
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel quinoxaline-3-propanamides as VGFR-2 inhibitors and apoptosis inducers†
Magda M. F. Ismail a,
Taghreed Z. Shawera,
Rabab S. Ibrahima,
Mostafa S. Abusaifb,
Mona M. Kamalc,
Rasha M. Allamd and
Yousry A. Ammarb
a,
Taghreed Z. Shawera,
Rabab S. Ibrahima,
Mostafa S. Abusaifb,
Mona M. Kamalc,
Rasha M. Allamd and
Yousry A. Ammarb
aDepartment of Pharmaceutical Medicinal Chemistry and Drug Design, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo, 11754, Egypt. E-mail: m.elalfy101@gmail.com; magdaismail@azhar.edu.eg
bDepartment of Chemistry, Faculty of Science, Al-Azhar University, Cairo, 11754, Egypt
cDepartment of Pharmacology, Faculty of Pharmacy (Girls), 11754 Al-Azhar University, Cairo, Egypt
dDepartment of Pharmacology, Medical and Clinical Research Institute, National Research Centre, 12622, Dokki, Cairo, Egypt
First published on 31st October 2023
Abstract
Vascular endothelial growth factor receptor-2 is a vital target for therapeutic mediation in various types of cancer. This study was aimed at exploring the cytotoxic activity of seventeen novel quinoxaline-3-propanamides against colon cancer (HCT-116) and breast cancer (MCF-7) using MTT assay. Results revealed that compounds 8, 9, and 14 elicited higher cytotoxicity than the reference drugs, doxorubicin (DOX) and sorafenib. Interestingly, they are more selective for HCT-116 (SI 11.98–19.97) and MCF-7 (SI 12.44–23.87) compared to DOX (SI HCT-116 0.72 and MCF-7 0.9). These compounds effectively reduced vascular endothelial growth factor receptor-2; among them, compound 14 displayed similar VEGFR-2 inhibitory activity to sorafenib (IC50 0.076 M). The ability of 14 to inhibit angiogenesis was demonstrated by a reduction in VEGF-A level compared to control. Furthermore, it induced a significant increase in the percentage of cells at pre-G1 phase by almost 1.38 folds (which could be indicative of apoptosis) and an increase in G2/M by 3.59 folds compared to the control experiment. A flow cytometry assay revealed that compound 14 triggered apoptosis via the programmed cell death and necrotic pathways. Besides, it caused a remarkable increase in apoptotic markers, i.e., caspase-3 p53 and BAX. When compared to the control, significant increase in the expression levels of caspase-3 from 47.88 to 423.10 and p53 from 22.19 to 345.83 pg per ml in MCF-7 cells. As well, it increased the proapoptotic protein BAX by 4.3 times while lowering the antiapoptotic marker BCL2 by 0.45 fold. Docking studies further supported the mechanism, where compound 14 showed good binding to the essential amino acids in the active site of VEGFR-2. Pharmacokinetic properties showed the privilege of these hits over sunitinib: they are not substrates of P-gp protein; this suggests that they have less chance to efflux out of the cell, committing maximum effect; and in addition, they do not allow permeation to the BBB.
1. Introduction
One may argue that abnormal angiogenesis is a crucial condition for the growth and spread of tumors. The growth and occurrence of numerous tumor types are strongly correlated with the aberrant expression of VEGFR-2 in neovascular tumor endothelial cells, according to mounting evidence.1,2 At the moment, blocking VEGFRs to target tumor angiogenesis is a successful oncotherapy method.3–5 All VEGFR-2 inhibitors, despite the fact that the majority of them lack specificity, provide differing degrees of therapeutic efficacy against various tumor types by inhibiting angiogenesis and lymphangiogenesis.6 Currently, there are three types of VEGFR inhibitors: type I inhibitors, type II inhibitors, and type III inhibitors.7 Sunitinib (SU11248), a type I inhibitor, is the first small molecule to contain the indolin-2-one template and is effective against a number of kinases, including the stem cell factor receptor (c-Kit), platelet-derived growth factor receptors A and B (PDGFRA and PDGFRB), colony-stimulating factor 1 receptor (CSF-1R), FMS-like tyrosine kinase 3. (FLT3)8 Type II inhibitors, e.g., sorafenib, are characterized by binding to the inactive “DFG-out” conformation of the kinase and occupying a hydrophobic pocket adjacent to the ATP-binding site.9,10 Structurally, the extension into the less conservative allosteric hydrophobic back pocket facilitates the affinity and selectivity of the type II inhibitors.11 Type III, e.g., vatalanib, could exert their pharmacological functions by irreversibly binding to cysteine at specific sites on the kinases.12Quinoxaline derivatives are a widespread class of heterocycles receiving the most attention, especially in the field of chemotherapy.13–16 Many drugs incorporating the quinoxaline scaffold have achieved promising results and have been submitted to clinical trials for anticancer therapeutic purposes.17,18
The therapeutic applicability of current VEGFR inhibitors, however, is constrained to some extent by their low clinical efficacy and probable toxicity. Based on the aforementioned findings (cf. Fig. 1) and as a continuation of our prior work in the field of the design and synthesis of new anticancer medicines,19–26 a new series of 6-chloroquinoxaline-3-propanamides have been designed as sunitinib's mimetic to optimize pharmacodynamics properties and to overcome certain pharmacokinetic problems demonstrated on sunitinib clinical use such as, BBB permeation and being P-gp substrate.
 | ||
| Fig. 1 Feauture similarities between ligand and target compounds. | ||
Herein, the hetero-aromatic system was designed to be 6-chloroquinoxalin-2-one as an isostere for 5-fluoroindoline-2-one, and the linker group was an aliphatic chain (–CH2CH2–) as a chain contraction to replace 2,4-dimethyl-1H-pyrrole of sunitinib. The pharmacophore moiety was kept in all the designed compounds as an amide group. The terminal hydrophobic moiety was an aryl moiety with a variation of substituents instead of an aliphatic amino side chain of sunitinib. These modifications were followed to optimize both the pharmacodynamics and pharmacokinetic criteria of sunitinib, (Fig. 1).
2. Discussion
2.1. Chemistry
The starting material, 3,27 was readily produced in nearly a quantitative yield (95%) by cyclocondensing 4-chloro-1,2-phenylenediamine, 1, with ketoglutaric acid, 2, in acetic acid/ethanol with stirring at room temperature. The acid derivative 3 was then treated with methanol and a few drops of concentrated H2SO4 in a reflux environment for three hours (Scheme 1) to produce the equivalent methyl ester 4.27 | ||
| Scheme 1 Reagents and conditions, (i) acetic acid/EtOH, stirr, r.t. 15 min (ii) CHsOH, dps conc. H2SO4, reflux, 3 h, (iii) aromatic amines, EtOH/DMF, reflux, 8–16 h. | ||
Additionally, aminolysis28 of ethyl propanoate ester 4 with substituted anilines at positions 2, 3, or 4 was carried out to yield the corresponding quinoxaline-3-propanamides 5–17. A single molecule of methanol was eliminated through reflux in a catalytic amount of DMF in ethanol solvent over the course of eight to sixteen hours, successfully completing the reaction despite aniline's very weak nucleophilicity towards the carbonyl carbon of the ester group, (Scheme 1).
The spectroscopic and micro-analytical data from all quinoxaline-3-propanamides 5–17 were used to elucidate them. All of our series are characterized by the presence of two triplets in the 1H NMR spectra that indicate linker protons (–CH2–CH2–CO) at δ (ppm): 1.63–1.75 for CH2 and 2.25–2.38 for CH2CO protons. Besides, deshielded signals for three quinox-Hs and four aryl-Hs are appeared at their appropriate aromatic region. It is important to note several distinctive signals in the 1H NMR spectra of our series; for example, the 4-CH3 protons of compound 11 appeared as a singlet at around δ 2.40 ppm, while the 4-OCH3 protons of compound 14 appeared at about δ 3.90 ppm. Compound 15's 1H NMR spectrum showed a singlet signal at about δ 2.67 ppm that was attributed to COCH3 protons. In the 1H NMR spectrum of the ethyl ester 17, a triplet–quartet pattern was seen at δ 1.34 and 4.36 ppm respectively.
Hydrazinolysis of the ester 4 with hydrazine hydrate using absolute ethanol produced a good yield of the corresponding acid hydrazide derivative, 18.29 Nucleophilic substitution reaction of compound 18 with 2,4-dichlorobenzoyl chloride was achieved by heating under reflux condition in ethanol with DMF drops30 to furnish the corresponding 2,4-dichlorobenzoyl hydrazide, 19 (Scheme 2). Furthermore, synthesis of the target compounds 20 and 21 was achieved by treatment of the intermediate 18 with the appropriate acid anhydrides namely; succinic anhydride or phthalic anhydride respectively via ring opening amidation reaction in ethanol with catalytic amount of DMF.31 Compound 20 was verified based on spectral data; its IR spectrum displayed extra bands at 3360 and 1706 cm−1 contributed to OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of carboxyl group respectively. 1H NMR spectrum displayed extra two triplets at δ 2.38 and 2.59 ppm contributed to CH2CH2 protons of succinic acid moiety. As mentioned in literature,32 reaction of the starting material 18 with phthalic anhydride is a solvent dependent. Thus, when the reaction was carried out in ethanol; the product was formulated as carboxylic acid derivative 21. While performing the reaction in glacial acetic acid as a solvent, the corresponding isoindoline derivative 22 was obtained via ring closure due to condensation reaction. The isoindoline 22 was further confirmed by refluxing compound 21 in glacial acetic for 2 h to afford one and the same product (22). The structures of these compounds were consistent with spectral analysis. IR of compound 21 showed stretching bands around 3339 and 1705 cm−1 pointing to hydroxyl and carbonyl of COOH respectively. 1H NMR spectrum showed a characteristic singlet at δ 10.80 ppm attributed to carboxylic proton; also 4 extra signals (4 Ar–H) at the range from δ 7.44 to 8.74 ppm were observed. Mass spectrum exhibited a peak at m/z 414 referred to molecular ion peak (M+). Concerning IR spectrum of compound 22, lack of broad band of OH and NH is observed due to removal of water molecule. In addition, the frequency of carbonyl band is increased due to ring closure, where 1H NMR spectrum displayed only two signals at 12.22 and 12.54 ppm for two NH protons with lack of COOH and one NH signals ensures the cyclization (Scheme 2).
O of carboxyl group respectively. 1H NMR spectrum displayed extra two triplets at δ 2.38 and 2.59 ppm contributed to CH2CH2 protons of succinic acid moiety. As mentioned in literature,32 reaction of the starting material 18 with phthalic anhydride is a solvent dependent. Thus, when the reaction was carried out in ethanol; the product was formulated as carboxylic acid derivative 21. While performing the reaction in glacial acetic acid as a solvent, the corresponding isoindoline derivative 22 was obtained via ring closure due to condensation reaction. The isoindoline 22 was further confirmed by refluxing compound 21 in glacial acetic for 2 h to afford one and the same product (22). The structures of these compounds were consistent with spectral analysis. IR of compound 21 showed stretching bands around 3339 and 1705 cm−1 pointing to hydroxyl and carbonyl of COOH respectively. 1H NMR spectrum showed a characteristic singlet at δ 10.80 ppm attributed to carboxylic proton; also 4 extra signals (4 Ar–H) at the range from δ 7.44 to 8.74 ppm were observed. Mass spectrum exhibited a peak at m/z 414 referred to molecular ion peak (M+). Concerning IR spectrum of compound 22, lack of broad band of OH and NH is observed due to removal of water molecule. In addition, the frequency of carbonyl band is increased due to ring closure, where 1H NMR spectrum displayed only two signals at 12.22 and 12.54 ppm for two NH protons with lack of COOH and one NH signals ensures the cyclization (Scheme 2).
 | ||
| Scheme 2 Reagents and conditions, (i) hydrazine hydrate, EtOH, reflux, 5 h. (ii) 2,4-dichlorobenzoyi chloride, EtOH/DME, reflux, 3 h. (iii) sucdnic anhydride, EtOH/DMF, reflux, 4 h, (iv) phthalic anhydride, EtOH/DME, reflux, 5 h, (v) phthalic anhydride, gl. CH3COOH, reflux, 4 h, (vi) gl. CH3COOH, reflux, 2 h. | ||
2.2. Biological evaluation
| Compd. no. | HCT-116 | MCF-7 | Compd. no. | HCT-116 | MCF-7 |
|---|---|---|---|---|---|
| a IC50 (μM): 1–10 (very strong). 11–20 (strong). 21–50 (moderate). 51–100 (weak) and above 100 (non-cytotoxic).b DOX: Doxorubicin. | |||||
| DOX | 9.27 ± 0.3 | 7.43 ± 0.2 | 12 | 28.35 ± 2.2 | 24.56 ± 1.9 |
| Sorafenib | 18.60 ± 1.9 | 16.0 ± 3.6 | 13 | 22.19 ± 1.9 | 18.88 ± 1.4 |
| Sunitinib | 3.42 ± 0.5 | 4.77 ± 0.2 | 14 | 5.81 ± 0.5 | 4.61 ± 1.1 |
| 5 | 45.52 ± 2.7 | 39.03 ± 2.4 | 15 | 84.46 ± 3.9 | 76.73 ± 3.8 |
| 6 | 62.21 ± 3.3 | 54.73 ± 3.2 | 16 | 75.06 ± 3.6 | 67.23 ± 3.5 |
| 7 | 93.14 ± 4.6 | 80.39 ± 4.1 | 17 | 36.02 ± 2.4 | 31.17 ± 2.2 |
| 8 | 4.72 ± 0.8 | 3.95 ± 0.2 | 19 | 6.18 ± 2.1 | 5.11 ± 0.8 |
| 9 | 7.19 ± 0.7 | 6.92 ± 0.5 | 20 | 26.41 ± 3.8 | 10.17 ± 3.3 |
| 10 | 19.41 ± 1.5 | 11.93 ± 0.9 | 21 | 48.17 ± 2.7 | 41.47 ± 2.5 |
| 11 | 61.35 ± 3.4 | 53.95 ± 3.1 | 22 | 37.2 ± 2.3 | 25.92 ± 1.9 |
| Compd no. | WI-38 | HCT-116 | SI | MCE-7 | SI | VEGFR-2 |
|---|---|---|---|---|---|---|
| 8 | 94.27 ± 3.5 | 4.72 ± 0.8 | 19.97 | 3.95 ± 0.2 | 23.87 | 0.109 |
| 9 | 86.11 ± 4.1 | 7.19 ± 0.7 | 11.98 | 6.92 ± 0.5 | 12.44 | 0.131 |
| 14 | 88.72 ± 3.9 | 5.81 ± 0.5 | 15.27 | 4.61 ± 1.1 | 19.25 | 0.076 |
| 19 | 71.60 ± 2.8 | 6.18 ± 2.1 | 11.58 | 5.11 ± 0.8 | 14.01 | — |
| 20 | 93.04 ± 2.2 | 26.41 ± 3.8 | 3.52 | 10.17 ± 3.3 | 9.15 | — |
| DOX | 6.72 ± 0.5 | 9.27 ± 0.3 | 0.72 | 7.43 ± 0.2 | 0.90 | — |
| Sunitinib | — | 3.42 ± 0.5 | — | 4.77 ± 0.2 | — | 0.139 (ref. 35) |
| Sorafenib | — | 18.60 ± 1.9 | — | 16.0 ± 3.6 | — | 0.076 |
In regard to positional isomers, the activity's order was 13 (2-CH3) > 12 (3-CH3) > 11 (4-CH3). The high potency is correlated with electron-releasing by +I of CH3 at p-2 (the preferred position), thus the electron-releasing is a beneficial criteria for cytotoxicity. Conversely, it appeared from the IC50s of compounds 8 (4-OH) and 9 (2-OH) that 8 was slightly more active; this may be attributed to the fact that 4-OH affects the activity via both +I and +M effects rather than 2-OH, which shows only +I effects.
Another polar but highly electron-withdrawing compound is compound 10 (4-NO2), which displayed strong cytotoxicity (IC50 19.41, 11.93 μM), towards HCT116 and MCF-7, respectively. Additionally, moderate activities appeared for compounds 5 (4-H) and 17 (4-COOEt), while both the analogues 15 (4-COCH3) and 16 (4-COOH) bearing strong electron-withdrawing groups are weak cytotoxic agents. Regarding compound 19, very strong cytotoxicity results (IC50 6.18, 5.11 μM) against HCT116 and MCF-7, respectively was noticed. Finally, the cyclized phthalimide derivative 22 exhibited better antitumor activity than the open-chain one, 21 (Table 1).
2.2.3.1 In vitro evaluation of VEGFR-2 inhibitory activity (IC50)37. The most selective cytotoxic derivatives (8, 9, and 14) exhibiting the greatest SIs were further investigated in vitro for their inhibitory activities against VEGFR-2 as a possible mechanism of their antitumor activity. In the meantime, sorafenib was used as a positive control. Half-maximal inhibitory concentration (IC50) values as presented in Table 2 were calculated from the obtained concentration–inhibition response curves. As can be seen from Table 2, the inhibitory effects of VEGFR-2 by our hits (8, 9, and 14) are ranged from IC50s of 0.076–0.131 μM. It became evident that compound 14 had the highest VEGFR-2 inhibitor potency (IC50 0.076 μM), making it equivalent to the reference standard sorafenib. It was interesting to see that it had 1.83 times the potency of sunitinib (IC50 0.139 μM) (Table 2 and Fig. 2).
 | ||
| Fig. 2 The inhibitory effect of quinoxaline derivatives 8, 9 and 14 against VEGFR-2. | ||
2.2.3.2 Quantitative determination of VEGFR-2 in MCF-7 cell line:38. According to research, VEGF/VEGFR-2 activation is essential for the angiogenesis process and has been identified as a key factor in tumor vascularization.39 So, we measured the VEGFR-2 concentrations in MCF-7 cell line. Our findings showed that compound 14 reduces VEGFR-2 production in medium from MCF-7 cells (170.2 4 pg ml−1) in comparison to untreated cells (330.8 4.1 pg ml−1) (Fig. 3). These outcomes support the therapeutic targeting of compound 14 as a VEGFR2 inhibitor.
 | ||
| Fig. 3 Secretion of VEGFR2 in cell culture media of MCF-7 cells treated with IC50 of compound 14 for 24 h, was measured by ELISA kits. Results were expressed as means ± SD of samples performed in triplicates. | ||
2.2.3.3 Effect of compound 14 on angiogenesis (VEFG-A) level40. It is known that angiogenesis plays a crucial role in the progression and advancement of tumors in different cancer types, including breast cancer. Among pro-angiogenic factors, vascular endothelial growth factor (VEGF-A) is a prominent target that encourages tumor growth and metastasis. Therefore, we also evaluated the concentration of VEGF in culture media of MCF-7 after treatment with 14 (Fig. 4). ELISA analysis showed that 14 suppressed VEGF-A secretion in media of MCF-7 when compared to the control group. These findings suggested that 14 could be valuable in vivo tumor suppression via inhibition of angiogenesis.
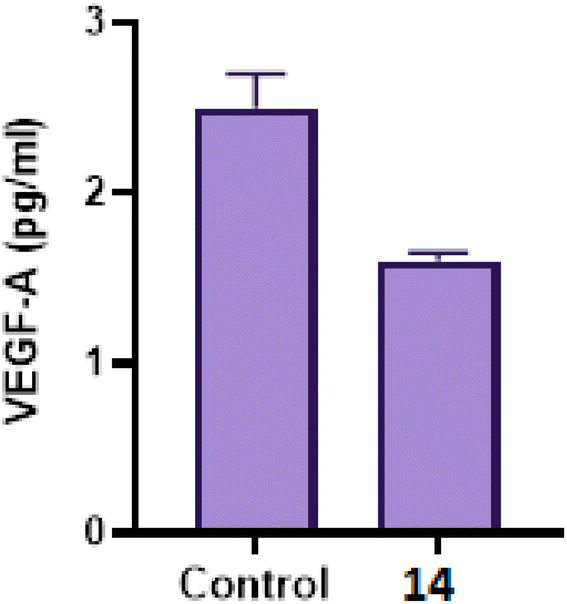 | ||
| Fig. 4 VEGF-A concentrations in cell culture media of MCF-7 breast cancer cell line using ELISA assay following treatment with 14. Results were expressed as means ± SD of samples performed in triplicates. | ||
 | ||
| Fig. 5 (a) Cell cycle analysis of control and compound 14 in MCF-7 cell line. (b) Cell cycle analysis of MCF-7 cells treated with DMSO, (c) cell cycle analysis of MCF-7 cells treated with compound 14. | ||
| Sample | Total | Early apoptosis | Late apoptosis | Necrosis |
|---|---|---|---|---|
| 14/MCF-7 | 22.03 | 2.05 | 6.25 | 12.73 |
| Control/MCF-7 | 2.17 | 0.15 | 0.73 | 1.29 |
 | ||
| Fig. 6 (a) Effect of control on apoptosis of MCF-7 cells. (b) Effect of 14 on apoptosis of MCF-7 cells. | ||
The quadrants in the cytograms represent the following: Necrotic cells (higher left quadrant of the cytogram); late apoptotic cells (higher right quadrant of the cytogram); non-apoptotic and non-necrotic cells (living cells) (lower left quadrant of the cytogram); early apoptotic cells (lower right quadrant of the cytogram).
| Sample | Caspase 3 (pg ml−1) | Fld | p53 (pg ml−1) | Fld | BAX fold change | BCL2 fold change |
|---|---|---|---|---|---|---|
| 14 | 423.10 ± 11.24 | 8.836 | 345.83 ± 7.44 | 15.585 | 4.336 | 0.448 |
| Sunitinib | 384.53 ± 8.40 | 8.031 | 278.34 ± 5.27 | 12.543 | — | — |
| Staurosporine | — | — | — | — | 7.5517 | 0.3248 |
| Control | 47.88 ± 13.15 | 1 | 22.19 ± 8.39 | 1 | 1 | 1 |
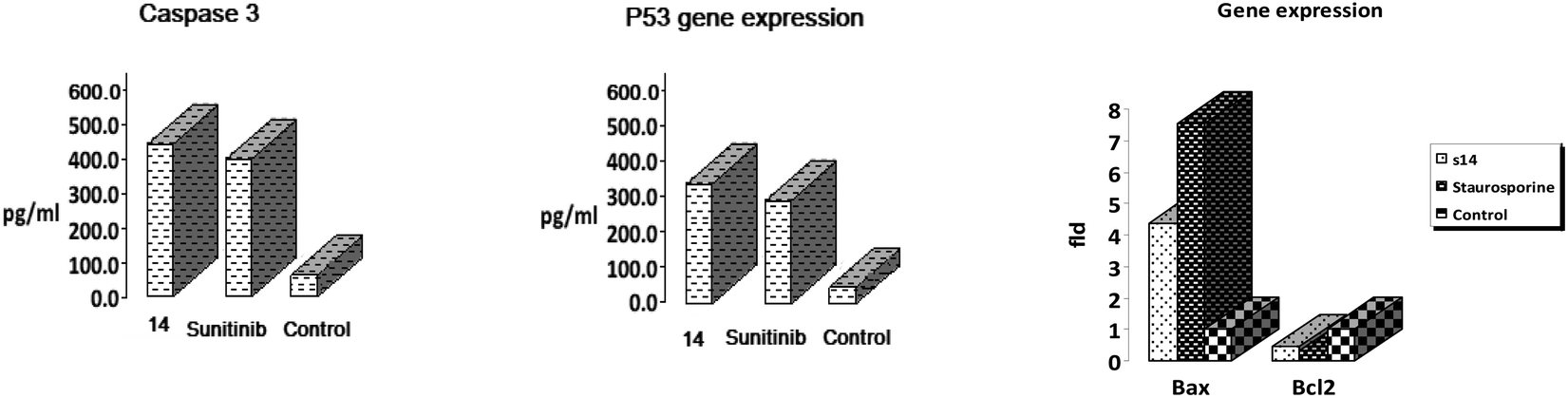 | ||
| Fig. 7 The effect of 14 on (a), caspase-3. (b) p53, (c) BAX and BCL2. | ||
2.3. Molecular modeling study
Here, using the online version of SwissADME, a computational assessment of the synthesized compounds was carried out to assess the physicochemical and ADME properties.47 Regarding Lipinski's rule for oral medications, which indicates that a molecule is more likely to be absorbed or permeated if its molecular weight is less than 500 g mol−1, its M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP value is lower than 5, and it has at least 5 H-donor and 10 H-acceptor atoms.48 While Veber's rule49 specifies polar surface area (PSA) 140 and rotatable bond count 10 as drug-likeness restrictions, respectively.50 It was discovered that all of the synthesized compounds have Lipinski zero violations in their physicochemical parameters and every chemical has rotatable bonds between 5 and 8 that indicate molecular flexibility for its bio target (Table 5). In addition, all hits pass muster under the Veber guidelines used for screening.
logP value is lower than 5, and it has at least 5 H-donor and 10 H-acceptor atoms.48 While Veber's rule49 specifies polar surface area (PSA) 140 and rotatable bond count 10 as drug-likeness restrictions, respectively.50 It was discovered that all of the synthesized compounds have Lipinski zero violations in their physicochemical parameters and every chemical has rotatable bonds between 5 and 8 that indicate molecular flexibility for its bio target (Table 5). In addition, all hits pass muster under the Veber guidelines used for screening.
| Cpd. no. | HBD | HBA | MlogP |
MWt | No. of Rot. bonds | Lipinski's violations | Veber's violations |
|---|---|---|---|---|---|---|---|
| 5 | 2 | 3 | 2.39 | 327.76 | 5 | 0 | 0 |
| 6 | 2 | 3 | 2.89 | 362.21 | 5 | 0 | 0 |
| 7 | 2 | 4 | 2.78 | 345.76 | 5 | 0 | 0 |
| 8 | 3 | 4 | 1.85 | 343.76 | 5 | 0 | 0 |
| 9 | 3 | 4 | 1.85 | 343.76 | 5 | 0 | 0 |
| 10 | 2 | 5 | 1.40 | 372.76 | 6 | 0 | 0 |
| 11 | 2 | 3 | 2.63 | 341.79 | 5 | 0 | 0 |
| 12 | 2 | 3 | 2.63 | 341.79 | 5 | 0 | 0 |
| 13 | 2 | 3 | 2.63 | 341.79 | 5 | 0 | 0 |
| 14 | 2 | 4 | 2.08 | 357.79 | 6 | 0 | 0 |
| 15 | 2 | 4 | 1.97 | 369.80 | 6 | 0 | 0 |
| 16 | 3 | 5 | 2.02 | 371.77 | 6 | 0 | 0 |
| 17 | 2 | 5 | 2.47 | 399.83 | 8 | 0 | 0 |
| 19 | 3 | 4 | 3.15 | 439.68 | 7 | 0 | 0 |
| 20 | 4 | 6 | 0.57 | 366.76 | 9 | 0 | 1 |
| 21 | 4 | 6 | 1.83 | 414.80 | 8 | 0 | 1 |
| 22 | 2 | 5 | 1.93 | 396.78 | 5 | 0 | 0 |
| Sorafenib | 3 | 7 | 2.91 | 464.82 | 9 | 0 | 0 |
| Sunitinib | 3 | 4 | 2.06 | 398.47 | 8 | 0 | 0 |
The topological polar surface area (TPSA) is known to be a reliable indication of drug penetration through the blood–brain barrier (TPSA less than 60 Å2) and intestinal drug absorption (TPSA less than 140 Å2). All compounds exhibit computational TPSA values in the appropriate range for permeating cell membranes so they obey Veber's rule except compounds 20 and 21 (TPSA 141 Å2). Moreover, absorption (% ABS) was calculated using the formula% ABS = 109 − (0.345 × TPSA), and it was discovered that the calculated % ABS of all these hits ranged between 60.26% and 83.17%, indicating that these synthetic derivatives may have the necessary cell membrane permeability and bioavailability (Table 6).
| Comp. no. | TPSA | ABS% |
|---|---|---|
| 5 | 74.85 | 83.17 |
| 6 | 74.85 | 83.17 |
| 7 | 74.85 | 83.17 |
| 8 | 95.08 | 76.19 |
| 9 | 95.08 | 76.19 |
| 10 | 120.67 | 67.36 |
| 11 | 74.85 | 83.17 |
| 12 | 74.85 | 83.17 |
| 13 | 74.85 | 83.17 |
| 14 | 84.08 | 79.99 |
| 15 | 91.92 | 77.28 |
| 16 | 112.15 | 70.30 |
| 17 | 101.15 | 74.10 |
| 19 | 103.95 | 73.13 |
| 20 | 141.25 | 60.26 |
| 21 | 141.25 | 60.26 |
| 22 | 112.23 | 70.28 |
| Sorafenib | 92.35 | 77.13 |
| Sunitinib | 77.23 | 108.92 |
The synthesized compounds' pharmacokinetic and medicinal chemistry parameters (Table 7) made it clear that all of the derivatives had high gastrointestinal absorption and that the majority of them did not cross the blood–brain barrier, guaranteeing that these systemically targeted molecules would have few to no CNS side effects. Analyzing the P-glycoprotein (P-gp) non-substrate candidature during preclinical analysis trials was another crucial factor. P-gp performs the function of an efflux transporter, expelling medicines, other substances, and its substrate from the cell. This sounds like an explanation for why anticancer chemotherapeutic drugs like Imatinib, Lonafarnib, and Taxanes are ineffective. The fact that not all hits are P-gp protein substrates (Table 7) suggests that these hits have a reduced likelihood of effluxing out of the cell and committing maximal effect.
| Compd no. | GI absorption | BBB permeation | P-gp substrate | Bioavailability score | PAINS alerts | Synthetic accessibility |
|---|---|---|---|---|---|---|
| 5 | High | Yes | No | 0.55 | 0 | 2.58 |
| 6 | High | Yes | No | 0.55 | 0 | 2.60 |
| 7 | High | Yes | No | 0.55 | 0 | 2.59 |
| 8 | High | No | No | 0.55 | 0 | 2.57 |
| 9 | High | No | No | 0.55 | 0 | 2.61 |
| 10 | High | No | No | 0.55 | 0 | 2.68 |
| 11 | High | Yes | No | 0.55 | 0 | 2.63 |
| 12 | High | Yes | No | 0.55 | 0 | 2.65 |
| 13 | High | Yes | No | 0.55 | 0 | 2.66 |
| 14 | High | No | No | 0.55 | 0 | 2.66 |
| 15 | High | No | No | 0.55 | 0 | 2.69 |
| 16 | High | No | No | 0.56 | 0 | 2.60 |
| 17 | High | No | No | 0.55 | 0 | 2.85 |
| 19 | High | No | No | 0.55 | 0 | 2.96 |
| 20 | High | No | Yes | 0.56 | 0 | 2.91 |
| 21 | High | No | Yes | 0.56 | 0 | 2.98 |
| 22 | High | No | No | 0.55 | 0 | 2.83 |
| Sorafenib | Low | No | No | 0.55 | 0 | 2.87 |
| Sunitinib | High | Yes | Yes | 0.55 | 0 | 3.58 |
The amount of drug present in the plasma is measured by bioavailability, which is regarded as the most important factor influencing absorption. It's interesting to note that all of the synthetic compounds have high bioavailability ratings on par with sorafenib and sunitinib.
Chemical substances known as pan-assay interference chemicals (PAINS) frequently produce false-positive findings in high-throughput screening. Instead of directly affecting one intended target, PAINS typically respond non-specifically with many biological targets. Checking any PAINS alert of the recently created derivatives is crucial. SwissADME did a PAIN that showed no alarms for any of the hits. The SwissADME Synthetic Accessibility (SA) Score is based mainly on the supposition that the frequency of molecular fragments in ‘really’ attainable molecules correlates with the ease of synthesis; the score is normalized to range from 1 (very easy) to 10 (very difficult). SA scores of all the analogues were found to be between 2.57 and 2.85, indicating that they can be easily synthesized on a large scale.
The in silico ADME prediction analysis results showed that the synthesized compounds exhibit the computational assessment and are therefore viewed as a pharmacologically active framework that should be taken into consideration when moving forward with potential hits.
Using MOE 2014 software, it is possible to investigate the binding interaction of the target compounds using the known crystal structure of VEGFR-2 (PDB ID: 4ASD).52 Reference molecules were the co-crystallized ligands sorafenib and sunitinib. The results of the docking studies demonstrated that our hit 14 had a high affinity for VEGFR-2 in comparison to the reference molecules. Sorafenib, a co-crystallized ligand, was re-docked against VEGFR-2 to verify the docking techniques. The applied docking methodology was validated by the resulting RMSD of 0.88 (Fig. 8).
 | ||
| Fig. 8 The proposed 2D (left) and 3D (right) binding interaction of co-crystalized sorafenib with 4ASD. | ||
Sorafenib's docking score was −10.74 kcal mol−1, and the urea moiety's three hydrogen bonds with the critical amino acids Glu885, Asp1046, and Cys1045 allowed it to bind to the receptor. The hinge region, where the pyridine moiety established a hydrogen link with Cys919 and an arene–H bond with Leu840, was also occupied by the N-methylpicolinamide moiety. Additionally, Leu840, Val916, Leu889, Phe1047, and Leu1035 all showed five hydrophobic contacts (Fig. 8).
Sunitinib inhibits the ATP pocket by an H-bond acceptor/donor interaction between two HB of the indolin-2-one core and Cys919 and Glu917 in the hinge region, together with the pyrrole group as a linker connected to Gly922 via arene-H. Docking experiments showed that sunitinib had a binding energy of −5.76 kcal mol−1. Leu840, Phe1047, Leu1035, Gly922, Val916, and Phe918 were among the hydrophobic interactions that the sunitinib tail was involved in (Fig. 9).
 | ||
| Fig. 9 The proposed 2D (left) and 3D (right) binding interaction of the parent sunitinib with 4ASD. | ||
Our hit 14 had an incredibly low free binding energy of −11.58 kcal mol−1 when compared to sorafenib and sunitinib. It showed 2HBA between the quinox-CO, Phe918, and Cys919 in the hinge region. Quinox-NH was also shared by 1HBD with Glu917. On the other hand, 1HBD also provided CH2 of the linker to the critical amino acid Cys919. Unexpectedly, the gate area's Asp1046 bound to the chloro functionality by HBD, therefore it is supposed that 14 may act as type II inhibitor of VEGFR-2. Additionally, several hydrophobic interactions involving the methoxyl group, terminal phenyl, and aliphatic linker helped the compound attach to VEGFR-2 (Fig. 10). These interactions included Gly922, Asn923, Val916, Phe1047, Le840, and Leu1035. Overlay docking alignment was also carried out between sorafenib (yellow), sunitinib (blue), and 14 (green) in the active site of VEGFR-2 to verify the target chemical 14's mode of action as a VEGFR-2 inhibitor (Fig. 11).
 | ||
| Fig. 10 The proposed 2D (left) and 3D (right) binding interaction of compound 14. | ||
 | ||
| Fig. 11 Overlay docking alignment of sorafenib (yellow), sunitinib (blue), and 14 (green) in the active site of VEGFR-2. | ||
3. Conclusion
Thirteen novel quinoxaline-3-propamide derivatives were created using various substitutions, and their SARs as potential anticancer agents that target the VEGFR-2 enzyme were examined. Eight substances had cytotoxic effects on HCT-116 and MCF-7 that ranged from extremely strong to mild. The top three substances (8, 9 and 14) were then further examined as potential VEGFR-2 inhibitors. Intriguingly, 14 was equivalent to sorafenib (IC50 0.076 M) and 1.83 folds more potent (IC50 0.076 M) than the reference medication, sunitinib (IC50 0.139 M). It reduces VEGFR-2 production in MCF-7 cells when compared to untreated cells. What's more, it lowers VEGF-A level more than control in MCF-7 cells which suggested that 14 could be valuable in vivo tumor suppression via inhibition of angiogenesis. Cell cycle analysis was used to conduct a more detailed assessment of the most active compound, 14, utilizing the MCF-7 cell line. In comparison to the control, it markedly boosted the pre-G phase while halting the G2/M phase of the cell cycle. Additionally, a flow cytometry assay utilizing Annexin V was performed on it, and the outcomes demonstrated an increase in the overall apoptosis percentage compared to the control. Once more, it increased the expression of caspase-3, p53, and BAX; while reduced the BCL2 level than the control experiment; both of which are apoptosis inducers. Docking studies supported our proposed mode of action for 14 as a VEGFR-2 inhibitor and demonstrated nearly identical binding characteristics to the co-crystallized inhibitor (TSA). Because it is not a P-gp substrate and does not permeate the BBB, our hit 14 had more favorable pharmacokinetic characteristics than sunitinib.4. Experimental section
4.1. Chemistry
Reagents and solvents were ordered from regular commercial suppliers and used without further purification. The reported yields apply to distilled products. All reactions were routinely checked with thin-layer chromatography (TLC) of Merck Silica Gel 60 F254 (0.25 mm thick) and visualization with a UV lamp. The melting points were measured in open capillary tubes using the Stuart SMP3 apparatus. IR spectra (KBr) were measured on a Shimadzu FT/IR 1650 (PerkinElmer) spectrometer. 1H and 13C NMR spectra were recorded on a Brüker Advance-400 instrument (400 MHz for 1H and 100 MHz for 13C) in DMSO-d6 Chemical shifts (δ) are reported in ppm relative to TMS as an internal standard, or to the solvent in which the spectrum was recorded. Mass spectra were performed on a Shimadzu GS/MS-QP 2010 plus spectrometer at 70 eV.4.2 General procedure
Equimolar amounts of ethyl propanoate ester 4 and different substituted anilines were refluxed in ethanol/DMF (15/5) for 8–16 h. The reaction medium was cooled and poured into ice water. The precipitate was filtered, dried and crystalized from ethanol to afford the corresponding anilides 5–17.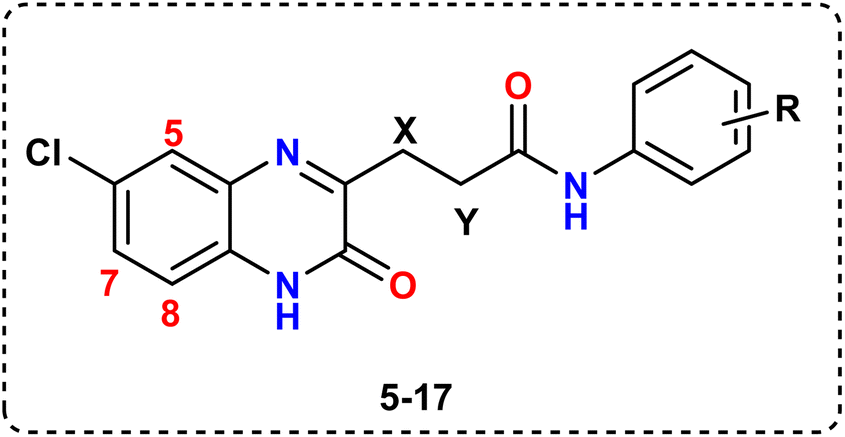 O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.68 (t, 2H, CH2x, J = 7.6 Hz), 2.34 (t, 2H, CH2y, J = 7.6 Hz), 6.87 (t, 1H, Ar–H4), 7.07 (t, 2H, Ar–H3,5, J = 8 Hz), 7.41 (d, 2H, Ar–H2,6, J = 8 Hz), 7.60 (d, 1H, quinoxaline-H7, J = 8 Hz), 8.23 (s, 1H, quinoxaline-H5), 8.33 (d, 1H, quinoxaline-H8, J = 8 Hz), 10.31, 12.59 (2s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.34 (CH2), 31.50 (CH2), 119.12, 120.34, 120.50, 120.57, 125.45, 125.75, 126.12, 126.19, 126.45, 128.34, 129.50, 130.57, 131.12 (13 Ar–C), 167.12, 170.34 (2CO). MS m/z: 327.12 (21.53%, M+), 329.14 (7.16%, M+2), 76.09 (100). Anal. calcd for: C17H14ClN3O2 (327.76): C, 62.30; H, 4.31; N, 12.82%, found: C, 62.73; H, 4.67; N, 12.40%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.66 (t, 2H, CH2x, J = 8 Hz), 2.33 (t, 2H, CH2y, J = 8 Hz), 7.14 (d, 1H, quinoxaline-H7, J = 7.6 Hz), 7.64 (d, 2H, Ar–H3,5, J = 8 Hz), 7.83 (d, 2H, Ar–H2,6, J = 8 Hz), 7.96 (s, 1H, quinoxaline-H5), 8.02 (d, 1H, quinoxaline-H8, J = 7.6 Hz), 11.51, 11.64 (2s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.03 (CH2), 30.81 (CH2), 121.68, 128.62, 128.69, 129.29, 129.34, 134.35, 134.36, 135.62, 136.28, 137.69, 145.29, 147.27, 148.21 (13 Ar–C), 164.09, 165.69 (2CO). MS m/z: 361.53 (62.18%, M+), 363.20 (20.46%, M+2), 365.14 (8.06%, M+4), 74.03 (100). Anal. calcd for: C17H13Cl2N3O2 (362.21): C, 56.37; H, 3.62; N, 11.60%, found: C, 56.81; H, 3.93; N, 11.15%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.72 (t, 2H, CH2x, J = 8 Hz), 2.32 (t, 2H, CH2y, J = 8 Hz), 6.85 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 6.99 (d, 2H, Ar–H2,6, J = 8 Hz), 7.40 (t, 2H, Ar–H3,5, J = 8 Hz), 7.60 (s, 1H, quinoxaline-H5), 7.80 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 11.17, 11.32 (2 s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.46 (CH2), 31.28 (CH2), 151.57, 115.68, 116.70, 116.78, 118.78, 121.02, 121.78, 122.02, 122.25, 123.27, 131.18, 136.21, 143.32 (13 Ar–C), 166.10, 170.13 (2CO). MS m/z: 345.04 (18.43%, M+), 347.11 (5.96%, M+2), 77.14 (100). Anal. calcd for: C17H13ClFN3O2 (345.46): C, 59.05; H, 3.79; N, 12.15%, found: C, 59.55; H, 3.23; N, 12.62%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.69 (t, 2H, CH2x, J = 7.2 Hz), 2.34 (t, 2H, CH2y, J = 7.2 Hz), 6.74 (d, 2H, Ar–H3,5, J = 8 Hz), 7.00 (d, 1H, quinoxaline-H7, J = 7.6 Hz), 7.37 (s, 1H, quinoxaline-H5), 7.50 (d, 2H, Ar–H2,6, J = 8 Hz), 7.78 (d, 1H, quinoxaline-H8, J = 7.6 Hz), 9.83, 12.37 and 12.48 (3s, OH, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.87 (CH2), 30.69 (CH2), 126.52, 127.33, 128.42, 129.87, 130.34, 132.37, 133.50, 134.24, 137.28, 137.29, 137.30, 140.71, 150.37 (13 Ar–C), 164.32, 168.33 (2CO). MS m/z: 343.19 (27.20%, M+), 345.09 (9.56%, M+2), 71.93 (100). Anal. calcd for: C17H14ClN3O3 (343.76): C, 59.40; H, 4.10; N, 12.22%, found: C, 59.82; H, 4.43; N, 12.63%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.66 (t, 2H, CH2x, J = 7.6 Hz), 2.35 (t, 2H, CH2y, J = 7.6 Hz), 6.93 (t, 1H, Ar–H5, J = 8 Hz), 7.05 (d, 1H, Ar–H3, J = 8 Hz), 7.14 (t, 1H, Ar–H4, J = 8 Hz), 7.43 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.63 (s, 1H, quinoxaline-H5), 7.64 (d, 1H, Ar–H6, J = 8 Hz), 7.83 (d, 1H, quinoxaline-H8, J = 8 Hz), 9.85, 11.09 and 11.12 (3 s, OH, 2NH D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.48 (CH2), 30.32 (CH2), 120.12, 126.32, 128.20, 129.40, 130.47, 134.60, 134.77, 135.47, 135.60, 135.78, 137.37, 137.38, 138.90 (13 Ar–C), 165.47, 170.50 (2CO). MS m/z: 343.23 (31.89%, M+), 345.14 (10.03%, M+2), 89.1 (100). Anal. calcd for: C17H14ClN3O3 (343.76): C, 59.40; H, 4.10; N, 12.22%, found: C, 59.67; H, 4.38; N, 12.42%.O), 1508, 1369 (NO2); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.75 (t, 2H, CH2x, J = 6.4 Hz), 2.35 (t, 2H, CH2y, J = 6.4 Hz), 7.39 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.46 (s, 1H, quinoxaline-H5), 7.78 (d, 2H, Ar–H2,6, J = 8 Hz), 8.15 (d, 2H, Ar–H3,5, J = 8 Hz), 8.31 (d, 1H, quinoxaline-H8, J = 8 Hz), 11.46, 11.57 (2 s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.11 (CH2), 31.28 (CH2), 122.75, 122.83, 126.37, 127.32, 127.67, 128.11, 131.28, 131.75, 133.14, 134.95, 142.36, 143.04, 144.11 (13 Ar–C), 163.04, 164.11 (2CO). MS m/z: 372.11 (37.12%, M+), 374.03 (12.53%, M+2), 89.07 (100). Anal. calcd for: C17H13ClN4O4 (372.76): C, 54.78; H, 3.52; N, 15.03%, found: C, 54.33; H, 3.83; N, 15.43%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.74 (t, 2H, CH2x, J = 6.4 Hz), 2.32 (t, 2H, CH2y, J = 6.4 Hz), 2.40 (s, 3H, CH3), 7.02 (d, 2H, Ar–H3,5, J = 8.4 Hz), 7.22 (s, 1H, quinoxaline-H5), 7.49 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 7.68 (d, 2H, Ar–H2,6, J = 8.4 Hz), 8.03 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 11.74, 11.86 (2 s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 24.55 (CH3), 29.75 (CH2), 31.19 (CH2), 123.52, 126.25, 126.44, 127.59, 130.55, 130.63, 130.67, 131.83, 132.23, 133.26, 134.26, 151.10, 151.15 (13 Ar–C), 164.44, 172.17 (2CO). MS m/z: 341.25 (8.04%, M+), 343.09 (2.66%, M+2), 87.24 (100). Anal. calcd for: C18H16ClN3O2 (341.79): C, 63.25; H, 4.72; N, 12.29%, found: C, 63.66; H, 4.31; N, 12.53%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.72 (t, 2H, CH2x, J = 8 Hz), 2.32 (t, 2H, CH2y, J = 8 Hz), 2.41 (s, 3H, CH3), 6.65 (d, 1H, Ar–H4, J = 8 Hz), 7.21 (t, 1H, Ar–H5, J = 7.6 Hz), 7.49 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 7.71 (s, 1H, Ar–H2), 7.82 (d, 1H, Ar–H6, J = 8 Hz), 7.96 (s, 1H, quinoxaline-H5), 8.09 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 11.20, 11.21 (2 s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 24.17 (CH3), 29.30 (CH2), 31.26 (CH2), 123.52, 126.23, 126.43, 127.59, 130.43, 130.54, 130.97, 131.51, 132.10, 132.15, 134.31, 151.29, 151.31 (13 Ar–C), 164.15, 172.24 (2CO). MS m/z: 341.12 (9.03%, M+), 343.40 (2.98%, M+2), 87.15 (100). Anal. calcd for: C18H16ClN3O2 (341.79): C, 63.25; H, 4.72; N, 12.29%, found: C, 63.71; H, 4.45; N, 12.66%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.73 (t, 2H, CH2x, J = 8 Hz), 2.32 (t, 2H, CH2y, J = 8 Hz), 2.40 (s, 3H, CH3), 6.77 (t, 1H, Ar–H4, J = 8 Hz), 7.83 (d, 1H, Ar–H3, J = 8.4 Hz), 7.07 (t, 1H, Ar–H5, J = 8.4 Hz), 7.13 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 7.50 (d, 1H, Ar–H 6, J = 8.4 Hz), 7.53 (s, 1H, quinoxaline-H5), 8.26 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 11.67, 11.77 (2s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 24.90 (CH3), 29.37 (CH2), 31.46 (CH2), 123.24, 124.35, 128.03, 129.40, 129.47, 129.60, 134.20, 134.40, 135.47, 135.60, 135.78, 137.40, 137.49 (13 Ar–C), 164.60, 168.78 (2CO). MS m/z: 341.28 (17.51%, M+), 343.20 (5.88%, M+2), 87.04 (100). Anal. calcd for: C18H16ClN3O2 (341.79): C, 63.25; H, 4.72; N, 12.29%, found: C, 63.55; H, 4.31; N, 12.44%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.77 (t, 2H, CH2x, J = 8 Hz), 2.38 (t, 2H, CH2y, J = 8 Hz), 3.90 (s, 3H, OCH3), 7.22 (d, 2H, Ar–H3,5, J = 8.4 Hz), 7.70 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 7.82 (s, 1H, quinoxaline-H5), 7.97 (d, 2H, Ar–H2,6, J = 8.4 Hz), 8.46 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 11.77, 11.87 (2s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.10 (CH2), 31.15 (CH2), 55.39 (OCH3), 118.22, 119.67, 120.10, 120.46, 128.22, 128.82, 130.35, 131.15, 131.68, 132.01, 134.26, 145.03, 145.05 (13 Ar–C), 168.93, 170.68 (2CO). MS m/z: 357.01 (12.03%, M+), 359.40 (4.11%, M+2), 82.17 (100). Anal. calcd for: C18H16ClN3O3 (357.79): C, 60.42; H, 4.51; N, 11.74%, found: C, 60.71; H, 4.82; N, 11.52%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.65 (t, 2H, CH2x, J = 8 Hz), 2.35 (t, 2H, CH2y, J = 8 Hz), 2.67 (s, 3H, COCH3), 7.21 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.42 (d, 1H, quinoxaline-H8, J = 8 Hz), 7.47 (s, 1H, quinoxaline-H5), 7.76 (d, 2H, Ar–H 2,6, J = 8 Hz), 8.00 (d, 2H, Ar–H3,5, J = 8 Hz), 12.23, 12.37 (2 s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 28.22 (CO CH3), 29.82 (CH2), 31.15 (CH2), 118.03, 119.05, 120.19, 120.20, 128.39, 128.67, 130.10, 131.01, 131.26, 132.18, 134.22, 145.46, 145.55, (13 Ar–C), 167.35, 171.15 and 188.93 (3CO). MS m/z: 369.13 (39.02%, M+), 371.17 (12.98%, M+2), 82.29 (100). Anal. calcd for: C19H16ClN3O3 (369): C, 61.71; H, 4.36; N, 11.36%, found: C, 61.35; H, 4.62; N, 11.68%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.62 (t, 2H, CH2x, J = 7.6 Hz), 2.24 (t, 2H, CH2y, J = 7.6 Hz), 7.27 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.43 (d, 1H, quinoxaline-H8, J = 8 Hz), 7.47 (s, 1H, quinoxaline-H5), 7.97 (d, 2H, Ar–H2,6, J = 8 Hz), 8.46 (d, 2H, Ar–H 3,5, J = 8 Hz), 10.83 (s, 1H, COOH), 12.75, 12.82 (br, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.95 (CH2), 31.61 (CH2), 123.21, 125.24, 126.92, 127.30, 127.31, 127.50, 128.23, 129.66, 130.05, 130.80, 131.26, 131.27, 132.56 (13 Ar–C), 166.21, 169.31, 171.93 (3CO). MS m/z: 371.37 (45.11%, M+), 373.17 (15.07%, M+2), 84.19 (100). Anal. calcd for: C18 H14ClN3O4 (371.77): C, 58.15; H, 3.80; N, 11.30%, found: C, 58.46; H, 3.43; N, 11.63%.O); 1 H NMR (400 MHz, DMSO-d6) δ (ppm): 1.34 (t, 3H, OCH2CH3, J = 7.2 Hz),1.73 (t, 2H, CH2x, J = 7.2 Hz), 2.34 (t, 2H, CH2y, J = 7.2 Hz), 4.36 (q, 2H, OCH2CH3, J = 7.2 Hz), 6.47 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 7.41 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 7.49 (d, 2H, Ar–H2,6, J = 8.4 Hz), 7.94 (s, 1H, quinoxaline-H5), 8.09 (d, 2H, Ar–H 3,5, J = 8.4 Hz), 11.89, 11.92 (2s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 14.11 (CH3), 29.40 (CH2), 31.49 (CH2), 60.03 (OCH2CH3), 117.91, 118.03, 128.24, 129.40, 129.47, 129.60, 134.11, 134.12, 135.13, 135.38, 135.39, 137.78, 137.90 (13 Ar–C), 164.07, 168.16 (2CO of amide), 172.39 (CO of ester). MS m/z: 399.06 (62.14%, M+), 401.19 (20.94%, M+2), 88.13 (100). Anal. calcd for: C20H18ClN3O4 (399.83): C, 60.08; H, 4.54; N, 10.51%, found: C, 60.41; H, 4.71; N, 10.77%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 2.74 (t, 2H, CH2, J = 4 Hz), 3.09 (t, 2H, CH2, J = 4 Hz), 6.86 (s, 1H, quinoxaline-H5), 6.98 (d, 1H, quinoxaline-H7, J = 4 Hz), 7.28 (d, 1H, Ar–H5, J = 8 Hz), 7.43 (s, 1H, Ar–H3), 7.62 (d, 1H, quinoxaline-H8, J = 8 Hz), 7.80 (d, 1H, Ar–H6, J = 8 Hz), 12.42, 12.43 and 12.60 (s, 3NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.75 (CH2), 31.02 (CH2), 119.12, 120.19, 120.67, 120.92, 125.45, 125.75, 126.12, 126.19, 126.67, 128.34, 129.50, 130.57, 131.02, 162.70, 167.67, 170.34 (3CO). MS m/z (%): 438 (20.41, M+), 440.86 (6.5, M+2), 185.91 (100). Anal. calcd for: C18H13N4O3Cl3 (438): C, 49.17; H, 2.98; N, 12.74; Cl, 24.19%, found: C, 49.52; H, 2.48; N, 12.43; Cl, 24.55%.O); 1H NMR (400 MHz, DMSO- d6) δ (ppm): 2.38 (t, 2H, CH2CH2COOH), 2.59 (t, 2H, CH2CH2COOH, J = 4 Hz), 2.64 (t, 2H, CH2, J = 4 Hz), 3.04 (t, 2H, CH2, J = 4 Hz), 7.30 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.33 (s, 1H, quinoxaline-H5), 7.75 (d, 1H, quinoxaline-H8, J = 8 Hz), 10.85 (s, OH, D2O exchangeable), 12.38, 12.40 and 12.41 (s, 3NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.40 (CH2), 31.14 (CH2), 32.47 (CH2), 32.68 (CH2), 120.74, 121.01, 121.27, 124.13, 124.46, 131.20, 131.96, 161.13, 166.65, 167.04, 167.31 (4CO). MS m/z (%): 366 (14.41, M+), 368 (4.56, M+2), 185.02 (100). Anal. calcd for: C15H15N4O5Cl (366): C, 49.12; H, 4.12; N, 15.28; Cl, 9.67%, found: C, 49.53; H, 4.37; N, 15.71; Cl, 9.32%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 2.85 (t, 2H, CH2, J = 4 Hz), 3.09 (t, 2H, CH2, J = 4 Hz), 6.84 (s, 1H, quinoxaline-H5), 6.94 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.28 (d, 1H, quinoxaline-H8, J = 8 Hz), 7.44 (t, 1H, Ar–H4, J = 12 Hz), 7.57 (t, 1H, Ar–H5, J = 12 Hz), 8.20 (d, 1H, Ar–H6, J = 8 Hz), 8.74 (d, 1H, Ar–H3, J = 8 Hz), 10.80 (s, OH, D2O exchangeable), 12.43, 12.44 and 12.83 (s, 3NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.66 (CH2), 31.95 (CH2), 110.45, 112.32, 117.36, 119.70, 120.73, 121.26, 124.45, 131.95, 139.66, 140.57, 142.43, 142.87, 151.11, 161.11, 168.88, 170.26 and 173.45 (4CO). MS m/z (%): 414 (20.41, M+), 416.06 (6.5, M+2), 184.91 (100). Anal. calcd for: C19H15N4O5Cl (414): C, 55.02; H, 3.64; N, 13.51; Cl, 8.55%, found: C, 55.41; H, 3.22; N, 13.82; Cl, 8.16%.
O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.68 (t, 2H, CH2x, J = 7.6 Hz), 2.34 (t, 2H, CH2y, J = 7.6 Hz), 6.87 (t, 1H, Ar–H4), 7.07 (t, 2H, Ar–H3,5, J = 8 Hz), 7.41 (d, 2H, Ar–H2,6, J = 8 Hz), 7.60 (d, 1H, quinoxaline-H7, J = 8 Hz), 8.23 (s, 1H, quinoxaline-H5), 8.33 (d, 1H, quinoxaline-H8, J = 8 Hz), 10.31, 12.59 (2s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.34 (CH2), 31.50 (CH2), 119.12, 120.34, 120.50, 120.57, 125.45, 125.75, 126.12, 126.19, 126.45, 128.34, 129.50, 130.57, 131.12 (13 Ar–C), 167.12, 170.34 (2CO). MS m/z: 327.12 (21.53%, M+), 329.14 (7.16%, M+2), 76.09 (100). Anal. calcd for: C17H14ClN3O2 (327.76): C, 62.30; H, 4.31; N, 12.82%, found: C, 62.73; H, 4.67; N, 12.40%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.66 (t, 2H, CH2x, J = 8 Hz), 2.33 (t, 2H, CH2y, J = 8 Hz), 7.14 (d, 1H, quinoxaline-H7, J = 7.6 Hz), 7.64 (d, 2H, Ar–H3,5, J = 8 Hz), 7.83 (d, 2H, Ar–H2,6, J = 8 Hz), 7.96 (s, 1H, quinoxaline-H5), 8.02 (d, 1H, quinoxaline-H8, J = 7.6 Hz), 11.51, 11.64 (2s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.03 (CH2), 30.81 (CH2), 121.68, 128.62, 128.69, 129.29, 129.34, 134.35, 134.36, 135.62, 136.28, 137.69, 145.29, 147.27, 148.21 (13 Ar–C), 164.09, 165.69 (2CO). MS m/z: 361.53 (62.18%, M+), 363.20 (20.46%, M+2), 365.14 (8.06%, M+4), 74.03 (100). Anal. calcd for: C17H13Cl2N3O2 (362.21): C, 56.37; H, 3.62; N, 11.60%, found: C, 56.81; H, 3.93; N, 11.15%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.72 (t, 2H, CH2x, J = 8 Hz), 2.32 (t, 2H, CH2y, J = 8 Hz), 6.85 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 6.99 (d, 2H, Ar–H2,6, J = 8 Hz), 7.40 (t, 2H, Ar–H3,5, J = 8 Hz), 7.60 (s, 1H, quinoxaline-H5), 7.80 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 11.17, 11.32 (2 s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.46 (CH2), 31.28 (CH2), 151.57, 115.68, 116.70, 116.78, 118.78, 121.02, 121.78, 122.02, 122.25, 123.27, 131.18, 136.21, 143.32 (13 Ar–C), 166.10, 170.13 (2CO). MS m/z: 345.04 (18.43%, M+), 347.11 (5.96%, M+2), 77.14 (100). Anal. calcd for: C17H13ClFN3O2 (345.46): C, 59.05; H, 3.79; N, 12.15%, found: C, 59.55; H, 3.23; N, 12.62%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.69 (t, 2H, CH2x, J = 7.2 Hz), 2.34 (t, 2H, CH2y, J = 7.2 Hz), 6.74 (d, 2H, Ar–H3,5, J = 8 Hz), 7.00 (d, 1H, quinoxaline-H7, J = 7.6 Hz), 7.37 (s, 1H, quinoxaline-H5), 7.50 (d, 2H, Ar–H2,6, J = 8 Hz), 7.78 (d, 1H, quinoxaline-H8, J = 7.6 Hz), 9.83, 12.37 and 12.48 (3s, OH, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.87 (CH2), 30.69 (CH2), 126.52, 127.33, 128.42, 129.87, 130.34, 132.37, 133.50, 134.24, 137.28, 137.29, 137.30, 140.71, 150.37 (13 Ar–C), 164.32, 168.33 (2CO). MS m/z: 343.19 (27.20%, M+), 345.09 (9.56%, M+2), 71.93 (100). Anal. calcd for: C17H14ClN3O3 (343.76): C, 59.40; H, 4.10; N, 12.22%, found: C, 59.82; H, 4.43; N, 12.63%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.66 (t, 2H, CH2x, J = 7.6 Hz), 2.35 (t, 2H, CH2y, J = 7.6 Hz), 6.93 (t, 1H, Ar–H5, J = 8 Hz), 7.05 (d, 1H, Ar–H3, J = 8 Hz), 7.14 (t, 1H, Ar–H4, J = 8 Hz), 7.43 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.63 (s, 1H, quinoxaline-H5), 7.64 (d, 1H, Ar–H6, J = 8 Hz), 7.83 (d, 1H, quinoxaline-H8, J = 8 Hz), 9.85, 11.09 and 11.12 (3 s, OH, 2NH D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.48 (CH2), 30.32 (CH2), 120.12, 126.32, 128.20, 129.40, 130.47, 134.60, 134.77, 135.47, 135.60, 135.78, 137.37, 137.38, 138.90 (13 Ar–C), 165.47, 170.50 (2CO). MS m/z: 343.23 (31.89%, M+), 345.14 (10.03%, M+2), 89.1 (100). Anal. calcd for: C17H14ClN3O3 (343.76): C, 59.40; H, 4.10; N, 12.22%, found: C, 59.67; H, 4.38; N, 12.42%.O), 1508, 1369 (NO2); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.75 (t, 2H, CH2x, J = 6.4 Hz), 2.35 (t, 2H, CH2y, J = 6.4 Hz), 7.39 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.46 (s, 1H, quinoxaline-H5), 7.78 (d, 2H, Ar–H2,6, J = 8 Hz), 8.15 (d, 2H, Ar–H3,5, J = 8 Hz), 8.31 (d, 1H, quinoxaline-H8, J = 8 Hz), 11.46, 11.57 (2 s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.11 (CH2), 31.28 (CH2), 122.75, 122.83, 126.37, 127.32, 127.67, 128.11, 131.28, 131.75, 133.14, 134.95, 142.36, 143.04, 144.11 (13 Ar–C), 163.04, 164.11 (2CO). MS m/z: 372.11 (37.12%, M+), 374.03 (12.53%, M+2), 89.07 (100). Anal. calcd for: C17H13ClN4O4 (372.76): C, 54.78; H, 3.52; N, 15.03%, found: C, 54.33; H, 3.83; N, 15.43%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.74 (t, 2H, CH2x, J = 6.4 Hz), 2.32 (t, 2H, CH2y, J = 6.4 Hz), 2.40 (s, 3H, CH3), 7.02 (d, 2H, Ar–H3,5, J = 8.4 Hz), 7.22 (s, 1H, quinoxaline-H5), 7.49 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 7.68 (d, 2H, Ar–H2,6, J = 8.4 Hz), 8.03 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 11.74, 11.86 (2 s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 24.55 (CH3), 29.75 (CH2), 31.19 (CH2), 123.52, 126.25, 126.44, 127.59, 130.55, 130.63, 130.67, 131.83, 132.23, 133.26, 134.26, 151.10, 151.15 (13 Ar–C), 164.44, 172.17 (2CO). MS m/z: 341.25 (8.04%, M+), 343.09 (2.66%, M+2), 87.24 (100). Anal. calcd for: C18H16ClN3O2 (341.79): C, 63.25; H, 4.72; N, 12.29%, found: C, 63.66; H, 4.31; N, 12.53%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.72 (t, 2H, CH2x, J = 8 Hz), 2.32 (t, 2H, CH2y, J = 8 Hz), 2.41 (s, 3H, CH3), 6.65 (d, 1H, Ar–H4, J = 8 Hz), 7.21 (t, 1H, Ar–H5, J = 7.6 Hz), 7.49 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 7.71 (s, 1H, Ar–H2), 7.82 (d, 1H, Ar–H6, J = 8 Hz), 7.96 (s, 1H, quinoxaline-H5), 8.09 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 11.20, 11.21 (2 s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 24.17 (CH3), 29.30 (CH2), 31.26 (CH2), 123.52, 126.23, 126.43, 127.59, 130.43, 130.54, 130.97, 131.51, 132.10, 132.15, 134.31, 151.29, 151.31 (13 Ar–C), 164.15, 172.24 (2CO). MS m/z: 341.12 (9.03%, M+), 343.40 (2.98%, M+2), 87.15 (100). Anal. calcd for: C18H16ClN3O2 (341.79): C, 63.25; H, 4.72; N, 12.29%, found: C, 63.71; H, 4.45; N, 12.66%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.73 (t, 2H, CH2x, J = 8 Hz), 2.32 (t, 2H, CH2y, J = 8 Hz), 2.40 (s, 3H, CH3), 6.77 (t, 1H, Ar–H4, J = 8 Hz), 7.83 (d, 1H, Ar–H3, J = 8.4 Hz), 7.07 (t, 1H, Ar–H5, J = 8.4 Hz), 7.13 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 7.50 (d, 1H, Ar–H 6, J = 8.4 Hz), 7.53 (s, 1H, quinoxaline-H5), 8.26 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 11.67, 11.77 (2s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 24.90 (CH3), 29.37 (CH2), 31.46 (CH2), 123.24, 124.35, 128.03, 129.40, 129.47, 129.60, 134.20, 134.40, 135.47, 135.60, 135.78, 137.40, 137.49 (13 Ar–C), 164.60, 168.78 (2CO). MS m/z: 341.28 (17.51%, M+), 343.20 (5.88%, M+2), 87.04 (100). Anal. calcd for: C18H16ClN3O2 (341.79): C, 63.25; H, 4.72; N, 12.29%, found: C, 63.55; H, 4.31; N, 12.44%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.77 (t, 2H, CH2x, J = 8 Hz), 2.38 (t, 2H, CH2y, J = 8 Hz), 3.90 (s, 3H, OCH3), 7.22 (d, 2H, Ar–H3,5, J = 8.4 Hz), 7.70 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 7.82 (s, 1H, quinoxaline-H5), 7.97 (d, 2H, Ar–H2,6, J = 8.4 Hz), 8.46 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 11.77, 11.87 (2s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.10 (CH2), 31.15 (CH2), 55.39 (OCH3), 118.22, 119.67, 120.10, 120.46, 128.22, 128.82, 130.35, 131.15, 131.68, 132.01, 134.26, 145.03, 145.05 (13 Ar–C), 168.93, 170.68 (2CO). MS m/z: 357.01 (12.03%, M+), 359.40 (4.11%, M+2), 82.17 (100). Anal. calcd for: C18H16ClN3O3 (357.79): C, 60.42; H, 4.51; N, 11.74%, found: C, 60.71; H, 4.82; N, 11.52%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.65 (t, 2H, CH2x, J = 8 Hz), 2.35 (t, 2H, CH2y, J = 8 Hz), 2.67 (s, 3H, COCH3), 7.21 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.42 (d, 1H, quinoxaline-H8, J = 8 Hz), 7.47 (s, 1H, quinoxaline-H5), 7.76 (d, 2H, Ar–H 2,6, J = 8 Hz), 8.00 (d, 2H, Ar–H3,5, J = 8 Hz), 12.23, 12.37 (2 s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 28.22 (CO CH3), 29.82 (CH2), 31.15 (CH2), 118.03, 119.05, 120.19, 120.20, 128.39, 128.67, 130.10, 131.01, 131.26, 132.18, 134.22, 145.46, 145.55, (13 Ar–C), 167.35, 171.15 and 188.93 (3CO). MS m/z: 369.13 (39.02%, M+), 371.17 (12.98%, M+2), 82.29 (100). Anal. calcd for: C19H16ClN3O3 (369): C, 61.71; H, 4.36; N, 11.36%, found: C, 61.35; H, 4.62; N, 11.68%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.62 (t, 2H, CH2x, J = 7.6 Hz), 2.24 (t, 2H, CH2y, J = 7.6 Hz), 7.27 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.43 (d, 1H, quinoxaline-H8, J = 8 Hz), 7.47 (s, 1H, quinoxaline-H5), 7.97 (d, 2H, Ar–H2,6, J = 8 Hz), 8.46 (d, 2H, Ar–H 3,5, J = 8 Hz), 10.83 (s, 1H, COOH), 12.75, 12.82 (br, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.95 (CH2), 31.61 (CH2), 123.21, 125.24, 126.92, 127.30, 127.31, 127.50, 128.23, 129.66, 130.05, 130.80, 131.26, 131.27, 132.56 (13 Ar–C), 166.21, 169.31, 171.93 (3CO). MS m/z: 371.37 (45.11%, M+), 373.17 (15.07%, M+2), 84.19 (100). Anal. calcd for: C18 H14ClN3O4 (371.77): C, 58.15; H, 3.80; N, 11.30%, found: C, 58.46; H, 3.43; N, 11.63%.O); 1 H NMR (400 MHz, DMSO-d6) δ (ppm): 1.34 (t, 3H, OCH2CH3, J = 7.2 Hz),1.73 (t, 2H, CH2x, J = 7.2 Hz), 2.34 (t, 2H, CH2y, J = 7.2 Hz), 4.36 (q, 2H, OCH2CH3, J = 7.2 Hz), 6.47 (d, 1H, quinoxaline-H7, J = 8.4 Hz), 7.41 (d, 1H, quinoxaline-H8, J = 8.4 Hz), 7.49 (d, 2H, Ar–H2,6, J = 8.4 Hz), 7.94 (s, 1H, quinoxaline-H5), 8.09 (d, 2H, Ar–H 3,5, J = 8.4 Hz), 11.89, 11.92 (2s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 14.11 (CH3), 29.40 (CH2), 31.49 (CH2), 60.03 (OCH2CH3), 117.91, 118.03, 128.24, 129.40, 129.47, 129.60, 134.11, 134.12, 135.13, 135.38, 135.39, 137.78, 137.90 (13 Ar–C), 164.07, 168.16 (2CO of amide), 172.39 (CO of ester). MS m/z: 399.06 (62.14%, M+), 401.19 (20.94%, M+2), 88.13 (100). Anal. calcd for: C20H18ClN3O4 (399.83): C, 60.08; H, 4.54; N, 10.51%, found: C, 60.41; H, 4.71; N, 10.77%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 2.74 (t, 2H, CH2, J = 4 Hz), 3.09 (t, 2H, CH2, J = 4 Hz), 6.86 (s, 1H, quinoxaline-H5), 6.98 (d, 1H, quinoxaline-H7, J = 4 Hz), 7.28 (d, 1H, Ar–H5, J = 8 Hz), 7.43 (s, 1H, Ar–H3), 7.62 (d, 1H, quinoxaline-H8, J = 8 Hz), 7.80 (d, 1H, Ar–H6, J = 8 Hz), 12.42, 12.43 and 12.60 (s, 3NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.75 (CH2), 31.02 (CH2), 119.12, 120.19, 120.67, 120.92, 125.45, 125.75, 126.12, 126.19, 126.67, 128.34, 129.50, 130.57, 131.02, 162.70, 167.67, 170.34 (3CO). MS m/z (%): 438 (20.41, M+), 440.86 (6.5, M+2), 185.91 (100). Anal. calcd for: C18H13N4O3Cl3 (438): C, 49.17; H, 2.98; N, 12.74; Cl, 24.19%, found: C, 49.52; H, 2.48; N, 12.43; Cl, 24.55%.O); 1H NMR (400 MHz, DMSO- d6) δ (ppm): 2.38 (t, 2H, CH2CH2COOH), 2.59 (t, 2H, CH2CH2COOH, J = 4 Hz), 2.64 (t, 2H, CH2, J = 4 Hz), 3.04 (t, 2H, CH2, J = 4 Hz), 7.30 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.33 (s, 1H, quinoxaline-H5), 7.75 (d, 1H, quinoxaline-H8, J = 8 Hz), 10.85 (s, OH, D2O exchangeable), 12.38, 12.40 and 12.41 (s, 3NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.40 (CH2), 31.14 (CH2), 32.47 (CH2), 32.68 (CH2), 120.74, 121.01, 121.27, 124.13, 124.46, 131.20, 131.96, 161.13, 166.65, 167.04, 167.31 (4CO). MS m/z (%): 366 (14.41, M+), 368 (4.56, M+2), 185.02 (100). Anal. calcd for: C15H15N4O5Cl (366): C, 49.12; H, 4.12; N, 15.28; Cl, 9.67%, found: C, 49.53; H, 4.37; N, 15.71; Cl, 9.32%.O); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 2.85 (t, 2H, CH2, J = 4 Hz), 3.09 (t, 2H, CH2, J = 4 Hz), 6.84 (s, 1H, quinoxaline-H5), 6.94 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.28 (d, 1H, quinoxaline-H8, J = 8 Hz), 7.44 (t, 1H, Ar–H4, J = 12 Hz), 7.57 (t, 1H, Ar–H5, J = 12 Hz), 8.20 (d, 1H, Ar–H6, J = 8 Hz), 8.74 (d, 1H, Ar–H3, J = 8 Hz), 10.80 (s, OH, D2O exchangeable), 12.43, 12.44 and 12.83 (s, 3NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.66 (CH2), 31.95 (CH2), 110.45, 112.32, 117.36, 119.70, 120.73, 121.26, 124.45, 131.95, 139.66, 140.57, 142.43, 142.87, 151.11, 161.11, 168.88, 170.26 and 173.45 (4CO). MS m/z (%): 414 (20.41, M+), 416.06 (6.5, M+2), 184.91 (100). Anal. calcd for: C19H15N4O5Cl (414): C, 55.02; H, 3.64; N, 13.51; Cl, 8.55%, found: C, 55.41; H, 3.22; N, 13.82; Cl, 8.16%.4.3 Another procedure
Compound 21 (0.01 mol) was refluxed in 15 ml of glacial acetic acid for 2 h, and the resulting product was filtered and recrystallized from the ethanol to afford compound 22. Light brown powder; yield (78%); mp. 264–266 °C; IR (KBr, cm−1): 3327 and 3189 (2 NH), 1751, 1669, 1635 and 1623 (4CO); 1H NMR (400 Mz, DMSO-d6) δ (ppm): 2.82 (t, 2H, CH2, J = 4), 3.03 (t, 2H, CH2, J = 4 Hz), 6.89 (s, 1H, quinoxaline-H5), 6.97 (d, 1H, quinoxaline-H7, J = 8 Hz), 7.32 (d, 1H, quinoxaline-H8, J = 8 Hz), 7.48 (t, 1H, Ar–H4, J = 8 Hz), 7.60 (t, 1H, Ar–H5, J = 8 Hz), 8.28 (d, 1H, Ar–H6, J = 8 Hz), 8.78 (d, 1H, Ar–H3, J = 8 Hz), 12.22 and 12.54 (s, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ (ppm): 29.33 (CH2), 31.54 (CH2), 115.66, 115.72, 115.85, 115.94, 117.59, 119.53, 120.41, 121.23, 121.31, 124.07, 131.38, 135.81, 135.84 (13 Ar–C), 159.68, 165.73, 166.70 and 169.74 (4CO). MS m/z (%): 396 (9.05, M+), 398.52 (2.98, M+2), 102.34 (100). Anal. calcd for: C19H13N4O4Cl (396): C, 57.51; H, 3.30; N, 14.12; Cl, 8.94%, found: C, 57.04; H, 3.72; N, 14.43; Cl, 8.56%.
4.4. Biological evaluation
Antiproliferative assay: the prepared compounds were estimated for their antiproliferative activity against two human breast cancer cell lines, MCF-7 and HCT-116 in comparison with doxorubicin as a reference standard at concentrations 100, 50, 25, 12.5, 6.25, 3.125, and 1.56 μM utilizing MTT assay guided by the reported procedure.33VEGFR-2 inhibition estimation: VEGFR-2 inhibition estimation was executed utilizing VEGFR-2 (KDR) Kinase Assay Kit of Biosciences and the IC50 values were detected according to the manufacturer's instructions and the reported method.37
Quantitative determination of VEGFR2 in MCF-7 cell line: ELISA analysis to determine VEGFR2 quantitatively in MCF-7 cell line is following the reported procedure38
Effect of 14 on angiogenesis (VEFG-A) level: ELISA analysis to determine VEGF-A secretion is following the reported procedure.40
Cell cycle analysis: FACS Caliber flow cytometer was utilized to determine the effect of 14 on the cell cycle of breast tumor NCF-7 guided by the previously reported method.39
Annexin-V-FITC apoptosis assay: the apoptosis of 14 was detected through Annexin V-FITC/PI apoptosis detection kit utilizing FACS Caliber flow cytometer following the reported procedure.40
Effect of 14 on active caspase-3 level
Caspase-3 is evaluated by using the reported method.41
Evaluation of apoptosis regulator p53
P53 expression is measured in MCF-7 by the reported method.42
Evaluation of BAX and BCL2 levels43,44
Author contributions
Magda M. F. Ismail (corresponding author)-conceived and designed the analysis and wrote the article, Taghreed Z. Shawer-revised the manuscript and collected the data, Rabab S. Ibrahim-performed the synthetic work and collected the data, Mostafa S. Abusaif-contributed in synthetic part, Mona M. Kamal-contributed data and analysis, Rasha M. Allam-performed some analysis, Yousry A. Ammar-designed the chemistry part and revised the manuscript.Conflicts of interest
The authors declare that they have no conflicts of interest. The authors alone are responsible for the content and writing of this manuscript.References
- L. Zhao, H. Y. Chen, L. Lu, L. Wang, X. K. Zhang and X. L. Guo, New insights into the role of co-receptor neuropilins in tumour angiogenesis and lymphangiogenesis and targeted therapy strategies, J. Drug Targeting, 2021, 29, 155–167 CrossRef CAS PubMed.
- C. B. Prasad, D. Singh, L. K. Pandey, S. Pradhan, S. Singh and G. Narayan, VEGFa/VEGFR2 autocrine and paracrine signaling promotes cervical carcinogenesis via β-catenin and snail, Int. J. Biochem. Cell Biol., 2022, 142, 106122 CrossRef CAS PubMed.
- Y. Liu, L. Yang, Y. Wang, C. Lin, D. Zhang, J. Chen, L. Ouyang, F. Wu, J. Zhang and C. Lei, Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy, J. Hematol. Oncol., 2022, 15, 89, DOI:10.1186/s13045-022-01310-7.
- C. C. Estrada, A. Maldonado and S. K. Mallipattu, Therapeutic inhibition of VEGF signaling and associated nephrotoxicities, J. Am. Soc. Nephrol., 2019, 30, 187–200 CrossRef CAS PubMed.
- P. Zhu, C. Hu, K. Hui and X. Jiang, The role and significance of VEGFR2 regulatory T cells in tumor immunity, OncoTargets Ther., 2017, 10, 4315–4319 CrossRef PubMed.
- F. Gao and C. Yang, Anti-VEGF/VEGFR2 monoclonal antibodies and their combinations with PD-1/PD-L1 inhibitors in clinic, Curr. Cancer Drug Targets, 2020, 20, 3–18 CrossRef CAS PubMed.
- R. Jr Roskoski, Vascular endothelial growth factor (VEGF) and VEGF receptor inhibitors in the treatment of renal cell carcinomas, Pharmacol. Res., 2017, 120, 116–132 CrossRef CAS PubMed.
- M. Yousefian and R. Ghodsi, Structure–activity relationship studies of indolin-2-one derivatives as vascular endothelial growth factor receptor inhibitors and anticancer agents, Arch. Pharm., 2020, 353, e2000022, DOI:10.1002/ardp.202000022.
- K. K. Bhanumathy, A. Balagopal, F. S. Vizeacoumar, F. J. Vizeacoumar, A. Freywald and V. Giambra, Protein tyrosine kinases: their roles and their targeting in leukemia, Cancers, 2021, 13(2), 184, DOI:10.3390/cancers13020184.
- M. Liao, J. Zhang, G. Wang, L. Wang, J. Liu, L. Ouyang and B. Liu, Small-moleculedrug discovery in triple negative breast cancer: current situation and future directions, J. Med. Chem., 2021, 64, 2382–2418 CrossRef CAS PubMed.
- S. T. Staben, J. A. Feng, K. Lyle, M. Belvin, J. Boggs, J. D. Burch, C. C. Chua, H. Cui, A. G. DiPasquale, L. S. Friedman, C. Heise, H. Koeppen, A. Kotey, R. Mintzer, A. Oh, D. A. Roberts, L. Rouge, J. Rudolph, C. Tam, W. Wang, Y. Xiao, A. Young, Y. Zhang and K. P. Hoeflich, Back pocket flexibility provides group II p21-activated kinase (PAK) selectivity for type I 1/2 kinase inhibitors, J. Med. Chem., 2014, 57, 1033–1045 CrossRef CAS PubMed.
- A. Abdeldayem, Y. S. Raouf, S. N. Constantinescu, R. Moriggl and P. T. Gunning, Advances in covalent kinase inhibitors, Chem. Soc. Rev., 2020, 49, 2617–2687 RSC.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, H. M. Alkahtani, M. M. Alanazi, M. A. Alharbi, I. H. Eissa and M. A. Dahab, New quinoxaline-based VEGFR-2 inhibitors: design, synthesis, and antiproliferative evaluation with in silico docking, ADMET, toxicity, and DFT studies, RSC Adv., 2021, 11, 30315 RSC.
- M. Montana, V. Montero, O. Khoumeri and P. Vanelle, Molecules, 2020, 25, 2784 CrossRef CAS PubMed.
- M. Montana, F. Mathias, T. Terme and P. Vanelle, Eur. J. Med. Chem., 2019, 163, 136–147 CrossRef CAS PubMed.
- E. M. Abbass, A. K. Khalil, M. M. Mohamed, I. H. Eissa and A. M. El-Naggar, Bioorg. Chem., 2020, 104, 104255 CrossRef CAS PubMed.
- M. A. Naylor, M. A. Stephens, J. Nolan, B. Sutton, J. H. Tocher, E. M. Fielden, G. E. Adams and I. J. Stratford, Anti-Cancer Drug Des., 1993, 8, 439–461 CAS.
- T. H. Corbett, P. Lorusso, L. Demchick, C. Simpson, S. L. Pugh, K. White, J. Kushner, L. L. Polin, J. A. Meyer, J. Czarnecki, L. K. Heilbrun, J. P. Horwitz, J. L. Gross, C. H. Behrens, B. A. Harrison, R. J. Mcripley and G. L. Trainor, Invest. New Drugs, 2004, 16, 129–139 CrossRef PubMed.
- M. M. F. Ismail, D. H. Soliman, A. M. Farrag and R. Sabour, Synthesis, antitumor activity, pharmacophore modeling and QSAR studies of novel pyrazoles and pyrazolo [1, 5-a] pyrimidines against breast adenocarcinoma mcf-7 cell line, Int. J. Pharm. Pharm. Sci., 2016, 8, 434–442 CAS.
- M. M. F. Ismail, D. H. Soliman, R. Sabour and A. M. Farrag, Synthesis of new arylazopyrazoles as apoptosis inducers: Candidates to inhibit proliferation of MCF-7 cells, Arch. Pharm., 2020, 354, 2000214, DOI:10.1002/ardp.202000214.
- M. M. F. Ismail, A. M. Farrag and M. F. Harras, Novel 1,3,4-Triaryl Pyrazoles: Synthesis, QSAR Studies and Cytotoxicity against Breast Cancer, Anti-Cancer Agents Med. Chem., 2019, 19, 948–959 CrossRef CAS PubMed.
- M. M. F. Ismail, H. S. El-Zahabi, R. S. Ibrahim and A. B. Mehany, Design and synthesis of novel tranilast analogs: Docking, antiproliferative evaluation and in-silico screening of TGFβR1 inhibitors, Bioorg. Chem., 2020, 105, 104368, DOI:10.1016/j.bioorg.2020.104368.
- A. M. Farrag, M. H. Ibrahim, A. B. Mehany and M. M. F. Ismail, New cyanopyridine-based scaffold as PIM-1 inhibitors and apoptotic inducers: Synthesis and SARs study, Bioorg. Chem., 2020, 105, 104378 CrossRef CAS PubMed.
- M. M. F. Ismail, A. M. Farrag, M. F. Harras, M. H. Ibrahim and A. B. Mehany, Apoptosis: A target for anticancer therapy with novel cyanopyridines, Bioorg. Chem., 2020, 94, 103481 CrossRef CAS PubMed.
- A. M Farrag, M. F Harras and M. M. F. Ismail, Design, Synthesis and QSAR Studies of Novel 1,3,4-Triarylpyrazoles as Anti-breast Cancer Agents, J. Appl. Pharm. Sci., 2017, 3(1), 63–74 Search PubMed.
- M. M. F. Ismail, A. M. Farrag and D. Abou-El Ella, Synthesis, anticancer screening, and in silico ADME prediction of novel 2-pyridones as Pim inhibitors, J. Heterocycl. Chem., 2020, 57, 1–19, DOI:10.1002/jhet.4064.
- S. Tariq, O. Alam and M. Amir, Synthesis, anti-inflammatory, p38α MAP kinase inhibitory activities and molecular docking studies of quinoxaline derivatives containing triazole moiety, Bioorg. Chem., 2018, 76, 343–358, DOI:10.1016/j.bioorg.2017.12.003.
- https://www.masterorganicchemistry.com/reaction-guide/formation-of-amides-from-esters/Biology.
- Y. A. Ammar, M. M. F. Ismail, M. K. Ibrahim and H. S. A. El-Zahaby, 3-ethoxycarbonylmethylenequinoxalin-2-one in heterocyclic synthesis. Part1: synthesis of new substituted and condensed quinoxalines, Afinidad, 2005, 516, 151 Search PubMed.
- S. El-Hashash, A. Maher, D. B. Guirguis, N. A. M. AbdEl-Wahed and M. A. Kadhim, Synthesis of novel series of phthalazine derivatives with antibacterial and mantifungal evaluation, J. Chem. Eng. Process Technol., 2014, 5(4), 1000191–1000196 Search PubMed.
- L. A. Shemchuk, V. P. Chernykh, P. S. Arzumanov and I. L. Starchikova, Synthesis of 3-amino-3, 4-dihydroquinazolin-4-one derivatives from anthranilic acid hydrazide and dicarboxylic acids, Russ. J. Org. Chem., 2007, 43(12), 1830–1835 CrossRef CAS.
- H. El-Sabbagh, I. Osama, M. E. El-Sadek, S. M. Lashine, S. H. Yassin and S. M. El-Nabtity, Synthesis of new 2 (1H)-quinoxalinone derivatives for antimicrobial and anti-inflammatory evaluation, Med. Chem. Res., 2009, 18(9), 782–797 CrossRef.
- L. Freimoser, C. A. Jakob, M. Aebi and U. Tuor, Appl. Environ. Microbiol., 1999, 65(8), 3727–3729 CrossRef PubMed.
- T. H. Yang, C. I. Lee, W. H. Huang and A. R. Lee, Synthesis and evaluation of novel 2-pyrrolidone-fused (2-oxoindolin-3-ylidene) methylpyrrole derivatives as potential multi-target tyrosine kinase receptor inhibitors, Molecules, 2017, 22(6), 913 CrossRef PubMed.
- M. A. Abdelgawad, A. M. Hayallah, S. N. A. Bukhari, A. Musa, M. Elmowafy, H. M. Abdel-Rahman and K. Mohammed, Design, Synthesis, Molecular Modeling, and Anticancer Evaluation of New VEGFR-2 Inhibitors Based on the Indolin-2-One Scaffold, Pharma, 2022, 15(11), 1416 CAS.
- A. Shindikar, G. Deshpande, U. Chaudhari, D. Bhatia and K. Joshi, Comparative effect of tyrosine kinase inhibitors in human cancer cell lines, J. Pharm. Clin. Res., 2016, 1(2), 555557 Search PubMed.
- VEGFR-2 (KDR), kinase assay kit catalog, 40325, https://bpsbioscience.com/.
- T. Atakul, serum levels of angiogenic factors distinguish between women with preeclampsia and normotensive pregnant women but not severity of preeclampsia in an obstetric center in Turkey, Med. Sci. Monit., 2019, 25, 6935–6942, DOI:10.12659/MSM.915092.
- P. Mabeta and V. Steenkamp, The VEGF/VEGFR Axis Revisited: implications for cancer therapy, Int. J. Mol. Sci., 2022, 23, 15585, DOI:10.3390/ijms232415585.
- B. Y. Jeong, K. H. Cho, K. J. Jeong, Y. Y. Park, J. M. Kim, S. Y. Rha, C. G. Park, G. B. Mills, J. H. Cheong and H. Y. Lee, Rab25 augments cancer cell invasiveness through a β1 integrin/EGFR/VEGF-A/Snail signaling axis and expression of fascin, Exp. Mol. Med., 2018, 50, e435, DOI:10.1038/emm.2017.248.
- Y. Wang, P. Ji, J. Liu, R. R. Broaddus, F. Xue and W. Zhang, Mol. Cancer, 2009, 8, 8, DOI:10.1186/1476-4598-8-8.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516, DOI:10.1080/01926230701320337.
- P. J. Lakshmi, B. V. S. S. Kumar, R. S. Nayana, M. S. Mohan, R. Bolligarl, S. K. Das, M. U. Bhanu, A. K. Kondapi and M. Ravikumar, Design, synthesis, and discovery of novel non-peptide inhibitor of Caspase-3 using ligand based and structure based virtual screening approach, Bioorg. Med. Chem., 2009, 17, 6040–6047 CrossRef PubMed.
- J. T. Zilfou and S. W. Lowe, Tumor suppressive functions of p53, Cold Spring Harb. Perspect. Biol., 2009, 1(5), 001883, DOI:10.1101/cshperspect.a001883.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed.
- S. Salakou, D. Kardamakis, A. C. Tsamandas, V. Zolota, E. Apostolakis, V. Tzelepi, P. Papathanasopoulos, D. S. Bonikos, T. Papapetropoulos and T. Petsas, Increased Bax/Bcl-2 Ratio Up-regulates Caspase-3 and Increases Apoptosis in the Thymus of Patients with Myasthenia Gravis, In Vivo, 2007, 21, 123–132 CAS.
- D. Antoine, M. Olivier and Z. Vincent, SwissADME: a free web tool to evaluate pharmacokinetics, drug likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7(1), 42717 CrossRef PubMed.
- A. L. Christopher, L. Franco, W. D. Beryl and J. F. Paul, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45(12), 2615–2623 CrossRef CAS PubMed.
- M. d. S. Marciane, C. Marina, S. D. Thiago, A. F. Mary, E. d. C. João, d. C. V. Maria and S. N. F. Anelise, Synthesis, Antiproliferative Activity and Molecular Properties Predictions of Galloyl Derivatives, Molecules, 2015, 20, 5360–5373 CrossRef PubMed.
- S. Rampogu, A. Baek, A. Zeb and K. W. Lee, Exploration for novel inhibitors showing back-to-front approach against VEGFR-2 kinase domain (4AG8) employing molecular docking mechanism and molecular dynamics simulations, BMC Cancer, 2018, 18(1), 1–21 CrossRef PubMed.
- A. E. Abdallah, R. R. Mabrouk, M. M. S. Al Ward, S. I. Eissa, E. B. Elkaeed, A. B. Mehany and M. A. El-Zahabi, Synthesis, biological evaluation, and molecular docking of new series of antitumor and apoptosis inducers designed as VEGFR-2 inhibitors, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 573–591 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05066a |
| This journal is © The Royal Society of Chemistry 2023 |