
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt-assisted route to rare carbocyclic C-ribonucleosides†
A. C. Ojeda-Porras,
V. Roy*,
O. Bourzikat,
P. Favetta and
L. A. Agrofoglio *
*
Université d'Orléans et CNRS, ICOA, UMR 7311, F-45067, Orléans, France. E-mail: luigi.agrofoglio@univ-orleans.fr
First published on 20th October 2023
Abstract
(Re)emerging RNA viruses have been major threats to public health in the past years, and from the few drugs available, nucleoside analogues are still at the cornerstone of the antiviral therapy. Among them, the synthesis of carbocyclic C-nucleosides is suffering from long syntheses and poor yields. Herein we report a concise stereoselective synthesis of rare carbocyclic C-nucleosides (11a–l) bearing non-canonical nucleobases through a cobalt-assisted-route as key step starting from the optically pure (−)-cyclopentenone 1. This approach paves the route for novel carbocyclic C-nucleoside discovery.
Introduction
The continuous growth of the human population as well as human interaction with wild environments have shown several emerging and re-emerging RNA viruses responsible of highly fatal viral diseases and threatened public health with deadly outbreaks of global concern,1–4 the majority of RNA viruses are zoonotic or vector-borne infectious agents5–7 and are considered as the most common class of pathogens (2 to 3 novel viruses discovered by year);8 they represent a challenge for global disease control9 because of their faster evolutionary rates and increased mutability compared to some DNA viruses.10 Except for the SARS-CoV-2 for which vaccines have been developed at an unprecedented speed, the biological diversity and rapid adaptive rates of RNA viruses make that no effective commercial vaccine is available for any of the above-mentioned viruses, and we are still awaiting the first effective antiviral drugs to be approved. Nucleoside analogues, with more than 40 derivatives marketed to treat viral DNA infections as well as certain cancers or bacterial infections,11 are the most promising and leading-class of antivirals against RNA emerging viruses.12 On one hand, the replacement of the oxygen in the furanose ring with methylene group led to carbocyclic nucleosides which are chemically more stable towards the enzymatic cleavage of the N-glycosidic bond and have an increased cell permeability because of its higher lipophilicity than their nucleoside parents. The broad spectrum of biological activities of carbocyclic nucleosides and their pharmaceutical importance have generated considerable research interest in their design and synthesis. Many reviews by us13 and others14 have been published on this topic, as well as on their biological applications.15 On the other hand, C-nucleosides,16 in which the ribofuranosyl moiety is linked to a heterocyclic non-canonical base (such as bearing hydrophobic aryl group)17 through a C–C bond instead of C–N-bond found in natural nucleosides, have been reported for their antiviral and/or anticancer potential. So far, no natural carbocyclic C-nucleosides have been found in nature and only a few have been synthesized to date (Fig. 1), such as the inhibitor of histone methyltransferase DOT1L recently developed by Paruch et al.,18 the carbocyclic formycin19 analog by Leahy et al., and the compound reported by Fletcher et al.20 bearing a difluoro pyridine non-canonical nucleobase.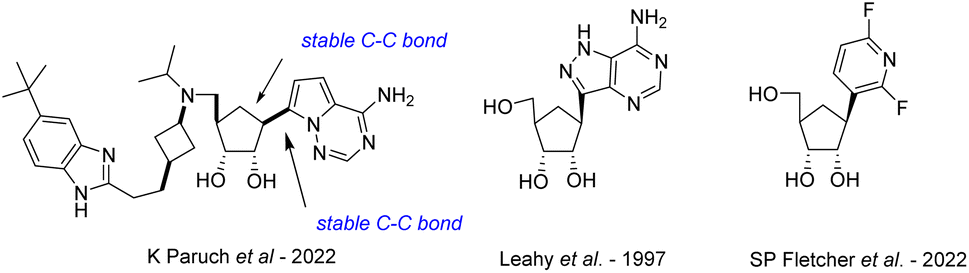 | ||
| Fig. 1 Structure of synthetic carbocyclic C-nucleosides. | ||
A first approach was reported in 2001 by Chu and co-workers21 from 1,4-γ-ribonolactone as chiral starting material (Scheme 1A). The key intermediate is obtained by Bu3SnH–AIBN reduction of an olefinic intermediate obtained by treating the protected ribonolactone with ethyl cyanoacetate and tBuOK and then it is converted to final carbocyclic C-nucleosides by the full construction of the pyrazole non-canonical nucleobases.
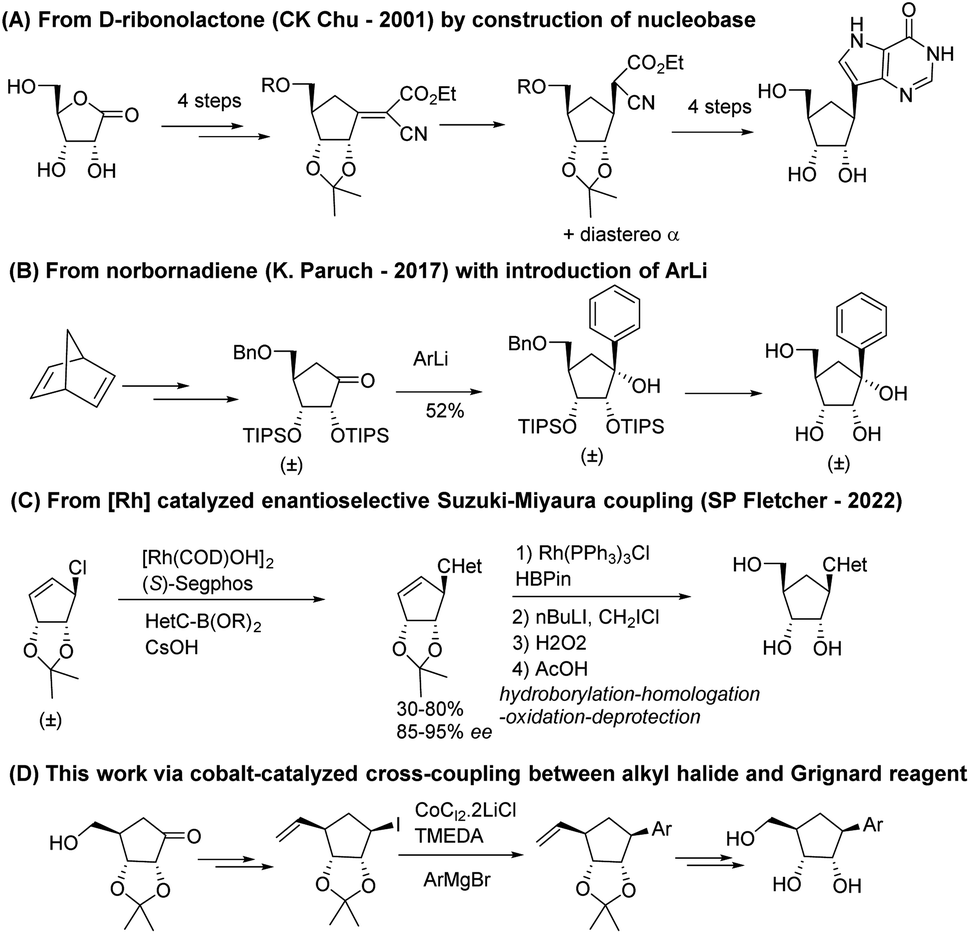 | ||
| Scheme 1 Comparison of previous syntheses and our synthesis of carbocyclic C-ribonucleosides. | ||
Paruch and co-workers developed a diastereoselective, racemic procedure from norbornadiene through the TIPS-protected cyclopentanone which underwent diastereoselective addition of various lithiated (hetero)aryl moieties (Scheme 1B).22 A recent publication by Fletcher group20 reported the Rh-catalytic asymmetric synthesis of carbocyclic C-nucleosides using an asymmetric Suzuki–Miyaura type reaction as the key C–C bond forming step, followed by late-stage addition of the hydroxymethyl group to ribose analogs (Scheme 1C).
Even if several efforts towards the synthesis of enantio-enriched carbocyclic nucleosides have been reported in the past years, the multi-step sequences combined with the difficulty associated to the installation of stereocenters make these approaches long, tedious, and not suitable for the pharmaceutical industry. Thus, the synthetic difficulties have probably hampered the production of those very promising carbocyclic C-nucleoside analogs.
Hence, we focused on designing a new, versatile, robust and stereo-controlled synthetic route to diverse carbocyclic C-nucleosides without the use chiral catalyst but taking advantage of stereocenters previously set in the molecule (Scheme 1D). Several non-canonical aromatic bases were installed.23 This (hetero)aryl moieties included electron-donating and electron-withdrawing substituents, and the presence of heteroatoms in the ring, while maintaining π-stacking and Watson–Crick hydrogen bond interactions.
Results and discussion
We began the synthesis from the commercially available optically pure (−)-cyclopentenone 1 which is easily obtained in 3 steps from D-ribose.24 Following a reported procedure by Schneller et al.,25a the diastereoselective Grignard addition of vinyl magnesium25b bromide on 1 afforded the ketone 2 as one diastereomer in 63% yield (Scheme 2). 1H NMR and NOESY experiments revealed that H3 correlates only with H2 (d, J(H3,H2) = 5.2 Hz) and not with H4 corresponding to a dihedral angle close to 90° according to Karplus equation. This angle is attainable with H3 and H4 trans to each other (but not when they are cis), proving that the vinyl substituent of the organocuprate added to the less hindered face resulting in 2 with the depicted stereochemistry, as illustrated by molecular models (see ESI†). The use of TBAF in the quenching of the Grignard addition step is vital since the yield of 2 can be negatively affected by the formation and isolation of silyl ether 2′. Reduction of resulting cyclopentanone 2 with AlLiH4 afforded alcohol 3 in 97% yield. The compound was diastereomerically pure according to 1H and 13C NMR. 1H NMR and NOESY analysis, and Karplus equation indicate that compound 3 has H1 and H2 in a cis relationship.25b This is in agreement with molecular models (see ESI†). With alcohol 3 in hand, an Appel-type reaction (proceeding with the inversion of configuration) using iodine and triphenylphosphine in the presence of imidazole was intended. This reaction was initially performed using dichloromethane as solvent, but unfortunately, just starting material was recovered. Finally when toluene was used as solvent and the reaction was done under reflux conditions, the desired iodohydrin 4 was obtained in 90% yield. Interestingly, 1H NMR clearly shows a van der Waals deshielding effect of the iodine atom on the vinylic C–H proton.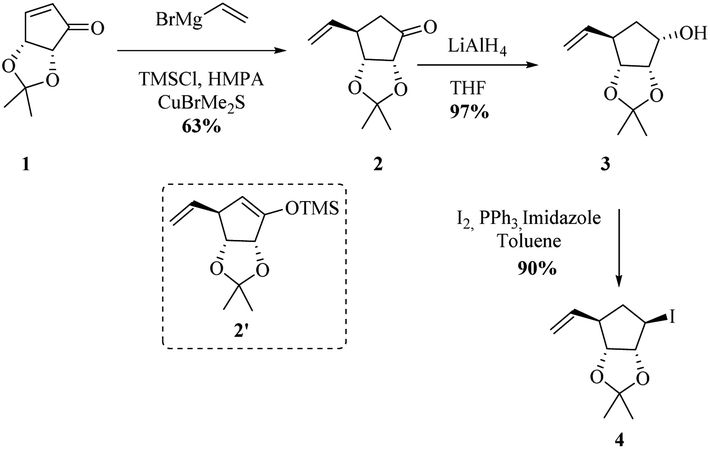 | ||
| Scheme 2 Synthesis of enantiomerically pure halohydrin 4. | ||
Inspired by the cobalt-mediated diastereoselective cross coupling reaction reported by Knochel and co-workers,26 we decided to apply this methodology as key step of the synthesis of carbocyclic C-nucleosides. According to the authors, the presence of the neighbouring TBS-ether group is responsible for the high diastereoselectivity observed because the silyl ether and the newly inserted aromatic group adopt an anti-configuration. We sought the isopropylidene motive at C2–C3 would have a similar effect by blocking the α-phase of the cyclopentane nucleoside. The cobalt solution can be prepared in a day using the protocol reported by Knochel. Before trying the conditions in the previously synthesized iodohydrin 4, we decided to test them on the model substrate 7 (Scheme 3). Starting from epoxide 5, iodohydrin 6 was obtained in 72% yield. Further protection of the alcohol as a TBS ether afforded the model substrate 7 in 84% yield as a racemate.
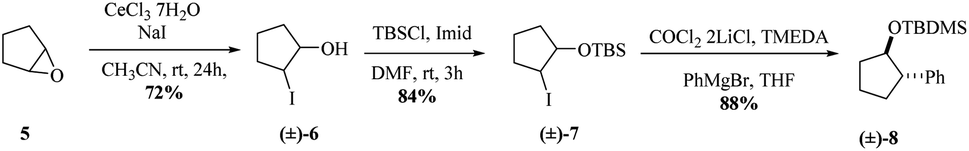 | ||
| Scheme 3 Cobalt-assisted cross-coupling on a model compound. | ||
With 7 in hand, we tested the cobalt-catalysed cross coupling conditions. Thus, to a cooled solution of the iodohydrin 7 and TMEDA in dry THF was slowly added the Grignard reagent. To our delight, we were able to isolate the desired compound 8 in 88% yield as only relative trans stereochemistry, when the reaction was allowed to stir at room temperature overnight. The aryl group was trans to the silylated ether. We did notice that shorter reaction times resulted in a decreased reaction yield.
We then applied the above-mentioned conditions for the cross-coupling reaction between halohydrin 4 and different Grignard reagents (Scheme 4). The desired products (9a–l) were obtained in good yields in the presence of activating groups, such as methoxide 9c and trifluoro-methoxide 9d. Fluorinated compound 9b was isolated in 68% yield. Ortho-substituted product 9e was successfully obtained in moderate yield despite its steric hindrance and the presence of a nitrogenated base.
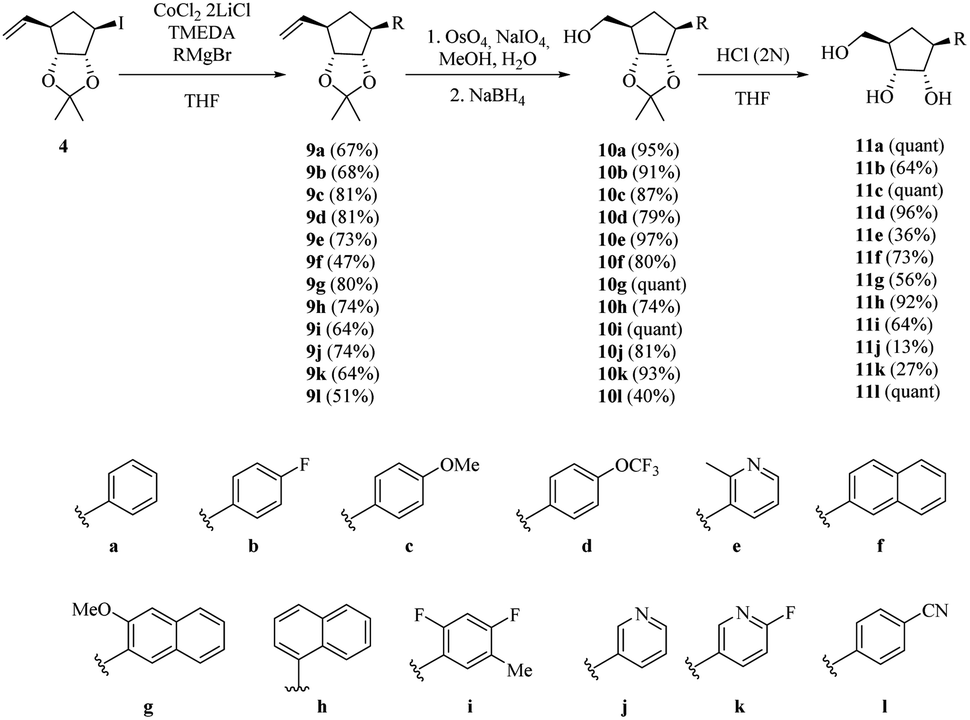 | ||
| Scheme 4 Synthesis of carbocyclic C-nucleosides 11a–l. | ||
Binaphthyl substituted products 9f–h were also were also isolated, although in poor to moderate yields. Only one isomer is produced and its formation proceeds through a proposed radical mechanism (Scheme 5).27
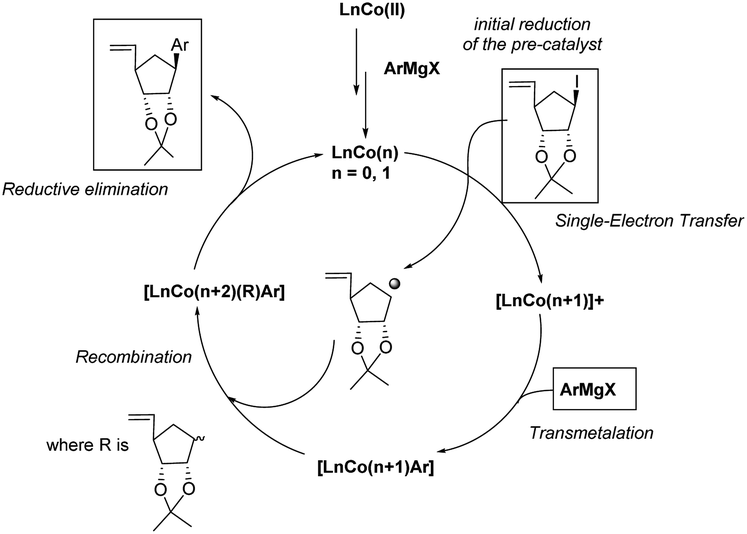 | ||
| Scheme 5 Proposed mechanism (adapted from Guérinot and Cossy27). | ||
With alkenes 9a–l in hand, an oxidative cleavage of the double bond was attempted. Initially, a ruthenium catalysed reaction was performed on 9a furnishing 10a in just 25% yield. To our delight, the use of osmium tetroxide afforded alcohols 10a–l in excellent yields (Scheme 4). Later deprotection of the isopropylidene ketal in acidic media yielded the desired carbocyclic C-nucleoside analogues 11a–l. It should be noticed that the 1H and 13C NMR spectra of 11a are consistent with those reported by Fletcher et al. for the same compound obtained by a different way.20
Conclusions
In summary, a series of hitherto unknown rare carbocyclic C-nucleosides (11a–l) were successfully synthesized in a highly efficient manner from protected ribonolactone 1 through a cobalt-catalyzed cross-coupling between the enantiopure halohydrine 4 and various aryl Grignard reagents. To the best of our knowledge, the proposed synthetic procedure is far superior to any other route reported to date. It is believed that this study will contribute to explore this promising class of carbocyclic C-nucleosides.Experimental section
General methods
Commercially available chemicals were of reagent grade and used as received. The reactions were monitored by thin layer chromatography (TLC) analysis using silica gel plates (Kieselgel 60F254, E. Merck). Column chromatography was performed on Silica Gel 60 M (0.040 × 0.063 mm, E. Merck). Optical rotations were measured with a PerkinElmer 341 polarimeter in appropriate solvent, at the indicated temperature (20 °C) and at 589 nm sodium line, in a 1 dm cell. Concentrations are given in g/100 mL. The 1H and 13C NMR spectra were recorded on a Varian InovaUnity 400 spectrometer (400 MHz) in (d4) methanol, CDCl3, shift values in parts per million relative to SiMe4 as internal reference. High Resolution Mass spectra were performed on a Bruker maXis mass spectrometer by the “Federation de Recherche” ICOA/CBM (FR2708) platform.Preparation of the CoCl2·2LiCl (1.0 M) solution in THF
The CoCl2·2LiCl (1.0 M) solution in THF was prepared following the reported experimental procedure.24 A dried two neck 50 mL flask equipped with a stirring bar a septum was charged with anhydrous LiCl (2.12 g) and heated at 130 °C under high vacuum for 2 h. After cooling to room temperature under argon, CoCl2 (3.24 g) was added. The resulting mixture was heated to 130 °C for 5 h under high vacuum, cooled down to room temperature under argon and charged with dry THF (25 mL). The resulting solution was vigorously stirred at room temperature overnight. The dark blue solution was used within 1 month.Synthesis of Grignard reagents
All non-commercially available aryl Grignard reagents were prepared following the following experimental procedure. To a flask containing a stirring bar was added Mg turnings (240 mg, 10 mmol, 2.0 equiv.) and LiCl (230 mg, 5.5 mmol, 1.1 equiv.). The flask was flame dried under vacuum and flushed with argon three times. THF (5 mL) and dibromoethane (25 μL) were then added, followed by the dropwise addition of 1-bromo-4-methoxybenzene (935 mg, 5 mmol). The resulting solution was stirred at room temperature for two hours and used immediately in the cross coupling reaction.General procedure A: cobalt mediated cross coupling reaction
To a stirred solution of iodohydrin (1.0 equiv.) in dry THF (1 M) was added TMEDA (0.3 equiv.) and a 1.0 M solution of CoCl2·2LiCl in THF (0.85 equiv.). The resulting blue solution was cooled down to −78 °C before the Grignard reagent (1.7 equiv.) was slowly added. The reaction mixture was stirred at room temperature overnight. A saturated aqueous solution of NH4Cl was added to quench the reaction. The aqueous layer was extracted with AcOEt, and the combined organic extracts were washed with brine, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The crude mixture was then purified by flash chromatography to afford the desired product.General procedure B: oxidative cleavage of double bond catalysed by osmium
To a stirred solution of alkene (1.0 equiv.) in a mixture of methanol: water (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) (0.11 M) was added NaIO4 (2.07 equiv.). The reaction mixture was cooled down to 0 °C and a 2.5 wt% solution of OsO4 in t-BuOH (0.08 equiv.) was slowly added. The resulting mixture was stirred 1 h at 0 °C and 2 h at room temperature. The newly formed white solid was removed by filtration and washed with a mixture of methanol water (2:1). The filtrate was then cooled down to 0 °C and NaBH4 (6.4 equiv.) was added portion wise, turning the reaction black. The resulting mixture was allowed to stir at room temperature for 30 minutes, after which the volatiles were removed under reduced pressure. Brine was then added and the aqueous layer was extracted with CH2Cl2. The combined organic extracts were washed with brine, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure afford the desired product.
:3) (0.11 M) was added NaIO4 (2.07 equiv.). The reaction mixture was cooled down to 0 °C and a 2.5 wt% solution of OsO4 in t-BuOH (0.08 equiv.) was slowly added. The resulting mixture was stirred 1 h at 0 °C and 2 h at room temperature. The newly formed white solid was removed by filtration and washed with a mixture of methanol water (2:1). The filtrate was then cooled down to 0 °C and NaBH4 (6.4 equiv.) was added portion wise, turning the reaction black. The resulting mixture was allowed to stir at room temperature for 30 minutes, after which the volatiles were removed under reduced pressure. Brine was then added and the aqueous layer was extracted with CH2Cl2. The combined organic extracts were washed with brine, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure afford the desired product.
General procedure C: deprotection of the isopropylidene acetal
To a stirred solution of isopropylidene acetal (1.0 equiv.) in THF (0.4 M) at 0 °C was added dropwise a 2.0 M aqueous solution of HCl (0.2 M final concentration). The reaction mixture was stirred at room temperature for 16 h. The resulting solution was neutralized by the addition of a saturated aqueous solution of NaHCO3. The aqueous layer was abundantly extracted with AcOEt, and the combined organic extracts were washed with brine, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure to afford the desired product.:AcOEt to afford the desired product 1 as a white solid (109 mg, 84% yield). 1H NMR (400 MHz, CDCl3) δ ppm: 7.57–7.63 (m, 1H), 6.21 (d, J = 5.9 Hz, 1H), 5.34–5.18 (m, 1H), 4.46 (d, J = 5.5 Hz, 1H), 1.41 (s, 6H). 13C NMR (100 MHz, CDCl3) δ ppm: 202.9, 159.5, 134.3, 115.6, 78.6, 76.5, 27.4, 26.2.:AcOEt (5:1) to afford the desired product 2 as a pale yellow oil (3.54 g, 65% yield).25a [α]20D = −95.1 (c = 1.2, CH3OH). 1H NMR (400 MHz, CDCl3) δ ppm: 5.83 (ddd, J = 17.2, 10.6, 6.5 Hz, 1H), 5.20–5.02 (m, 2H), 4.64 (d, J = 5.2 Hz, 1H), 4.20 (d, J = 5.2 Hz, 1H), 3.11 (t, J = 7.3 Hz, 1H), 2.83 (dd, J = 18.3, 8.6 Hz, 1H), 2.28 (d, J = 18.3 Hz, 1H), 1.44 (s, 3H), 1.35 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 213.2, 137.1, 116.4, 112.4, 81.4, 77.9, 39.8, 38.6, 26.9, 24.9.:CH2Cl2 (8:2) to (1:1) to afford the desired product 4 as a pale yellow solid (4.67 g, 86% yield). [α]20D = −2.7 (c = 0.60, CH3OH). 1H NMR (400 MHz, CDCl3) δ ppm: 5.97 (ddd, J = 17.2, 10.3, 6.9 Hz, 1H), 5.15–5.05 (m, 2H), 4.97–4.81 (m, 1H), 4.41 (dd, J = 6.7, 4.0 Hz, 1H), 4.11–3.98 (m, 1H), 2.77–2.57 (m, 2H), 2.05–2.13 (m, 1H), 1.50 (s, 3H), 1.30 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 138.1, 115.8, 113.2, 90.5, 85.4, 49.9, 43.2, 27.3, 24.9, 24.8. HRMS (ESI): calcd for C10H16IO2+ [M + H]+, 295.0189; found, 295.0190.:CH2Cl2 (7:3) to (1:1). [α]20D = −8.4 (c = 15.3, CH3OH). 1H NMR (400 MHz, CDCl3) δ ppm: 7.37–7.23 (m, 5H), 5.96 (ddd, J = 17.3, 10.3, 7.1 Hz, 1H), 5.28–5.10 (m, 2H), 4.61–4.57 (m, 1H), 4.49–4.43 (m, 1H), 3.27 (dt, J = 12.6, 6.3 Hz, 1H), 2.81 (td, J = 12.7, 6.3 Hz, 1H), 2.36 (dt, J = 12.7, 6.3 Hz, 1H), 1.87–1.76 (m, 1H), 1.61 (s, 3H), 1.35 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 142.3, 139.0, 128.5, 127.1, 126.5, 115.2, 113.4, 87.0, 85.3, 50.5, 49.5, 38.4, 27.7, 25.2. HRMS (ESI): calcd for C16H21O2+ [M + H]+, 245.1536; found, 245.1535.:CH2Cl2 (1:2). [α]20D = −22.9 (c = 5.10, CH3OH). 1H NMR (400 MHz, CDCl3) δ ppm: 7.29–7.23 (m, 2H), 6.99–7.06 (m, 2H), 6.02–5.87 (m, 1H), 5.20 (d, J = 17.2 Hz, 1H), 5.11 (d, J = 10.4 Hz, 1H), 4.54–4.40 (m, 2H), 3.31–3.16 (m, 1H), 2.81–2.76 (m, 1H), 2.36–2.31 (m, 1H), 1.83–1.69 (m, 1H), 1.60 (s, 3H), 1.34 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 161.6 (d, J = 245 Hz), 138.8, 137.9 (d, J = 3.03 Hz), 128.5 (d, J = 7.07 Hz), 115.3, 115.2 (d, J = 21.2), 113.5, 87.0, 85.2, 49.9, 49.3, 38.4, 27.6, 25.1. 19F NMR (376 MHz, CDCl3) δ ppm: −116.69. HRMS (ESI): calcd for C16H20FO2+ [M + H]+, 263.1442; found, 263.1443.:CH2Cl2 (6:4) to (4:6). 1H NMR (400 MHz, CDCl3) δ ppm: 7.19 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 5.92 (ddd, J = 17.3, 10.3, 7.1 Hz, 1H), 5.19–5.07 (m, 2H), 4.52–4.49 (m, 1H), 4.43–4.39 (m, 1H), 3.79 (s, 3H), 3.19 (dt, J = 12.6, 6.2 Hz, 1H), 2.81–2.69 (m, 1H), 2.35–2.25 (m, 1H), 1.79–1.71 (m, 1H), 1.57 (s, 3H), 1.31 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 158.3, 139.0, 134.4, 128.0, 115.1, 113.9, 113.3, 87.2, 85.3, 55.3, 49.7, 49.5, 38.6, 27.7, 25.1. HRMS (ESI): calcd for C17H23O3+ [M + H]+, 275.1645; found, 275.1642. [α]20D = 18.23 (c 1.0, CH2Cl2).:CH2Cl2 (7:3) to (1:1). [α]20D = −43.5 (c = 1.40, CH3OH). 1H NMR (400 MHz, CDCl3) δ ppm: 7.40–7.14 (m, 4H), 5.95 (ddd, J = 17.3, 10.3, 7.2 Hz, 1H), 5.20 (d, J = 17.8 Hz, 1H), 5.12 (d, J = 10.3 Hz, 1H), 4.44–4.58 (m, 2H), 3.25 (dt, J = 12.7, 6.3 Hz, 1H), 2.83–2.78 (m, 1H), 2.38–2.33 (m, 1H), 1.87–1.70 (m, 1H), 1.60 (s, 3H), 1.34 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 147.8 (q, J = 147 Hz), 141.0, 138.7, 128.4, 121.1, 120.5 (q, J = 258 Hz), 115.3, 113.6, 86.9, 85.2, 50.0, 49.3, 38.2, 27.6, 25.1. 19F NMR (376 MHz, CDCl3) δ ppm: −57.95. HRMS (ESI): calcd for C17H20F3O3+ [M + H]+, 329.1359; found, 329.1361.:AcOEt:Et3N (9:1:0.1) to (8:2:0.1). [α]20D = −3.7 (c = 5.30, CH3OH). 1H NMR (400 MHz, CDCl3) δ ppm: 8.35 (d, J = 4.6 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.14–7.07 (m, 1H), 5.91 (ddd, J = 17.1, 9.7, 7.4 Hz, 1H), 5.17 (d, J = 17.2 Hz, 1H), 5.09 (d, J = 10.3 Hz, 1H), 4.56–5.50 (m, 1H), 4.47–4.42 (m, 1H), 3.45 (dt, J = 12.3, 6.1 Hz, 1H), 2.84–2.78 (m, 1H), 2.65 (s, 3H), 2.29–2.21 (m, 1H), 1.79–1.67 (m, 1H), 1.57 (s, 3H), 1.29 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 157.2, 146.9, 138.6, 135.6, 133.3, 121.3, 115.4, 113.4, 86.8, 85.2, 49.6, 46.4, 38.2, 27.7, 25.2, 22.9. HRMS (ESI): calcd for C16H22NO2+ [M + H]+, 260.1645; found, 260.1649.:CH2Cl2 (7:3) to (3:7). [α]20D = −20.3 (c = 1.00, CH3OH). 1H NMR (400 MHz, CDCl3) δ ppm: 7.83–7.79 (m, 3H), 7.70 (s, 1H), 7.48–7.42 (m, 3H), 6.04–5.89 (m, 1H), 5.21 (d, J = 17.2 Hz, 1H), 5.11 (d, J = 10.3 Hz, 1H), 4.68–4.62 (m, 1H), 4.53–4.44 (m, J = 6.8 Hz, 1H), 3.41 (dt, J = 12.6, 6.2 Hz, 1H), 2.86–2.79 (m, 1H), 2.46–2.39 (m, 1H), 1.97–1.87 (m, 1H), 1.61 (s, 3H), 1.34 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 139.7, 139.0, 133.5, 132.4, 128.2, 127.7, 127.6, 126.1, 125.8, 125.5, 125.3, 115.3, 113.5, 86.9, 85.4, 50.7, 49.6, 38.4, 27.7, 25.2. HRMS (ESI): calcd for C20H23O2+ [M + H]+, 295.1693; found, 295.1686.:CH2Cl2 (6:4) to (4:6). 1H NMR (400 MHz, CDCl3) δ ppm: 7.78 (t, J = 8.1 Hz, 2H), 7.72 (s, 1H), 7.47 (t, J = 7.5 Hz, 1H), 7.39 (t, J = 7.4 Hz, 1H), 7.17 (s, 1H), 6.01 (ddd, J = 17.3, 10.2, 7.3 Hz, 1H), 5.25 (d, J = 17.2 Hz, 1H), 5.15 (d, J = 10.3 Hz, 1H), 4.97 (t, J = 5.9 Hz, 1H), 4.58 (t, J = 6.8 Hz, 1H), 3.98 (s, 3H), 3.68 (dt, J = 11.7, 5.8 Hz, 1H), 2.98–2.81 (m, 1H), 2.48–2.29 (m, 1H), 2.01 (q, J = 12.3 Hz, 1H), 1.67 (s, 3H), 1.41 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 156.2, 139.3, 133.6, 132.1, 128.8, 127.6, 127.3, 126.3, 125.9, 123.8, 115.0, 112.8, 105.6, 85.7, 85.4, 55.3, 50.4, 47.0, 38.4, 28.0, 25.5. HRMS (ESI): calcd for C21H25O3+ [M + H]+, 325.1798; found, 325.1804.:CH2Cl2 (7:3) to (1:1). 1H NMR (400 MHz, CDCl3) δ ppm: 8.40 (d, J = 8.5 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.66–7.46 (m, 4H), 6.03 (ddd, J = 17.3, 10.3, 7.1 Hz, 1H), 5.29 (d, J = 17.2 Hz, 1H), 5.19 (d, J = 10.3 Hz, 1H), 4.84–4.72 (m, 1H), 4.57 (t, J = 6.8 Hz, 1H), 4.20–4.05 (m, 1H), 3.10–2.89 (m, 1H), 2.56–2.38 (m, 1H), 2.10 (q, J = 12.1 Hz, 1H), 1.74 (s, 3H), 1.39 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 139.1, 138.4, 134.1, 132.3, 128.8, 127.3, 126.1, 125.7, 125.4, 124.2, 122.9, 115.4, 113.1, 86.7, 85.4, 49.9, 46.1, 38.2, 27.9, 25.3. HRMS (ESI): calcd for C20H23O2+ [M + H]+, 295.1693; found, 295.1694.:CH2Cl2 (8:2) to (6:4). 1H NMR (400 MHz, CDCl3) δ ppm: 7.05 (t, J = 8.4 Hz, 1H), 6.76 (t, J = 10.0 Hz, 1H), 5.93 (ddd, J = 17.3, 10.3, 7.1 Hz, 1H), 5.18 (d, J = 17.2 Hz, 1H), 5.10 (d, J = 10.3 Hz, 1H), 4.69 (t, J = 6.5 Hz, 1H), 4.47 (t, J = 6.8 Hz, 1H), 3.33 (dt, J = 12.5, 6.3 Hz, 1H), 2.78 (dq, J = 13.0, 6.6 Hz, 1H), 2.35–2.17 (m, 4H), 1.83 (q, J = 12.6 Hz, 1H), 1.58 (s, 3H), 1.33 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 160.6 (dd, J = 60.9, 11.8 Hz), 158.2 (dd, J = 60.4, 11.8 Hz), 138.7, 130.9 (t, J = 6.4 Hz), 124.2 (dd, J = 14.1, 4.1 Hz), 120.4 (dd, J = 17.1, 3.9 Hz), 115.3, 113.3, 103.6 (t, J = 26.3 Hz), 85.6 (d, J = 1.3 Hz), 85.2, 49.7, 45.3, 38.2 (d, J = 2.2 Hz), 27.7, 25.2, 13.9 (d, J = 3.0 Hz). 19F NMR (376 MHz, CDCl3) δ ppm: −116.16 to −116.38 (m), −117.60 (dd, J = 16.9, 8.4 Hz). HRMS (ESI): calcd for C17H21F2O2+ [M + H]+, 295.1504; found, 295.1505.:AcOEt:Et3N (9:1:0.1) to (8:2:0.1). 1H NMR (400 MHz, CDCl3) δ ppm: 8.66–8.38 (m, 2H), 7.58 (d, J = 7.8 Hz, 1H), 7.31–7.12 (m, 1H), 5.91 (ddd, J = 17.3, 10.2, 7.1 Hz, 1H), 5.13 (dd, J = 33.0, 13.8 Hz, 2H), 4.53 (t, J = 6.9 Hz, 1H), 4.44 (t, J = 6.7 Hz, 1H), 3.22 (dt, J = 12.7, 6.3 Hz, 1H), 2.89–2.70 (m, 1H), 2.45–2.25 (m, 1H), 1.77 (q, J = 12.7 Hz, 1H), 1.56 (s, 3H), 1.31 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 148.8, 148.0, 138.5, 137.5, 134.6, 123.4, 115.4, 113.7, 86.5, 85.2, 49.3, 48.3, 38.0, 27.6, 25.1. HRMS (ESI): calcd for C15H20NO2+ [M + H]+, 246.1489; found, 246.1492.:AcOEt:Et3N (50:5:0.5) 1H NMR (400 MHz, CDCl3) δ ppm: 8.11 (s, 1H), 7.71 (td, J = 8.3, 1.8 Hz, 1H), 6.89 (dd, J = 8.3, 2.3 Hz, 1H), 5.92 (ddd, J = 17.3, 10.1, 7.2 Hz, 1H), 5.19 (d, J = 17.2 Hz, 1H), 5.11 (d, J = 10.3 Hz, 1H), 4.55–4.40 (m, 2H), 3.31–3.14 (m, 1H), 2.81 (td, J = 12.0, 6.0 Hz, 1H), 2.36 (dt, J = 12.6, 6.2 Hz, 1H), 1.76 (q, J = 12.6 Hz, 1H), 1.58 (s, 3H), 1.32 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 162.7 (d, J = 237.9 Hz), 145.9 (d, J = 14.5 Hz), 139.9 (d, J = 7.8 Hz), 138.4, 135.2 (d, J = 4.5 Hz), 115.6, 113.8, 109.2 (d, J = 37.5 Hz), 86.6, 85.1, 49.2, 47.5, 37.9, 27.6, 25.1. 19F NMR (376 MHz, CDCl3) δ ppm: −71.17. HRMS (ESI): calcd for C15H19FNO2+ [M + H]+, 264.1394; found, 264.1394.:CH2Cl2 (6:4) to (0:1). 1H NMR (400 MHz, CDCl3) δ ppm: 7.63 (d, J = 8.3 Hz, 2H), 7.41 (d, J = 8.1 Hz, 2H), 6.01–5.84 (m, 1H), 5.24–5.10 (m, 2H), 4.50 (dt, J = 13.3, 7.3 Hz, 2H), 3.29 (dt, J = 12.7, 6.3 Hz, 1H), 2.82 (td, J = 12.5, 6.3 Hz, 1H), 2.37 (dt, J = 12.7, 6.3 Hz, 1H), 1.79 (q, J = 12.7 Hz, 1H), 1.59 (s, 3H), 1.34 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 147.9, 138.6, 132.4, 128.1, 119.0, 115.7, 113.9, 110.6, 86.6, 85.3, 50.8, 49.3, 37.9, 27.7, 25.2. HRMS (ESI): calcd for C17H20NO2+ [M + H]+, 270.1489; found, 264.1491.:MeOH (50:5) to (45:5). [α]20D = −70.4 (c = 1.00, CH3OH). 1H NMR (400 MHz, MeOD) δ ppm: 7.80–7.63 (m, 3H), 7.36 (t, J = 7.5 Hz, 1H), 7.28 (t, J = 7.5 Hz, 1H), 7.18 (s, 1H), 4.29 (dd, J = 7.5, 5.7 Hz, 1H), 4.02 (t, J = 5.0 Hz, 1H), 3.90 (s, 3H), 3.75–3.51 (m, 3H), 2.36–2.16 (m, 2H), 1.44 (dt, J = 10.4, 8.0 Hz, 1H). 13C NMR (100 MHz, MeOD) δ ppm: 156.6, 133.5, 132.8, 129.0, 126.9, 126.3, 126.0, 125.3, 123.2, 104.9, 76.3, 74.2, 63.7, 54.4, 46.7, 44.8, 31.0. HRMS (ESI): calcd for C17H21O4+ [M + H]+, 289.1434; found, 289.1432.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by European funding in Region Centre-Val de Loire (FEDER EURO-FéRI EX010351), as well as by GAVO project (MERSI-CNRS). General functioning of ICOA comes from CHemBio (FEDER-FSE 2014-2020-EX003677), Techsab (FEDER-FSE 2014-2020-EX011313), RTR Motivhealth (2019-00131403) and Labex programs SYNORG (ANR-11-LABX-0029) and IRON (ANR-11-LABX-0018-01).References
- S. Roychoudhury, A. Das, P. Sengupta, S. Dutta, S. Roychoudhury, A. P. Choudhury, A. B. F. Ahmed, S. Bhattacharjee and P. Slama, Int. J. Environ. Res. Public Health, 2020, 15, 9411 CrossRef PubMed.
- WHO, Report of the WHO/FAO/OIE joint consultation on emerging zoonotic diseases, World Health Organization, 2004 Search PubMed.
- S. T. Nichol, J. Arikawa and Y. Kawaoka, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12411–12412 CrossRef CAS PubMed.
- Y. K. Choi, Exp. Mol. Med., 2021, 53, 711–712 CrossRef CAS PubMed.
- M. E. J. Woolhouse, K. Adair and L. Brierley, Microbiol. Spectrum, 2013, 1, 1 CAS.
- L. Wang, Y. Wang, D. Ye and Q. Liui, Int. J. Antimicrob. Agents, 2020, 55, 105948 CrossRef CAS PubMed.
- A. J. Bennett, T. Bushmaker, K. Cameron, A. Ondzie, F. R. Niama, H. J. Parra, J. V. Mombouli, S. H. Olson, V. J. Munster and T. L. Goldberg, Virology, 2019, 528, 64–72 CrossRef CAS PubMed.
- R. Rosenberg, Cell. Mol. Life Sci., 2015, 72, 1115–1125 CrossRef CAS PubMed.
- R. Carrasco-Hernandez, R. Jacome, Y. Lopez Vidal and S. Ponce de Leon, ILAR J., 2017, 58, 343–358 CrossRef CAS PubMed.
- S. Duffy, L. Schackelton and E. C. Holmes, Nat. Rev. Genet., 2008, 9, 267–276 CrossRef CAS PubMed.
- M. Slusarczyk, M. Serpi and F. Pertusati, Antiviral Chem. Chemother., 2018, 26, 2040206618775243 CAS.
- V. Roy and L. A. Agrofoglio, Drug Discovery Today, 2022, 27, 1945–1953 CrossRef CAS PubMed.
- (a) L. A. Agrofoglio and S. R. Challand, in Acyclic, Carbocyclic and L-Nucleosides, Kluwer Academic Publishers, Dordrecht, 1998 CrossRef; (b) M. Bessières, F. Chevrier, V. Roy and L. A. Agrofoglio, Future Med. Chem., 2015, 7, 1809–1828 CrossRef PubMed; (c) A. Mieczkowski and L. A. Agrofoglio, in Modified Nucleosides in Biochemistry, Biotechnology and Medecine, P. Herdewijn, Wiley-VCH GmbH & Co. KgaA, Weinheim. 2008, pp. 393–424 Search PubMed; (d) L. A. Agrofoglio, E. Suhas, A. Farese, R. Condom, S. R. Challand, R. A. Earl and R. Guedj, Tetrahedron, 1994, 50, 10611–10670 CrossRef CAS; (e) A. C. Ojeda-Porras, V. Roy and L. A. Agrofoglio, Chem. Rec., 2022, 22, e202100307 CrossRef CAS PubMed.
- O. Boutureira, M. I. Mathieu, Y. Diaz and S. Castillon, Chem. Soc. Rev., 2013, 42, 5056–5072 RSC.
- (a) R. K. Rawal, J. Bariwal and V. Singh, Curr. Top. Med. Chem., 2016, 16, 3258–3273 CrossRef CAS PubMed; (b) G. J. Yuen, S. Weller and G. E. Pakes, Clin. Pharmacokinet., 2008, 47, 351–371 CrossRef CAS PubMed; (c) M. Lu, Q. B. Lu and J. F. Honek, Bioorg. Med. Chem. Lett., 2017, 27, 282–287 CrossRef CAS PubMed; (d) L. S. Jeong and J. A. Lee, Antiviral Chem. Chemother., 2004, 15, 235–250 CrossRef CAS PubMed.
- (a) J. Štambaský, M. Hocek and P. Kočovský, Chem. Rev., 2009, 109, 6729–6764 CrossRef PubMed; (b) K. Temburnikar and K. L. Seley-Radtke, Beilstein J. Org. Chem., 2018, 14, 772–785 CrossRef CAS PubMed.
- (a) L. Wang and P. G. Schultz, Chem. Commun., 2002, 1–11 RSC; (b) A. A. Henry and F. E. Romesberg, Curr. Opin. Chem. Biol., 2003, 7, 727–733 CrossRef CAS PubMed; (c) E. T. Kool, J. C. Morales and K. M. Guckian, Angew. Chem., Int. Ed., 2000, 39, 990–1009 CrossRef CAS; (d) E. T. Kool, Acc. Chem. Res., 2002, 35, 936–943 CrossRef CAS PubMed; (e) M. Stefko, R. Pohl, B. Klepetarova and M. Hocek, Eur. J. Org Chem., 2008, 1689–1704 CrossRef CAS.
- P. Khirsariya, P. Pospíšil, L. Maier, M. Boudný, M. Babáš, O. Kroutil, M. Mráz, R. Vácha and K. Paruch, J. Med. Chem., 2022, 65, 5701–5723 CrossRef CAS PubMed.
- S. J. Boyer and J. W. Leahy, J. Org. Chem., 1997, 62, 3976–3980 CrossRef CAS.
- S. Mishra, F. C. T. Modicom, C. L. Dean and S. P. Fletcher, Commun. Chem., 2022, 5, 154 CrossRef CAS PubMed.
- B. K. Chun, G. Y. Song and C. K. Chu, J. Org. Chem., 2001, 66, 4852–4858 CrossRef CAS PubMed.
- L. Maier, P. Khirsariya, O. Hylse, S. K. Adla, L. Cernova, M. Poljak, S. Krajcovicova, E. Weis, S. Drapela, K. Soucek and K. Paruch, J. Org. Chem., 2017, 82, 3382–3402 CrossRef CAS PubMed.
- (a) T. Carell, C. Brandmayr, A. Hienzsch, M. Muller, D. Pearson, V. Reiter, I. Thoma, P. Thumbs and M. Wagner, Angew. Chem., Int. Ed., 2012, 51, 7110–7131 CrossRef CAS PubMed; (b) A. Marx and K. Betz, Chem.–Eur. J., 2020, 26, 3446–3463 CrossRef CAS PubMed; (c) G. T. Hwang, A. M. Leconte and F. E. Romesberg, ChemBioChem, 2007, 8, 1606–1611 CrossRef CAS PubMed; (d) M. Stefko, L. Slavetinska, B. Klepetarova and M. Hocek, J. Org. Chem., 2011, 76, 6619–6635 CrossRef CAS PubMed.
- (a) S. M. Ali, K. Ramesh and R. T. Borchardt, Tetrahedron Lett., 1990, 31, 1509–1512 CrossRef CAS; (b) M. S. Wolfe, B. L. Anderson, D. R. Borcherding and R. T. Borchardt, J. Org. Chem., 1990, 55, 4712–4717 CrossRef CAS.
- (a) M. Yang, W. Ye and S. W. Schneller, J. Org. Chem., 2004, 69, 3993–3996 CrossRef CAS PubMed; (b) C. R. Johnson and Y. F. Chen, J. Org. Chem., 1991, 56, 3344–3351 CrossRef CAS.
- J. M. Hammann, A. K. Steib and P. Knochel, Org. Lett., 2014, 16(24), 6500–6503 CrossRef CAS PubMed.
- A. Guérinot and J. Cossy, Acc. Chem. Res., 2020, 53, 1351–1363 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C data of selected compounds. See DOI: https://doi.org/10.1039/d3ra04937j |
| This journal is © The Royal Society of Chemistry 2023 |