
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, molecular docking and DFT studies on novel melatonin and isatin based azole derivatives†
Keshav Kumar Saini ab,
Ravindra Kumar Upadhyayac,
Ravi Kantd,
Arpita Vajpayeee,
Kalpana Jainf,
Amit Kumarb,
Lalita S. Kumarg and
Rakesh Kumar*a
ab,
Ravindra Kumar Upadhyayac,
Ravi Kantd,
Arpita Vajpayeee,
Kalpana Jainf,
Amit Kumarb,
Lalita S. Kumarg and
Rakesh Kumar*a
aDepartment of Chemistry, University of Delhi, Delhi 110007, India. E-mail: rakeshkp@email.com
bDepartment of Chemistry, Dyal Singh College, University of Delhi, Lodhi Road, New Delhi 110003, India
cDepartment of Chemistry, Sri Venkateswara College, University of Delhi, New Delhi 110021, India
dDepartment of Chemistry, Government Post Graduate College, G.B. Nagar, Noida, UP 201301, India
eDepartment of Physics, Dyal Singh College, University of Delhi, Lodhi Road, New Delhi 110003, India
fDepartment of Physics, D. J. College, Baraut, UP 250611, India
gChemistry Discipline, School of Sciences, Indira Gandhi National Open University, New Delhi 110068, India
First published on 14th September 2023
Abstract
In order to address the pressing demand for newer broad-spectrum antifungal medicines with enhanced activity, computer modelling was utilised to rationally develop newer antifungal azole-based drugs. Based on the drug and active sites of the Lanosterol 14 alpha-Demethylases (LAD) of the prominent fungal pathogen Candida albicans interaction, Novel triazole-linked melatonin and isatin derivatives 7a–d and 8a–d were synthesised using bioisosterism. Besides the experimental synthesis and subsequent characterization, the present study focused on obtaining optimised geometries, frequency calculations, and TD-DFT studies of the synthesised molecules. We also performed molecular docking studies to explore the inhibitory ability of the synthesised compounds against the active sites of the Lanosterol 14 alpha-Demethylases (LAD) of the prominent fungal pathogen Candida albicans. The binding interactions resulted in positive findings, demonstrating the involvement of the synthesised compounds in the suppression of fungal growth. Comparative analysis of the binding potential of the synthesised molecules and commercially available drug fluconazole revealed a remarkable note: the docking scores for the designed drugs 7b, 7c, and 8c are much greater than those of the fluconazole molecule. The in silico study of the designed series of drug molecules serves as an important guideline for further exploration in the quest for potent antifungal agents.
Introduction
Heterocycles are an immensely important class of organic compounds, accounting for more than half of all documented organic molecules. They serve as an important core moiety in a wide range of natural products, including haemoglobin, biomolecules, RNA, DNA, proteins, vitamins, and biologically active compounds.1 Heterocyclic scaffolds are important synthetic precursors for the synthesis of a variety of biologically active compounds. According to recent surveys, an enormous number of molecules are under investigation by researchers, including nitrogen-containing heterocycles such as indoles, pyrimidyl, pyrazolyl, thiazolyl, and pyridyl indoles, for their applications in the field of pharmacological and therapeutic effects.2–4 Naturally occurring melatonin belongs to the privileged class of indoleamine, which is synthesised and discovered in a variety of species, including bacteria and eukaryotes.5 Melatonin is a pineal gland hormone produced by serotonin that governs the cycle of sleep and wakefulness, the circadian rhythm, cycles of menstruation, ageing, immunity, and antioxidants in the body, among other things. Melatonin production and release are more intense at night, whereas light exposure suppresses them.6 Previous research has documented that melatonin has a wide range of physiological effects, including antioxidant,7,8 cardiovascular,9,10 anti-inflammatory,11,12 neuroprotective,13,14 stroke protective,15 pain modulatory,16 antitumor,17 antibacterial,18,19 liver injury protective properties,20 retinal,21 as well as effects on offspring metabolism,22 etc. 1H-indole-2,3-dione, which is commonly referred to as Isatin or indenedione, is a versatile moiety with a broad range of biological activity. Isatin is the precursor for a number of derivatives that possess a broad range of potential biological and pharmaceutical properties23 such as anticancer properties in various types of cancers,24–26 antibiotics,27 anxiogenic,28 antibacterial,29 antidiabetic,30 anticonvulsant,31 sedative,32 carboxylesterases,33 antidepressants,34 antifungals,35 antimicrobial36 etc. Triazoles, namely 1,2,3-triazole and 1,2,4-triazole, are among the most significant groups of nitrogen-containing heterocycles. Triazole may increase solubility and binding to bimolecular targets via a variety of non-covalent interactions.37 Triazoles are known to possess a wide number of biological activities, such as antimicrobial, antihistaminic, analgesic, anti-inflammatory, antimycotic, anticancer, antiprotozoal, insecticidal, antimalarial, anticonvulsant, antimycobacterial, and anti-ulcer activity.38,39 The 14α-demethylase enzyme (CYP51), also known as lanosterol, is essential for the biosynthesis of ergosterol. By inhibiting ergosterol production, fungal growth is inhibited.40The incorporation of two or more pharmacophores into a single hybrid molecule via triazole link delivers an appealing approach to facilitating the development of newer drugs with the capacity to overcome cross-resistance and enhance potency in comparison to the individual moiety. Based on the aforementioned details and our keen interest in discovering newer, more potent antifungal agents to overcome drug resistance, we are reporting a simple technique for the synthesis of novel N-(2-(1-((1-(2-(2,3-dioxoindolin-1-yl)ethyl)-1H-1,2,3-triazol-5-yl)methyl)-5-methoxy-1H-indol-3-yl)ethyl)acetamide and its derivatives via alkyne-azide cycloaddition catalysed by Cu(I) (CuAAC) reaction. In the present investigation, we also reported the molecular docking study of all synthesised molecules in order to forecast probable binding modalities with Lanosterol 14 alpha-Demethylases (LAD) of prominent fungal pathogens Candida albicans to block sites responsible for ergosterol biosynthesis. The outcomes seem to strongly indicate that the synthesised compounds hold promise as potential antifungal drug molecules, thus, the outcome of the present study serve as important guidelines for further scientific exploration of these potential candidates for use in in vitro and in vivo studies for the development of efficient antifungal agents.
Design strategy
Furthermore, numerous clinically approved and under-trial drugs with robust antifungal characteristics are based on melatonin, isatin, and 1,2,3-triazole rings, making the melatonin, isatin, and 1,2,3-triazole core a fascinating and understudied pharmacophore. In continuation of our keen interest in the designing of single drug with multiple targets, we have synthesised new hybrids by inclusion of melatonin, isatin, and triazole scaffolds into a single target molecule (Fig. 1). | ||
| Fig. 1 Melatonin, triazole, and isatin-based antifungal agents, mapped to the designed target compound. | ||
Result discussion
Methods and materials
All the reagents required for the current study were analytical reagent grade and were used as such without further purification. All the reagents and materials were procured from Sigma Aldrich, Merk, and Spectrochem from commercial suppliers. All the solvents were purchased from Finar Ltd. All the reactions were monitored with thin layer chromatography silica gel plates (TLC) Kieselgel 60 F254 (Merk) using ethyl acetate/petrol or methanol/chloroform as mobile phases and visualised in UV light. The purification of all synthesised molecules was carried out using silica gel 100–200 Mesh (Qualikems). The melting points of all synthesised molecules were determined by the electrothermal melting point apparatus Buchi instrument (M-560) and were uncorrected. Infrared spectra were recorded using KBr pellets on a SHIMADZU FT-IR affinity spectrophotometer, and the recordings are expressed as υmax cm−1. The formation of the synthesised molecule was confirmed by 1H NMR and 13C NMR on a JEOL, ECX-400P spectrometer USA at 400 MHz and 100 MHz using tetramethylsilane (TMS) as an internal reference standard. The chemical shift (δ), coupling constant, and absorption frequency for the NMR spectra were reported as parts per million, J (Hz), and ν (cm−1), respectively in dimethyl sulfoxide (DMSO-d6) as the solvent. The spectral data uses the notation s, d, t, q, dd, and m for singlet, doublet, triplet, quartet, doublet of doublet, and multiplate, respectively. All mass spectrometry readings for all synthesised compounds were taken using a 6530 Accurate-Mass Q-TOF LC/MS instrument.Method of preparations
The synthesised target molecules 7a–d and 8a–d in the present work include three fundamental structural elements: (i) N-substituted melatonin; (ii) a substituted 1,2,3-triazole moiety; and (iii) a substituted isatin moiety. The procedure for the synthesis of triazole-linked melatonin isatine derivatives was accomplished in four steps, beginning with the reaction of melatonin (1) with one equivalent of propargyl bromide and NaH in DMF at RT to get N-(2-(5-methoxy-1-(prop-2-yn-1-yl)-1H-indol-3-yl)ethyl) acetamide (2) in 91% yield (Scheme 1). Following that, a variety of nonpolar, polar aprotic, and polar protic solvents (entries 1–12) were tested with K2CO3 and NaH bases, and the experimental findings suggested that reaction with NaH in DMF (entry 10) was the most appropriate choice for the current chemical reaction under investigation (Table 1). | ||
| Scheme 1 Synthesis of propargylated derivative of melatonin 1 1(1 eq), NaH (1 eq). | ||
| Entry | Solvent | Base | Number of equivalents (base) | Temp (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | Toluene | K2CO3 | 1 | RT | 8 | No reaction |
| 2 | Toluene | NaH | 1 | RT | 8 | No reaction |
| 3 | Xylene | K2CO3 | 1 | RT | 8 | No reaction |
| 4 | Xylene | NaH | 1 | RT | 8 | No reaction |
| 5 | THF | K2CO3 | 1 | RT | 8 | 23 |
| 6 | THF | NaH | 1 | RT | 8 | 25 |
| 7 | ACN | K2CO3 | 1 | RT | 6 | 36 |
| 8 | ACN | NaH | 1 | RT | 6 | 38 |
| 9 | DMF | K2CO3 | 1 | RT | 3.0 | 85 |
| 10 | DMF | NaH | 1 | RT | 1.5 | 91 |
| 11 | t-Butanol | K2CO3 | 1 | RT | 5 | 42 |
| 12 | t-Butanol | NaH | 1 | RT | 5 | 42 |
In step 2 azide derivatives of isatin (5a–d)were synthesised according to the reported methods.41,42 Alkylation of substituted isatin (3a–d) with 1,2-dibromoethane yielded 4a–d in presence of NaH and DMF Scheme 2. 3 and 4. The resulting N-alkylbromoisatins (4a–d) were subsequently treated with sodium azide in DMF at 80 °C to give the corresponding N-alkylazido-isatins (5a–d).
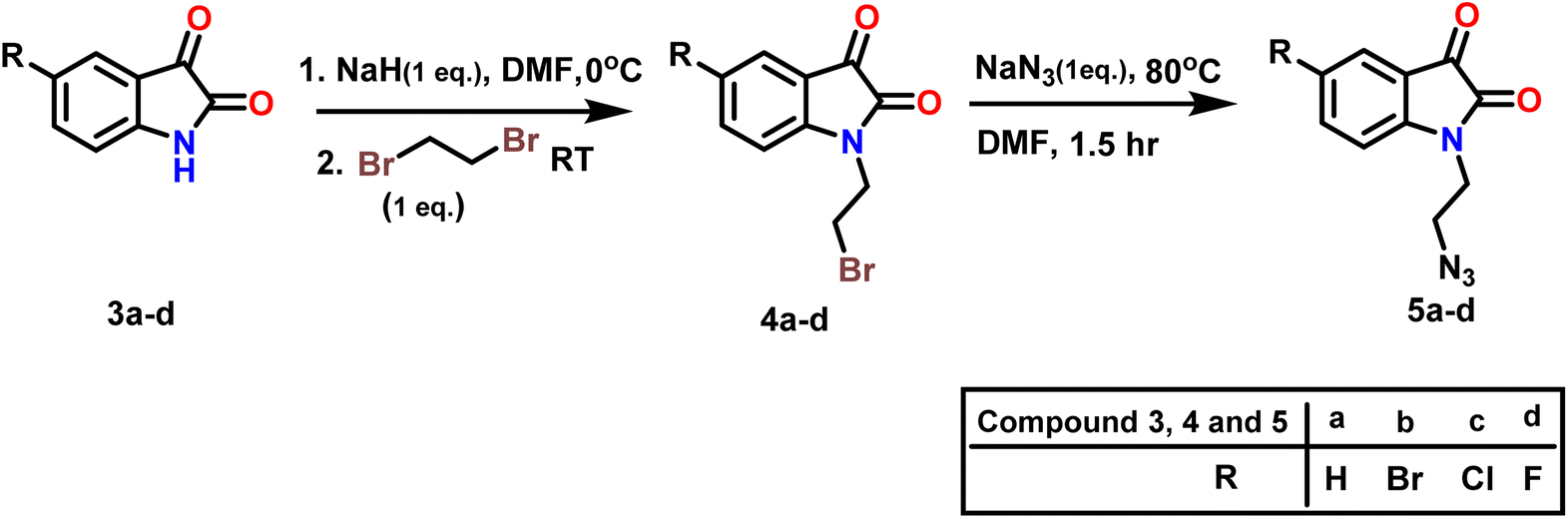 | ||
| Scheme 2 Synthesis of 2-azidoethyl-Isatins derivatives (5a–d). | ||
 | ||
| Scheme 3 Synthesis of N-alkylazido-hydxoxyiminoisatins derivatives (6a–d). | ||
 | ||
| Scheme 4 Synthesis of target compound 7a–d and 8a–d using click reaction. | ||
The precursor, 5a–d were further refluxed with hydroxyl amine in pryridine and ethanol for 1 h to give a N-alkylazido-hydxoxyiminoisatins43 (6a–d).
In an attempt to adopt an environmentally friendly green method for the synthesis of the target compound 7a–d and 8a–d. We used Cu(I) catalysed click reaction by reported methods44,45 (Table 2). But these reactions have been going on for a long time at high temperatures, and the yield in response to them is also discouraging (Table 2 entries no. 1, 4, and 5). Therefore, target compounds were prepared by Cu(I)-catalyzed click reactions via 1,3-dipolar cycloaddition in t-BuOH-H2O (entry 3: Table 2) to get better yield. Thus, N-propargylated melatonin (2) was allowed to react with isatin and substitute isatin azides 5a–d and 6a–d in t-BuOH-H2O (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) (1:1) using CuSO4 and sodium ascorbate as catalysts at 45 °C for 12–18 minutes to afford corresponding 1,4-disubstituted 1,2,3-triazoles (7a–d, 8a–d) in excellent yield (Table 3).
:2) (1:1) using CuSO4 and sodium ascorbate as catalysts at 45 °C for 12–18 minutes to afford corresponding 1,4-disubstituted 1,2,3-triazoles (7a–d, 8a–d) in excellent yield (Table 3).
| S No. | Catalyst | Solvent | Reaction condition | Yield (%) |
|---|---|---|---|---|
| 1 | CuSO4/Sod ascorbate | Water | 120 °C 24 h | 33 |
| 2 | CuSO4/Sod ascorbate | DMF + H2O (1:1) |
60 °C 15 min | 86 |
| 3 | CuSO4/Sod ascorbate | t-BuOH/H2O (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2) :2) |
45 °C 12 min | 90 |
| 4 | CuSO4/Sod ascorbate | Ethanol | 60 °C | No reaction |
| 5 | CuSO4/Sod ascorbate | Ethanol/H2O | 60 °C | No reaction |
| S No. | Compound no. | Structure | Melting point (°C) | Yield (%) |
|---|---|---|---|---|
| 1 | 7a |  |
141–143 | 87 |
| 2 | 7b |  |
133–135 | 86 |
| 3 | 7c | 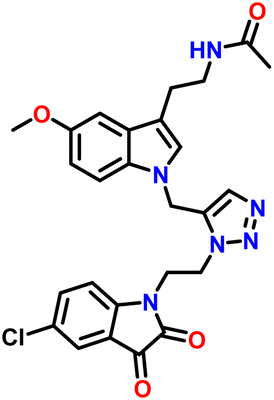 |
138–140 | 88 |
| 4 | 7d |  |
131–133 | 85 |
| 5 | 8a |  |
182–184 | 85 |
| 6 | 8b |  |
181–183 | 84 |
| 7 | 8c | 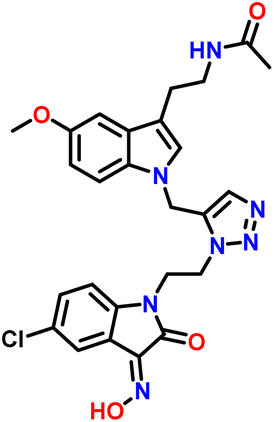 |
185–187 | 84 |
| 8 | 8d |  |
180–182 | 84 |
The Structure of synthesised compounds were characterised using 1H NMR, 13C NMR, FTIR and HRMS analytical techniques. In the 1H NMR of compound 7b, a singlet was observed at δ 1.8 ppm for 3 protons of the CH3 group attached to the amide group of melatonin. A singlet for 3 protons of methyl of the methoxy group attached to the aromatic ring of the melatonin core was observed at δ 3.76 ppm. A characteristic triplet was observed at δ 7.96 ppm for 1 proton, corresponding to the NH of the amide group of melatonin. A singlet appeared at δ 8.04 ppm for one proton for the triazole hydrogen. The 13C NMR spectrum of compound 7b showed a peak at δ 182.20 ppm, which was assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (keto) carbon of the five-membered ring of isatin. The peak at δ 131.61 ppm corresponds to the carbon of the triazole ring. The peaks at δ 126.97 ppm were assigned to the carbon of the five-membered ring of melatonin.
O (keto) carbon of the five-membered ring of isatin. The peak at δ 131.61 ppm corresponds to the carbon of the triazole ring. The peaks at δ 126.97 ppm were assigned to the carbon of the five-membered ring of melatonin.
In the 1H NMR of compound 8c, a singlet was observed at δ 1.80 ppm for 3 protons of the CH3 group attached to the amide group of melatonin. A singlet for 3 protons of methyl of the methoxy group attached to the aromatic ring of the melatonin core was observed at δ 3.76 ppm. A characteristic triplet was observed at δ 7.93 ppm for 1 proton, corresponding to the NH of the amide group of melatonin. A singlet appeared as a singlet at δ 8.00 ppm for the triazole hydrogen. A singlet observed at δ 8.00 ppm, corresponding to the hydrogen of the hydroxy group of isatinoximes. The 13C NMR spectrum of compound 8c showed a peak at δ 163.16 ppm, which was assigned to the CN carbon of the five-membered ring of isatin. The peak that appeared at δ 131.61 ppm corresponds to the carbon of the triazole ring. The peaks that appeared at δ 126.56 ppm were assigned to the carbon of the five-membered ring of melatonin.
Computational study
 | ||
| Fig. 2 Optimised geometrical structures of the designed molecules; 7b, 7c and 8c. | ||
| S No. | Drug name (ligand) | Binding energy (kcal mol−1) | Hydrogen bond interaction | Hydrostatic interaction with amino acids |
|---|---|---|---|---|
| 1 | 6 | −7.3 | — | Gly 65(A), Leu 87(A), Leu 88(A), Lys 90(A), Tyr A: 122, Pro 230(A), Phe 233(A), Ile 231(A), His 377(A), Phe 380(A), Ser 507(A), Met 508(A) |
| 2 | 7a | −10.1 | His 377(A) 3.25 Å, Met 508(A) 2.91 Å | Gly 65(A), Leu 87(A), Leu 88(A), Tyr 118(A), Thr 122(A), Phe 228(A), Pro 230(A), Phe 233(A), Leu 376(A), Ser 378(A), Phe 380 (A), Tyr 505(A), Ser 506(A), Val 509(A) |
| 3 | 7b | −11.1 | Ser 378(A) 2.65 Å | Leu 87(A), Tyr 118(A), Thr 122(A), Tyr 132(A), Pro 230(A), Phe 233(A), Gly 307(A), His 310(A), Thr 311(A), Leu 376(A), Ile 379(A), Phe 380(A), Ser 507(A), Met 508(A), Val 509(A) |
| 4 | 7c | −11.0 | Ser 378(A) 3.08 Å, Met 508(A) 2.92 Å | Leu 87(A), Tyr 118(A), Leu 121(A), Thr 122(A), Phe 126(A), Tyr 132(A), Phe 228(A), Pro 230(A), Phe 233(A), Gly 307(A), Thr 311(A), Leu 376(A), His 377(A), Phe 380(A), Ser 507(A) |
| 5 | 7d | −10 | Ser 378(A) 3.18 Å, Met 508(A) 3.09 Å | Gly 65(A), Leu 87(A), Leu 88(A), Tyr 118(A), Thr 122(A), Phe 228(A), Pro 230(A), Phe 233(A), Leu 376(A), His 377(A), Phe 380(A), Tyr 505(A), Ser 506(A), Val 509(A) |
| 6 | 8a | −9.1 | Asp 225(A) 2.87 Å, Met 189(A) 3.06 Å, His 310(A) 2.88 Å | Glu 194(A), Ile 197(A), Pro 193(A), Phe 213(A), Ala 218(A), Tyr 221(A), Ser 222(A), Phe 228(A), Gln 309(A), Ser 507(A), Met 508 (A), Val 509(A), Leu 511(A) |
| 7 | 8b | −8.9 | Asp 225(A) 2.70 Å | Glu 194(A), Met 189(A), Pro 193(A), Ile 197(A), Phe 198(A), Ala 218(A), Tyr 221(A), Ser 222(A), Phe 228(A), His 310(A), Gln 309(A), Ala 313(A), Ser 314(A), Ser 507(A), Met 508 (A), Leu 511(A) |
| 8 | 8c | −11.6 | His 377(A) 2.94 Å, Ser 378(A) 2.92 Å | Leu 87(A), Tyr 118(A), Tyr 132(A), Phe 228(A), Pro 230(A), Phe 233(A), 307(A), Thr 311(A), Leu 376(A), Phe 380(A), Tyr 505(A), Ser 507(A), Met 508(A) |
| 9 | 8d | −9.2 | Asp 225(A) 2.80 Å | Met 189(A), Pro 193(A), Glu 194(A), Ile 197(A), Phe 198(A), Phe 213(A), Ala 218(A), Tyr 221(A), Ser 222(A), Phe 228(A), Gln 309(A), His 310 (A), Ala 313(A), Ser 314(A), Ser 507(A), Met 508 (A), Val 509(A), Leu 511(A) |
| 10 | Fluconazole | −8.2 | — | Tyr 118(A), Leu 121(A), Thr 122(A), Ile 131(A), Tyr 132(A), Hem O(B), Phe 228(A), Gly 303(A), Gly 307(A), Leu 376(A), Met 508(A), Val 509(A) |
 | ||
| Fig. 3 The best docked molecules 7b, 7c, and 8c to Lanosterol 14 alpha-Demethylases protein with the highest docking score of −11.1 kcal mol−1 (7b), −11.0 kcal mol−1 (7c) and −11.6 kcal mol−1 (8c) as compared with standard drug fluconazole (−8.2 kcal mol−1). | ||
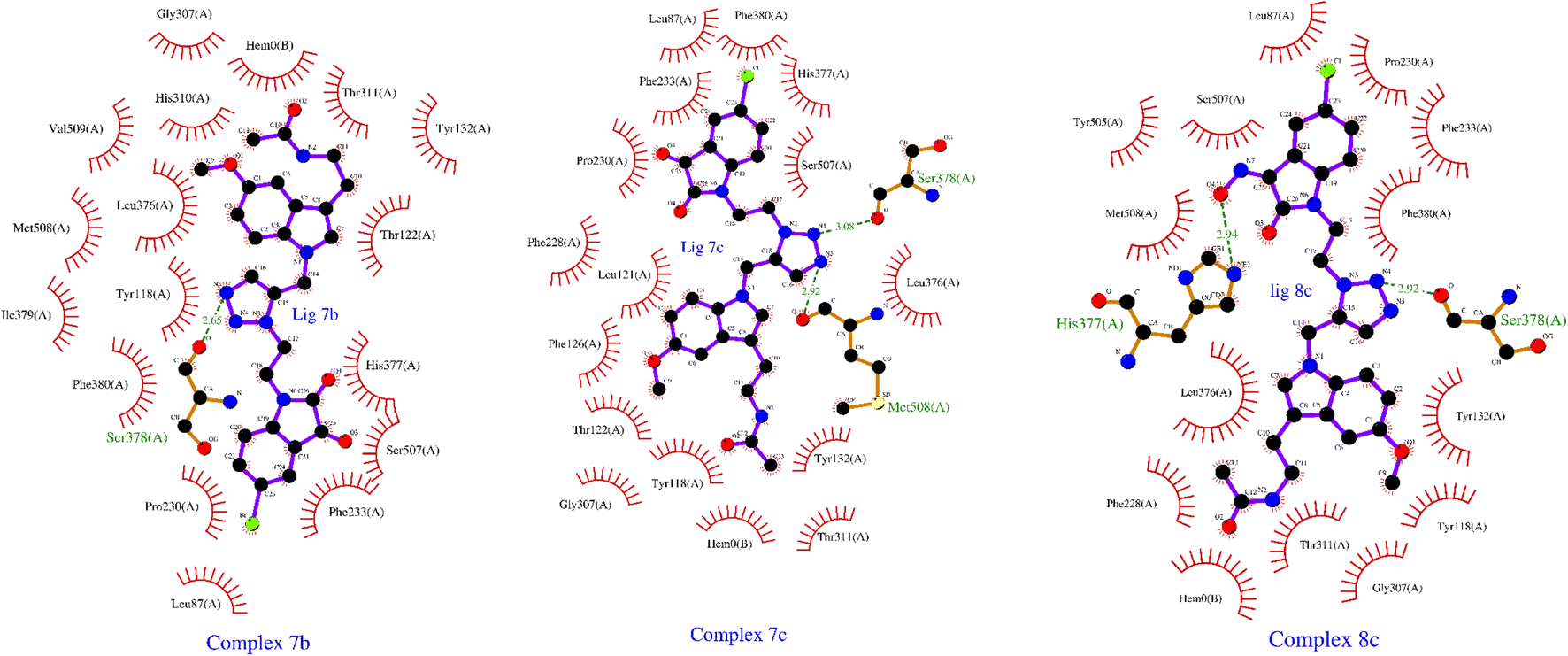 | ||
| Fig. 4 Detailed depiction of molecular interaction of the ligand molecules (7b, 7c, and 8c) having highest binding energy, with various amino acid residue in the binding pocket of the protein Lanosterol 14 alpha-Demethylases of prominent fungal pathogens Candida albicans. | ||
 | ||
| Fig. 5 The docking score for the synthesised molecules i.e., 6, 7a–d, 8a–d and commercially available fluconazole drug with reference to their binding energy. | ||
Conclusion
In conclusion, a targeted library of structurally varied substituted 1,2,3-triazoles comprising melatonin and isatin cores was mapped and effectively synthesised by Cu(I)-sodium ascorbate catalysed click reaction. A broad range of 1,2,3-triazoles has been synthesised in excellent yield and characterised by 1H NMR and 13C NMR, FT-IR, and HRMS analytical techniques. Following the synthesis of the targeted hybrid compounds, molecular docking assesses with the Lanosterol 14 alpha-Demethylases have been performed to validate the binding ability of synthesised compounds to inhibit Lanosterol 14 alpha-Demethylases active sites. Among all docked molecules, the oxime substituted isatin derivative molecules 8c followed by 7b and 7c compounds significantly block the active site of targeted enzyme. The concept of combining physiologically active moieties in a single hybrid molecule prove to be a workable strategy to design novel strategic drug molecules for facilitating for design and synthesis of potential antifungal therapeutic candidates. These results are important guideline for further exploration and testing for potent antifungal agents.Conflicts of interest
The authors affirm that there are no conflicts of interest in this article's content.Acknowledgements
KKS is grateful to the Institutions of Eminence (IoE) for providing funds to carry out the present research work. KKS also grateful to the University of Delhi's USICU (University Science Instrumentation Centre), IIT Delhi for providing the required instrumentation facilities and access to the Supercomputing Facility for Bioinformatics and Computational Biology Center, IIT, Delhi for carrying out electronic structure calculations on the studied molecules. Authors also gratefully acknowledge CSIR for financial support for this work.References
- B. Eftekhari-Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958–3043 CrossRef CAS PubMed.
- H. Mizoguchi, H. Oikawa and H. Oguri, Nat. Chem., 2014, 6, 57–64 CrossRef CAS PubMed.
- H. Liu and A. Dömling, J. Org. Chem., 2009, 74, 6895–6898 CrossRef CAS PubMed.
- A. Kumari and R. K. Singh, Bioorg. Chem., 2019, 89, 103021 CrossRef CAS PubMed.
- N. Ferlazzo, G. Andolina, A. Cannata, M. G. Costanzo, V. Rizzo, M. Currò, R. Ientile and D. Caccamo, Antioxidants, 2020, 9, 1–29 CrossRef PubMed.
- D. Acuña-Castroviejo, G. Escames, C. Venegas, M. E. Díaz-Casado, E. Lima-Cabello, L. C. López, S. Rosales-Corral, D. X. Tan and R. J. Reiter, Cell. Mol. Life Sci., 2014, 71, 2997–3025 CrossRef PubMed.
- M. S. Estevão, L. C. Carvalho, D. Ribeiro, D. Couto, M. Freitas, A. Gomes, L. M. Ferreira, E. Fernandes and M. M. B. Marques, Eur. J. Med. Chem., 2010, 45, 4869–4878 CrossRef PubMed.
- Z. A. Velkov, Y. Z. Velkov, B. T. Galunska, D. N. Paskalev and A. V. Tadjer, Eur. J. Med. Chem., 2009, 44, 2834–2839 CrossRef CAS PubMed.
- A. Lochner, E. Marais and B. Huisamen, J. Pineal Res., 2018, 65, 1–22 CrossRef PubMed.
- H. Sun, A. M. Gusdon and S. Qu, Curr. Opin. Lipidol., 2016, 27, 408–413 CrossRef CAS PubMed.
- X. Chen, C. Sun, P. Laborda, Y. He, Y. Zhao, C. Li and F. Liu, Plant Pathol., 2019, 68, 288–296 CrossRef CAS.
- X. Chen, C. Sun, P. Laborda, Y. Zhao, I. Palmer, Z. Q. Fu, J. Qiu and F. Liu, Front. Microbiol., 2018, 9, 1–14 CrossRef PubMed.
- D. Alonso-Alconada, A. Álvarez, O. Arteaga, A. Martínez-Ibargüen and E. Hilario, Int. J. Mol. Sci., 2013, 14, 9379–9395 CrossRef PubMed.
- B. S. Alghamdi, J. Neurosci. Res., 2018, 96, 1136–1149 CrossRef CAS PubMed.
- H. W. Lin and E. J. Lee, Neuropsychiatr. Dis. Treat., 2009, 5, 157–162 CrossRef CAS PubMed.
- S. A. zeved de Zanette, R. Vercelino, G. Laste, J. R. ipol. Rozisky, A. Schwertner, C. B. uzzatt. Machado, F. Xavier, I. C. ristin, C. de Souza, A. Deitos, I. L. S. Torres and W. Caumo, BMC Pharmacol. Toxicol., 2014, 15, 40 CrossRef PubMed.
- G. Di Bella, F. Mascia, L. Gualano and L. Di Bella, Int. J. Mol. Sci., 2013, 14, 2410–2430 CrossRef CAS PubMed.
- L. Xu, W. Zhang, M. Kwak, L. J. Zhang, P. C. W. Lee and J. O. Jin, Front. Immunol., 2019, 10, 1–11 CrossRef PubMed.
- F. He, X. Wu, Q. Zhang, Y. Li, Y. Ye, P. Li, S. Chen, Y. Peng, R. Hardeland and Y. Xia, Front. Immunol., 2021, 12, 1–15 Search PubMed.
- F. Ye, J. He, X. Wu, J. Xie, H. Chen, X. Tang, Z. Lai, R. Huang and J. Huang, Biomed. Pharmacother., 2019, 117, 109141 CrossRef CAS PubMed.
- G. Tosini, K. Baba, C. K. Hwang and P. M. Iuvone, Exp. Eye Res., 2012, 103, 82–89 CrossRef CAS PubMed.
- D. S. Ferreira, F. G. Amaral, C. C. Mesquita, A. P. L. Barbosa, C. Lellis-Santos, A. O. Turati, L. R. Santos, C. S. Sollon, P. R. Gomes, J. A. Faria, J. Cipolla-Neto, S. Bordin and G. F. Anhê, PLoS One, 2012, 7(6), e38795 CrossRef CAS PubMed.
- P. Brandão, C. Marques, A. J. Burke and M. Pineiro, Eur. J. Med. Chem., 2021, 5(211), 113102 CrossRef PubMed.
- C. Liang, J. Xia, D. Lei, X. Li, Q. Yao and J. Gao, Eur. J. Med. Chem., 2014, 74, 742–750 CrossRef CAS PubMed.
- Z. Ding, M. Zhou and C. Zeng, Arch. Pharm., 2020, 353, 1–13 CrossRef PubMed.
- S. Kumar, S. T. Saha, L. Gu, G. Palma, S. Perumal, A. Singh-Pillay, P. Singh, A. Anand, M. Kaur and V. Kumar, ACS Omega, 2018, 3, 12106–12113 CrossRef CAS PubMed.
- C. Wu, C. Du, J. Gubbens, Y. H. Choi and G. P. Van Wezel, J. Nat. Prod., 2015, 78, 2355–2363 CrossRef CAS PubMed.
- A. Medvedev, A. Kopylov, O. Buneeva, L. Kurbatov, O. Tikhonova, A. Ivanov and V. Zgoda, Int. J. Mol. Sci., 2020, 21, 1–23 Search PubMed.
- H. Guo, Eur. J. Med. Chem., 2019, 164, 678–688 CrossRef CAS PubMed.
- M. Solangi, Kanwal, K. M. Khan, S. Chigurupati, F. Saleem, U. Qureshi, Z. Ul-Haq, A. Jabeen, S. G. Felemban, F. Zafar, S. Perveen, M. Taha and S. Bhatia, Arch. Pharm., 2022, 355, 1–15 CrossRef PubMed.
- H. M. Osman, T. Elsaman, B. A. Yousef, E. Elhadi, A. A. E. Ahmed, E. M. Eltayib, M. S. Mohamed and M. A. Mohamed, J. Chem., 2012, 2021, 1–11 Search PubMed.
- A. Mesripour, E. Jafari, M. R. Hajibeiki and F. Hassanzadeh, Iran. J. Basic Med. Sci., 2023, 26, 438–444 Search PubMed.
- S. M. Davis and T. J. Eckroat, Med. Chem. Res., 2021, 30, 2289–2300 CrossRef CAS.
- M. Valdés-Tovar, R. Estrada-Reyes, H. Solís-Chagoyán, J. Argueta, A. M. Dorantes-Barrón, D. Quero-Chávez, R. Cruz-Garduño, M. G. Cercós, C. Trueta, J. Oikawa-Sala, M. L. Dubocovich and G. Benítez-King, Br. J. Pharmacol., 2018, 175, 3200–3208 CrossRef PubMed.
- R. K. Upadhyay, K. K. Saini, N. Deswal, T. Singh, K. P. Tripathi, P. Kaushik, N. A. Shakil, A. C. Bharti and R. Kumar, RSC Adv., 2022, 12, 24412–24426 RSC.
- V. K. R. Tangadanchu, Y. F. Sui and C. H. Zhou, Bioorg. Med. Chem. Lett., 2021, 41, 128030 CrossRef CAS PubMed.
- X. M. Chu, C. Wang, W. L. Wang, L. L. Liang, W. Liu, K. K. Gong and K. L. Sun, Eur. J. Med. Chem., 2019, 166, 206–223 CrossRef CAS PubMed.
- S. Zhang, Z. Xu, C. Gao, Q. C. Ren, L. Chang, Z. S. Lv and L. S. Feng, Eur. J. Med. Chem., 2017, 138, 501–513 CrossRef CAS PubMed.
- S. Sathish Kumar and H. P. Kavitha, Mini-Rev. Org. Chem., 2013, 10, 40–65 CrossRef.
- G. I. Lepesheva, R. D. Ott, T. Y. Hargrove, Y. Y. Kleshchenko, I. Schuster, W. D. Nes, G. C. Hill, F. Villalta and M. R. Waterman, Chem. Biol., 2007, 14, 1283–1293 CrossRef CAS PubMed.
- Y. Wang, R. Ding, Z. Tai, H. Hou, F. Gao and X. Sun, Arabian J. Chem., 2022, 15, 103639 CrossRef CAS.
- H. Singh, J. V. Singh, M. K. Gupta, A. K. Saxena, S. Sharma, K. Nepali and P. M. S. Bedi, Bioorg. Med. Chem. Lett., 2017, 27, 3974–3979 CrossRef CAS PubMed.
- S. K. Kancharla, S. Birudaraju, A. Pal, L. Krishnakanth Reddy, E. R. Reddy, S. K. Vagolu, D. Sriram, K. B. Bonige and R. B. Korupolu, New J. Chem., 2022, 46, 2863–2874 RSC.
- Z. Wang and H. Qin, Chem. Commun., 2003, 2, 2450–2451 RSC.
- P. M. Chaudhary, S. R. Chavan, F. Shirazi, M. Razdan, P. Nimkar, S. P. Maybhate, A. P. Likhite, R. Gonnade, B. G. Hazara, M. V. Deshpande and S. R. Deshpande, Bioorg. Med. Chem., 2009, 17, 2433–2440 CrossRef CAS PubMed.
- A. Singh, K. Kaur, H. Kaur, P. Mohana, S. Arora, N. Bedi, R. Chadha and P. M. S. Bedi, J. Mol. Struct., 2023, 1274, 134456 CrossRef CAS.
- M. H. Shaikh, D. D. Subhedar, V. M. Khedkar, P. C. Jha, F. A. K. Khan, J. N. Sangshetti and B. B. Shingate, Chin. Chem. Lett., 2016, 27, 1058–1063 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05531k |
| This journal is © The Royal Society of Chemistry 2023 |