
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of unnatural coordination glycopolymer particles (CGPs): unleashing the potential of catechol-saccharide derivatives†
Celina Bideplán-Moyanoa,
Marcos J. Lo Fiego b,
Juan José Calmelsa,
Belén Alonsoa,
Gabriel Radivoya,
Daniel Ruiz-Molinac,
Juan Mancebo-Aracil*a and
Fabiana Nador*a
b,
Juan José Calmelsa,
Belén Alonsoa,
Gabriel Radivoya,
Daniel Ruiz-Molinac,
Juan Mancebo-Aracil*a and
Fabiana Nador*a
aInstituto de Química del Sur (INQUISUR-CONICET) – NANOSYN, Departamento de Química, Universidad Nacional del Sur (UNS), Av. Alem 1253, 8000 Bahía Blanca, Buenos Aires, Argentina. E-mail: juan.mancebo@uns.edu.ar; fabiana.nador@uns.edu.ar
bInstituto de Química del Sur (INQUISUR-CONICET) – GIQOS. Departamento de Química, Universidad Nacional del Sur (UNS), Av. Alem 1253, 8000 Bahía Blanca, Buenos Aires, Argentina
cCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and The Barcelona Institute of Science and Technology (BIST), Campus UAB, Bellaterra, 08193 Barcelona, Spain
First published on 13th September 2023
Abstract
Our study unveils an innovative methodology that merges catechols with mono- and disaccharides, yielding a diverse array of compounds. This strategic fusion achieves robust yields and introduces ligands with a dual nature: encompassing both the chelating attributes of catechols and the recognition capabilities of carbohydrates. This synergistic design led us to couple one of the novel ligands with an Fe(III) salt, resulting in the creation of Coordination Glycopolymer Particles (CGPs). These CGPs demonstrate remarkable qualities, boasting outstanding dispersion in both aqueous media and Phosphate Buffered Saline (PBS) solution (pH ∼7.4) at higher concentrations (0.26 mg μL−1). Displaying an average Z-size of approximately 55 nm and favourable polydispersity indices (<0.25), these particles exhibit exceptional stability, maintaining their integrity over prolonged periods and temperature variations. Notably, they retain their superior dispersion and stability even when subjected to freezing or heating to 40 °C, making them exceptionally viable for driving biological assays. In contrast to established methods for synthesizing grafted glycopolymers, where typically a glycopolymer is doped with catechol derivatives to create synergy between chelating properties and those inherent to the saccharide, our approach provides a more efficient and versatile pathway for generating CGPs. This involves combining catechols and carbohydrates within a single molecule, enabling the fine-tuning of organic structure from a monomer design step and subsequently transferring these properties to the polymer.
Introduction
Carbohydrates are involved in many essential biological functions, such as adhesion, growth, signal transduction, and recognition, through specific carbohydrate-mediated interactions.1–3 Consequently, these biomolecules actively participate in controlling various physiological and pathological processes, including bacterial and virus infection, immune responses, tumor formation, and metastasis, among others.4 These properties have led to the design and production of various carbohydrate-based materials for use in imaging, diagnosis, and therapy.1,3 A particular characteristic of carbohydrate-based interactions is their extremely weak and low affinity.5,6 Therefore, it is essential to design materials that can create multivalent systems with proper orientation and spacing in order to achieve high affinity.Glyconanostructures have emerged as promising tools, leveraging the unique properties of carbohydrates, along with enhanced biocompatibility, bioavailability, and biodegradability.2,5,7 So far, glyconanoparticles,8 glycodendrimers9 and glycoliposomes10 have been reported. Of special relevance are glycopolymers,11 commonly prepared using polymerizable saccharides derivatives or by incorporating saccharides into already formed polymer backbones.12 Several successful examples of glycopolymers and their applications in direct therapeutic methods, medical adhesives, and biosensors have been reported.13,14 However, despite its interest, there are still severe limitations to its successful implementation, among them the nonbiodegradable character of the carbon chain backbones, which have no other biological functions than to serve as spacer units. So, the development of alternative approaches to obtain glycopolymers still represents a challenge nowadays.
Catechol-based derivatives have been shown to be one of the key functionalities in the formation of highly complex macromolecules and organic compounds due to their adhesive properties, metal coordination capabilities, and interaction with nearly any surface.15 They can coordinate with different metal ions, with catechol/Fe(III) complexes being the most extensively studied.16 Different types of materials, including adhesives,17 self-repairing hydrogels,18 dyes,19 coatings,20 and coordination polymers,21 have been developed using the coordination of catechol to Fe(III). In our search to generate catechol-based structures with versatile properties applicable to diverse materials, we have developed a new methodology for synthesising thiol catechol-based derivatives. This involves a conjugate addition reaction of functional mono-, di- and trithiols to freshly prepared o-benzoquinone (Scheme 1).18b,22 Using this technique, a wide variety of products have been successfully synthesised with moderate to good yields.
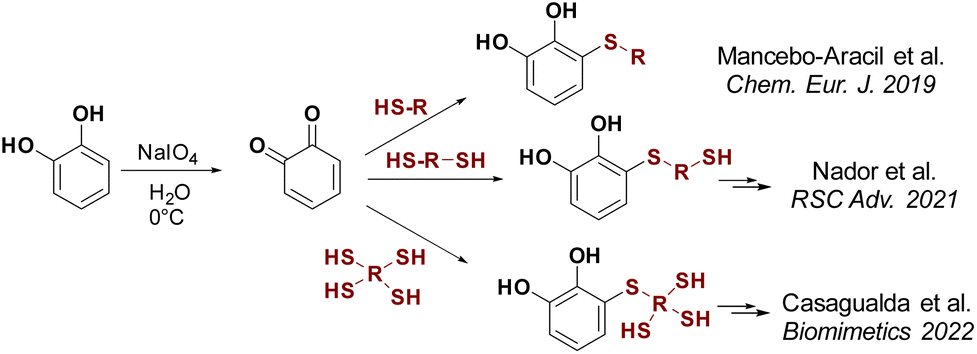 | ||
| Scheme 1 Synthesis of thiol-catecol based derivatives by thiol conjugate addition. | ||
In this study, we present the organic synthesis of monomers incorporating both catechol and a carbohydrate unit. Our aim is to subsequently generate the glycopolymer through controlled polymerisation in the presence of metal salts, capitalising on the chelating properties of the catechol functionality. To achieve this, we employed a commercial trithiol A following the aforementioned technique, to facilitate the synthesis of derivative 1, which possesses two additional thiols capable of reacting with other substrates (Scheme 2). Compound 1 was then reacted with various peracetylated carbohydrates using a thioglycosylation reaction facilitated by an acidic catalyst.23 This reaction led to the formation of novel catechol–sugar structures after deprotection. We envision that these compounds could be excellent candidates for generating new materials as they leverage the chelating and adhesion properties provided by catechols, as well as the stability, targeting and recognition properties offered by sugars.
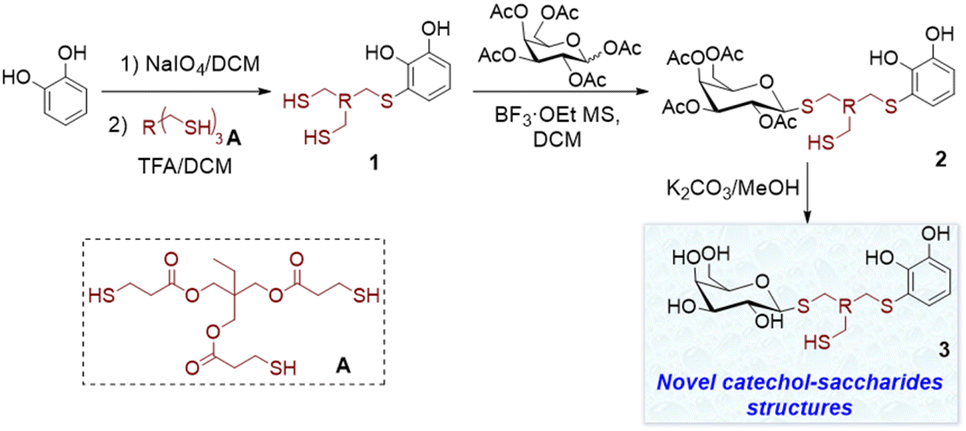 | ||
| Scheme 2 Synthesis of catechol–sugar structures 3 by thiol conjugate addition followed by thioglycosylation reaction. | ||
Results and discussion
The starting substrates 1 were prepared by using the procedure previously reported by some of us,22a which involved the oxidation of pyrocatechol to quinone, followed by the nucleophilic attack of a dithiol or trithiol (Scheme 3). Through this procedure, the Michael adducts 1a and 1b were obtained with yields of 35% and 40% respectively. Disulphide or double catechol addition by-products were also found although in very low proportions so they can be easily separated during the purification step.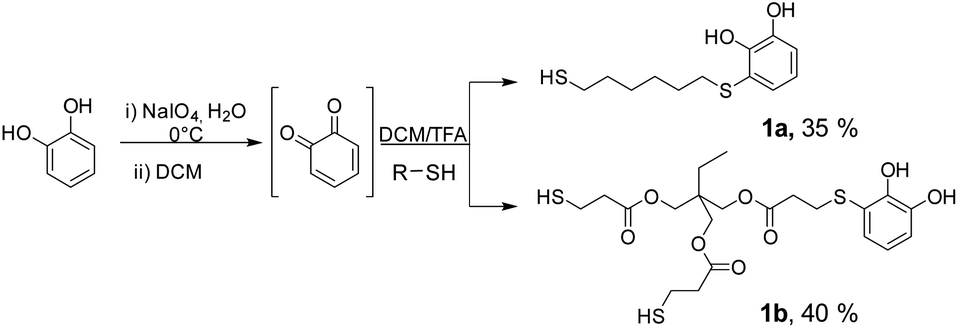 | ||
| Scheme 3 Syntheses of Michael adducts 1. | ||
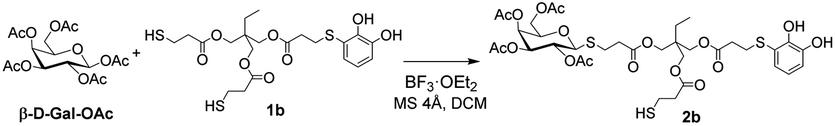
One of the most widely used and efficient ways to generate thioglycosides, especially on a large scale, is by using the corresponding peracetylated monosaccharide dissolved in DCM and a slight excess of a thiol, in the presence of a strong Lewis acid such as BF3·OEt2. By means of this methodology and using simple thiols, the β-acetates reacted faster compared to the α-anomers, resulting in the formation of the corresponding 1,2-trans products due to neighbouring group participation, with yields ranging from good to excellent.23 Following this protocol, Michael adducts 1a and 1b were utilised to carry out the thioglycosylation of peracetylated saccharides such as β-D-glucose pentaacetate (β-D-Glu-OAc), β-D-galactose pentaacetate (β-D-Gal-OAc), α-D-manose pentaacetate (α-D-Man-OAc) and α/β-D-lactose octaacetate (α/β-D-Lac-OAc). To optimise the glycosylation conditions, β-D-Gal-OAc, a slight excess of Michael adduct 1b, and molecular sieves were mixed in DCM under inert atmosphere (Table 1). Then, BF3·OEt2 was added, and the reaction progress was monitored by TLC. The optimisation of reactions conditions mainly focused on varying the β-D-Gal-OAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Michael adduct 1b ratio to determine the optimal proportion for obtaining 2b. In entry 1, a 20% excess of 1b over β-D-Gal-OAc allowed us to exclusively obtain the β-glycosylated product 2b with a yield of 20%. Higher excesses of 1b improved the yield of 2b (entries 2 and 3). This last result may be attributed to the presence of two thiols in the molecule. Therefore, increasing the concentration of 1b would prevent the formation of by-products, such as the addition of two β-D-Gal-OAc units. It is important to note that in all the cases studied, a significant percentage of the excess of 1b could be recovered, reaching nearly 100% recovery when considering the excess of thiol used. Finally, under the same conditions, but using higher amount of the starting substrates, the yield of 2b significantly increased (entries 3 to 5). Furthermore, despite employing nearly twice the amount of reagents as in entry 5, the reaction yield decreased to 41% (entry 6). However, we successfully obtained product 2b on a gram scale, which is highly attractive when considering the potential applications of this compound in functional materials.
:Michael adduct 1b ratio to determine the optimal proportion for obtaining 2b. In entry 1, a 20% excess of 1b over β-D-Gal-OAc allowed us to exclusively obtain the β-glycosylated product 2b with a yield of 20%. Higher excesses of 1b improved the yield of 2b (entries 2 and 3). This last result may be attributed to the presence of two thiols in the molecule. Therefore, increasing the concentration of 1b would prevent the formation of by-products, such as the addition of two β-D-Gal-OAc units. It is important to note that in all the cases studied, a significant percentage of the excess of 1b could be recovered, reaching nearly 100% recovery when considering the excess of thiol used. Finally, under the same conditions, but using higher amount of the starting substrates, the yield of 2b significantly increased (entries 3 to 5). Furthermore, despite employing nearly twice the amount of reagents as in entry 5, the reaction yield decreased to 41% (entry 6). However, we successfully obtained product 2b on a gram scale, which is highly attractive when considering the potential applications of this compound in functional materials.
|
||||
|---|---|---|---|---|
| Entry | β-D-Gal-OAc:1b |
β-D-Gal-OAc (mmol) | 2b yield (%) | 1b recoveryb (%) |
| a Conditions: Gal-OAc, adduct Michael 1b and MS 4 Å were mixed in DCM under N2 atmosphere. Then BF3·OEt2 was added and the reaction monitored by TLC.b Percentage by mass calculated with respect to 1b. | ||||
| 1 | 1:1.2 |
0.60 | 20 | 45 |
| 2 | 1:1.6 |
0.70 | 24 | 48 |
| 3 | 1:2.0 |
0.66 | 37 | 50 |
| 4 | 1:2.0 |
0.91 | 41 | 30 |
| 5 | 1:2.0 |
1.85 | 50 | 39 |
| 6 | 1:2.0 |
3.00 | 41 | 40 |
Under the optimised conditions shown in Table 1, the products depicted in Fig. 1 were obtained. Glycosylation of β-D-Gal-OAc with both Michael adducts 1a and 1b yielded the corresponding β-anomers 2a and 2b after 4 h, with yields of 62% and 50%, respectively. Other peracetylated monosaccharides such as β-D-Glu-OAc and α-D-Man-OAc were also glycosylated, resulting in products 2c and 2d albeit with lower yields. To expand our approximation, the peracetylated disaccharide α/β-D-Lac-OAc was also tested, resulting in the corresponding 2e product with a yield of 19% of β-anomer. In all examples, 1,2-trans glycosides were obtained over the 1,2-cis due to the participatory effect of the neighboring 2-acetyl substituent.24 Furthermore, when α-anomers such as α-D-glucose pentaacetate or α-D-lactose octaacetate were used, no reaction was observed. The addition reaction was also carried out using the thiol saccharide I (Fig. 1) previously synthesised and reported by Lo Fiego et al.25 In this case, product 2f was obtained with a yield of 40%, which is comparable to the yields of the Michael adducts 1.
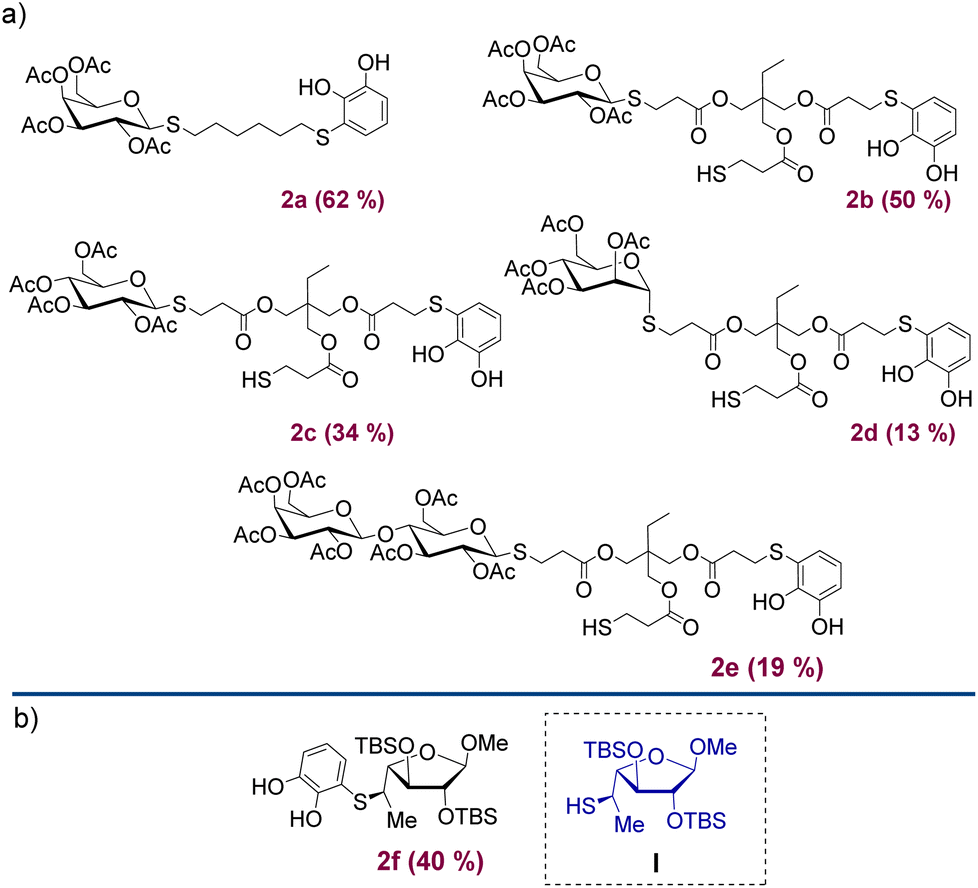 | ||
| Fig. 1 (a) Structure of products 2a–2e obtained after glycosylation of Michael adducts 1. (b) Structure of product 2f obtained after thiol–sugar conjugate addition of 1,2-benzoquinone with the thiol saccharide I. | ||
Moreover, through this reaction, we demonstrated that the methodology was also compatible with silylated groups, which are frequently susceptible to TFA conditions, as well as with more hindered thiols. This result validates that the products can be obtained by forming the Michael adduct with free thiols, and subsequently reacting with the saccharide in a second step through an addition reaction. Alternatively, if the sugar already contains a thiol in its structure, it can directly attack the quinone to form product 2.
To obtain the corresponding deacetylated products, we decided to perform the O-deacetylation of sugars under basic conditions, using either MeONa/MeOH or K2CO3/MeOH. The substrate 2b was dissolved in anhydrous MeOH under an inert atmosphere, followed by the addition of a base such as MeONa or K2CO3. The reaction conditions using K2CO3 as base were found to be the most compatible with the presence of the catechol functionality, which is susceptible to oxidation under basic conditions in presence of mild oxidant like oxygen. Initially, following previously reported saccharides deprotection procedures,26 a deficiency of base with respect to 2b was used, but no reaction progress was observed, leaving the starting substrate intact. Therefore, an excess of base was required for successful deprotection. This could be attributed to the presence of catechols and thiols that can undergo deprotonation in the reaction medium, resulting in an increased consumption of K2CO3. In Table 2 the results of the deprotection of 2b using K2CO3 are presented. In entry 1, the compound 2b was initially dissolved in anhydrous MeOH under an inert atmosphere, and then K2CO3 was added in a 1:1.8 ratio (2b:K2CO3). After a few minutes, both the desired product 3b and a secondary by-product resulting from the hydrolysis of the ester functionalities present in 2b, were detected (see ESI S1†). To prevent this, the conditions of entry 1 were repeated with a lower amount of K2CO3 which was added in two portions (entry 2). This simple strategy significantly improved the deprotection process, resulting in a 57% yield of product 3b after 1.5 h. Although no hydrolysis by-products were observed, a small amount of substrate 2b remained intact. In entry 3, the conditions of entry 2 were replicated, but the total amount of base was added from the beginning of the reaction. After completion of the reaction, product 3b was purified by washing with diethyl ether, resulting in a 68% yield. At this stage of the optimisation process, we obtained two products: the desired product 3b from the methanolysis of the sugar esters, and undesired by-products involving the methanolysis of the esters present in the starting trithiol backbone (see Fig. S1 in the ESI†). Based on this, we decided to employ two strategies to prevent the undesired methanolysis reactions. Thus, the reaction was carried out by lowering the temperature of the reaction medium to 0 °C (entry 4) or using a less polar solvent (entry 5) to reduce the solubility of K2CO3. However, both reactions were equally disfavoured, and even after 6 h, a significant percentage of the acetylated product 2b remained unreacted, especially under the conditions of entry 5. Based on these observations, the conditions of entry 3 were deemed the most suitable.
|
|||||
|---|---|---|---|---|---|
| Entry | 2b:K2CO3 |
Solvent | Temp. (°C) | Time (h) | 3b yield (%) |
| a Conditions: Substrate 2 (0.22 mmol) was dissolved in anhydrous MeOH (12 mL) under N2 atmosphere and K2CO3 (0.40 mmol) was added. The reaction was monitored by TLC until starting material was consumed. After that, the reaction mixture was passed through a column filled with Dowex 50×, which acted as acid source, and washed with MeOH. The final residue was purified by chromatographic column unless otherwise stated.b 0.20 mmol of K2CO3 were added and the reaction was monitored by TLC. After 0.5 h additional 0.2 mmol K2CO3 were added.c The product was isolated after reverse phase filtration and washed with diethyleter. | |||||
| 1 | 1:1.8 |
MeOH | 25 | 2.5 | Traces |
| 2b | 1:1.6 |
MeOH | 25 | 1.5 | 57 |
| 3 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1.6 :1.6 |
MeOH | 25 | 1.25 | 68c |
| 4 | 1:1.6 |
MeOH | 0 | 6 | <20 |
| 5 | 1:1.6 |
DCM:MeOH |
25 | 6 | <20 |
| 4:1 |
|||||
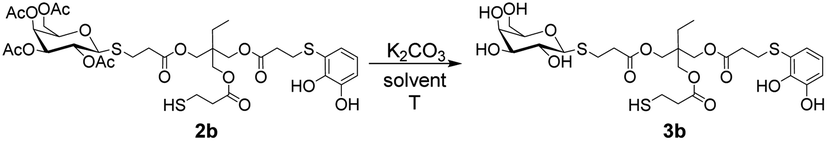
Next, different acids for neutralise the basic reaction medium were tested. When a methanolic solution of HCl (0.34 M) was used, it was observed by 1H NMR that the signals coming from the saccharide started to unfold, possibly due to the formation of new by-products related to ester hydrolysis. We also tested the addition of p-toluensulphonic acid, without obtaining encouraging results. The best conditions found were the use of trifluoroacetic acid or the addition of a Dowex ion exchange resin. Fig. 2 shows the products obtained after O-deacetilation of products 2, with yields ranging from moderate to very good.
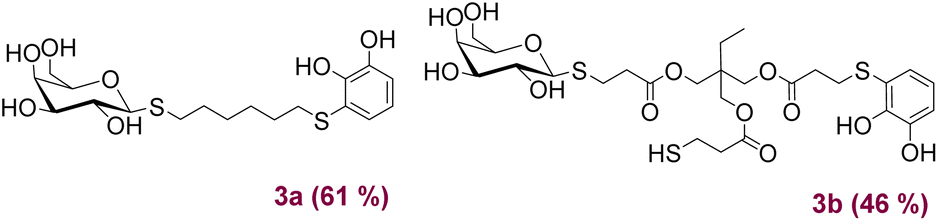 | ||
| Fig. 2 Structure and yields of products 3 obtained after deprotection of 2. | ||
Following a methodology previously reported,22a compound 2b was selectively dimerised using I2 to form the disulphide 4a (Scheme 4) with quantitative yield. These novel structures, incorporating both catechols and sugars, offer chelating and coordination properties with metal ions. Additionally, the presence of carbohydrates could provide stabilisation in biological environments, as well as recognition and targeting capabilities. Based on these considerations, the ligand 4a in combination with Fe(III) were appropriated candidates for the preparation of novel CGPs (Fig. 3a). The process began by dissolving ligand 4a in a mixture of DMSO and MeOH, followed by the gradual addition of an aqueous solution of FeCl3·6H2O under magnetically stirring. This resulted in the formation of a green-blue suspension with a slight precipitation of a solid material. Subsequently, a 0.1 M aqueous solution of NaOH was added until a deep violet-blue colour was achieved. The solution was then stirred for 3 h, followed by centrifugation, washing with water and MeOH, and finally freeze-dried. Regarding the ligand 4a:Fe ratios, we decided to conduct several tests by varying the proportions and determining the mass of the precipitated solid. The best condition was found to be a 1:1 ratio, resulting in a solid yield between of 60–75%. The solid was then analysed by UV-Vis, IR, SEM, EDX, DLS and atomic absorption techniques.
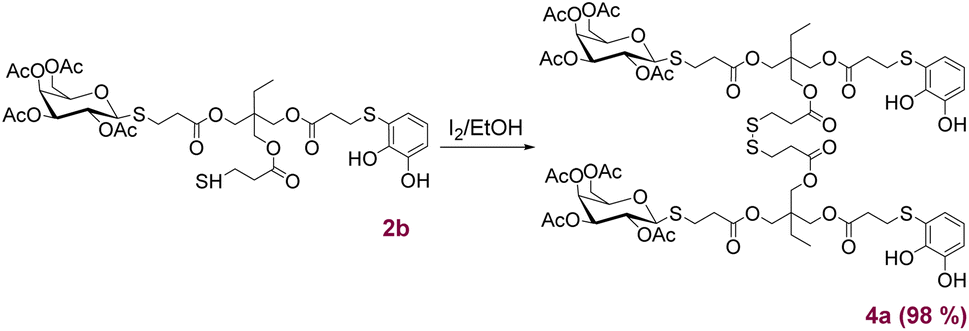 | ||
| Scheme 4 Syntheses of product 4a by oxidation of 2b. | ||
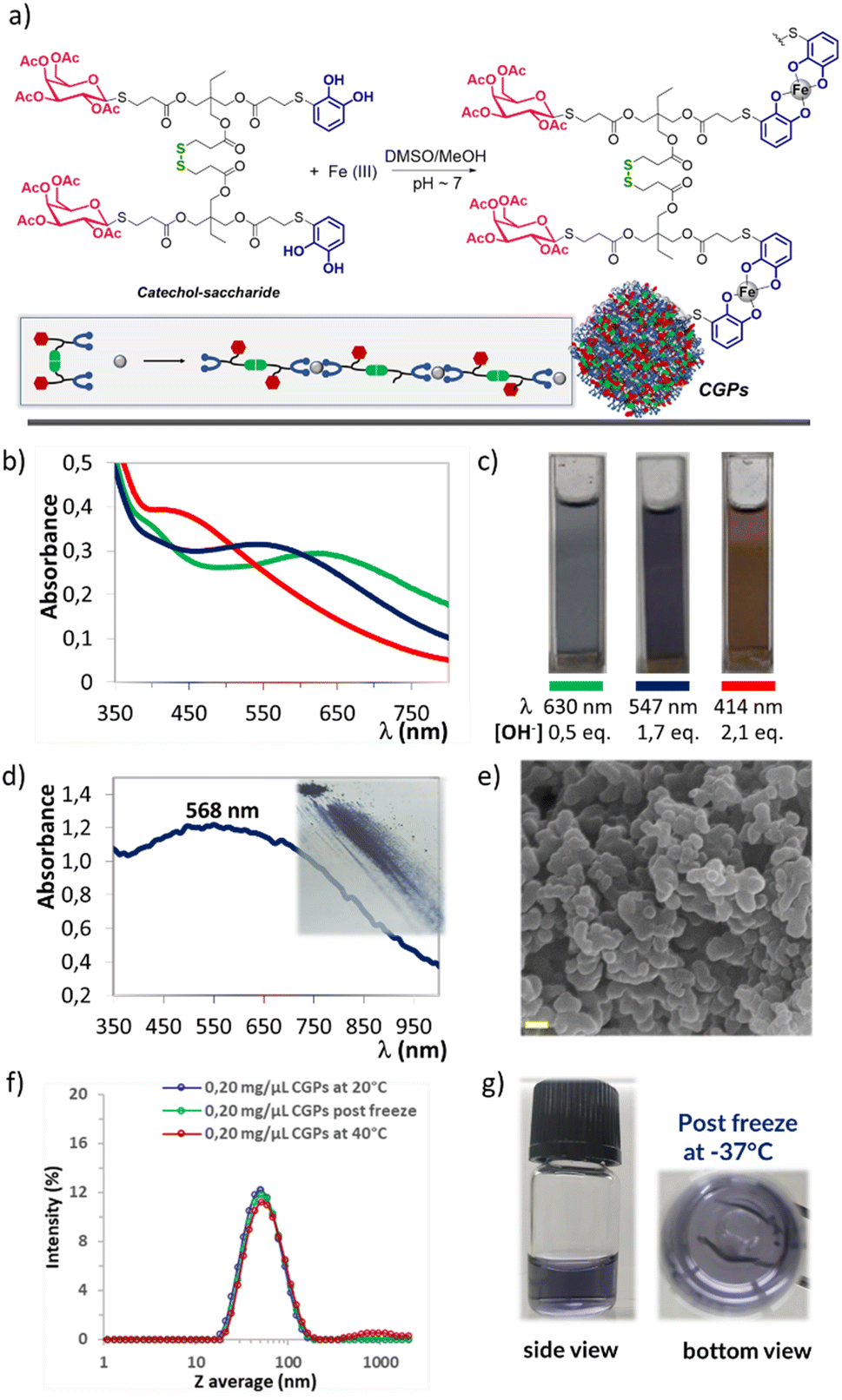 | ||
| Fig. 3 (a) Schematic representation of the preparation of CGPs. (b) UV-Vis spectra of 4a and Fe(III) solutions at differents NaOH concentrations. (c) Suspensions of CGPs at different NaOH concentrations. (d) UV-Vis and image of solid CGPs. (e) SEM image of CGPs. The scale bar corresponds to 500 nm. (f) DLS of resuspended CGPs in water at different temperatures. (g) Images of CGPs in water solution after freezing at −37 °C. | ||
Firstly, to determine the type of interaction between Fe(III) and 4a, we analysed both the obtained suspension and the precipitated using UV-Vis. According to the literature,27 iron solutions containing catechol moieties adjusted to a pH value of 6–7 result in a 2:1 catechol:iron bis species, which exhibits a UV-Vis maximum at 570 nm, corresponding to a deep blue colour. Alternatively, at pH values above 9.5, tris species (3:1, catechol:iron) are formed, with a UV-Vis maximum at 490 nm corresponding to a red colour. Based on these data, we conducted the synthesis under three different conditions (Fig. 3b and c), as follows. (A) Mixing only ligand 4a in DMSO/MeOH and adding an aqueous iron solution resulted in a green-blue coloured solution displaying an absorption band at 630 nm. However, no solid formation was observed. (B) Mixing 4a, dissolved in DMSO/MeOH, with an aqueous iron solution and adding 0.1 M NaOH until the solution change to violet-blue. This solution exhibited an absorption band at 547 nm, and a significant amount of violet-blue precipitate, displaying an absorption band at 568 nm (see Fig. 3d). These results indicated that both the solid and the suspension predominantly consisted of bis-catecholate species. (C) Mixing 4a, dissolved in DMSO/MeOH, with an aqueous iron solution and adding 0.1 M NaOH until a red-purple coloured solution appeared with an absorption band at 440 nm. Additionally, a significant amount of violet-blue precipitate formed, corresponding to an absorption band at 568 nm. These last results suggested that the species present in the suspension were different from those found in the precipitate. In summary, the addition of NaOH solution was necessary to promote the formation of the solid CGPs, we assumed that this base would be assisting in the deprotonation of the catechol and consequently in the coordination with iron.27,28 Moreover, the analysis of conditions (B) and (C) showed that regardless of the change in pH, once the violet-blue solid was formed, further increases in the amount of base did not change its colouration and consequently its structure. We obtained CGPs corresponding to the interaction of two catecholates per Fe ion, indicating a ligand:Fe ratio of 1:1, considering the ligand's bidentate nature (Fig. 3a). These data correlate perfectly with the observations made during the initial optimisation, where an excess of ligand 4a relative to Fe at ratios of 2:1 or even 3:1 did not result in an increased amount of precipitated solid. Furthermore, the analysis by atomic absorption of the product dissolved in concentrated acid mixture yielded a percentage of Fe consistent with the expected ligand–metal ratio (see Experimental section).
To exclude the possibility that the observed precipitate may originate from the oxidation and precipitation of ligand 4a, it was subjected to the same optimised reaction conditions, but in the absence of the metal. It was observed that the solution remained colourless for 20 minutes, but thereafter it started to change its colour, acquiring a yellow/brownish appearance, which is typical of polymers and oligomers generated by the oxidation of the catechol molecules. However, even after 24 h of reaction, no precipitation was observed. This solution was also analysed using UV-Vis spectroscopy to confirm the absence of any absorption band above 400 nm (Fig. S3†).
The characterisation of CGPs was also performed using IR spectroscopy with KBr pellets, confirming the presence of the main bands of ligand 4a in the solid (Fig. S4†). Even when a decrease in the intensity of the O–H stretching band (3550–3320 cm−1) of the free catechols present in the starting ligand 4a was observed compared to the CGPs, their complete disappearance was not detected. This could be attributed to partial deacetylation of the acetyl groups present in the sugar, which can readily hydrolyse in the light basic media. Additionally, a band centred in 620 cm−1 was observed, which could be assigned to the Fe–O interaction resulting from the coordination of the catecholate with Fe.
The CGPs sample was also characterised using SEM microscopy, which revealed a homogeneous composition consisting primarily of oval-shaped nanoparticles arranged in chains and clusters. The average diameter ranged between 400 ± 150 nm (Fig. S6†). Additionally, EDX spectrum confirmed the presence of S, C, O and Fe elements throughout the entire analysed surface (Fig. S7†).
The freeze-dried solid was then studied using DLS to evaluate the average particle size in solution and assess the dispersion and stability of the system in different media. The first assay we conducted was an attempt to redisperse the CGPs solid in different organic solvents. DMSO, which is a commonly used solvent in biological assays, resulted in the most efficient due to this solution quickly acquired a violet-blue colour without any suspended solids or precipitates (see Fig. S8†). Additionally, we attempted to disperse the solid directly in water or a PBS buffer (pH ∼ 7.4), but the process was not successful, resulting in a significant amount of precipitated solid. Based on these preliminary results and with the intention of using our CGPs in future bioassays, we decided to prepare solutions of different concentrations of CGPs in DMSO and gradually add them to water or PBS buffer (see Fig. S9a†). Our goal with these assays was to prepare CGPs/DMSO solutions with the highest possible concentration, enabling the addition of a significant amount of CGPs to aqueous media while minimising the required volume of DMSO. Typically, the limit of DMSO for biological assays is around 4% or less in H2O or physiological media. With this in mind, the best results obtained were those corresponding to concentrations of 0.25 mg μL−1 of CGPs in H2O. These solutions revealed a Z average mean size around 35–65 nm and low PdI values (<0.25) (Fig. S9b†). The same analysis was carried out with PBS solutions (pH ∼ 7.4) with good quality measurements but with a larger Z average distribution around 55–105 nm (see Fig. S10†). The more concentrated samples were also analysed over time in aqueous media, revealing a slight increase in Z average mean size and very good stability even after 7 days without the presence of sedimented sample (see Fig. S11†). Due to the excellent stability results obtained for the CGPs in an aqueous medium, at this point, we decided to take it one step further and exposed the samples to two temperature conditions. One of them was frozen at −37 °C, and the other was heated to 40 °C for 1 hour. Both samples showed the same quality measurements by DLS as at room temperature, revealing the suitability of these samples to proceed to a phase of biological testing (Fig. 3f and g).
Conclusions
In summary, our study has made a significant advancement in glycopolymer synthesis by successfully thioglycosylating peracetylated mono- and disaccharides using Michael adducts containing both catechols and thiols (1). This methodology selectively yielded 1,2-trans glycosides (2) with moderate to good yields, even at gram-scale production. Furthermore, in cases where the saccharide already possessed a thiol group, direct reaction with the quinone from pyrocatechol resulted in the formation of the thiol–sugar conjugate addition product (2f). Additionally, the corresponding O-deacetylated products (3) were obtained with very good yields under basic conditions, even in the presence of other ester functionalities in the thiol backbone. A notable accomplishment in our work was the synthesis of a dimer (4a) through a simple and clean oxidation reaction promoted by I2. This novel structure, incorporating both catechol and sugar moieties, was utilised in combination with Fe(III) to prepare novel CGPs where two catecholates interacted with each Fe ion. Characterisation of the CGPs revealed that the solid samples consisted of chains and clusters of oval-shaped nanoparticles primarily composed of S, C, and Fe. Of particular significance is the excellent stability of the CGPs in aqueous media (H2O and PBS buffer solution) as indicated by DLS analysis showing an average Z size of approximately 55 nm, low PDI values, and remarkable stability even after 7 days. These samples exhibited consistent measurements even after undergoing freeze or heat treatments at 40 °C.In conclusion, our research has accomplished the development of novel glycopolymer compounds with exceptional stability in water, indicating their potential applications in various fields. Additionally, our advances in organic synthesis have allowed the production of pure products through efficient and few-step synthetic routes. We believe that this work establishes a solid foundation for future investigations in the field of glycopolymer research and their applications in diverse scientific and technological domains.
Experimental
Materials and methods
DCM, MeOH, EtOAc and hexane solvents were previously distilled. All starting materials were of the best available grade (Merck, Fluka, Anedra, AK Scientific, Alpha Aesar) and were used without further purification unless otherwise stated. Pyrocatechol was recrystallised from hexane before use it. Nitric acid 65% and hydrochloric acid 37% used for analysis of the metal traces were purchased from Cicarelli and Anedra respectively. Column chromatography was performed with Merck silica gel 60 (0.040–0.063 μm, 240–400 mesh) and hexane/EtOAc as eluent. Reactions were monitored by thin-layer chromatography on silica gel plates (60F-254) visualised under UV light and/or using FeCl3 in water for staining.NMR spectra were recorded on a Bruker ARX-300 spectrometer using CDCl3 or CD3OD as solvents. High-resolution mass spectra were recorded using a Bruker micrOTOF-Q II mass spectrometer under ESI ionisation mode. IR spectra were recorded on a Nicolet Nexus FT spectrophotometer instrument in the range of 4000–400 cm−1 using KBr pellets. UV-Vis spectra of liquid samples were obtained on a Cary 60 UV-Vis Spectrophotometer from Agilent, equipped with a Xenon flash lamp (80 Hz), double beam photometric system and 1.5 nm spectral bandwidth. Spectra were collected on a range wavelength from 200 to 800 nm. UV-Vis spectra of CGPs were registered on a Thermo Scientific™ ISA-220 with Integrating Sphere Accessory in reflectance configuration. Spectra were collected on a range wavelength from 300 to 1100 nm.
CGPs samples were lyophilised with a Rificor L-A-B4 freeze dry acrylic chamber. Temperature range −40 °C to 40 °C. Vacuum power: 0.001 mm Hg. SEM images were performed in a LEO EVO 40XVP system operated at 10 kV coupled to an EDX detector Oxford X-Max 50. Samples were prepared by drop casting of the corresponding dispersion on an aluminium tape followed by evaporation of the solvent under room conditions, then metallised with gold in a sputter coater.
The CGPs iron content was determined by using an Atomic Absorption Spectrometer, AAnalyst200 from PerkinElmer equipped with a Fe hollow cathode lamp (HCLs). Before its use, all glass material was washed with HNO3 and rinsed with distilled water. The standard solution from Merck Certipur of 1000 ppm Fe in 5% HNO3 was diluted up to prepare the calibration curves (1, 3 and 6 ppm). Size distribution and electrophoretic mobilities were measured using a Malvern Zetasizer Nano ZS90 equipment.
Preparation of Michael adducts 1
For the synthesis of adducts 1, 2 mmol of the corresponding dithiol (or trithiol) were dissolved in 3 mL of CH2Cl2 in a Schlenk flask under a nitrogen atmosphere. To this solution, 460 μL of trifluoroacetic acid (TFA, 2 mmol) were added with stirring. In a separate flask, a solution of 468 mg of NaIO4 (2.2 mmol) in 80 mL of H2O was prepared and cooled in an ice bath. 220 mg of pyrocatechol (2 mmol) dissolved in 1 mL of Et2O were added to the aqueous solution and vigorously stirred for 15 min. The resulting orange-reddish o-benzoquinone was extracted with 4 × 15 mL of DCM, and the organic phase was dried over anhydrous Na2SO4, filtered, and immediately added to the dithiol solution. The reaction mixture was stirred in the dark at room temperature for 6 h. After this, the solvent and TFA were evaporated under reduced pressure and the crude product was purified by flash column chromatography (hexane–EtOAc) to obtain the corresponding Michael adducts. Compounds 1a and 1b were characterised by comparing their physical and spectroscopic data with those described in the literature (see ESI S2†).22a:AcOEt, 70:30). 1H NMR (300 MHz, CDCl3) δ 7.00 (dd, J = 7.8, 1.6 Hz, 1H), 6.92 (dd, J = 8.1, 1.6 Hz, 1H), 6.79 (deform. dd, J = 8.1, 7.8 Hz, 1H), 6.56 (br. s, 1H, OH), 5.40 (br. s, 1H, OH), 2.70 (t, J = 7.3 Hz, 2H), 2.51 (deform. dt, J = 7.8, 7.3 Hz, 2H), 1.63–1.51 (m, 4H), 1.40–1.36 (m, 4H), 1.33 (t, J = 7.8 Hz, 1H, SH). 13C NMR (75 MHz, CDCl3) δ 143.9 (C); 143.6 (C); 126.2 (CH); 120.5 (CH); 119.4 (C); 116.1 (CH); 36.1 (CH2); 33.5 (CH2); 29.2 (CH2); 27.7 (CH2); 27.5 (CH2); 24.2 (CH2).:AcOEt, 60:40). 1H NMR (300 MHz, CDCl3) 7.00–6.92 (m, 3H, OH), 6.79 (deform. dd, J = 7.8 Hz, 1H), 5.96 (s, 1H, OH), 4.09 (s, 6H), 2.97 (t, J = 6.6 Hz, 2H), 2.82–2.72 (m, 4H), 2.70–2.65 (m, 4H), 2.56 (t, J = 6.6 Hz, 2H), 1.64 (t, J = 8.2 Hz, 2H, SH), 1.51 (q, J = 7.5 Hz, 2H), 0.91 (t, J = 7.6 Hz, 3H). 13C NMR (75.5 MHz, CDCl3) δ 172.0 (CO), 171.7 (2× CO); 145.1 (C); 144.6 (C); 127.0 (CH); 121.2 (CH); 118.0 (C); 117.0 (CH); 64.3 (CH2); 64.0 (2× CH2); 40.9 (C); 38.6 (2× CH2); 34.0 (CH2); 31.4 (CH2); 23.1 (CH2); 19.9 (2× CH2), 7.6 (CH3).Preparation of thiol–catechol compounds 2
To a solution of compound 1 (1.11 mmol) and peracetylated sugar (0.07 mmol) in dry CH2Cl2 (18 mL) under nitrogen atmosphere, freshly activated powdered molecular sieves (4 Å) were added. After 30 minutes, the mixture was cooled using an ice bath, and BF3·OEt2 (1.11 mmol, 136.8 μL) was added. The reaction was stirred at room temperature for 4 h. The crude mixture was filtered through a pad of Celite and washed with a saturated NaHCO3 solution. Finally, the solvent was evaporated under reduced pressure, and the crude product was purified by flash column chromatography (hexane–EtOAc) to obtain the corresponding thiol–catechol compounds.:AcOEt, 60:40). 1H NMR (300 MHz, CDCl3) δ 6.96 (dd, J = 7.7, 1.5 Hz, 1H), 6.87 (dd, J = 8.1, 1.5 Hz, 1H), 6.75 (deform. dd, J = 8.1, 7.7 Hz, 1H), 6.58 (br. s, 1H, OH), 5.74 (br. s, 1H, OH), 5.41 (d, J = 3.1 Hz, 1H), 5.21 (deform. dd, J = 10.0, 9.9 Hz, 1H), 5.03 (dd, J = 9.9, 3.1 Hz, 1H), 4.44 (d, J = 10.0 Hz, 1H), 4.15 (dd, J = 11.3, 6.7 Hz, 1H), 4.08 (dd, J = 11.3, 6.6 Hz, 1H), 3.90 (deform. dd, J = 6.7, 6.6 Hz, 1H), 2.71–2.60 (m, 4H), 2.12, 2.04, 2.02, 1.97 (s, 12H, 4× CH3CO), 1.66–1.47 (m, 4H), 1.46–1.26 (m, 4H). 13C NMR (75.5 MHz, CDCl3) δ 170.4, 170.2, 170.1, 169.6 (4× CO); 144.2 (C); 143.8 (C); 126.3 (CH); 120.6 (CH); 119.2 (C); 116.2 (CH); 84.0 (CH); 74.3 (CH); 71.8 (CH); 67.2 (CH); 67.2 (CH); 61.4 (CH2); 36.3 (CH2); 29.9 (CH2); 29.3 (CH2), 29.3 (CH2); 28.0 (CH2); 27.9 (CH2); 20.7 (CH3), 20.6 (2× CH3), 20.5 (CH3). HRMS (ESI) m/z calcd for C26H36NaO11S2+, [M + Na]+: 611.1597; found 611.,1591.:AcOEt, 50:50). 1H NMR (300 MHz, CDCl3) 6.99 (dd, J = 7.8, 1.4 Hz, 1H), 6.94 (dd, J = 7.9, 1.4 Hz, 1H), 6.79 (deform. dd, J = 7.9, 7.8 Hz, 1H), 5.43 (dd, J = 3.3, 1.1 Hz, 1H), 5.23 (deform. dd, J = 10.1, 9.9 Hz, 1H), 5.05 (dd, J = 10.1, 3.3 Hz, 1H), 4.51 (d, J = 9.9 Hz, 1H), 4.18–4.03 (m, 8H), 3.94 (td, J = 6.6, 1.1 Hz, 1H), 3.04–2.85 (m, 4H), 2.81–2.66 (m, 6H), 2.53 (t, J = 6.6 Hz, 2H), 2.16, 2.07, 2.05, 1.99 (4s, 12H, 4× CH3CO), 1.65 (t, J = 8.1 Hz, 1H, SH), 1.50 (q, J = 7.5 Hz, 2H), 0.90 (t, J = 7.5 Hz, 3H), OH n.d. 13C NMR (75.5 MHz, CDCl3) δ 171.6, 171.5, 171.4, 170.4, 170.2, 170.0, 169.6 (7× CO); 145.0 (C); 144.5 (C); 126.8 (CH); 121.0 (CH); 117.9 (C); 116.9 (CH); 84.4 (CH); 74.5 (CH); 71.9 (CH); 67.3 (CH); 67.1 (CH); 64.2 (CH2); 64.0 (CH2); 63.9 (CH2); 61.5 (CH2); 40.8 (C); 38.4 (CH2); 35.3 (CH2), 33.8 (CH2); 31.3 (CH2); 25.3 (CH2); 23.0 (CH2); 20.9 (CH3); 20.7 (2× CH3); 20.7 (CH3); 19.7 (CH2); 7.4 (CH3). HRMS (ESI) m/z calcd for C35H48NaO17S3+, [M + Na]+: 859.1951; found 859.1946.:AcOEt, 50:50). 1H NMR (300 MHz, CDCl3) 6.98 (dd, J = 7.8, 1.6 Hz, 1H), 6.92 (dd, J = 7.9, 1.6 Hz, 1H), 6.78 (deform. dd, J = 7.9, 7.8 Hz, 1H), 5.21 (deform. dd, J = 9.8, 9.7 Hz, 1H), 5.12–4.98 (m, 2H), 4.51 (d, J = 10.0 Hz, 1H), 4.23 (dd, J = 12.4, 4.8 Hz, 1H), 4.10–4.04 (m, 7H), 3.70 (ddd, J = 10.1, 4.9, 2.5 Hz, 1H), 2.97 (t, J = 6.5 Hz, 2H), 2.96 (t, J = 7.0 Hz, 1H), 2.88 (t, J = 6.9 Hz, 1H), 2.80–2.63 (m, 6H), 2.54 (t, J = 6.5 Hz, 2H), 2.07, 2.04, 2.02, 2.00 (4s, 12H, 4× CH3CO), 1.63 (t, J = 8.2 Hz, 1H, SH), 1.49 (q, J = 7.5 Hz, 2H), 0.89 (t, J = 7.5 Hz, 3H), OH n.d. 13C NMR (75.5 MHz, CDCl3) δ 171.7, 171.4, 171.4, 170.6, 170.1, 169.4, 169.4 (7× CO); 144.9 (C); 144.4 (C); 126.7 (CH); 120.9 (CH); 117.8 (C); 116.8 (CH); 83.9 (CH); 76.1 (CH); 73.9 (CH); 69.8 (CH); 68.4 (CH); 64.2 (CH2); 64.0 (CH2); 63.9 (CH2); 62.2 (CH2); 40.8 (C); 38.3 (CH2); 35.2 (CH2), 33.7 (CH2); 31.3 (CH2); 25.1 (CH2); 22.9 (CH2); 20.7 (2× CH3); 20.6 (2× CH3); 19.6 (CH2); 7.5 (CH3). HRMS (ESI) m/z calcd for C35H48NaO17S3+, [M + Na]+: 859.1951; found 859.1946.:AcOEt, 50:50). 1H NMR (300 MHz, CDCl3) 6.93 (br. s, 1H, OH), 6.91 (d, J = 7.8 Hz, 1H), 6.85 (dd, J = 8.0 Hz, 1H), 6.72 (deform. dd, J = 7.9, 7.8 Hz, 1H), 5.78 (br. s, 1H, OH), 5.30–5.22 (m, 2H), 5.16 (dd, J = 10.1, 3.2 Hz, 1H), 4.33–4.15 (m, 2H), 4.10–3.97 (m, 8H), 2.90 (t, J = 6.6 Hz, 2H), 2.82 (deform. dd, J = 6.8, 6.6 Hz, 1H), 2.75–2.56 (m, 7H), 2.48 (t, J = 6.6 Hz, 2H), 2.10, 2.04, 1.98, 1.92 (4s, 12H, 4× CH3CO), 1.57 (t, J = 8.2 Hz, 1H, SH), 1.43 (q, J = 7.4 Hz, 2H), 0.84 (t, J = 7.4 Hz, 3H). 13C NMR (75.5 MHz, CDCl3) δ 171.8, 171.6, 171.3, 170.8, 170.1, 170.0, 169.9 (7× CO); 145.0 (C); 144.5 (C); 126.9 (CH); 121.1 (CH); 117.9 (C); 116.9 (CH); 85.8 (CH); 71.0 (CH); 69.5 (CH); 69.3 (CH); 66.3 (CH); 64.1 (CH2); 64.0 (CH2); 63.9 (CH2); 62.5 (CH2); 40.9 (C); 38.5 (CH2); 34.6 (CH2); 33.9 (CH2); 31.5 (CH2); 26.4 (CH2); 23.0 (CH2); 21.0 (CH3); 20.9 (CH3); 20.8 (2× CH3); 19.8 (CH2); 7.5 (CH3). HRMS (ESI) m/z calcd for C35H48NaO17S3+, [M + Na]+: 859.1951; found 859.1949.:AcOEt, 50:50). 1H NMR (300 MHz, CDCl3) 7.11 (d, J = 7.7, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.77 (deform. dd, J = 8.0, 7.7 Hz, 1H), 6.56 (br. s, 1H, OH), 5.36–5.21 (m, 2H), 5.19–5.08 (m, 2H), 4.97–4.89 (m, 2H), 4.53–4.39 (m, 2H), 4.18–3.98 (m, 10H), 3.92–3.73 (m, 2H), 3.07 (t, J = 7.1 Hz, 2H), 2.79–2.71 (m, 4H), 2.66–2.62 (m, 4H), 2.58 (t, J = 7.1 Hz, 2H), 2.14, 2.11, 2.10, 2.07, 2.05, 2.04, 1.96 (7s, 21H, 7× CH3CO), 1.61 (t, J = 8.1 Hz, 1H, SH), 1.48 (q, J = 7.3 Hz, 2H), 0.89 (t, J = 7.3 Hz, 3H). 13C NMR (75.5 MHz, CDCl3) δ 171.5, 171.4, 170.5, 170.4, 170.2× 3, 169.7, 169.3, 169.2 (10× CO); 144.7 (C); 144.3 (C); 129.1 (CH); 120.9 (CH); 120.2 (C); 118.4 (CH); 101.2 (CH); 101.0 (CH); 76.2 (CH); 73.1 (CH); 72.4 (CH); 71.6 (CH); 71.0 (CH); 70.9 (CH); 69.2 (CH); 68.2 (CH); 66.7 (CH); 64.1 (CH2); 64.0 (CH2); 61.9 (CH2); 60.9 (CH2); 60.8 (CH2); 40.8 (C); 38.5× 2 (CH2); 38.4 (CH2); 34.4 (CH2), 28.9 (CH2); 23.1 (CH2); 21.1 (CH3); 21.0 (2× CH3); 20.9 (2× CH2); 20.8 (CH3); 20.6 (CH3); 19.8 (CH2); 7.5 (CH3).:AcOEt, 30:70). 1H NMR (300 MHz, CDCl3) δ 7.28 (s, 1H, OH), 7.01 (dd, J = 7.8, 1.6 Hz, 1H), 6.93 (J = 8.0, 1.6 Hz, 1H), 6.76 (deform. dd, J = 8.0, 7.8 Hz, 1H), 5.45 (s, 1H, OH), 4.74 (d, J = 1.2 Hz, 1H), 4.00 (d, J = 1.3 Hz, 1H), 3.93–3.88 (m, 2H), 3.39 (s, 3H), 3.28–3.08 (qd, J = 7.1 Hz, 1.3 Hz, 1H), 1.28 (d, J = 7.1 Hz, 3H), 0.90 (s, 9H, (CH3)3CSiMe2), 0.83 (s, 9H, (CH3)3CSiMe2), 0.10, 0.09, 0.07, 0.03 (4s, 12H, 4× CH3, (CH3)2SiBut). 13C NMR (75 MHz, CDCl3) δ 145.4 (C); 144.3 (C); 128.0 (CH); 120.5 (CH); 117.6 (C); 116.5 (CH); 109.4 (CH); 85.5 (CH); 84.1 (CH); 80.7 (CH); 55.0 (CH3O); 46.3 (CH); 25.8 ((CH3)3CSiMe2); 25.7 ((CH3)3CSiMe2); 17.9 (C, Me3CSiMe2); 17.8 (C, Me3CSiMe2); 15.5 (CH3); −4.1, −4.5, −4.6, −4.9 (4× Me, (CH3)2SiBut). HRMS (ESI) m/z calcd for C25H46NaO6SSi2+, [M + Na]+: 553.2451; found 553.2446.Preparation of products 3
Compound 2 (0.22 mmol) was dissolved in anhydrous MeOH (12 mL) under a nitrogen atmosphere, and K2CO3 (0.40 mmol) was added. The reaction progress was monitored by TLC until starting material was fully consumed. Subsequently, the reaction mixture was passed through a column packed with Dowex 50×, serving as an acid source, and washed with MeOH. The resulting product was dried and characterised without undergoing further purification.:MeOH, 80:20). 1H NMR (300 MHz, CD3OD) δ 6.84 (dd, J = 7.5, 1.8 Hz, 1H), 6.74 (dd, J = 8.0, 1.8 Hz, 1H), 6.67 (deform. dd, J = 8.0, 7.5 Hz, 1H), 4.32 (d, J = 9.3 Hz, 1H), 3.91 (d, J = 3.2 Hz, 1H), 3.77 (dd, J = 11.4, 6.9 Hz, 1H), 3.71 (dd, J = 11.4, 5.4 Hz, 1H), 3.57 (deform. dd, J = 9.3, 9.2 Hz, 1H), 3.56–3.52 (m, 1H), 3.46 (dd, J = 9.2, 3.2 Hz, 1H), 2.80 (t, J = 7.2 Hz, 2H), 2.78–2.64 (m, 2H), 1.68–1.55 (m, 4H), 1.50–1.36 (m, 4H), OH n.d. 13C NMR (75 MHz, CD3OD) δ 146.3 (C); 146.2 (C); 124.7 (CH); 122.4 (C); 120.7 (CH); 115.7 (CH); 87.6 (CH); 80.4 (CH); 76.2 (CH); 71.4 (CH); 70.4 (CH); 62.5 (CH2); 34.7 (CH2); 30.8 (CH2); 30.7 (CH2); 30.3 (CH2); 29.3 (CH2); 29.2 (CH2).:MeOH, 70:30). 1H NMR (300 MHz, CD3OD) 6.84 (d, J = 7.7 Hz, 1H), 6.76 (dd, J = 7.8 Hz, 1H), 6.66 (deform. dd, J = 7.8, 7.7 Hz, 1H), 4.35 (d, J = 9.2 Hz, 1H), 4.09–4.04 (m, 6H), 3.88 (d, J = 3.1 Hz, 1H), 3.78–3.64 (m, 2H), 3.58–3.44 (m, 3H), 3.06 (t, J = 6.9 Hz, 2H), 3.00–2.85 (m, 2H), 2.78–2.71 (m, 4H), 2.69–2.64 (m, 2H), 2.59 (t, J = 6.8 Hz, 2H), 1.52 (q, J = 7.6 Hz, 2H), 0.92 (t, J = 7.4 Hz, 3H), OH n.d. 13C NMR (75.5 MHz, CD3OD) δ 173.4, 173.2, 173.1 (3× CO); 146.8 (C); 146.5 (C); 125.7 (CH); 120.8 (C); 120.8 (CH); 116.4 (CH); 87.8 (CH); 80.6 (CH); 76.2 (CH); 71.3 (CH); 70.5 (CH); 65.1 (CH2); 65.0 (CH2); 65.0 (CH2); 62.6 (CH2); 42.1 (C); 39.5 (CH2); 36.5 (CH2); 35.3 (CH2); 29.9 (CH2); 26.3 (CH2); 24.0 (CH2); 20.3 (CH2); 7.7 (CH3).Preparation of product 4a
Compound 2b (0.73 mmol, 610 mg) was dissolved in anhydrous EtOH (4 mL) and an I2 solution (0.37 mmol in 5.4 mL of EtOH) was added dropwise until a persistent yellow/orange colour was observed. The reaction mixture was then evaporated, and the resulting residue was redissolved in AcOEt. It was extracted with a 10% Na2S2O3 solution (2 × 12 mL) and washed with water. The organic phase was separated and dried over Na2SO4, resulting in the quantitative yield of disulphide 4a as a dense, pale yellow foam.:AcOEt, 30:70). 1H NMR (300 MHz, CDCl3) δ 7.02 (s, 2H, OH), 6.97 (d, J = 7.9 Hz, 2H), 6.93 (d, J = 8.0 Hz, 2H), 6.78 (deform. dd, J = 8.0, 7.9 Hz, 2H), 5.99 (s, 2H, OH), 5.42 (d, J = 3.3 Hz, 2H), 5.22 (deform. dd, J = 10.0, 9.9 Hz, 2H), 5.04 (dd, J = 10.0, 3.3 Hz, 2H), 4.51 (d, J = 9.9 Hz, 2H), 4.15–4.01 (m, 16H), 3.93 (t, J = 6.5 Hz, 2H), 3.02–2.84 (m, 12H), 2.78–2.68 (m, 8H), 2.54 (t, J = 6.5 Hz, 4H), 2.14, 2.06, 2.03, 1.98 (4s, 24H, 8× CH3CO), 1.49 (q, J = 7.4 Hz, 4H), 0.89 (t, J = 7.4 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 171.7, 171.5, 171.4, 170.4, 170.2, 170.0, 169.6 (14× CO); 144.9 (2× C); 144.4 (2× C); 126.7 (2× CH); 120.9 (2CH); 117.8 (2× C); 116.8 (2× CH); 84.3 (2× CH); 74.4 (2× CH); 71.7 (2× CH); 67.1 (2× CH); 67.0 (2× CH); 64.1 (2× CH2); 63.9 (2× CH2); 63.8 (2× CH2); 61.3 (2× CH2); 40.7 (2× C); 35.2 (2× CH2); 33.9 (2× CH2); 33.7 (2× CH2); 32.7 (2× CH2); 31.3 (2× CH2); 25.2 (2× CH2); 22.8 (2× CH2); 20.8 (2× CH3); 20.7 (2× CH3); 20.7 (2× CH3); 20.6 (2× CH3); 7.3 (2× CH3). HRMS (ESI) m/z calcd for C70H94NaO34S6+, [M + Na]+: 1693.3848; found 1693.3843.Preparation of CGPs
Ligand 4a (40 mg, 0.024 mmol) was placed in a vial equipped with magnetic stirrer and dissolved in DMSO (250 μL). After that, 2 mL of MeOH was added. On the other hand, FeCl3·6H2O (6.5 mg, 0.024 mmol) was dissolved in 100 μL of water. This aqueous solution was added dropwise to the organic solution of 4a under stirring at 1000 rpm. Then, a 0.1 M NaOH solution was added until a violet-blue colouration appeared. Finally, an aqueous solution of 0.1 M NaOH was added until a deep violet-blue colour was achieved (approximately 0.3 mL of the solution). The solution was left under agitation for 3 hours, and then the precipitate was centrifuged, washed with water (×2) and MeOH (×3). The obtained yield ranged from 60–75% by mass.Quantification of Fe in CGPs by atomic absorption
To begin the procedure, 8 mg of the solid CGPs were weighed into a vial equipped with a magnetic stirrer. Then, 2 mL of concentrated HNO3 was added slowly while stirring for 24 h, resulting in an orange-red coloration. Following that, 2 mL of concentrated HCl was added, and the mixture was stirred for an additional 24 h. The resulting solution was translucent and yellow in colour, which was then diluted with water. This entire procedure was performed in triplicate. After the atomic absorption analysis, the average Fe concentration was determined to be 3.1% by mass. This value is very close to the theoretical value of 3.2%.Author contributions
C. B.-M., J. J. C. and B. A. performed the experiments and collected data. M. L. F. conducted the experiments, discussed the results, and participated in the writing. G. R. wrote the manuscript and funded the present work. D. R.-M. participated in the supervision of the writing as mentorship external to the core team. J. M.-A. and F. N. were responsible for the conceptualization, direction and writing of the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Eliana Pecini from Universidad Nacional del Sur for her assistance in the realization of the DLS measurements and analysis. CBM thanks CIN for his student grant. JJC and BA thank CONICET for supporting his doctoral work. This work was supported by the FONCyT (Projects PICT 2018-02471 and PICT 2020-03931), SGCyT-UNS (Project PGI 24/Q106) and CONICET (Project PIP 11220200101665CO).References
- X. Zhang, G. Huang and H. Huang, The glyconanoparticle as carrier for drug delivery, Drug Delivery, 2018, 25, 1840–1845 CrossRef CAS PubMed.
- S. Ganda, Y. Jiang, D. S. Thomas, J. Eliezar and M. H. Stenzel, Biodegradable glycopolymeric micelles obtained by RAFT-controlled radical ring-opening polymerization, Macromolecules, 2016, 49, 4136–4146 CrossRef CAS.
- I. García, M. Marradi and S. Penadés, Glyconanoparticles: multifunctional nanomaterials for biomedical applications, Nanomedicine, 2010, 5, 777–792 CrossRef PubMed.
- J. M. de la Fuente and S. Penadés, Understanding carbohydrate-carbohydrate Interactions by means of glyconanotechnology, Glycoconjugate J., 2004, 21, 149–163 CrossRef CAS PubMed.
- M. Marradi, M. Martín-Lomas and S. Penadés, in Glyconanoparticles polyvalent tools to study carbohydrate-based interactions, Advances in carbohydrate chemistry and biochemistry, ed., D. Horton, Academic Press, 2010, Vol. 64, pp. 211–290 Search PubMed.
- B. T. Houseman and M. Mrksich, in Host-Guest Chemistry, Topics in Current Chemistry, ed., S. Penadés, Springer, Berlin, Heidelberg, Germany, 2002, Vol. 218 Search PubMed.
- (a) J. Rieger, H. Freichels, A. Imberty, J.-L. Putaux, T. Delair, Ch. Jérôme and R. Auzély-Velty, Polyester nanoparticles presenting mannose residues: toward the development of new vaccine delivery systems combining biodegradability and targeting properties, Biomacromolecules, 2009, 10(3), 651–657 CrossRef CAS PubMed; (b) M. N. Jones, Carbohydrate-mediated liposomal targeting and drug delivery, Adv. Drug Delivery Rev., 1994, 13, 215–249 CrossRef CAS; (c) X. Li, X. Rao, L. Cai, X. Liu, H. Wang, W. Wu, Ch. Zhu, M. Chen, P. G. Wang and W. Yi, Targeting Tumor Cells by Natural Anti-Carbohydrate Antibodies Using Rhamnose-Functionalized Liposomes, ACS Chem. Biol., 2016, 11(5), 1205–1209 CrossRef CAS PubMed.
- (a) J. M. de la Fuente, A. G. Barrientos, T. C. Rojas, J. Rojo, J. Cañada, A. Fernández and S. Penadés, Gold Glyconanoparticles as Water-Soluble Polyvalent Models to Study Carbohydrate Interactions, Angew. Chem., Int. Ed., 2001, 40, 2257–2261 CrossRef; (b) J. M. de la Fuente and S. Penadés, Glyconanoparticles: types, synthesis and applications in glycoscience, biomedicine and material science, Biochim. Biophys. Acta, 2006, 1760, 636–651 CrossRef CAS PubMed; (c) A. Carvalho de Souza, K. M. Halkes, J. D. Meeldijk, A. J. Verkleij, J. F. G. Vliegenthart and J. P. Kamerling, Gold Glyconanoparticles as Probes to Explore the Carbohydrate-Mediated Self-Recognition of Marine Sponge Cells, ChemBioChem, 2005, 6, 828–831 CrossRef CAS PubMed.
- (a) R. Yadav and R. Kikkeri, Carbohydrate functionalized iron(iii) complexes as biomimetic siderophores, Chem. Commun., 2012, 48, 1704–1706 RSC; (b) R. Kikkeri, D. Grünstein and P. H. Seeberger, Lectin Biosensing Using Digital Analysis of Ru(II)-Glycodendrimers, J. Am. Chem. Soc., 2010, 132(30), 10230–10232 CrossRef CAS PubMed; (c) J.-F. Nierengarten, J. Lehl, V. Oerthel, M. Holler, B. M. Illescas, A. Muñoz, N. Martín, J. Rojo, M. Sánchez-Navarro, S. Cecioni, S. Vidal, K. Buffet, M. Durka and S. P. Vincent, Fullerene sugar balls, Chem. Commun., 2010, 46, 3860–3862 RSC.
- (a) J. E. Kingery-Wood, K. W. Williams, G. B. Sigal and G. M. Whitesides, The agglutination of erythrocytes by influenza virus is strongly inhibited by liposomes incorporating an analog of sialyl gangliosides, J. Am. Chem. Soc., 1992, 114(18), 7303–7305 CrossRef CAS; (b) J. D. Lewicky, N. L. Fraleigh, A. Boraman, A. L. Martel, T. M.-D. Nguyen, P. W. Schiller, T. Ch. Shiao, R. Roy, S. Montaut and H.-T. Le, Mannosylated glycoliposomes for the delivery of a peptide kappa opioid receptor antagonist to the brain, Eur. J. Pharm. Biopharm., 2020, 154, 290–296 CrossRef CAS; (c) B. Wu, W. Ndugire, X. Chen and M. Yan, Maltoheptaose-Presenting Nanoscale Glycoliposomes for the Delivery of Rifampicin to E. coli, ACS Appl. Nano Mater., 2021, 4(7), 7343–7357 CrossRef CAS PubMed.
- (a) X. Li and G. Chen, Glycopolymer-based nanoparticles: synthesis and application, Polym. Chem., 2015, 6, 1417–1430 RSC; (b) I. Pramudya and H. Chung, Recent progress of glycopolymer synthesis for biomedical applications, Biomater. Sci., 2019, 7, 4848–4872 RSC; (c) A. Bukchin, G. Pascual-Pasto, M. Cuadrado-Vilanova, H. Castillo-Ecija, C. Monterrubio, N. G. Olaciregui, M. Vila-Ubach, L. Ordeix, J. Mora, A. M. Carcaboso and A. Sosnik, Glucosylated nanomicelles target glucose-avid pediatric patient-derived sarcomas, J. Controlled Release, 2018, 276, 59–71 CrossRef CAS PubMed.
- M. R. Thalji, A. A. Ibrahim, K. F. Chong, A. V. Soldatov and G. M. Ali, Glycopolymer Based Materials: Synthesis, Properties, and Biosensing Applications, Top. Curr. Chem., 2022, 45, 380 Search PubMed.
- I. Pramudya and H. Chung, Recent progress of glycopolymer synthesis for biomedical applications, Biomater. Sci., 2019, 7, 4848–4872 RSC.
- X. Li and G. Chen, Glycopolymer-based nanoparticles: synthesis and application, Polym. Chem., 2015, 6, 1417–1430 RSC.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, The Chemistry behind Catechol-Based Adhesion, Angew. Chem., Int. Ed., 2019, 58, 696–714 CrossRef CAS PubMed.
- J. Bijlsma, W. J. C. de Bruijn, J. A. Hageman, P. Goos, K. P. Velikov and J.-P. Vincken, Revealing the main factors and two-way interactions contributing to food discolouration caused by iron-catechol complexation, Sci. Rep., 2020, 10, 8288 CrossRef CAS PubMed.
- (a) J. J. Wilker, Marine bioinorganic materials: mussels pumping iron, Curr. Opin. Chem. Biol., 2010, 14, 276–283 CrossRef CAS PubMed; (b) D. Lee, H. Bae, J. Ahn, T. Kang, D.-G. Seo and D. S. Hwang, Catechol-thiol-based dental adhesive inspired by underwater mussel adhesion, Acta Biomater., 2020, 103, 92–101 CrossRef CAS PubMed.
- (a) B. P. Lee and S. Konst, Novel Hydrogel Actuator Inspired by Reversible Mussel Adhesive Protein Chemistry, Adv. Mater., 2014, 26, 3415–3419 CrossRef CAS PubMed; (b) K. Marcisz, J. Romanski, Z. Stojek and M. Karbarz, Environmentally sensitive hydrogel functionalized with electroactive and complexing-iron(III) catechol groups, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3236–3242 CrossRef CAS.
- (a) J. J. Calmels, L. Aguilar, J. Mancebo-Aracil, G. Radivoy, C. Domini, M. Garrido, M. D. Sánchez and F. Nador, Novel pH-sensitive catechol dyes synthesised by a three component one-pot reaction, Front. Chem., 2023, 10, 1116887 CrossRef PubMed; (b) C. Queirós, A. M. G. Silva, S. C. Lopes, G. Ivanova, P. Gameiro and M. Rangel, A novel fluorescein-based dye containing a catechol chelating unit to sense iron(III), Dyes Pigm., 2012, 93, 1447–1455 CrossRef.
- (a) Q. Wei, K. Achazi, H. Liebe, A. Schulz, P.-L. M. Noeske, I. Grunwald and R. Haag, Mussel-Inspired Dendritic Polymers as Universal Multifunctional Coatings, Angew. Chem., Int. Ed., 2014, 53, 11650–11655 CrossRef CAS PubMed; (b) M. Iacomino, J. Mancebo-Aracil, M. Guardingo, R. Martín, G. D'Errico, M. Perfetti, P. Manini, O. Crescenzi, F. Busqué, A. Napolitano, M. d'Ischia, J. Sedó and D. Ruiz-Molina, Replacing Nitrogen by Sulfur: From Structurally Disordered Eumelanins to Regioregular Thiomelanin Polymers, Int. J. Mol. Sci., 2017, 18, 2169 CrossRef PubMed.
- (a) F. Novio, J. Lorenzo, F. Nador, K. Wnuk and D. Ruiz-Molina, Carboxyl Group (–CO2H) Functionalized Coordination Polymer Nanoparticles as Efficient Platforms for Drug Delivery, Chem.–Eur. J., 2014, 20, 15443–15450 CrossRef CAS PubMed; (b) F. Nador, J. Mancebo-Aracil, D. Zanotto, D. Ruiz-Molina and G. Radivoy, Thiol-yne click reaction: an interesting way to derive thiol-provided catechol, RSC Adv., 2021, 11, 2074–2082 RSC; (c) J. García-Pardo, F. Novio, F. Nador, I. Cavaliere, S. Suárez-García, S. Lope-Piedrafita, A. P. Candiota, J. Romero-Gimenez, B. Rodríguez-Galván, J. Bové, M. Vila, J. Lorenzo and D. Ruiz-Molina, Bioinspired Theranostic Coordination Polymer Nanoparticles for Intranasal Dopamine Replacement in Parkinson's Disease, ACS Nano, 2021, 15(5), 8592–8609 CrossRef PubMed.
- (a) J. Mancebo-Aracil, C. Casagualda, M. A. Moreno-Villaécija, F. Nador, J. García-Pardo, A. Franconetti-García, F. Busqué, R. Alibés, M. J. Esplandiu, D. Ruiz-Molina and J. Sedó-Vegara, Bioinspired Functional Catechol Derivatives through Simple Thiol Conjugate Addition, Chem.–Eur. J., 2019, 25, 12367–12369 CrossRef CAS PubMed; (b) C. Casagualda, J. Mancebo-Aracil, M. Moreno-Villaécija, A. López-Moral, R. Alibés, F. Busqué and D. Ruiz-Molina, Mussel-Inspired Lego Approach for Controlling the Wettability of Surfaces with Colourless Coatings, Biomimetics, 2023, 8, 3 CrossRef CAS PubMed.
- S. Oscarson, in Glycoscience: Chemistry and Chemical Biology I–III, ed., B. O. Fraser-Reid, K. Tatsuta and J. Thiem, Springer, Berlin, Heidelberg, Germany, 2001, Ch. 3.5 Search PubMed.
- S. S. Nigudkar and A. V. Demchenko, Stereocontrolled 1,2-cis glycosylation as the driving force of progress in synthetic carbohydrate chemistry, Chem. Sci., 2015, 6, 2687–2704 RSC.
- M. J. Lo Fiego, C. Marino and O. Varela, Synthesis of galactofuranosyl-(1 → 5)-thiodisaccharide glycomimetics as inhibitors of a β-D-galactofuranosidase, RSC Adv., 2015, 5, 45631–45640 RSC.
- (a) A. W. Hobsteter, M. A. Badajoz, M. J. Lo Fiego and G. F. Silbestri, Galactopyranoside-Substituted N-Heterocyclic Carbene Gold(I) Complexes: Synthesis, Stability, and Catalytic Applications to Alkyne Hydration, ACS Omega, 2022, 7, 21788–21799 CrossRef CAS PubMed; (b) B. Wang, D. D. McClosky, Ch. T. Anderson and G. Chen, Synthesis of a suite of click-compatible sugar analogs for probing carbohydrate metabolism, Carbohydr. Res., 2016, 433, 54–62 CrossRef CAS PubMed; (c) B. Ren, M. Wang, J. Liu, J. G. Xiaoling Zhang and H. Dong, Zemplén transesterification: a name reaction that has misled us for 90 years, Green Chem., 2015, 17, 1390–1394 RSC.
- M. J. Sever and J. J. Wilker, Visible absorption spectra of metal–catecholate and metal–tironate complexes, Dalton Trans., 2004, 1061–1072 RSC.
- (a) J. Bijlsma, W. J. C. de Bruijn, J. A. Hageman, P. Goos, K. P. Velikov and J.-P. Vincken, Revealing the main factors and two-way interactions contributing to food discolouration caused by iron-catechol complexation, Sci. Rep., 2020, 10, 8288 CrossRef CAS PubMed; (b) J. Oh, D. Kang, S. Hong, S. H. Kim, J.-H. Choi and J. Seo, Formation of a tris(catecholato) iron(III) complex with a nature-inspired cyclic peptoid ligand, Dalton Trans., 2021, 50, 3459–3463 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, UV-Vis, IR and EDX spectra, DLS measurements and SEM images. See DOI: https://doi.org/10.1039/d3ra05316d |
| This journal is © The Royal Society of Chemistry 2023 |