
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrrolizine/indolizine-bearing (un)substituted isoindole moiety: design, synthesis, antiproliferative and MDR reversal activities, and in silico studies †
Amr L. AbdelSamada,
Mohammed T. El-Saadibc,
Ahmed M. Goudab and
Asmaa M. AboulMagd *a
*a
aDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Nahda University in Beni-Suef (NUB), Beni-Suef 62513, Egypt. E-mail: asmaa.aboulmaged@nub.edu.eg
bDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Beni-Suef University, Beni-Suef 62514, Egypt
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Sinai University-Kantra Branch, Ismailia, Egypt
First published on 20th October 2023
Abstract
Two new series of pyrrolizine/indolizine derivative-bearing (un)substituted isoindole moiety were designed and synthesized. The anticancer potential of the new compounds was evaluated against hepatocellular carcinoma (HepG-2), colorectal carcinoma, colon cancer (HCT-116), and breast cancer (MCF-7) cell lines. Compounds 6d and 6o were the most potent derivatives with IC50 values ranging from 6.02 to 13.87 μM against HePG-2, HCT-116, and MCF-7 cell lines. Moreover, methyl analog of the fluoro-substituted indolizine derivative 6m revealed significant antiproliferative activity against HePG-2, HCT-116, and MCF-7 cancer cell lines with IC50 values of 11.97, 28.37, and 19.87 μM, respectively. The most active anticancer analogs, 6d, 6m, and 6o, were inspected for their putative mechanism of action by estimating their epidermal growth factor receptor (EGFR) and cyclin-dependent kinase (CDK 2) inhibitory activities. Thus, compound 6o displayed the most inhibitory activity against EGFR and CDK 2 with IC50 values of 62 and 118 nM, respectively. Additionally, the quantitative real-time PCR analysis for the P-glycoprotein effect of compounds 6d, 6m, and 6o was performed, in which compound 6o illustrated significant down-regulation of P-gp against the HepG-2 cell line by 0.2732 fold. Mechanistic studies for the most active compounds involving the reversal doxorubicin (DOX) effect of compounds 6d, 6m, and 6o were performed, which illustrated cytotoxic activity with IC50 22.27, 3.88, and 8.79 μM, respectively. Moreover, the apoptotic activity of the most active derivative 6o on HCT-116 cancer cells showed accumulation in the G1 and S phases of the cell cycle.
1. Introduction
Cancer is one of the most prevalent conditions affecting public health, and it is the second-leading cause of mortality after cardiovascular diseases.1 The clinical efficacy of anticancer drugs that are currently in use has decreased, which may be attributed to certain factors such as increased drug resistance, establishment of tumor resistance, and the severe adverse effects of chemotherapeutic agents.2,3 In addition, multi-drug resistance of cancerous tumors is one of the primary factors contributing to the clinical progression of these tumors.4,5 Although targeted medications and immunotherapy for cancer are becoming common, there is still an urgent need to develop more effective and less resistant anticancer agents.6,7Overexpression of ATP-binding cassette (ABC) transporters is a common cause of multi-drug resistance (MDR), which is the leading cause of chemotherapy failure.8 ABC transporters have been researched extensively; those related to MDR in cancer cells include ABCB1, ABCC1, and ABCG2. ABCB1, often referred to as P-glycoprotein (P-gp), has a high expression in several vital organs, such as the liver, kidney, lung, placenta, and intestines, for the purposes of protection and detoxification.9,10 According to the literature, verapamil (I), febuxostat (II), and the aminopyrimidine-based drug (III) exhibit potential P-glycoprotein inhibitory activity (Fig. 1).11–13
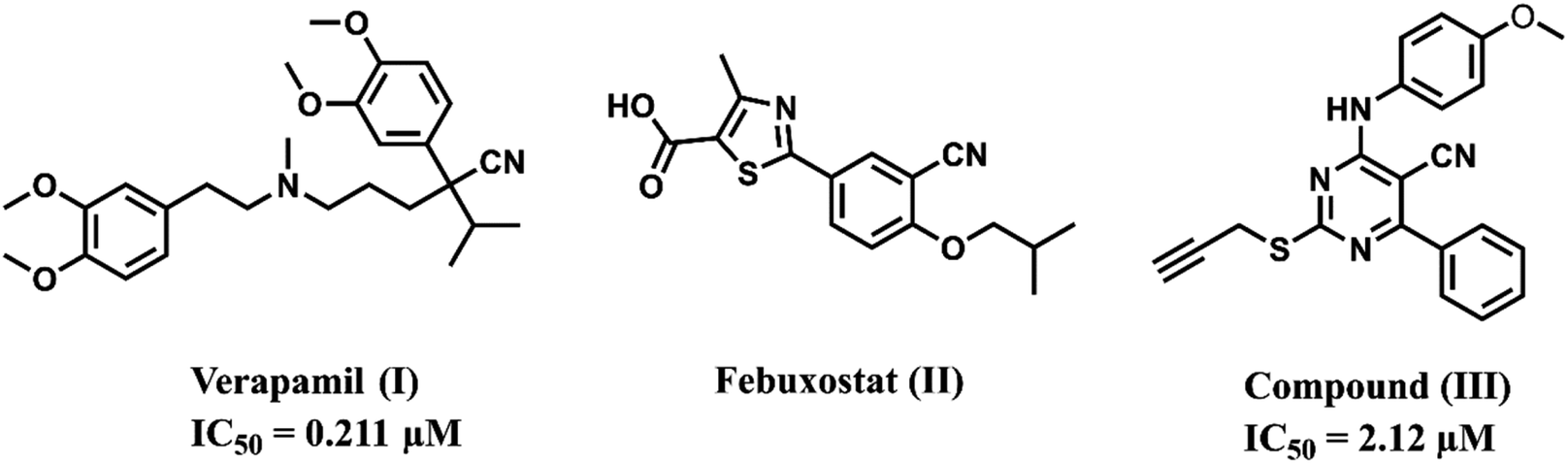 | ||
| Fig. 1 Examples of compounds showing P-glycoprotein inhibitory activity. | ||
Recent studies showed that isoindole derivatives might be potential anticancer agents. For example, compound (IV) exhibited good activity against A549 lung cancer cell lines at IC50 = 19 μM when compared to 5-fluorouracil (V) with IC50 < 5 μM.14 Moreover, derivative (VI) showed significant activity against S1-B1-20 cell lines with IC50 = 0.81 μM; interestingly, the latter derivative was designed as an MDR reversal agent, which showed promising findings, and the mechanism of action of this compound is believed to involve down regulation of the drug efflux pump, P-glycoprotein.15 Furthermore (VII), produced a potentiation effect in the MDR cell line in combination with VIN (60.0%) and PTX (80.5%) (Fig. 2).16
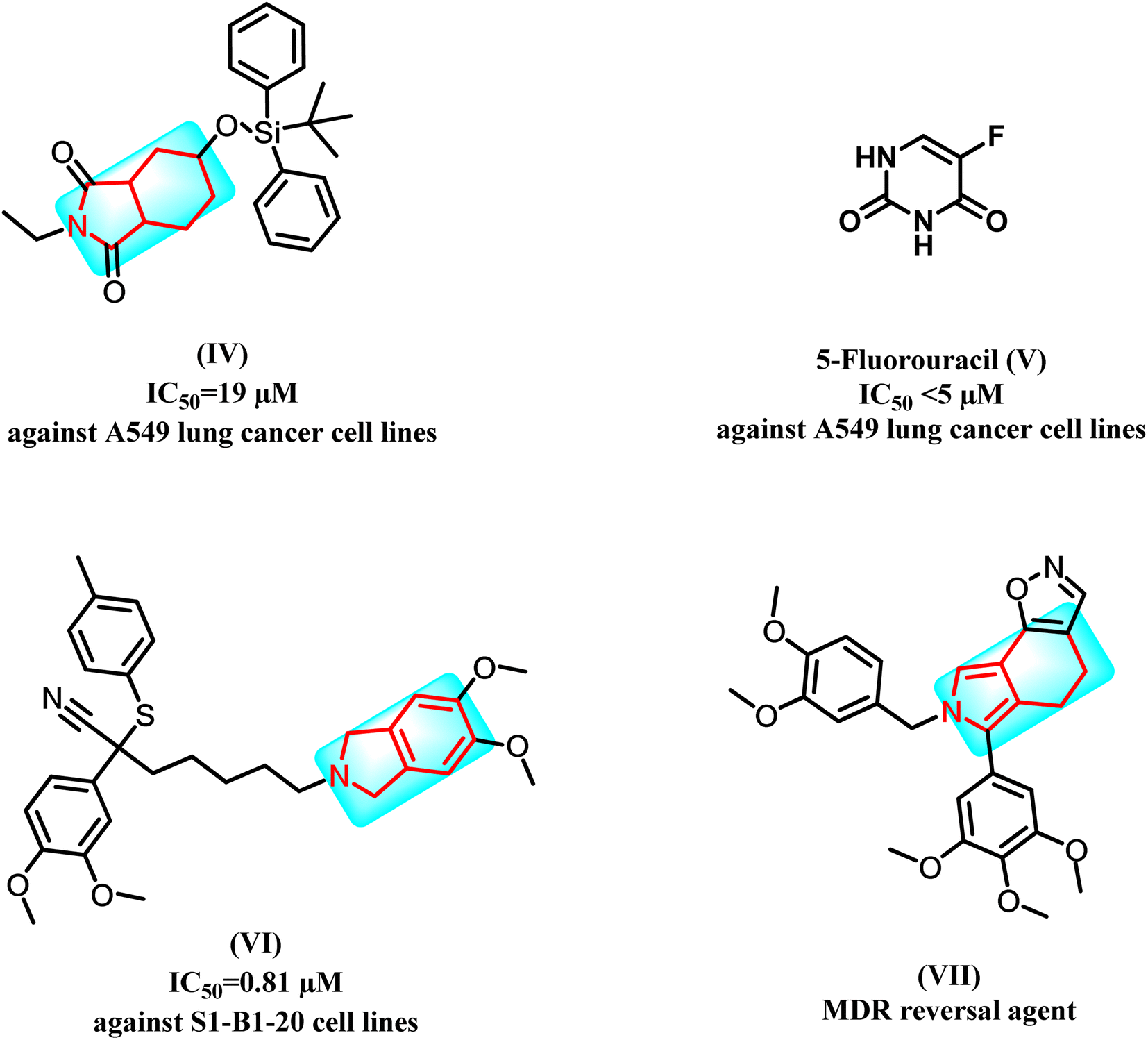 | ||
| Fig. 2 Examples of anticancer agents bearing isoindole moiety. | ||
From another point of view, the pyrrolizine ring comprises the basic skeleton in some compounds with diverse pharmacological activities such as antiviral, antimicrobial, anti-inflammatory, and anticancer activities. It was reported that pyrrolizine-based compounds have the ability to exhibit antitumor activities as a result of their ability to cross-link DNA.17–19 Interestingly, licofelone (VIII) is a pyrrolizine-based compound that exhibited significant cytotoxic activity with an IC50 value of 5.5 μM on breast cancer cell line (MCF-7) by indirect blocking of the EGFR kinase.20–22 In addition, MR22388 (IX) exhibited notable anticancer activity against leukemia L1210 cells, with an IC50 value of 15 nM.23 Pyrrolizine-containing compounds have been reported to show potent inhibition of the mitogen-activated protein kinase (MAP) pathway regulated by the conserved receptor tyrosine kinase (EGFR/MAPK) signaling mechanism. Thus, these compounds displayed strong inhibition of several protein kinases, mainly EGFR and CDK-2, in addition to their ability to inhibit tubulin polymerization, which allowed it to trigger apoptosis in leukemia cell lines (Fig. 3).24,25
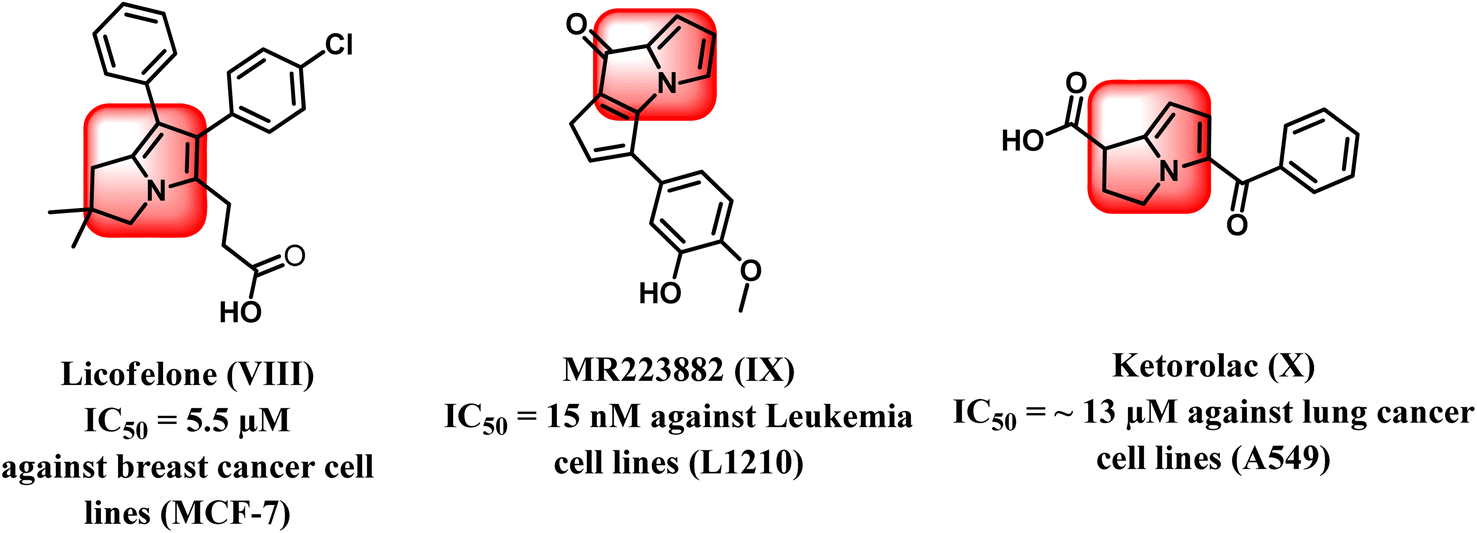 | ||
| Fig. 3 Pyrrolizine-based compounds as anticancer agents. | ||
Also, Ketorolac (X), another drug based on pyrrolizine, had significant anticancer activity against lung cancer cell lines A549 with an IC50 value of about 13 μM26 (Fig. 3).27
Modern bio-/chemoinformatics methods can effectively support the estimation of the most likely targets for small compounds. Thus, the SwissTarget web tool makes predictions about the likeliest protein targets for our designed compounds, resulting in high predictions towards kinase proteins.28 So, this encouraged us to target kinase proteins as epidermal growth factor receptor tyrosine kinases (EGFR TKs) and cyclin-dependent kinases (CDKs) due to their wide carcinogenic signaling pathways.29
Increasing levels of epidermal growth factor receptor EGFR and its endogenous ligands (EGF) have been shown to be a keystone in carcinogenesis as well as playing the most important role in cancer growth.30 Therefore, EGFR is regarded as an applicable target for the improvement of novel anticancer drugs. According to the literature, several pyrrolizine derivatives were developed and synthesized with the belief of more binding moieties, following the pharmacophoric features of EGFR TKIs overcoming the multi-resistance that may have emerged. For example, compound (XI) exhibited EGFR TK percent inhibition of 97.6% at 10 μM and showed anti-proliferative activity against MCF-7 breast cancer cell lines with IC50 value 8.6 μM (Fig. 4).31
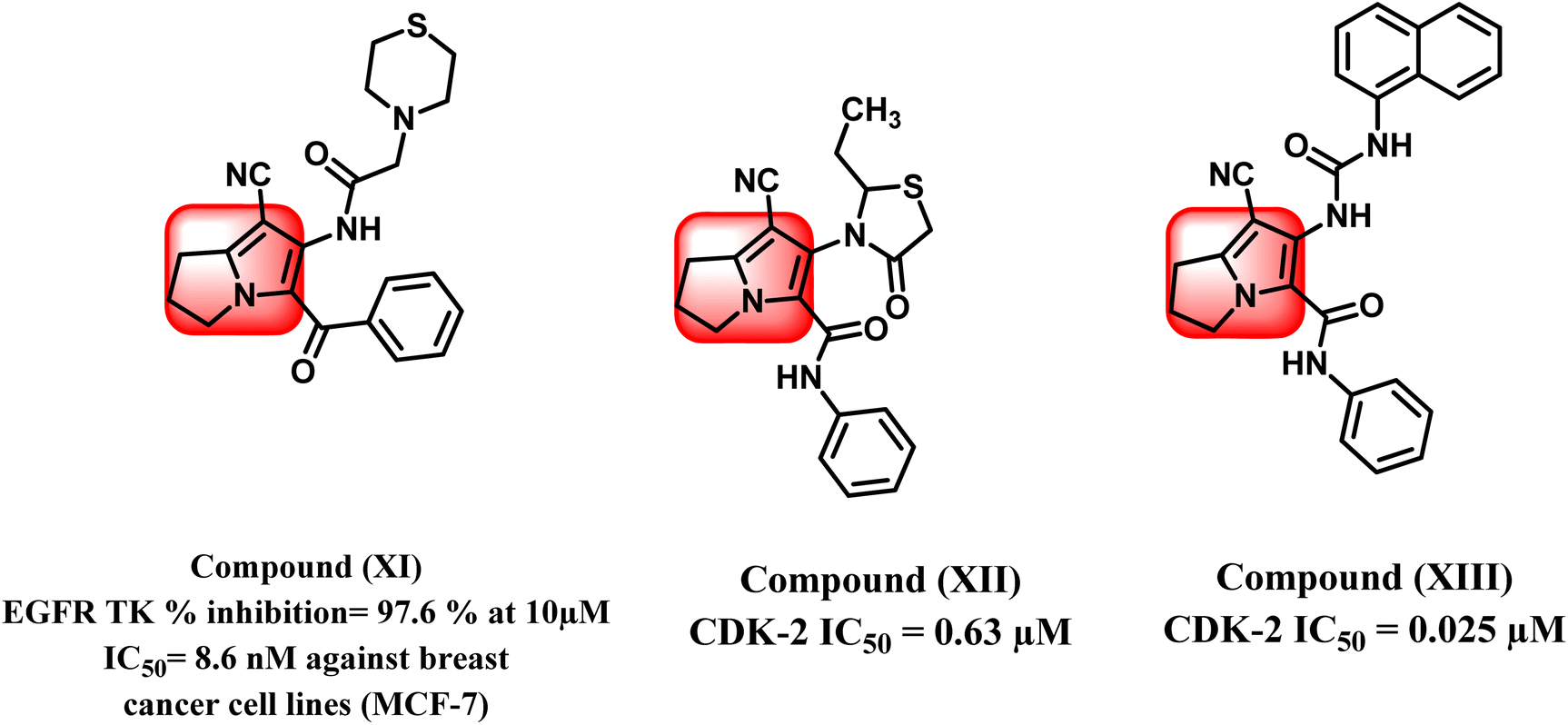 | ||
| Fig. 4 Pyrrolizine as EGFR TK and CDK-2 inhibitors. | ||
In the same context, cell cycle progression and cellular proliferation are both regulated by a family of enzymes known as cyclin-dependent kinases (CDKs).32 They act by phosphorylating crucial serine and threonine residues in host proteins, allowing those proteins to be activated.32 It is widely accepted that blocking CDKs could aid in preventing the unchecked cellular growth observed in some cancers.33
Recent research studies have demonstrated that pyrrolizine scaffold can also participate in designing anticancer agents via inhibition of CDK-2 enzyme. Compounds (XII) and (XIII) showed inhibition of CDK-2 enzyme at IC50 of 0.63 μM and 0.025 μM, respectively.34,35 (Fig. 4).36
Recently, a novel form of cancer treatment called multi-target anticancer drugs has surfaced.37 This method garnered a lot of interest since it might offer a better alternative for combination therapy with less toxicity and drug–drug interaction issues.38 Literature has many examples of multi-target molecules, such as EGFR/CDK-2 dual inhibitors as potent anticancer agents.36,39,40
1.1. Design and rationale
Based on the findings that we retrieved from the literature, hybridization between isoindoline nucleus and pyrrolizine moiety could serve as good anti-proliferative agents via dual inhibition to EGFR TK and CDK-2.41,42 The shared pharmacophoric binding moieties include: (a) hybrid isoindoline-pyrrolizine moiety acts as main scaffold providing the ATP binding site moiety; (b) cyano group may form strong binding interactions with the key amino acids at the active domains of EGFR TK or CDK-2 enzymes and also, considered as an important pharmacophore among P-glycoprotein inhibitors against MDR cancer cell lines; and (c) one or two amide linker between the pyrrolizine main scaffold and the hydrophobic tail I. Regarding the above-mentioned pharmacophore, the newly synthesized compounds were designed as EGFR TK and CDK-2 dual inhibitors by maintaining the essential moieties and applying some lead optimization strategies, which can be summarized in (a) substitution on the hydrophobic tail (phenyl ring) with small and large lipophilic groups with electron donating and electron withdrawing properties to evaluate the structure–activity relationship of this position; (b) ring expansion of pyrrolizine ring to indolizine to assess its possible effect on the anti-proliferative activity, all shown at Fig. 5. The synthesized compounds were confirmed by different spectroscopic techniques to ensure their chemical structures and purity. All the compounds were evaluated for the anti-proliferative activity against HePG2 liver cancer, HCT-116 colon cancer, and MCF-7 breast cancer cell lines. Then, the effect of the most potent compounds on EGFR TK and CDK-2 was also evaluated. Additionally, the most active compounds were tested for their activity against MDR cancer cell lines.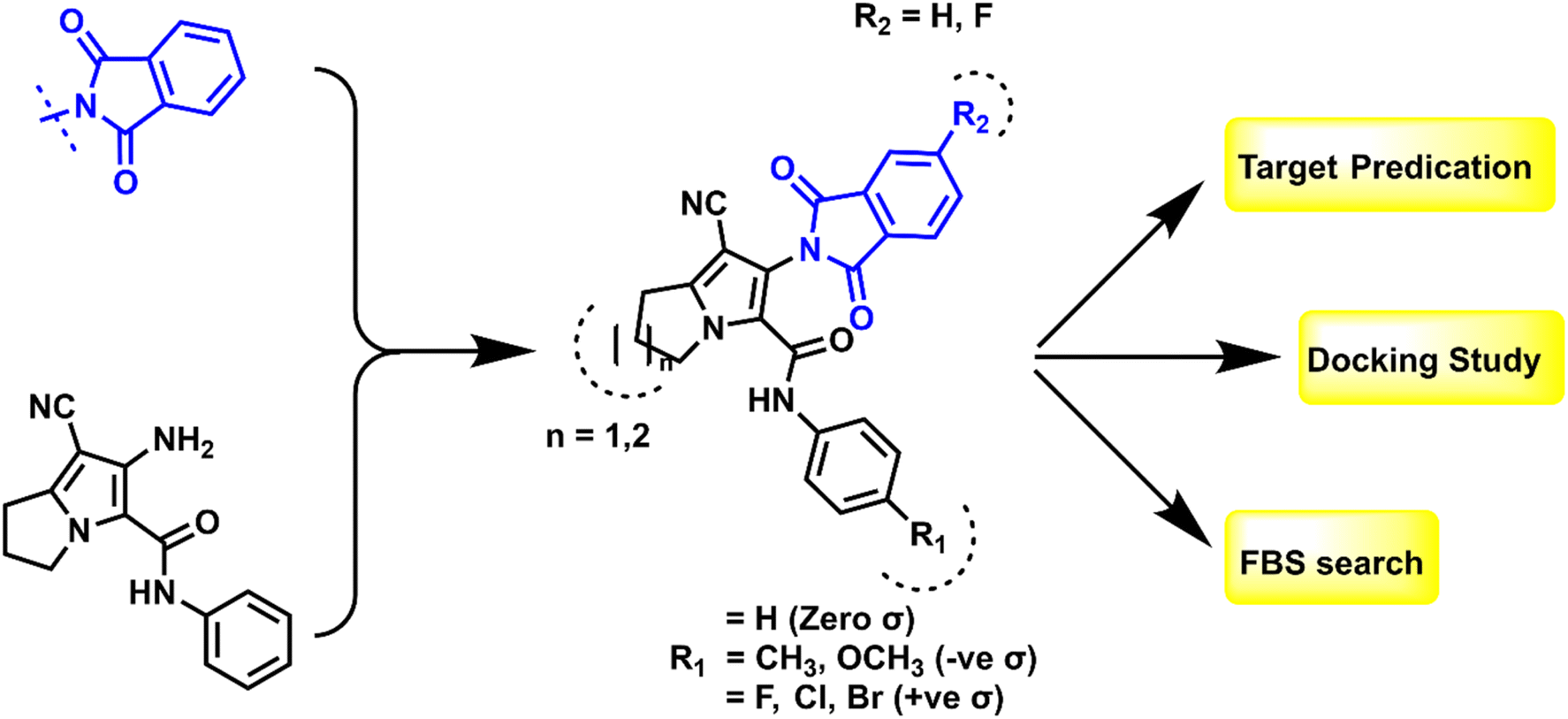 | ||
| Fig. 5 Rationale design of the final compounds 6a–q. | ||
Furthermore, the promising candidates were subjected to apoptosis assay and cell cycle analysis to determine their apoptotic mechanism. Finally, in silico study was performed to elucidate the binding of the active compounds inside their active sites.
2. Results and discussion
2.1. Chemistry
The designed compounds were synthesized according to the chemical pathway outlined in Scheme 1. A literature survey showed that the acetanilide derivatives 2a–f were obtained through acylation of the appropriate amines with chloroacetyl chloride using different reaction conditions such as triethylamine, potassium carbonate, sodium acetate or pyridine in different solvents such as glacial acetic acid, dichloromethane, DMF or acetone.43–46 In this study, the acetanilide derivatives 2a–f were synthesized with good yield using acetic acid and sodium acetate according to the method by Kumar et al.47 On the other hand, intermediates 4a and b were synthesized by reacting 2-pyrrolidinone 3a or 2-piperidinone 3b with malononitrile in dry benzene via condensation reaction.48 These intermediates were confirmed by their melting points, as reported. In this work, two series of the target compounds were obtained via the reaction of intermediates 4a and b with the appropriate anilides 2a–f in acetone catalyzed by potassium carbonate to afford the corresponding key intermediates pyrrolizine derivatives 5a–f or indolizine derivatives 5g–j, respectively.48 The reported intermediates, pyrrolizine derivatives 5a–f and indolizine derivatives 5g and h, were confirmed by their reported melting points and different spectral data according to the literature.27,48 However, two newly synthesized indolizine derivatives 5i and 5j were synthesized and confirmed by different spectroscopic techniques. IR spectra of the latter compounds 5i and 5j revealed absorption bands in the range of 3210–3320 cm−1 (forked band), indicating the presence of –NH2 (primary amine). Also, other absorption bands in the range of 2212–2215 cm−1 were assigned to the cyano group and stretching bands in the range of 1665–1675 cm−1 were attributed to the amidic carbonyl group. Moreover, the 1H NMR spectrum of these compounds revealed four signals in the range of δ 2.51–4.23 ppm, attributed to the aliphatic methylene groups of indolizine nucleus as well as a singlet signal at δ 5.24–5.76 ppm assigned to amino (–NH2) group. In addition to the appearance of two doublets of doublet signals at the aromatic range assigned to four aromatic protons of para-substituted phenyl ring and the appearance of singlet signal at δ 9.97–10.20 ppm indicating the –NH– of the amidic moiety. Furthermore, the intermediate 5j was characterized by the appearance of a singlet signal at δ 3.79 ppm, attributed to three aliphatic protons of the methoxy group (–OCH3). Finally, the target compounds 6a–q were obtained via condensation of intermediates 5a–j with phthalic anhydride or 4-fluoro phthalic anhydride using glacial acetic acid as a dehydrating agent and solvent. These compounds were confirmed by several spectroscopic methods. IR spectra of the mentioned compounds revealed the disappearance of the forked peak assigned to –NH2 of the intermediates, and the appearance of additional intense peaks between 1697 and 1778 cm−1 attributed to two carbonyl groups of the isoindolin-1,3-dione moiety as well as, sharp absorption bands at 2210–2237 cm−1, indicating the cyano group. Moreover, absorption bands at 2910–2960 cm−1 and 3030–3070 cm−1 were assigned to aliphatic (–C–H) and aromatic (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H), respectively. Additionally, sharp absorption bands at 3282–3329 cm−1 indicate the –NH of the amidic group. Also, 1H-NMR spectrum of compounds 6a–i revealed three signals in the range of δ 1.92–4.39 ppm, attributed to the aliphatic methylene groups of the pyrrolizine ring, while the 1H-NMR spectrum of compounds 6j–q showed four signals in the range of δ 1.86–4.23 ppm assigned to indolizine nucleus. The 1H-NMR spectrum of compounds 6a–q showed the disappearance of the –NH2 signal and increasing intensity of aromatic proton at the aromatic range δ 6.57–8.12 ppm due to additional phenyl moiety, in addition to a singlet signal at the range of δ 9.10–9.99 ppm assigned to amidic –NH– which was exchanged with D2O. Moreover, all synthesized compounds were confirmed by 13C-NMR, which revealed three signals in the range of δ 165.02–167.88 ppm, indicating the carbonyl carbons of each derivative. Additionally, a distinctive signal at δ 117.3–120.2 was attributed to the carbon of the cyano group. Moreover, derivatives 6a–i showed three signals in the range of δ 25.2–49.4 ppm, attributed to the aliphatic carbons of the pyrrolizine ring, while derivatives 6j–q showed four signals in the range of δ 20.3–47.6 ppm. Finally, mass spectroscopy revealed the molecular mass of each compound in m/z.
C–H), respectively. Additionally, sharp absorption bands at 3282–3329 cm−1 indicate the –NH of the amidic group. Also, 1H-NMR spectrum of compounds 6a–i revealed three signals in the range of δ 1.92–4.39 ppm, attributed to the aliphatic methylene groups of the pyrrolizine ring, while the 1H-NMR spectrum of compounds 6j–q showed four signals in the range of δ 1.86–4.23 ppm assigned to indolizine nucleus. The 1H-NMR spectrum of compounds 6a–q showed the disappearance of the –NH2 signal and increasing intensity of aromatic proton at the aromatic range δ 6.57–8.12 ppm due to additional phenyl moiety, in addition to a singlet signal at the range of δ 9.10–9.99 ppm assigned to amidic –NH– which was exchanged with D2O. Moreover, all synthesized compounds were confirmed by 13C-NMR, which revealed three signals in the range of δ 165.02–167.88 ppm, indicating the carbonyl carbons of each derivative. Additionally, a distinctive signal at δ 117.3–120.2 was attributed to the carbon of the cyano group. Moreover, derivatives 6a–i showed three signals in the range of δ 25.2–49.4 ppm, attributed to the aliphatic carbons of the pyrrolizine ring, while derivatives 6j–q showed four signals in the range of δ 20.3–47.6 ppm. Finally, mass spectroscopy revealed the molecular mass of each compound in m/z.
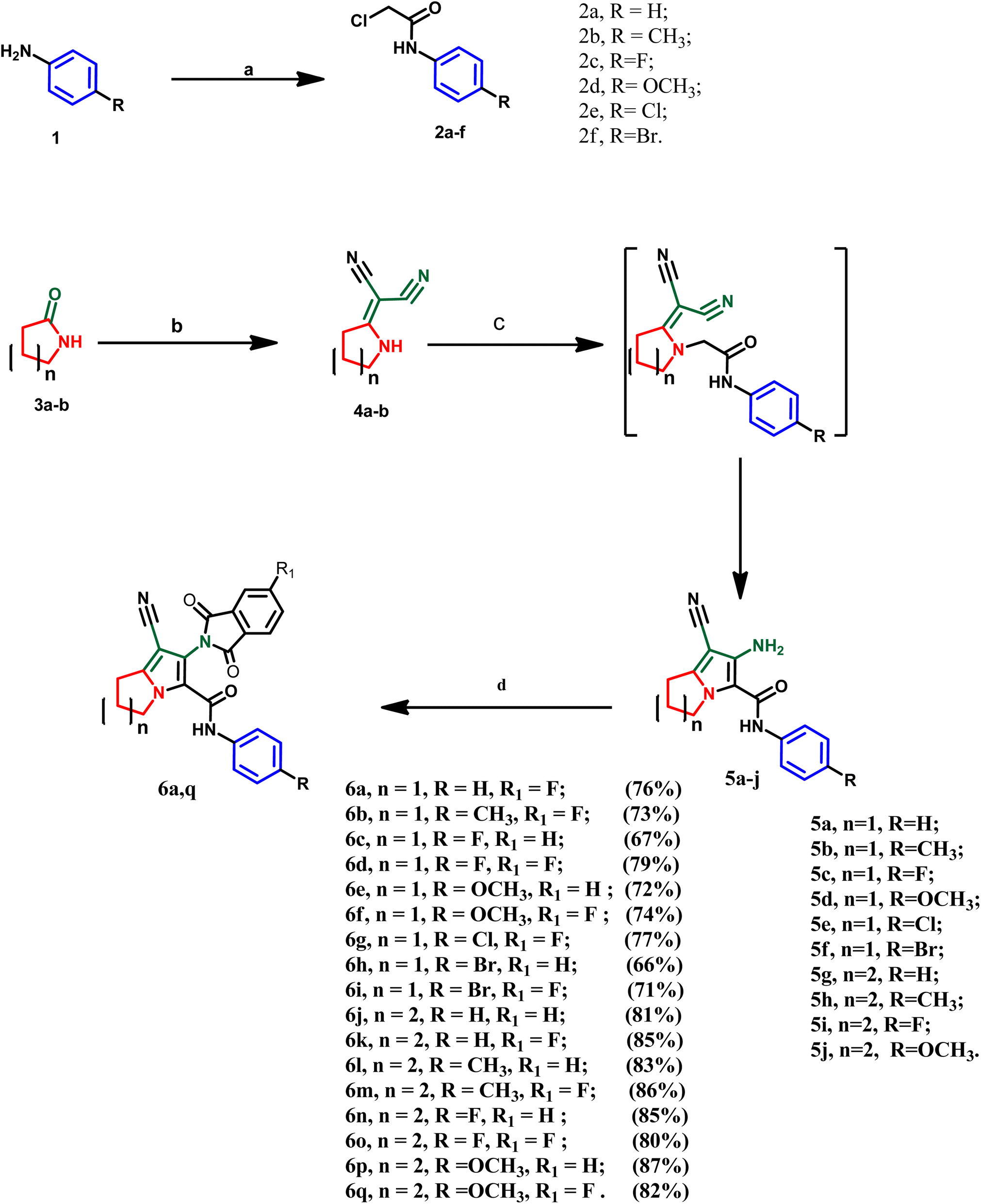 | ||
| Scheme 1 Synthesis pathway of the final compounds 6a–q. Reagents and conditions: (a) ClCH2COCl, acetic acid, CH3COONa, stirring, 1 h; (b) (CH3O)2SO2, dry benzene, CH2(CN)2, reflux, 8 h; (c) compounds 2a–f, acetone, K2CO3, reflux, 24 h; (d) un/substituted phthalic anhydride, glacial acetic acid, reflux, 4–6 h. | ||
2.2. Biological evaluation
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In vitro cytotoxicity IC50 (μM) | |||||||||||||
| Compound No | Substitution | HePG2 | HCT-116 | MCF-7 | Compound no | Substitution | HePG2 | HCT-116 | MCF-7 | ||||
| n | R | R1 | n | R | R1 | ||||||||
| a DOX: Doxorubicin. | |||||||||||||
| DOXa | — | — | — | 4.50 ± 0.2 | 5.23 ± 0.3 | 4.17 ± 0.2 | 6i | 1 | Br | F | 20.44 ± 1.5 | 14.27 ± 1.1 | 25.32 ± 2.0 |
| 6a | 1 | H | F | 85.14 ± 4.1 | >100 | 93.55 ± 4.7 | 6j | 2 | H | H | 34.13 ± 1.9 | 22.09 ± 1.6 | 31.26 ± 2.3 |
| 6b | 1 | CH3 | F | 55.38 ± 3.2 | 66.33 ± 3.1 | 58.11 ± 3.3 | 6k | 2 | H | F | 78.24 ± 3.8 | 83.65 ± 4.0 | 80.62 ± 4.1 |
| 6c | 1 | F | H | 49.08 ± 2.9 | 47.87 ± 2.6 | 54.65 ± 3.1 | 6l | 2 | CH3 | H | 61.62 ± 3.5 | 57.51 ± 3.1 | 63.22 ± 3.5 |
| 6d | 1 | F | F | 7.97 ± 0.5 | 9.49 ± 0.6 | 13.87 ± 0.9 | 6m | 2 | CH3 | F | 11.97 ± 0.8 | 28.37 ± 1.9 | 19.87 ± 1.4 |
| 6e | 1 | OCH3 | H | 45.38 ± 2.5 | 38.14 ± 2.4 | 42.71 ± 2.5 | 6n | 2 | F | H | 65.44 ± 3.7 | 71.27 ± 3.5 | 75.84 ± 3.9 |
| 6f | 1 | OCH3 | F | 88.31 ± 4.3 | >100 | >100 | 6o | 2 | F | F | 6.02 ± 0.3 | 5.84 ± 0.2 | 8.89 ± 0.5 |
| 6g | 1 | Cl | F | 52.04 ± 2.8 | 60.19 ± 3.3 | 69.44 ± 3.7 | 6p | 2 | OCH3 | H | 73.33 ± 3.9 | 79.63 ± 3.7 | 86.89 ± 4.3 |
| 6h | 1 | Br | H | 91.36 ± 4.8 | >100 | >100 | 6q | 2 | OCH3 | F | 41.20 ± 2.3 | 32.19 ± 2.2 | 37.44 ± 2.6 |
The relationship between cytotoxic activity and the structure of the synthesized compounds revealed that the substitution of the isoindolinedione moiety with a fluoro group remarkably showed higher anti-proliferative activity than the unsubstituted isoindolinedione moiety in all pyrrolizine and indolizine derivatives. As well as, substitution with a small electron withdrawing group, such as fluoro atom on the phenyl ring in pyrrolizine and indolizine derivatives, increases the cytotoxic activity against the three cancer cell lines. Also, methylphenyl or bromophenyl with fluoroisoindolinedione series showed potent anti-proliferative activity, unlike the remaining tested derivatives chloro, methoxy, and the unsubstituted derivatives. Finally, the ring expansion of the pyrrolizine ring to the indolizine ring significantly decreased the anti-proliferative activity of the tested compounds.
2.2.2.1 EGFR inhibition assay. Depending on the anti-proliferative screening of the synthesized compounds, the most active compounds 6o, 6d, and 6m were subjected to epidermal growth factor activity.36 The results revealed that the difluoro indolizine derivative (6o) showed the most inhibitory activity against the EGFR enzyme with an IC50 value of 62 nM in comparison to the positive control lapatinib (IC50 = 69 nM). Also, methylphenyl fluoroisoindolinedione derivative (6m) and difluoropyrrolizine derivative (6d) exhibited EGFR inhibitory activity with IC50 values of 130 and 362 nM, respectively (Table 2).
2.2.2.2 CDK-2 inhibition assay. Moreover, these three compounds 6o, 6d, and 6m were evaluated for cyclin-dependent kinase 2 inhibitory activity.36 The results revealed that the difluoro indolizine derivative (6o) revealed the most inhibitory activity against CDK-2 enzyme with an IC50 value of 118 nM in comparison to roscovitine (IC50 = 206 nM). Also, difluoropyrrolizine derivative (6d) and methylphenyl fluoroisoindolinedione derivative (6m) exhibited CDK-2 inhibitory activity with IC50 values of 174 and 402 nM, respectively (Table 2). These results adhere with the anti-proliferative activity results in that the difluoro indolizine derivative 6o was the most potent derivative against the test cell lines.
| Compound | P-gp RT-PCR fold change |
|---|---|
| 6d/HepG2 | 0.4097 |
| 6m/HepG2 | 0.6754 |
| 6o/HepG2 | 0.2732 |
| Tamoxifen/HepG2 | 0.2176 |
| Control HepG2 | 1 |
| Compound | HepG2/ADR inhibition IC50 μM |
|---|---|
| 6d/Dox | 22.27 |
| 6m/Dox | 3.88 |
| 6o/Dox | 8.79 |
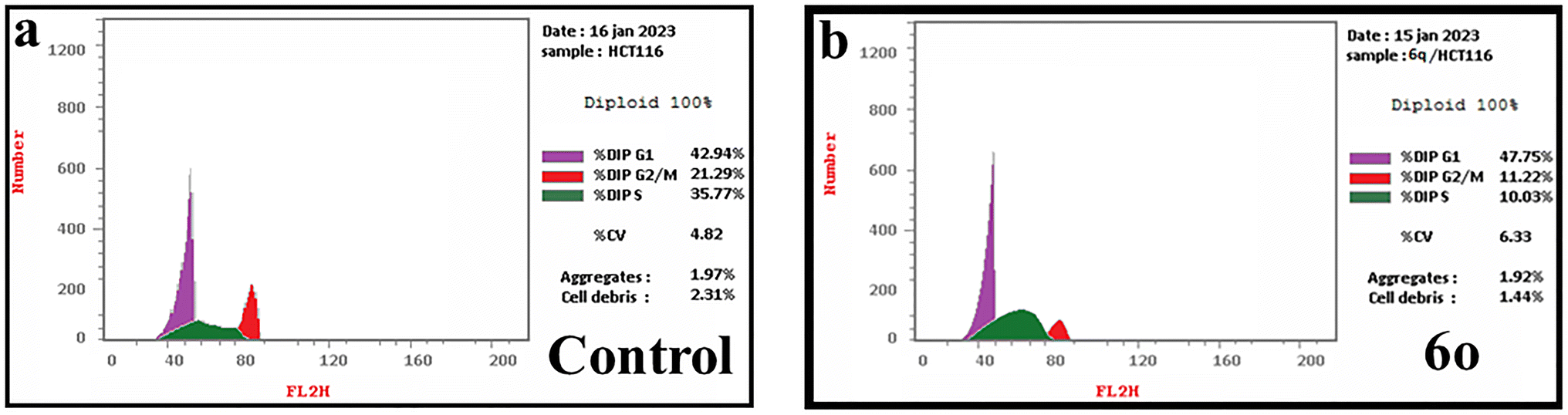 | ||
| Fig. 6 Cell cycle analysis of HCT-116 cells treated with (a) DMSO as a control; (b) compound 6o. | ||
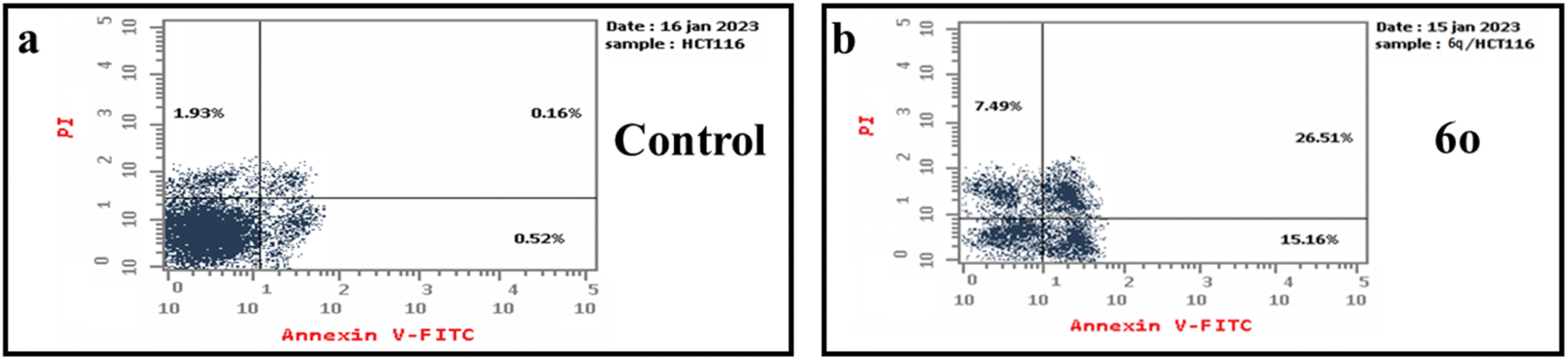 | ||
| Fig. 7 Apoptosis effect in HCT-116 cancer cell line detected with annexin V-FITC/PI double staining post treatment with IC50 of the control (a) and the tested compound 6o (b); lower left part represents live cells, lower right part represents early/primary apoptotic cells, upper right part represents late/secondary apoptotic cells, and upper left part represents the necrotic cells. | ||
2.3. Molecular docking study
Molecular Operating Environment (MOE) software (version 09.2022) was used to conduct molecular modeling experiments to explore several structural possibilities regarding the potential binding mode and interactions of the most active compounds 6d, 6m, and 6o into EGFR TK and CDK-2 active sites. Additionally, the EGFR kinase domain (PDB ID: 1XKK) was used to validate the docking protocol and re-dock the co-crystallized ligand quinazoline inhibitor—GW572016—into the active site.55 On the other hand, the structure of phosphorylated Thr160 CDK-2 in complex with the inhibitor roscovitine was used to validate the docking protocol for CDK-2 (PDB ID: 3DDQ).56 With a docking score (S) of −16.7402, the re-docked ligand displayed an rmsd of 1.6928 between the docked pose and the co-crystallized ligand for EGFR TK. When the co-crystallized ligand was docked, the rmsd between the two was 0.6221 (docking score (S) = −8.6888) for the CDK-2 enzyme (Fig. 8). Recent research revealed that the EGFR TK and CDK-2 inhibitors were positioned inside the binding site, forming intermolecular interactions with essential residues, as reported.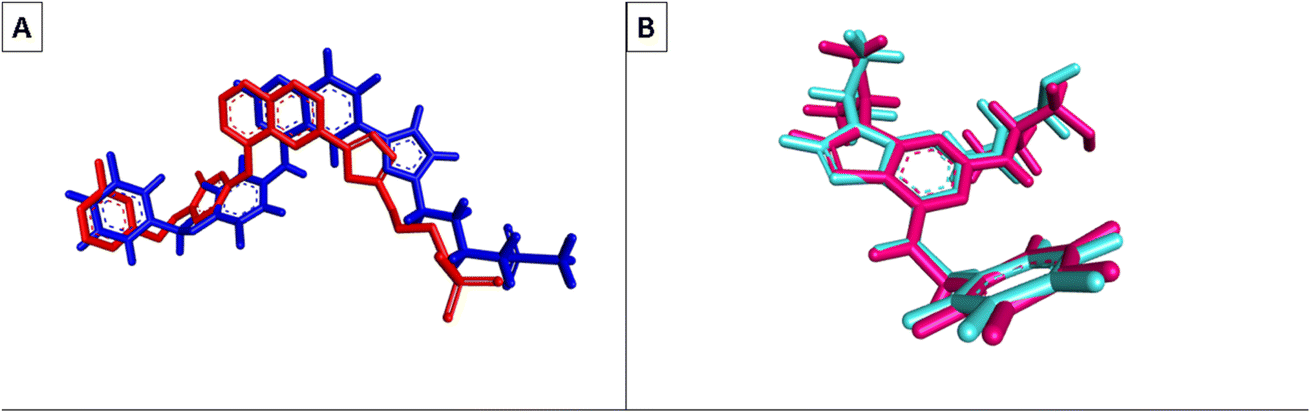 | ||
| Fig. 8 (A) Superimposition of the co-crystallized (red) and re-docked ligands (blue) during validation of EGFR TK docking; (B) superimposition of the co-crystallized (purple) and re-docked ligands (cyan blue) during validation of CDK-2 docking. | ||
Lapatinib (as a positive control) and the most effective compounds 6d, 6m, and 6o were docked into the EGFR's active site. The findings of the docking studies are shown in Tables 5 and 6, which also provide the type of interactions, distances (in Å) from the interacting residues, and binding affinity in kcal mol−1. Comparing compounds 6d, 6m, and 6o to co-docked ligand lapatinib as a positive control, these compounds showed strong interaction with Met793 and other essential interacting amino acids required for inhibition of EGFR TK. Notably, compound 6o showed a strong interaction score with a binding affinity of −12.6356 kcal mol−1 compared to lapatinib binding affinity of −13.0626 kcal mol−1. These strong binding affinities can be explained by the numerous binding interactions with the key amino acid within the active site of the EGFR-TK, especially the gate-keeper Met793 and Lys 745 residues (Fig. 9 and 10).
| Compound | EGFR TK (PDB: 1XKK) | |||
|---|---|---|---|---|
| Binding affinity (kcal mol−1) | Distance from main residue (in Å) | Interaction type | ||
| 6d | −9.2503 | 2.54 | Met793 | Halogen bond |
| 3.89 | Leu718 | Hydrophobic | ||
| 4.12 | Leu844 | Hydrophobic | ||
| 3.43 | Lys745 | Hydrophobic | ||
| 2.39 | Asn842 | Hydrophobic | ||
| 6m | −11.2312 | 3.34 | Met793 | H-acceptor |
| 4.39 | Leu718 | Hydrophobic | ||
| 4.11 | Leu844 | Hydrophobic | ||
| 3.90 | Lys745 | H-acceptor | ||
| 4.42 | Cys797 | Hydrophobic | ||
| 6o | −12.6356 | 2.61 | Met793 | Halogen bond |
| 2.72 | Leu718 | Hydrophobic | ||
| 4.09 | Leu844 | Hydrophobic | ||
| 2.90 | Lys745 | H-acceptor | ||
| 4.15 | Cys797 | Hydrophobic | ||
| 3.51 | Arg841 | Hydrophobic | ||
| Lapatinib | −14.0626 | 2.96 | Met793 | H-acceptor |
| 4.55 | Leu718 | Hydrophobic | ||
| 3.30 | Leu844 | Hydrophobic | ||
| 4.66 | Lys745 | Hydrophobic | ||
| 3.49 | Cys775 | Halogen bond | ||
| 3.10 | Leu788 | Halogen bond | ||
| 3.13 | Thr790 | Halogen bond | ||
| 3.12 | Arg776 | Halogen bond | ||
| Compound | CDK-2 (PDB: 3DDQ) | |||
|---|---|---|---|---|
| Binding affinity (kcal mol−1) | Distance from main residue (in Å) | Interaction type | ||
| 6d | −7.9959 | 1.98 | Leu83 | H-acceptor |
| 2.54 | Lys33 | Halogen bond | ||
| 2.49 | Lys89 | H-acceptor | ||
| 4.64 | Ala144 | Hydrophobic | ||
| 4.25 | Val18 | Hydrophobic | ||
| 4.61 | Leu134 | Hydrophobic | ||
| 4.19 | Ile10 | Hydrophobic | ||
| 6m | −5.5006 | 2.46 | Leu83 | H-acceptor |
| 4.48 | Leu134 | Hydrophobic | ||
| 4.27 | Ile10 | Hydrophobic | ||
| 4.54 | Val18 | Hydrophobic | ||
| 4.61 | Phe80 | Hydrophobic | ||
| 6o | −8.0611 | 2.15 | Leu83 | H-acceptor |
| 2.10 | Lys33 | Halogen bond | ||
| 3.35 | Glu51 | Halogen bond | ||
| 3.61 | Leu134 | Hydrophobic | ||
| 3.72 | Ala144 | Hydrophobic | ||
| 3.70 | Val18 | Hydrophobic | ||
| 3.17 | Ile10 | Hydrophobic | ||
| Roscovitine | −7.70482 | 2.23 | Leu83 | H-donor |
| 2.74 | Glu12 | H-donor | ||
| 4.24 | Ala144 | Hydrophobic | ||
| 4.01 | Ala31 | Hydrophobic | ||
| 4.44 | Lys33 | Hydrophobic | ||
| 3.79 | Val64 | Hydrophobic | ||
| 3.98 | Val18 | Hydrophobic | ||
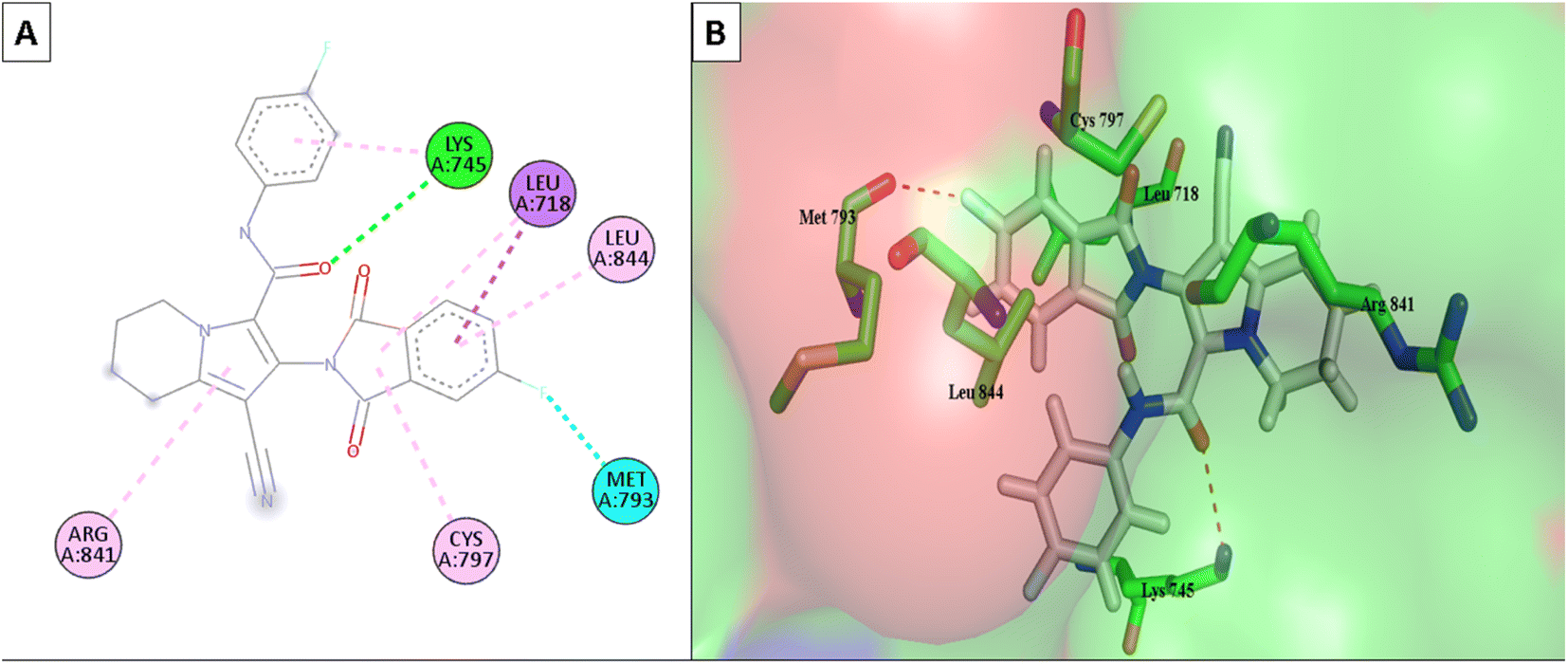 | ||
| Fig. 9 (A) The 2D interaction diagram of the top docking pose of 6o to EGFR active domain; (B) the 3D interaction diagram of the top docking pose of 6o to EGFR active domain. | ||
 | ||
| Fig. 10 (A) The 2D interaction diagram of the top docking pose of lapatinib (positive control) to EGFR active domain; (B) the 3D interaction diagram of the top docking pose of lapatinib (positive control) to EGFR active domain. | ||
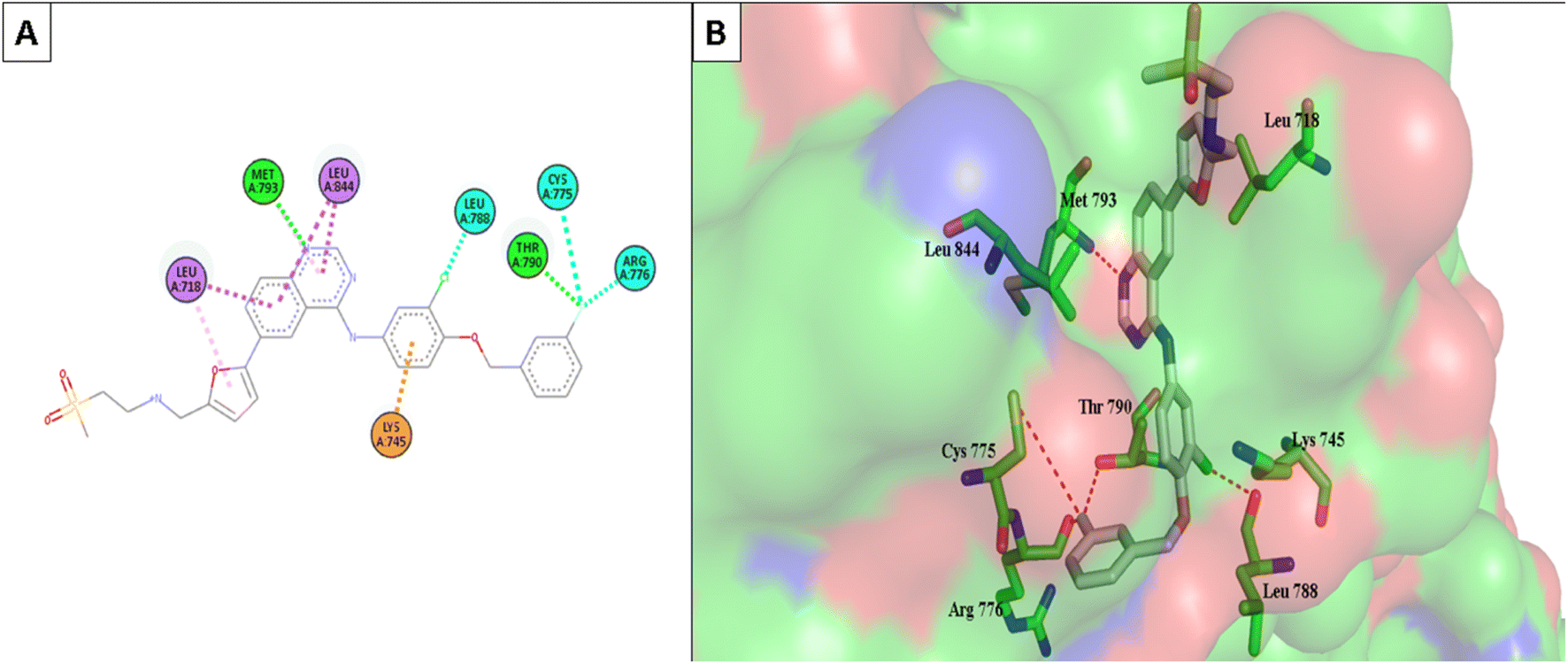
Furthermore, the most active compounds, 6d, 6m, and 6o, were docked into the CDK-2's active site along with roscovitine as a positive control. Tables 5 and 6 summarize the results of the docking study and include the type of interactions, distances (in Å) from the interacting residues, and binding affinity (in kcal mol−1). Roscovitine, a co-docked ligand, was used as a positive control when comparing compounds 6d, 6m, and 6o. These compounds demonstrated strong interactions with Leu83 and other crucial interacting amino acids necessary for CDK-2 inhibition. In particular, compounds 6d and 6o had a significant interaction score with a binding affinity of −7.9959 and −8.0611 kcal mol−1, respectively, compared to Roscovitine binding affinity of −7.70482 kcal mol−1. The strong interaction of the synthesized compound with the key amino acid with the active site of CDK-2 enzyme, mainly the –CN moiety with Leu83, was responsible for the high binding score of these compounds (Fig. 11 and 12).
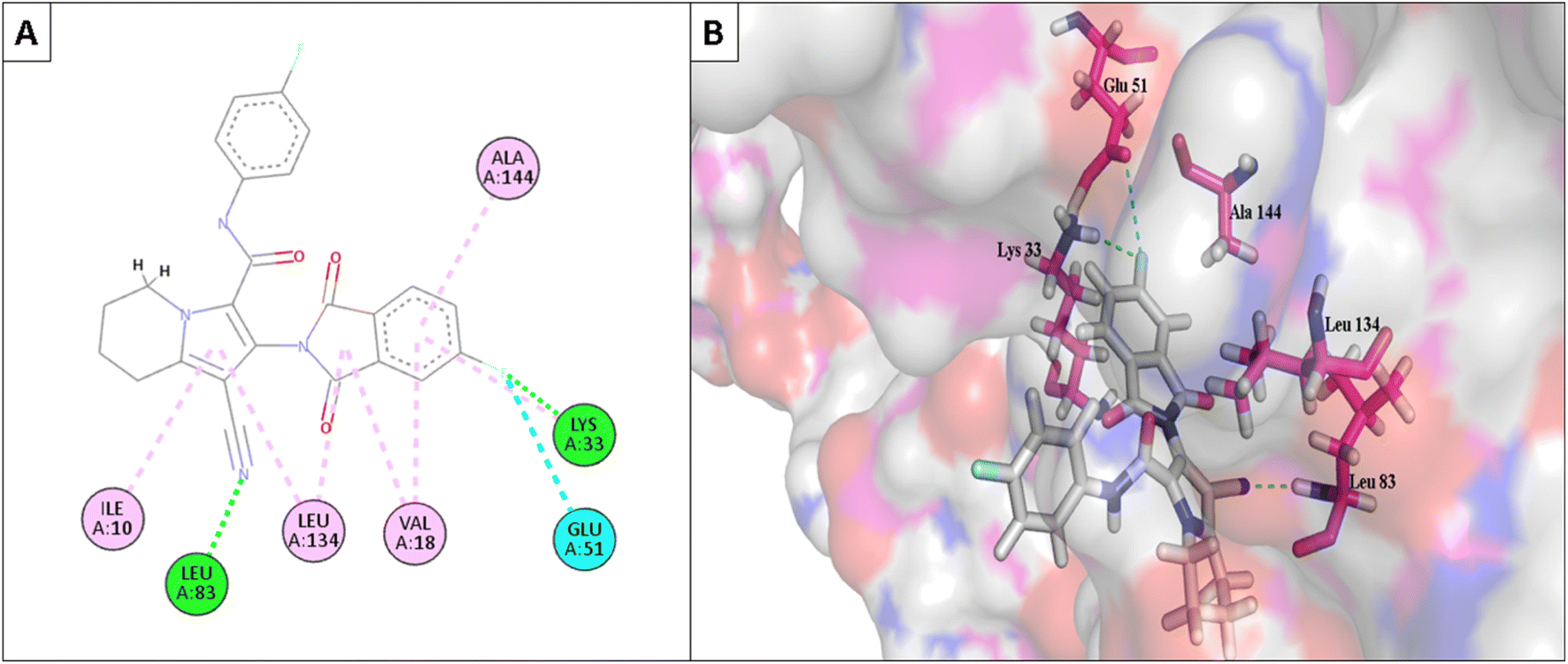 | ||
| Fig. 11 (A) The 2D interaction diagram of the top docking pose of 6o to CDK-2 active domain; (B) the 3D interaction diagram of the top docking pose of 6o to CDK-2 active domain. | ||
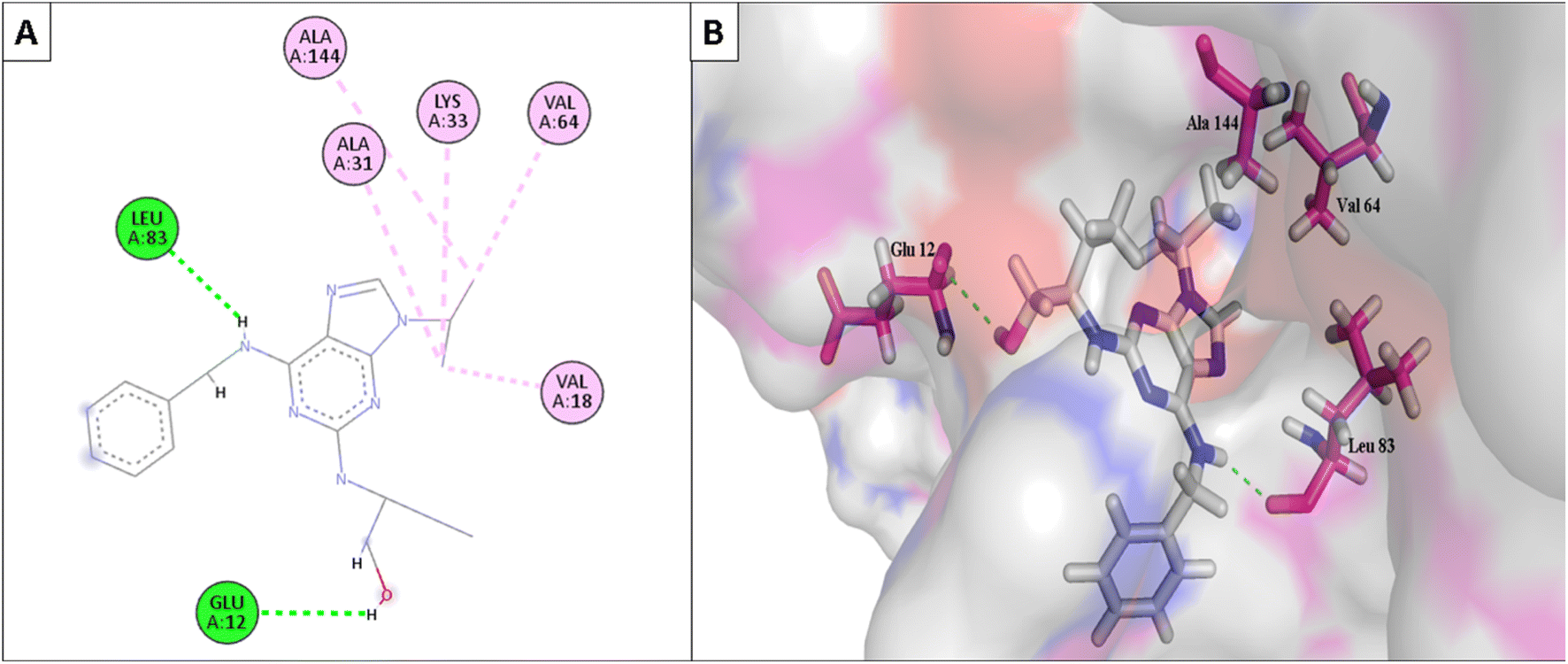 | ||
| Fig. 12 (A) The 2D interaction diagram of the top docking pose of roscovitine (positive control) to CDK-2 active domain; (B) the 3D interaction diagram of the top docking pose of roscovitine (positive control) to CDK-2 active domain. | ||
2.4. In silico ADMET studies
Using the PKCSM online software pkCSM (https://biosig.lab.uq.edu.au/pkcsm/), it was possible to estimate the physiochemical properties, pharmacokinetics, and drug-likeness profile of compounds 6d, 6m, and 6o.57,58The results suggested that all compounds under investigation had considerable oral absorption values. Among the analogs, compound 6d had the highest water solubility value. It was predicted that all the assessed compounds would have high cellular permeability, particularly for intestinal cells (95.31%, 96.20%, and 96.14%) (Table 7).
| Property | Model name | Unit | 6d | 6m | 6o |
|---|---|---|---|---|---|
| Absorption | Water solubility | Numeric (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mol L−1) mol L−1) |
−4.864 | −5.02 | −5.046 |
| Caco2 permeability | Numeric (logPapp in 10−6 cm s−1) |
0.571 | 0.504 | 0.57 | |
| Intestinal absorption (human) | Numeric (% absorbed) | 95.315 | 96.201 | 96.147 | |
| P-glycoprotein substrate | Categorical (yes/no) | Yes | Yes | Yes | |
| P-glycoprotein I inhibitor | Categorical (yes/no) | Yes | Yes | Yes | |
| P-glycoprotein II inhibitor | Categorical (yes/no) | Yes | Yes | Yes | |
| Distribution | Volume of distribution (human) | Numeric (logL kg−1) |
−0.193 | −0.076 | −0.138 |
| Fraction unbound (human) | Numeric (Fu) | 0.095 | 0.054 | 0.085 | |
| BBB permeability | Numeric (logBB) |
−0.614 | −0.384 | −0.592 | |
| CNS permeability | Numeric (logPS) |
−2.107 | −1.856 | −2.015 | |
| Metabolism | CYP2D6 substrate | Categorical (yes/no) | No | No | No |
| CYP3A4 substrate | Categorical (yes/no) | Yes | Yes | Yes | |
| CYP1A2 inhibitor | Categorical (yes/no) | No | No | No | |
| CYP2C19 inhibitor | Categorical (yes/no) | Yes | Yes | Yes | |
| CYP2C9 inhibitor | Categorical (yes/no) | Yes | Yes | Yes | |
| CYP2D6 inhibitor | Categorical (yes/no) | No | No | No | |
| CYP3A4 inhibitor | Categorical (yes/no) | Yes | Yes | Yes | |
| Excretion | Total clearance | Numeric (logmL min−1 kg−1) |
0.079 | −0.138 | 0.079 |
| Renal OCT2 substrate | Categorical (yes/no) | No | No | No | |
| Toxicity | AMES toxicity | Categorical (yes/no) | No | No | No |
| Max. tolerated dose (human) | Numeric (logmg per kg per day) |
−0.48 | −0.6 | −0.479 | |
| Oral rat acute toxicity (LD50) | Numeric (mol kg−1) | 3.091 | 3.129 | 3.141 | |
| Oral rat chronic toxicity (LOAEL) | Numeric (logmg per kg_bw per day) |
1.361 | 1.695 | 1.382 | |
| T. pyriformis toxicity | Numeric (logμg L−1) |
0.395 | 0.5 | 0.39 | |
| Minnow toxicity | Numeric (logmM) |
0.411 | 0.757 | 0.294 |
It was well recognized that a molecule to be absorbed orally should obey both Lipinski and Veber'ss rules reflecting the lipophilicity–hydrophilicity equilibrium. As shown in Table 8, compounds 6d, 6m, and 6o were drug-like because they adhered to Lipinski and Veber's principles.59,60 Additionally, the determined lipophilicity was positively correlated with Caco-2 permeability but negatively correlated with the targeted compound's ability to dissolve in water. This was based on the finding that there was no correlation between drug permeability as evaluated by the human colon adenocarcinoma (Caco-2) cell line test and lipophilicity.57
Compound 6m showed the most extensive BBB permeability and unbound fraction among the other analogs, according to the prediction of distribution properties. Additionally, the parameters linked to metabolism and excretion did not reveal appreciable variations among the tested molecules. Additionally, the toxicological properties of the compounds 6d, 6m, and 6o were investigated. Herein, the tested compounds showed no toxicity to AMES, an in silico test to determine the toxicity of the synthesized compounds via bacterial reverse mutation assay. In addition, they showed no or minimum values for oral rat acute toxicity (LD50) and Tetrahymena Pyriformis toxicity test. These results showed that the compounds 6d, 6m, and 6o are predicted to be safe for human use.
3. Conclusion
Two new series of pyrrolizine-5-carboxamides (6a–i) and indolizine-6-carboxamide (6j–q) were designed and synthesized. IR, 1H NMR, 13C NMR, and mass spectroscopy confirmed the chemical structures of target compounds. Screening of the cytotoxic effect of target compounds against HePG-2, HCT-116, and MCF-7 cell lines revealed that compound 6d was the most potent compound among its series, with IC50 values of 7.97, 9.49, and 13.87 μM against HePG-2, HCT-116, and MCF-7, respectively. While compound 6o exhibited the most anti-proliferative activity with IC50 values of 6.02, 5.84, and 8.89 μM against HePG-2, HCT-116, and MCF-7. EGFR and CDK-2 inhibitory effects of the most active compounds, 6d, 6m, and 6o, showed that compound 6o displayed the most inhibitory activity against EGFR with IC50 value of 0.062 μM and CDK-2 with IC50 value of 0.118 μM in comparison with Imatinib and Roscovitine, respectively. Annexin V-FITC apoptosis assay exposed that compound 6o accumulated HCT-116 cells in the G1 and S phases and induced apoptosis.4. Experimental
4.1. Chemistry
Melting points were detected by the Tomas-Hoover capillary melting apparatus without any correction. All solvents, chemicals, and reagents were supplied from Aldrich chemical company (Milwaukee, WI) and El Nasr pharmaceutical chemical companies, Cairo, Egypt. Infrared (IR) spectra were monitored as films on KBr discs using Schimadzu FT-IR 8400S spectrophotometer and values were presented as cm−1. The purity of the synthesized compounds and the reaction progress were checked by using precoated thin layer chromatography (TLC) silica gel plates 60F254 with a thickness of 0.25 supplied from MERCK, Darmstadt, Germany. A UV lamp was used to monitor the reaction process. 13C NMR and 1H NMR spectra were carried out on a Bruker Avance III 400 MHz spectrophotometer, Faculty of Pharmacy, Benisuef University, Egypt, in dimethyl sulfoxide (DMSO-d6) or D2O as a solvent. Chemical shift was estimated in ppm on δ scale and J (Coupling constant) was estimated in Hertz. Microanalysis for C, H, and N were performed on a PerkinElmer 2400 analyzer (PerkinElmer, Norwalk, CT. USA) at the regional center for Mycology and Biotechnology, Al-Azhar University, Egypt. All results were within ±0.4% of the theoretical values.4.1.3.1 6-Amino-7-cyano-N-phenyl-2,3-dihydro-1H-pyrrolizine-5-carboxamide 5a. Yellow; yield 77%; mp 180–182 °C; IR: (KBr, cm−1) 3303 (NH), 2227 (CN), 1712 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.34 (m, 2H, CH2), 2.91 (m, 2H, CH2), 3.47 (t, 2H, CH2), 6.27 (s, 2H, NH2), 7.02 (t, 1H, Ar–H), 7.43 (d, 2H, J = 8.9 Hz, Ar–H), 7.61 (d, 2H, J = 8.9 Hz, Ar–H), 9.15 (s, 1H, NH D2O exchangeable).
4.1.3.2 6-Amino-7-cyano-N-(p-tolyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 5b. White; yield 68%; mp 232–234 °C; IR: (KBr, cm−1) 3307 (NH), 2230 (CN), 1715 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.23 (s, 3H, CH3), 2.43 (m, 2H, CH2), 2.89 (m, 2H, CH2), 3.47 (t, 2H, CH2), 5.56 (s, 2H, NH2), 7.43 (d, 2H, J = 8.9 Hz, Ar–H), 7.92 (d, 2H, J = 8.9 Hz, Ar–H), 9.82 (s, 1H, NH D2O exchangeable).
4.1.3.3 6-Amino-7-cyano-N-(4-fluorophenyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 5c. Pale yellow; yield 85%; mp 271–273 °C; IR: (KBr, cm−1) 3305 (NH), 2232 (CN), 1716 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.44 (m, 2H, CH2), 2.98 (m, 2H, CH2), 3.67 (t, 2H, CH2), 5.45 (s, 2H, NH2), 7.23 (d, 2H, J = 8.9 Hz, Ar–H), 7.78 (d, 2H, J = 8.9 Hz, Ar–H), 9.95 (s, 1H, NH D2O exchangeable).
4.1.3.4 6-Amino-7-cyano-N-(4-methoxyphenyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 5d. Yellow; yield 84%; mp 211–213 °C; IR: (KBr, cm−1) 3311 (NH), 2234 (CN), 1711 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.20 (m, 2H, CH2), 2.83 (m, 2H, CH2), 3.72 (t, 2H, CH2), 3.83 (s, 3H, OCH3) 5.76 (s, 2H, NH2), 6.99 (d, 2H, J = 8.9 Hz, Ar–H), 7.60 (d, 2H, J = 8.9 Hz, Ar–H), 9.23 (s, 1H, NH D2O exchangeable).
4.1.3.5 6-Amino-N-(4-chlorophenyl)-7-cyano-2,3-dihydro-1H-pyrrolizine-5-carboxamide 5e. White; yield 72%; mp 238–240 °C; IR: (KBr, cm−1) 3308 (NH), 2231 (CN), 1709 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.02 (m, 2H, CH2), 2.98 (m, 2H, CH2), 3.78 (t, 2H, CH2), 5.77 (s, 2H, NH2), 7.44 (d, 2H, J = 8.9 Hz, Ar–H), 7.69 (d, 2H, J = 8.9 Hz, Ar–H), 9.43 (s, 1H, NH D2O exchangeable).
4.1.3.6 6-Amino-N-(4-bromophenyl)-7-cyano-2,3-dihydro-1H-pyrrolizine-5-carboxamide 5f. White; yield 78%; mp 220–222 °C; IR: (KBr, cm−1) 3313 (NH), 2227 (CN), 1714 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.86 (m, 2H, CH2), 3.23 (m, 2H, CH2), 4.41 (t, 2H, CH2), 6.24 (s, 2H, NH2), 7.63 (d, 2H, J = 8.9 Hz, Ar–H), 7.88 (d, 2H, J = 8.9 Hz, Ar–H), 9.70 (s, 1H, NH D2O exchangeable).
4.1.3.7 2-Amino-1-cyano-N-phenyl-5,6,7,8-tetrahydroindolizine-3-carboxamide 5g. Pale yellow; yield 76%; mp 214–216 °C; IR: (KBr, cm−1) 3304 (NH), 2222 (CN), 1718 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.88 (m, 2H, CH2), 1.97 (m, 2H, CH2), 2.75 (t, 2H, CH2), 4.13 (t, 2H, CH2), 5.74 (s, 2H, NH2), 7.12 (t, 1H, Ar–H) 7.63 (d, 2H, J = 8.9 Hz, Ar–H), 7.89 (d, 2H, J = 8.9 Hz, Ar–H), 9.14 (s, 1H, NH D2O exchangeable).
4.1.3.8 2-Amino-1-cyano-N-(p-tolyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide 5h. Yellow; yield 84%; mp 234–236 °C; IR: (KBr, cm−1) 3306 (NH), 2223 (CN), 1712 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.75 (m, 2H, CH2), 1.87 (m, 2H, CH2), 2.23 (s, 3H, CH3), 2.77 (t, 2H, CH2), 4.31 (t, 2H, CH2), 6.14 (s, 2H, NH2), 7.23 (d, 2H, J = 8.9 Hz, Ar–H), 7.43 (d, 2H, J = 8.9 Hz, Ar–H), 9.61 (s, 1H, NH D2O exchangeable).
4.1.3.9 2-Amino-1-cyano-N-(4-fluorophenyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide 5i. White; yield 82%; mp 242–244 °C; IR: (KBr, cm−1) 3301 (NH), 2224 (CN), 1706 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.78 (m, 2H, CH2), 1.88 (m, 2H, CH2), 2.78 (t, 2H, CH2), 4.11 (t, 2H, CH2), 5.24 (s, 2H, NH2), 7.13 (d, 2H, J = 8.9 Hz, Ar–H), 7.59 (d, 2H, J = 8.9 Hz, Ar–H), 9.50 (s, 1H, NH D2O exchangeable).
4.1.3.10 2-Amino-1-cyano-N-(4-methoxyphenyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide 5i. Yellow; yield 75%; mp 237–239 °C; IR: (KBr, cm−1) 3305 (NH), 2346 (CN), 1772 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.78 (m, 2H, CH2), 1.87 (m, 2H, CH2), 2.78 (t, 2H, CH2), 3.79 (s, 3H, OCH3), 4.12 (t, 2H, CH2), 5.16 (s, 2H, NH2), 6.90 (d, 2H, J = 9.1 Hz, Ar–H), 7.50 (d, 2H, J = 9.1 Hz, Ar–H), 9.35 (s, 1H, NH D2O exchangeable).
4.1.3.11 7-Cyano-6-(1,3-dioxoisoindolin-2-yl)-N-phenyl-2,3-dihydro-1H-pyrrolizine-5-carboxamide (6a–q). The phthalic anhydride (0.56 g, 3.75 mmol) was combined with the carboxamide derivatives 5a–j (3.75 mmol) in glacial acetic acid (20 mL), which was then refluxed for 3 h. The solution was then evaporated under reduced pressure. The product was dried after being washed with water. Then, they were crystallized from a mixture of acetone/ethanol (1
:3) to afford final compounds 6a–q.61
4.1.3.12. 7-Cyano-6-(5-fluoro-1,3-dioxoisoindolin-2-yl)-N-phenyl-2,3-dihydro-1H-pyrrolizine-5-carboxamide 6a. Green; yield 76%; mp 184–186 °C; IR: (KBr, cm−1) 3302 (NH), 2233 (CN), 1720 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.52 (m, 2H, CH2), 3.11 (t, 2H, CH2), 4.39 (t, 2H, CH2), 7.08 (t, 1H, Ar–H), 7.28 (d, 2H, J = 8.00 Hz, Ar–H), 7.44 (d, 2H, J = 8.00 Hz, Ar–H), 7.78 (d, 1H, J = 5.9 Hz, Ar–H), 7.94 (d, 1H, J = 5.6 Hz, Ar–H) 8.09 (s, 1H, Ar–H) 9.97 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz DMSO-d6): δ 24.96, 25.91, 49.90, 84.63, 112.07, 112.32, 114.50, 119.78, 120.35, 121.32, 122.67, 124.54, 127.13, 127.23, 127.96, 129.03, 138.62, 146.84,157.77, 164.95, 166.18; EIMS for C23H15FN4O3 m/z (100): 414.41 [M+] (23.98), 407.12 (100).
4.1.3.13 7-Cyano-6-(5-fluoro-1,3-dioxoisoindolin-2-yl)-N-(p-tolyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 6b. Pale yellow; yield 73%; mp 233–235 °C; IR: (KBr, cm−1) 3290 (NH), 2229 (CN), 1728 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.92 (m, 2H, CH2), 2.23 (s, 3H, CH3), 3.09 (t, 2H, CH2), 4.37 (t, 2H, CH2), 7.06 (d, 2H, J = 8.10 Hz, Ar–H), 7.30 (d, 2H, J = 8.20 Hz, Ar–H), 7.67 (d, 1H, J = 7.00 Ar–H), 7.92 (d, 1H, J = 7.40 Hz, Ar–H) 8.07 (s, 1H, Ar–H) 9.86 (s, 1H, NH, D2O exchangeable): δ EIMS for C24H17FN4O3 m/z (100): 428.73 [M+] (32.72), 134.54 (100).
4.1.3.14 7-Cyano-6-(1,3-dioxoisoindolin-2-yl)-N-(4-fluorophenyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 6c. Pale yellow; yield 67%; mp 273–275 °C; IR: (KBr, cm−1) 3294 (NH), 2229 (CN), 1712 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.51 (m, 2H, CH2), 3.10 (t, 2H, CH2), 4.33 (t, 2H, CH2), 7.07 (d, 2H, J = 8.9 Hz, Ar–H), 7.39 (d, 2H, J = 8.9 Hz, Ar–H), 7.88 (t, 2H, J = 6.4 Hz, Ar–H), 8.08 (d, 2H, J = 8.4 Hz, Ar–H), 9.99 (s, 1H, NH D2O exchangeable), 13C NMR (100 MHz DMSO-d6): δ 24.96, 25.92, 49.86, 84.66, 114.53, 115.55, 115.77, 120.06, 120.19, 123.21, 123.29, 124.31, 131.87, 135.64, 146.84, 157.81, 166.17; EIMS m/z for C23H15FN4O3 (100): 414.13 [M+] (29.29), 92.33 (100).
4.1.3.15 7-Cyano-6-(5-fluoro-1,3-dioxoisoindolin-2-yl)-N-(4-fluorophenyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 6d. Yellow; yield 79%; mp 245–247 °C; IR: (KBr, cm−1) 3298 (NH), 2233 (CN), 1720 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.51 (m, 2H, CH2), 3.10 (t, 2H, CH2), 4.38 (t, 2H, CH2), 7.11 (d, 2H, J = 8.9 Hz, Ar–H), 7.44 (d, 2H, J = 8.7 Hz, Ar–H), 7.75 (d, 1H, J = 8.2 Hz, Ar–H), 7.94 (d, 1H, J = 7.3 Hz, Ar–H), 8.09 (s, 1H, Ar–H), 10.00 (s, 1H, NH, D2O exchangeable): δ EIMS for C23H15FN4O3 m/z (100): 432.41 [M+] (9.67), 322.17 (100).
4.1.3.16 7-Cyano-6-(1,3-dioxoisoindolin-2-yl)-N-(4-methoxyphenyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 6e. Yellowish white; yield 72%; mp 209–211 °C; IR: (KBr, cm−1) 3282 (NH), 2210 (CN), 1697 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 3.09 (m, 2H, CH2), 3.71 (s, 3H, OCH3), 4.10 (t, 2H, CH2), 4.37 (t, 2H, CH2), 6.84 (d, 2H, J = 8.9 Hz, Ar–H), 7.31 (d, 2H, J = 8.9 Hz, Ar–H), 7.94 (t, 2H, J = 6.4 Hz, Ar–H), 8.01 (d, 2H, J = 8.4 Hz, Ar–H), 9.79 (s, 1H, NH, D2O exchangeable): δ EIMS for C24H18N4O4 m/z (100): 426.91 [M+] (30.86), 52.23 (100).
4.1.3.17 7-Cyano-6-(5-fluoro-1,3-dioxoisoindolin-2-yl)-N-(4-methoxyphenyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 6f. Green; yield 74%; mp 223–225 °C; IR: (KBr, cm−1) 3286 (NH), 2229 (CN), 1728 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.09 (m, 2H, CH2), 3.18 (t, 2H, CH2), 3.71 (s, 3H, OCH3), 4.37 (t, 2H, CH2), 6.85 (d, 2H, J = 7.80 Hz Ar–H), 7.31 (d, 2H, J = 9.10 Hz, Ar–H), 7.76 (d, 1H, J = 7.30 Hz, Ar–H), 7.94 (d, 1H, J = 7.60 Hz, Ar–H), 8.08 (s, 1H, Ar–H) 9.8 (s, 1H, NH, D2O exchangeable): δ EIMS for C24H17FN4O4 m/z (100): 444.22 [M+] (19.65), 110.32 (100).
4.1.3.18 N-(4-Chlorophenyl)-7-cyano-6-(5-fluoro-1,3-dioxoisoindolin-2-yl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 6g. Brown; yield 77%; mp 239–241 °C; IR: (KBr, cm−1) 3296 (NH), 2233 (CN), 1720 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.97 (m, 2H, CH2), 3.10 (t, 2H, CH2), 4.38 (t, 2H, CH2), 7.33 (d, 2H, J = 8.80, Ar–H), 7.47 (d, 2H, J = 8.80 Hz, Ar–H), 7.79 (d, 1H, J = 8.5 Hz, Ar–H),7.94 (d, 1H, J = 8.7 Hz, Ar–H), 8.08 (s, 1H, Ar–H), 10.09 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz DMSO-d6): δ 24.97, 25.71, 50.02, 84.89, 87.83, 93.93, 106.64, 112.15, 118.92, 119.83, 121.33, 122.63, 122.66, 127.91, 128.16, 129.07, 129.21, 137.45, 147.04, 157.84, 165.11; EIMS for C23H14ClFN4O3 m/z (100): [M+], (100).
4.1.3.19 N-(4-Bromophenyl)-7-cyano-6-(1,3-dioxoisoindolin-2-yl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 6h. Yellow; yield 66%; mp 221–223 °C; IR: (KBr, cm−1) 3329 (NH), 2233 (CN), 1732 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.51 (m, 2H, CH2), 3.10 (t, 2H, CH2), 4.38 (t, 2H, CH2), 7.41 (d, 2H, J = 9.00 Hz, Ar–H), 7.46 (d, 2H, J = 9.1 Hz, Ar–H), 7.94 (t, 2H, J = 6.4 Hz, Ar–H), 8.00 (d, 2H, J = 8.7 Hz, Ar–H), 10.09 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz DMSO-d6): δ 24.97, 25.91, 49.87, 84.71, 114.51, 116.18, 120.03, 120.23, 122.96, 124.32, 131.74, 131.88, 135.65, 138.15, 146.99, 157.88, 166.12; EIMS for C23H15BrN4O3 m/z (100): 475.29 [M+] (7.58), 165.26 (100).
4.1.3.20 N-(4-Bromophenyl)-7-cyano-6-(5-fluoro-1,3-dioxoisoindolin-2-yl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide 6i. Brown; yield 71%; mp 234–236 °C; IR: (KBr, cm−1) 3302 (NH), 2233 (CN), 1778 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.51 (m, 2H, CH2), 3.10 (t, 2H, CH2), 4.38 (t, 2H, CH2), 7.42 (d, 2H, J = 9.00, Ar–H), 7.46 (d, 2H, J = 8.80 Hz, Ar–H), 7.78 (d, 1H, J = 8.2 Hz, Ar–H) 7.93 (d, 1H, J = 5.40 Hz, Ar–H), 8.08 (s, 1H, Ar–H), 10.09 (s, 1H, NH, D2O exchangeable): EIMS for C23H14BrFN4O3 m/z (100): 493.52 [M+] (25.04), 102.19 (100).
4.1.3.21 1-Cyano-2-(1,3-dioxoisoindolin-2-yl)-N-phenyl-5,6,7,8-tetrahydroindolizine-3-carboxamide 6j. Green; yield 81%; mp 213–215 °C; IR: (KBr, cm−1) 3302 (NH), 2225 (CN), 1708 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.86 (m, 2H, CH2), 1.94 (m, 2H, CH2), 2.94 (t, 2H, CH2), 4.19 (t, 2H, CH2), 7.05 (t, 1H, Ar–H), 7.25 (t, 2H, Ar–H), 7.40 (d, 2H, J = 8.7 Hz, Ar–H) 7.94 (m, 4H, Ar–H), 10.12 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz DMSO-d6): δ 19.00, 22.76, 45.92, 53.11, 88.45, 117.20, 121.19, 123.35, 124.27, 125.73, 128.99, 131.62, 136.63, 138.63, 140.80, 157.86, 166.04: EIMS for C24H18N4O3 m/z (100): 410.74 [M+] (12.07), 57.36 (100).
4.1.3.22 1-Cyano-2-(5-fluoro-1,3-dioxoisoindolin-2-yl)-N-phenyl-5,6,7,8-tetrahydroindolizine-3-carboxamide 6k. Pale yellow; yield 85%; mp 218–220 °C; IR: (KBr, cm−1) 3309 (NH), 2225 (CN), 1728 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.92 (m, 4H, CH2, CH2), 2.95 (t, 2H, CH2), 4.23 (t, 2H, CH2), 7.06 (t, 1H, J = 6.5 Hz, Ar–H) 7.26 (d, 2H, J = 8.9 Hz, Ar–H), 7.43 (d, 2H, J = 11.3 Hz, Ar–H), 7.74 (d, 1H, J = 8.2 Hz, Ar–H), 7.91 (d, 1H, J = 7.3 Hz, Ar–H), 8.05 (s, 1H, Ar–H), 10.13 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz DMSO-d6): δ 18.92, 22.55, 45.96, 53.22, 88.41, 112.04, 112.29, 114.38, 116.96, 121.20, 122.66, 123.30, 124.51, 125.71, 127.19, 127.86, 128.99, 129.37, 138.67, 140.83, 157.79, 165.02; EIMS for C24H17FN4O3 m/z (100): 428.91 [M+] (9.84), 204.93 (100).
4.1.3.23 1-Cyano-2-(1,3-dioxoisoindolin-2-yl)-N-(p-tolyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide 6l. Brown; yield 83%; mp 236–238 °C; IR: (KBr, cm−1) 3313 (NH), 2222 (CN), 1716 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.87 (m, 2H, CH2), 2.01 (m, 2H, CH2), 2.20 (s, 3H, CH3), 2.98 (t, 2H, CH2), 4.11 (t, 2H, CH2), 7.04 (d, 2H, J = 8.4 Hz, Ar–H), 7.28 (m, 4H, Ar–H), 7.95 (d, 2H, J = 7.3 Hz, Ar–H) 10.02 (s, 1H, NH, D2O exchangeable): EIMS for C25H20N4O3 m/z (100): 424.09 [M+] (14.71), 57.36 (100).
4.1.3.24 1-Cyano-2-(5-fluoro-1,3-dioxoisoindolin-2-yl)-N-(p-tolyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide 6m. Dark brown; yield 86%; mp 242–244 °C; IR: (KBr, cm−1) 3294 (NH), 2222 (CN), 1732 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.87 (m, 2H, CH2), 1.96 (m, 2H, CH2), 2.23 (s, 3H, CH3), 2.92 (t, 2H, CH2), 4.19 (t, 2H, CH2) 7.05 (d, 2H, J = 7.7 Hz, Ar–H), 7.27 (d, 2H, J = 8.0 Hz, Ar–H), 7.75 (d, 1H, J = 8.2 Hz, Ar–H), 7.90 (d, 1H, J = 7.2 Hz, Ar–H), 8.05 (s, 1H, Ar–H), 10.02 (s, 1H, NH, D2O exchangeable).
4.1.3.25 1-Cyano-2-(1,3-dioxoisoindolin-2-yl)-N-(4-fluorophenyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide 6n. Brown; yield 85%; mp 243–245 °C; IR: (KBr, cm−1) 3303 (NH), 2225 (CN), 1708 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.87 (m, 2H, CH2), 1.96 (m, 2H, CH2), 2.95 (t, 2H, CH2), 4.21 (t, 2H, CH2), 7.09 (d, 2H, J = 8.8 Hz, Ar–H), 7.36 (d, 2H, J = 8.8 Hz, Ar–H), 7.88 (t, 2H, J = 6.8 Hz, Ar–H), 7.98 (d, 2H, J = 7.5 Hz, Ar–H), 10.14 (s, 1H, NH, D2O exchangeable), 13C NMR (100 MHz DMSO-d6): δ 18.91, 22.60, 45.93, 88.44, 114.41, 115.51, 115.73, 117.24, 123.08, 123.16, 124.27, 131.65, 134.98, 135.68, 140.88, 157.86, 160.19, 166.01; EIMS for C24H17FN4O3 m/z (100): 428.67 [M+] (12.03), 195.22 (100).
4.1.3.26 1-Cyano-2-(5-fluoro-1,3-dioxoisoindolin-2-yl)-N-(4-fluorophenyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide 6o. Gray; yield 80%; mp 252–254 °C; IR: (KBr, cm−1) 3309 (NH), 2225 (CN), 1728 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.87 (m, 2H, CH2), 1.96 (m, 2H, CH2), 2.94 (t, 2H, CH2), 4.22 (t, 2H, CH2), 7.10 (d, 2H, J = 8.5 Hz, Ar–H), 7.39 (d, 2H, J = 8.5 Hz, Ar–H), 7.74 (d, 1H, J = 8.3 Hz, Ar–H) 7.90 (d, 1H, J = 7.3 Hz, Ar–H), 8.05 (s, 1H, Ar–H), 10.14 (s, 1H, NH, D2O exchangeable)13C NMR (100 MHz DMSO-d6): 18.90, 22.53, 45.96, 88.38, 112.03, 112.23, 114.36, 115.51, 115.74, 116.99, 123.01, 123.11, 123.19, 127.87, 134.94, 134.96, 140.96, 157.80, 160.21, 165.00, 165.29, 167.88; EIMS for C24H16F2N4O3 m/z (100): 446.52 [M+] (23.02), 339.28 (100).
4.1.3.27 1-Cyano-2-(1,3-dioxoisoindolin-2-yl)-N-(4-methoxyphenyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide 6p. Green; yield 87%; mp 227–229 °C; IR: (KBr, cm−1) 3305 (NH), 2237 (CN), 1770 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1.86 (m, 2H, CH2), 1.96 (m, 2H, CH2), 2.98 (t, 2H, CH2), 3.78 (s, 3H, OCH3), 4.21 (t, 2H, CH2), 6.80 (d, 2H, J = 8.8 Hz, Ar–H) 7.00 (d, 2H, J = 8.8 Hz, Ar–H), 7.33 (t, 2H, Ar–H), 7.95 (d, 2H, J = 7.4 Hz, Ar–H) 9.93 (s, 1H, NH, D2O exchangeable): EIMS for C25H20N4O4 m/z (100): 440.20 [M+] (9.06), 195.22 (100).
4.1.3.28 1-Cyano-2-(5-fluoro-1,3-dioxoisoindolin-2-yl)-N-(4-methoxyphenyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide 6q. Brown; yield 82%; mp 238–240 °C; IR: (KBr, cm−1) 3306 (NH), 2345 (CN), 1770 (C
O); 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.00 (m, 2H, CH2), 2.09 (m, 2H, CH2), 3.02 (t, 2H, CH2), 3.78 (s, 3H, OCH3), 4.17 (t, 2H, CH2), 6.99 (d, 2H, J = 8.8 Hz, Ar–H), 7.33 (d, 2H, J = 8.80 Hz, Ar–H), 7.69 (d, 1H, J = 7.9 Hz, Ar–H) 7.91 (d, 1H, J = 7.2 Hz, Ar–H), 8.01 (s, 1H, Ar–H), 9.93 (s, 1H, NH, D2O exchangeable): EIMS for C25H19FN4O4 m/z (100): 458.94 [M+] (40.95), 80.37 (100).
4.2. Biological activity
The aforementioned cell lines were used to examine the drug's inhibitory effects on cell growth using the MTT assay. In RPMI-1640 media with 10% foetal bovine serum, cell lines were grown. Antibiotics of 100 units per mL penicillin and 100 g mL−1 streptomycin were introduced at 37 °C in a 5% CO2 incubator. The cell lines were seeded at a density of 1.0 × 104 cells per well in a 96-well plate and incubated for 48 h at 37 °C with 5% CO2.49
000 events (Becton Dickinson, USA).51In vitro enzyme test for cyclin-dependent kinase/cyclin 2A inhibition. The ADP-GloTM Kinase assay methodology was used for the CDK2/Cyclin2A enzyme inhibition assay. The manufacturer's instructions were followed while using the ADP-GloTM Kinase Promega Corporation test kit (2800 Woods Hollow Road, Madison, WI 53711–5399, USA). ADP-GloTM reagent was introduced to the CDK2 reaction after it had finished using ATP and producing ADP in order to stop the kinase process and exhaust any remaining ATP. ADP was afterwards transformed to ATP using the kinase detection reagent, and the recently formed ATP was then used to produce light via a luciferase reaction. DMSO was applied as a solvent for the investigated compound, which was introduced to the mixture at various quantities during the assay, which was carried out at room temperature. On the Tecan-spark READER, continuous kinetic monitoring of enzyme activity was finished. The enzyme inhibition percentages of all the compounds at each concentration were calculated, and Graphpad was used to get the IC50 values.36
4.3. Molecular docking study
Using MOE 2022.09.01 (Molecular Operating Environment, Chemical Computing Group, Canada) software, docking studies, investigations, and calculations were performed for the molecular modeling of EGFR TK (PDB ID: 1XKK) and CDK-2 (PDB ID: 3DDQ).55,56After docking by placement utilizing the Triangle Matcher protocol and post-placement refinement using Rigid Receptor in the case of EGFR TK docking, poses for docked compounds were scored by the initial and final rescoring methodologies (London dG). After docking by placement using the Triangle Matcher procedure, poses for docked compounds were scored using the initial rescoring methodology (London dG) and the final rescoring methodology (GBVI/WSA dG), and post-placement refinement was Rigid Receptor in the case of the CDK-2 study. A combination of Discovery Studio Client Visualizer 2021 and Pymol 2.5 was used to generate 2D and 3D captions.49,62,63
4.4. In silico ADMET studies
The molecular structures of the synthesized pyrrolizine and indolizine derivatives 6d, 6m, and 6o were uploaded into the ADME/Tox web tools SwissADME and pkCSM-pharmacokinetics using the simplified molecular-input line-entry specification (SMILES) nomenclature. To depict the ADME/Tox profile, we picked the key ADME/Tox attributes from the available web resources.Conflicts of interest
There are no conflicts to declare.References
- M. C. Hulvat, Cancer incidence and trends, Surgical Clinics, 2020, 100(3), 469–481 Search PubMed.
- S. T. Pan, et al., Molecular mechanisms for tumour resistance to chemotherapy, Clin. Exp. Pharmacol. Physiol., 2016, 43(8), 723–737 CrossRef CAS PubMed.
- A.-M. Florea and D. Büsselberg, Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects, Cancers, 2011, 3(1), 1351–1371 CrossRef CAS PubMed.
- Q. Wu, et al., Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches, Cancer Lett., 2014, 347(2), 159–166 CrossRef CAS PubMed.
- C. M. Neophytou, et al., Apoptosis deregulation and the development of cancer multi-drug resistance, Cancers, 2021, 13(17), 4363 CrossRef CAS PubMed.
- Q. Gao, et al., Opportunities and challenges for co-delivery nanomedicines based on combination of phytochemicals with chemotherapeutic drugs in cancer treatment, Adv. Drug Delivery Rev., 2022, 114445 CrossRef CAS PubMed.
- R. van der Meel, et al., Smart cancer nanomedicine, Nat. Nanotechnol., 2019, 14(11), 1007–1017 CrossRef CAS PubMed.
- Z. Chen, et al., Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade, Cancer Lett., 2016, 370(1), 153–164 CrossRef CAS PubMed.
- A. Lazarowski, ABC-transporters and drug efflux in hematologic cancers, in Drug Efflux Pumps in Cancer Resistance Pathways: From Molecular Recognition and Characterization to Possible Inhibition Strategies in Chemotherapy, Elsevier, 2020, pp. 149–195 Search PubMed.
- J. Veiga-Matos, F. Remião and A. Motales, Pharmacokinetics and toxicokinetics roles of membrane transporters at kidney level, J. Pharm. Pharm. Sci., 2020, 23, 333–356 CAS.
- X. Gu, et al., Synthesis and biological evaluation of bifendate–chalcone hybrids as a new class of potential P-glycoprotein inhibitors, Bioorg. Med. Chem., 2012, 20(8), 2540–2548 CrossRef CAS PubMed.
- A. M. Shawky, et al., Discovery of new pyrimidopyrrolizine/indolizine-based derivatives as P-glycoprotein inhibitors: Design, synthesis, cytotoxicity, and MDR reversal activities, Eur. J. Med. Chem., 2021, 218, 113403 CrossRef CAS PubMed.
- B. Wang, et al., Exploration of 1, 2, 3-triazole-pyrimidine hybrids as potent reversal agents against ABCB1-mediated multidrug resistance, Eur. J. Med. Chem., 2018, 143, 1535–1542 CrossRef CAS PubMed.
- A. Tan, et al., Evaluation of cytotoxic potentials of some isoindole-1, 3-dione derivatives on HeLa, C6 and A549 cancer cell lines, Med. Chem., 2020, 16(1), 69–77 CrossRef CAS PubMed.
- D. Berger, et al., Novel multidrug resistance reversal agents, J. Med. Chem., 1999, 42(12), 2145–2161 CrossRef CAS PubMed.
- M. Labbozzetta, et al., Novel insights on [1, 2] oxazolo [5, 4-e] isoindoles on multidrug resistant acute myeloid leukemia cell line, Drug Dev. Res., 2022, 83(6), 1331–1341 CrossRef CAS PubMed.
- J. Tamariz, et al., Pyrrolizidine alkaloids, The alkaloids: chemistry and biology, 2018, vol. 80, pp. 1–314 Search PubMed.
- X. Wei, W. Ruan and K. Vrieling, Current knowledge and perspectives of pyrrolizidine alkaloids in pharmacological applications: A mini-review, Molecules, 2021, 26(7), 1970 CrossRef CAS PubMed.
- A. M. Gouda, et al., Design, synthesis and pharmacological evaluation of novel pyrrolizine derivatives as potential anticancer agents, Bioorg. Chem., 2014, 53, 1–7 CrossRef CAS PubMed.
- S. Tavolari, et al., Licofelone, a dual COX/5-LOX inhibitor, induces apoptosis in HCA-7 colon cancer cells through the mitochondrial pathway independently from its ability to affect the arachidonic acid cascade, Carcinogenesis, 2008, 29(2), 371–380 CrossRef CAS PubMed.
- W. Liu, et al., Investigations on cytotoxicity and anti-inflammatory potency of licofelone derivatives, Eur. J. Med. Chem., 2011, 46(3), 907–913 CrossRef CAS PubMed.
- A. M. Gouda, et al., Antitumor activity of pyrrolizines and their Cu (II) complexes: Design, synthesis and cytotoxic screening with potential apoptosis-inducing activity, Eur. J. Med. Chem., 2018, 145, 350–359 CrossRef CAS PubMed.
- V. Lisowski, et al., Design, synthesis and antiproliferative activity of tripentones: a new series of antitubulin agents, Bioorg. Med. Chem. Lett., 2001, 11(16), 2205–2208 CrossRef CAS PubMed.
- C. Rochais, et al., MR22388, a novel anti-cancer agent with a strong FLT-3 ITD kinase affinity, Cancer Lett., 2013, 331(1), 92–98 CrossRef CAS PubMed.
- A. M. Gouda, et al., Pyrrolizines: design, synthesis, anticancer evaluation and investigation of the potential mechanism of action, Bioorg. Med. Chem., 2017, 25(20), 5637–5651 CrossRef CAS PubMed.
- B. C. Q. Nguyen, et al., 1, 2, 3-Triazolyl ester of Ketorolac: A “Click Chemistry”-based highly potent PAK1-blocking cancer-killer, Eur. J. Med. Chem., 2017, 126, 270–276 CrossRef CAS PubMed.
- M. A. Abourehab, et al., Pyrrolizine/indolizine-cinnamaldehyde Schiff bases: Design, synthesis, biological evaluation, ADME, and molecular docking study, Eur. J. Med. Chem. Rep., 2022, 4, 100036 CAS.
- C. Lima, S. F. Eto and M. Lopes-Ferreira, Shedding Light on the Drug–Target Prediction of the Anti-Inflammatory Peptide Tn P with Bioinformatics Tools, Pharmaceuticals, 2022, 15(8), 994 CrossRef CAS PubMed.
- L. H. Al-Wahaibi, et al., Synthesis and Biological Evaluation of Indole-2-Carboxamides with Potent Apoptotic Antiproliferative Activity as EGFR/CDK2 Dual Inhibitors, Pharmaceuticals, 2022, 15(8), 1006 CrossRef CAS PubMed.
- M. A. Mansour, et al., Insights into fourth generation selective inhibitors of (C797S) EGFR mutation combating non-small cell lung cancer resistance: a critical review, RSC Adv., 2023, 13(27), 18825–18853 RSC.
- A. Belal, Synthesis, molecular docking and antitumor activity of novel pyrrolizines with potential as EGFR-TK inhibitors, Bioorg. Chem., 2015, 59, 124–129 CrossRef CAS PubMed.
- Y. Wang, et al., Design, synthesis and biological evaluation of pyrimidine derivatives as novel CDK2 inhibitors that induce apoptosis and cell cycle arrest in breast cancer cells, Bioorg. Med. Chem., 2018, 26(12), 3491–3501 CrossRef CAS PubMed.
- T. Otto and P. Sicinski, Cell cycle proteins as promising targets in cancer therapy, Nat. Rev. Cancer, 2017, 17(2), 93–115 CrossRef CAS PubMed.
- H. A. Abd El-wahab, et al., New cell cycle checkpoint pathways regulators with 2-Oxo-indoline scaffold as potential anticancer agents: Design, synthesis, biological activities and in silico studies, Bioorg. Chem., 2022, 120, 105622 CrossRef CAS PubMed.
- A. M. Shawky, et al., Pharmacophore-based virtual screening, synthesis, biological evaluation, and molecular docking study of novel pyrrolizines bearing urea/thiourea moieties with potential cytotoxicity and CDK inhibitory activities, J. Enzyme Inhib. Med. Chem., 2021, 36(1), 15–33 CrossRef CAS PubMed.
- A. Belal, et al., Design, synthesis and molecular docking of new fused 1 H-pyrroles, pyrrolo [3, 2-d] pyrimidines and pyrrolo [3, 2-e][1, 4] diazepine derivatives as potent EGFR/CDK2 inhibitors, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 1884–1902 CrossRef CAS PubMed.
- X.-J. Liu, et al., Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy, Bioorg. Chem., 2023, 106425 CrossRef CAS PubMed.
- J. Tang and T. Aittokallio, Network pharmacology strategies toward multi-target anticancer therapies: from computational models to experimental design principles, Curr. Pharm. Des., 2014, 20(1), 23–36 CrossRef CAS PubMed.
- I. Shawish, et al., Pyrazolyl-s-triazine with indole motif as a novel of epidermal growth factor receptor/cyclin-dependent kinase 2 dual inhibitors, Front. Chem., 2022, 10, 1078163 CrossRef CAS PubMed.
- M. S. Altowyan, et al., Synthesis, characterization, and cytotoxicity of new spirooxindoles engrafted furan structural motif as a potential anticancer agent, ACS Omega, 2022, 7(40), 35743–35754 CrossRef CAS PubMed.
- H. A. Gomaa, et al., Optimization and SAR investigation of novel 2, 3-dihydropyrazino [1, 2-a] indole-1, 4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities, Bioorg. Chem., 2022, 120, 105616 CrossRef CAS PubMed.
- M. H. Saad, et al., Discovery of new symmetrical and asymmetrical nitrile-containing 1, 4-dihydropyridine derivatives as dual kinases and P-glycoprotein inhibitors: synthesis, in vitro assays, and in silico studies, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 2489–2511 CrossRef CAS PubMed.
- W. S. Hamama, et al., Recent advances in the chemistry of 2-chloroquinoline-3-carbaldehyde and related analogs, RSC Adv., 2018, 8(16), 8484–8515 RSC.
- M. A. Metwally and E. Abdel-Latif, Thiohydantoins: synthetic strategies and chemical reactions, J. Sulfur Chem., 2012, 33(2), 229–257 CrossRef CAS.
- S. Kumari, S. Paliwal and R. Chauhan, Synthesis of pyrazole derivatives possessing anticancer activity: Current status, Synth. Commun., 2014, 44(11), 1521–1578 CrossRef CAS.
- E. Abdel-Latif, M. M. Fahad and M. A. Ismail, Synthesis of N-aryl 2-chloroacetamides and their chemical reactivity towards various types of nucleophiles, Synth. Commun., 2020, 50(3), 289–314 CrossRef CAS.
- D. Kumar, et al., Synthesis and antimicrobial investigation of some novel phenyl pyrazole, azetidinone and diazenyl ethanone derivatives of benzofurans, 2008 Search PubMed.
- K. M. Attalah, et al., Ethyl benzoate bearing pyrrolizine/indolizine moieties: Design, synthesis and biological evaluation of anti-inflammatory and cytotoxic activities, Bioorg. Chem., 2020, 94, 103371 CrossRef CAS PubMed.
- A. M. Shaker, et al., Novel 1, 3-diaryl pyrazole derivatives bearing methylsulfonyl moiety: Design, synthesis, molecular docking and dynamics, with dual activities as anti-inflammatory and anticancer agents through selectively targeting COX-2, Bioorg. Chem., 2022, 129, 106143 CrossRef CAS PubMed.
- L. Sun, et al., Icaritin reverses multidrug resistance of HepG2/ADR human hepatoma cells via downregulation of MDR1 and P-glycoprotein expression, Mol. Med. Rep., 2013, 8(6), 1883–1887 CrossRef CAS PubMed.
- H. S. Mohamed, et al., Design, synthesis, biological assessment, and in-silico studies of 1, 2, 4-triazolo [1, 5-a] pyrimidine derivatives as tubulin polymerization inhibitors, Bioorg. Chem., 2022, 121, 105687 CrossRef CAS PubMed.
- A. F. Zaher, et al., Synthesis, antitumor screening and cell cycle analysis of novel benzothieno [3, 2-b] pyran derivatives, J. Enzyme Inhib. Med. Chem., 2016, 31(sup4), 145–153 CrossRef CAS PubMed.
- B. Xie, et al., Synthesis, characterization and anticancer efficacy studies of iridium (III) polypyridyl complexes against colon cancer HCT116 cells, Molecules, 2022, 27(17), 5434 CrossRef CAS PubMed.
- I. A. Abdel-Azziz, et al., Design, synthesis and mechanistic studies of benzophenones hydrazone derivatives as cathepsin inhibitors, J. Mol. Struct., 2023, 1274, 134583 CrossRef CAS.
- M. A. Abourehab, et al., Globally approved EGFR inhibitors: Insights into their syntheses, target kinases, biological activities, receptor interactions, and metabolism, Molecules, 2021, 26(21), 6677 CrossRef CAS PubMed.
- K. H. Oudah, et al., Design, synthesis and molecular docking of novel pyrazolo [1, 5-a][1, 3, 5] triazine derivatives as CDK2 inhibitors, Bioorg. Chem., 2019, 92, 103239 CrossRef CAS PubMed.
- M. A. Mansour, A. M. AboulMagd and H. M. Abdel-Rahman, Quinazoline-Schiff base conjugates: in silico study and ADMET predictions as multi-target inhibitors of coronavirus (SARS-CoV-2) proteins, RSC Adv., 2020, 10(56), 34033–34045 RSC.
- D. E. Pires, T. L. Blundell and D. B. Ascher, pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures, J. Med. Chem., 2015, 58(9), 4066–4072 CrossRef CAS PubMed.
- C. A. Lipinski, et al., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25 CrossRef CAS.
- T. Hou, et al., ADME evaluation in drug discovery. 6. Can oral bioavailability in humans be effectively predicted by simple molecular property-based rules?, J. Chem. Inf. Model., 2007, 47(2), 460–463 CrossRef CAS PubMed.
- S. E. Abbas, et al., Novel substituted and fused pyrrolizine derivatives: Synthesis, anti-inflammatory and ulcerogenecity studies, Eur. J. Med. Chem., 2010, 45(2), 482–491 CrossRef CAS PubMed.
- S. S. El-Hawary, et al., Characterization of Promising Cytotoxic Metabolites from Tabebuia guayacan Hemsl.: Computational Prediction and In Vitro Testing, Plants, 2022, 11(7), 888 CrossRef CAS PubMed.
- Y. O. Mekhlef, A. M. AboulMagd and A. M. Gouda, Design, Synthesis, Molecular docking, and biological evaluation of novel 2, 3-diaryl-1, 3-thiazolidine-4-one derivatives as potential anti-inflammatory and cytotoxic agents, Bioorg. Chem., 2023, 133, 106411 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05310e |
| This journal is © The Royal Society of Chemistry 2023 |