
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic dehydration of glycerol over Cu–Fe–Al-based oxides: understanding changes in active sites throughout the reaction†
Felipe Fernandes Barbosaa,
João Edson Tavares b,
Anderson dos Reis Albuquerqueb,
Marco Antonio Morales Torresc,
Enrique Rodríguez-Castellónd,
Sibele B. C. Perghera and
Tiago Pinheiro Braga*a
b,
Anderson dos Reis Albuquerqueb,
Marco Antonio Morales Torresc,
Enrique Rodríguez-Castellónd,
Sibele B. C. Perghera and
Tiago Pinheiro Braga*a
aLaboratório de Peneiras Moleculares, Instituto de Química, Universidade Federal do Rio Grande do Norte, 59078-970, Natal, RN, Brazil. E-mail: tiago.braga@ufrn.br; Tel: +55-84-3342-2323
bInstituto de Química, Universidade Federal do Rio Grande do Norte, 59078-970, Natal, RN, Brazil
cDepartamento de Física, Universidade Federal do Rio Grande do Norte, 59078-970, Natal, RN, Brazil
dDepartamento de Química Inorgánica, Cristalografía y Mineralogía, Facultad de Ciencias, Universidad de Málaga, 29071, Málaga, Spain
First published on 24th October 2023
Abstract
The glycerol conversion into acetol using Fe, Al and Cu-based oxides was investigated. XRD results indicate the formation of nanosized particles with high phase dispersion, however, Raman, Mössbauer, 27Al NMR and XPS spectroscopies suggest the presence of iron(III) oxide, Al2O3 and CuO phases. The FTIR with pyridine adsorption revealed high Lewis acidity. The TPR profile showed the reduction temperature range for the Fe3+ and Cu2+ sites, indicating the suitable condition for pretreatment. The N2 adsorption–desorption isotherms indicated the presence of micro–mesopores with interesting textural properties and specific area varying between 71 and 220 m2 g−1, while the porous morphology was observed by SEM and TEM images. The optimized catalytic tests showed glycerol conversion of 60% and acetol selectivity of 92% with 17% of coke according to TG profile. The recycling tests confirmed the efficiency of the solid, reaching 28% conversion and 91% acetol selectivity after four reuses and, after reactivation in an oxidizing atmosphere, the catalytic performance obtained results close to the second reuse. The interaction between the different Lewis acid sites involved in the mechanisms for the acetol and coke formation on the catalyst surface is discussed. The charge distribution represented by colors which indicates the acid–base surface was evaluated by a simple theoretical–computational study based on the DFT approach. The synergy between the active sites indicates that the presence of Cu0/Cu+ drastically increases the acetol selectivity which is a more important characteristic than the high Lewis acidity of Fen+ and Al3+.
1 Introduction
Biodiesel is already a consolidated biofuel since the beginning of the 21st century. Despite the advantages compared to fossil fuels, mainly because it has renewable raw materials (oils and fats), the transesterification reaction for biodiesel production still has some challenges such as the generation of the by-product glycerol.1 Currently, about 10% of glycerol is generated as a by-product of produced biodiesel.2 Therefore, it is very importance to find new applications for refined and crude glycerol, since it has been traditionally used as animal feed or low-grade burning fuel, limiting the commercial value and compromising the sustainability of biodiesel process.3 On the other hand, the glycerol molecule is quite versatile and reactive, and can be reused in several reaction pathways using appropriate catalysts,4 adding value to the by-product of the transesterification reaction and generating new markets for the biodiesel industry.Thus, the main reaction pathways for the glycerol conversion are reforming,5 hydrogenolysis,6 dehydration,7 cyclization,8 oligomerization,9 hydrogenation,10 acetylation,11 carboxylation,12 selective oxidation13 in the presence of specific catalysts.14 Regarding the products generated, acrolein, 1,2-propanediol, synthesis gas (syngas), propene, polyglycerol, glycerol carbonate, glyceraldehyde and acetol stand out.15 Particularly, acetol (hydroxyacetone) is a valuable and important chemical intermediate in the production of propylene glycol, propionaldehyde, acetaldehyde and furan derivatives.14 The food, cosmetics, textile and disposable industries use acetol more frequently in the manufacture of their products.16 In addition, the acetol production obtained by glycerol conversion from biodiesel industry using heterogeneous catalysis is gaining prominence due to the less expensive operation of the process compared to the traditional petrochemical process.17
It is well known that glycerol reaction processes involving heterogeneous catalysis are more advantageous compared to homogeneous processes and the main concern is the easy separation of the catalyst from the reaction medium, favoring its recyclability.14 However, for the reaction in the presence of heterogeneous catalysis to have high performance, it is necessary that the solid had specific characteristics such as adequate acidity, porosity-accessibility, high dispersion of active sites, strong metal–support interaction (SMSI) regarding resistance to sintering and high tolerance to coke deposition.18
In this sense, the catalyst must have an extended lifetime related to resistance to coke formation and sintering over long reaction times.19 Although the carbon deposits on the active sites is inevitable, the possibility of regenerating a catalyst is also an advantage. However, the discussion about the mechanisms that involve the coke formation and the consequent deactivation are poor explored in the literature and needs additional studies for the design of more resistant catalysts.20 In addition, the interactions of the molecules present in the reaction with the metallic active sites may be the key point for understanding the triggering of coke formation. It is reported the occurrence of soft and hard coke in glycerol conversion,21 where the soft coke comprises the formation of oxygenated molecules, intermediates which adsorb on the surface, while the hard coke consists of polyglycols with greater complexity.22
This work describes the activity of solids containing Cu–Fe–Al in the glycerol conversion. Initially, the synthesis of different catalysts was investigated, followed by characterization to evaluate structural, redox, textural, acidic and morphological properties. Catalytic, recycle and reactivation tests were used to confirm the stability of the synthesized solids. Furthermore, the synergistic and mechanistic interactions on the surface chemistry of the different phases present in the material during the glycerol dehydration reaction to acetol and the catalytic deactivation by coke formation and sintering, based on experimental and theoretical results (electrostatic potential maps), are discussed.
2 Experimental
2.1 Synthesis of Cu, Fe and Al-based oxides
The synthesis route used is based on the polymeric precursor method according to the flowchart presented in Scheme S1.† Aluminum nitrate nonahydrate (Al(NO3)3·9H2O), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O), citric acid monohydrate (AC) (C6H8O7·H2O) and ethylene glycol (EG) (C2H6O2) were used as a starting chemical reagents. An AC/metal ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (mol) was maintained for all samples. The amount of metal is the sum of Fe and Al. The AC/EG molar ratio was 2:3. Initially, Fe and Al precursors were dissolved in water and mixed with citric acid for 60 min at 60 °C for the initial step of metal complexation. Then, ethylene glycol was added to the AC–metals mixture at 100–120 °C for 2 h until the formation of the polymeric resin through the polyesterification reaction. The formed resin was pre-calcined at 250 °C for 2 h in air flow and calcined at 500 °C for 2 h under air atmosphere, thus, obtaining the catalyst containing Fe and Al. The two prepared solids were designated as Fe10Al90 (10% of Fe and 90% of Al) and Fe20Al80 (20% of Fe and 80% of Al). According to the synthesis stoichiometry, it is expected to obtain iron(III) oxide (Fe2O3) and aluminum oxide (Al2O3).
:1 (mol) was maintained for all samples. The amount of metal is the sum of Fe and Al. The AC/EG molar ratio was 2:3. Initially, Fe and Al precursors were dissolved in water and mixed with citric acid for 60 min at 60 °C for the initial step of metal complexation. Then, ethylene glycol was added to the AC–metals mixture at 100–120 °C for 2 h until the formation of the polymeric resin through the polyesterification reaction. The formed resin was pre-calcined at 250 °C for 2 h in air flow and calcined at 500 °C for 2 h under air atmosphere, thus, obtaining the catalyst containing Fe and Al. The two prepared solids were designated as Fe10Al90 (10% of Fe and 90% of Al) and Fe20Al80 (20% of Fe and 80% of Al). According to the synthesis stoichiometry, it is expected to obtain iron(III) oxide (Fe2O3) and aluminum oxide (Al2O3).
Copper(II) oxide was prepared by the incipient impregnation method of the two samples described in the previous step. The solution containing copper(II) nitrate trihydrate (Cu(NO3)2·3H2O) in ethanol was dropwise added to the Fe10Al90 and Fe20Al80 materials. Subsequently, the obtained solids were placed in an oven at 60 °C during 1 h for drying. Finally, the materials were calcined at 500 °C for 2 h under air atmosphere, obtaining the samples named as Fe10Al90Cu and Fe20Al80Cu. It is desired to obtain copper oxide (CuO) containing 5% by mass according to the stoichiometry used. In this synthesis method, the copper phase is randomly dispersed in the Fe–Al matrix.
2.2 Characterizations of catalysts
Structural properties were evaluated by X-ray diffraction. The equipment used was a Bruker D2 Phaser diffractometer using CuKα radiation (λ = 1.54 Å) with Ni filter, 0.02° step, 10 mA current and 30 kV voltage using a Lynxeye detector. Phase identification was carried out using X-Pert HighScore Panalytical software and the JCPDS-ICDD 2003 database.23 Rietveld refinement was performed using the GSAS software24 and the EXPGUI interface.25 The peaks was used to calculate the crystallite size using the Scherrer equation.26 Furthermore, the crystallite size was also calculated using the size–strain plot (SSP) method.27 This method is more suitable for samples with a size of up to 20 nm and that have a negative slope when solving the Williamsom–Hall equation.28The composition of Fe, Al and Cu was determined by the inductively coupled plasma optical emission spectroscopy technique (ICP-OES). The analysis was performed using the ICAP 6300 Duo equipment (Thermo Fisher Scientific), with axial and radial incidence, equipped with a CID detector (Charge Injection Device) simultaneously. The gas used was Argon (99.996% purity) with the following parameters: RF power supply of 1150 W, nebulizer gas flow rate of 0.75 L min−1, auxiliary gas rate of 0.5 L min−1 and stabilization time of 15 s.
Raman spectroscopy was used to identify the chemical environment of the oxide phases present in the catalysts. The analyzes were carried out at room temperature with the confocal Raman microscope equipment, model LabRAM HR Evolution from the manufacturer HORIBA Scientific. The parameters used were: 532 nm Ar+ laser (100 mW), laser intensity of 10%, acquisition time of 40 s and accumulations 10.
To analyze the chemical structure of Al-containing catalysts, 27Al-NMR spectroscopy was performed. The equipment used was the model Avance III HD NMR Spect 300 from the manufacturer Bruker with a superconducting magnetic system with a 5.4 cm hole, operating field at 7.0463 Tesla, CP/MAS Probehead, BB, 15N-31P + 1H and rotor size of 4 mm.
Mössbauer Spectroscopy analyzes were performed to evaluate the chemical environment of Fe before and after the reaction by ex situ experiments. 57Fe Mössbauer analyzes were carried out at 300 K in the transmission mode using a spectrometer from SEECO with triangular velocity sweep. The γ-radiation with energy 14.4 keV was provided by a radioactive source of 57Co in Rh matrix with an activity of 25 mCi. Isomer shifts are reported relative to α-Fe at 300 K. Spectra were fitted using Normos90 software.
XPS analyzes were used to study the surface chemical composition and the chemical state of the elements on the solid surface. XPS analysis was performed with a Physical Electronics PHI 5700 spectrometer (Physical Electronic, Minneapolis, MN, USA). Non-monochromatic Al Kα radiation and a multichannel detector were used. Multipack software version 9.6.0.15 was used to analyze the formed peaks.
Temperature programmed-reduction analysis was applied to investigate the redox properties and verify the best pretreatment conditions. Initially, the pre-treatment step took place at a temperature of 350 °C with a heating rate of 10 °C min−1 and N2(g) flow rate of 30 mL min−1 for 1 h. Then, the analysis was recorded in the range of 50–700 °C, where material (60 mg) was disposed in a quartz reactor under a flow rate of 20 mL min−1 using a gaseous mixture of 8% H2/N2 with a heating rate of 10 °C min−1. Hydrogen consumption was monitored by a thermal conductivity detector (TCD).
The N2 adsorption–desorption isotherms at −196 °C were used to investigate the textural parameters, obtaining the specific surface area, volume and pore size values. The analyzes were carried out in a Micromeritics adsorption analyzer model ASAP 2020. The samples were previously degassed at 200 °C for 2 h to remove adsorbed impurities. The specific surface area (SBET) were obtained by the Brunauer, Emmett and Teller (BET) method.29 Based on the adsorption data, the type of isotherm was indicated, taking into account the IUPAC recommendations.30 The total pore volume (Vp) was obtained by Gurvich rule at a relative pressure of 0.98.31 The mesopores diameter (Dp) was obtained by the Barrett–Joyner–Halenda (BJH) model.32
Morphological characteristics were visualized by images obtained by scanning electron microscopy (SEM). The equipment used was the Zeiss Auriga 40, with an energy dispersive X-ray spectrometer (EDS). The accelerating voltage was 5 kV and different magnifications (30.00 to 80.00 KX) were applied. Morphological properties were also obtained through images obtained by transmission electron microscopy (TEM). The TEM technique also allows the identification of exposed planes via selected area electron diffraction (SAED), complementing the XRD and spectroscopy results. The TEM is JEOL model JEM-2100 (Tokyo, Japan) equipped with EDS from Thermo scientific, (Waltham, MA, USA) was used. The solids were prepared by placing a drop of the ketone-solid dispersion on a carbon-coated copper support (300 mesh).
Infrared technique was used with pyridine adsorption as a probe molecule to evaluate the surface acidity and identify the presence of Lewis or Brønsted sites. A Nicolet 5700 equipment with an optical resolution of 2 cm−1 was used. The sample was previously pressed (1 ton) in a self-supporting pellet (2 cm2) and pre-treated from 20 to 450 °C under dry air flow rate of 100 mL min−1. Afterwards, the sample was degassed (10−5 bar) for 1 h at 200 °C under vacuum. For pyridine adsorption, the probe was adsorbed at 150 °C, around 2 mBar for 5 min. The sample was degassed under vacuum for 1 h to remove the physissorbed pyridine. Then, an infrared spectrum was recorded. For the acidity estimation, the Beer–Lambert–Bouguer rule33 was considered, where the areas under the peaks were used to quantify the Lewis acid sites around 1450 cm−1 according to eqn (1).
 | (1) |
2.3 Computational procedure
For the computational simulation present in the Fe–Al–Cu material, the following crystallographic planes were considered: (001) Al2O3, (001) Fe2O3, (111) FeO, (001) and (111) Cu2O and (001) CuO. The computational model adopted for the surface construction was the slabs model in a three-dimensional orthorhombic box under periodic boundary conditions, which consists of orthogonally sectioning the crystal bulk oriented along the vector [hkl] normal to the plane (hkl). The atomic terminations on the slab faces and their thickness are adjusted and, in all cases, an empty region of 15 Å is created at the interface with the slabs along the z axis, taking into account that the upper and lower faces are not influenced by their images under periodic boundary conditions. Thus, the surfaces can be considered as two-dimensional. The crystallographic planes chosen for the preparation of the slabs were preferably symmetrical and with the largest interplanar distances (dhkl), following the Bravais–Friedel–Donnay–Harker (BFDH) law, which establishes that, due to the symmetry of the spatial groups, the crystal morphology more important will have the largest interplanar spacings.35 In addition, the surfaces were modeled with low oxygen saturation in the outermost layers in order to simulate the reducing conditions of the experimental results.
2.4 Catalytic test for glycerol conversion
The catalytic tests were carried out using a fixed bed reactor in gas-phase at atmospheric pressure. In the first test, the best pre-treatment condition (H2/350 °C) was used for 30 min. Then, the reaction took place for 4 h using 30 mL min−1 of N2 as carrier gas, glycerol flow rate of 3.6 mL h−1 (10% m V−1) and 200 mg of catalyst. The catalytic performance was monitored by gas chromatography and a fraction of 2 μL was injected into the gas chromatograph from PerkinElmer, model Clarus 680, FID detector, with an intermediate polar column of fused silica (30 m × 0.25 mm × 0.25 μm), filling from the poly column (5% diphenyl/95% dimethyl siloxane). The injector temperature was 220 °C in splitless injection mode, with column flow of 2 mL min−1 for carrier gas N2. Initially the column temperature was 30 °C increasing to 70 °C (rate of 8 °C min−1), then heated to 220 °C (rate of 30 °C min−1). Glycerol conversion (Cglycerol) and product selectivity (Sacetol) were calculated according to eqn (2) and (3), respectively. The quantification of the products was obtained by the internal standard method and n-butanol was used as internal standard.
 | (2) |
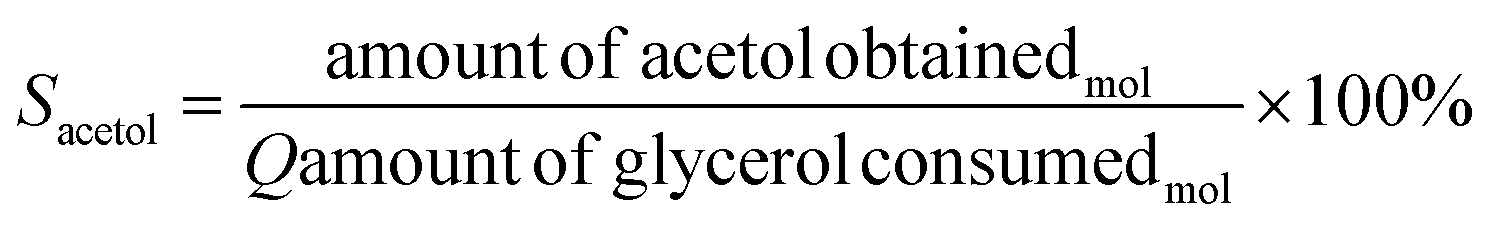 | (3) |
In addition, a mass detector chromatograph (GC-MS) was used to identify the main reaction products. The GC-MS used is a GC-2010 Plus model with a QP 2020 mass spectrometer from Shimadzu, equipped with a polar column (30 m × 0.25 mm × 0.25 μm) and automatic injection model AOC-20i. The library present in the software for composition identification is NIST (National Institute of Standards and Technology).
3 Results and discussion
3.1 Structural properties and chemical environment of fresh metal oxides (XRD, Raman, 27Al-NRM, Mössbauer and XPS analysis)
Initially, it is worth mentioning that the elemental composition was determined by the ICP-OES, shown in Table S1.† The obtained experimental values are close to nominal values used in the synthesis. The samples were analyzed by X-ray diffraction and the results are shown in Fig. 1a. Although the Fe and Al-based phases are not observed in the diffractograms, it may be the result of highly dispersed phases with very small crystallites. In fact, the XRD technique has limitations when dealing with highly dispersed and extremely small crystallites.41 In addition, depending on the orientation, symmetry, presence of amorphous phase or even overlapping peaks, thus, other techniques to identify the phases are required.42 These features are common for this synthesis methodology as previously described.43 However, after adding copper to the Fe–Al-based sample, reflections are observed referring to copper(II) oxide, CuO, (ICDD 01-089-5898) with peak positions (2θ) at 35° and 38°, and Miller indices (−111) and (111), for the samples Fe10Al90Cu and Fe20Al80Cu, respectively.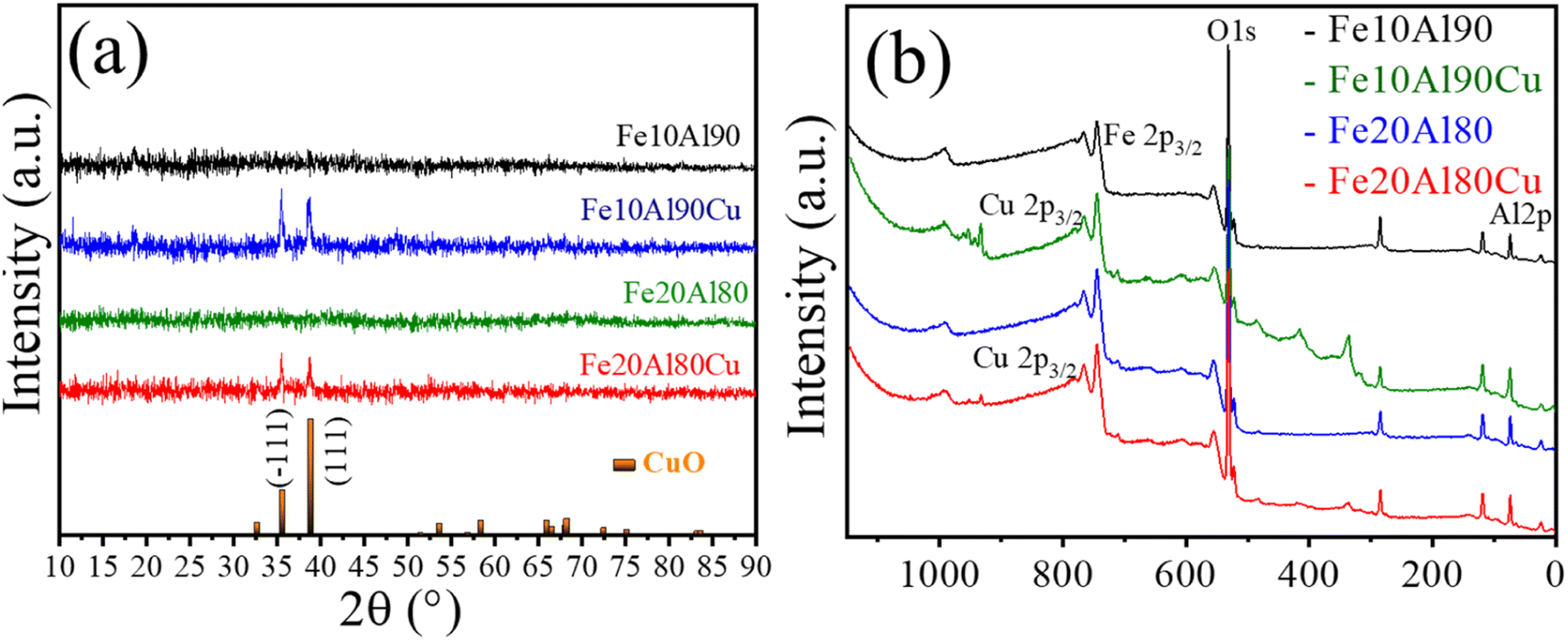 | ||
| Fig. 1 Characterization of fresh catalyst: (a) X-ray diffractograms and (b) wide-scan XPS spectra for Fe10Al90, Fe10Al90Cu, Fe20Al80 and Fe20Al80Cu samples. | ||
Rietveld refinement was performed for the CuO phase identified in Fe10Al90Cu and Fe20Al90Cu catalysts. It was possible to calculate the crystallite size from the Scherrer equation as well as by using Williamsom–Hall (WH) equation in the size strain plot (SSP), the microstrain was also extracted from the SSP. The crystallite size and microstrain values are shown in Table 1. The CuO phase has crystallite size of 24 and 18 nm for the Fe10Al90Cu and Fe20Al80Cu samples, respectively.
| Sample | Average crystallite size (nm) | Microstrain (ε) | |
|---|---|---|---|
| Scherrer | SSP | ||
| Fe10Al90Cu | 24.1 ± 4.2 | 29.54 | 2.04 × 10−3 |
| Fe20Al80Cu | 18.5 ± 3.3 | 22.8 | 2.75 × 10−3 |
The composition of the chemical surface for all the catalysts was analyzed using the X-ray photoelectron spectroscopy technique. Fig. 1b and S1† presents the wide-scan spectra and Table S2† shows the observed binding energies. The assigned species of Al 2p (∼74 eV), O 1s (∼530 eV) and Fe 2p3/2 (∼710 eV) were identified in all spectra. The Cu 2p3/2 (∼933 eV) was identified for the Cu-based catalysts. The O 1s region is associated with the binding energy peak corresponding to the lattice oxygen present in the different Fe2O3 (Fe3+), Al2O3 and CuO oxides.44
The typical Fe 2p3/2 spectrum is related to Fe3+-based phase with binding energy of 710 eV.45 Furthermore, the Cu 2p3/2 signal has energy of 933 eV and is ascribed to Cu2+ in the CuO phase46 as already observed in the diffractograms. Finally, Al 2p spectrum with binding energy of 74 eV, present in Al2O3 oxide,47 corroborating the 27Al-NMR spectra. Thus, it was possible to identify the presence of all oxides present in the surface of the Fe–Al–Cu-based catalysts.
Raman spectroscopy characterization was carried out to show the chemical environment of the formed oxides. The spectra for all samples are shown Fig. S2a and Table S3.† It is possible to suggest the presence of iron and aluminum-based phases in addition to the CuO phase. In this sense, all samples were analyzed by 27Al-NMR spectroscopy to investigate the nature of the Al species, these spectra are shown in Fig. S2b.† The results indicate the presence of the Al2O3 phase with octahedral coordination for the Al species. Detailed description of Raman and 27Al-NMR results are present in the ESI.†
3.2 Redox properties (H2-TPR results)
TPR-H2 analysis was used to study the redox property and the reduction behavior for the different species present in the catalysts. The result is presented in Fig. 2, showing the H2 consumption profile, which exhibits one or three main events depending on the sample. The literature48,49 describes that the peaks in the range of approximately 230 to 380 °C can be attributed to the gradual reduction of CuO, i.e. Cu2+ to Cu+ at low temperature and represented by α1 and α2, and Cu+ to Cu0 at higher temperature and indicated as β. The intensity of the peaks can be modified due to the H2 flow rate and the type of support compared to the standard containing pure copper oxide (unsupported). Fig. 2 shows the peaks α1 and α2 and the Cu reduction step represented by β coincides with the Fe3+ reduction step, which occurs at temperatures between 350 and 550 °C. | ||
| Fig. 2 TPR-H2 profile of Fe10Al90, Fe10Al90Cu, Fe20Al80, Fe20Al80Cu fresh catalysts and pure CuO and Fe2O3 standards. | ||
The reduction of iron(III) oxide is reported in two main events, the first with low intensity in the range of 290–320 °C, which reflects the first reduction stage of Fe3+ → Fe3+/Fe2+, and the second highest temperature peak in the range of 400–550 °C represents the reduction of Fe3+/Fe2+ → Fe0.50 Regarding the phase containing Al2O3, the literature reports that this Al-based oxide is metastable.51
On the other hand, the Fe10Al90Cu sample containing copper(II) oxide exhibits three events, the first two peaks at temperatures between 190 and 330 °C refer to the CuO reduction, represented by α1 and α2, respectively. The β step referring to the reduction to Cu0 is also in the reduction range for the Fe-containing phase. The Fe20Al80 solid exhibits a reduction profile of typical Fe-based materials, in the range between 340 and 580 °C. A very broad peak is also observed, similar to Fe10Al90 sample, indicating high dispersion of the Fe phase on the aluminium support, corroborating the proposition that iron oxide may be compromised in the Al2O3 structure.
Thus, the copper oxide phase produced a synergistic interaction with the other species, shifting the Cu reduction event to slightly lower temperatures when compared to the pure CuO. Similarly, concerning the Fe3+-based species, present in the different catalysts and compared to the pure Fe3+ standard, significant changes occurred in samples containing Cu, probably due to interactions among different species, which can cause significant changes in the catalytic performance.
3.3 Textural properties (N2 physisorption)
N2 adsorption/desorption analysis was performed to investigate textural properties for all solids. The obtained isotherms are present in Fig. S3† and the textural properties are shown in Table S4.† All materials have a similar type-IV isotherm and H3-type hysteresis curve according to the IUPAC classification,30 where the adsorption curve is very sharp and the desorption curve is steeper for Fe10Al90 and Fe10Al90Cu solids, however, the hysteresis loop is less wide for Fe20Al80 and Fe20Al80Cu catalysts.52 This type of hysteresis and isotherm profile are characteristic of mesopores solid, slit-shaped pores and particle aggregates.53 The changes between Fe10Al90 and Fe10Al90Cu solids when compared to Fe20Al80 and Fe20Al80Cu are related to the type of pore and the mechanisms involved in N2 adsorption/desorption process. Furthermore, the regions with low relative pressure values confirms the presence of micropores.54Table S4† presents the textural properties concerning the specific surface area (SBET), total pore volume (Vp), pore diameter (Dp) and micropore volume (Vμp). It is observed that the catalysts based on Fe and Al without copper, Fe10Al90 and Fe20Al80, have similar specific area, however, the Fe20Al80 material presents a relative increase in surface area, probably due to the greater amount of precursor used, considering that when it is degraded during the calcination step leads to better textural properties (larger area and pore size).
On the other hand, the insertion of copper considerably decreases the specific surface area and the pore volume, which is related to the partial filling of the pores after the addition of copper by impregnation. The materials have pore diameter values mostly in the mesopore range as indicated by the BJH method, which can be confirmed by the pore size distribution curve. A slight increase in pore diameter is observed for both samples after adding the Cu-based phase. The pore diameter is larger for the Fe20Al80 and Fe20Al80Cu material, indicating that the greater iron content used must be filling most of the micropores, as observed in the micropore volume, causing an increase in the average pore size and, consequently, a smaller specific surface area for the Fe20Al80Cu catalyst. It is important to mention that the surface area changes observed between the Fe, Al and Cu-based solids are mainly influenced by the calcination temperature and the interaction between the formed phases.
3.4 Morphological properties (SEM and TEM results)
Morphological properties were investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The SEM results are shown in Fig. 3. Sponge-like morphology was observed for all samples, which indicates a solid with high porosity. In addition, several cavities with different sizes were seen, which is an excellent property for the catalyst, avoiding diffusion limitations and greater resistance to pore blockage by coke.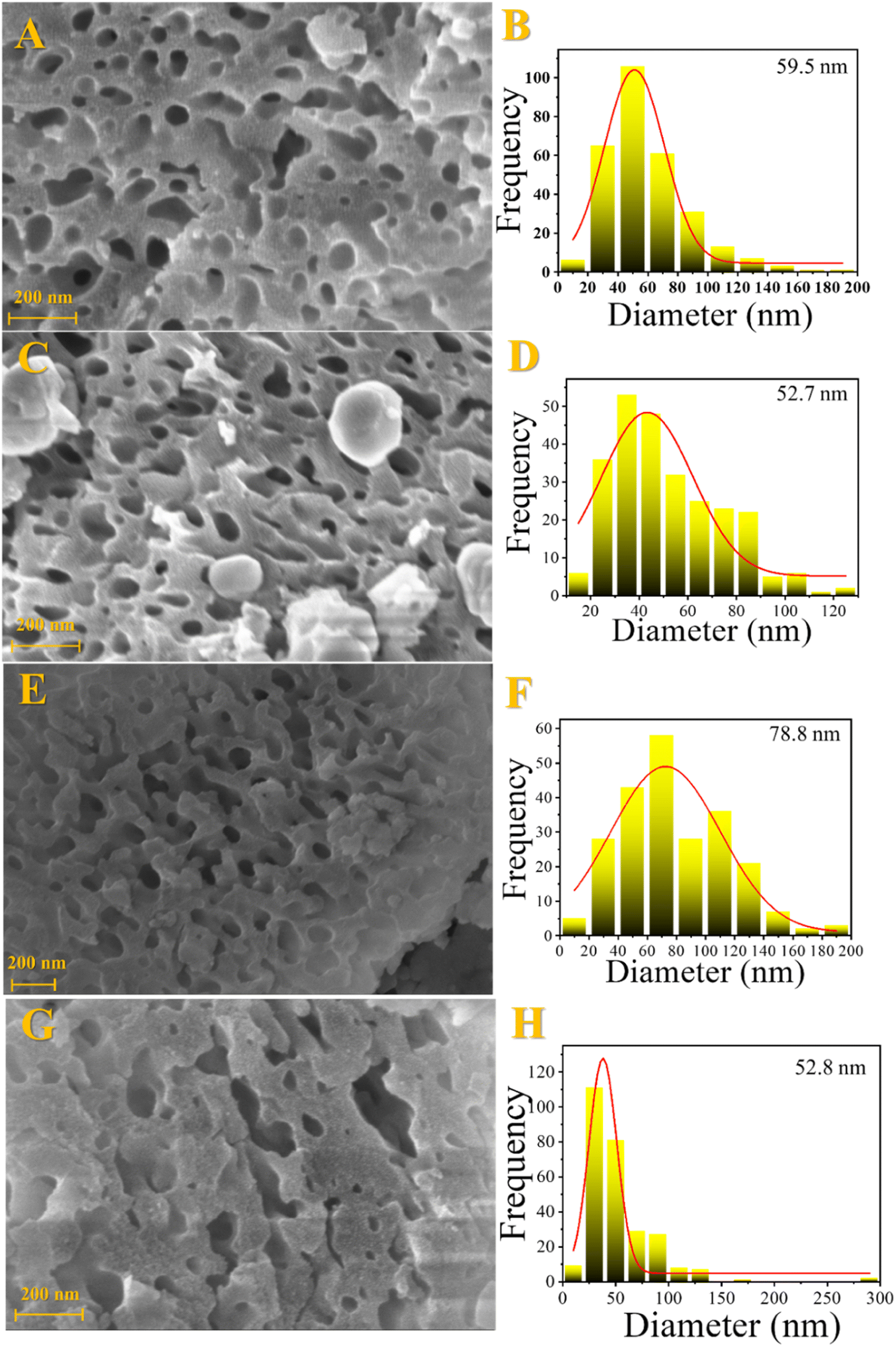 | ||
| Fig. 3 Scanning electron microscopy images and cavity size distribution for the fresh catalysts: Fe10Al90 (A and B), Fe10Al90Cu (C and D), Fe20Al80 (E and F) and Fe20Al80Cu (G and H). | ||
Fig. 4 presents the results obtained by the TEM analysis and their respective SAED images which are identified crystalline planes. Similar to SEM analyses, it is possible to identify a sponge-like morphology typical of a highly porous solid, Fig. 4A.
 | ||
| Fig. 4 Images obtained by transmission electron microscopy of the Fe10Al90Cu sample with pretreatment at 350 °C/H2 (A and B), 300 °C/H2 (C and D) and 350 °C/N2 (E and F). | ||
In addition, the planes of the oxide phases in different pretreatment conditions were identified via SAED and compared with the planes observed by XRD and suggested by spectroscopy. For the samples treated at 350 °C/H2 and 300 °C/H2, the (111) and (200) planes of the Cu0 phase were identified, also the (111) plane for Cu2O which is similar to Cu0 according to the interplanar distance found in the diffractograms. Regarding the condition at 350/N2, the planes (003) and (214) are related to Al2O3, and the planes (111) and (−202) are referred to CuO phase. It is important to highlight that no specific planes were observed for iron-based phases, which suggests that the Fe3+ is highly dispersed and may be embedded within the alumina structure, corroborating with the Raman results.
The vacancies size was calculated from SEM images and the results are shown in Table S5.† The materials have cavities with sizes greater than 50 nm. It is noticed that the process of introducing copper by impregnation followed by recalcination leads to a slight decrease in vacancies size. This fact is due to the sintering process, in addition to the introduction of CuO, which occupies previously unoccupied spaces. It is noticed that the Fe20Al80 catalyst has a larger cavities size than the Fe10Al80, while the Fe10Al90Cu and Fe20Al80Cu samples have similar values. It is worth emphasizing that it was possible to visualize the larger cavities by SEM and TEM images, which was not possible by the N2 physisorption analyses, considering that the N2 isotherms only show the formation of micro–mesopores.
3.5 Acidic properties
Infrared analysis with pyridine adsorption was performed to identify the nature of the acid sites. The results are shown in Fig. 5. It is known that when pyridine is adsorbed on strong Lewis acid sites (LPy) are observed bands in the range of 1445–1460 and 1610–1620 cm−1 related to the vibrations of the ν19b and ν8a ring, respectively. However, when pyridine is adsorbed on Brønsted sites (BPy) are observed stretches at 1540–1500 (ν19b) and 1640–1630 cm−1 (ν8a).55–57 In this work are observed Lewis acid bands at 1450 and 1610 cm−1 and a broad band concerning the Lewis and Brønsted contribution (L + B) at 1550 cm−1. Therefore, it is possible to notice that no well-defined stretches referring exclusively to the Brønsted sites were observed, suggesting that the catalysts contain mostly Lewis sites as expected for Cu–Fe–Al-based oxides.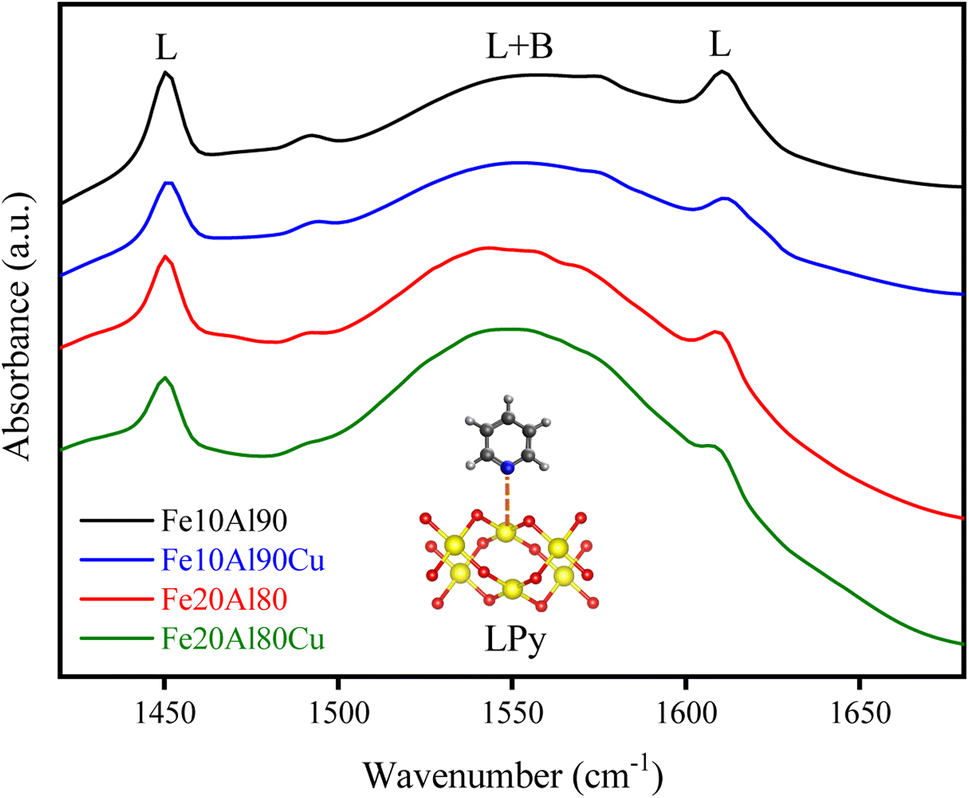 | ||
| Fig. 5 FTIR spectra of pyridine adsorbed at 150 °C for the fresh catalysts. | ||
Table S6† presents the values for the most intense acid sites referring to stretching at 1450 cm−1, obtained through the Beer–Lambert–Bouguer equation. It was observed that the insertion of CuO in the Fe–Al-based solid considerably reduced the acidity. The metallic sites related to Fe2+, Al3+ and Cu2+ have Lewis acid characteristics, which aluminum is in greater quantity, thus, the presence of Cu2+ phase decreases the surface acidity analyzed by pyridine adsorption. The addition of copper by impregnation can cover parts of the Fe3+ and Al3+ Lewis acid sites. This fact can be explained by the characteristic acidity of Al3+ sites, which is higher compared to Cu2+ sites. Furthermore, Al is octahedrally coordinated in the Al2O3 oxide for Fe–Al-based solids, as discussed in the NMR results. However, the addition of the CuO phase can modify this coordination, which was also observed in the 27Al-NMR for the Fe20Al80Cu sample, identifying Al-tetrahedral, which decreases its Lewis acidity.58 The Fe10Al90 material has a higher acidity than the Fe20Al80 solid, and this is due to the fact that the first one has a higher amount of Al than the second one, decreasing its acidity.
3.6 Catalytic performance
Thus, the Fe10Al90Cu material was analyzed by XRD after each pretreatment condition to identify phase variations and investigate the SMSI interaction based on the species formed. The result is shown in Fig. 6. The phases identified were CuO (ICDD 01-089-2530), Cu2O (ICDD 00-034-1354), Cu0 (ICDD 01-085-1326) and Al2O3 (ICDD 00-001-1303). Reflections related to CuO phase were also identified in the 350°/N2 condition, whereas Cu2O and Cu0 phases were observed in the 300 °C/H2 and 350 °C/H2 conditions. The phases observed in the XRD after pre-treatment are corroborating with the electron diffraction results seen in the TEM images (Fig. 4). The condition of 350 °C/H2 is already sufficient to reduce almost completely CuO phase into Cu0, as predicted by the TPR result. Despite the great similarity between the Cu2O and Cu0 patterns, the Cu0 phase is clearly predominant, as indicated by the TPR-H2 profiles. The fresh catalyst has a crystallite size of approximately 24 nm according to the Rietveld refinement using the Scherrer equation, however, after pre-treatment under H2 atmosphere the values decrease to 20 (H2/300 °C) and 19 nm (H2/350 °C) due to the reduction of copper oxide to metallic copper. Regarding the N2/350 °C condition, the material remains with a similar crystallite size of 24 nm, since there is no considerable structural change, only the catalyst activation. It was possible to observe peaks referring to Al2O3 at 37, 45 and 66° 2θ after copper phase reduction for all pretreatment conditions, which are in agreement with the NMR spectra and SAED results. It is important to highlight that alumina is a metastable phase and does not undergo reduction in the studied temperature range.
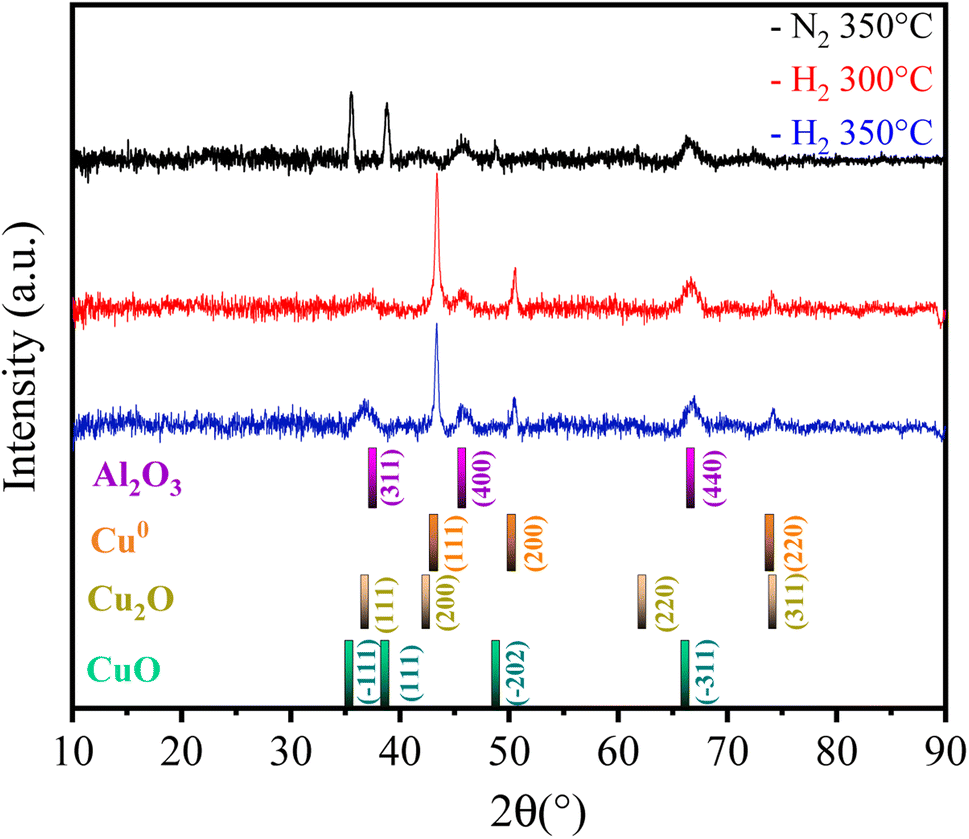 | ||
| Fig. 6 X-ray diffractograms of Fe10Al90Cu catalyst after pre-treatment. | ||
The catalytic tests results under the different pre-treatment conditions studied are shown in Table 2. Moderate to high glycerol conversion and acetol selectivity were obtained for samples treated using H2, while the catalytic performance was lower with pretreatment in presence of N2. The acetol selectivity value was even higher in the 350 °C/H2 condition, thus, this was the pre-treatment condition chosen for all other tests. 1,2-propanediol, propanoic acid and acetic acid were obtained as minor by-products.
| Condition | Conversiona (%) | Selectivitya (%) | |||
|---|---|---|---|---|---|
| Glycerol | Acetic acid | Propanoic acid | 1,2-Propanediol | Acetol | |
| a The error reported was based on the duplicate experiment. | |||||
| 300 °C/H2 | 18 ± 0.8 | 3 ± 0.4 | 18 ± 0.6 | 7 ± 0.8 | 72 ± 0.4 |
| 350 °C/H2 | 60 ± 0.6 | 1 ± 0.8 | 2 ± 0.6 | 4 ± 0.4 | 93 ± 0.7 |
| 350 °C/N2 | 9 ± 0.6 | 5 ± 0.5 | 16 ± 0.8 | 22 ± 0.6 | 57 ± 0.8 |
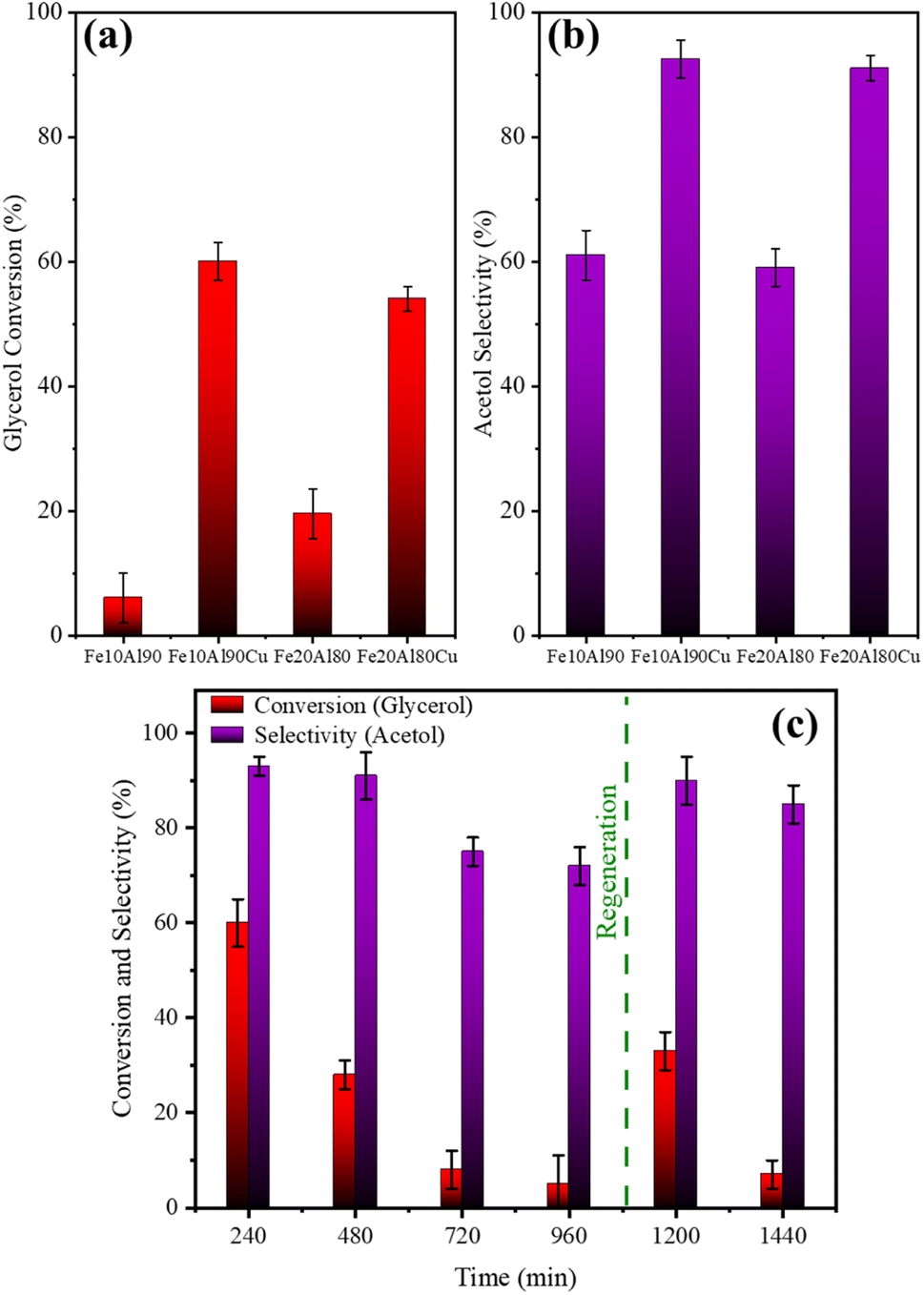 | ||
| Fig. 7 Average (1–4 h) conversion (a) and selectivity (b) for materials in the presence and absence of copper. Reuse and regeneration tests for Fe10Al90Cu catalyst (c). | ||
On the other hand, these results suggest that only the presence of high Lewis acidity from Fe and Al oxides is not enough to obtain high acetol selectivity. The catalysts with Cu active phase (Fe10Al90Cu and Fe20Al80Cu) show higher glycerol conversion and acetol selectivity, confirming the essential role of copper in the reaction pathway and the larger acetol selectivity and glycerol conversion.
The spent catalysts were evaluated by thermogravimetric analysis to obtain the amount of coke deposited through mass loss due to carbon oxidation. Fig. S4† shows the obtained profile. Two main mass loss events are observed for each sample. The first event is in the range from room temperature to approximately 200 °C and is related to physisorbed water. The second event is at a higher temperature (400–800 °C) and is due to the combustion of carbonaceous compounds.61 The burning event of carbonaceous compounds occurs at temperatures larger than 200–600 °C which indicates the presence of soft coke related to short-chain intermediates, while the temperature event above 600 °C is related to the formation of hard coke concerning compounds containing polyaromatics species.62
The DSC thermal analysis results, shown in Fig. S4E,† indicate that the observed events are exothermic, which are related to the carbon burning (coke). The main DTG event occurs at 503 °C for Fe10Al90 and Fe10Al90Cu catalysts, while at 522 °C for sample Fe20Al80 and, finally, at 526 °C for sample Fe20Al80Cu.
Table S8† shows the comparison of the values obtained from the TG result and the mass balance obtained from the mass of each sample after the reaction. Thus, mostly soft-type coke and a greater amount of coke are observed for copper-based catalysts, since they are more active.
Four points referring to the FTIR-py analysis were used to qualitatively observe the dependence between the conversion/selectivity and the strength of the Lewis acid sites, as shown in Fig. 8. An approximate linear inverse correlation is observed between glycerol conversion and Lewis acidity (R2 = 0.9), Fig. 8a. As previously discussed, acidity is related to conversion, thus, for higher values of Lewis acidity, greater glycerol conversion is observed. Fig. 8b similarly presents an inverse weak linear relationship (R2 = 0.7) concerning acetol selectivity. This observation is directly related to the fact that high Lewis acidity alone is not enough to obtain high glycerol conversion and acetol selectivity, confirming the essential role of copper sites.
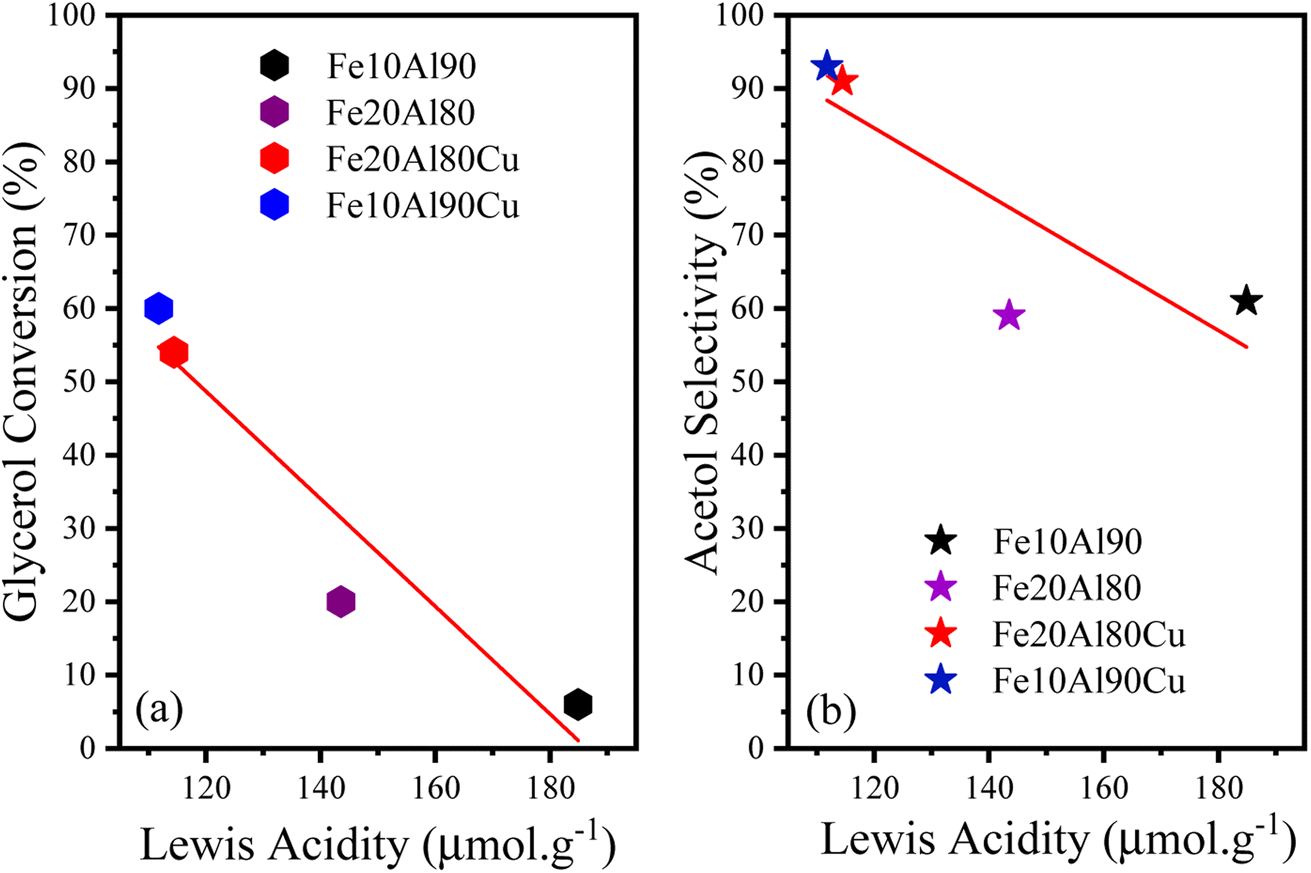 | ||
| Fig. 8 (a) Relationship between conversion and Lewis acidity and (b) acetol selectivity and Lewis acidity for catalysts based on Fe, Al or Fe, Al and Cu. | ||
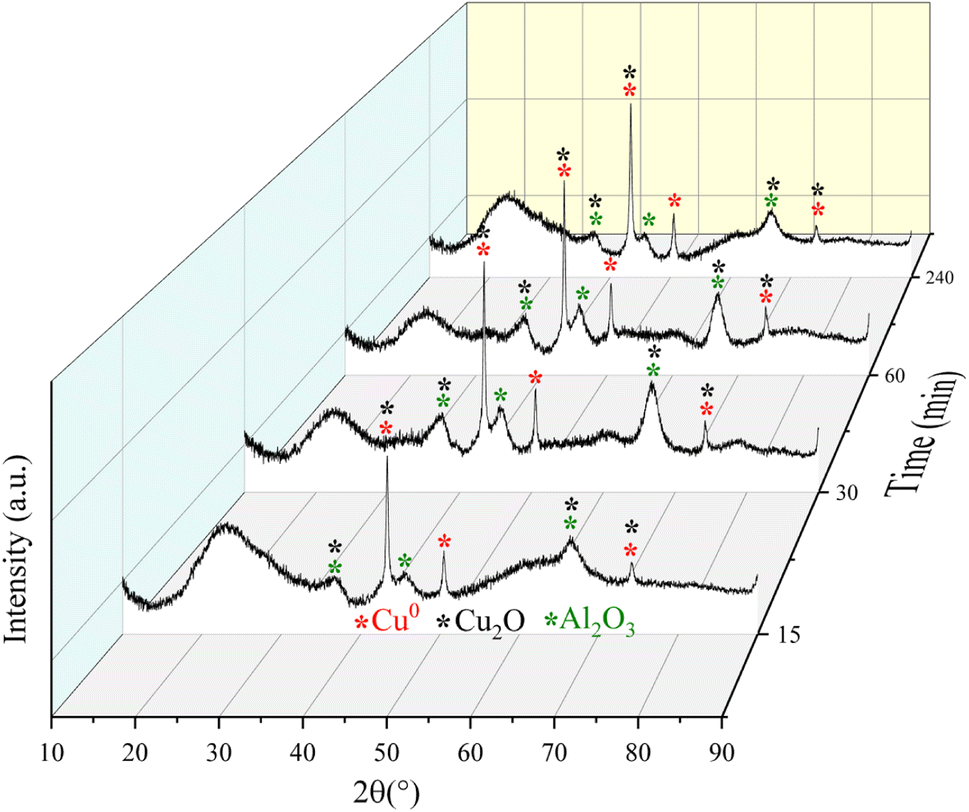 | ||
| Fig. 9 Diffractograms for the Fe10Al90Cu catalyst obtained from the catalytic tests during 15, 30, 60 and 240 min of reaction through ex situ characterization. | ||
More intense peaks were observed for the Cu0 phase throughout the reaction; thus, the crystallite size was calculated specifically from these reflections. The crystallite size of pre-treated sample was 19 nm. After 15 min of reaction, the size changes to 18 nm. However, for the reaction times of 30 and 60 min, the crystallite sizes of both samples changed to 14 nm, and after a reaction of 240 min it increased to 24 nm. The variation in crystallite size between the fourth reaction time and the fresh solid, is related to interactions based on the SMSI effect that is because the complex reaction environment and the drastic reaction conditions, where different reaction pathways occur, including deactivation of the active phases by coke formation and sintering. Despite the variation in crystallite size for the Cu0 species, this phase remained without oxidizing up to 240 min of reaction, confirming the structural stability of the phases although the aggressive reaction atmosphere.
Furthermore, it is worth emphasizing that the presence of Cu+ and Cu0 sites is directly related to synergy and cooperative effects in the catalytic cycle, improving performance in the glycerol conversion.59,63,64 As identified in pre-treatment tests, ex situ characterizations and subsequent application (Table 2), Cu+/Cu0 species are more active than Cu2+.
Mössbauer spectroscopy analysis was carried out for the sample Fe10Al90Cu, to evaluate the chemical environment of iron in the iron-based phases, as shown in Fig. 10A. The spectrum recorded at 300 K, at a maximum velocity of 4.39 mm s−1, shows an asymmetric doublet with isomer shift (IS) and quadrupole splitting (QS) typical of iron(III).65 The spectrum is related to Fe3+ embedded within the alumina structure, as suggested in a previous work on a high-energy-ball-milled (α-Fe2O3)x(Al2O3)1−x system for x ranging from 0.02 to 0.30 and with milling time of 24 h.66 In fact, Cótica et al. found doublets with IS in the range from 0.32 to 0.39 mm s−1 and QS values in the range between 0.70–1.18 mm s−1. According to Cótica et al. the doublet is due to iron substituting aluminum in the alumina matrix.66 In the present work, to fit the spectrum of sample Fe10Al90Cu we considered a distribution of doublets. The obtained average isomer shift and quadrupole splitting are 〈IS〉 = 0.41 mm s−1 and 〈QS〉= 1.17 mm s−1, as shown in Table S9.† The asymmetric doublet may be due to the stress caused in the crystalline structure by the difference of ionic radii between Al3+ = 0.39 Å and Fe3+ = 0.55 Å (low spin) or Fe3+ = 0.645 Å (high spin) in the octahedral coordination.67
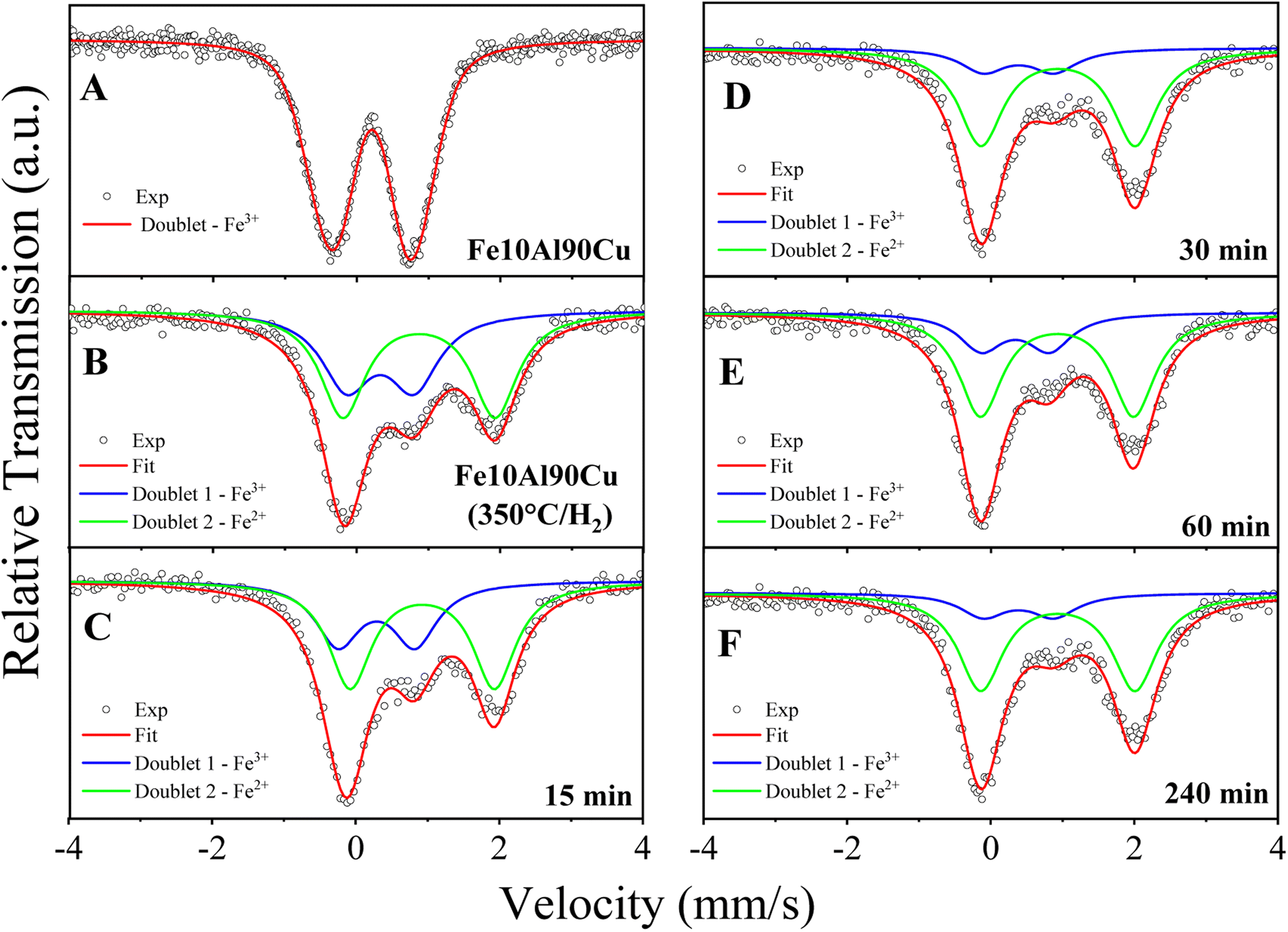 | ||
| Fig. 10 Mössbauer spectroscopy performed at 300 K (A) for fresh Fe10Al90Cu catalyst; (B) after pre-treatment at 350 °C/H2; after reactions time of (C) 15 min; (D) 30 min; (E) 60 min and (F) 240 min by ex situ characterization. | ||
Ex situ Mössbauer spectroscopy measurements were also performed at 300 K to observe structural changes related to the Fe-based species for the sample pre-treated at 350 °C/H2, and the samples at reaction times of 15, 30, 60 and 240 min. The results are shown in Fig. 10B–F and Table S10.† The spectrum of the H2 reduced sample (Fig. 10B) was analysed considering two paramagnetic components with IS and QS values whiting the range of Fe3+ and Fe2+ species,65 both Fe species are embedded in the alumina matrix. The doublet with smaller IS and QS values is due to Fe3+ species and is similar to the doublet found in the spectrum for sample Fe10Al90Cu. The doublet with larger IS and QS is due to Fe2+ species in the alumina matrix and is a result of the reduction step at 350 °C/H2, as discussed in the TPR results. The reduction condition is insufficient to fully reduce the Fe3+-based phase, thus, it formed two Lewis acid sites, Fe3+ and Fe2+, already described in the TPR-H2 profiles. It is worth noting that the synergy between these sites may have positive effects on the reaction mechanisms.
Table S10† shows the hyperfine parameters obtained after fitting the spectra for the samples after several reaction times in the catalytic test. From the area of Fe2+ specie, it is noticed an increasing presence of Fe2+ with the period during the glycerol dehydration reaction process. The sample obtained after reduction of Fe10Al90Cu has 53.9 wt% Fe2+, and the samples obtained after 15, 30, 60 and 240 min of reaction have 61.1, 71.3, 73.1 and 79.8 wt% Fe2+, respectively. As expected for a spectrum with two components, the increase in the spectral area of Fe2+ is accompanied by a decrease of areas for Fe3+ ions. These results indicate that the gases released during the reaction tests have the ability to partially reduce the Fe3+ species to Fe2+, it is because the glycerol dehydrogenation can produce H2 in situ and cause the reduction of iron oxide.
Ex situ TG analyses were performed for the fresh solid Fe10Al90Cu and for the sample after the reaction using the same periods as the XRD and Mössbauer analyses. The results are shown in Fig. 11 and Table S10.† It was observed, as expected, that the amount of coke increases as the reaction progresses, considering that the amount of species adsorbed on the solid surface increases during the reaction. For samples treated at times of 15, 30 and 60 min, the burning profile of the deposited carbon are similar and not very intense, referring mainly to the deposition of water and soft coke. On the other hand, there is a change in the DTG profile for the reaction at 240 min, which is possible to identify hard coke.
 | ||
| Fig. 11 Mass variation and DTG profiles for Fe10Al90Cu catalyst after 15 (a), 30 (b), 60 (c) and 240 min (d) of reaction by ex situ characterization. Black line is related to mass loss and red line is for DTG profile. | ||
DSC results were also recorded to observe more clearly the burning region and the type of coke. Fig. S5† shows the main mass loss events referring to soft coke and hard coke specifically, these signals are stronger for the reaction along 240 min, in fact, it was the only result which indicated an event at elevated temperatures of 807 °C. For the samples obtained at 15, 30 and 60 min, the amount of soft coke is evidenced by the low intensity peaks at around 252 °C.
Thus, the ex situ characterizations were important since they show the changes in the Cu sites and its possible sintering, the changes in the Fe sites after reduction and during the reaction as well as the amount and type of coke deposited during the glycerol conversion. In this sense, from the ex situ results obtained from Mössbauer spectroscopy, TG and Rietveld refinement, it was possible to correlate the different information, as shown in Fig. S6.† This correlation helped to understand the structural variations that occur during the glycerol conversion reaction related to the Fe3+/Fe2+ phases. The effect of copper sintering due to the increase in crystallite size was also confirmed by comparing the value obtained after pre-treatment (∼19 nm) with the 4 h of reaction (∼24 nm). In addition, the deposited coke content also increased over time, reaching 17% of carbon in the last hour. Table S11† presents the catalytic performance for the glycerol conversion and amount of coke for different mixed oxide catalysts similar to those synthesized in this work and compared with the Fe10Al90Cu sample, confirming the viability of the synthesized material.
3.7 Simple theoretical–computational study (surface acid–base distribution)
Electron density map of the reagent and the main active sites were calculated through a theoretical–computational study using DFT methodology to complement the characterizations and obtain additional information about the Cu, Fe and Al sites. The results are shown in Fig. 12 and S7,† in which is possible to visually observe the acidic and basic sites in the different phases identified in the experimental characterizations and estimate possible interactions with the functional groups present in the glycerol structure. The glycerol molecule presents sites susceptible to electrophilic and nucleophilic reaction, consequently, sites vulnerable to acid and basic attack as shown in Fig. 12a, which it is possible to observe the regions with higher negative electron density with red color around the hydroxyl groups, being prone to attack by an acidic site (Mn+). However, the region with positive electron density in blue color around the hydrogens is prone to attack by a basic site (lattice oxygen), considering that they are electron deficient.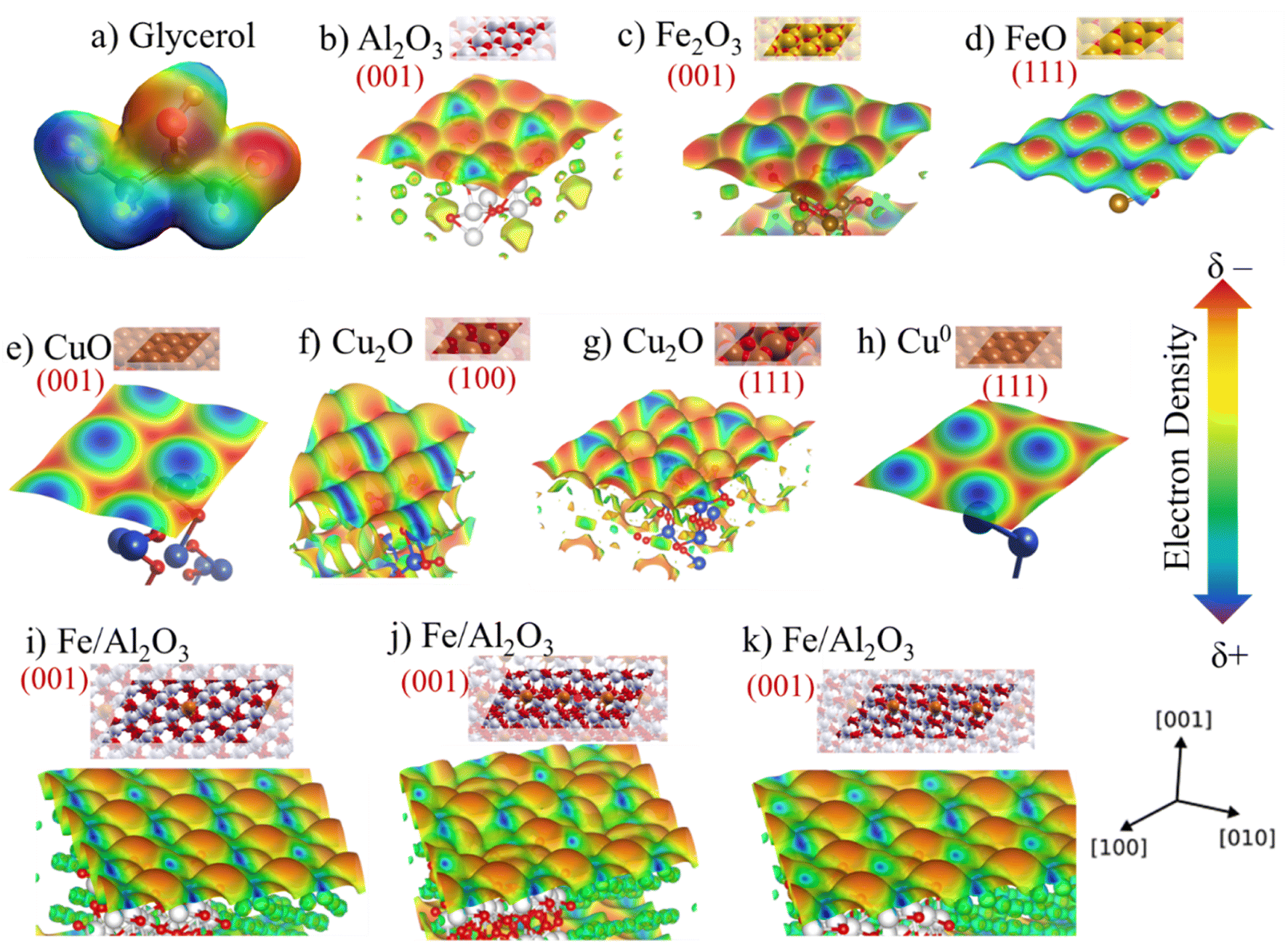 | ||
| Fig. 12 (a) Electronic density map of the glycerol molecule, (b) and for the phases (001) Al2O3, (c) (001) Fe2O3; (d) (111) FeO; (e) (001) CuO, (f) (100) Cu2O, (g) (111) Cu2O; (h) Cu0 and (i) Fe3+ embedded in the structure of the (001) Al2O3 phase; (j) Fe subsurface doped in the (001) alumina vacancy, generating Fe3+/Fe2+ mixture; (k) Fe subsurface doped in the (001) alumina. | ||
It is worth mentioning that surface electrostatic potential maps have been extensively studied to assess the acidity of metallic oxides by visualizing the surface charge distribution, which is related to the change of colors concerning more positive (blue) or negative regions (red).68,69 Thus, Fig. 12b presents the electron density for the Al2O3 phase (001). Fig. 12c and d exhibits the density for Fe2O3 (001) and FeO (111) phases, respectively. Fig. 12e–h, displays the density for copper phases, CuO (001), Cu2O (100), Cu2O (111) and Cu0 (111), respectively. Furthermore, taking into account that some experimental techniques suggested that the iron species may be embedded in the alumina crystal lattice, a map was also obtained simulating this experimental result and the result is shown in Fig. 12i and j.
The map for the aluminum oxide present in Fig. 12b indicates a high electronic density of the sites exposing the lattice oxygen, in addition, DFT studies show that the Al–O bond is very polar, causing greater electron density on the oxygen.70 It is also noted that the volume of Al3+ sites is much smaller than O2−, this effect occurs due to its polarizability, where O2− attracts the net electron density of Al3+.71
Fig. 12c and d displays the maps for Fe2O3 (Fe3+) and FeO (Fe2+) species, which it can be seen from the density on the plane (111) of the wüstite a considerable increase in the Lewis acidity compared to the plane (001) of the hematite. This effect is related to the fact that the hematite structure has more oxygens in the crystal lattice, which increases the electronic density, while the region close to Fe3+ is electron deficient.72 Hence, the change of oxidation state from Fe3+ to Fe2+ due to reduction during pretreatment occurs and the number of oxygens in the lattice decreases. It is worth mentioning once again that the Mössbauer spectroscopy results indicate the presence of both species (Fe3+ and Fe2+), which is a positive feature due to the synergistic interaction effect of these sites involving the glycerol dehydration mechanism. Previous studies have described that the synergistic interaction between metal oxides with different oxidation states directly affects the catalytic performance for glycerol conversion.59
The density of negatively charged electrons is greater for the Cu2+ sites present in the CuO structure, since lattice oxygen is tetrahedrally coordinated by Cu2+. On the other hand, in the Cu2O phase, only one oxygen is coordinated by Cu+, having a positive polarity with electron deficiency, since the species becomes more electrophilic and its Lewis acidity is also increased. The absence of lattice oxygen in the Cu0 species leads to a greater deficiency of electrons and, consequently, increasing its Lewis acidity. In addition, Cu2O (100) and (111) have different electron densities compared to the Cu2+ and Cu0 phases, which has an adjacent combination between the (δ+/δ−) sites and can facilitate the interaction process with the reagent molecule. This factor is directly related to the structure of the Cu2O phase and bond lengths that reflect on the local electronic density and, consequently, on the interaction strength with the reagent.73 Thus, the synergy between the two Cu+/Cu0 phases, identified in the ex situ analysis, is even more evident (Fig. 9).
Furthermore, Fig. 12i shows the density map of Al2O3 (001) with 10% Fe3+ species embedded in its structure. Some theoretical studies involving DFT calculations have already discussed the possibility of Fe being inserted into the alumina structure.74 This fact reflects directly on the electronic density of the surface as observed in Fig. 12 comparing with pure Al2O3 and Fe2O3. Thus, the acid sites are more exposed, since the profile of the map shows a more positively charged surface represented by a more intense blue color compared to pure (001) Al2O3.
It is possible to notice that the insertion of Fe3+ in the alumina lattice, the oxidation state of the metal in this environment can also vary. Fig. 12j shows the possibility of inserting Fe species to generate an oxygen vacancy on the surface, generating Fe2+, while Fig. 12k shows the presence of Fe in the alumina subsurface, which simulates the process of partial reduction of Fe3+ observed after pre-treatment and during the reaction according to ex situ experimental results. The map in Fig. 12j shows that the charge distribution and color intensity change compared to the map in Fig. 12i containing only Fe3+ in alumina, which confirms that acid–base properties change when partially reducing Fe3+ to Fe2+ and alter the catalytic performance in the conversion of glycerol to acetol.
Corroborating the TPR-H2 results, the pre-treatment at 300 °C/H2 and the defined condition at 350 °C/H2 are sufficient to partially reduce the Fe phase. Thus, the presence of Fe2+ and Fe3+ sites due to partial reduction of iron, also indicated by Mössbauer spectroscopy, showed better conversion and selectivity performance compared to the fresh catalyst containing only the presence of Fe3+ with pre-treatment at 350 °C/N2, as indicated in Table 2.
3.8 Mechanism for the glycerol conversion to acetol
Acetol formation occurs preferentially after interacting with Lewis acid sites, however, it follows a complex reaction pathway, involving mechanisms for direct dehydration of the primary and/or secondary hydroxyl group or dehydrogenation followed by dehydration, depending on the type and strength of the acid sites present on the catalyst surface.75 It is known that the proper balance achieved between surface acidity and redox property contributes to the best catalytic performance.76 This evidence was observed by our catalytic test results according to FTIR-pyridine and TPR-H2 characterizations.The species observed in the glycerol conversion tests were identified by GC-MS, which are shown in Fig. S8.† Therefore, the glycerol conversion reactions generate acetol (Reaction (1)), 1,2-propanediol (Reaction (2)), propanoic acid (Reaction (3)) and acetic acid (Reaction (4)) as the main products. However, dehydration (Reaction (1) and (2)), hydrogenation (Reaction (2)) and cracking (Reaction (4)) reactions are also observed.
| C3H5(OH)3 → C3H5O(OH) + H2O | (Reaction 1) |
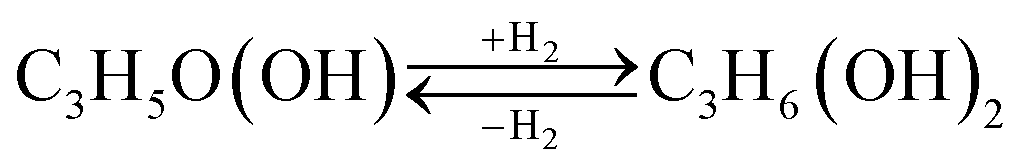 | (Reaction 2) |
| C3H5(OH)3 → C3H5OOH + H2O | (Reaction 3) |
| C3H5(OH)3 → C2H3OOH + CH3OH | (Reaction 4) |
The acetol and 1,2-propanediol formation occurs via the glycerol dehydration reaction, however, this balance becomes more favorable for the acetol formation. To obtain propanoic acid, glycerol is firstly dehydrated to form 3-hydroxypropanal, followed by another dehydration to form acrolein, therefore, a hydrogenation reaction occurs forming propanal, followed by an oxidation obtaining propanoic acid. Two routes concerning the acetic acid formation were proposed, in the first one, cracking of 1,2-propanediol forming formaldehyde and acetaldehyde occurs, which is oxidized into acetic acid. In the second, glycerol is initially dehydrated, forming 3-hydroxypropanal, which undergoes a cracking reaction, generating formaldehyde and acetaldehyde, and is subsequently oxidized into acetic acid.77
Due to the high acetol selectivity, a more detailed reaction mechanism was proposed showing the different steps and surface reactions during the catalytic cycle based on previous studies.59,78 The glycerol dehydration mechanism into acetol was described in 4 steps, as shown in Scheme 1, where the red spheres represent oxygen, the yellow spheres copper, the light gray smaller hydrogen, the dark gray larger carbon and the blue spheres aluminum/iron sites. CuO phases were reduced to Cu+/Cu0 and randomly dispersed in the iron and aluminum oxide matrix. It is worth emphasizing that the synergistic action of the reduced (and partially reduced) sites are associated with the interaction of the reagent and intermediates, facilitating the reaction process.63 In the first step, the glycerol vapor is adsorbed by the copper-containing catalyst, starting the interactions of the terminal hydroxyl group present in the glycerol with the active sites of solid, specifically at the activated Lewis acid site (Mn+), while the basic site (lattice oxygen, O2− from Cu+, Fe2+/3+ or Al3+ oxides) interacts with the H of the secondary carbon of glycerol. These interactions preferentially occur due to the different regions of electron densities (δ) for glycerol and catalyst with a higher δ for the terminal hydroxyl and a lower δ for the central H of glycerol, interacting in more favorable regions in the catalyst sites, Lewis acid and base, respectively, as already shown in the maps of the Fig. 12. The literature indicates,79,80 via optimized geometry DFT calculations, that the distance between the glycerol molecule and the Cu (111) metal site in the O–Cu interaction is approximately 2.23 Å, and the H–O interaction (H from glycerol with O from catalyst) is around 2.24 Å.
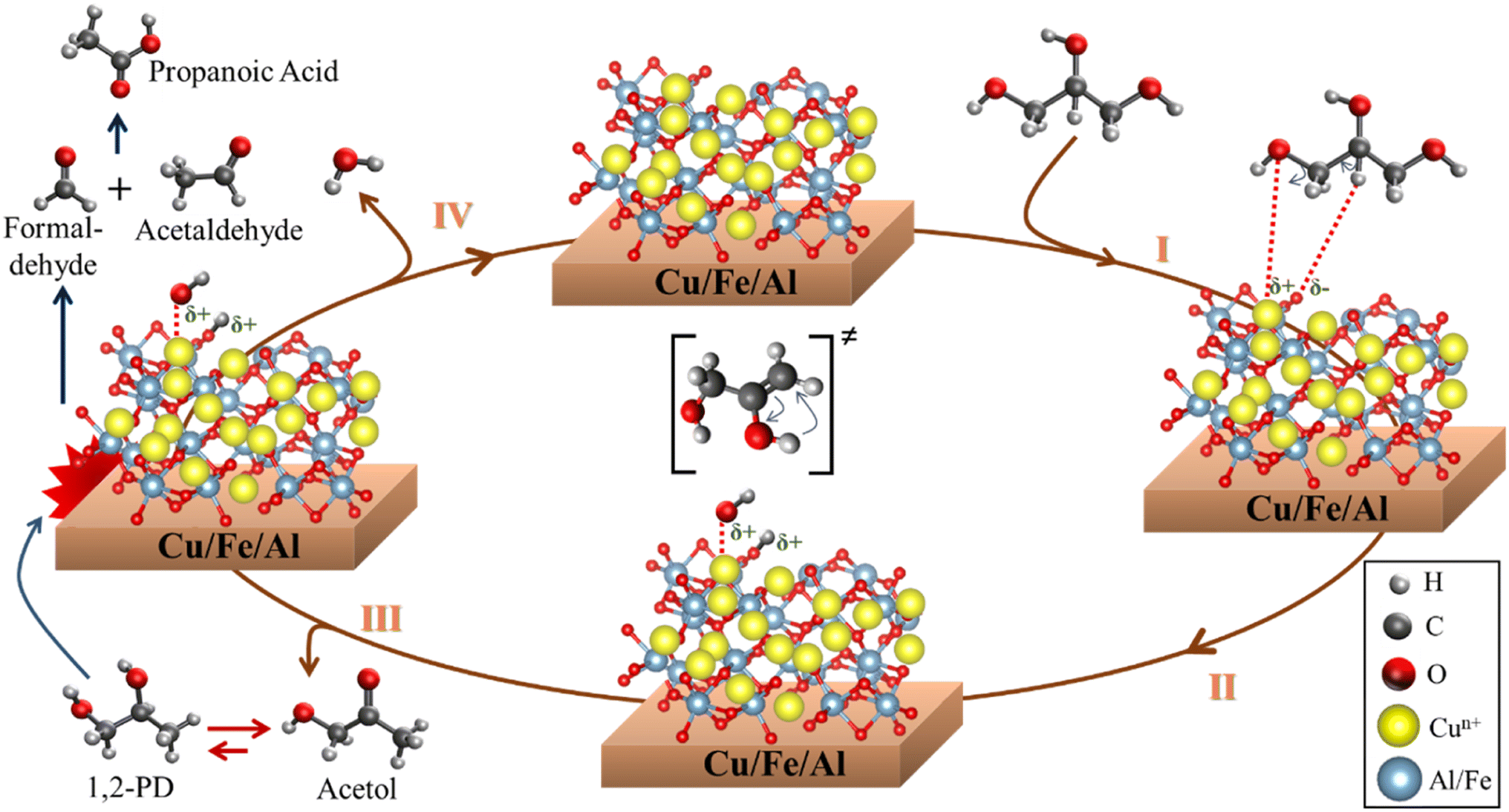 | ||
| Scheme 1 Surface reaction sequence for the glycerol conversion to acetol, 1,2-propanediol, formaldehyde, acetaldehyde and propanoic acid. | ||
In step II, the dehydration of the terminal hydroxyl occurs and the formation of the OH group referring to the interaction of the lattice oxygen in the solid with the H from glycerol, creating two Brønsted pseudo-acid sites. Then, the hydrogen from the OH group of glycerol interacts with the acid site, removing the hydroxyl and forming the 2,3-enol intermediate. Steps III and IV occur in parallel, however, for a better understanding of the catalytic cycle, it has been placed separately in the Scheme 1. Thus, in step III, the unstable molecule in its 2,3-enol intermediate state is reorganized by tautomerization, ensuring greater stability and, consequently, being converted into acetol. However, a part of the acetol is partially converted to 1,2-propanediol (Reaction (2)). The acetol conversion into 1,2-propanediol occurs through the hydrogenation reaction, on the other hand, the reverse conversion occurs by dehydrogenation which produces H2 in situ.81 Furthermore, the H2 source can be correlated to the beginning of the glycerol dehydration reaction in the transient regime, where several gases are formed,82 in addition to parallel reactions such as proton transfer,83 which generates a highly complex environment.
Step IV is related to the water formation, generated from the dehydration of the hydroxyl groups formed during the reaction, once the molecule is released, the lattice oxygen reorganizes with the acid site, reestablishing the initial oxidation state and regenerating the catalyst. Concomitantly, formaldehyde and acetaldehyde are formed by a cracking reaction of 1,2-propanediol, which is subsequently reorganized into propanoic acid. Finally, the products are desorbed and a new catalytic cycle is reestablished.
It is important to highlight that, despite the described mechanism focusing on the interaction between Cu+/Cu0 sites with glycerol, since they are more selective to acetol, the other sites (Fe3+/Fe2+ and Al3+) also contribute to the glycerol dehydration to acetol according to the maps in Fig. 12, although the samples containing iron and aluminum without copper were much less active in the reaction. Previous studies indicate that the Cu phase remains active after several reuses, regenerations and that the Cu+ species identified in spectroscopic studies is more active, although there are contributions from the Cu2+ and Cu0 sites,63 which are in agreement with the results observed in this work. In addition, the high dispersion and redox functionality with adequate Lewis acidity are essential to obtain high performance in the glycerol dehydration reaction into acetol.64
Thus, the synergism between the phases is related to adequate Lewis acid strength, as discussed in FTIR-Py, SMSI effects (bifunctionality between sites, electronic and geometric factors), adequate pretreatment, resulting in greater activity for the catalysts with Cu active centers compared to solids based on Fe–Al without copper and pure CuO phase. In addition to detailing the surface reactions regarding the products observed by GC-MS, it is also essential to describe the surface reactions sequence that lead to coke formation.
3.9 Mechanisms for coke formation from glycerol conversion
It is important to mention that coke deposition directly depends on some factors such as porosity, dispersion of active sites and the nature of acid–base sites (Lewis and/or Brønsted).84 Scheme 2 presents a route for the formation of these compounds identified by GC-MS (Fig. S8 and S9†), after extraction of the most superficial carbon from the spent catalyst. Possible routes for coke formation on the catalyst surface are dehydration, cracking, condensation, hydrogenation and dehydrogenation reactions. | ||
| Scheme 2 Possible routes for coke formation on the catalyst surface. | ||
However, the literature reports85 that the condensation reactions and formation of oxygen-containing aromatic compounds originate from the main products generated from the glycerol dehydration such as acetic acid, 1,2-propanediol, propanoic acid and acetol. The tendency is to form higher molecular weight compounds such as aromatics and cyclic compounds, which ultimately generate coke and partially obstruct the active sites and pores, causing their deactivation.
Considering that the catalyst contains mostly Lewis sites, the literature reports that this type of site interacts with less intensity with intermediate molecules and final products for coke formation during the catalytic cycle.62
4 Conclusions
The materials have micro-mesoporosity and textural properties varied depending on the iron content and he CuO phase has nanometer crystallite diameter (∼20 nm). The catalyst with the best performance was pre-treated under a H2 atmosphere at 350 °C, almost completely reducing the Cu species and partially the Fe species. Fe and Al-based materials have a higher Lewis acidity than copper-containing solids and the presence of Cun+ (Cu+ and Cu0) sites in synergistic effect are more important than high Lewis acidity for the acetol selectivity. The Fe10Al90Cu catalyst showed the best glycerol conversion (60%) and acetol selectivity (92%) in 240 min. In reuse tests, the catalyst decreases the glycerol conversion, but the acetol selectivity remains high (72%). After regeneration, conversion and selectivity values are similar to the catalytic performance of the second reuse. The study of the ex situ characterizations at different reaction times showed a decrease in the intensity of the Cu0 phase, the variation on the composition of Fe3+/Fe2+ species, while the carbon deposition is related to the soft coke formation. The surface electrostatic potential maps expose the faces identified and were related to Lewis acidity and basicity. Finally, the present study showed several insights into the surface chemistry during the glycerol conversion to acetol in the presence of Fe, Al, Cu-based catalysts and can enkindle ideas for the development of new materials resistant to deactivation by coke and sintering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Brazilian CNPq and CAPES funding agencies for financial support. A. R. Albuquerque would like to thank CNPq for grant no. 312854/2021-8. The computational facilities were supported by resources supplied by the High-Performance Computing Center (NPAD) at UFRN. University of Poitiers for FTIR-pyridine analysis. Financial assistance received from Ministerio de Ciencia e Innovación, project no PID2021-126235OB-C32 and FEDER funds is also acknowledged.References
- J. Zhang, Y. Wang, V. L. Muldoon and S. Deng, Renewable Sustainable Energy Rev., 2022, 159, 112206 CrossRef CAS.
- Z. Pirzadi and F. Meshkani, Fuel, 2022, 329, 125044 CrossRef CAS.
- O. Valerio, T. Horvath, C. Pond, M. Misra and A. Mohanty, Ind. Crops Prod., 2015, 78, 141–147 CrossRef CAS.
- L. R. Kumar, S. K. Yellapu, R. D. Tyagi and X. Zhang, Bioresour. Technol., 2019, 293, 122155 CrossRef CAS PubMed.
- N. A. Roslan, S. Z. Abidin, A. Ideris and D.-V. N. Vo, Int. J. Hydrogen Energy, 2020, 45, 18466–18489 CrossRef CAS.
- Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2011, 1, 179 RSC.
- T. Laino, C. Tuma, A. Curioni, E. Jochnowitz and S. Stolz, J. Phys. Chem. A, 2011, 115, 3592–3595 CrossRef CAS PubMed.
- F. Adam, H. E. Hassan and K. M. Hello, J. Taiwan Inst. Chem. Eng., 2012, 43, 619–630 CrossRef CAS.
- N. Galy, R. Nguyen, P. Blach, S. Sambou, D. Luart and C. Len, J. Ind. Eng. Chem., 2017, 51, 312–318 CrossRef CAS.
- Z. Tang, H. Cao, Y. Tao, H. J. Heeres and P. P. Pescarmona, Appl. Catal., B, 2020, 263, 118273 CrossRef CAS.
- P. S. Kong, M. K. Aroua, W. M. A. W. Daud, H. V. Lee, P. Cognet and Y. Pérès, RSC Adv., 2016, 6, 68885–68905 RSC.
- P. U. Okoye and B. H. Hameed, Renewable Sustainable Energy Rev., 2016, 53, 558–574 CrossRef CAS.
- G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem., 2011, 123, 10318–10321 CrossRef.
- N. Tabassum, R. Pothu, A. Pattnaik, R. Boddula, P. Balla, R. Gundeboyina, P. Challa, R. Rajesh, V. Perugopu, N. Mameda, A. B. Radwan, A. M. Abdullah and N. Al-Qahtani, Catalysts, 2022, 12, 767 CrossRef CAS.
- P. J. M. Lima, R. M. da Silva, C. A. C. G. Neto, N. C. Gomes e Silva, J. E. da S. Souza, Y. L. Nunes and J. C. Sousa dos Santos, Biotechnol. Appl. Biochem., 2022, 69(6), 2794–2818 CrossRef CAS PubMed.
- A. P. Pinheiro Pires, J. Arauzo, I. Fonts, M. E. Domine, A. Fernández Arroyo, M. E. Garcia-Perez, J. Montoya, F. Chejne, P. Pfromm and M. Garcia-Perez, Energy Fuels, 2019, 33, 4683–4720 CrossRef CAS.
- C. Len, F. Delbecq, C. Cara Corpas and E. Ruiz Ramos, Synthesis, 2018, 50, 723–741 CrossRef CAS.
- C.-J. Pan, M.-C. Tsai, W.-N. Su, J. Rick, N. G. Akalework, A. K. Agegnehu, S.-Y. Cheng and B.-J. Hwang, J. Taiwan Inst. Chem. Eng., 2017, 74, 154–186 CrossRef CAS.
- X. C. Jiang, C. H. Zhou, R. Tesser, M. Di Serio, D. S. Tong and J. R. Zhang, Ind. Eng. Chem. Res., 2018, 57, 10736–10753 CrossRef CAS.
- A. Talebian-Kiakalaieh, N. A. S. Amin and Z. Y. Zakaria, J. Ind. Eng. Chem., 2016, 34, 300–312 CrossRef CAS.
- M.-L. Gou, J. Cai, W. Song, Z. Liu, Y.-L. Ren, B. Pan and Q. Niu, Catal. Commun., 2017, 98, 116–120 CrossRef CAS.
- S. He, H. R. Goldhoorn, Z. Tegudeer, A. Chandel, A. Heeres, M. C. A. Stuart and H. J. Heeres, Chem. Eng. J., 2022, 434, 134620 CrossRef CAS.
- G. R. Meima and P. G. Menon, Appl. Catal., A, 2001, 212, 239–245 CrossRef CAS.
- R. B. Von Dreele and A. C. Larson, General Structure Analysis System (GSAS), Los Alamos Natl, Lab. Rep., 2004, vol. 86, DOI:10.4135/9781483371283.n174.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- A. V. Azaroff, Elements of X-ray crystallography, 1968, vol. 26 Search PubMed.
- K. Maniammal, G. Madhu and V. Biju, Phys. E, 2017, 85, 214–222 CrossRef CAS.
- A. R. Stokes and A. J. C. Wilson, Proc. Phys. Soc., 1944, 56, 174–181 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- J. Rouquerol and F. Rouquerol, in Adsorption by Powders and Porous Solids, ed. F. Rouquerol, J. Rouquerol, K. S. W. Sing, P. Llewellyn and G. Maurin, Academic Press, Oxford, second edn, 2014, pp. 57–104 Search PubMed.
- R. Bardestani, G. S. Patience and S. Kaliaguine, Can. J. Chem. Eng., 2019, 97, 2781–2791 CrossRef CAS.
- V. Zholobenko, C. Freitas, M. Jendrlin, P. Bazin, A. Travert and F. Thibault-Starzyk, J. Catal., 2020, 385, 52–60 CrossRef CAS.
- A. Bonnin, J.-D. Comparot, Y. Pouilloux, V. Coupard, D. Uzio and L. Pinard, Appl. Catal., A, 2021, 611, 117974 CrossRef CAS.
- R. Docherty, G. Clydesdale, K. J. Roberts and P. Bennema, J. Phys. D Appl. Phys., 1991, 24, 89–99 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens.Matter, 2009, 21, 395502 CrossRef PubMed.
- A. Kokalj, J. Mol. Graphics Modell., 1999, 17, 176–179 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed.
- H. Khan, A. S. Yerramilli, A. D'Oliveira, T. L. Alford, D. C. Boffito and G. S. Patience, Can. J. Chem. Eng., 2020, 98, 1255–1266 CrossRef CAS.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- J. Baltrusaitis, D. M. Cwiertny and V. H. Grassian, Phys. Chem. Chem. Phys., 2007, 9, 5542 RSC.
- Z. Jiang, D. Jiang, W. Wei, Z. Yan and J. Xie, J. Mater. Chem. A, 2015, 3, 23607–23620 RSC.
- Y. Liu, X. Cao, D. Jiang, D. Jia and J. Liu, J. Mater. Chem. A, 2018, 6, 10474–10483 RSC.
- Z. Jiang, K. Chen, Y. Zhang, Y. Wang, F. Wang, G. Zhang and D. D. Dionysiou, Sep. Purif. Technol., 2020, 236, 116272 CrossRef CAS.
- Z. Wang, Z. Niu, Q. Hao, L. Ban, H. Li, Y. Zhao and Z. Jiang, Catalysts, 2019, 9(1), 35 CrossRef.
- C. Huo, J. Ouyang and H. Yang, Sci. Rep., 2014, 4, 3682 CrossRef PubMed.
- W. K. Jozwiak, E. Kaczmarek, T. P. Maniecki, W. Ignaczak and W. Maniukiewicz, Appl. Catal., A, 2007, 326, 17–27 CrossRef CAS.
- S. M. Pudi, P. Biswas, S. Kumar and B. Sarkar, J. Braz. Chem. Soc., 2015, 26, 1551–1564 CAS.
- J. L. Gunjakar, T. W. Kim, H. N. Kim, I. Y. Kim and S.-J. Hwang, J. Am. Chem. Soc., 2011, 133, 14998–15007 CrossRef CAS PubMed.
- Z. Li, X. Chen, J. Chen, H. Chang, L. Ma and N. Yan, Catalysts, 2022, 12, 527 CrossRef CAS.
- Z. ALOthman, Materials, 2012, 5, 2874–2902 CrossRef CAS.
- Z. Wu, A. K. P. Mann, M. Li and S. H. Overbury, J. Phys. Chem. C, 2015, 119, 7340–7350 CrossRef CAS.
- R. Buzzoni, S. Bordiga, G. Ricchiardi, C. Lamberti, A. Zecchina and G. Bellussi, Langmuir, 1996, 12, 930–940 CrossRef CAS.
- G. Varvoutis, M. Lykaki, E. Papista, S. A. C. Carabineiro, A. C. Psarras, G. E. Marnellos and M. Konsolakis, J. CO2 Util., 2021, 44, 101408 CrossRef CAS.
- A. Penkova, L. F. Bobadilla, F. Romero-Sarria, M. A. Centeno and J. A. Odriozola, Appl. Surf. Sci., 2014, 317, 241–251 CrossRef CAS.
- S. Sato, M. Akiyama, R. Takahashi, T. Hara, K. Inui and M. Yokota, Appl. Catal., A, 2008, 347, 186–191 CrossRef CAS.
- T. P. Braga, N. Essayem, S. Prakash and A. Valentini, J. Braz. Chem. Soc., 2016, 27(12) DOI:10.5935/0103-5053.20160134.
- H. Zhang, Y. Wang, S. Shao and R. Xiao, Sci. Rep., 2016, 6, 37513 CrossRef CAS PubMed.
- A. L. Folkard, M. D. Farahani, A. S. Mahomed and H. B. Friedrich, ChemCatChem, 2022, 14(20), e202200602 CrossRef CAS.
- J. Mazarío, P. Concepción, M. Ventura and M. E. Domine, J. Catal., 2020, 385, 160–175 CrossRef.
- J. Mazarío, J. A. Cecilia, E. Rodríguez-Castellón and M. E. Domine, Appl. Catal., A, 2023, 652, 119029 CrossRef.
- P. Gütlich, E. Bill and A. X. Trautwein, Mössbauer Spectroscopy and Transition Metal Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011 Search PubMed.
- L. F. Cótica, A. Paesano, S. C. Zanatta, S. N. de Medeiros and J. B. M. da Cunha, J. Alloys Compd., 2006, 413, 265–272 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751–767 CrossRef.
- T. C. Dinadayalane, A. Hassan and J. Leszczynski, Theor. Chem. Acc., 2012, 131, 1131 Search PubMed.
- M. Da Costa Borges Soares, F. F. Barbosa, M. A. M. Torres, A. Valentini, A. Dos Reis Albuquerque, J. R. Sambrano, S. B. C. Pergher, N. Essayem and T. P. Braga, Catal. Sci. Technol., 2019, 9, 2469–2484 RSC.
- M. Yazdanmehr, S. J. Asadabadi, A. Nourmohammadi, M. Ghasemzadeh and M. Rezvanian, Nanoscale Res. Lett., 2012, 7, 488 CrossRef PubMed.
- E. Menéndez-Proupin and G. Gutiérrez, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 035116 CrossRef.
- L. Zhou, H. Zhu and W. Zeng, Inorganics, 2021, 9, 80 CrossRef CAS.
- R. J. Chimentão, P. Hirunsit, C. S. Torres, M. B. Ordoño, A. Urakawa, J. L. G. Fierro and D. Ruiz, Catal. Today, 2021, 367, 58–70 CrossRef.
- S. Debnath, M. Jorewitz, K. R. Asmis, F. Müller, J. B. Stückrath, F. A. Bischoff and J. Sauer, Phys. Chem. Chem. Phys., 2022, 24, 20913–20920 RSC.
- J. P. Lourenço, A. Fernandes, R. A. Bértolo and M. F. Ribeiro, RSC Adv., 2015, 5, 10667–10674 RSC.
- H. Wang, Q. Zhang, T. Zhang, J. Wang, G. Wei, M. Liu and P. Ning, Appl. Surf. Sci., 2019, 485, 81–91 CrossRef CAS.
- A. K. Kinage, P. P. Upare, P. Kasinathan, Y. K. Hwang and J.-S. Chang, Catal. Commun., 2010, 11, 620–623 CrossRef CAS.
- R. B. Mane, A. Yamaguchi, A. Malawadkar, M. Shirai and C. V Rode, RSC Adv., 2013, 3, 16499–16508 RSC.
- X. Zhang, G. Cui, H. Feng, L. Chen, H. Wang, B. Wang, X. Zhang, L. Zheng, S. Hong and M. Wei, Nat. Commun., 2019, 10, 5812 CrossRef CAS PubMed.
- Z. Babaei, A. Najafi Chermahini and M. Dinari, J. Colloid Interface Sci., 2020, 563, 1–7 CrossRef CAS PubMed.
- J. B. Restrepo, J. A. Bustillo, A. J. Bula and C. D. Paternina, Processes, 2021, 9, 479 CrossRef CAS.
- E. D'Hondt, S. Van de Vyver, B. F. Sels and P. A. Jacobs, Chem. Commun., 2008, 6011 RSC.
- R. Rousseau, D. A. Dixon, B. D. Kay and Z. Dohnálek, Chem. Soc. Rev., 2014, 43, 7664–7680 RSC.
- A. Talebian-Kiakalaieh and N. A. S. Amin, Chin. J. Catal., 2017, 38, 1697–1710 CrossRef CAS.
- Y. Hu, Q. He and C. Xu, Catalysts, 2021, 11, 1455 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05454c |
| This journal is © The Royal Society of Chemistry 2023 |