
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of benzo[a]carbazole derivatives via intramolecular cyclization using Brønsted acidic carbonaceous material as the catalyst†
Hai Truong Nguyen ab,
Phat Ngoc Nguyenab,
Tan Van Leab,
Trinh Hao Nguyenab,
Linh Dieu Nguyenab and
Phuong Hoang Tran*ab
ab,
Phat Ngoc Nguyenab,
Tan Van Leab,
Trinh Hao Nguyenab,
Linh Dieu Nguyenab and
Phuong Hoang Tran*ab
aDepartment of Organic Chemistry, Faculty of Chemistry, University of Science, Vietnam National University, Ho Chi Minh City 700000, Vietnam. E-mail: thphuong@hcmus.edu.vn
bVietnam National University, Ho Chi Minh City 700000, Vietnam
First published on 29th September 2023
Abstract
In this work, a new procedure for the synthesis of benzo[a]carbazole from 1,3-diketones, primary amines, phenylglyoxal monohydrate, and malononitrile employing a solid acidic catalyst has been developed. The multicomponent reaction provided 3-cyanoacetamide pyrrole as an intermediate and then the formation of benzo[a]carbazole via intramolecular ring closure. The reaction was carried out for 2 h at 240 °C, resulting in the desired product with 73% yield. Acidic sites on the solid acid catalyst, made from rice husk-derived amorphous carbon with a sulfonic acid core (AC-SO3H), provided the best activity. Acidic sites on the surface of the catalyst, including carboxylic, phenolic, and sulfonic acids, were 4.606 mmol g−1 of the total acidity. AC-SO3H demonstrated low cost, low toxicity, porosity, stability, and flexibility of tuning and reusability.
Introduction
Carbazole frames are a peculiar type of N-heterocyclic compound. These compounds typically appear as structural motifs in both naturally occurring alkaloids and other synthetic chemicals. The physical properties of carbazoles are similar to those of synthetic dyes,1 conducting polymers,2 and optoelectronic components.3 Recently, several polymer derivatives have been incorporated into solar cells made of polymers (2,7-carbazole). Additionally, these compounds are frequently used as white, red, and green emitters in organic light-emitting diodes. By changing the structural characteristics of carbazoles at the C-2, -3, -6, -7, and -9 positions, it is possible to change their molecular and optical properties.4 Inorganic light-emitting systems have utilized a few benzo[a]carbazole, benzo[c]carbazole, and indolo[3,2-b]carbazole frames as molecular platforms, hole transport, and host materials. Recent research has revealed the usefulness of carbazole-containing ligands as anion receptors. For instance, derivatives of 3,6-dichlorocarbazole-1,8-diamide have the ability to hydrogen bond with anions. Notably, carbazoles are a diverse group of organic plant chemicals. Many of them possess strong biological properties, such as anticancer,5 antiepileptic,6 antibacterial,7 anti-inflammatory,8 antioxidative,9 analgesic,10 antidiarrheal,11 antihistaminic,12 neuroprotective,13 and pancreatic lipase inhibitory (Scheme 1).14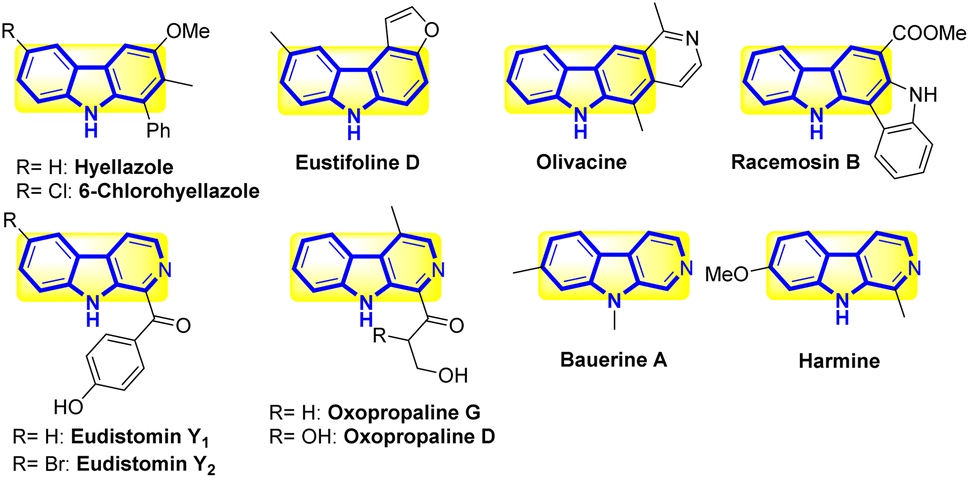 | ||
| Scheme 1 Chemical structure of benzo[a]carbazole frames and their bioactive. | ||
Recently, Knölker et al. thoroughly investigated the production of these beneficial carbazole alkaloids.15 Historically, carbazoles were made via processes like insertion of nitrene,16 indolization of Fischer,17 cyclization by Pummerer,18 Diels–Alder technique,19 cyclization via dehydrogenation of diarylamines.20 Studies in more recent years have concentrated on transition metal-mediated processes, including ring-closing metathesis,21 cyclotrimerization,22 benzannulation,23 Suzuki-Miyaura coupling,24 and the creation of C–C and C–N bonds.25–27 To address issues with regiochemical selectivity and efficacy, many of these methods were continuously refined. This article's discussion of the most recent synthetic methods was organized according to how they create rings through the use of strategic bonds.
Maji et al. used Brønsted acid/base catalyzed for one-pot sequential-triple-relay benzannulation of 2-alkenyl indoles with low-cost aldehydes, a wide variety of structurally distinct carbazoles can be synthesized.23 Deng et al. developed a straightforward three-component approach to produce diversely doped carbazoles through thermodynamically induced [2 + 2 + 2] annulation of indole, ketone, and nitroalkene.28 Liu et al. proposed a metal-free, sequential triple C–C coupling-radical-cyclization procedure to regioselectively generate substituted carbazole compounds using DMAP as the only reagent.29 Through the transition-metal and exogenous-oxidant-free C–H bond amination approach, P. Zhang et al. also produced the carbazole-derived alkaloids clausine and glycozoline.30 They also developed a technique for an environmentally secure electrochemical C–H bond dehydrogenative amination. This technique may be used to create preferred carbazole moieties, which were advantageous in a variety of situations. It was emphasized that this method's scalability, relatively mild conditions, and universal applicability made it more environmentally friendly and sustainable. A general technique for creating the carbazol-4-amine motif had been devised by D. Cao et al. by a vinylogous Michael addition, cyclization, isomerization, and elimination reaction of 3-nitroindoles with alkylidene malononitriles. The process of this method did not involve the usage of transition metals at any point.31
An innovative kind of solid Brønsted acid catalyst was amorphous carbon-carrying sulfonic acid groups. In recent years, organic synthesis, adsorbents, and electrochemistry had all researched novel forms of carbon materials in detail. Using readily accessible agricultural by-products, it was inexpensive and simple to create the sulfonated amorphous carbon, which has special physical and chemical characteristics including strong Brønsted acid, large surface area, high stability, reactivity, and cyclability. By incompletely carbonizing sulfopolycyclic aromatic compounds in concentrated H2SO4 or sulfonating incompletely carbonized natural organic materials, carbon-based solid acids can be made easily. Three distinct functional groups including –SO3H, –COOH, and –OH groups, which containing in the material with carbon frames.32–34 The usual solid acids tested; however, all have a single functional group. The strong catalytic activity of the carbon material may be due to this. After the reaction, the carbon catalyst can be easily removed from the saccharide solution and reused without losing any activity.
Nowadays, the synthesis of carbonaceous framework materials from agricultural waste sources has received much attention and popular research around the world. Because of its exceptional characteristics, amorphous carbon materials are now the subject of a significant amount of study all around the globe in a variety of applications. These applications include biomedical, energy systems, and environmental remediation. To possibly produce amorphous carbon compounds, carbonaceous feedstocks such as rice byproducts (straw and husk), wood fiber, sugarcane bagasse, wood, sawdust, etc., may be used.35–37
In our previous work, effective sulfonated amorphous carbon from rice husk carbonation is reported. By utilizing sulfuric acid for sulfonation, the AC-SO3H was created. The AC-SO3H catalyst activity was evaluated through the synthesis of benzo[a]carbazoles from 3-cyanoacetamide pyrroles through intramolecular cyclization. This method was considered a novel and effective method for synthesizing benzo[a]carbazole frames through intramolecular cyclization. Additionally, it attempted to explore the recycling of catalysts.
Experimental
Chemical and analytical techniques
All chemicals with high purity were purchased from Sigma-Aldrich and Acros. Analytical thin-layer chromatography (TLC) results were gathered on Merck F-254 silica gel-coated aluminum plates. Column chromatography using Merck silica gel (60, 230–400 mesh) was done. With the use of a Bruker Avance 500 MHz, the 1H and 13C NMR spectra were recorded. Internal standards were either TMS or solvent peaks, and the solvent was DMSO-d6. Determining melting point included using the Buchi melting point B-545. ATR-FTIR spectra were sustained using a Bruker E400 FT-IR spectrometer. ATR-FTIR spectra in the region of 4000–600 cm−1. TGA was measured using the Q-500 thermal gravimetric analyzer under airflow with a temperature gradient 5 °C min−1. Powder X-ray diffraction (P-XRD) data for refinement were collected on a Bruker D8 Advance utilizing Ni-filtered Cu K (λ = 1.54059) radiation. Using the Hitachi S-4800 scanning electron microscope and the XZS-107T digital microscope connected to NHV-CAM through the program eScope, the materials' morphology was examined (SEM). The Quantachrome NOVA 3200e system was used to quantify the N2 isotherm at 77 K. To ascertain the elemental composition of sorbents, energy-dispersive X-ray spectroscopy (EDX) examination was realized utilizing an EMAX energy EX-400 EDX instrument. The Bruker micrOTOF-QII MS operating at 80 eV was used to collect data for HRMS (ESI).Typical procedure for the synthesis of amorphous carbon immobilized sulfonic acid group (AC-SO3H)
Under N2 atmosphere, at a temperature of 400 °C prolong 10 hours, rice husks were partly carbonized. After heating the combination for ten hours at 150 °C while it was being purged with nitrogen, powerful sulfuric acid was incorporated to the mixture. The dark material was washed many times with deionized water hot at 80 °C until the presence of sulfate anions was no longer detectable in the filtrate. The sulfonated amorphous carbon catalyst subsequently went through a drying process that lasted for two hours and was carried out at a temperature of 100 °C in air. FT-IR, XRD, TGA, EDS, and SEM were the instruments that were used, the catalyst's purity and structural integrity were verified.General procedure for measuring the acidity of the AC-SO3H catalyst
0.25 g of AC or AC-SO3H was added to a sodium hydroxide aqueous solution (0.05 N, 30 mL) (NaOH was calibrated with oxalic acid). At room temperature, the mixture was stirred for 60 min. Following centrifugal separation, hydrochloric acid (0.05 N) aqueous solution was used to titrate the supernatant solution using phenolphthalein as an indicator. Record the pH of the solution every 0.1 mL of 0.05 N HCl.Methodological outline for the synthesis of benzo[a]carbazoles
In a 25 mL flask, a mixture of 3-cyanoacetamide pyrrole scaffolds (0.25 mmol) and AC-SO3H (6 mg) in the presence of DMSO (5 mL). The reaction was stirred and refluxed for 2 h (the temperature of the reaction was controlled by a sand bath heated at different temperatures). The reaction mixture was monitored by TLC, then filtered to separate the catalyst from the solution. After washing the organic layer with water (3 × 5 mL) and ethyl acetate (25 mL), then making it anhydrous with sodium sulfate, and allowed to crystallize at room temperature. The crystal was filtered and washed with cold ethyl acetate to obtain a pure product. The structure and purity of the product were determined by 1H, 13C NMR, HRMS, and melting point.Recycling of AC-SO3H
After the reaction was finished, the product was collected from the reaction mixture using ethyl acetate extraction, and the catalyst was taken out of the solvent. Ethyl acetate was used to wash the recovered catalyst. The model reaction was restarted after it had been dried for two hours at 100 °C. A mixture of (1a) (0.25 mmol) and AC-SO3H (6 mg) in the presence of DMSO (5.0 mL). The reaction was stirred and refluxed at 240 °C for 2 hours.Results and discussion
Amorphous carbon-immobilized sulfonic acid characterization (AC-SO3H)
From the previous research results, AC-SO3H was constructed using the described method.38,39 Rice husk was employed as the starting material, which goes through partial carbonization in an inert atmosphere (known as the amorphous carbon = AC) before being further sulfonated to create AC-SO3H, which was used as a catalyst. The properties of AC-SO3H materials were determined by modern technology namely FTIR, P-XRD, TGA, SEM, and EDS. The FTIR spectra of AC-SO3H and AC are shown in Fig. S1.† The results revealed numerous identifying peaks at 3425 cm−1 and 1700 cm−1, corresponding to O–H bonds, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively, which was identified as a signal of the phenolics, carboxylic acids and sulfonic acids site. The signals of the polyaromatic structure, including CC pairs, were represented by the stretching vibration peak at 1625 cm−1. The AC-SO3H line exhibits prominent absorption bands of about 1101 cm−1 representing the sulfonic acid's SO stretching vibrations. The results of this study were compared with published studies.40
O bonds, respectively, which was identified as a signal of the phenolics, carboxylic acids and sulfonic acids site. The signals of the polyaromatic structure, including CC pairs, were represented by the stretching vibration peak at 1625 cm−1. The AC-SO3H line exhibits prominent absorption bands of about 1101 cm−1 representing the sulfonic acid's SO stretching vibrations. The results of this study were compared with published studies.40
There were three steps in the thermal stabilities of AC shown in Fig. S2,† the first one for the remaining water and finishing at 350 °C. The second stage began at 350 °C and finished at 500 °C due to the decarbonation of amorphous carbon, and the last step began at 550 °C and terminated almost immediately at 800 °C due to the catalyst's evaporation of carbon dioxide gas with a 30% residual. Similar to this, the dehydration of the AC-SO3H structure was estimated to occur at 500 °C, and the 60% decrease of organic functional groups was estimated to occur at 600 °C. The remaining 35% was kept at a temperature between 600 °C and 800 °C. Thermal gravimetric analysis was used to investigate the thermal stabilities of the waste base material in an N2 atmosphere.
The SEM image results show that the amorphous carbon material before sulfonation (AC) and after sulfonation (AC-SO3H) has an agglomerated form (Fig. S3a and b†). Based on EDX analysis of AC and AC-SO3H materials (Fig. S3d and e†), the structure of AC material contains elements such as C, O, Si, and S with 56.25%, 36.17%, 6.92%, and 0.66% about atomic%, respectively. Besides, AC-SO3H material also contains elements C (65.56%), O (27.70%), Si (5.19%), and S (1.55%). From the results, the atomic% of the silicon element in AC-SO3H decreased compared to AC, and the atomic% of the sulfur element in AC-SO3H increased compared to that of AC-SO3H with AC. The result showed that the pore structure of AC-SO3H partially collapsed after sulfonation,41 and successfully sulfonated AC-SO3H material. Elemental mapping analysis was also used to confirm the outcomes of the EDX test (Fig. S4†). This technique also makes it possible to see elemental scattering clearly.
P-XRD analysis was used to characterize the structural characteristics of AC and AC-SO3H. As can be seen in Fig. S5,† the wide peak C(002), which is associated with the graphitic reflection plane amorphous carbon, can be seen in the diffraction pattern of AC and AC-SO3H, which is represented by the broad peaks in the XRD pattern, in the 2θ range of 15–35°. The modest and broad C(101) diffraction signal which was observed makes it quite evident that is evidently detected 2θ between 40 and 50° is affected by the graphite structure's a axis.42
The titration method was applied to determine the total acid concentration of AC and AC-SO3H (Fig. S6†).
Table S1† displays the number of surface functional groups determined via Boehm titration.43 Using the acidification solution of AC increased the number of total acidic functional groups from an initial value of 0.828 mmol g−1 for the AC to 4.606 mmol g−1 for the AC-SO3H. Specifically, greater carboxylic (–COOH) and phenolic (–OH) groups were favored, corresponding with the findings of Mena Aguilar et al.44 Similar trends were seen between the concentrations of sulfuric acid and –SO3H groups. Inductively Coupled Plasma Mass Spectrometry (ICP-MS) determined that the amount of element S present in the AC-SO3H catalyst was 2.185 mmol.g−1.
Synthesis of benzo[a]carbazole frames
AC-SO3H catalyst activity was evaluated through the synthesis of 3-cyanoacetamide pyrrole from 1,3-diketones, primary amines, phenylglyoxal monohydrate, and malononitrile under acetone, which has been published in our study results.45 Continuing to develop from that research result, we investigated the activity of AC-SO3H as a catalyst for benzo[a]carbazole synthesis through the intramolecular cyclization of 3-cyanoacetamide pyrrole scaffolds to form benzo[a]carbazole frames (Scheme 2).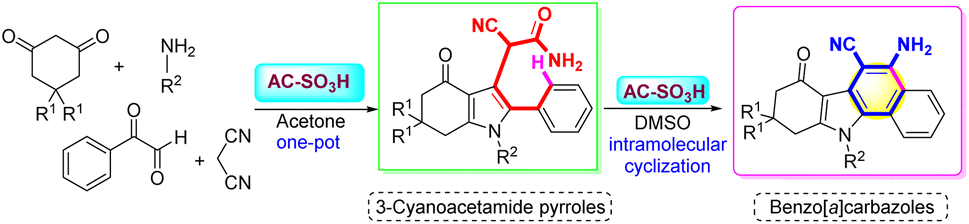 | ||
| Scheme 2 Synthesis of benzo[a]carbazole frames from 3-cyanoacetamide pyrrole scaffolds using AC-SO3H. | ||
The parameters of the reaction that are investigated when the intramolecular cyclization reaction occurs between 2-cyano-2-(6,6-dimethyl-4-oxo-1,2-diphenyl-4,5,6,7-tetrahydro-1H-indol-3-yl)acetamide (1) and DMSO to form 5-amino-9,9-dimethyl-7-oxo-11-phenyl-8,9,10,11-tetrahydro-7H-benzo[a]carbazole-6-carbonitrile (1a) include reaction temperature, reaction time, and amount of AC-SO3H (Table 1). In the presence of 10 mg AC-SO3H used as a catalyst in DMSO for 4 hours, the reaction was investigated for the influence of reaction temperature with different temperatures extending from 160 °C to 280 °C (entries 1–8, Table 1). The outcomes of the investigation indicate that when the temperature increases, the product (1a) also increases, but this process increases slowly and achieves high results at 240 °C with 30%. From the best result, the process of intramolecular cyclization requires high temperature. Next, the reaction time was also surveyed to choose the best time to form the product (1a). The reaction time was investigated at 240 °C with different time intervals, including 1 hour, 2 hours, 3 hours, 4 hours, 5 hours, and 6 hours in the presence of 10 mg AC-SO3H (entries 9–13, Table 1). The results of the trial showed that the best reaction time was two hours. The loading of the AC-SO3H catalyst added to the reaction mixture was also investigated, consisting of 0, 2, 4, 6, 8, 10, 12, and 14 mg (entries 14–20, Table 1). In cases where the reaction was conducted without a catalyst, the reaction did not occur at all to form the desired product, which is explained by the absence of a source of H+ for the oxygen of the carbonyl group, resulting in a reaction that did not occur. Next, when the amount of catalyst is increased to 2 mg, the reaction was also not released to form a major product (1a). However, when AC-SO3H was used in larger quantities, product formation (1a) also increased, and the highest efficiency was achieved when using 6 mg of AC-SO3H with 73%.
| Entry | Temp. (°C) | Time (h) | Loading of AC-SO3H (mg) | Yieldsb (%) |
|---|---|---|---|---|
| a Reaction condition: 1 (0.25 mmol, 99.29 mg), AC-SO3H under DMSO (5 mL). The temperature of the reaction was controlled by a sand bath heated at different temperatures.b Yields were isolated yields by crystallization in ethanol (10–15 mL). | ||||
| 1 | 160 | 4 | 10 | 16 |
| 2 | 180 | 4 | 10 | 14 |
| 3 | 200 | 4 | 10 | 22 |
| 4 | 220 | 4 | 10 | 23 |
| 5 | 240 | 4 | 10 | 30 |
| 6 | 250 | 4 | 10 | 33 |
| 7 | 260 | 4 | 10 | 29 |
| 8 | 280 | 4 | 10 | 30 |
| 9 | 240 | 1 | 10 | 34 |
| 10 | 240 | 2 | 10 | 41 |
| 11 | 240 | 3 | 10 | 38 |
| 12 | 240 | 5 | 10 | 33 |
| 13 | 240 | 6 | 10 | 36 |
| 14 | 240 | 2 | 0 | 0 |
| 15 | 240 | 2 | 2 | 0 |
| 16 | 240 | 2 | 4 | 58 |
| 17 | 240 | 2 | 6 | 73 |
| 18 | 240 | 2 | 8 | 74 |
| 19 | 240 | 2 | 12 | 69 |
| 20 | 240 | 2 | 14 | 75 |
From the results close to the factors affecting the major product formation, the influence of catalyst, solvent, and solvent volume was also evaluated when the intramolecular cyclization reaction occurred with (1) (Table 2). In the presence of DMSO as a solvent for the reaction, the activity of the AC-SO3H catalyst was studied and compared with other catalysts with similar properties to AC-SO3H. When applying AC as a catalyst for the process, intramolecular cyclization of (1) proceeds with poor yield; this is hypothesized to be due to AC only containing –OH and –COOH groups in its structure, which makes its catalytic activity less than AC-SO3H. IL1-SO3H was prepared from imidazole,46 which was used as the catalyst for the synthesis of (1a) with 67%. BAIL gel was synthesized from 1-(4-sulfonic acid)butyl-3-methylimidazolium hydrogen sulfate and tetraethoxysilane,47 which was employed to yield of (1a) with an isolation yield of 64%. And, SiO2–H2SO4 (ref. 48) was also selected as a catalyst for the reaction because of its similar structure to AC-SO3H, the reaction yield was as low as 40%. The major product (1a) was formed with low yield when using TsOH or H2SO4 as catalysts for the intramolecular cyclization reaction of (1). At 240 °C, for 2 hours, the reaction did not occur when carried out in the absence of a catalyst or solvent. Under the same reaction conditions, DMSO was replaced by DMF as the solvent for the synthesis reaction (1a) with a much lower yield than using DMSO as the reaction solvent, which indicates that DMSO was the best solvent-favorable environment for the reaction due to its ability to be difficult to evaporate and can provide good heat for the reaction. The volume of DMSO solvent was also evaluated for product formation, the volume value investigated included 0, 4, 5, and 6 mL of DMSO solvent. The best results were obtained using 5 mL of DMSO.
| Entry | Catalysts | Solvents | Volume of solvent (mL) | Yieldsb (%) |
|---|---|---|---|---|
| a Reaction condition: 1 (0.25 mmol, 99.29 mg), catalyst, and solvents.b Yields were isolated yields by crystallization in ethanol (10–15 mL).c IL1-SO3H = [SiO2(CH2)3Im(CH2)4SO3H]HSO4; BAIL gel = Brønsted-acidic ionic liquid gel.d H2SO4 (98.0 wt% in water): 13.0 μL. | ||||
| 1 | AC | DMSO | 5 | 34 |
| 2 | AC-SO3H | DMSO | 5 | 73 |
| 3 | IL1-SO3Hc | DMSO | 5 | 67 |
| 4 | BAIL gelc | DMSO | 5 | 64 |
| 5 | SiO2–H2SO4 | DMSO | 5 | 39 |
| 6 | TsOH | DMSO | 5 | 25 |
| 7 | H2SO4d | DMSO | 5 | 10 |
| 8 | None | DMSO | 5 | 0 |
| 9 | AC-SO3H | None | 0 | 0 |
| 10 | AC-SO3H | DMF | 5 | 48 |
| 11 | AC-SO3H | DMSO | 4 | 64 |
| 12 | AC-SO3H | DMSO | 6 | 58 |
The optimal conditions for synthesis, which were established to evaluate the role of the substrate in the synthesis of benzo[a]carbazoles, were 3-cyanoacetamide pyrroles (0.5 mmol), 6 mg of the AC-SO3H catalyst, the temperature at 240 °C, and DMSO (5 mL) for 2 h. The best result was shown in Scheme 3. By using (1) as the survey object, from the survey results, the isolated yield of forming (1a) reached the highest at 73%. When using (1) carrying electron-donating substituents at the papa position, namely –Me and –OMe groups, the major product was low yields 36% and 12%, respectively (Scheme 3, compounds 2a, 3a). However, when the electron-withdrawing substituent was present at the para position of (1), the reaction occurred better with over 44% yields, namely –Br, –Cl groups (Scheme 3, compounds 4a, 5a). The pyrrole substrate carries a 1H-benzo[d]imidazol-2-yl substituent, the reaction was difficult due to the resonance of the strong group, reducing the electron density on the carbonyl group of (1), leading to the yield of a major product as low as 23% (Scheme 3, compound 6a). The stereochemistry of the substrate, where the electrons of the –Me group were strongly attracted to the carbonyl group on the active substrate, explained why the electron-donating group at the ortho position generated a larger intramolecular cyclization yield than the non-substituent substrate (Scheme 3, compound 7a).
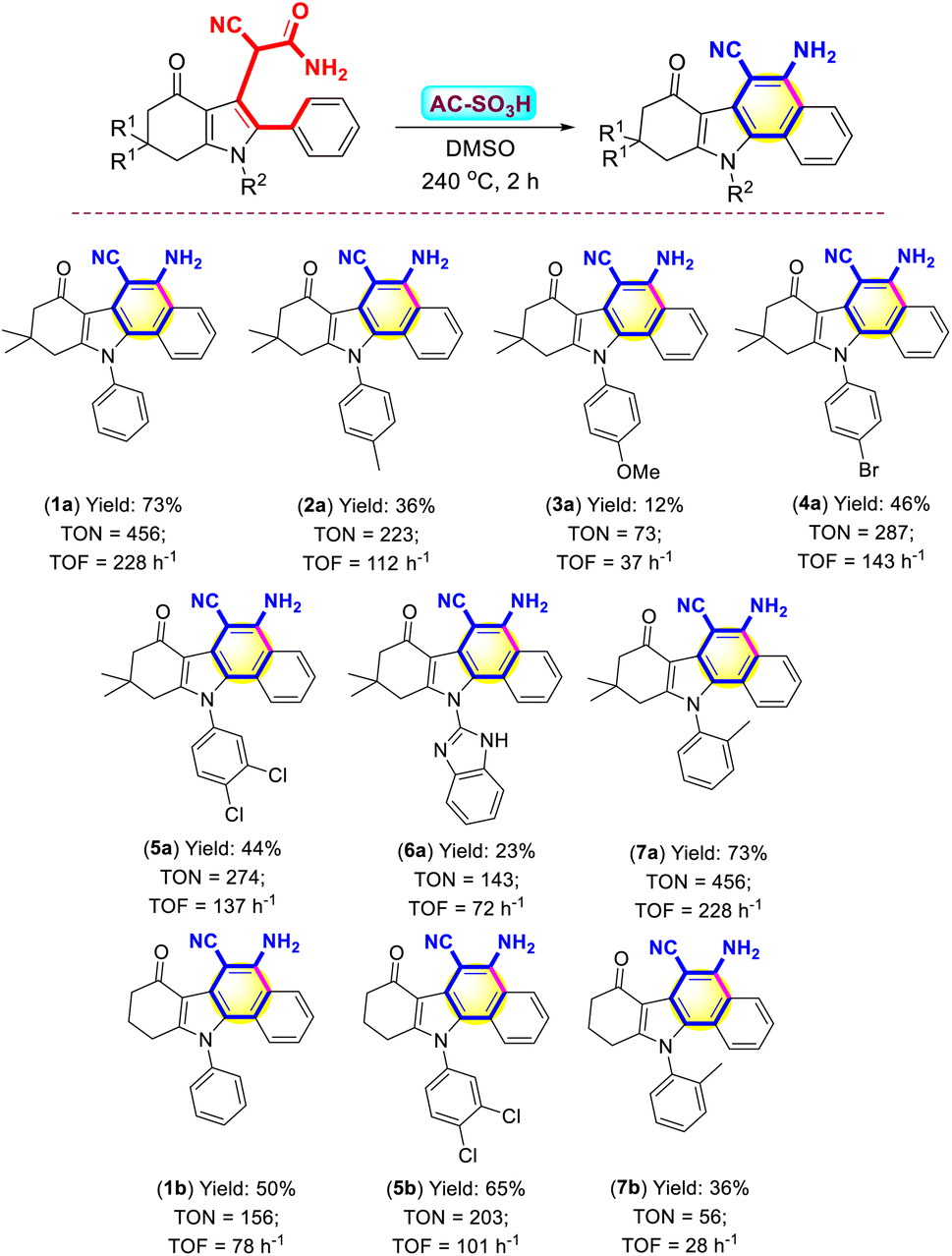 | ||
| Scheme 3 Synthesis of benzo[a]carbazole frames in the presence of AC-SO3H catalyst. Reaction condition: 3-cyanoacetamide pyrroles (0.25 mmol), AC-SO3H (6 mg) under DMSO (5 mL) at 240 °C for 2 hours. Yields were isolated yields by crystallization in ethanol (10–15 mL). TON = (moles of product)/(moles of catalyst); TOF = TON/hour (h−1). | ||
In Scheme 4, the proposed mechanistic pathway for the synthesis of benzo[a]carbazole derivative was shown. For the one-pot reaction of (1), the expected result was attained. 3-Cyanoacetamide pyrrole was hydrolyzed with AC-SO3H, and the resulting hydrolysis produced an intermediate termed azaniumylidene (A). Through the interaction of the azaniumylidene group with the H of ring B, the intermediate (B) was produced by the intramolecular cyclocondensation of intermediate (A). A water molecule was broken down in the reaction to create an intermediate (C). Then protonation to create the major product (1a).
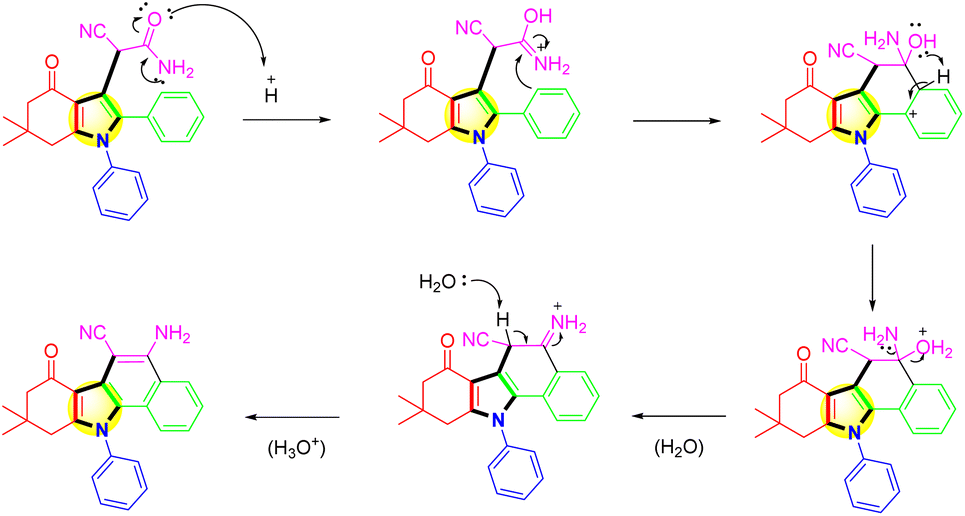 | ||
| Scheme 4 Mechanism of the intramolecular cyclization of 3-cyanoacetamide pyrrole over the AC-SO3H catalyst. | ||
The recyclability of AC-SO3H was evaluated in the intramolecular cyclization of 3-cyanoacetamide pyrrole at 240 °C for 2 hours in the presence of DMSO (Fig. 1). The finished product was separated with ethyl acetate, and the AC-SO3H was then isolated from the reaction mixture. The recovered catalyst dried for 2 hours at 100 °C and was utilized in the subsequent cycle of the model reaction. Even after five recycling experiments, under optimal conditions, the percentage conversion efficiency increased from 71% to 74%. The composition of the recovered catalyst was determined by SEM, EDX, and FTIR. As can be seen in Fig. S3,† the agglomeration structure of the AC-SO3H catalyst after recovery and reuse was still maintained. The collapsible structure of the AC-SO3H catalyst collapsed after being used three times, and the amount of element sulfur was drastically reduced. From the FTIR spectrum results, the functional group structure in the catalyst was still maintained (Fig. 2).
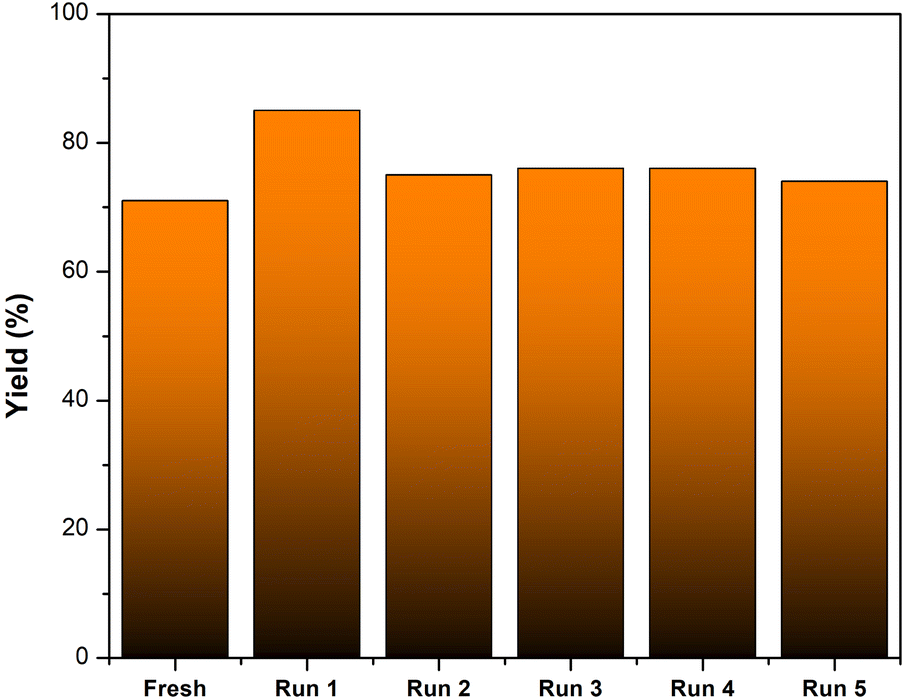 | ||
| Fig. 1 Recyclability of AC-SO3H in the synthesis of (1a). | ||
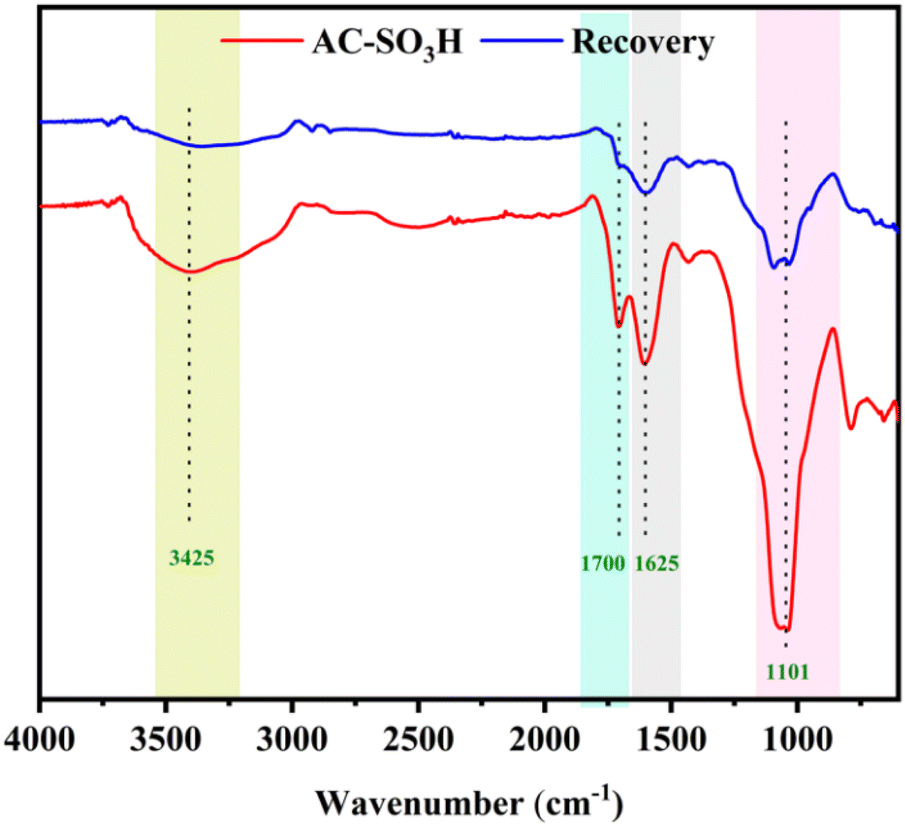 | ||
| Fig. 2 FT-IR spectrum of AC-SO3H (fresh) and AC-SO3H (after 3 times). | ||
Table 3 presents a comparison of the benzo[a]carbazole framework synthesis method of the study with other studies. The results show that the study's process uses a simpler chemical with a shorter reaction time. However, the reaction requires high temperature.
| Entry | Substrates | Conditions | Product | Yields (%) | Ref. |
|---|---|---|---|---|---|
| 1 |  |
PIFA |  |
43 | 49 |
| BF3·Et2O | |||||
| CH2Cl2 | |||||
| −40 °C | |||||
| 2 | 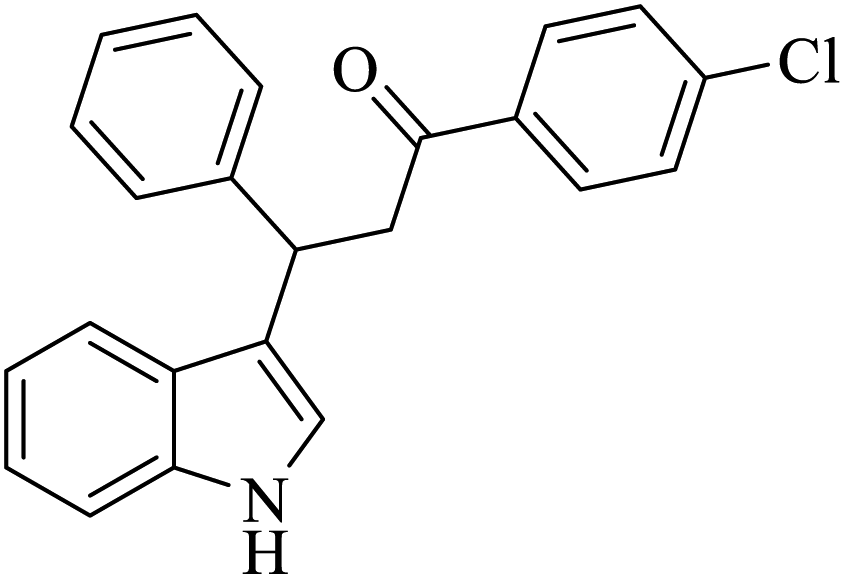 |
Benzoquinone (5 eq.) | 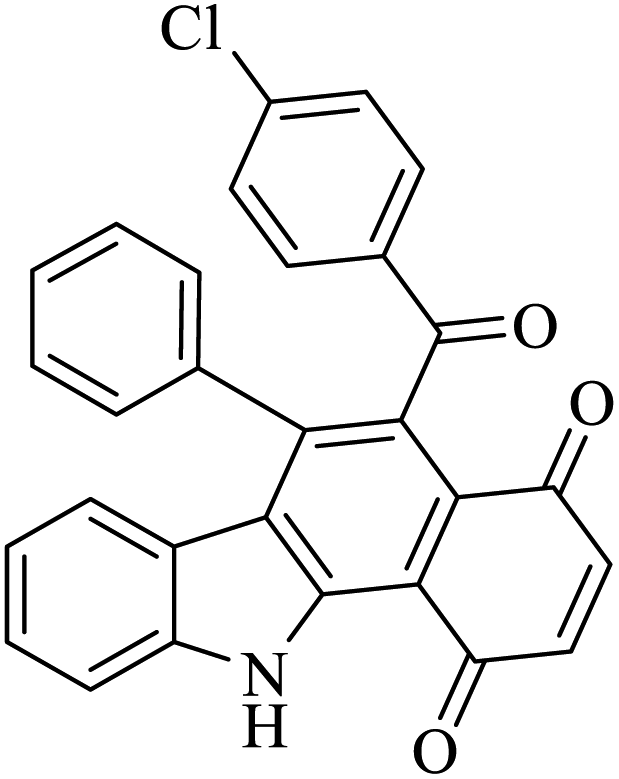 |
76 | 50 |
| Toluene/acid acetic (4°:°1, v/v) (5 mL) | |||||
| 100 °C, 8 h | |||||
| 3 | 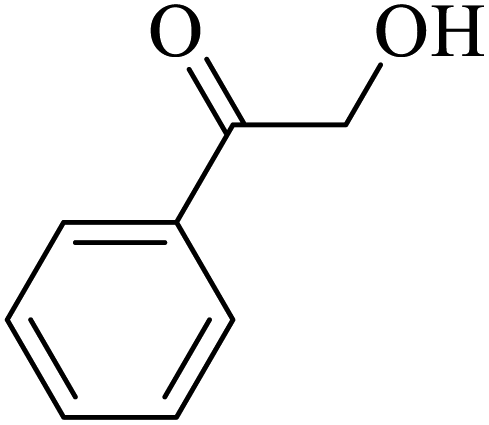 |
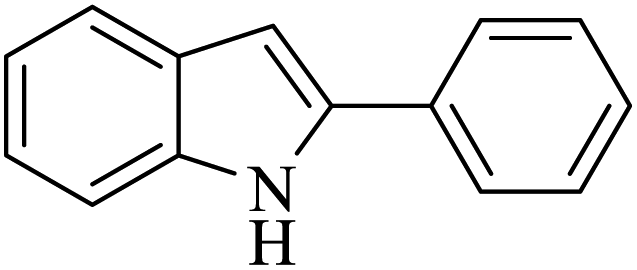 |
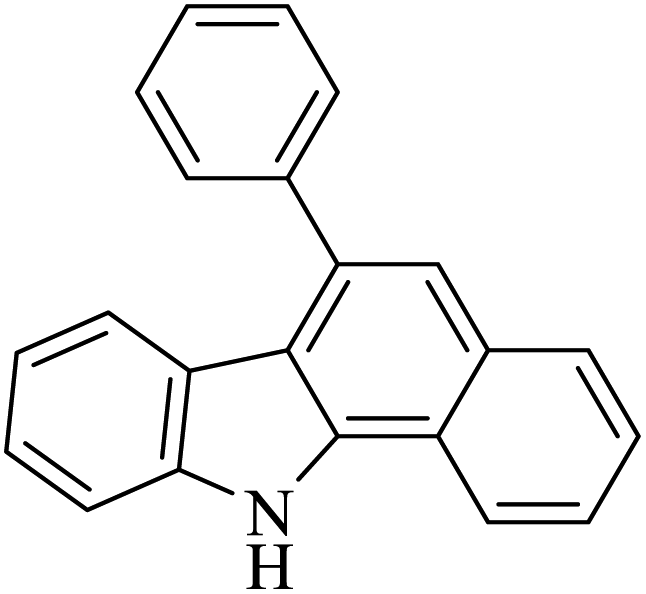 |
50 | 51 |
| TfOH (10% mol) | |||||
| EtNO2, ionic liquid | |||||
| H2O, 95 °C, 4 h | |||||
| 4 | 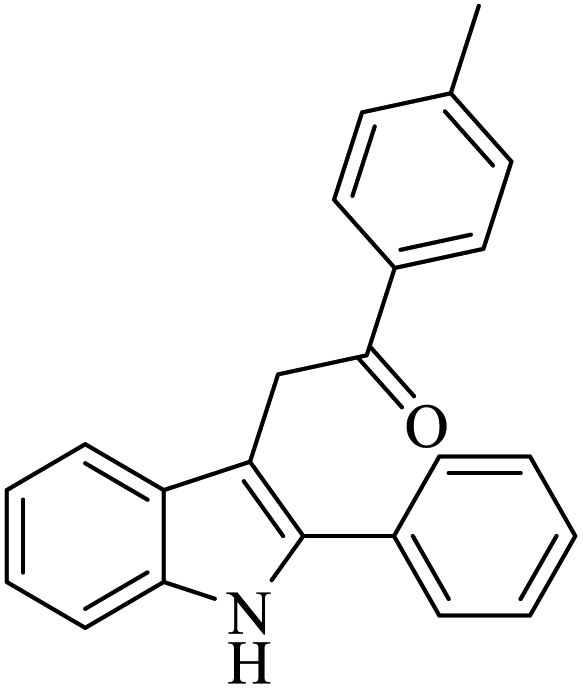 |
HBr/acid acetic (5% v/v) | 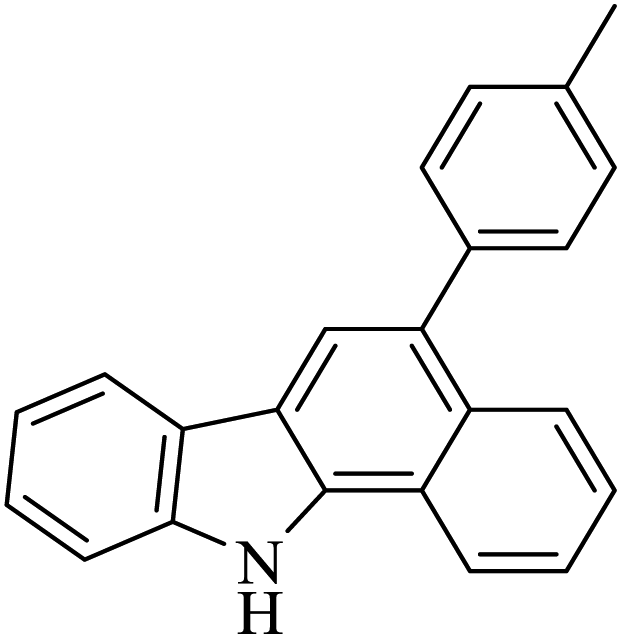 |
69 | 52 |
| Argon, 125 °C | |||||
| 5 | 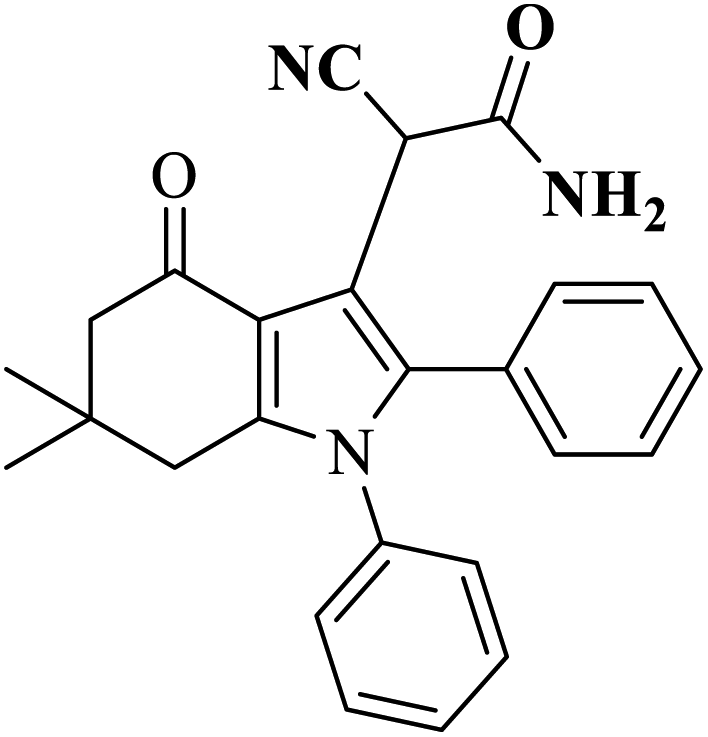 |
AC-SO3H (6 mg) | 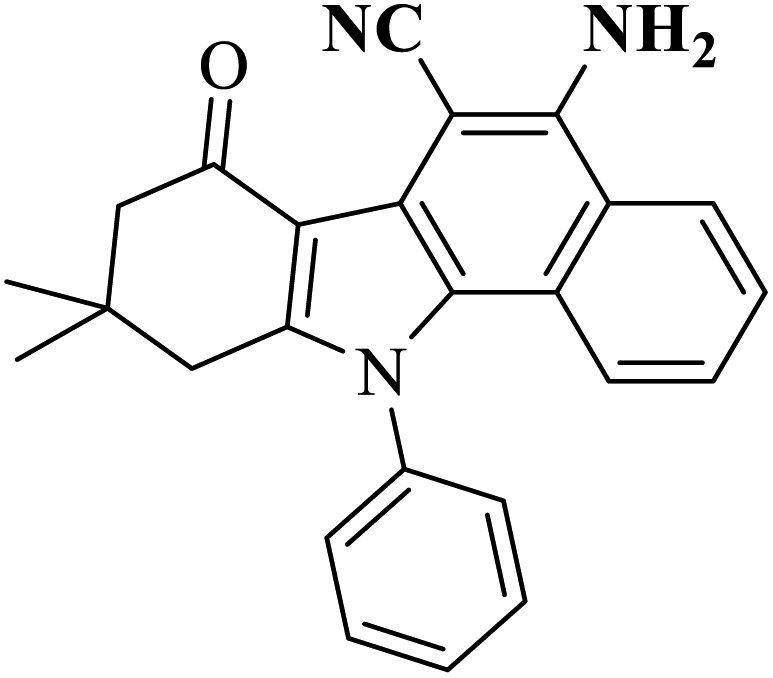 |
73 | Present work |
| DMSO (5 mL) | |||||
| 240 °C, 2 h |
Conclusions
In conclusion, we have developed an eco-friendly and effective catalyst that is made from rice husk. In N2 atmosphere, carbonization and sulfonation by sulfonic acid were conducted. Immobilizing the sulfonic acid functional group on the carbon framework improves the chemical properties of amorphous carbon compounds. The substance has an aggregated structure according to analytical procedures FT-IR, TGA, XRD, SEM, EDX, and the total acid concentration was found to be 4.606 mmol g−1 with elements C (65.56%), O (27.70%), Si (5.19%), and S (1.55%). Through the intramolecular cyclization reaction of 3-cyanoacetamide pyrrole in DMSO at 240 °C for 2 hours, the activity of the AC-SO3H catalyst was assessed. This study looked at the reaction conditions, and it found that 6 mg of AC-SO3H increased the intended product yield to 73%. After use, the catalyst was collected and repurposed five times with no structural changes. In addition, compounds containing the benzo[a]carbazole framework have many important biological potentials such as glucosidase enzyme inhibition and antibacterial properties.Author contributions
Hai Truong Nguyen: investigation, methodology, resources, formal analysis, validation, data curation, writing – original draft. Phat Ngoc Nguyen: investigation, methodology, resources, formal analysis, validation. Tan Van Le: investigation, methodology, resources, formal analysis, validation. Trinh Hao Nguyen: investigation, methodology, resources, formal analysis, validation. Linh Dieu Nguyen: investigation, methodology, resources, formal analysis, validation. Phuong Hoang Tran: methodology, resources, formal analysis, validation, data curation, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from Vietnam National University, Ho Chi Minh City (VNU-HCM) under grant number 562-2022-18-03.Notes and references
- H.-Y. Wang, F. Liu, L.-H. Xie, C. Tang, B. Peng, W. Huang and W. Wei, J. Phys. Chem. C, 2011, 115, 6961–6967 CrossRef CAS.
- O. D. Is, F. B. Koyuncu, S. Koyuncu and E. Ozdemir, Polymer, 2010, 51, 1663–1669 CrossRef CAS.
- E. M. Barea, C. Zafer, B. Gultekin, B. Aydin, S. Koyuncu, S. Icli, F. F. Santiago and J. Bisquert, J. Phys. Chem. C, 2010, 114, 19840–19848 CrossRef CAS.
- R. M. Adhikari, D. C. Neckers and B. K. Shah, J. Org. Chem., 2009, 74, 3341–3349 CrossRef CAS PubMed.
- G. Wang, S. Sun and H. Guo, Eur. J. Med. Chem., 2022, 229, 113999 CrossRef CAS PubMed.
- X. Zhu, Y. He, Z. Liu, Z. Zhu, Y. He, J. Qiu, D. Liu, M. Mo, P. Wang, X. Tian and P. Xu, Appl. Mater. Today, 2020, 19, 100559 CrossRef.
- Y. J. Xue, M. Y. Li, X. J. Jin, C. J. Zheng and H. R. Piao, J. Enzyme Inhib. Med. Chem., 2021, 36, 295–306 CAS.
- Y. Chen, N. Cao, H. Lv, K. Zeng, J. Yuan, X. Guo, M. Zhao, P. Tu and Y. Jiang, Phytochemistry, 2020, 170, 112186 CrossRef CAS PubMed.
- Y. Tachibana, H. Kikuzaki, N. H. Lajis and N. Nakatani, J. Agric. Food Chem., 2003, 51, 6461–6467 CrossRef CAS PubMed.
- J. A. Scatina, D. R. Hicks, M. Kraml and M. N. Cayen, Xenobiotica, 1989, 19, 991–1002 CrossRef CAS PubMed.
- S. Mandal, A. Nayak, M. Kar, S. K. Banerjee, A. Das, S. N. Upadhyay, R. K. Singh, A. Banerji and J. Banerji, Fitoterapia, 2010, 81, 72–74 CrossRef CAS PubMed.
- J. H. Burckhalter, V. C. Stephens and L. A. Hall, J. Am. Pharm. Assoc., 1950, 39, 271–273 CrossRef CAS PubMed.
- Y.-P. Liu, J.-M. Guo, Y.-Y. Liu, S. Hu, G. Yan, L. Qiang and Y.-H. Fu, J. Agric. Food Chem., 2019, 67, 5764–5771 CrossRef CAS PubMed.
- R. Birari, S. K. Roy, A. Singh and K. K. Bhutani, Nat. Prod. Commun., 2009, 4, 1089–1092 CrossRef CAS PubMed.
- H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4428 CrossRef PubMed.
- X. Tian, L. Song and A. S. K. Hashmi, Angew Chem. Int. Ed. Engl., 2020, 59, 12342–12346 CrossRef CAS PubMed.
- C. Bosch, P. López-Lledó, J. Bonjoch, B. Bradshaw, P. J. Nieuwland, D. Blanco-Ania and F. P. J. T. Rutjes, J. Flow Chem., 2016, 6, 240–243 CrossRef CAS.
- S. K. Bur and A. Padwa, Chem. Rev., 2004, 104, 2401–2432 CrossRef CAS PubMed.
- M. F. Martínez-Esperón, D. Rodríguez, L. Castedo and C. Saá, Tetrahedron, 2008, 64, 3674–3686 CrossRef.
- E. Barrera, R. I. Hernández-Benitez, C. A. González-González, C. H. Escalante, A. Fuentes-Benítes, C. González-Romero, E. Becerra-Martínez, F. Delgado and J. Tamariz, Eur. J. Org Chem., 2022, 2022, e202200364 CrossRef CAS.
- T. Mandal and J. Dash, Org. Biomol. Chem., 2021, 19, 9797–9808 RSC.
- K. Kirchner, M. J. Calhorda, R. Schmid and L. F. Veiros, J. Am. Chem. Soc., 2003, 125, 11721–11729 CrossRef CAS PubMed.
- A. Banerjee, S. Sahu and M. S. Maji, Adv. Synth. Catal., 2017, 359, 1860–1866 CrossRef CAS.
- A. F. Saber, S. U. Sharma, J.-T. Lee, A. F. M. El-Mahdy and S.-W. Kuo, Polymer, 2022, 254, 125070 CrossRef CAS.
- S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef CAS PubMed.
- W. C. P. Tsang, N. Zheng and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560–14561 CrossRef CAS PubMed.
- S. Maiti and P. Mal, Org. Lett., 2017, 19, 2454–2457 CrossRef CAS PubMed.
- S. Chen, Y. Li, P. Ni, H. Huang and G.-J. Deng, Org. Lett., 2016, 18, 5384–5387 CrossRef CAS PubMed.
- X. Liu, D. Du, S. Li, X. Wang, C. Xu and M. Wang, Adv. Synth. Catal., 2020, 362, 5135–5140 CrossRef CAS.
- P. Zhang, B. Li, L. Niu, L. Wang, G. Zhang, X. Jia, G. Zhang, S. Liu, L. Ma and W. Gao, Adv. Synth. Catal., 2020, 362, 2342–2347 CrossRef CAS.
- D. Cao, A. Ying, H. Mo, D. Chen, G. Chen, Z. Wang and J. Yang, J. Org. Chem., 2018, 83, 12568–12574 CrossRef CAS PubMed.
- S. H. Y. S. Abdullah, N. H. M. Hanapi, A. Azid, R. Umar, H. Juahir, H. Khatoon and A. Endut, Renewable Sustainable Energy Rev., 2017, 70, 1040–1051 CrossRef CAS.
- L. J. Konwar, P. Mäki-Arvela and J.-P. Mikkola, Chem. Rev., 2019, 119, 11576–11630 CrossRef CAS PubMed.
- Q. Xie, X. Yang, K. Xu, Z. Chen, B. Sarkar and X. Dou, Environ. Res., 2020, 188, 109887 CrossRef CAS PubMed.
- S. Ravi and S. Vadukumpully, J. Environ. Chem. Eng., 2016, 4, 835–856 CrossRef CAS.
- H. Jirimali, J. Singh, R. Boddula, J. K. Lee and V. Singh, Materials, 2022, 15, 3969 CrossRef CAS PubMed.
- N. Karić, A. S. Maia, A. Teodorović, N. Atanasova, G. Langergraber, G. Crini, A. R. L. Ribeiro and M. Đolić, Chem. Eng. J. Adv., 2022, 9, 100239 CrossRef.
- H. T. Nguyen, T. V. Le and P. H. Tran, J. Environ. Chem. Eng., 2021, 9, 105228 CrossRef CAS.
- H. T. Nguyen, M.-N. H. Truong, T. V. Le, N. T. Vo, H. D. Nguyen and P. H. Tran, ACS Omega, 2022, 7, 17432–17443 CrossRef CAS PubMed.
- N. Nagasundaram, M. Kokila, P. Sivaguru, R. Santhosh and A. Lalitha, Adv. Powder Technol., 2020, 31, 1516–1528 CrossRef CAS.
- S. Na, Z. Minhua, D. Xiuqin and W. Lingtao, RSC Adv., 2019, 9, 15941–15948 RSC.
- M. Hara, T. Yoshida, A. Takagaki, T. Takata, J. N. Kondo, S. Hayashi and K. Domen, Angew. Chem., Int. Ed., 2004, 43, 2955–2958 CrossRef CAS PubMed.
- P. V. Rathod and V. H. Jadhav, ACS Sustainable Chem. Eng., 2018, 6, 5766–5771 CrossRef CAS.
- K. M. Mena Aguilar, Y. Amano and M. Machida, J. Environ. Chem. Eng., 2016, 4, 4644–4652 CrossRef CAS.
- H. T. Nguyen, T. Van Le, P. N. Nguyen, K. H. Nguyen, L. H. T. Nguyen, T. Le Hoang Doan and P. H. Tran, New J. Chem., 2023, 47, 14733–14745 RSC.
- A. S. Amarasekara and O. S. Owereh, Catal. Commun., 2010, 11, 1072–1075 CrossRef CAS.
- P. H. Tran, X.-T. T. Nguyen and D.-K. N. Chau, Asian J. Org. Chem., 2018, 7, 232–239 CrossRef CAS.
- D. Kumar, M. Sonawane, B. Pujala, V. K. Jain, S. Bhagat and A. K. Chakraborti, Green Chem., 2013, 15, 2872–2884 RSC.
- N. J. Truax, F. Banales Mejia, D. O. Kwansare, M. M. Lafferty, M. H. Kean and E. T. Pelkey, J. Org. Chem., 2016, 81, 6808–6815 CrossRef CAS PubMed.
- C.-W. Kuo, A. Konala, L. Lin, T.-T. Chiang, C.-Y. Huang, T.-H. Yang, V. Kavala and C.-F. Yao, Chem. Commun., 2016, 52, 7870–7873 RSC.
- M. Li, F. Wu and Y. Gu, Chin. J. Catal., 2019, 40, 1135–1140 CrossRef CAS.
- J. Jeon and C.-H. Cheon, Org. Chem. Front., 2019, 6, 456–467 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04943d |
| This journal is © The Royal Society of Chemistry 2023 |