
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitron-derivative-based palladium carbene complexes: structural characterization, theoretical calculations, and catalytic applications in the Mizoroki–Heck coupling reaction†
Ming-Yi Lee,
Chih-Hsiang Liao,
Hsiu-Yu Hung ,
Jhen-Yi Lee* and
Hon Man Lee*
,
Jhen-Yi Lee* and
Hon Man Lee*
Department of Chemistry, National Changhua University of Education, Changhua 500, Taiwan. E-mail: leehm@cc.ncue.edu.tw
First published on 13th September 2023
Abstract
New palladium(0) and palladium(II) complexes with N-heterocyclic carbene (NHC) ligands derived from nitron and its derivatives were synthesized. The structures of most of these complexes were established by single-crystal X-ray diffraction studies. Among the new complexes, the palladium complex with a monodentate NHC ligand derived from nitron demonstrated the highest efficacy as a catalyst precursor in the Mizoroki–Heck coupling reaction of aryl chlorides with alkenes. Theoretical calculations provide valuable insights into the electronic parameters of both the ligands and the palladium complexes, highlighting the significance of a robust Pd–C bond and the π-accepting property of the NHC ligand in achieving enhanced catalytic activity. Notably, catalyst activation occurred much more rapidly with the preformed palladium(0) complex compared to its palladium(II) counterpart.
Introduction
The development of efficient palladium complexes for catalyzing the Mizoroki–Heck coupling reaction of unreactive aryl halide substrates has garnered significant interest.1–9 Various preformed palladium(II) complexes,10 encompassing a wide range of ligand types such as cyclometallated ligands,11 pincer ligands,12 and electron-rich tertiary phosphines,6,13 etc., have been employed as pre-catalysts. Nevertheless, harnessing the potential of cost-effective aryl chlorides as substrates has been a challenging endeavor, largely due to the formidable strength of the C–Cl bonds. Over the past two decades, palladium complexes featuring N-heterocyclic carbene (NHC) ligands have emerged as highly versatile pre-catalysts for a wide array of cross-coupling reactions such as Suzuki–Miyaura, Sonogashira, Negishi, and Buchwald–Hartwig coupling reactions.14–25 Some prominent examples of such palladium NHC complexes are shown in Scheme 1. Despite their extensive applicability, there remains a relative scarcity of literature regarding palladium NHC complexes capable of effectively activating C–Cl bonds in the Mizoroki–Heck coupling reaction, leading to a limited exploration of their potential in enabling the utilization of cost-effective aryl chlorides as substrates in this coupling process.26–29 The ligated zero-valent palladium complex is widely accepted as the active species in Pd-catalyzed transformations.30–32 Consequently, preformed palladium(0) NHC complexes are anticipated to offer expedited catalyst activation compared to their palladium(II) NHC counterparts. Nevertheless, the literature on palladium(0) NHC complexes is still relatively limited.33–40 As pre-catalysts, the preformed palladium(0) NHC complexes should exhibit ample stability for convenient handling and resist decomposition into palladium black when in solution.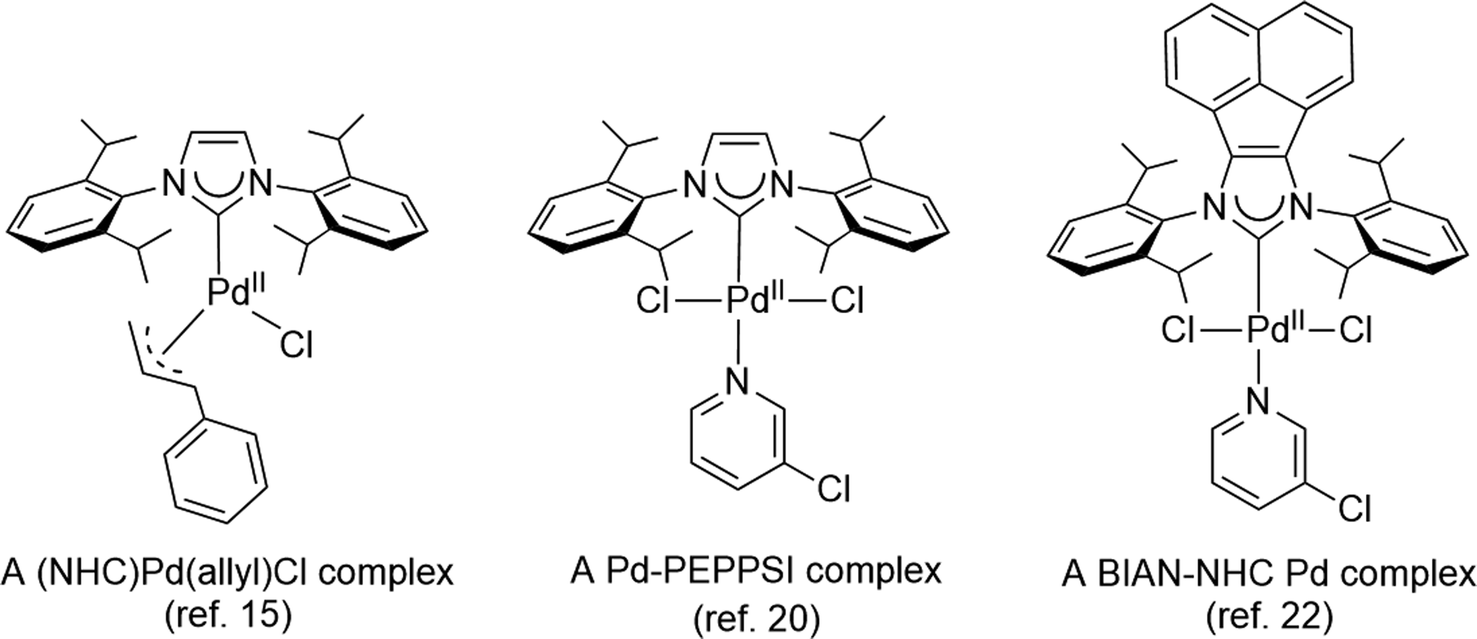 | ||
| Scheme 1 Representative palladium NHC complexes as pre-catalysts in cross-coupling reactions. | ||
Nitron, a mesoionic triazolium compound commercially known as 1,4-diphenyl-3-(phenylamino)-1H-1,2,4-triazolium inner salt, has been extensively utilized as an analytical compound for several decades.41–44 Its tautomer form in solution assumes the form of a free NHC ligand which readily reacts with different transition metal precursors.45–48 Previous studies have documented several palladium(II) NHC complexes derived from nitron (Scheme 2).48,49 However, to our knowledge, there is no literature on their corresponding palladium(0) complexes incorporating nitron-based NHC ligands. In this study, we present new palladium(0) complexes with NHC ligands derived from nitron and its derivatives. Significantly, these new palladium(0) complexes exhibit excellent air stability and certain compounds exhibit exceptional efficiency in the Mizoroki–Heck coupling reaction, particularly when employing aryl chlorides as coupling partners. In-depth theoretical calculations have been performed to elucidate the electronic properties of these new ligands and their palladium(0) complexes, thereby providing insights into the design of effective precatalysts in the catalytic reaction.
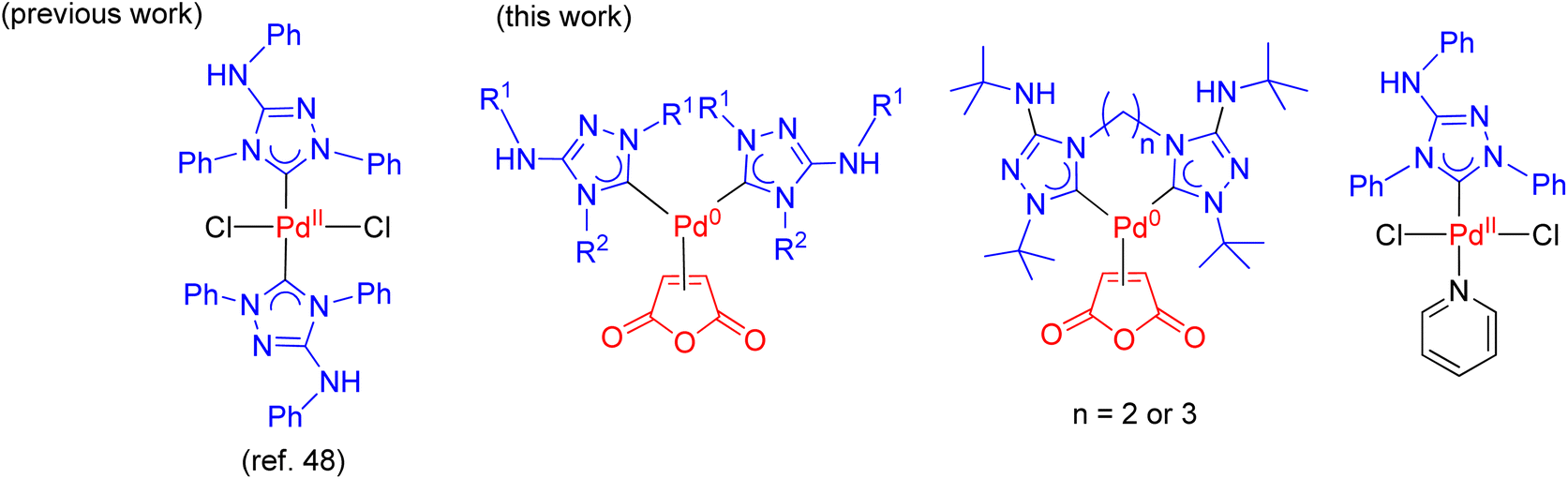 | ||
| Scheme 2 Palladium NHC complexes derived from nitron. | ||
Results and discussion
Synthesis of palladium(0) and palladium(II) NHC complexes
The synthesis of all new palladium complexes is depicted in Scheme 3. Nitron and other monodentate ligand precursors were obtained from commercial sources. To synthesize PdL12(ma) (1a) (ma = maleic anhydride), the reaction between nitron and the palladium(0) precursor, Pd(dbd)(ma) (dbd = N,N′-ditert-butylethane-1,2-diimine),50 was carried out in DMF at ambient temperature (Scheme 3a). In this particular reaction, the use of a base was unnecessary as the tautomer of nitron, which is the free carbene ligand L1, was captured by Pd(dbd)(ma) in the solution. As a result, complex 1a was obtained in a favorable yield of 90%. On the other hand, the synthesis of palladium(II) complex 4 involved the reaction between nitron and PdI2 in neat pyridine, leading to the isolation of the pure product with a yield of 58% (Scheme 3d). In contrast, the preparation of PdL22(ma) (1b) and PdL32(ma) (1c) from triazolium precursors [L2H]BF4 and [L3H]Br required the addition of potassium carbonate for the initial deprotonation of the acidic NH protons as depicted in Scheme 3b. Subsequently, tautomerizations occurred, resulting in the generation of free carbenes L2 and L3. Additionally, a higher temperature ranging from 50–80 °C was necessary for the formation of 1b and 1c, and the yields achieved were 84% and 66%, respectively.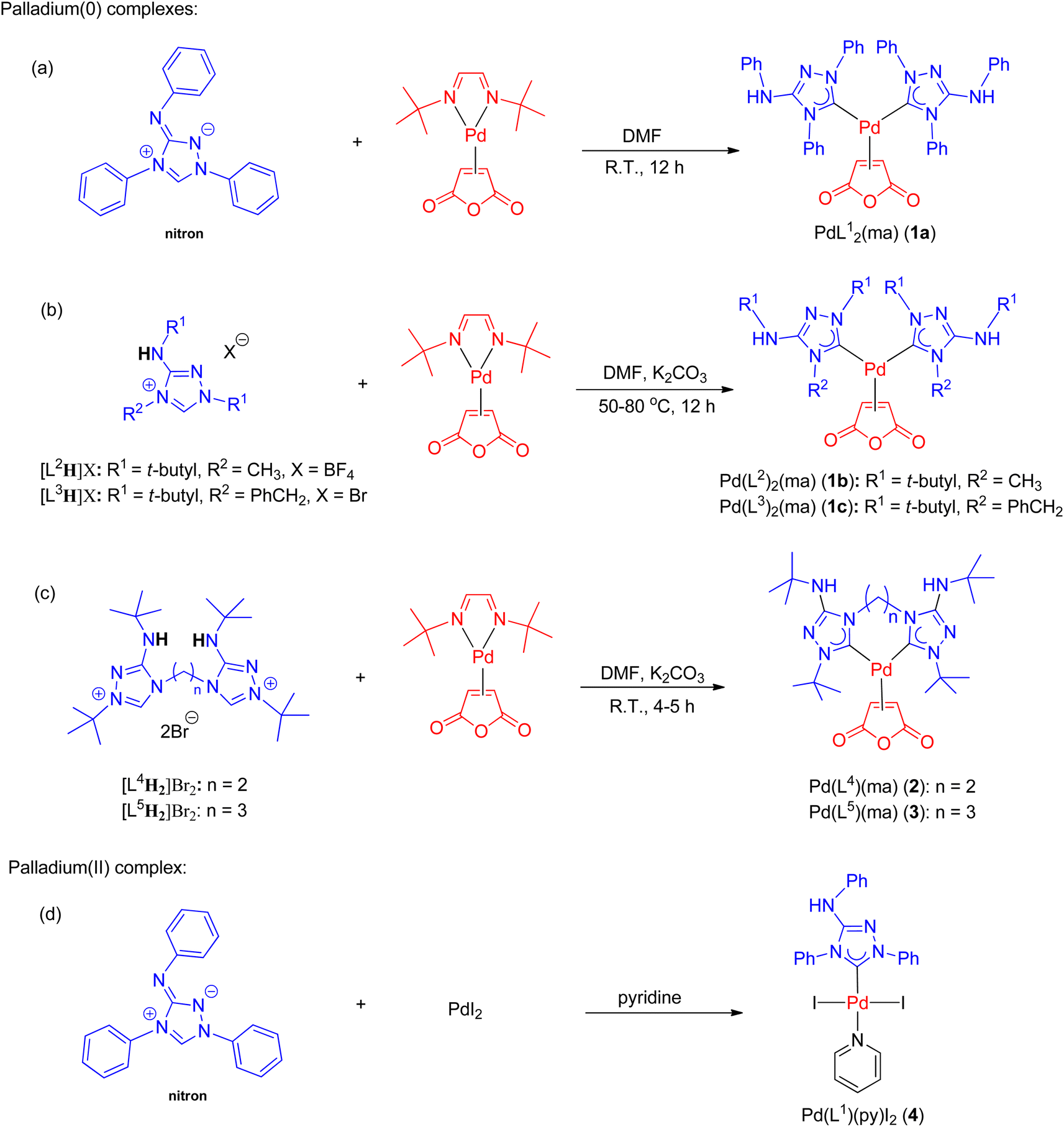 | ||
| Scheme 3 Synthesis of palladium(0) NHC (a–c) and palladium(II) NHC complexes (d). | ||
The synthesis of palladium(0) complexes with bidentate carbene ligands followed a similar procedure as depicted in Scheme 3c. The ligand precursors [L4H2]Br2 and [L5H2]Br2, featuring bis(carbene) ligands with ethylene and propylene bridges (L4 and L5, respectively), were previously unknown. Detailed information regarding their preparation and characterization can be found in the ESI.† In the reactions involving Pd(dbd)(ma) and [L4H2]Br2 or [L5H2]Br2, K2CO3 was added as a base at room temperature. This resulted in the formation of palladium(0) complexes, namely PdL4(ma) (2) and PdL5(ma) (3), with moderate yields of 56% and 64%, respectively.
The successful formation of metal carbene complexes was confirmed by the presence of downfield carbene signals in their 13C{1H} NMR spectra. The carbene signals for the palladium monocarbene complexes 1a–c were observed within a narrow range at 187.9, 184.6, and 185.6 ppm, respectively. Notably, complex 1a with N-phenyl groups, exhibited a slightly more downfield carbene resonance compared to complexes 1b–c, which had N-t-butyl groups. Interestingly, the carbene signal for palladium complex 3, with propylene-linked carbene moieties, appeared at the typical chemical shift of 184.3 ppm, while the corresponding signal for palladium complex 2, with ethylene-linked carbene ligands, showed an upfield shift at 177.9 ppm. This difference suggests the interplay of steric and electronic effects. In contrast, the carbene signal in palladium(II) complex 4 appeared very upfield at 139.7 ppm compared to the palladium(0) complexes. Importantly, all the palladium(0) and palladium(II) complexes were highly stable and could be easily handed in air.
Structural description
The structures of all new complexes, except 3, were successfully determined through single-crystal diffraction studies (Fig. 1). The crystallographic data can be found in Table S1 in the ESI.† In an asymmetric unit, there were two independent molecules of 1c, but only one of them was utilized for structural discussion. In each of the palladium(0) complexes, the coordination geometry around the metal center is formally trigonal planar, however, significant distortions were observed. For instance, in complex 1a, the three angles between the Pd–carbene bonds and the centroid of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond were measured to be 137.95°, 104.14°, 102.86°, deviating substantially from the ideal 120°. The coordination geometry around the palladium(II) center in complex 4 was a distorted square planar, with the two iodide ligands positioned in a trans configuration (∠C–Pd–N = 174.4(2)°; ∠I–Pd–I = 175.59(2)°). Remarkedly, the Pd–C bonds in palladium(0) complexes were much longer compared to the Pd–C bond in palladium(II) complex 4 (1a: 2.073(3) and 2.038(3); 1b: 2.074(5) and 2.088(5); 1c: 2.087(5) and 2.109(5); 2
C bond were measured to be 137.95°, 104.14°, 102.86°, deviating substantially from the ideal 120°. The coordination geometry around the palladium(II) center in complex 4 was a distorted square planar, with the two iodide ligands positioned in a trans configuration (∠C–Pd–N = 174.4(2)°; ∠I–Pd–I = 175.59(2)°). Remarkedly, the Pd–C bonds in palladium(0) complexes were much longer compared to the Pd–C bond in palladium(II) complex 4 (1a: 2.073(3) and 2.038(3); 1b: 2.074(5) and 2.088(5); 1c: 2.087(5) and 2.109(5); 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.101(2) and 2.078(2) Å, while 4: 1.954(5) Å). Additionally, a wide range of C–Pd–C angles was observed in the palladium(0) complexes. The angles in the complexes with monodentate NHC ligands (1a: 102.86(11)°; 1b: 99.78(18)°; 1c: and 111.13(19)°) were significantly smaller than the angle in the complex with the bidentate bis(NHC) ligand (2: 154.28(10)°).
:2.101(2) and 2.078(2) Å, while 4: 1.954(5) Å). Additionally, a wide range of C–Pd–C angles was observed in the palladium(0) complexes. The angles in the complexes with monodentate NHC ligands (1a: 102.86(11)°; 1b: 99.78(18)°; 1c: and 111.13(19)°) were significantly smaller than the angle in the complex with the bidentate bis(NHC) ligand (2: 154.28(10)°).
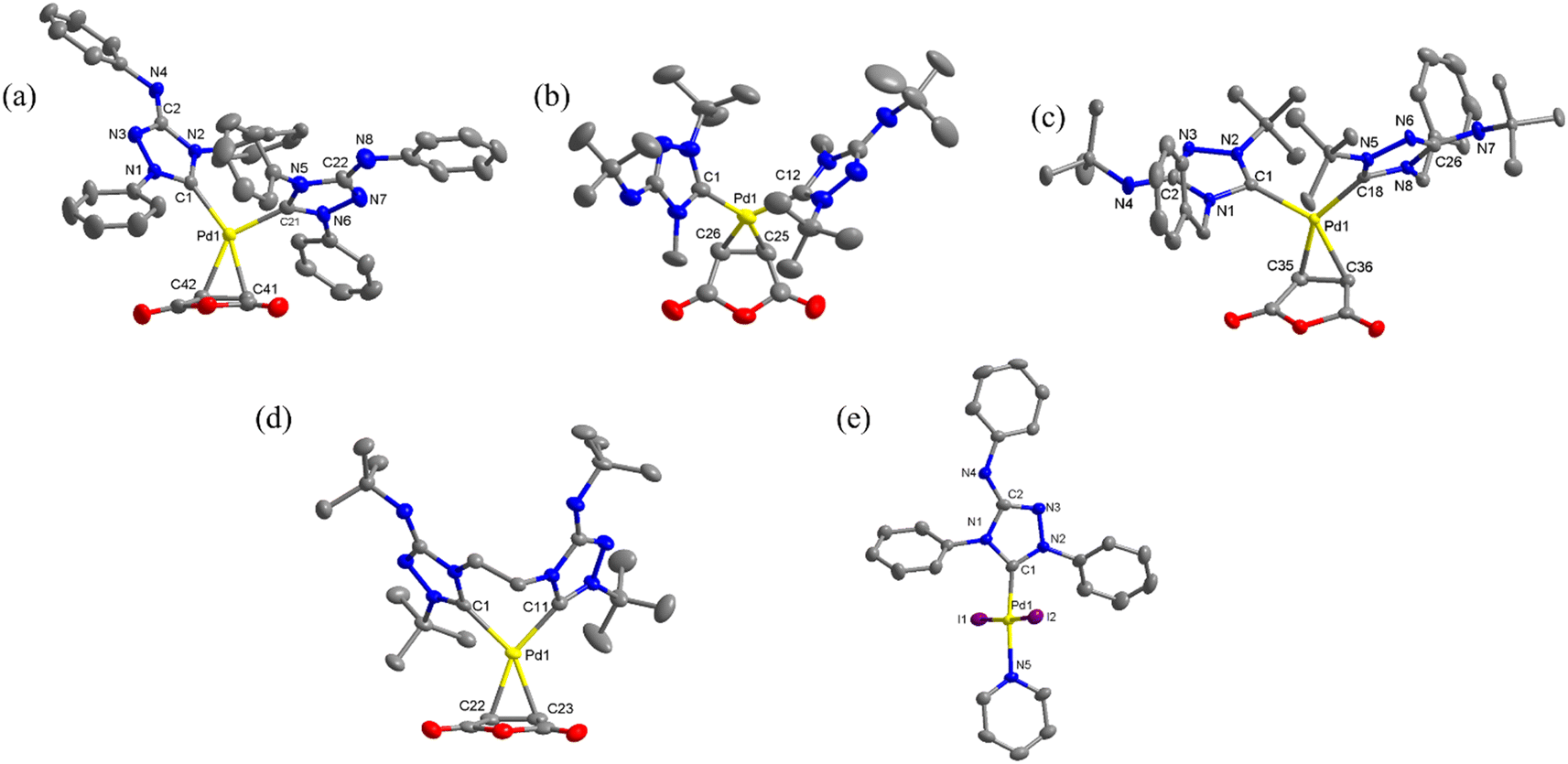 | ||
| Fig. 1 The new molecular structures with thermal ellipsoids drawn at 50% probability level. Hydrogen atoms are omitted for clarity. (a) The molecular structure of 1a; selected bond distances and angles (Å and °): Pd1–C1, 2.073(3); Pd1–C21, 2.038(3); Pd1–C41, 2.078(3); Pd1–C42, 2.162(3); C41–C42, 1.437(4); C1–Pd1–C21, 102.86(11); C1–Pd1–C42, 118.59(11); C21–Pd1–C41, 98.96(12); C1–Pd1–C41, 158.15(11); C21–Pd1–C42, 138.46(12). (b) The molecular structure of 1b; selected bond distances and angles (Å and °): Pd1–C1, 2.074(5); Pd1–C12, 2.088(5); Pd1–C25, 2.090(5); Pd1–C26, 2.120(5); C25–C26, 1.424(7); C1–Pd1–C12, 99.78(18); C1–Pd1–C26, 113.8(2); C12–Pd1–C25, 106.77(19); C1–Pd1–C25, 153.3(2); C12–Pd1–C26, 146.20(19). (c) The molecular structure of 1c; only one of the two independent molecules in an asymmetric unit is shown; selected bond distances and angles (Å and °): Pd1–C1, 2.087(5); Pd1–C18, 2.109(5); Pd1–C35, 2.098(5); Pd1–C36, 2.127(5); C35–C36, 1.447(8); C1–Pd1–C18, 111.13(19); C1–Pd1–C35, 99.8(2); C18–Pd1–C36, 108.7(2); C1–Pd1–C36, 139.8(2); C18–Pd1–C35, 148.4(2). (d) The molecular structure of 2; selected bond distances and angles (Å and °): Pd1–C1, 2.101(2); Pd1–C11, 2.078(2); Pd1–C22, 2.113(2); Pd1–C23, 2.114(2); C22–C23, 1.444(4); C1–Pd1–C11, 89.59(9); C1–Pd1–C22, 114.37(9); C11–Pd1–C23, 116.04(10); C1–Pd1–C23, 154.28(10); C11–Pd1–C22, 155.96(10). (e) The molecular structure of 4; selected bond distances and angles (Å and °): Pd1–C1, 1.954(5); Pd1–N5, 2.077(4); Pd1–I1, 2.6067(6); Pd–I2, 2.6000(6); C1–Pd1–I1, 88.91(16); C1–Pd1–I2, 88.11(17); N5–Pd1–I1, 92.97(13); N5–Pd1–I2, 90.30(13), C1–Pd1–N5, 174.4(2); I1–Pd1–I2, 175.59(2). | ||
Notable structural changes in the ligands upon metal coordination were observed. The CC bond distances in the maleic anhydride ligands (1a: 1.437(4) Å; 1b: 1.424(7) Å; 1c: 1.447(8) Å; 2: 1.444(4) Å) were found to be longer than the typical CC bond length of 1.35 Å, indicating significant π-back bonding interactions between the dπ orbitals the Pd(0) centers and the π* orbitals of the CC bonds. In all the palladium(0) complexes, the triazolium rings exhibited unsymmetrical bond lengths between the carbon and nitrogen atoms. Specifically, the NN–C bond was consistently shorter than the CN–C bonds by 0.23–0.53 Å. DFT calculations demonstrated that the shorter NN–C bonds possess a predominate double bond character (vide infra). Interestingly, a linear correlation was observed between the Pd–C bond length and the CN–C distance (Fig. 2a). Complex 1a exhibited the shortest CN–C bond and Pd–C bond among all the palladium(0) complexes, while complex 1c displayed the longest bonds of both types. This correlation can be explained by the π electron density originating from the nitrogen atom of the CN–C bond, as well as the palladium(0) center, which donate into the vacant p-orbital of the carbenic carbon, resulting in shorter bonds.
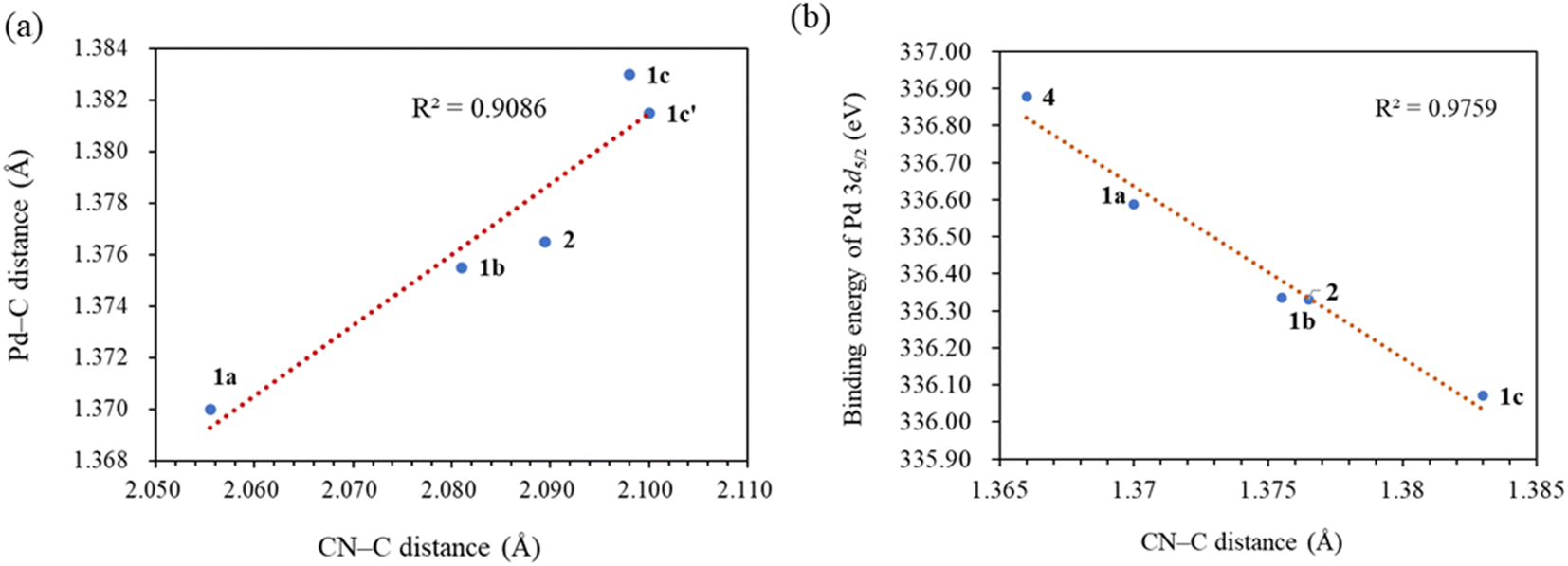 | ||
| Fig. 2 (a) The correlation between Pd–carbene and CN–C distances in the X-ray structures. Complexes 1c and 1c′ are the two independent molecules in an asymmetric unit. (b) The correlation between Pd 3d5/2 binding energies and CN–C distances. | ||
X-ray photoelectron spectroscopy (XPS)
XPS spectra were utilized to investigate the electronic properties of the newly synthesized complexes, Table 1 presents the Pd 3d5/2 binding energies of the complexes. Notably, Pd(II) complex 4 exhibited the highest binding energy of 336.88 eV, attributed to its higher nuclear charge, whereas all the other Pd(0) complexes have binding energies at least 0.29 eV lower. Among the Pd(0) complexes with monodentate NHC ligands, complex 1a, featuring three N-phenyl substituents, displayed the highest binding energy, while complex 1c, containing an electron-donating N-tert-butyl and N-benzyl groups, exhibited the lowest energy.Interestingly, there exists a correlation between the increase in Pd 3d5/2 binding energies and the decrease in CN–C bond distances in the heterocyclic rings (R2 = 0.9759) (Fig. 2b). Subsequent computational studies supported these findings, revealing that ligand L1 in complex 1a possesses weaker σ-donating capabilities, resulting in a higher binding energy of the Pd 3d core levels. However, L1 acts as a superior π-acceptor, reinforcing the coordination bond through π-backbonding, which leads to the shortest CN–C distance. Conversely, ligand L3 in complex 1c exhibits good σ-donating abilities but poor π-acceptor characteristics. Consequently, complex 1c displays the lowest binding energy but the longest CN–C bond. This correlation demonstrates that the CN–C distance obtained from X-ray structural data serves as a convenient probe for determining the Pd 3d5/2 binding energy.
Computational studies
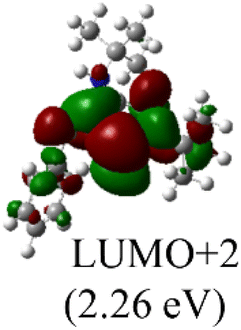
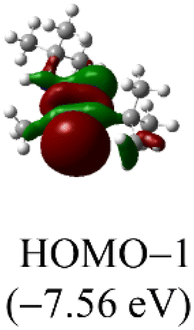
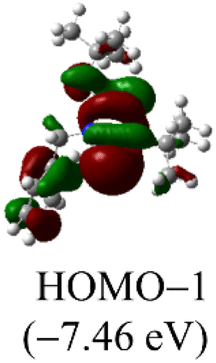
DFT calculations were conducted using the Gaussian 09W program to gain more chemical insights into the new ligands and metal complexes.51 Geometry optimizations and energy calculations for the free ligands were performed at the M06-2X/6-31g* level of theory. To account for the influence of the DMF solvent, the polarized continuum model (PCM) was employed.52 The energies of carbene orbitals involved in metal coordination for the new monodentate carbene ligands are presented in Table 2. Among the three new carbene ligands, L3 possesses the highest energy for the σ-donating orbital (−7.46 eV). This energy surpasses that of the commonly used IMes and IPr ligands. Conversely, L1 is the least electron-donating carbene ligand, exhibiting the lowest energy for the σ-donating orbital (HOMO-2) at −8.09 eV. However, L1 serves as an excellent π-acceptor due to its low-lying π-accepting orbital (LUMO) with an energy of only 0.18 eV. In contrast, the π-accepting orbitals in L2 and L3 possess significantly higher energies, indicating that these ligands primarily act as σ-donors.| Ligand | L1 | L2 | L3 | IMes | IPr |
|---|---|---|---|---|---|
| π-accepting orbitals | 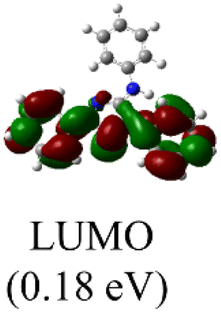 |
 |
 |
 |
 |
| σ-donating orbitals |  |
 |
 |
 |
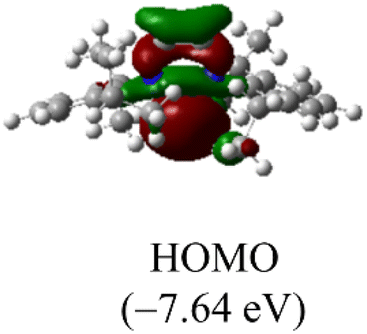 |
Geometry optimizations for metal complexes were carried out using the B3LYP53,54/LANL2DZ55 level of theory, based on the coordinates of the X-ray structural data. Energy calculations were performed using M06L functional with the def2-TZVP56 basis set. The PCM was again employed to account for the solvent effect of DMF.52 The Pd–C bond distances obtained from the X-ray structures and the optimized structures of the palladium complexes were compared, and the results are presented in Table 3. Overall, the optimized geometries closely agree with the experimental data. The Pd–C bond distances obtained from the optimized geometries are slightly larger, with deviations ranging from 0.008–0.046 Å. Notably, the computational method demonstrates high accuracy in reproducing the X-ray data for the palladium complex 2, which contains a bis(NHC) ligand, yielding a very small error of 0.008 Å. For palladium complexes with monodentate NHC ligands, a positive and linear relationship (R2 = 0.9999) is observed between the experimental and computational Pd–C bond distances (Fig. 3a).
| Complexes | Pd–C distances from the X-ray data (Å) | Pd–C distances from the optimized structures (Å) | Difference between structural and computational data (Å) |
|---|---|---|---|
| a There are two Pd–C distances in the Pd(0) complexes. An average distance is calculated. | |||
| 1aa | 2.056 | 2.092 | –0.036 |
| 1ba | 2.081 | 2.113 | –0.032 |
| 1ca | 2.100 | 2.131 | –0.031 |
| 2a | 2.090 | 2.098 | –0.008 |
| 4 | 1.954 | 2.000 | –0.046 |
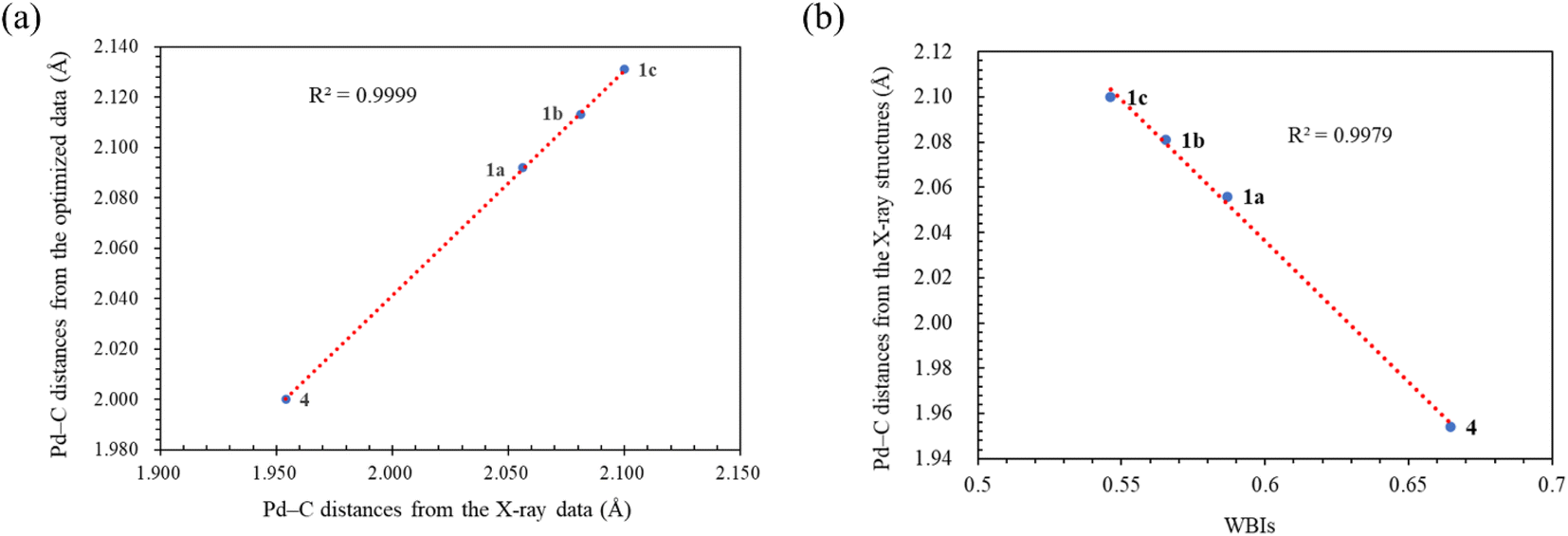 | ||
| Fig. 3 (a) The linear correlation of the Pd–C distances between the X-ray and optimized structural data. (b) The correlation between Pd–C distances in the X-ray structures and WBIs. | ||
The Frontier orbitals and their energies of palladium complexes 1a and 4 are presented in Fig. 4. In complex 1a–c, the HOMOs are distributed over the Pd atoms and carbene ligands, while the LUMOs are primarily localized on the coordinated maleic anhydride. This arrangement allows for facile nucleophilic attacks on the maleic anhydride moiety. In contrast, the HOMO of palladium(II) complex 4 is concentrated along the I–Pd–I axis, indicating a strong interaction between the iodine atoms and the palladium center. The LUMO, on the other hand, is found along both the I–Pd–I and C–Pd–N axes (Fig. 4).
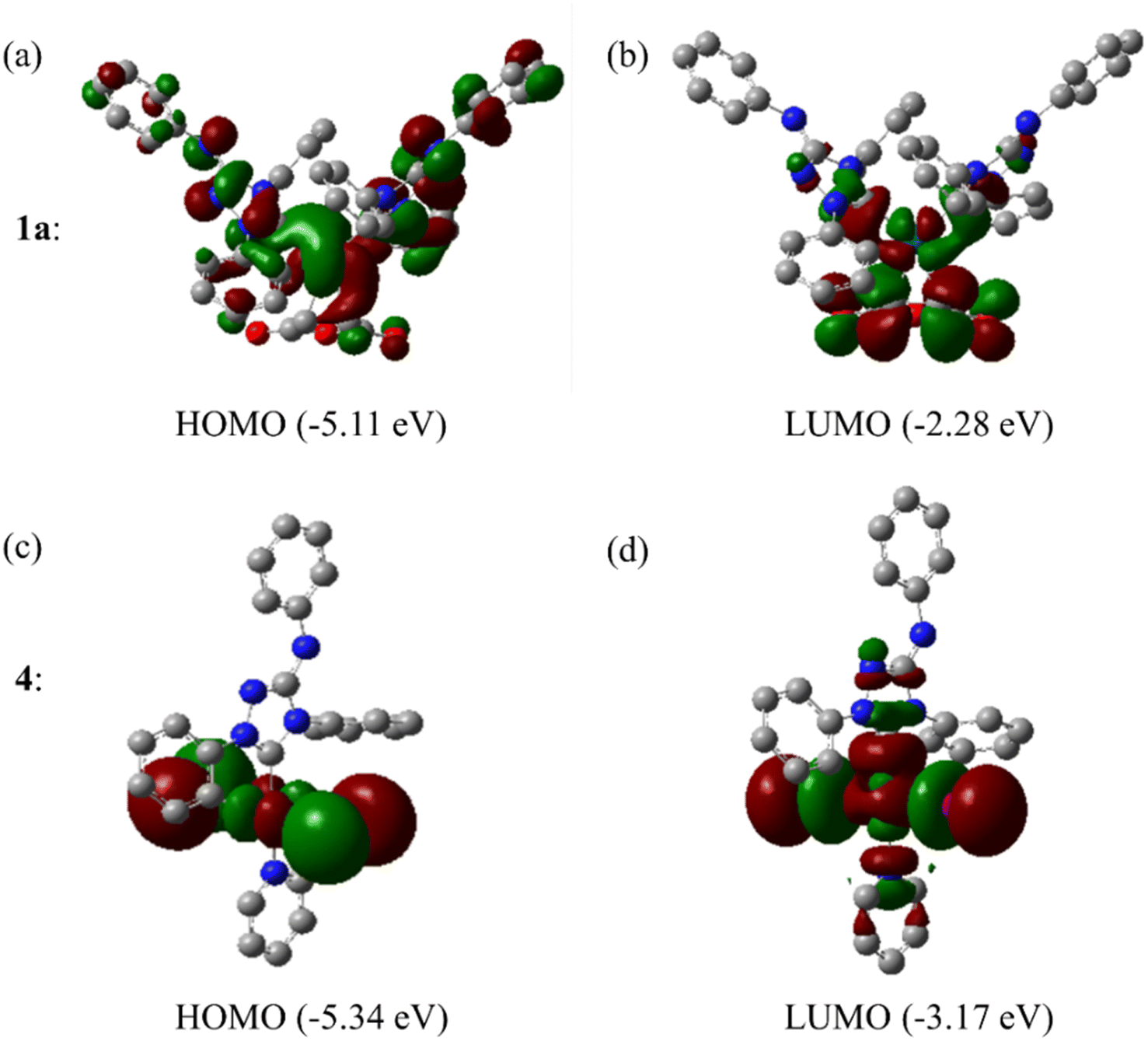 | ||
| Fig. 4 Frontier orbitals in Pd(0) complex 1a and Pd(II) complex 4. | ||
The Wiberg bond indices (WBIs) were calculated using natural bond orbital (NBO) analysis57 to determine the bond order between the palladium and carbene bonds. The average WBIs of the Pd–carbene bonds are presented in Table 1. The WBI of the well-known PEPPSI complex [trans-Pd(IPr)(3-Cl-py)Cl2] was previously estimated to be 0.6871.58 Notably, the Pd–carbene bonds in all the palladium(0) complexes are weaker compared to the Pd–carbene bond in the palladium(II) complex 4, which is consistent with their bond distances observed in the structural studies (vide supra). Two factors contribute to these weaker bonds. Firstly, the maleic anhydride ligand is a strong π-acceptor, drawing electron density away from the palladium(0) centers. This significantly reduces the extent of π-backbonding between the Pd(0) center and the NHC ligand. For instance, in complex 1a, the CC π* orbital in the maleic anhydride ligand is highly occupied (0.68751 electrons), indicating strong electron withdrawal. Secondly, the N–C bonds flanking the carbenic carbon are unequal in length. NBO analysis reveals that the shorter NN–C bonds possess significant double bond characters. For example, in complex 1a, the average occupation of the σ and π bonding electrons between the C and N atoms are 1.98013 and 1.88249 electrons, respectively. Consequently, π-donation from the lone pair on the N atom of the longer CN–C bond to the substantially filled pπ orbital on the carbenic carbon, as well as the π-backdonation from the Pd center, are significantly reduced. Consistently, there is an inverse correlation between the experimental Pd–carbene bond distances and the WBIs (R2 = 0.9979), indicating that shorter bonds exhibit higher WBIs and vice versa. A positive correlation exists between Wiberg Bond Indices (WBIs) and Pd 3d5/2 binding energies within the trio palladium(0) complexes 1 with monodentate NHC ligands. Despite being a weaker σ-donating ligand, the carbene ligand in complex 1a exhibits strong π-backbonding, resulting in its highest WBI (0.58675) and Pd 3d5/2 binding energy (336.59 eV) among the complexes. The CN–C bond distances from X-ray structures also align with WBIs; complex 1a, having the highest WBI (0.58675), also features the shortest CN–C bond (1.370 A) among the trio.
The NBO second-order perturbation theory analysis provided average stabilizing energies of π-backdonation from the lone pairs of Pd atoms to the π* orbital of the C–N bonds. The values obtained were 8.49, 7.96, and 7.70 kcal mol−1 for complexes 1a to 1c, respectively. These values indicate the extent of π-backdonation from the Pd centers to the carbene ligands. To further confirm the different abilities of the new monodentate carbene ligands as π-acceptors in receiving backdonation from Pd centers, a charge decomposition analysis (CDA) was performed on LPd(0) (L = L1, L2, L3).59 The CDA results confirmed that L1 is the most efficient π-acceptor, exhibiting the highest back-donation ratio (Table 4). Conversely, the extent of backdonation in L2 and L3 ligands was found to be similar. These CDA results are consistent with the π-accepting orbital energies of the ligands, as discussed earlier.
| Complexes | db | bc | rd | (d + b) | b/(d + b) |
|---|---|---|---|---|---|
| a At the M06-L/def2TZVP level; at the PBE1PBE/def2-TZVP level in brackets.b Number of electrons donated from the ligand to the Pd atom.c Number of electrons donated from the Pd atom to the ligand.d Closed-shell interaction. | |||||
| L1Pd | 0.184444 (0.187785) | 0.214079 (0.175547) | −0.228735 (−0.079429) | 0.398523 (0.363332) | 53.7% (48.3%) |
| L2Pd | 0.189166 (0.197697) | 0.204760 (0.169322) | −0.241592 (−0.094406) | 0.393926 (0.367019) | 52.0% (46.1%) |
| L3Pd | 0.185302 (0.187185) | 0.208898 (0.171341) | −0.223542 (−0.088492) | 0.394200 (0.358526) | 51.9% (47.8%) |
The analysis conducted in this part reveals important insights into the nature of the Pd–carbene bonds in the palladium(0) complexes. It is observed that these bonds are weaker compared to those found in Pd(II) complexes, primarily due to the presence of the strong π-accepting maleic anhydride (MA) ligand in the palladium(0) complexes. Despite their weakened nature, significant backdonation from the Pd center to the carbene ligands is still observed, indicating the presence of a bonding interaction. Among the monodentate carbene ligands studied, L1, which bears three N-phenyl rings, demonstrates the highest π-acceptor ability, while L3, with three N-tert-butyl and one N-benzyl groups, exhibits the best σ-donor characteristics. The shorter NN–C bonds within the heterocyclic rings show significant double bond character, indicating a strong bonding interaction. Conversely, the longer CN–C bond distances in the carbene ligands correlate well with various electronic and structural parameters, including Pd 3d5/2 binding energies, WBIs, and Pd–carbene bond distances. These findings provide valuable insights into the electronic and structural properties of the Pd–carbene bonds in the studied complexes, contributing to a better understanding of their reactivity and catalytic behavior.
Mizoroki–Heck coupling reaction
The newly synthesized palladium(0) complexes were evaluated as catalyst precursors in the Mizoroki–Heck coupling reaction. The coupling of 4-chloroacetophenone with styrene in neat tetrabutylammonium bromide (TBAB) was taken as the benchmark, following previously established conditions.26,27 The catalyst screening results are summarized in Table 5. Remarkably, when a 0.2 mol% of Pd loading was employed, complex 1a, which featured the monodentate L1, as well as complexes 2 and 3, containing bidentate bis(NHC) ligands L4 and L5, respectively, exhibited excellent yields of the coupling product (entries 1, 4, and 5). Interestingly, complexes 1b and 1c, which feature more electron-donating ligands L2 and L3, exhibited significantly lower effectiveness in the Mizoroki–Heck coupling reaction (entries 2 and 3). Typically, the rate-determining step (rds) involves the oxidative addition of the strong C–Cl bond in the aryl chloride to the Pd(0) species. Consequently, one would expect that the more electron-rich complexes 1b and 1c would exhibit higher activities compared to complex 1a. However, experimental observations suggest that the actual rds might be different in the catalytic cycle, such as the reductive elimination. Furthermore, the stability of the palladium(0)–carbene bond might play a more crucial role in the catalytic reaction than solely the electron-rich nature of the complex itself. Complex 1a, which possesses the strongest Pd–C bond among complexes 1a–c due to enhanced π-back-bonding interactions (vide supra), was also compared to complex 2 featuring a bidentate bis(NHC) ligand. For this comparative study, the reaction time was shortened to 1 h. Remarkedly, complex 1a demonstrated superior performance with a yield of 94%, while complex 2 yielded only 49% (entries 6 vs. 7). The time–yield curves of these two complexes are presented in Fig. S1 in the ESI.† It was determined that the optimized Pd loading for the reaction was 0.2 mol%, as reducing the loading to 0.1 mol% resulted in a significant decrease in product yield to 17% (entry 8).
|
|||||
|---|---|---|---|---|---|
| Entry | Cat | Pd mol% | Time (h) | Yield (%) | TON |
| a Reaction conditions: 1.4 mmol of styrene, 1 mmol of aryl halide, 1.1 mmol of NaOAc, 2 g of TBAB, Pd cat., 120 °C. Yield determined by using 1,3,5-trimethoxy-benzene as internal standard. | |||||
| 1 | 1a | 0.2 | 2 | 98 (95:5) |
490 |
| 2 | 1b | 0.2 | 2 | 5 | 25 |
| 3 | 1c | 0.2 | 2 | 54 (95:5) |
270 |
| 4 | 2 | 0.2 | 2 | 98 (95:5) |
490 |
| 5 | 3 | 0.2 | 2 | 97 (94:6) |
485 |
| 6 | 1a | 0.2 | 1 | 94 (95:5) |
470 |
| 7 | 2 | 0.2 | 1 | 49 (95:5) |
245 |
| 8 | 1a | 0.1 | 1 | 17 (97:3) |
170 |

Following the identification of the most promising catalyst precursor 1a, the substrate scope was further investigated (Table 6). The reaction time was partially optimized to achieve the best yields. Entries 1–6 clearly demonstrate that the catalyst system based on 1a exhibited remarkable effectiveness in utilizing electron-deficient aryl chlorides in combination with styrene, resulting in isolated yields ranging from 62% to quantitative yield. The reactions were conducted at a temperature of 120 °C for 2 h. In contrast, the system exhibited lower efficiency when utilizing electron-neutral and electron-rich aryl chloride substrates, requiring a prolonged reaction time of 12 h to achieve reasonable yields of coupling products (entries 7 and 8). However, when electron-rich aryl bromide substrates were employed, excellent yields of the products were obtained. Notably, 97 and 99% product yields were achieved using 4-bromo- and 3-bromoanisole, respectively (entries 9 and 10). Furthermore, even sterically hindered 2-bromoanisole readily underwent coupling with styrene, yielding a 69% product yield (entry 11). The substituted 4-methoxystyrene was also tested as a substrate, and once again, activated aryl chloride substrates exhibited smooth reactivity, resulting in high product yields (entries 12–14). However, the coupling reaction with 4-chloroanisole showed a poor yield (entry 15). Additionally, the applicability of n-butylacrylate as a coupling partner was explored (entries 16–23). The reaction with 4-chloroacetophenone yielded a mediocre yield of 54% in 6 h (entry 16). Significantly improved results were obtained when the reaction was conducted at 140 °C, leading to an 81% yield of the coupling product in 2 h (entry 17). Electron-deficient aryl chlorides provided yields ranging from 40% to 96% (entries 18–21). However, the reactivity with electron-neutral or electron-rich aryl chloride substrates was less promising (entries 22 and 23).
|
|||||||
|---|---|---|---|---|---|---|---|
| entry | R1 | X | R2 | Product | Time | Yield (%) | TON |
| a Reaction conditions (unless otherwise specified): 1.0 mmol aryl halide, 1.4 mmol alkene, 1.1 mmol NaOAc, 2 g of TBAB, 0.2 mol% of 1a, 140 °C unless specified otherwise., 2–12 h. Isolated yield.b 120 °C. | |||||||
| 1 | 4-CH3C(O) |
Cl | Ph | 5a | 2 | 99b | 495 |
| 2 | 4-NO2 | Cl | Ph | 5b | 2 | 97b | 485 |
| 3 | 4-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N N |
Cl | Ph | 5c | 2 | 76b | 380 |
| 4 | 4-HC(O) |
Cl | Ph | 5d | 2 | 82b | 410 |
| 5 | 4-CF3 | Cl | Ph | 5e | 2 | 72b | 360 |
| 6 | 4-CH3OC(O) |
Cl | Ph | 5f | 2 | 62b | 310 |
| 7 | H | Cl | Ph | 5g | 12 | 38 | 190 |
| 8 | 4-CH3O | Cl | Ph | 5h | 12 | 52 | 260 |
| 9 | 4-CH3O | Br | Ph | 5h | 12 | 97 | 485 |
| 10 | 3-CH3O | Br | Ph | 5i | 12 | 99 | 495 |
| 11 | 2-CH3O | Br | Ph | 5j | 12 | 69 | 345 |
| 12 | 4-CH3C(O) |
Cl | 4-CH3OPh | 5k | 2 | 99, 90b | 495, 490b |
| 13 | 4-NO2 | Cl | 4-CH3OPh | 5l | 2 | 99 | 495 |
| 14 | 4-HC(O) |
Cl | 4-CH3OPh | 5m | 2 | 99 | 495 |
| 15 | 4-CH3O | Cl | 4-CH3OPh | 5n | 12 | 17 | 85 |
| 16 | 4-CH3C(O) |
Cl | CO2nBu | 5o | 6 | 54b | 270 |
| 17 | 4-CH3C(O) |
Cl | CO2nBu | 5o | 2 | 81 | 405 |
| 18 | 4-NO2 | Cl | CO2nBu | 5p | 2 | 96 | 480 |
| 19 | 4-CN |
Cl | CO2nBu | 5q | 2 | 72 | 360 |
| 20 | 4-HC(O) |
Cl | CO2nBu | 5r | 2 | 67 | 335 |
| 21 | 4-CF3 | Cl | CO2nBu | 5s | 2 | 40 | 200 |
| 22 | H | Cl | CO2nBu | 5t | 12 | 11 | 55 |
| 23 | 4-CH3O | Cl | CO2nBu | 5u | 12 | 10 | 50 |

Next, we compare the catalytic activities of palladium(0) complex 1a with relevant Pd(II) NHC complexes (Table 7). A comparison of entries 1 and 2 clearly indicates that the palladium(0) complex 1a functions as a significantly better precatalyst than the palladium(II) complex 4. The time–yield curves for 1a and 4 in the benchmark reaction are presented in Fig. 5, highlighting that complex 4 requires an activation time of 30 min to transform into an active Pd(0) species, while complex 1a essentially completes the reaction within that time frame. Additionally, complex 1a demonstrates superior catalytic activity compared to simple Pd(OAc)2 and the PEPPSI complex, trans-Pd(IPr)(3-Cl-py)Cl2 (entries 1 versus 3 and 4). Tetranuclear palladium(II) complexes with abnormal NHC ligands have exhibited high activity in the Mizoroki–Heck coupling reaction.27 Furthermore, the activity of 1a is comparable to multinuclear catalyst systems when utilizing the electron-poor 4-chloroacetophenone substrate (entries 1 versus 5 and 6). However, it exhibits lower efficiency in the utilization of unreactive 4-chloroanisole compared to the tetranuclear catalyst systems (entries 7 versus 8 and 9).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pre-catalyst | Pd mol% | T (oC) | Time | Yield (%) | Reference |
| a Reaction conditions: 1.4 mmol of styrene, 1 mmol of aryl halide, 1.1 mmol of NaOAc, 2 g of TBAB, monopalladium loading, 140 °C.b Yield determined by using 1,3,5-trimethoxy-benzene as internal standard.c Isolated yield. | ||||||
| 1 | 1a | 0.2 | 140 | 2 | 99b (94:6) |
This work |
| 2 | 4 | 0.2 | 140 | 2 | 43b (93:7) |
This work |
| 3 | Pd(OAc)2 | 0.2 | 140 | 2 | 76b (97:3) |
Ref. 27 |
| 4 | Pd(IMes)Cl2(3-Clpy) | 0.2 | 140 | 2 | 26b (92:8) |
Ref. 27 |
| 5 | Tetranuclear Pd(aNHC) | 0.2 | 140 | 2 | >99b (95:5) |
Ref. 27 |
| 6 | Tetranuclear Pd(aNHC) | 0.2 | 140 | 2 | >99b (96:4) |
Ref. 26 |
 |
||||||
| 7 | 1a | 0.2 | 140 | 12 | 52c | This work |
| 8 | Tetranuclear Pd(aNHC) | 0.2 | 140 | 12 | 64c | Ref. 27 |
| 9 | Tetranuclear Pd(aNHC) | 0.2 | 140 | 12 | >99c | Ref. 26 |

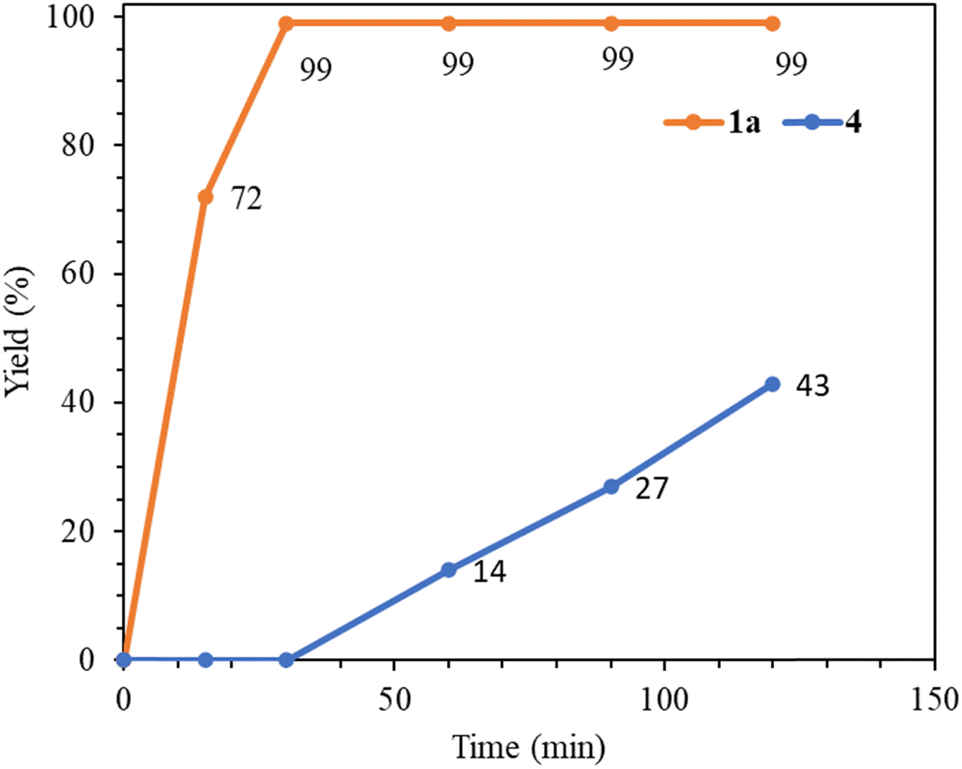 | ||
| Fig. 5 Time-yield curves of complexes 1a and 4 in the catalytic reaction between 4-chloroacetophenone and styrene carried out at 140 °C. | ||
In order to determine the nature of the catalytic species involved in the reaction, a Hg drop test was performed. Excess Hg (Hg:Pd = 100:1) was added to the benchmark reaction, and a slightly lower yield of 74% was obtained compared to the original reaction. This result suggests that the major catalytic species in the reaction is likely to be homogeneous, indicating that the palladium complex is predominantly responsible for the observed catalytic activity. In the Mizoroki–Heck reaction, the C–Cl bond in an aryl chloride undergoes oxidative addition to Pd(0) to form a Pd(II) intermediate.32 To confirm the presence of this Pd(II) species in the catalytic cycle, a solution of Pd(0) complex 1a and 4-chloroacetophenone in a 1:1 ratio in DMF was heated to 140 °C for 2 h. After the reaction, the solution was removed, and XPS analysis of the solid remained revealed a 3d5/2 binding energy of 336.868 eV for the Pd residue. This value closely resembles the 3d5/2 binding energy of the Pd(II) complex 4 (336.878 eV), indicating a change in valency from 0 to +2 and supporting the conversion of Pd(0) to Pd(II) species in the catalytic cycle.
Conclusions
The synthesis of stable palladium(0) complexes with monodentate and bis(carbene) ligands based on nitron and its derivatives was successfully achieved. The Pd–carbene bonds in these complexes are weaker compared to Pd(II) complexes due to the presence of strong π-accepting maleic anhydride (MA) ligands. However, significant back-donation still occurs, with the best π-acceptor being L1 with three N-phenyl rings, and the best σ-donor being L3 with three N-tert-butyl and one N-benzyl groups.The shorter NN–C bonds in the heterocyclic rings exhibit double bond character, while longer CN–C bond distances correlate with various electronic and structural parameters, such as Pd 3d5/2 binding energies, Wiberg bond indices (WBIs), and Pd–carbene bond distances.
Among the complexes, 1a with monodentate ligand L1 and a strong Pd–C bond, attributed to enhanced π-back-bonding, was identified as the best catalyst precursor for the Mizoroki–Heck coupling reaction between alkenes and aryl chloride substrates. The catalytic system showed excellent performance with activated aryl chloride substrates, yielding high coupling product yields. However, complexes 1b and 1c with more electron-donating ligands (L2 and L3) were found to be less effective, emphasizing the importance of the stability of the palladium(0) precatalyst over the electron-richness of the complex in the catalytic reaction which necessitate a high heating temperature. In comparison, palladium(0) complex 1a outperformed palladium(II) complex 4, with a significantly shorter activation time required for complex 1a to generate an active species. This study highlights the significance of a strong metal–carbene bond, crucial for palladium complexes to serve as efficient precatalysts in the Mizoroki–Heck coupling reaction.
Conflicts of interest
No competing financial interests are declared by the authors.Acknowledgements
We thank the National Science and Technology Council, Taiwan for sponsoring this work (111-2113-M-018-002).References
- M. Portnoy, Y. Ben-David, I. Rousso and D. Milstein, Organometallics, 1994, 13, 3465–3479 CrossRef CAS.
- W. A. Herrmann, C. Brossmer, C.-P. Reisinger, T. H. Riermeier, K. Öfele and M. Beller, Chem. - Eur. J., 1997, 3, 1357–1364 CrossRef CAS.
- M. T. Reetz, G. Lohmer and R. Schwickardi, Angew Chem Int. Ed., 1998, 37, 481–483 CrossRef CAS PubMed.
- M. Beller and A. Zapf, Synlett, 1998, 1998, 792–793 CrossRef.
- A. F. Littke and G. C. Fu, J. Org. Chem., 1999, 64, 10–11 CrossRef CAS PubMed.
- A. F. Littke and G. C. Fu, Angew Chem Int. Ed., 2002, 41, 4176–4211 CrossRef CAS PubMed.
- R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283–2321 CrossRef CAS.
- A. Zapf and M. Beller, Chem. Commun., 2005, 431–440 RSC.
- K. R. Balinge and P. R. Bhagat, C. R. Chim., 2017, 20, 773–804 CrossRef CAS.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS.
- V. K. Jain, Coord. Chem. Rev., 2021, 427, 213546 CrossRef CAS.
- L. González-Sebastián and D. Morales-Morales, J. Organomet. Chem., 2019, 893, 39–51 CrossRef.
- C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694–711 RSC.
- W. A. Herrmann, K. Öfele, D. v. Preysing and S. K. Schneider, J. Organomet. Chem., 2003, 687, 229–248 CrossRef CAS.
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS PubMed.
- E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew Chem Int. Ed., 2007, 46, 2768–2813 CrossRef CAS PubMed.
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew Chem Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- C. Chen, F.-S. Liu and M. Szostak, Chem. - Eur. J., 2021, 27, 4478–4499 CrossRef CAS PubMed.
- J. Li, D. He, Z. Lin, W. Wu and H. Jiang, Org. Chem. Front., 2021, 8, 3502–3524 RSC.
- S. J. Firsan, V. Sivakumar and T. J. Colacot, Chem. Rev., 2022, 122, 16983–17027 CrossRef CAS PubMed.
- G. R. Peddiahgari Vasu, K. R. Motakatla Venkata, R. R. Kakarla, K. V. S. Ranganath and T. M. Aminabhavi, Environ. Res., 2023, 225, 115515 CrossRef CAS PubMed.
- J.-Y. Lee, Y.-S. Su, Y.-S. Wang and H. M. Lee, Adv. Synth. Catal., 2019, 361, 4714–4726 CrossRef CAS.
- C.-H. Hung, W.-Y. Zheng and H. M. Lee, Organometallics, 2021, 40, 702–713 CrossRef CAS.
- P. J. Anju, M. Neetha and G. Anilkumar, ChemistrySelect, 2022, 7, e202103564 CrossRef CAS.
- Z. Zeng, Y. Chen, X. Zhu and L. Yu, Chin. Chem. Lett., 2023, 34, 107728 CrossRef CAS.
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS PubMed.
- J. P. Knowles and A. Whiting, Org. Biomol. Chem., 2007, 5, 31–44 RSC.
- C. S. Wei, G. H. M. Davies, O. Soltani, J. Albrecht, Q. Gao, C. Pathirana, Y. Hsiao, S. Tummala and M. D. Eastgate, Angew Chem Int. Ed., 2013, 52, 5822–5826 CrossRef CAS PubMed.
- V. P. W. Böhm, C. W. K. Gstöttmayr, T. Weskamp and W. A. Herrmann, J. Organomet. Chem., 2000, 595, 186–190 CrossRef.
- N. D. Clement, K. J. Cavell and L.-l. Ooi, Organometallics, 2006, 25, 4155–4165 CrossRef CAS.
- S. Fantasia and S. P. Nolan, Chem. - Eur. J., 2008, 14, 6987–6993 CAS.
- J.-Y. Lee, P.-Y. Cheng, Y.-H. Tsai, G.-R. Lin, S.-P. Liu, M.-H. Sie and H. M. Lee, Organometallics, 2010, 29, 3901–3911 CrossRef CAS.
- S. N. Sluijter, S. Warsink, M. Lutz and C. J. Elsevier, Dalton Trans., 2013, 42, 7365–7372 RSC.
- Y.-M. Jhou, D. Nandi, J.-Y. Lee, R.-J. Tzeng and H. M. Lee, Polyhedron, 2015, 100, 28–35 CrossRef CAS.
- T. Scattolin, C. Santo, N. Demitri, L. Canovese and F. Visentin, Dalton Trans., 2020, 49, 5684–5694 RSC.
- A. F. Henwood, M. Lesieur, A. K. Bansal, V. Lemaur, D. Beljonne, D. G. Thompson, D. Graham, A. M. Z. Slawin, I. D. W. Samuel, C. S. J. Cazin and E. Zysman-Colman, Chem. Sci., 2015, 6, 3248–3261 RSC.
- W. C. Cope and J. Barab, J. Am. Chem. Soc., 1917, 39, 504–514 CrossRef CAS.
- R. C. Young and P. M. Hernays, Ind. Eng. Chem., Anal. Ed., 1940, 12, 90 CrossRef CAS.
- M. Bobtelsky and J. Eisenstaedter, Anal. Chim. Acta, 1959, 20, 216–227 CrossRef CAS.
- A. Hulanicki and M. Maj, Talanta, 1975, 22, 767–769 CrossRef CAS PubMed.
- C. Färber, M. Leibold, C. Bruhn, M. Maurer and U. Siemeling, Chem. Commun., 2012, 48, 227–229 RSC.
- S. Hitzel, C. Färber, C. Bruhn and U. Siemeling, Organometallics, 2014, 33, 425–428 CrossRef CAS.
- C. Thie, S. Hitzel, L. Wallbaum, C. Bruhn and U. Siemeling, J. Organomet. Chem., 2016, 821, 112–121 CrossRef CAS.
- P. J. Quinlivan, A. Loo, D. G. Shlian, J. Martinez and G. Parkin, Organometallics, 2021, 40, 166–183 CrossRef CAS.
- A. Y. Chernenko, A. V. Astakhov, V. V. Kutyrev, E. G. Gordeev, J. V. Burykina, M. E. Minyaev, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Inorg. Chem. Front., 2021, 8, 3382–3401 RSC.
- K. J. Cavell, D. J. Stufkens and K. Vrieze, Inorg. Chim. Acta, 1981, 47, 67–76 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- T. H. Dunning Jr and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1977, vol. 3, pp. 1–28 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, TCI, University of Wisconsin, Madison, 1998 Search PubMed.
- M. M. Rahman, J. Zhang, Q. Zhao, J. Feliciano, E. Bisz, B. Dziuk, R. Lalancette, R. Szostak and M. Szostak, Organometallics, 2022, 41, 2281–2290 CrossRef CAS.
- S. Dapprich and G. Frenking, J. Phys. Chem., 1995, 99, 9352–9362 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; NMR spectra of ligand precursors, complexes, and catalytic products; crystallographic data. CCDC 2277731, 2277757–2277760. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra05016e |
| This journal is © The Royal Society of Chemistry 2023 |