
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-terminal conjugation of small interfering RNAs with spermine improves duplex binding and serum stability with position-specific incorporation†
Keisuke Koyasua,
Akash Chandelab and
Yoshihito Ueno*abc
aDepartment of Life Science and Chemistry, Graduate School of Natural Science and Technology, Gifu University, Japan. E-mail: ueno.yoshihito.e7@f.gifu-u.ac.jp; Fax: +81-58-293-2919; Tel: +81-58-293-2919
bCourse of Applied Life Science, Faculty of Applied Biological Sciences, Gifu University, Japan
cUnited Graduate School of Agricultural Science, Gifu University, 1-1 Yanagido, Gifu, 501-1193, Japan
First published on 23rd August 2023
Abstract
The conjugation of small interfering RNAs (siRNAs) has been studied using lipid and ligand conjugates for efficient delivery. However, most conjugates have been inserted at the terminal position; very few have been inserted at non-terminal positions. Herein, we synthesized a 4′-C-propyllevulinate-2′-O-methyluridine analog for non-terminal conjugation of spermine into the passenger strand of siRNA. Solid-phase oligonucleotide synthesis using this analog was successful, with the conjugation of one or two spermine molecules. The siRNAs conjugated with spermine displayed improved thermodynamic stability and resistance against nucleases, which depended on the site of conjugation in each case. Circular dichroism spectroscopy revealed that the A-type helical structure of the RNA duplex was not altered by these modifications. However, the gene-silencing activity of conjugated siRNAs was reduced and further decreased when the number of spermine molecules was increased. Hence, this work supplies valuable information and provides scope for the further development of drug-delivery systems through non-terminal conjugation.
Introduction
Post-transcriptional gene silencing can be administered with short, double-stranded RNAs (viz. small interfering RNAs, or siRNAs), which target complementary mRNA, leading to its degradation in a sequence-specific manner.1,2 This suppression of gene expression is an enzyme-mediated process known as RNA interference (RNAi).3,4 In a typical process, upon cell internalization, the siRNAs are recognized by the Argonaute 2 protein, forming an RNA-induced silencing complex (RISC), which, upon activation, is guided towards the cleavage of the target mRNA.5,6 This technological tool of RNAi is helpful to develop therapeutics for diseases that lack effective drug treatments. However, the administration of naked siRNAs as drugs is challenging because they are vulnerable to hydrolytic degradation by extracellular and intracellular nucleases and have poor cell permeability owing to their negatively charged phosphodiester backbone, which limits their pharmaceutical applications.7–9Despite these limitations, constant advances in this field have led to the approval of five siRNA drugs by the Food and Drug Administration (FDA). Four of these drugs were conjugated to the N-acetylgalactosamine (GalNAc) ligand, which specifically binds to the asialoglycoprotein receptor (ASGPR) expressed on the liver surface.10–13 Additionally, conventional modifications with 2′-methoxy or fluoro groups have been introduced to improve the stability of siRNA.14–17 Despite these developments, targeted delivery is only possible using these ligand-conjugated siRNA systems.18–22 Therefore, there is scope for the exploitation of drug-delivery systems to enhance the expansion of cell targets, which is critical for the development of oligonucleotide therapeutics.
Numerous delivery systems for the efficient transport of synthetic siRNAs have been developed; however, the search for potential candidates is ongoing.23–27 From this background research, we previously conjugated cRGD, a cyclic peptide that specifically binds to tumor cells, and spermine, a cationic polyamine, at the terminal positions of siRNA.28 The results were promising for gene silencing in a carrier-free system. However, most of the previous studies reported the conjugation at terminal position while some including lipid conjugation at non-terminal positions, to obtain effective gene silencing in carrier-free mode.29–33 Therefore, we envisioned spermine modification at the non-terminal position by conjugation with a linker at the 4′-C position of the modified nucleoside. We hypothesized that such a conjugated system would not alter the overall physical structure of the duplex and would exhibit conserved gene silencing with improved stability.
To test our hypothesis, we designed and synthesized a nucleoside analog, 4′-C-propyllevulinate-2′-O-methyluridine (4′-PLev-U), with a linker at 4′-C and an orthogonal protecting group to introduce spermine at the non-terminal position after oligonucleotide synthesis using a solid support. A schematic of the strategy used in this study is presented in Fig. 1, it depicts the reverse-tracking from the conjugated oligonucleotide to identify the target modified nucleoside phosphoramidite required for the synthesis. The synthesized spermine-conjugated siRNAs were evaluated for thermodynamic stability, resistance to nucleases, and gene-inhibition activity to acquire insights into the function of this novel siRNA as a potential drug.
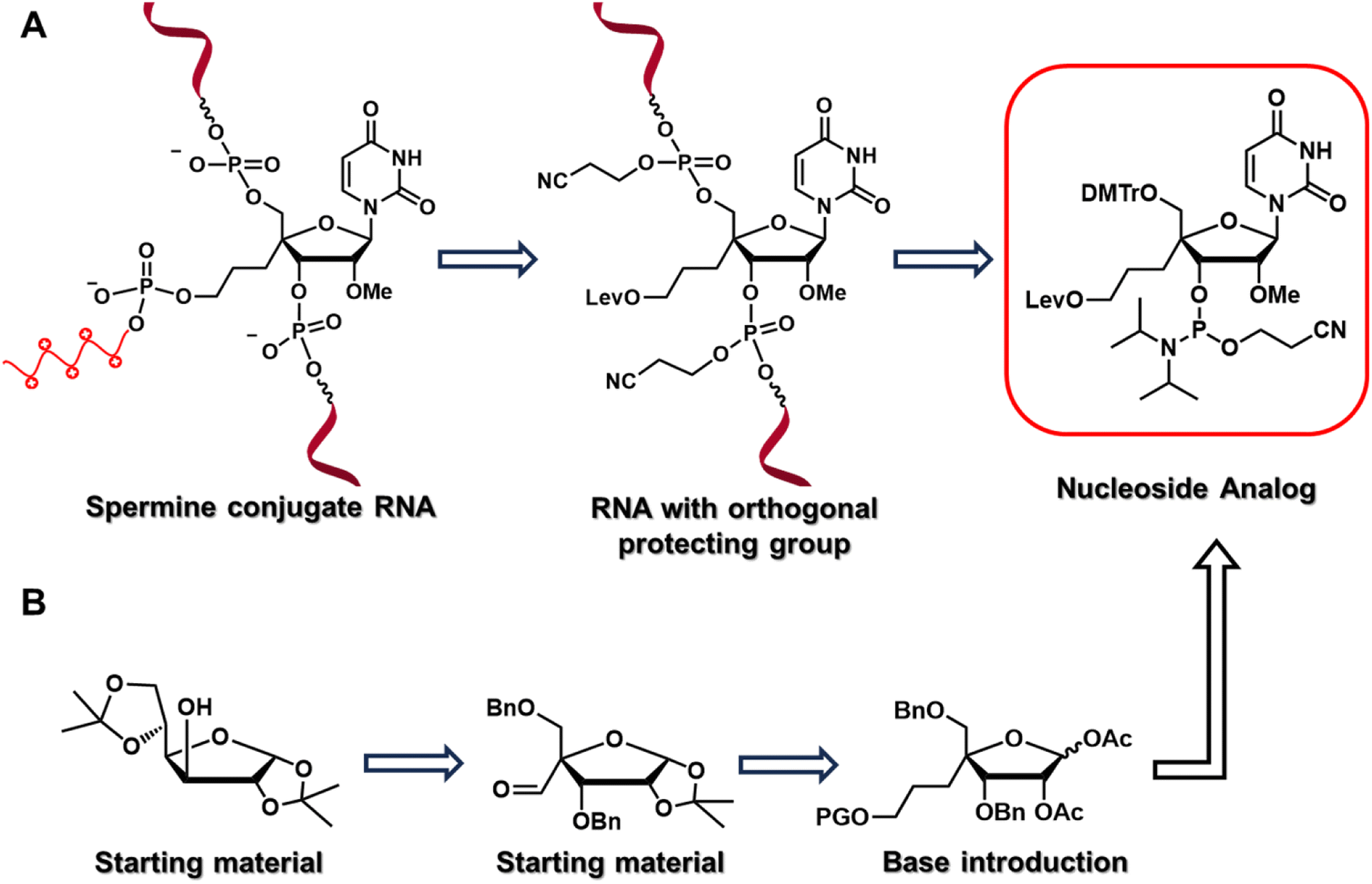 | ||
| Fig. 1 (A) Retrosynthetic analysis for identification of nucleoside analog by backtracking from target oligonucleotide to recognize the simpler precursor. (B) Representation of organic synthesis strategy for obtaining target nucleoside phosphoramidite. | ||
Results and discussion
Synthesis of nucleoside analog
The nucleoside analog 4′-C-propyllevulinate-2′-O-methyluridine (4′-PLev-U) was synthesized as a phosphoramidite and introduced into the oligonucleotide during automated solid-phase synthesis. The synthesis route was based on a previously reported method to obtain compound 2 which is the starting material for the synthesis of the desired phosphoramidite as shown in Scheme 1.34 The 4′-position was functionalized with the linker according to our previously reported method for 4′-C-aminoalkyl modifications.35,36 However, to incorporate spermine, the terminal hydroxyl group was protected with a levulinoyl orthogonal protecting group instead of an amine group.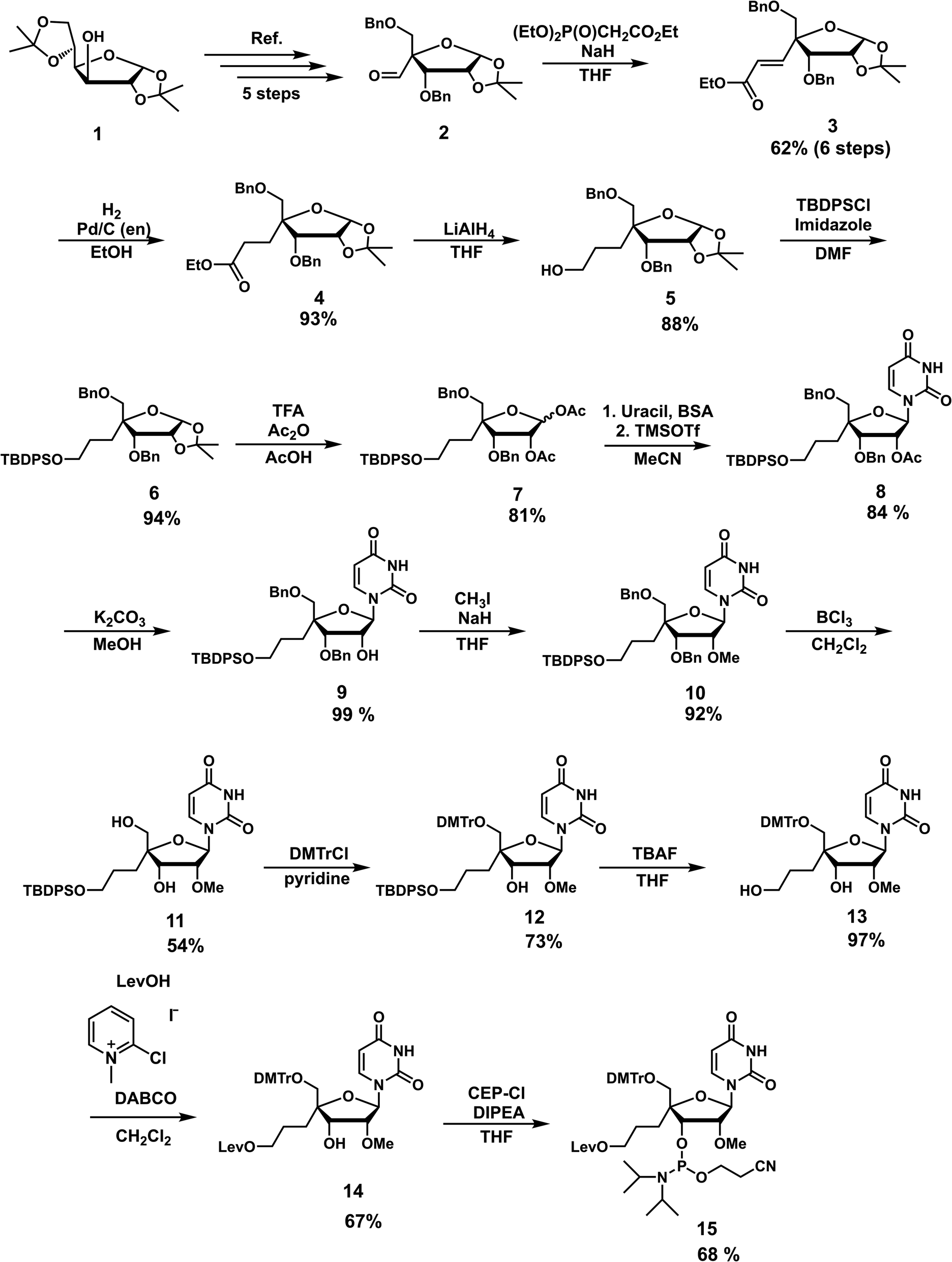 | ||
| Scheme 1 Synthesis of 4′-C-propyllevulinate-2′-O-methyluridine (4′-PLev-U) phosphoramidite. | ||
We used 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose (1) as the starting material to obtain 2 using a previously reported method. Using the Horner–Wadsworth–Emmons reaction, compound 2 was reacted with the corresponding phosphonate carbanion to yield 3, an α,β-unsaturated ester, at the 4-α position in 62% yield from 1. Subsequently, 4 was synthesized via the catalytic reduction of 3 with Pd/C(en) to afford the product in 93% yield. Compound 4 was reacted with LiAlH4 to reduce the ester to alcohol, affording 5 in 88% yield. Next, 5 was protected with a tert-butyldiphenylsilyl (TBDPS) group to afford 6 in 94% yield. Compound 7 was obtained in 81% yield by the acetolysis of 6 using acetic acid (AcOH) and trifluoroacetic acid (CF3CO2H) for acid hydrolysis and acetic anhydride (Ac2O) for acetylation. The glycosylation of 7 with uracil under Vorbrüggen conditions provided 8 in 84% yield. The acetyl group at the 2′-position of compound 8 was selectively removed using K2CO3, affording 9 in 99% yield. Next, the 2′-hydroxy group of 9 was methylated with methyl iodide in the dark to obtain 10 in 92% yield. The benzyl group of 10 was removed in the presence of BCl3 to produce 11 in 54% yield, and then the 5′-hydroxy function was protected with a dimethoxytrityl (DMTr) group to afford 12 in 73% yield. Subsequently, we obtained 13 in 97% yield by deprotection of the TBDPS group with tetra-n-butylammonium fluoride (TBAF), which was reacted with levulinic acid in the presence of 2-chloro-1-methylpyridinium iodide (Mukaiyama reagent) to obtain 14 in 67% yield. The phosphitylation of 14 afforded the desired phosphoramidite 15 in 67% yield. The total yield of this synthesis over 18 steps was 5%.
Synthesis of oligonucleotide
RNA oligomers containing 4′-PLev-U analogs were obtained by solid-phase synthesis using a DNA/RNA automated synthesizer and the phosphoramidite method. Here, in the first step, the oligonucleotide was conventionally synthesized using a universal solid support and respective nucleoside phosphoramidites, but the 5′-terminus of the modified RNA oligomers was capped after the synthesis was complete. Then, the levulinoyl group was selectively deprotected on the solid phase using 0.5 M hydrazine hydrate in pyridine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetic acid (3:2), which was freshly prepared immediately before the reaction following the procedure reported by Damha et al.37 Afterwards, the solid phase was washed with acetonitrile and then coupled to commercially available spermine phosphoramidite with the oligonucleotide to obtain a spermine-conjugated RNA oligomer (Scheme 2). After synthesis, the RNA oligomers (unmodified and modified) were treated with methylamine and 28% ammonia solution for 10 min at 65 °C to cleave the solid support and deprotect the acyl groups. The cyanoethyl group was selectively removed from the oligomers containing spermine by treatment with 10% diethylamine in MeCN for 5 min before treatment with ammonia solution. Next, the 2′-O-TBDMS group was removed from the oligomers by treatment with Et3N·3HF in DMSO for 90 min at 65 °C. The reaction mixture was then quenched and desalted using a Sep-Pak C18 cartridge. The obtained crude product was purified using 20% polyacrylamide gel electrophoresis (PAGE), extracted from the gel, and desalted using a Sep-Pak C18 cartridge to afford the desired oligonucleotides. The RNA oligomers were quantified and characterized based on molecular weight.
:acetic acid (3:2), which was freshly prepared immediately before the reaction following the procedure reported by Damha et al.37 Afterwards, the solid phase was washed with acetonitrile and then coupled to commercially available spermine phosphoramidite with the oligonucleotide to obtain a spermine-conjugated RNA oligomer (Scheme 2). After synthesis, the RNA oligomers (unmodified and modified) were treated with methylamine and 28% ammonia solution for 10 min at 65 °C to cleave the solid support and deprotect the acyl groups. The cyanoethyl group was selectively removed from the oligomers containing spermine by treatment with 10% diethylamine in MeCN for 5 min before treatment with ammonia solution. Next, the 2′-O-TBDMS group was removed from the oligomers by treatment with Et3N·3HF in DMSO for 90 min at 65 °C. The reaction mixture was then quenched and desalted using a Sep-Pak C18 cartridge. The obtained crude product was purified using 20% polyacrylamide gel electrophoresis (PAGE), extracted from the gel, and desalted using a Sep-Pak C18 cartridge to afford the desired oligonucleotides. The RNA oligomers were quantified and characterized based on molecular weight.
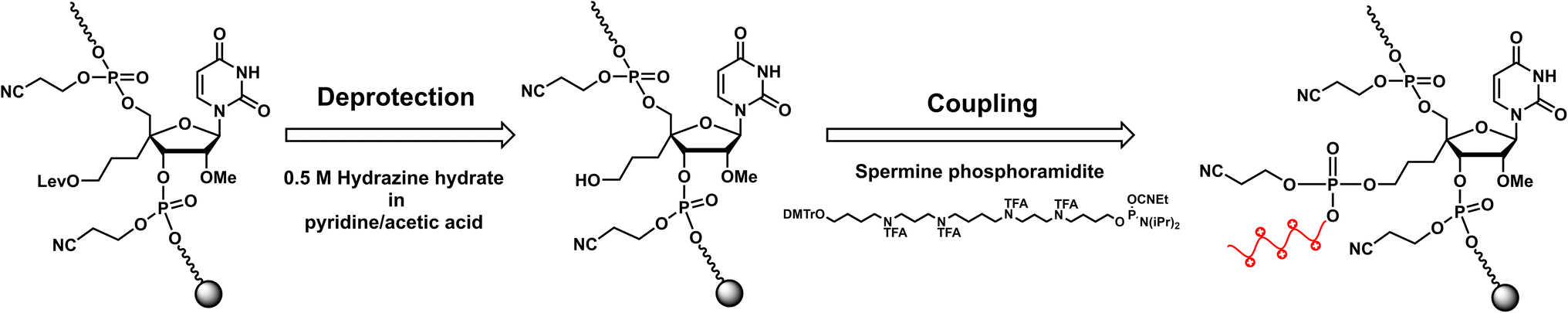 | ||
| Scheme 2 Synthesis of spermine conjugated oligonucleotide. | ||
Evaluation of thermodynamic stability of conjugated duplexes and siRNAs
Initially, the synthesized oligonucleotides were assessed for thermodynamic stability as short RNA duplexes of 11 mer. Duplexes containing no modification, a single modification with a nucleoside analog, and conjugation of spermine with an oligonucleotide at the analog were compared (Table 1). As shown in the table, the modifications were introduced into only one strand, as we planned to conjugate with the sense strand of the siRNA. The annealed duplexes were thermally denatured, and the absorbance of the samples at 260 nm was measured using a UV-visible spectrophotometer at the corresponding temperature. The melting temperature of each sample was calculated from a sigmoid curve (Fig. S1†). The results did not show a substantial decrease in the melting temperature (Tm) of the modified duplexes, exhibiting only a slight reduction in duplex binding and thermal stability. The Tm value of modified duplex 2 and 3 dropped by 2.8 °C and 1.5 °C, respectively, with respect to unmodified duplex 1. In previous reports, 4′-C aminoalkyl modifications shifted the sugar puckering equilibrium to the C2′-endo conformation, causing thermal destabilization of the RNA duplex. Therefore, we believe that similar effects result in reduced duplex binding.| Abbreviation of duplex | Abbreviation of RNA | Sequenceb | Tm (°C) | ΔTmc (°C) |
|---|---|---|---|---|
| a The Tm values were determined using 3 μM RNA duplex in a buffer containing 10 mM sodium phosphate (pH 7.0) and 100 mM NaCl.b U (bold) and s (italic) denote 4′-PLev-U and spermine conjugate, respectively.c ΔTm represents [Tm (duplex 2 or 3) − Tm (duplex 1)]. All experiments were performed in triplicates, and data are presented as the mean ± SD. | ||||
| Duplex 1 | RNA 1 | 5′-UUCUUCUUCUU-3′ | 41.7 ± 0.7 | — |
| RNA 4 | 3′-AAGAAGAAGAA-5′ | |||
| Duplex 2 | RNA 2 | 5′-UUCUUCUUCUU-3′ | 38.9 ± 0.6 | −2.8 |
| RNA 4 | 3′-AAGAAGAAGAA-5′ | |||
| Duplex 3 | RNA 3 | 5′-UUCUUsCUUCUU-3′ | 40.2 ± 0.6 | −1.5 |
| RNA 4 | 3′-AAGAAGAAGAA-5′ | |||
Next, we investigated the spermine-conjugated siRNAs with incorporations at different positions of the sense strand. Melting temperatures were calculated using the sigmoid curve obtained by thermal denaturing the siRNAs in buffer and measuring the absorbance at 260 nm. The determined Tm values were remarkable and comparable to those of unmodified siRNA (Table 2, Fig. S2†). The modified siRNAs 2, 3, and 4, which were conjugated at positions 5, 11, and 17 of the sense strand, respectively, had Tm values of 78.1, 77.3 and 77.1 °C, respectively, which are similar to that of unmodified siRNA 1 (77.3 °C). Interestingly, the deviations in the Tm values from the unmodified siRNA were +0.8 and −0.2 °C for siRNA 2 and 4, respectively. However, the Tm value of siRNA 3 was similar to that of the unmodified siRNA. These results indicated that the incorporation of spermine into the non-terminal positions of the sense strand of siRNA did not affect duplex binding or thermal stability. Such an effect could be position-specific or dependent on the nearest neighbor (adjacent nucleoside), as can be seen by the change in the base from pyrimidine (siRNA 2) to purine (siRNAs 3 and 4).
| Abbreviation of siRNA | Abbreviation of RNA | Sequenceb | Tm (°C) | ΔTmc (°C) |
|---|---|---|---|---|
| a The Tm values were determined using 3 μM RNA duplex in a buffer containing 10 mM sodium phosphate (pH 7.0) and 100 mM NaCl.b U (bold) and s (italic) denote 4′-PLev-U and spermine conjugate, respectively.c ΔTm represents [Tm (siRNA 2, 3 or 4) − Tm (siRNA 1)]. All experiments were performed in triplicates, and data are presented as the mean ± SD. | ||||
| siRNA 1 | RNA 5 | Sense strand 5′-GGCCUUUCACUACUCCUACUU-3′ | 77.3 ± 0.1 | — |
| RNA 9 | 3′-UUCCGGAAAGUGAUGAGGAUG-5′ antisense strand | |||
| siRNA 2 | RNA 6 | 5′-GGCCUsUUCACUACUCCUACUU-3′ | 78.1 ± 0.1 | 0.8 |
| RNA 9 | 3′-UUCCGGAAAGUGAUGAGGAUG-5′ | |||
| siRNA 3 | RNA 7 | 5′-GGCCUUUCACUsACUCCUACUU-3′ | 77.3 ± 0.1 | — |
| RNA 9 | 3′-UUCCGGAAAGUGAUGAGGAUG-5′ | |||
| siRNA 4 | RNA 8 | 5′-GGCCUUUCACUACUCCUsACUU-3′ | 77.1 ± 0.1 | −0.2 |
| RNA 9 | 3′-UUCCGGAAAGUGAUGAGGAUG-5′ | |||
On the basis of these results, positions 5 and 11 were selected for further evaluation. To this end, we designed siRNAs containing two spermine molecules at a single position or one spermine molecule at two positions. All the modified siRNAs had higher melting temperatures than unmodified siRNA 1, with siRNA 5 denatured at 2.3 °C higher and siRNAs 6 and 7 at 0.4 °C higher than unmodified siRNA (Table 3, Fig. S3†). These findings also support our hypothesis that nearest neighbors affect the thermodynamic stability of conjugated siRNAs as siRNA 7 with combination of pyrimidine and purine as adjacent base showing intermediate change in Tm value, in contrast to siRNA 2 and 3. Hence, the conjugation of spermine molecules at the non-terminal positions of siRNA comparatively improves thermal stability and is dependent on the position, nearest neighbor, and number of spermine molecules.
| Abbreviation of siRNA | Abbreviation of RNA | Sequenceb | Tm (°C) | ΔTmc (°C) |
|---|---|---|---|---|
| a The Tm values were determined using 3 μM RNA duplex in a buffer containing 10 mM sodium phosphate (pH 7.0) and 100 mM NaCl.b U (bold) and s (italic) denote 4′-PLev-U and spermine conjugate, respectively.c ΔTm represents [Tm (siRNA 2, 3 or 4) − Tm (siRNA 1)]. All experiments were performed in triplicates, and data are presented as the mean ± SD. | ||||
| siRNA 1 | RNA 5 | Sense strand 5′-GGCCUUUCACUACUCCUACUU-3′ | 77.3 ± 0.1 | — |
| RNA 9 | 3′-UUCCGGAAAGUGAUGAGGAUG-5′ antisense strand | |||
| siRNA 5 | RNA 10 | 5′-GGCCUssUUCACUACUCCUACUU-3′ | 79.6 ± 0.1 | 2.3 |
| RNA 9 | 3′-UUCCGGAAAGUGAUGAGGAUG-5′ | |||
| siRNA 6 | RNA 11 | 5′-GGCCUUUCACUssACUCCUACUU-3′ | 77.7 ± 0.2 | 0.4 |
| RNA 9 | 3′-UUCCGGAAAGUGAUGAGGAUG-5′ | |||
| siRNA 7 | RNA 12 | 5′-GGCCUsUUCACUsACUCCUACUU-3′ | 77.7 ± 0.1 | 0.4 |
| RNA 9 | 3′-UUCCGGAAAGUGAUGAGGAUG-5′ | |||
Circular dichroism spectra
Circular dichroism (CD) measurements were performed to elucidate the structure of the spermine-conjugated RNA duplexes. We studied duplexes 1–3 (Table 1) and calculated their CD spectra, as shown in Fig. 2. RNA duplexes form an A-type helical structure with a maximum at 269 nm and a minimum at 210 nm in the spectra. Notably, all duplexes featured similar waveforms, particularly spermine-conjugated duplex 3, which showed a CD spectrum similar to that of unmodified duplex 1. Therefore, it was evident that the introduction of spermine molecules at the non-terminal position of the RNA duplex did not alter its form.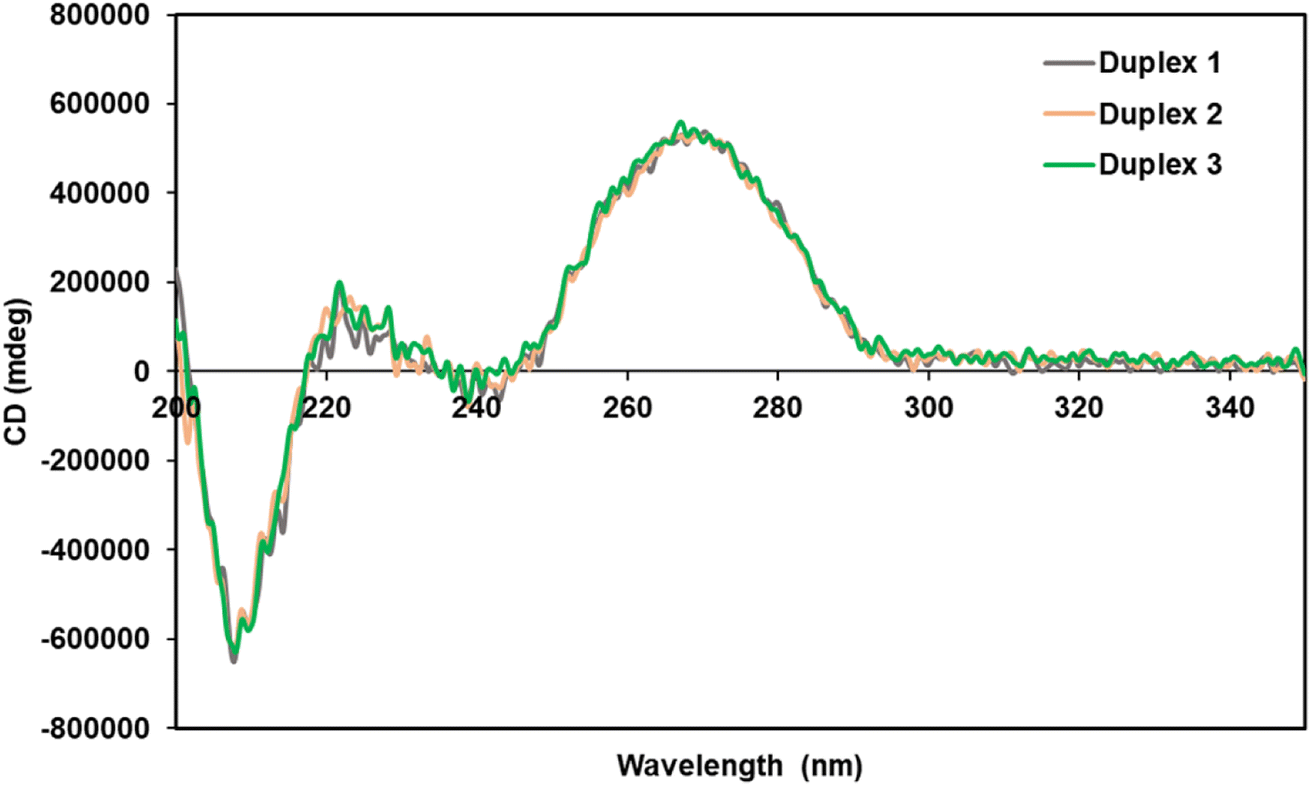 | ||
| Fig. 2 CD spectra of the unmodified and modified RNA duplexes in a buffer containing 10 mM sodium phosphate (pH 7.0) and 100 mM NaCl at 25 °C with duplex concentration of 4 μM. Duplex 1 is unmodified, Duplex 2 contains 4′-PLev-U and Duplex 3 contains analog conjugated with spermine at central position. | ||
Conjugated siRNAs for resistance against nucleases
Because RNAs are labile to degradation by nucleases, we analyzed the stability of fluorescein-labeled conjugated siRNAs following treatment with bovine serum (BS). The spermine-conjugated oligonucleotide was annealed to RNA 13, labeled with fluorescein at the 5′-terminus to obtain the modified siRNAs for serum stability (Table S1†). The annealed siRNAs were suspended in buffer and Opti-MEM, then treated with BS, and aliquots were obtained at predetermined time intervals. These aliquots were applied to a polyacrylamide gel, and the electrophoresis profile showed the extent of degradation at various time points for respective siRNAs (Fig. 3). The residual ratio of remaining intact siRNA which is the proportion of band for intact siRNA to the sum of intact and degraded siRNA band, was measured using ImageJ software, it depicts the degree of stability against nuclease attacks. In this manner, the nuclease resistance has been quantified as the percent of remaining intact siRNA. Unmodified siRNA 8 remained intact (17.6%), whereas spermine-modified siRNAs 9 and 10 were slightly more stable (22.1% and 38.2%, respectively) after 3 h of incubation, higher degree of intactness is proportional to the stability against serum. However, the stability of siRNA 11 acutely decreased, leading to its rapid decomposition. Hence, this conjugation imparted serum stability in a position-specific manner.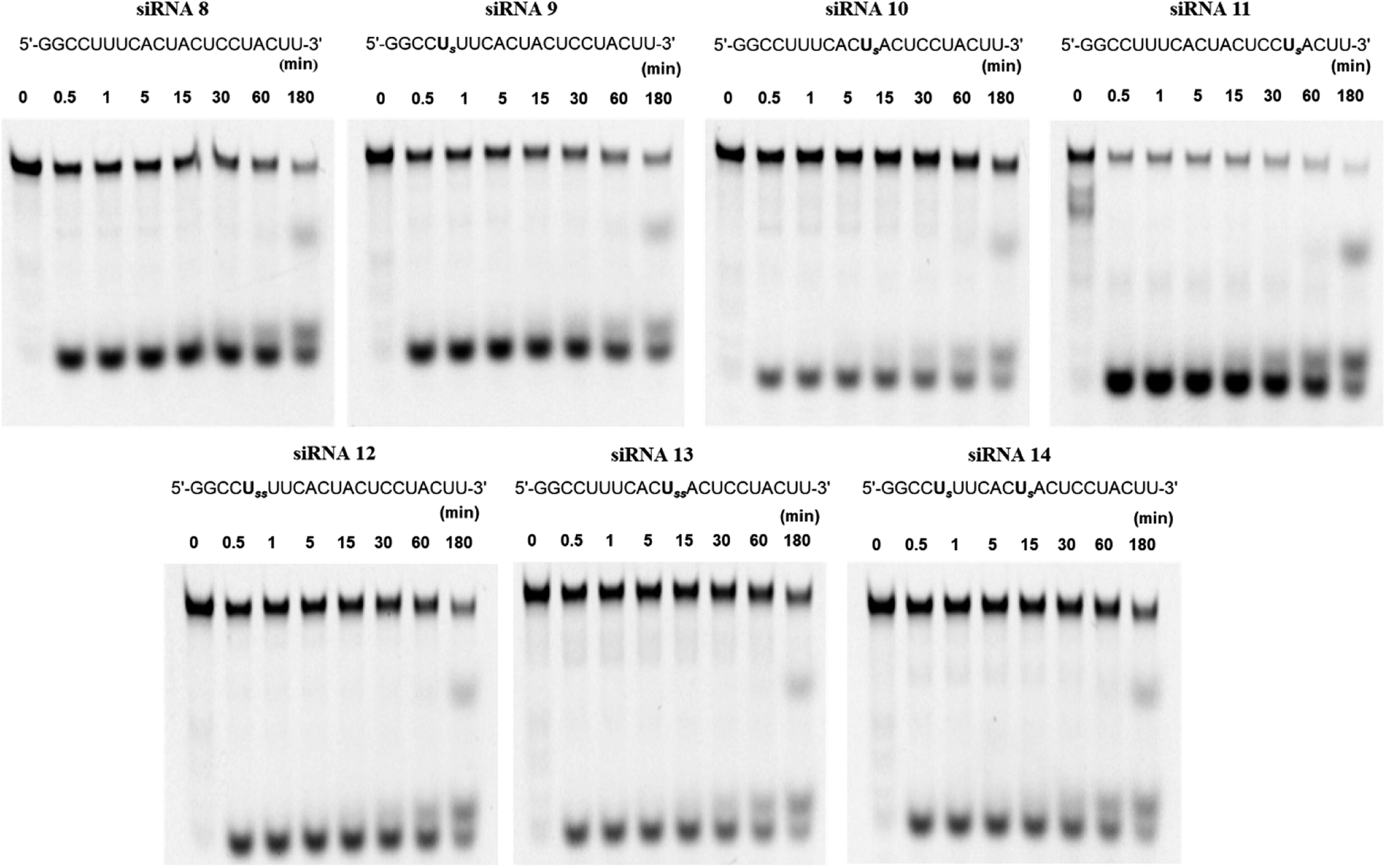 | ||
| Fig. 3 Degradation profile of siRNAs treated with 5% bovine serum on 15% polyacrylamide gel upon electrophoresis. Fluorescein labeled complementary RNA 13; 3′-UUCCGGAAAGUGAUGAGGAUG-F-5′ was annealed with modified RNAs shown above, to obtain corresponding siRNAs as mentioned. U denotes 4′-PLev-U analog, s denotes spermine and F denotes fluorescein. | ||
Further investigations involving double insertions of spermine molecules were carried out, either at a single position or a single insertion at two positions, as previously discussed. Here, we found that the percentages of intact siRNAs 12, 13, and 14 were 26.2%, 47.2%, and 35.4%, respectively, indicating their superior stability to unmodified siRNA. These results strongly suggest that the conjugation of spermine molecules with siRNA enhances the resistance against nucleases; however, this effect depends on the position of insertion.
Gene inhibition with conjugated siRNAs
The gene-silencing activity of spermine-conjugated and unmodified siRNAs was evaluated using a dual-luciferase reporter assay in HeLa cells, in which the target luciferase genes were constitutively expressed. The firefly luciferase gene was used as a control, and all siRNAs targeted the Renilla luciferase genes. Unmodified and modified siRNAs at concentrations of 1 and 10 nM, respectively, were transfected into confluent cells using the RNAiMAX transfection reagent and then analyzed for the expression of both luciferase genes after 24 h of incubation. The values for Renilla luciferase relative to firefly luciferase were normalized to determine gene silencing. As discussed earlier, the sense strand was selected for inserting spermine at non-terminal positions, as conjugation with the antisense strand greatly disturbs gene silencing.Interestingly, this type of conjugation resulted in a modest reduction in RNAi activity (Fig. 4). The results for modified siRNAs 2 and 3 clearly indicate that the gene-silencing activity depends on the position of the spermine conjugate, as incorporations near the 5′-terminus of the passenger strand give a relatively better response than those at the center. In addition, increasing the number of spermine molecules adversely affected gene inhibition, as indicated by lower activity of modified siRNAs 5, 6, and 7. Comparing siRNA 2 with siRNA 5 and siRNA 3 with siRNA 6 notably shows the effect of increase in the number of spermine molecules as both the cases involve conjugation at the identical position. However, siRNA 7 exhibits a collective modification of siRNA 2 and 3 which further consolidates our hypothesis that conjugation near the center decreases the gene silencing activity. Furthermore, with an increase in the number of spermine molecules, activity was significantly reduced at a higher concentration of 10 nM. Therefore, these non-terminal modifications of spermine must be limited in number and requires further investigations in terms of position and quantity of insertions to enhance their effectiveness for efficient silencing activity.
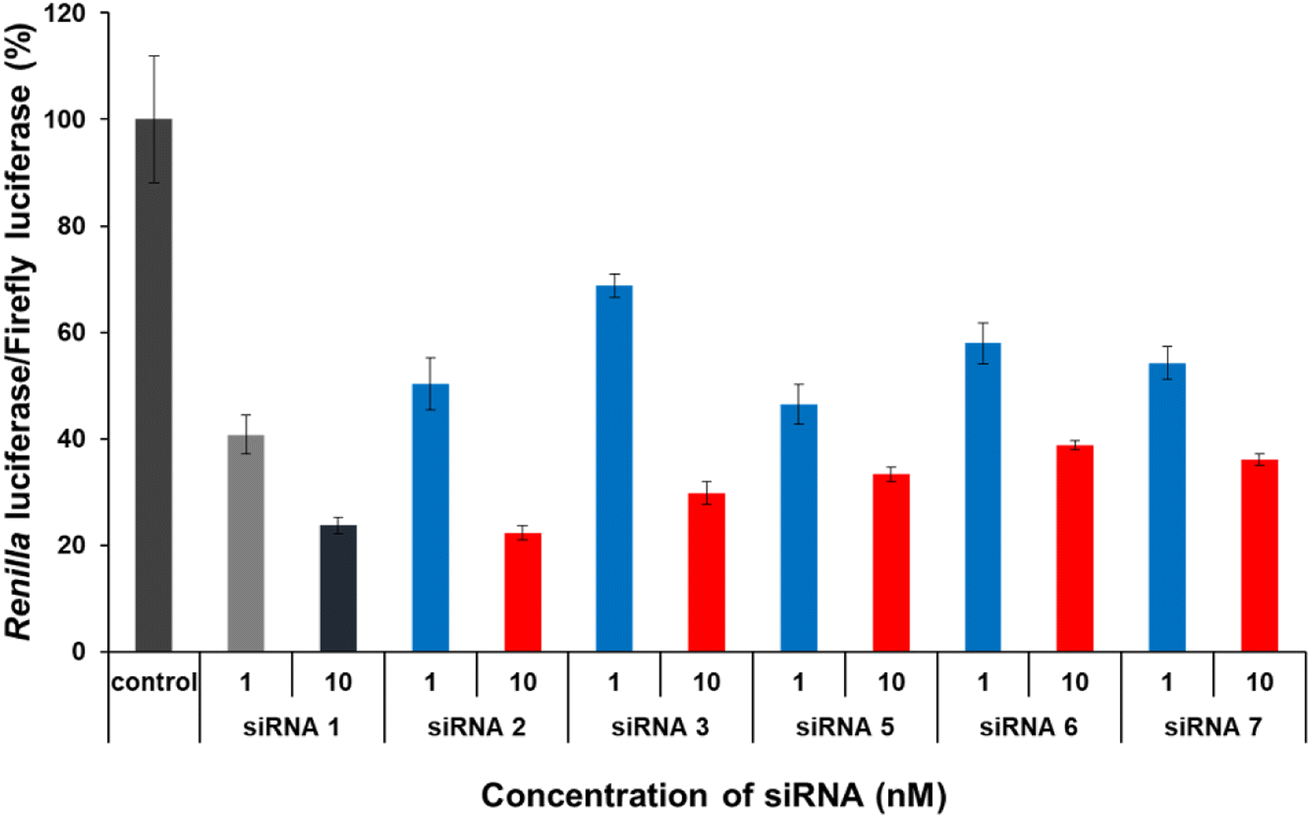 | ||
| Fig. 4 RNAi activity of siRNAs transfected into HeLa cells at concentrations of 1 and 10 nM. After 24 h of treatment, the activities of Renilla and firefly luciferases in the cells were determined using the dual luciferase reporter assay system. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average of four experiments as the mean ± SD. | ||
Conclusion
In this study, we successfully synthesized a 4′-C-propyllevulinate-2′-O-methyluridine (4′-PLev-U) analog and incorporated a spermine conjugate at non-terminal positions through deprotection of the orthogonal protecting group. The conjugated siRNAs exhibited comparably enhanced thermal stability and improved serum stability in a position-dependent manner. Insertion of a spermine molecule near the 3′-end of the passenger strand was found to be detrimental. In addition, structural analysis using circular dichroism revealed a spectrum consistent with that of an A-type helix-like RNA duplex. Lastly, the gene-inhibition activity was reduced in conjugated siRNAs. The effect was dependent on the position as well as the number of spermine molecules introduced.To the best of our knowledge, this is the first successful attempt to conjugate spermine to the non-terminal position of siRNA using solid-phase synthesis. This novel study analyzed conjugated siRNAs with notably improved functions. However, our approach is not limited to these conjugates; other conjugates can be added to the non-terminal positions of the analog used in this study. In subsequent studies, we intend to exploit this method using a larger number of spermine molecules to attain carrier-free delivery, as earlier reports state that an N/P ratio exceeding 1.3 for siRNA promotes cell permeability. Therefore, this study provides scope for extending research on the drug-delivery-system-free administration of siRNAs.
Experimental section
General remarks
All reagents and dry solvents (CH2Cl2, DMF, MeCN, THF, and pyridine) were obtained from commercial sources and used without any further purification. Thin layer chromatography (TLC) was performed on silica gel plates precoated with fluorescent indicator and visualized under UV light or stained with a solution of 5% (v/v) concentrated H2SO4 in a mixture of p-anisaldehyde and methanol, and then heated. Column chromatography for isolation of compounds was done using Silica gel (63–210 mesh). Samples were prepared in CDCl3 as solvent to obtain NMR spectra and chemical shifts (δ) are given in parts per million (ppm) and coupling constant (J) in Hz, with reference at 7.26 ppm for 1H NMR spectra and 77.16 ppm for 13C NMR spectra. H3PO4 was used as internal standard for 31P NMR with reference at 0 ppm. The abbreviations s, d, t and m signify singlet, doublet, triplet and multiplet, respectively. High resolution mass spectra (HRMS) were obtained with electrospray ionization (ESI-TOF) in positive mode.Synthesis of nucleoside analog
:1) to give compound 3 as a yellow oil (5.02 g, 10.7 mmol, 91%). 1H NMR (400 MHz, CDCl3) δ 7.38–7.21 (m, 10H), 6.25 (d, J = 16.0 Hz, 1H), 5.76 (d, J = 3.66 Hz, 1H), 4.78 (d, J = 12.4 Hz, 1H), 4.60 (d, J = 11.9 Hz, 1H), 4.57 (d, J = 4.58 Hz, 1H), 4.50 (d, J = 12.4, 1H), 4.41 (d, J = 12.0 Hz, 1H), 4.28 (d, J = 4.58 Hz, 1H), 4.23–4.15 (m, 2H), 3.34 (t, J = 11.0 Hz, 2H), 1.48 (s, 3H), 1.28 (s, 3H), 1.27 (s, 3H); 13C {1H } NMR (101 MHz, CDCl3) δ 166.3, 145.5, 137.8, 137.6, 128.5, 128.4, 128.1, 127.8, 127.8, 122.8, 113.5, 104.0, 85.9, 77.8, 73.6, 72.6, 71.6, 60.4, 26.0, 25.5, 14.3; HRMS (ESI) m/z: calcd for C27H32NaO7 [M + Na]+ 491.2046, found 491.2028.:1) to afford compound 5 as a yellow oil (1.59 g, 3.71 mmol, 88%). 1H NMR (400 MHz, CDCl3) δ 7.35–7.23 (m, 10H), 5.76 (d, J = 4.12 Hz, 1H), 4.76 (d, J = 12.4 Hz, 1H), 4.63 (dd, J = 12.4 Hz, 4.12 Hz, 1H), 4.57 (d, J = 12.4 Hz, 1H), 4.52 (d, J = 12.4, 1H), 4.40 (d, J = 11.9 Hz, 1H), 4.17 (d, J = 5.50 Hz, 1H), 3.72–3.60 (m, 2H), 3.51 (d, J = 10.1, 2H), 3.31 (d, J = 10.1, 1H), 2.27–2.18 (m, 2H), 1.78–1.68 (m, 2H), 1.62 (s, 3H), 1.33 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 138.0, 128.4, 127.9, 127.9, 127.7, 127.7, 113.1, 104.1, 87.1, 79.0, 78.4, 73.6, 72.4, 62.7, 27.8, 26.9, 26.5, 26.0; HRMS (ESI) m/z: calcd for C25H32NaO6 [M + Na]+ 451.20996, found 451.20675.:1) to provide compound 6 as a yellow oil (4.42 g, 6.63 mmol, 94%). 1H NMR (400 MHz, CDCl3) δ 7.66–7.63 (m, 4H), 7.40–7.22 (m, 16H), 5.73 (d, J = 4.12 Hz, 1H), 4.74 (d, J = 12.4 Hz, 1H), 4.61 (dd, J = 9.16 Hz, 4.12 Hz, 1H), 4.53 (d, J = 12.4, 1H), 4.45 (d, J = 11.9 Hz, 1H), 4.38 (d, J = 12.4 Hz, 1H), 4.13 (d, J = 5.04, 1H), 3.70–3.57 (m, 2H), 3.44 (d, J = 10.1, 1H), 3.25 (d, J = 10.1, 1H), 2.17–2.09 (m, 1H), 1.85–1.77 (m, 1H), 1.68–1.52 (m, 2H), 1.48 (s, 3H), 1.30 (s, 3H), 1.03 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 138.4, 138.3, 135.7, 134.2, 134.1, 130.0, 128.5, 128.4, 127.8, 127.8, 127.8, 113.2, 104.2, 87.0, 79.6, 78.6, 73.6, 73.1, 72.4, 64.7, 28.4, 27.0, 26.9, 26.4, 19.3; HRMS (ESI) m/z: calcd for C41H50NaO6Si [M + Na]+ 689.32743, found 689.32845.:1) to obtain compound 7 as a colorless oil (0.40 g, 0.56 mmol, 81%). 1H NMR (400 MHz, CDCl3) δ 7.67–7.65 (m, 4H), 7.41–7.25 (m, 16H), 6.11 (s, 1H), 5.34 (d, J = 5.04 Hz, 1H), 4.60 (d, J = 11.9 Hz, 1H), 4.52 (d, J = 11.9 Hz, 1H), 4.44 (t, J = 11.9 Hz, 2H), 4.34 (d, J = 5.04 Hz, 1H), 3.68–3.61 (m, 2H), 3.42 (d, J = 9.62, 1H), 3.37 (d, J = 9.62, 1H), 2.03 (s, 3H), 1.88 (s, 3H), 1.87–1.70 (m, 3H), 1.64–1.57 (m, 1H), 1.04 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 170.0, 169.6, 138.2, 138.1, 135.7, 135.7, 134.2, 134.2, 129.6, 128.6, 128.4, 127.8, 127.8, 127.7, 127.7, 127.5, 97.8, 87.4, 79.4, 75.1, 73.4, 73.3, 73.2, 64.6, 29.2, 27.0, 26.4, 21.2, 20.9, 19.4; HRMS (ESI) m/z: calcd for C42H50KO8Si [M + K]+ 749.29120, found 749.28944.:1) to give compound 12 (0.36 g, 0.47 mmol, 84%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.99 (d, J = 1.83 Hz 1H), 7.73 (d, J = 8.24 Hz 1H), 7.67–7.65 (m, 4H), 7.44–7.25 (m, 16H), 6.17 (d, J = 4.58, Hz, 1H), 5.35–5.29 (m, 2H), 4.61 (d, J = 11.9 Hz, 1H), 4.44 (d, J = 11.9 Hz, 1H), 4.39 (d, J = 11.0 Hz, 1H), 4.33 (d, J = 5.95 Hz, 1H), 3.69–3.61 (m, 3H), 3.35 (d, J = 10.5 Hz, 1H), 2.05 (s, 3H), 1.96–1.92 (m, 1H), 1.76–1.61 (m, 2H), 1.57–1.47 (m, 1H), 1.06 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 170.1, 163.2, 150.4, 140.3, 137.7, 137.2, 135.7, 134.0, 129.7, 128.8, 128.6, 128.4, 128.1, 128.0, 127.8, 102.5, 88.0, 86.6, 77.4, 75.5, 74.3, 73.7, 72.6, 64.1, 28.8, 27.0, 26.5, 20.8, 19.3, 19.4; HRMS (ESI) m/z: calcd for C44H50N2NaO8Si [M + Na]+ 785.32341, found 785.32110.:1) to yield compound 9 as a white solid (0.34 g, 0.47 mmol, 99%). 1H NMR (400 MHz, CDCl3) δ 9.26 (d, J = 1.37 Hz 1H), 7.66–7.64 (m, 5H), 7.64–7.24 (m, 16H), 5.91 (d, J = 5.95, 1H), 5.38 (dd, J = 1.83 Hz, 7.79 Hz 1H), 4.69 (d, J = 11.5 Hz, 1H), 4.61 (d, J = 11.5 Hz, 1H), 4.47 (s, 2H), 4.31–4.25 (m, 1H), 4.12 (d, J = 5.95 Hz, 1H), 3.70–3.59 (m, 3H), 3.40 (d, J = 10.1 Hz, 1H), 3.24 (d, J = 8.24 Hz, 1H), 2.06 (m, 1H), 1.72–1.63 (m, 2H), 1.58–1.49 (m, 1H), 1.04 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 163.4, 150.9, 140.5, 137.2, 137.1, 136.7, 133.9, 129.7, 128.8, 128.7, 128.4, 128.4, 128.2, 128.1, 127.8, 88.9, 87.7, 79.8, 75.3, 74.8, 73.9, 73.7, 64.2, 29.1, 27.0, 26.6, 19.3, 20.8, 19.3, 19.4; HRMS (ESI) m/z: calcd for C42H48N2NaO7Si [M + Na]+ 743.31285, found 743.31529.:2) to provide compound 10 as a white solid (0.32 g, 0.435 mmol, 92%). 1H NMR (400 MHz, CDCl3) δ 9.18 (d, J = 1.37 Hz, 1H), 7.86 (d, J = 8.24 Hz, 1H), 7.59–7.57 (m, 4H), 7.34–7.09 (m, 16H), 5.91 (d, J = 1.83, 1H), 5.05 (dd, J = 1.83 Hz, 8.24 Hz 1H), 4.65 (d, J = 12.4 Hz, 1H), 4.39 (d, J = 11.9 Hz, 1H), 4.34 (d, J = 10.5, 1H), 4.29 (d, J = 10.5, 1H), 4.15 (d, J = 5.95, 1H), 3.67 (d, J = 10.5 Hz, 1H), 3.61–3.54 (m, 2H), 3.42 (s, 3H), 3.34 (d, J = 10.1 Hz, 1H), 2.06–1.96 (m, 1H), 1.69–1.52 (m, 2H), 1.48–1.38 (m, 1H), 1.04 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 163.6, 150.2, 140.7, 140.7, 137.8, 137.4, 135.7, 134.1, 134.0, 130.0, 128.7, 128.6, 128.4, 128.1, 128.1, 127.9, 127.7, 101.8, 88.2, 87.8, 84.3, 75.9, 73.7, 72.9, 71.6, 64.2, 59.4, 28.4, 27.0, 26.5, 19.3; HRMS (ESI) m/z: calcd for C43H50N2NaO7Si [M + Na]+ 757.32850, found 757.32950.:1, 20 mL) was added to the mixture, extracted with saturated aq. NaHCO3 and CHCl3. The organic layer was washed with brine, dried over Na2SO4 and concentrated. The residue was purified by chromatography on silica gel (hexane/EtOAc = 1:7) to obtain compound 11 as white solid (0.34 g, 0.61 mmol, 54%). 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 7.67–7.62 (m, 4H), 7.44–7.36 (m, 6H), 5.75 (dd, J = 1.37 Hz, 8.93 Hz 1H), 5.67 (d, J = 5.04 Hz, 1H), 4.43 (t, J = 5.50 Hz, 10.1 Hz, 1H), 4.20 (t, J = 5.50 Hz, 10.1 Hz, 1H), 3.79–3.76 (m, 1H), 3.68–3.62 (m, 3H), 3.49 (s, 3H), 2.85 (dd, J = 6.87 Hz, 3.66 Hz, 1H), 2.76 (d, J = 5.50 Hz, 1H), 1.84–1.77 (m, 1H), 1.70–1.57 (m, 3H), 1.05 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 163.2, 150.4, 142.3, 135.7, 133.9, 129.7, 127.8, 102.8, 90.3, 89.4, 83.1, 70.1, 65.4, 64.1, 59.2, 28.2, 27.0, 26.7, 19.3; HRMS (ESI) m/z: calcd for C29H38N2NaO7Si [M + Na]+ 577.23460, found 577.23545.:3) to afford compound 12 as a yellow solid (0.1 g, 1.17 mmol, 73%). 1H NMR (400 MHz, CDCl3) δ 9.27 (s, 1H), 7.92 (d, J = 8.24 Hz, 1H) 7.61 (d, J = 7.79 Hz, 4H), 7.41–7.24 (m, 16H), 6.84 (d, J = 8.70 Hz 4H), 6.01 (d, J = 2.75, 1H), 5.20 (dd, J = 1.83 Hz, 8.24 Hz 1H), 4.65 (t, J = 6.87 Hz, 13.74 Hz, 1H), 3.85 (dd, J = 6.41 Hz, 5.95 Hz 1H), 3.79 (s, 6H), 3.61–3.55 (m, 5H), 3.36 (d, J = 10.5, 1H), 2.80 (d, J = 7.79 Hz, 1H), 1.87–1.81 (m, 1H), 1.58–1.45 (m, 2H), 1.33–1.29 (m, 1H), 1.02 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 163.4, 158.8, 150.3, 144.4, 140.5, 135.7, 135.3, 135.1, 134.0, 130.4, 130.3, 130.0, 128.4, 128.1, 127.7, 127.3, 113.4, 102.4, 88.2, 87.5, 86.9, 85.0, 70.3, 65.2, 63.9, 59.5, 55.4, 28.3, 27.0, 26.3, 19.3; HRMS (ESI) m/z: calcd for C50H56N2NaO9Si [M + Na]+ 879.36528, found 879.36319.:1) to give compound 13 as a white formed solid (0.07 g, 0.113 mmol, 97%). 1H NMR (400 MHz, CDCl3) δ 9.59 (s, 1H), 7.85 (d, J = 8.24 Hz, 1H) 7.36–7.22 (m, 9H), 6.84 (d, J = 9.16, 4H), 6.07 (d, J = 4.12 Hz 1H), 5.24 (d, J = 8.24, 1H), 4.62 (t, J = 6.41 Hz, 12.4 Hz 1H), 3.94 (dd, J = 5.95 Hz, 4.12 Hz, 1H), 3.79 (s, 6H), 3.53 (s, 5H), 3.36 (s, 2H), 3.16 (d, J = 6.41, 1H), 1.82–1.75 (m, 1H), 1.68–1.60 (m, 1H), 1.56–1.47 (m, 1H), 1.41–1.32 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 163.5, 158.8, 158.8, 150.5, 144.3, 140.4, 135.2, 135.0, 130.3, 130.2, 128.3, 128.1, 127.3, 113.4, 102.7, 88.2, 87.5, 86.5, 84.7, 70.5, 65.6, 62.9, 59.3, 55.3, 28.6, 26.4; HRMS (ESI) m/z: calcd for C34H38N2NaO9 [M + Na]+ 641.24750, found 641.24688.:4) to yield compound 14 as a white solid (0.294 g, 0.41 mmol, 67%). 1H NMR (400 MHz, CDCl3) δ 9.39 (d, J = 1.83 Hz 1H), 7.84 (d, J = 8.24 Hz, 1H), 7.37–7.22 (m, 9H), 6.84 (d, J = 9.16, 4H), 6.03 (d, J = 3.66 Hz 1H), 5.22 (dd, J = 1.83, 8.24, 1H), 4.61 (t, J = 6.41 Hz, 12.8 Hz 1H), 4.02–3.96 (m, 2H), 3.90 (dd, J = 5.95 Hz, 3.66 Hz, 1H), 3.79 (s, 6H), 3.55 (s, 3H), 3.34 (s, 2H), 2.94 (d, J = 6.87, 1H), 2.71 (t, J = 6.87 Hz, 5.95 Hz 2H), 2.53 (t, J = 6.41 Hz, 6.87 Hz 2H), 1.83–1.75 (m, 1H), 1.62–1.57 (m, 2H), 1.46–1.43 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 206.8, 172.8, 163.4, 158.8, 158.8, 150.4, 144.3, 140.4, 135.2, 134.9, 130.3, 130.2, 128.3, 128.1, 127.3, 113.4, 102.6, 87.9, 87.5, 86.8, 84.7, 70.5, 65.3, 64.8, 59.4, 55.4, 38.0, 30.0, 28.5, 28.0, 22.6; HRMS (ESI) m/z: calcd for C39H44KN2O11 [M + K]+ 755.25822, found 755.26061.:3) to obtain compound 15 as white solid (0.49 g, 0.534 mmol, 68%). 31P NMR (162 MHz, CDCl3) δ 151.2, 150.8; HRMS (ESI) m/z: Calcd for C48H61N4NaO12P [M + Na]+ 939.39213, found 939.39437.Synthesis of oligonucleotides
The synthesis was carried out with DNA/RNA synthesizer by the phosphoroamidite method. Native RNA, 4′-PLev-U analog and spermine phosphoramidites were prepared for 0.1 M, 0.15 M and 0.075 M concentrations respectively, in MeCN for use during synthesis. After the synthesis, 5′ terminus of the RNA oligomers containing 4′-PLev-U were capped by acetyl group and treated with 0.5 M hydrazine hydrate in pyridine/AcOH (3:2; v/v) for 25 min (5 min × 5 set) to deprotect levulinyl group and washed with MeCN. Next, the spermine phosphoramidites were coupled to the deprotected linker at 4′-C position of analog in RNA strand. CPG beads were cleaved and deprotected by treatment with concentrated NH3 solution/40% methylamine (1:1, v/v) for 10 min at 65 °C. In the case of oligomers containing spermine, cyanoethyl group was selectively removed by treatment with 10% diethylamine in MeCN for 5 min prior to NH3 solution treatment. The 2′-O-TBDMS protecting groups were removed using Et3N·3HF in DMSO for 90 min at 65 °C, the reaction was quenched with 0.1 M TEAA buffer (pH 7.0), and the mixture was desalted using Sep-pak C18 cartridge. The oligonucleotides were purified by 20% PAGE containing 7 M urea to give highly purified RNAs. The band for target sequence was extracted from gel and the isolated RNA oligomers were analyzed by MALDI-TOF/MS for characterization with molecular weight.
MALDI-TOF/MS
The spectra were obtained with a time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). A solution of 3-hydroxypicolinic acid (3-HPA) and diammonium hydrogen citrate in H2O was used as the matrix. Data of synthetic ONs: RNA 1: m/z = 3301.37 (calcd for C99H125N25O83P10 [M − H]− 3301.57), RNA 2: m/z = 3373.43 (calcd for C103H133N25O84P10 [M − H]− 3373.72), RNA 3: m/z = 3780.71 (calcd for C121H173N29O88P11 [M − H]− 3782.11), RNA 4: m/z = 3605.61 (calcd for C110H133N55O67P10 [M − H]− 3606.13), RNA 5: m/z = 6503.83 (calcd for C194H245N65O150P20 [M − H]− 6505.97), RNA 6: m/z = 6983.17 (calcd for C216H293N69O155P21 [M − H]− 6986.65), RNA 7: m/z = 6983.17 (calcd for C216H293N69O155P21 [M − H]− 6986.45), RNA 8: m/z = 6983.17 (calcd for C216H293N69O155P21 [M − H]− 6987.09), RNA 9: m/z = 6812.95 (calcd for C203H248N86O144P20 [M − H]− 6815.80), RNA 10: m/z = 7390.45 (calcd for C234H333N73O159P22 [M − H]− 7394.90). RNA 11: m/z = 7390.45 (calcd for C234H333N73O159P22 [M − H]− 7395.37). RNA 12: m/z = 7462.50 (calcd for C238H340N73O160P22 [M − H]− 7467.83). RNA 13: m/z = 7351.08 (calcd for C230H273N87O153P21 [M − H]− 7354.35).Thermal stability
600 pmol, each of passenger and guide strand were suspended in 200 μL buffer of 10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl, heated to 100 °C and then cooled gradually to room temperature providing the siRNA with final concentration of 3.0 μM for the UV melting experiment. Thermally induced transitions were recorded at 260 nm with a UV/vis spectrophotometer fitted with temperature controller in 8-series micro multi-cell with a path length of 1.0 cm. The sample temperature was increased by 0.5 °C min−1.CD spectra
All CD spectra were recorded at 25 °C. The following instrument settings were used: resolution, 0.1 nm; response, 1.0 s; speed, 50 nm min−1; accumulation, 10.Bovine serum stability
Fluorescein labeled siRNAs (600 pmol) were dissolved in 20 μL buffer of 10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl. The samples were hybridized by heating 100 °C and then cooling gradually to room temperature to use for the serum stability test. 106 μL of opti-MEM and 6 μL of bovine serum were added, and the solution was incubated at 37 °C for the required time. Aliquots of 6.7 μL were diluted with a stop solution (65 mM EDTA, 15% glycerol, 6.0 μL). Samples were subjected to electrophoresis in non-denaturing 15% polyacrylamide gel and analyzed by a Luminescent Image analyzer LAS-4000 (Fujifilm).RNAi activity
HeLa cells were transfected with the psiCHECK-2 vector (Promega) and the pcDNA3.1 containing a hygromycin resistance gene (Thermo Fisher Scientific). HeLa Cells were grown in the presence of 0.5 mg mL−1 hygromycin for 1 week. Stable HeLa-psiCHECK-2 cells expressing both firefly and Renilla luciferases were grown in Dulbecco's Modified Eagle Medium (D-MEM) supplemented with 0.25 mg mL−1 hygromycin and 10% bovine serum (BS) at 37 °C. 24 hours prior to transfection of siRNAs, HeLa-psiCHECK-2 cells (8.0 × 104 mL−1) were grown in a 96-well plate (100 μL per well). The cells were transfected with siRNAs targeting the Renilla luciferase gene using lipofectamine RNAiMAX in Opti-MEM I reduced serum medium. Transfection without siRNAs was used as a control. After 1 hour of transfection, each cell was seeded with D-MEM (50 μL) containing 10% BS and cells were further incubated for another 24 hours. The activities of firefly and Renilla luciferases in the cells were measured using the Dual-Luciferase Reporter Assay System (Promega) according to manufacturer's protocol. The activity of Renilla luciferase was normalized by the firefly luciferase activity. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average from four experiments as mean ± SD.Author contributions
K. K. – conceptualization, methodology, data curation. A. C. – data curation, formal analysis, visualization, writing original draft preparation. Y. U. – conceptualization, funding acquisition, supervision, writing-review, and editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Japan Agency for Medical Research and Development (AMED) through its funding program for the research project number: 21ae0121029h0001.References
- N. Schütze, siRNA technology, Mol. Cell. Endocrinol., 2004, 213(2), 115–119, DOI:10.1016/j.mce.2003.10.078.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Duplexes of 21 ± nucleotide RNAs mediate RNA interference in cultured mammalian cells, Nature, 2001, 411(6836), 494–498, DOI:10.1038/35078107.
- A. Fire, RNA-triggered gene silencing, Trends Genet., 1999, 15(9), 358–363, DOI:10.1016/S0168-9525(99)01818-1.
- P. A. Sharp, RNA interference - 2001, Genes Dev., 2001, 15(5), 485–490, DOI:10.1101/gad.880001.
- J. B. Ma, K. Ye and D. J. Patel, Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain, Nature, 2004, 429(6989), 318–322, DOI:10.1038/nature02519.
- B. Wang, S. Li, H. H. Qi, D. Chowdhury, Y. Shi and C. D. Novina, Distinct passenger strand and mRNA cleavage activities of human Argonaute proteins, Nat. Struct. Mol. Biol., 2009, 16(12), 1259–1266, DOI:10.1038/nsmb.1712.
- Z. Paroo and D. R. Corey, Challenges for RNAi in vivo, Trends Biotechnol., 2004, 22(8), 390–394, DOI:10.1016/j.tibtech.2004.06.004.
- C. V. Pecot, G. A. Calin, R. L. Coleman, G. Lopez-Berestein and A. K. Sood, RNA interference in the clinic: Challenges and future directions, Nat. Rev. Cancer, 2011, 11(1), 59–67, DOI:10.1038/nrc2966.
- D. Haussecker, Current issues of RNAi therapeutics delivery and development, J. Control. Release, 2014, 195, 49–54, DOI:10.1016/j.jconrel.2014.07.056.
- J. K. Nair, J. L. S. Willoughby and A. Chan, et al., Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing, J. Am. Chem. Soc., 2014, 136(49), 16958–16961, DOI:10.1021/ja505986a.
- K. G. Rajeev, J. K. Nair and M. Jayaraman, et al., Hepatocyte-Specific Delivery of siRNAs Conjugated to Novel Non-nucleosidic Trivalent N-Acetylgalactosamine Elicits Robust Gene Silencing in Vivo, ChemBioChem, 2015, 16(6), 903–908, DOI:10.1002/cbic.201500023.
- M. M. Janas, M. K. Schlegel and C. E. Harbison, et al., Selection of GalNAc-conjugated siRNAs with limited off-Target-driven rat hepatotoxicity, Nat. Commun., 2018, 9(1), 723, DOI:10.1038/s41467-018-02989-4.
- Y. N. Lamb, Inclisiran: First Approval, Drugs, 2021, 81(3), 389–395, DOI:10.1007/s40265-021-01473-6.
- M. Amarzguioui, T. Holen, E. Babaie and H. Prydz, Tolerance for mutations and chemical modifications in a siRNA, Nucleic Acids Res., 2003, 31(2), 589–595, DOI:10.1093/nar/gkg147.
- M. Robbins, A. Judge, L. Liang, K. McClintock, E. Yaworski and I. MacLachlan, 2′-O-methyl-modified RNAs act as TLR7 antagonists, Mol. Ther., 2007, 15(9), 1663–1669, DOI:10.1038/sj.mt.6300240.
- M. Manoharan, A. Akinc and R. K. Pandey, et al., Unique Gene-Silencing and Structural Properties of 2′-Fluoro-Modified siRNAs, Angew. Chem., Int. Ed., 2011, 50(10), 2284–2288, DOI:10.1002/anie.201006519.
- A. Patra, M. Paolillo, K. Charisse, M. Manoharan, E. Rozners and M. Egli, 2′-Fluoro RNA shows increased Watson-Crick H-bonding strength and stacking relative to RNA: evidence from NMR and thermodynamic data, Angew Chem. Int. Ed. Engl., 2012, 51(47), 11863–11866, DOI:10.1002/anie.201204946.
- P. Kumar, H. S. Ban and S. S. Kim, et al., T Cell-Specific siRNA Delivery Suppresses HIV-1 Infection in Humanized Mice, Cell, 2008, 134(4), 577–586, DOI:10.1016/j.cell.2008.06.034.
- Y. Tang, Y. B. Li and B. Wang, et al., Efficient in vitro siRNA delivery and intramuscular gene silencing using PEG-modified PAMAM dendrimers, Mol. Pharm., 2012, 9(6), 1812–1821, DOI:10.1021/mp3001364.
- M. G. Sebestyén, S. C. Wong, V. Trubetskoy, D. L. Lewis and C. I. Wooddell, Targeted In Vivo Delivery of siRNA and an Endosome-Releasing Agent to Hepatocytes, Methods Mol. Biol., 2015, 1218, 163–186, DOI:10.1007/978-1-4939-1538-5_10.
- R. A. Haraszti, R. Miller and M. C. Didiot, et al., Optimized Cholesterol-siRNA Chemistry Improves Productive Loading onto Extracellular Vesicles, Mol. Ther., 2018, 26(8), 1973–1982, DOI:10.1016/j.ymthe.2018.05.024.
- S. Gangopadhyay, R. R. Nikam and K. R. Gore, Folate Receptor-Mediated siRNA Delivery: Recent Developments and Future Directions for RNAi Therapeutics, Nucleic Acid Therapeut., 2021, 31(4), 245–270, DOI:10.1089/nat.2020.0882.
- K. T. Gagnon, J. K. Watts and H. M. Pendergraff, et al., Antisense and Antigene Inhibition of Gene Expression by Cell-Permeable Oligonucleotide–Oligospermine Conjugates, J. Am. Chem. Soc., 2011, 133(22), 8404–8407, DOI:10.1021/ja200312y.
- C. Paris, V. Moreau and G. Deglane, et al., Conjugating Phosphospermines to siRNAs for Improved Stability in Serum, Intracellular Delivery and RNAi-Mediated Gene Silencing, Mol. Pharm., 2012, 9(12), 3464–3475, DOI:10.1021/mp300278b.
- M. Elsayed, V. Corrand and V. Kolhatkar, et al., Influence of Oligospermines Architecture on Their Suitability for siRNA Delivery, Biomacromolecules, 2014, 15(4), 1299–1310, DOI:10.1021/bm401849d.
- M. Nothisen, J. Bagilet, J. P. Behr, J. S. Remy and M. Kotera, Structure Tuning of Cationic Oligospermine–siRNA Conjugates for Carrier-Free Gene Silencing, Mol. Pharm., 2016, 13(8), 2718–2728, DOI:10.1021/acs.molpharmaceut.6b00309.
- T. Wang, B. Hao and S. Xu, et al., Effective RNAi in leukemia cells is enhanced by spermine-modified pullulan combined with desloratadine, Carbohydr. Polym., 2022, 292, 119646, DOI:10.1016/j.carbpol.2022.119646.
- K. Nakamoto, Y. Akao, Y. Furuichi and Y. Ueno, Enhanced Intercellular Delivery of cRGD–siRNA Conjugates by an Additional Oligospermine Modification, ACS Omega, 2018, 3(7), 8226–8232, DOI:10.1021/acsomega.8b00850.
- D. Chu, W. Xu, R. Pan, Y. Ding, W. Sui and P. Chen, Rational modification of oligoarginine for highly efficient siRNA delivery: Structure-activity relationship and mechanism of intracellular trafficking of siRNA, Nanomed. Nanotechnol. Biol. Med., 2015, 11(2), 435–446, DOI:10.1016/j.nano.2014.08.007.
- A. Srimanee, M. Arvanitidou, K. Kim, M. Hällbrink and Ü. Langel, Cell-penetrating peptides for siRNA delivery to glioblastomas, Peptides, 2018, 104, 62–69, DOI:10.1016/j.peptides.2018.04.015.
- L. Salim, C. McKim and J. P. Desaulniers, Effective carrier-free gene-silencing activity of cholesterol-modified siRNAs, RSC Adv., 2018, 8(41), 22963–22966, 10.1039/C8RA03908A.
- L. Salim, G. Islam and J. P. Desaulniers, Targeted delivery and enhanced gene-silencing activity of centrally modified folic acid-siRNA conjugates, Nucleic Acids Res., 2020, 48(1), 75–85, DOI:10.1093/NAR/GKZ1115.
- C. Fernandez, I. Giorgees, E. Goss and J. P. Desaulniers, Effective carrier-free gene-silencing activity of sphingosine-modified siRNAs, Org. Biomol. Chem., 2023, 21(10), 2107–2117, 10.1039/D2OB02099H.
- K. Morita, M. Takagi and C. Hasegawa, et al., Synthesis and properties of 2′-O,4′-C-ethylene-bridged nucleic acids (ENA) as effective antisense oligonucleotides, Bioorg. Med. Chem., 2003, 11(10), 221–2226, DOI:10.1016/S0968-0896(03)00115-9.
- K. Koizumi, Y. Maeda and T. Kano, et al., Synthesis of 4′-C-aminoalkyl-2′-O-methyl modified RNA and their biological properties, Bioorg. Med. Chem., 2018, 26(12), 3521–3534, DOI:10.1016/J.BMC.2018.05.025.
- T. Kano, Y. Katsuragi, Y. Maeda and Y. Ueno, Synthesis and properties of 4′-C-aminoalkyl-2′-fluoro-modified RNA oligomers, Bioorg. Med. Chem., 2018, 26(15), 4574–4582, DOI:10.1016/j.bmc.2018.08.001.
- M. Hassler, Y. Q. Wu, N. Mallikarjuna Reddy, T. H. Chan and M. J. Damha, RNA synthesis via dimer and trimer phosphoramidite block coupling, Tetrahedron Lett., 2011, 52(20), 2575–2578, DOI:10.1016/j.tetlet.2011.03.042.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04918c |
| This journal is © The Royal Society of Chemistry 2023 |