.00 (3H, s, AcO-9), 2.35 (3H, s, AcO-15); δC (ppm) 21.4 and 169.6 (AcO-9), 22.1 and 171.1 (AcO-15); aromatic protons and carbons of two benzoyloxy groups: BzO-1: δH (ppm) 8.05 (2H, dd, 7.2, 1.1, H-2′ and H-6′), 7.53 (2H, dd, 7.7, 7.2, H-3′ and H-5′), 7.64 (1H, ddd, 7.7, 7.2, 1.1, H-4′); δC (ppm) 166.2, 130.2 (C-1′), 129.4 (C-2′ and C-6′), 129.6 (C-3′ and C-5′), 133.9 (C-4′); BzO-3: δH (ppm) 8.02 (2H, dd, 7.2, 1.0, H-2′′ and H-6′′), 7.49 (2H, dd, 7.7, 7.2, H-3′′ and H-5′′), 7.58 (1H, ddd, 7.7, 7.2, 1.0, H-4′′); δC (ppm) 165.4, 130.0 (C-1′′), 128.6 (C-2′′ and C-6′′), 128.8 (C-3′′ and C-5′′), 133.2 (C-4′′). Notably, the signal of a methoxy group was observed in the spectra of P2 (δH (ppm) 2.68 (3H, s); δC (ppm) 55.7 (OCH3-7)). The HSQC spectra of P2 allowed for the signal definition for its protons, while from the COSY and HMBC spectra, P2 was determined to be 2,6,10,13-methylated, 9,15-acetoxylated, 1,3-benzoyloxylated, and 7-methoxylated (Fig. 5). Finally, the stereochemical structure of P2 was elucidated from its NOESY spectra as shown in Fig. 9 and was named 9α,15β-diacetoxy-1α,3β-dibenzoyloxy-13α,14α-dihydroxy-7β-methoxyjatropha-5,11E-diene.
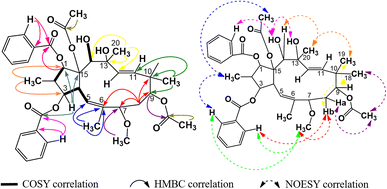 |
| | Fig. 5 Major COSY, HMBC, and NOESY correlations in P2. | |
As no referential publications were found for direct spectral data comparisons, P2 was compared with 9α,15β-diacetoxy-1α,3β,7β-tribenzoyloxy-13α,14α-trihydroxy-jatropha-5,11E-diene, which had the highest structural resemblance to that of P2 (Table 4). From this table, it was obvious that P2 had significant spectral similarities to those of 9α,15β-diacetoxy-1α,3β,7β-tribenzoyloxy-13α,14α-trihydroxy-jatropha-5,11E-diene, except for the replacement of the 7-benzoyloxy group with the 7-methoxy group. A reference on the SciFinder database indicated that the structure of P2 was new to science and this compound was named tithymal C.
Table 4 Spectral data comparison for P2 and 9α,15β-diacetoxy-1α,3β,7β-tribenzoyloxy-13α,14α-trihydroxy-jatropha-5,11E-diene
| Position |
P2 (CDCl3) |
9α,15β-Diacetoxy-1α,3β,7β-tribenzoyloxy-13α,14α-trihydroxy-jatropha-5,11E-diene (CDCl3)17 |
| 1H-NMR |
13C-NMR |
1H-NMR |
13C-NMR |
| 1 |
5.89 (1H, d, 11.7) |
87.2 |
5.88 (1H, d, 11.8) |
87.1 |
| 2 |
2.57 (1H, m) |
44.2 |
2.52 (1H, m) |
44.2 |
| 3 |
5.58 (1H, m) |
77.2 |
5.54 (1H, dd, 5.3, 4.3) |
77.2 |
| 4 |
4.13 (1H, dd, 9.8, 5.1) |
41.9 |
4.11 (1H, dd, 9.7, 5.1) |
41.9 |
| 5 |
5.78 (1H, d, 9.9) |
118.5 |
5.89 (1H, d, 9.7) |
119.3 |
| 6 |
— |
133.9 |
— |
134.4 |
| 7 |
2.68 (3H, s) |
55.7 |
5.30 (1H, s) |
74.2 |
| 8 |
1.87 (2H, m) |
33.7 |
2.08 (2H, m) |
32.4 |
| 9 |
4.97 (1H, t, 3.3) |
73.7 |
5.15 (1H, dd, 3.5, 2.9) |
73.9 |
| 10 |
— |
39.5 |
— |
39.6 |
| 11 |
5.46 (1H, d, 15.6) |
132.4 |
5.54 (1H, d, 15.5) |
132.1 |
| 12 |
5.27 (1H, d, 15.6) |
129.3 |
5.33 (1H, d, 15.5) |
129.9 |
| 13 |
— |
74.8 |
— |
74.7 |
| 14 |
4.86 (1H, m) |
72.3 |
4.86 (1H, d, 5.6) |
72.3 |
| 15 |
— |
90.0 |
— |
90.1 |
| 16 |
1.04 (3H, d, 6.8) |
11.5 |
0.93 (3H, d, 6.7) |
11.4 |
| 17 |
1.70 (3H, s) |
16.6 |
1.87 (3H, s) |
16.5 |
| 18 |
0.93 (3H, s) |
20.7 |
0.94 (3H, s) |
20.6 |
| 19 |
0.94 (3H, s) |
22.8 |
0.95 (3H, s) |
22.8 |
| 20 |
1.40 (3H, s) |
31.5 |
1.39 (3H, s) |
31.5 |
| AcO-9 |
2.00 (3H, s) |
21.4 |
2.04 (3H, s) |
21.3 |
| 169.6 |
171.1 |
| AcO-15 |
2.35 (3H, s) |
22.1 |
2.33 (3H, s) |
22.3 |
| 171.1 |
172.9 |
| BzO-1 |
|
166.2 |
— |
165.4 |
| 1′ |
|
130.2 |
— |
130.1 |
| 2′,6′ |
8.05 (2H, dd, 7.2, 1.1) |
129.4 |
8.02 (2H, dd, 7.3, 1.2) |
128.7 |
| 3′,5′ |
7.53 (2H, dd, 7.7, 7.2) |
129.6 |
7.50 (2H, dd, 7.7, 7.3) |
129.3 |
| 4′ |
7.64 (1H, ddd, 7.7, 7.2, 1.1) |
133.9 |
7.61 (1H, ddd, 7.7, 7.3, 1.2) |
133.2 |
| BzO-3 |
— |
165.4 |
— |
165.3 |
| 1′′ |
— |
130.0 |
— |
129.5 |
| 2′′,6′′ |
8.02 (2H, dd, 7.2, 1.0) |
128.6 |
7.60 (2H, dd, 7.3, 1.1) |
128.2 |
| 3′′,5′′ |
7.49 (2H, dd, 7.7, 7.2) |
128.8 |
7.13 (2H, dd, 7.8, 7.3) |
129.1 |
| 4′′ |
7.58 (1H, ddd, 7.7, 7.2, 1.0) |
133.2 |
7.31 (1H, ddd, 7.8, 7.3, 1.1) |
132.7 |
| BzO-7 |
— |
— |
— |
165.0 |
| 1′′′ |
— |
— |
— |
129.3 |
| 2′′′, 6′′′ |
— |
— |
7.58 (2H, dd, 7.2, 1.0) |
127.8 |
| 3′′′, 5′′′ |
— |
— |
7.00 (2H, dd, 7.8, 7.2) |
129.3 |
| 4′′′ |
— |
— |
7.28 (1H, ddd, 7.8, 7.2, 1.0) |
132.2 |
Compound P3 showed significant similarities in spectral characters to those of P1, with a diacetylated jatrophadiene skeleton (δH (ppm) 0.86 (3H, d, 6.6, H-16), 1.89 (3H, s, H-17), 0.98 (3H, s, H-18), 0.96 (3H, s, H-19), 1.35 (3H, s, H-20), 5.98 (1H, d, 10.2, H-5), 5.64 (1H, d, 15.6, H-11), 5.45 (1H, d, 15.6, H-12), 1.68 (3H, s, AcO-9); 2.34 (3H, s, AcO-15); δC (ppm) 86.9 (C-1), 78.3 (C-3), 75.2 (C-7), 74.5 (C-9), 75.4 (C-13), 73.2 (C-14), 91.4 (C-15), 11.8 (C-16), 16.7 (C-17), 23.4 (C-18), 21.1 (C-19), 31.4 (C-20), 121.2 (C-5), 134.8 (C-6), 132.3 (C-11), 132.2 (C-12), 20.9 and 170.2 (AcO-9), 22.6 and 172.8 (AcO-15)). However, signals of three benzoyloxy groups were observed in the spectra of P3, instead of two groups in P1, indicating that the structure of P3 was tribenzoyloxylated (complex signals of fifteen aromatic protons at δH (ppm) 8.21–7.08 and signals of oxygen-bonding aromatic carbons at δC (ppm) 166.5, (BzO-1); 165.7 (BzO-3); 165.3 (BzO-7)). The HSQC spectra of P3 allowed for the signal definition for its protons, while the COSY and HMBC spectra of P3 indicated that it was 2,6,10,13-methylated, 9,15-acetoxylated, and 1,3,7-benzoyloxylated (Fig. 6). Finally, the stereochemical structure of P3 was determined from its NOESY spectra as shown in Fig. 9. Comparison with a previous publication17 (Table 5) resulted in our conclusion for P3 to be 9α,15β-diacetoxy-1α,3β,7β-tribenzoyloxy-13β,14α-dihydroxy-jatropha-5,11E-diene.
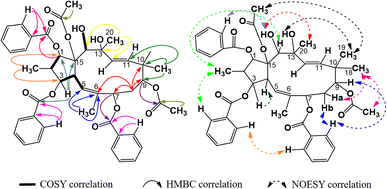 |
| | Fig. 6 Major COSY, HMBC, and NOESY correlations in P3. | |
Table 5 Spectral data comparison for P3 and 9β,15β-diacetoxy-3β,7β-dibenzoyloxy-1α,13β,14α-trihydroxy-jatropha-5,11E-diene
| Position |
P3 (acetone-d6) |
9α,15β-Diacetoxy-1α,3β,7β-tribenzoyloxy-13β,14α-dihydroxy-jatropha-5,11E-diene (CDCl3)17 |
| 1H-NMR |
13C-NMR |
1H-NMR |
13C-NMR |
| 1 |
5.87 (1H, d, 11.4) |
86.9 |
5.88 (1H, d, 11.8) |
87.1 |
| 2 |
2.34 (1H, m) |
44.2 |
2.52 (1H, m) |
44.2 |
| 3 |
5.51 (1H, dd, 5.4, 3.6) |
78.3 |
5.54 (1H, dd, 5.3, 4.3) |
77.2 |
| 4 |
4.25 (1H, dd, 9.6, 5.4) |
42.4 |
4.11 (1H, dd, 9.7, 5.1) |
41.9 |
| 5 |
5.98 (1H, d, 10.2) |
121.2 |
5.89 (1H, d, 9.7) |
119.3 |
| 6 |
— |
134.8 |
— |
134.4 |
| 7 |
5.27 (1H, s) |
75.2 |
5.30 (1H, s) |
74.2 |
| 8 |
2.21 (2H, m) |
33.3 |
2.08 (2H, m) |
32.4 |
| 9 |
5.22 (1H, dd, 3.6, 2.8) |
74.5 |
5.15 (1H, dd, 3.5, 2.9) |
73.9 |
| 10 |
— |
40.3 |
— |
39.6 |
| 11 |
5.64 (1H, d, 15.6) |
132.3 |
5.54 (1H, d, 15.5) |
132.1 |
| 12 |
5.45 (1H, d, 15.6) |
132.2 |
5.33 (1H, d, 15.5) |
129.9 |
| 13 |
— |
75.4 |
— |
74.7 |
| 14 |
4.67 (1H, d, 6.4) |
73.2 |
4.86 (1H, d, 5.6) |
72.3 |
| 15 |
— |
91.4 |
— |
90.1 |
| 16 |
0.86 (3H, d, 6.6) |
11.8 |
0.93 (3H, d, 6.7) |
11.4 |
| 17 |
1.89 (3H, s) |
16.7 |
1.87 (3H, s) |
16.5 |
| 18 |
0.98 (3H, s) |
23.4 |
0.94 (3H, s) |
20.6 |
| 19 |
0.96 (3H, s) |
21.1 |
0.95 (3H, s) |
22.8 |
| 20 |
1.35 (3H, s) |
31.4 |
1.39 (3H, s) |
31.5 |
| AcO-9 |
1.68 (3H, s) |
20.9 |
1.67 (3H, s) |
20.8 |
| 170.2 |
170.0 |
| AcO-15 |
2.34 (3H, s) |
22.5 |
2.38 (3H, s) |
22.1 |
| 171.1 |
170.9 |
| BzO-1 |
— |
166.5 |
— |
165.4 |
| 1′′′ |
— |
134.6 |
— |
133.2 |
| 2′′′, 6′′′ |
8.17 (2H, dd, 7.3, 1.2) |
133.1 |
8.02 (2H, dd, 7.3, 1.2) |
129.3 |
| 3′′′, 5′′′ |
7.58 (2H, dd, 8.4, 7.3) |
131.8 |
7.50 (2H, dd, 7.7, 7.3) |
128.7 |
| 4′′′ |
7.70 (1H, ddd, 8.4, 7.3, 1.2) |
133.6 |
7.61 (1H, ddd, 7.7, 7.3, 1.2) |
130.1 |
| BzO-3 |
— |
165.7 |
— |
165.3 |
| 1′ |
— |
133.7 |
— |
132.7 |
| 2′,6′ |
7.72 (2H, dd, 7.2, 1.1) |
130.1 |
7.60 (2H, dd, 7.3, 1.1) |
129.1 |
| 3′,5′ |
7.19 (2H, dd, 8.0, 7.2) |
130.0 |
7.13 (2H, dd, 7.8, 7.3 Hz) |
128.2 |
| 4′ |
7.38 (1H, ddd, 8.0, 7.2, 1.1) |
130.8 |
7.31 (1H, ddd, 7.8, 7.3, 1.1) |
129.5 |
| BzO-7 |
— |
165.3 |
— |
165.0 |
| 1′′ |
— |
132.3 |
— |
132.2 |
| 2′′,6′′ |
7.59 (2H, dd, 7.2, 1.0) |
130.7 |
7.58 (2H, dd, 7.2, 1.0) |
129.3 |
| 3′′,5′′ |
7.05 (2H, dd, 7.8, 1.0) |
129.2 |
7.00 (2H, dd, 7.8, 7.2) |
127.8 |
| 4′′ |
7.34 (1H, ddd, 7.8, 7.2, 1.0) |
130.0 |
7.28 (1H, ddd, 7.8, 7.2, 1.0) |
129.3 |
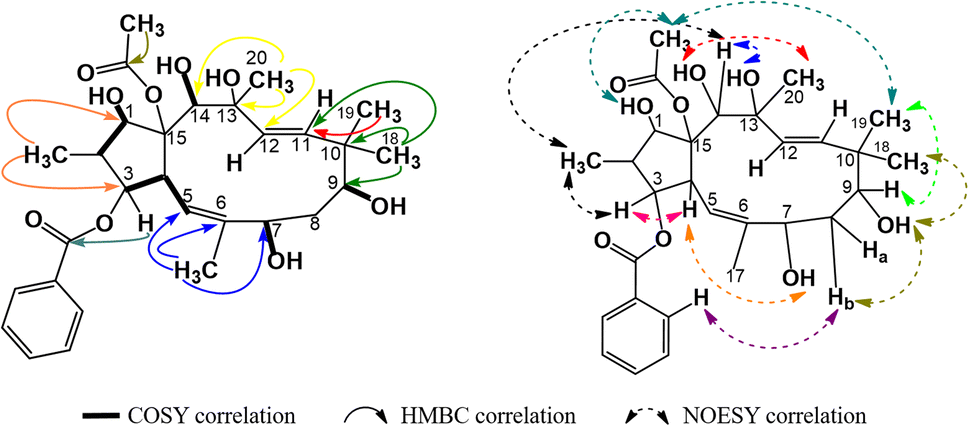
The spectral data of P4 showed similarities to those of P1, indicating that P4 was also acetylbenzoyloxymethyljatrophadiene. Specifically, from the 1H-NMR and 13C-NMR spectra, the signals of a jatrophane skeleton were observed: fifteen methyl protons (–CH3) at δH (ppm) 1.07 (3H, d, 6.5), 1.69 (3H, s), 1.11 (3H, s), 0.94 (3H, s), 1.33 (3H, s); three olefin protons at δH (ppm) 5.57 (1H, d, 10.0, H-5), 5.56 (1H, d, 15.5, H-11), 5.15 (1H, d, 15.5, H-12); three protons of an acetoxy group (AcO-15) at δH (ppm) 2.25 (3H, s); aromatic protons at δH (ppm) 7.94 (2H, dd, 7.5, 1.5, H-2′ and H-6′), 7.46 (2H, dd, 7.5, 7.5, H-3′ and H-5′), 7.59 (1H, ddd, 7.5, 7.5, 1.5, H-4′); seven C–OR carbons at δC (ppm) 87.2 (C-1), 78.8 (C-3), 72.6 (C-7), 72.5 (C-9), 74.6 (C-13), 72.4 (C-14), 91.5 (C-15); five methyl carbons at δC (ppm) 11.8 (C-16), 16.6 (C-17), 19.5 (C-18), 23.4 (C-19), 31.5 (C-20); the signal of carbons from an acetoxy group at δC (ppm) 22.6, 172.8 (AcO-15) and benzoyloxy group at δC (ppm) 165.9 (BzO-3); aromatic carbons at δC (ppm) 130.0 (C-1′), 128.7 (C-2′, C- 6′), 129.3 (C-3′, C-5′), 133.2 (C-4′); four olefin carbons at δC (ppm) 117.4 (C-5), 139.2 (C-6), 134.0 (C-11), 128.2 (C-1). Notably, the obtained 1H-NMR and 13C-NMR spectra of P4 showed that this compound possessed one acetoxy group and one benzoyloxy group less than P1. From the HSQC spectra of P4, the signal of each proton of its jatrophane skeleton was defined. The correlations between H-2/H-1, H-4/H-3 and H-5, OH-7/H-7 and H-17, OH-9/H-19, OH-14/H-14, OH-1/H-1, and H(AcO-15)/H-1 were observed in the COSY spectra of P4, while in its HMBC spectra, the correlations between H-16/C-1 and C-3; H-17/C-5, C-6, and C-7; H-18 and H-19/C-9, C-10, and C-11; H-20/C-12, C-13, and C-14 were observed (Fig. 7). Such spectral information indicated that P4 was methylated at the 16, 17, 18, 19, and 20 positions. However, H-3 correlated with the carboxyl carbon atom of the benzoyloxy group, indicating the presence of a 3-benzoyloxy moiety in the structure of P4. From all NMR spectral data above, P4 was considered to be 15-acetoxy-3-benzoyloxy-1,7,9,13,14-pentahydroxy-jatropha-5,11-diene. Finally, P4's NOESY spectra expressed the correlations between OH-13/H-8b, H-16/H-14 và OH-7, OH-14/H-20, H-19/OAc-15, H-9/H-19 (Fig. 7). Compound P4 was concluded to be 15α-acetoxy-3β-benzoyloxy-1α,7α,9β,13α,14β-pentahydroxy-jatropha-5,11E-diene (Fig. 9).
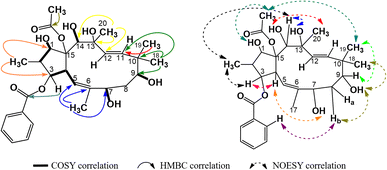 |
| | Fig. 7 Major COSY, HMBC, and NOESY correlations in P4. | |
As no referential data were found for P4, this compound was compared with peditithin G, to which P4 had the most similar structure. The comparative data are expressed in Table 6. It was obvious from this table that the structure of P4 and peditithin G were similar in the 15-acetoxy-3-benzoyloxyjatropha-5,11E-diene moiety, while the 9-acetoxy and 14-benzoyloxy groups of peditithin G were absent in P4. In addition, from the MS data, the molecular formula of P4 was revealed to be C29H40O9. This compound, therefore, was concluded to be 15α-acetoxy-3β-benzoyloxy-1α,7α,9β,13α,14β-pentahydroxy-jatropha-5,11E-diene. A reference on the SciFinder database indicated that the structure of P4 was new to science and this compound was named tithymal A.
Table 6 Spectral data comparison for P4 and peditithin G
| Position |
P4 (acetone-d6) |
Peditithin G (CDCl3)17 |
| 1H-NMR |
13C-NMR |
1H-NMR |
13C-NMR |
| 1 |
4.25 (1H, d, 12.0) |
87.2 |
4.12 (1H, d, 12.0) |
86.6 |
| 2 |
2.31 (1H, m) |
43.1 |
2.30 (1H, m) |
42.2 |
| 3 |
5.43 (1H, dd, 4.0, 4.0) |
78.8 |
5.49 (1H, dd, 4.5, 4.5) |
78.0 |
| 4 |
4.06 (1H, dd, 10.0, 4.5) |
41.4 |
4.43 (1H, dd, 9.6, 4.5) |
42.7 |
| 5 |
5.57 (1H, d, 10.0) |
117.4 |
5.73 (1H, d, 9.6) |
118.1 |
| 6 |
— |
139.2 |
— |
138.3 |
| 7 |
4.22 (1H, s) |
72.6 |
4.09 (1H, s) |
72.0 |
| 8 |
1.96 (Ha, m) |
36.7 |
2.00 (Ha, m) |
34.7 |
| 1.74 (Hb, m) |
1.86 (Hb, m) |
| 9 |
3.49 (1H, dd, 3.5, 3.5) |
72.5 |
4.77 (1H, dd, 3.2, 3.2) |
74.0 |
| 10 |
— |
39.3 |
— |
39.2 |
| 11 |
5.56 (1H, d, 15.5) |
134.0 |
5.51 (1H, d, 15.6) |
132.2 |
| 12 |
5.15 (1H, d, 15.5) |
128.2 |
5.31 (1H, d, 15.6) |
129.0 |
| 13 |
— |
74.6 |
— |
75.7 |
| 14 |
5.46 (1H, s) |
72.4 |
5.99 (1H, s) |
72.9 |
| 15 |
— |
91.5 |
— |
91.3 |
| 16 |
1.07 (3H, d, 6.5) |
11.8 |
0.84 (3H, d, 6.4) |
11.7 |
| 17 |
1.69 (3H, s) |
16.6 |
1.75 (3H, s) |
16.4 |
| 18 |
0.94 (3H, s) |
19.5 |
0.98 (3H, s) |
20.7 |
| 19 |
1.11 (3H, s) |
23.4 |
1.00 (3H, s) |
23.0 |
| 20 |
1.33 (3H, s) |
31.5 |
1.21 (3H, s) |
31.5 |
| AcO-9 |
— |
— |
2.04 (3H, s) |
21.3 |
| 171.1 |
| AcO-15 |
2.25 (3H, s) |
22.6 |
2.34 (3H, s) |
22.3 |
| 172.8 |
172.9 |
| BzO-3 |
— |
165.9 |
— |
166.2 |
| 1′ |
— |
130.0 |
— |
130.0 |
| 2′,6′ |
7.94 (2H, dd, 7.5, 1.5) |
128.7 |
7.93 (2H, dd, 7.8, 1.2) |
129.4 |
| 3′,5′ |
7.46 (2H, dd, 7.5, 7.5) |
129.3 |
7.43 (2H, dd, 7.8, 7.8) |
128.5 |
| 4′ |
7.59 (1H, ddd, 7.5, 7.5, 1.5) |
133.2 |
7.54 (1H, ddd, 7.8, 7.8, 1.2) |
133.0 |
| BzO-14 |
— |
— |
— |
165.0 |
| 1′′ |
— |
— |
— |
129.8 |
| 2′′,6′′ |
— |
— |
8.11 (dd, 7.8, 1.2) |
129.7 |
| 3′′,5′′ |
— |
— |
8.11, dd (7.8, 1.2) |
128.7 |
| 4′′ |
— |
— |
7.61, ddd (7.8, 7.2, 1.2) |
133.3 |
Compound P5 had significant spectral similarities to P4, indicating their structural resemblance. Specifically, P5 had an acetylbenzoyloxymethyljatrophadiene skeleton, with the signals of olefin protons and carbons at δH (ppm) 5.55 (1H, d, 9.6, H-5), 5.42 (1H, d, 15.6, H-11), 5.11 (1H, d, 15.6, H-12); δC (ppm) 117.4 (C-5), 139.2 (C 6), 134.0 (C-11), 128.2 (C-12); the protons and carbons of five methyl groups at δH (ppm) 1.66 (3H, s, CH3-17), 1.31 (3H, s, CH3-20), 1.04 (3H, d, 6.6, CH3-16), 1.09 (3H, s, CH3-19), 0.96 (3H, s, CH3-18); δC (ppm) 11.8 (C-16), 16.3 (C-17), 19.5 (C 18), 23.4 (C-19), 31.5 (C-20); the protons and carbons of an acetoxy group at δH (ppm) 2.23 (3H, d, 1.8); δC (ppm) 22.6, 172.8 (AcO-15); aromatic protons and carbons at δH (ppm) 7.91 (2H, dd, 7.2, 1.2, H-2′ and H-6′), 7.44 (2H, dd, 7.2, 7.2, H-3′ and H-5′), 7.54 (1H, ddd, 7.2, 7.2, 1.2, H-4′); δC (ppm) 130.0 (C-1′), 129.3 (C-2′ and 6′), 128.7 (C-3′ and 5′), 133.2 (C-4′), together with an oxygen-bearing benzoyl carbon at δC (ppm) 165.9 (BzO-3) and six other oxygen-bearing ones at δC (ppm) 87.2 (C-1), 78.8 (C-3), 72.5 (C-9), 74.6 (C-13), 72.4 (C-14), 91.5 (C-15). From the COSY spectra of P5, the correlations of H-2/H-1, H-4/H-3 and H-5, OH-9/H-8, OH-14/H-14, OH-1/H-1, H(CH3COO-15)/H-1 were observed, while those between H-16 and C-1, C-3; H-17 and C-5, C-6; H-18, H-19 and C-9, C-10, C-11; H-20 and C-12, C-13, C-14 were observed in the HMBC spectra (Fig. 8). Such data led to the consideration for P5's structure to be 15-acetoxy-3-benzoyloxy-1,9,13,14-tetrahydroxyjatropha-5,11E-diene. Finally, from the NOESY spectra of P5, the correlations of H-18/OH-9, H-8a/CH3COO-15, H-19/H-8b/H-3, OH-13/OH-1, OH-13/H-9/H-4, OH-14/H-2/H-3 were observed (Fig. 8), allowing for the stereochemical determination for this compound, as shown in Fig. 9, and P5 was revealed to be 15β-acetoxy-3β-benzoyloxy-1α,9α,13α,14β-tetrahydroxyjatropha-5,11E-diene.
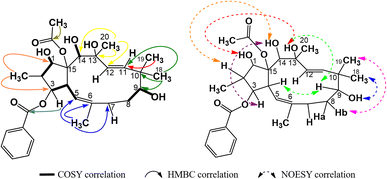 |
| | Fig. 8 Major COSY, HMBC, and NOESY correlations in P5. | |
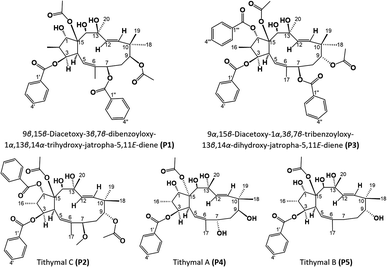 |
| | Fig. 9 Molecular structures of compounds P1–P5. | |
As no referential data were found for P5, this compound was compared with P4 to evaluate the spectral and structural similarities and differences. The comparative data are expressed in Table 7. It was obvious from this table that the structure of P5 is very similar to that of P4, except only for the absence of 7-OH group. However, from the MS data, the molecular formula of P5 was revealed to be C29H40O8. This compound, therefore, was concluded to be 15β-acetoxy-3β-benzoyloxy-1α,9α,13α,14β-tetrahydroxyjatropha-5,11E-diene. A reference on the SciFinder database indicated that the structure of P5 was new to science and this compound was named tithymal B.
Table 7 Spectral data comparison for P4 and P5
| Position |
P4 (acetone-d6) |
P5 (CDCl3) |
| 1H-NMR |
13C-NMR |
1H-NMR |
13C-NMR |
| 1 |
4.25 (1H, d, 12.0) |
87.2 |
4.22 (1H, d, 11.4) |
87.2 |
| 2 |
2.31 (1H, m) |
43.1 |
2.29 (1H, m) |
43.1 |
| 3 |
5.43 (1H, dd, 4.0, 4.0) |
78.8 |
5.49 (1H, dd, 3.8, 3.8) |
78.8 |
| 4 |
4.06 (1H, dd, 10.0, 4.5) |
41.4 |
4.04 (1H, m) |
41.4 |
| 5 |
5.57 (1H, d, 10.0) |
117.4 |
5.55 (1H, d, 9.6) |
117.4 |
| 6 |
— |
139.2 |
— |
139.2 |
| 7 |
4.22 (1H, s) |
72.6 |
2.17 (2H, d, 1.8) |
30.9 |
| 8 |
1.96 (Ha, m) |
36.7 |
1.72 (Ha, m) |
36.8 |
| 1.74 (Hb, m) |
1.94 (Hb, m) |
| 9 |
3.49 (1H, dd, 3.5, 3.5) |
72.5 |
4.77 (1H, dd, 3.0) |
72.5 |
| 10 |
— |
39.3 |
— |
39.3 |
| 11 |
5.56 (1H, d, 15.5) |
134.0 |
5.42 (1H, d, 15.6) |
134.0 |
| 12 |
5.15 (1H, d, 15.5) |
128.2 |
5.11 (1H, d, 15.6) |
128.2 |
| 13 |
— |
74.6 |
— |
74.6 |
| 14 |
5.46 (1H, s) |
72.4 |
4.28 (1H, s) |
72.4 |
| 15 |
— |
91.5 |
— |
91.5 |
| 16 |
1.07 (3H, d, 6.5) |
11.8 |
1.04 (3H, d, 6.6) |
11.8 |
| 17 |
1.69 (3H, s) |
16.6 |
1.66 (3H, s) |
16.3 |
| 18 |
0.94 (3H, s) |
19.5 |
0.96 (3H, s) |
19.5 |
| 19 |
1.11 (3H, s) |
23.4 |
1.09 (3H, s) |
23.4 |
| 20 |
1.33 (3H, s) |
31.5 |
1.31 (3H, s) |
31.5 |
| AcO-15 |
2.25 (3H, s) |
22.6 |
2.23 (3H, s) |
22.6 |
| 172.8 |
172.8 |
| BzO-3 |
— |
165.9 |
— |
165.9 |
| 1′ |
— |
130.0 |
— |
130.0 |
| 2′,6′ |
7.94 (2H, dd, 7.5, 1.5) |
128.7 |
7.91 (2H, dd, 7.2, 1.2) |
129.3 |
| 3′,5′ |
7.46 (2H, dd, 7.5, 7.5) |
129.3 |
7.44 (2H, dd, 7.2, 7.2) |
128.7 |
| 4′ |
7.59 (1H, ddd, 7.5, 7.5, 1.5) |
133.2 |
7.54 (1H, ddd, 7.2, 7.2, 1.2) |
133.2 |
Conclusions
In this study, the cytotoxicity and anti-diabetic activity of extracts and some isolated compounds of Vietnamese Euphorbia tithymaloides against human normal (fibroblasts) and cancer cell lines (HeLa, HepG2, Jurkat, MCF-7, and NCI-H460) were investigated. The obtained results indicated that this Euphorbia plant exhibited moderate toxicity to MCF-7 and NCI-H460, with the petroleum ether extract having the highest values. In addition, all extracts of the plant and tested isolated jatrophanes were non-toxic to normal cells. The aqueous extract of the plant exhibited the highest α-glucosidase inhibitory activity. From the petroleum ether extract, five jatrophane diterpenoids were isolated, three of which were new to science and respectively named tithymal A, tithymal B, and tithymal C. Interestingly, tithymal A exhibited significantly high α-glucosidase inhibitory activity. The obtained results contributed significantly to scientific knowledge on Vietnamese Euphorbia tithymaloides, reinforcing its potential to be exploited medicinally. In future studies, deeper steps will be taken on the remaining fractions of the petroleum ether extract, other extracts, as well as their other bioactivities and isolated compounds.
Author contributions
Nguyen Vu Duy Khang: methodology; validation; formal analysis; data curation; investigation; writing – original draft; visualization. Dinh Thi Hong Dao: methodology; data curation; investigation. Nguyen Thi Thanh Mai: conceptualization; methodology; validation. Tran Le Quan: conceptualization; methodology; supervision. Nguyen Thi Y. Nhi: conceptualization; methodology; validation; supervision; writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research is funded by Vietnam National University, Ho Chi Minh City (VNU-HCM) under grant number NCM2020-18-01.
References
- K. Douglas, P. Xolani, L. Moses and T. Jacqueline, Molecules, 2020, 35, 25174019 Search PubMed
 .
. - S. L. Gerson, B. C. Denis, C. M. Gerzia and A. M. M. Maria, Phytomedicine, 2015, 22, 1133–1137 CrossRef PubMed .
- T. Ju-Ying, R. Dóra, F. Peter, L. Yu, V. Andrea, H. Judit and W. Chin-Chung, J. Nat. Prod., 2016, 79, 2658–2666 CrossRef PubMed .
- N. P. Nelly, M. L. Emmanuel, D. Amogu, L. I. Clement, L. M. Christophe, Z. G. Benjamin, M. A. Colette, F. M. Clarisse, S. T. T. Damien, D. T. Dorothée, N. Koto-te-Nyiwa and T. M. Pius, Discovery Phytomedicine, 2020, 7(3), 186–194 Search PubMed .
- J. V. Arnold, P. Luc, A. Sandra, C. Kanyanga, M. Kahunu and T. Lutete, J. Ethnopharmacol., 2015, 174, 607–617 CrossRef PubMed .
- S. V. K. P. Shaik, B. Rajeswari, K. S. Pradeep and R. G. Terry, Biofuels, 2018, 12(5), 511–521 Search PubMed .
- Z. Ying, S. Yancai, D. Na, L. Bing-Bing and M. Jia, Mitochondrial DNA, Part B: Resources, 2019, 4(1), 1973–1974 CrossRef .
- H. A. Nguyen, P. SeonJu, T. T. Do, T. H. Y. Duong, H. Y. Bui, H. Y. Pham, V. K. Pham, T. C. Pham and X. N. Nguyen, Vietnam J. Sci. Technol., 2022, 60(2), 141–174 CrossRef PubMed .
- B. Rania, M. Abdelkarim, K. Neha, H. C. Eun, K. E. Abdel and K. K. Nagendra, Front. Plant Sci., 2022, 13, 1008881 CrossRef PubMed .
- B. T. Đậu and T. H. Hà, Hue University Journal of Science: Agriculture and Rural Development, 2019, 128(3D), 43–51 Search PubMed .
- T. L. Đỗ, Vietnamese Medicinal Plants and Remedies. Hong Duc, Ha Noi, 2022 Search PubMed .
- H. B. Pham and Q. C. Cao, Vinh University Journal of Science, 2019, 48(1A), 16–24 Search PubMed .
- A. D. Nguyễn and T. T. N. Nguyễn, The 4th National Scientific Conference of Ecology and Biological Resources, Ha Noi, Vietnam, 2011, pp. 74–78 Search PubMed .
- F. M. Charles, I. The Genera Pedilanthus and Cubanthus, and Other American Euphorbiaceae. Field Museum of Natural History, Chicago, 1913 Search PubMed .
- S. Rajani and S. Neetu, Pharma Innovation, 2019, 8(5), 109–115 CrossRef .
- T. I. Cầm, T. H. V. Nguyễn, M. Q. Phạm, T. Q. T. Trần, A. V. Trịnh, T. T. Nguyễn and T. T. Đỗ, Vietnam J. Chem., 2016, 54(3), 274–279 Search PubMed .
- Z. Jianyong, W. Ruimin, L. Lanlan, L. Wei, T. Guihua, B. Xianzhang and Y. Sheng, J. Med. Chem., 2016, 59(13), 6353–6369 CrossRef PubMed .
- P. S. Louis, J. F. Andrew, R. Joachim, A. Heidrun, O. Till and T. Eckhard, Tetrahedron Lett., 2012, 53, 2153–2156 CrossRef .
- M. Wantana and S. Somyote, J. Nat. Prod., 2007, 70(9), 1434–1438 CrossRef PubMed .
- P. N. K. Udaya, N. S. Sripriya, D. D. Raj, S. Deepa and S. Bhuvaneswari, Pak. J. Pharm. Sci., 2019, 32(5), 2117–2122 Search PubMed .
- M. T. Vu, T. H. Pham, H. N. Vu, T. S. Phan and M. G. Phan, Vietnam J. Chem., 2014, 52(2), 186–189 Search PubMed .
- T. N. My-Nuong and H. Thuy-Duong, BMC Complementary Altern. Med., 2016, 16(1), 202–236 CrossRef PubMed .
- T. H. T. Nguyễn, T. V. Nguyễn, T. T. Tất, T. T. G. Nguyễn, N. H. Nguyễn and H. T. D. Hồ, 2007 National Scientific Conference – Fundamental Researches in Life Science, Binh Dinh Province, Vietnam, 2007, p. 809 Search PubMed .
- W. Luo-sheng, C. Cui-ping, X. Zuo-qi, W. Yong-long, M. Qiu-xia, Y. Yuedong and C. Jiachun, J. Ethnopharmacol., 2013, 147(3), 622–630 CrossRef PubMed .
- V. D. K. Nguyen, T. Y. N. Nguyen and L. Q. Tran, Biofuels, Bioprod. Biorefin., 2023, 2023, bbb2472 Search PubMed .
- Y. Hequn, M. Aytilla, T. Dan and A. A. Haji, Bioorg. Chem., 2021, 112, 104989 CrossRef PubMed .
- Y. Yanlan, Z. Mi, W. Dongni, L. Xiangzhong, Y. Xiansheng, W. Guanghui, L. Ting, S. Cuiling, D. Rong, T. Wenjing and C. Haifeng, J. Nat. Prod., 2021, 84(2), 339–351 CrossRef PubMed .
- C. Yan-Ni, D. Xiao, L. Dong-Mei, L. Qing-Yun, L. Shuai, L. Ying-Yao, D. Ying-Tong, F. Xin and H. Xiao-Jiang, Nat. Prod. Bioprospect., 2021, 11, 357–364 CrossRef PubMed .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Dinh Thi Hong Daoab,
Nguyen Thi Thanh Maiabc,
Tran Le Quan
ab,
Dinh Thi Hong Daoab,
Nguyen Thi Thanh Maiabc,
Tran Le Quan



 : extract;
: extract;  : compound)
: compound)

 : extract;
: extract;  : compound)
: compound)