
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis, X-ray crystal structures, anticancer, DNA binding, and molecular modelling studies of pyrazole–pyrazoline hybrid derivatives†
Manish Ranaab,
Hungharla Hungyoc,
Palak Parasharc,
Shaban Ahmad d,
Rabiya Mehandia,
Vibha Tandonc,
Khalid Razad,
Mohammed A. Assirie,
Tarik E. Alie,
Zeinhom M. El-Bahyf and
Rahisuddin*a
d,
Rabiya Mehandia,
Vibha Tandonc,
Khalid Razad,
Mohammed A. Assirie,
Tarik E. Alie,
Zeinhom M. El-Bahyf and
Rahisuddin*a
aMolecular and Biophysical Research Lab (MBRL), Department of Chemistry, Jamia Millia Islamia, New Delhi 110025, India. E-mail: rahisuddin@jmi.ac.in; Tel: +91 9871460479
bDepartment of Chemistry, Ramjas College, University of Delhi, Delhi 110007, India
cSpecial Centre for Molecular Medicine, Jawaharlal Nehru University, New Delhi 110067, India
dDepartment of Computer Science, Jamia Millia Islamia, New Delhi 110025, India
eDepartment of Chemistry, Faculty of Science, King Khalid University, Abha, 61421, Saudi Arabia
fDepartment of Chemistry, Faculty of Science, Al-Azhar University, Nasr City 11884, Cairo, Egypt
First published on 6th September 2023
Abstract
We have designed and synthesized three pyrazole analogs (4, 5a, 5b), pyrazole-based chalcones (6a–6d) and (8a–8h), and N-formyl/acetyl 1,3,5-trisubstituted pyrazoline analogs (7a–7d), (9a–9d). FT-IR, 1H, 13C NMR, and mass spectrometry techniques were used to describe the structures of all the synthesized analogs. The single crystal X-ray method was used to identify the molecular structure of derivatives 4 and 5a. All synthesized analogs were screened by MTT assay on two cancer cell lines, the human lung cancer cell line (A549) and cervical cancer cell line (HeLa). Among all compounds, analog 9d demonstrates significant anticancer activity against HeLa (IC50 = 23.6 μM) and A549 (IC50 = 37.59 μM). The non-interactive interaction of active compound (9d) with Calf thymus DNA (Ct-DNA) has been investigated through various methods, such as UV-vis absorption, emission, cyclic voltammetry and circular dichroism. The DPPH (2,2-diphenyl-1-picrylhydrazyl) free radical has been used to measure the antioxidant capacity of the pyrazoline derivative (9d). The outcomes showed that active analog has significant antioxidant activity. In addition, MD simulation of the EGFR tyrosine kinase protein–ligand complex was performed at a time scale of 100 ns. The MMGBSA data of ligand–protein complex are showed stable interactions up to 100 ns.
1. Introduction
Cancer is a significant health issue in both developed and developing countries since it is the second most feared illness in the world that causes death after cardiovascular diseases.1–3 Heterocyclic compounds have exceptional medicinal utility due to their unique action and are utilised in treating serious infectious disorders. A broad spectrum of these heterocycles has been investigated to produce therapeutically important compounds including chalcone and its derivatives, such as pyrazoles.4,5 Commonly referred to as chalcones, α–β unsaturated ketones are a significant family of natural products as well as synthetic chemicals that have shown a variety of biological activity.6,7 However, not many ways for derivatizing flavonoids, particularly chalcones, under Suzuki cross-coupling conditions have been reported.8–10 As parent heterocycles of several commercially available medications, pyrazoline and pyrazole heterocycles have unique biological properties.11,12 Pyrazole dihydropyrazole is an active heterocycle that was initially discovered in watermelon seeds and is utilised as an antibacterial,13 antitubercular,14 antiviral,15 anti-malarial,16 anti-cancer,17,18 analgesic,19 and anti-inflammatory drug.20,21 In the framework of the hybrid pharmacophore method, which enables the achievement of new pharmacological profiles, potentiation of action, and decrease in toxicity, it is noteworthy that combining pyrazole fragments with other heterocycles is a recognised strategy to construct drug-like compounds. A viable direction for contemporary medicinal chemistry in this situation is the synthesis of novel drug-like compounds based on the pharmacologically appealing scaffolds of pyrazole/pyrazoline.22 Pyrazole and pyrazoline, working together synergistically, have produced compounds with enhanced anticancer activity.23 Here, we attempted to develop a variety of heterocyclic derivatives with potential anticancer action against human lung cancer cells (A549) and cervical cell line (HeLa). The current research investigates the properties of heterocyclic derivatives in terms of MD simulation, DNA-binding, antioxidant, and ADMET assay. The design strategy for synthesized compounds is:
2. Result and discussion
2.1 Chemistry
The intermediate 3 was synthesized by the condensation reaction of 4-bromo phenyl hydrazine and 4-methoxy acetophenone. To obtain pyrazole derivative 4, the intermediate 3 was cyclized with formylation by the Vilsmeier–Haack reaction. The chalcone derivatives 6a–6d were synthesized by the condensation reaction of pyrazole analog 4 with substituted acetophenone in the presence of 50% NaOH at room temperature. The derivatives 6a–6d further react with hydrazine hydrate in the presence of formic/acetic acid to obtain 7a–7d and 9a–9d, respectively. Analogs 4 and 6a–6d react with substituted boronic acid using Suzuki coupling reaction in the presence of Pd(PPh3)4 catalyst in solvent ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (H2O:THF:Toluene). The IR spectra of compound 4 showed a conspicuous, strong absorption band at 1678 cm−1 caused by the presence of the –C
:1:1 (H2O:THF:Toluene). The IR spectra of compound 4 showed a conspicuous, strong absorption band at 1678 cm−1 caused by the presence of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in carbaldehyde. At 3130 cm−1, a second sharp, strong absorption band attributed to the C–H stretching of the carbonyl group occurred. Compound 6a–6d has a –CO peak range of 1648–1663 cm−1 and a –CC– range of 1529–1596 cm−1. The IR spectra of the pyrazolo-pyrazoline derivatives (7a–7d) and (9a–9d) show distinctive (CO), (CN) bands that emerge at 1663–1719 and 1592–1663 cm−1, respectively. The characteristic peaks of heterocyclic analogs are given in Fig. S1.† A conventional ABX system, in which methylene proton resonates as a pair of doublets seen at the range 3.05–3.27 ppm (HA) and 3.58–3.78 ppm (HB) for compounds 7a–7d and 9a–9d respectively, indicated the development of pyrazoline ring in the target compounds. Due to its vicinal interaction with the magnetically non-equivalent proton of the methylene (–CH2) group, methine (–CH) proton was observed as a doublet of doublets at 5.78–5.85 ppm (HX) and a singlet of methoxy proton in the range of 3.80–3.91 ppm. In 13C NMR, compounds 4, 5a, and 5b show peaks in the range 185.09–185.27 ppm belonging to –CHO groups, confirming the formylation of the pyrazol ring. The compounds 6a–6d and 8a–8h show –CO groups at 181.40–191.14 ppm. The compounds 7a–7d exhibited an N-formyl peak in the range of 160.31–160.44 ppm and an N-acetyl peak for compounds 9a–9d in the range of 169.12–176.55 ppm. All the compounds show –OMe peak in the range of 55.26–55.61 ppm. The –CH protons of the pyrazoline ring for compounds 7a–7d and 9a–9d are in the range of 51.51–52.72 ppm, and –CH2 protons are at 42.06–44.90 ppm, which confirms the formation of pyrazoline ring. The 1H NMR and 13C NMR spectra of lead compound 9d are shown in Fig. 1 and remaining in Fig. S1.† The FT-IR and mass spectra of all heterocyclic analogs are given in Fig. S2 and S3.†
O group in carbaldehyde. At 3130 cm−1, a second sharp, strong absorption band attributed to the C–H stretching of the carbonyl group occurred. Compound 6a–6d has a –CO peak range of 1648–1663 cm−1 and a –CC– range of 1529–1596 cm−1. The IR spectra of the pyrazolo-pyrazoline derivatives (7a–7d) and (9a–9d) show distinctive (CO), (CN) bands that emerge at 1663–1719 and 1592–1663 cm−1, respectively. The characteristic peaks of heterocyclic analogs are given in Fig. S1.† A conventional ABX system, in which methylene proton resonates as a pair of doublets seen at the range 3.05–3.27 ppm (HA) and 3.58–3.78 ppm (HB) for compounds 7a–7d and 9a–9d respectively, indicated the development of pyrazoline ring in the target compounds. Due to its vicinal interaction with the magnetically non-equivalent proton of the methylene (–CH2) group, methine (–CH) proton was observed as a doublet of doublets at 5.78–5.85 ppm (HX) and a singlet of methoxy proton in the range of 3.80–3.91 ppm. In 13C NMR, compounds 4, 5a, and 5b show peaks in the range 185.09–185.27 ppm belonging to –CHO groups, confirming the formylation of the pyrazol ring. The compounds 6a–6d and 8a–8h show –CO groups at 181.40–191.14 ppm. The compounds 7a–7d exhibited an N-formyl peak in the range of 160.31–160.44 ppm and an N-acetyl peak for compounds 9a–9d in the range of 169.12–176.55 ppm. All the compounds show –OMe peak in the range of 55.26–55.61 ppm. The –CH protons of the pyrazoline ring for compounds 7a–7d and 9a–9d are in the range of 51.51–52.72 ppm, and –CH2 protons are at 42.06–44.90 ppm, which confirms the formation of pyrazoline ring. The 1H NMR and 13C NMR spectra of lead compound 9d are shown in Fig. 1 and remaining in Fig. S1.† The FT-IR and mass spectra of all heterocyclic analogs are given in Fig. S2 and S3.†
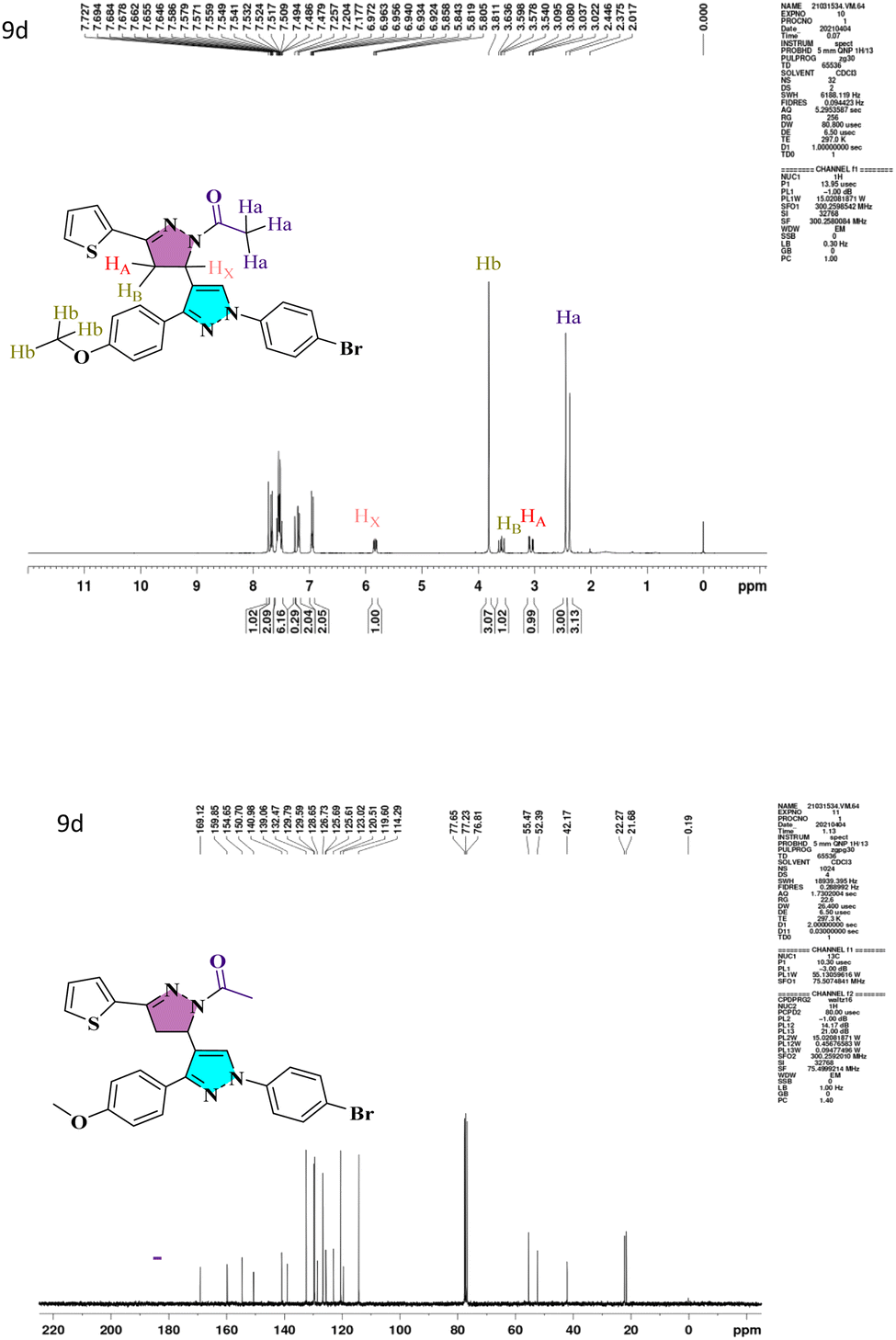 | ||
| Fig. 1 1H and 13C NMR spectra of lead compound 9d. | ||
2.2 Crystal structures
The pyrazol derivative structure and bonding characteristics were determined using X-ray crystallography.24 The X-ray crystal analyses demonstrate that the unit cell of compounds 4 and 5a was triclinic and monoclinic, respectively. Compound 4 has non-centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and 5a with P21/n space group, and z = 4, z = 8 correspond to compounds 4 and 5a based on single X-ray crystal studies. Lattice parameters for 4, a = 9.348(2) Å, b = 9.793(2) Å, c = 16.366(4) Å, α = 87.493(6)°, β = 87.318(6)°, γ = 84.676(6)°, and for 5a, a = 21.54552(17) Å, b = 7.38135(7) Å, c = 22.77667(19) Å, α = 90°, β = 101.0921(8)°, γ = 90° unit cell, as shown in Tables 1 and 2. The CCDC codes for the crystalline compounds 4 and 5a are 2196195 and 2235766, respectively. The ORTEP view of 4 and 5a is shown in Fig. 2.
and 5a with P21/n space group, and z = 4, z = 8 correspond to compounds 4 and 5a based on single X-ray crystal studies. Lattice parameters for 4, a = 9.348(2) Å, b = 9.793(2) Å, c = 16.366(4) Å, α = 87.493(6)°, β = 87.318(6)°, γ = 84.676(6)°, and for 5a, a = 21.54552(17) Å, b = 7.38135(7) Å, c = 22.77667(19) Å, α = 90°, β = 101.0921(8)°, γ = 90° unit cell, as shown in Tables 1 and 2. The CCDC codes for the crystalline compounds 4 and 5a are 2196195 and 2235766, respectively. The ORTEP view of 4 and 5a is shown in Fig. 2.
| Identification code | 4 |
|---|---|
| Empirical formula | C17H13BrN2O2 |
| Formula weight | 357.19 |
| Temperature/K | 150(2) K |
| Crystal system | Triclinic |
| Space group | P |
| a/Å | 9.348(2) Å |
| b/Å | 9.793(2) Å |
| c/Å | 16.366(4) Å |
| α/° | 87.493(6)° |
| β/° | 87.318(6)° |
| γ/° | 84.676(6)° |
| Volume/Å3 | 1489.0(6) Å3 |
| Z | 4 |
| ρcalcmg m−3 | 1.593 |
| μ/mm−1 | 2.769 |
| F(000) | 720.0 |
| Crystal size/mm3 | 0.06 × 0.05 × 0.04 |
| Radiation | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 2.090 to 25.062° |
| Index ranges | −11 ≤ h ≤ 11, −11 ≤ k ≤ 11, −19 ≤ l ≤ 19 |
| Reflections collected | 35618 |
| Independent reflections | 5267 [R(int) = 0.0586] |
| Data/restraints/parameters | 5267/0/399 |
| Goodness-of-fit on F2 | 1.024 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0277, wR2 = 0.0717 |
| Final R indexes [all data] | R1 = 0.0424, wR2 = 0.0781 |
| Largest diff. peak/hole/e Å−3 | 0.349 and −0.480 e Å−3 |
| Identification code | 5a |
|---|---|
| Empirical formula | C23H18N2O2 |
| Formula weight | 354.41 |
| Temperature/K | 293(2) |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a/Å | 21.54552(17) |
| b/Å | 7.38135(7) |
| c/Å | 22.77667(19) |
| α/° | 90 |
| β/° | 101.0921(8) |
| γ/° | 90 |
| Volume/Å3 | 3554.62(5) |
| Z | 8 |
| ρcalcmg m−3 | 1.3244 |
| μ/mm−1 | 0.683 |
| F(000) | 1492.6 |
| Crystal size/mm3 | 0.39 × 0.21 × 0.14 |
| Radiation | Cu Kα (λ = 1.54184) |
| 2Θ range for data collection/° | 5.18 to 136.32 |
| Index ranges | −23 ≤ h ≤ 25, −8 ≤ k ≤ 8, −27 ≤ l ≤ 21 |
| Reflections collected | 36401 |
| Independent reflections | 6446 [Rint = 0.0294, Rsigma = 0.0161] |
| Data/restraints/parameters | 6446/0/490 |
| Goodness-of-fit on F2 | 1.032 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0368, wR2 = 0.0989 |
| Final R indexes [all data] | R1 = 0.0426, wR2 = 0.1031 |
| Largest diff. peak/hole/e Å−3 | 0.18/−0.14 |
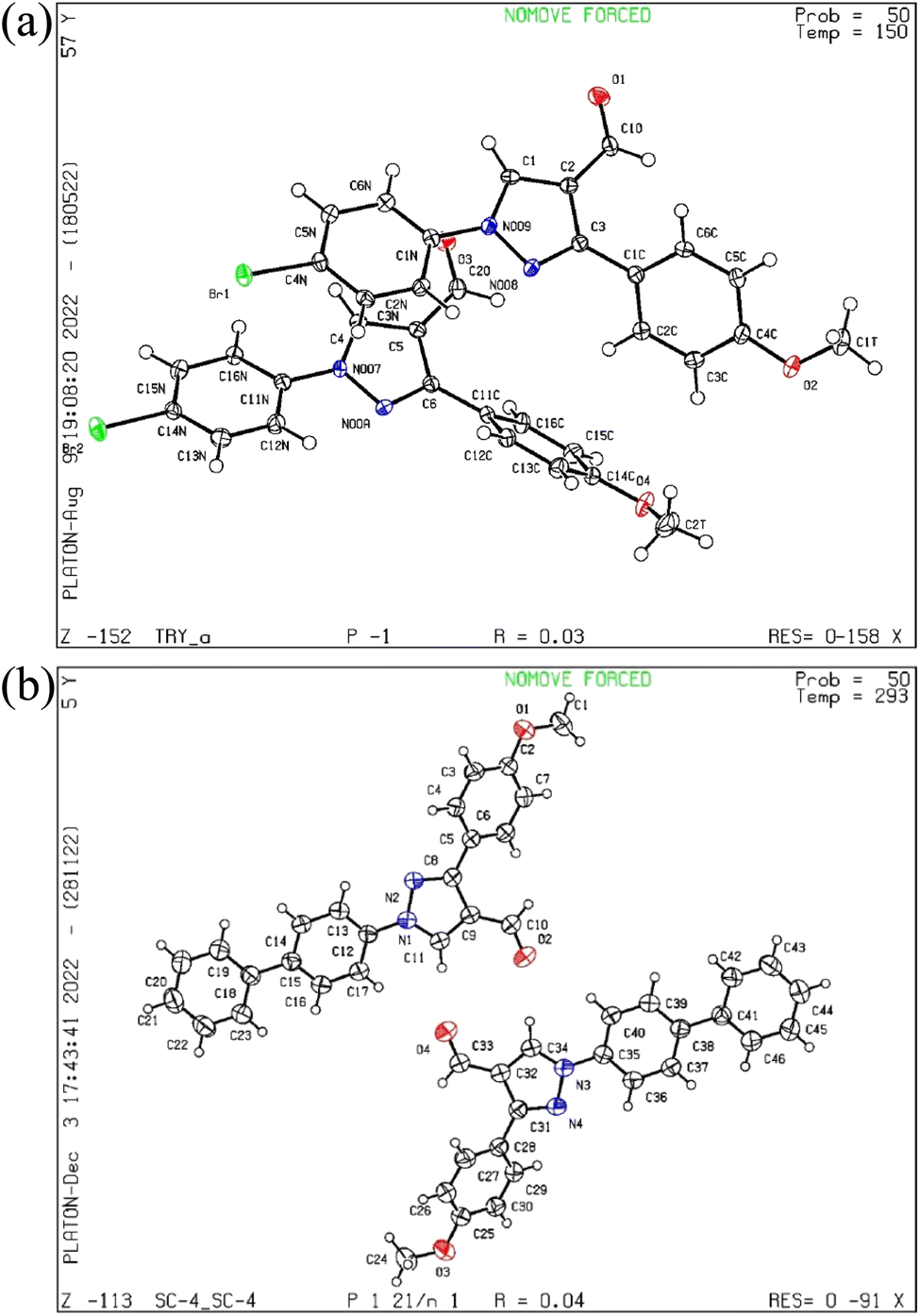 | ||
| Fig. 2 (a) The ORTEP view of compound 4, (b) the ORTEP view of compound 5a. | ||
2.3 Cytotoxicity
The cytotoxicity of synthesized compounds was evaluated against two cancer cell lines (A549 and HeLa) via MTT assay.25 It was found that the majority of chemical compounds synthesized in the laboratory showed no significant inhibitory activity. Compound 9d showed significant bioactivity as compared to standard drug nocodazole.26 As shown in Fig. 3 and Table 3, compound 9d exhibited significant inhibitory activities against HeLa and A549 cells at a dose concentration of 23.6 μM and 37.59 μM at 48 h, respectively.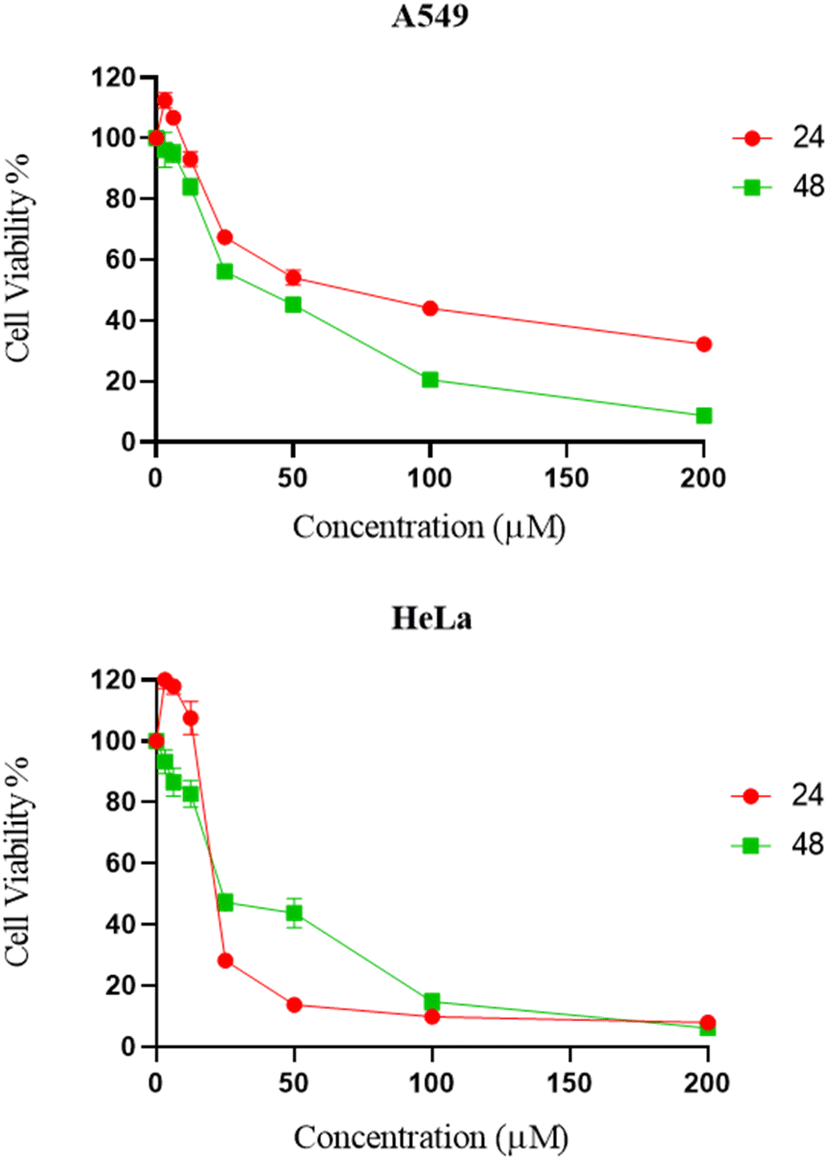 | ||
| Fig. 3 Dose response curves of compound 9d against A549 and HeLa cancer cell line. | ||
| Compounds | A549 | HeLa |
|---|---|---|
| 4 | 96.88 | 109.67 |
| 5a | 46.25 | >500 |
| 5b | 54.06 | 186.93 |
| 6a | >500 | >500 |
| 6b | >500 | >500 |
| 6c | >500 | >500 |
| 6d | >500 | >500 |
| 7a | >500 | >500 |
| 7b | >500 | >500 |
| 7c | >500 | >500 |
| 7d | 29.40 | 41.48 |
| 8a | >500 | >500 |
| 8b | >500 | >500 |
| 8c | >500 | >500 |
| 8d | >500 | >500 |
| 8e | >500 | >500 |
| 8f | >500 | >500 |
| 8g | >500 | >500 |
| 8h | >500 | >500 |
| 9a | 179.82 | >500 |
| 9b | 124.03 | 166 |
| 9c | >500 | >500 |
| 9d | 23.60 | 37.59 |
| Nocodazole26 | 1.87 ± 0.5 | 2.83 ± 0.3 |
2.4 Molecular docking and MD simulation
| PDB ID | PDB resolution | Docking score | MM/GBSA dG bind | Glide ligand efficiency sa | Glide ligand efficiency ln |
|---|---|---|---|---|---|
| 4HJO | 2.75 | −4.32 | −43.65 | −0.42 | −0.961 |
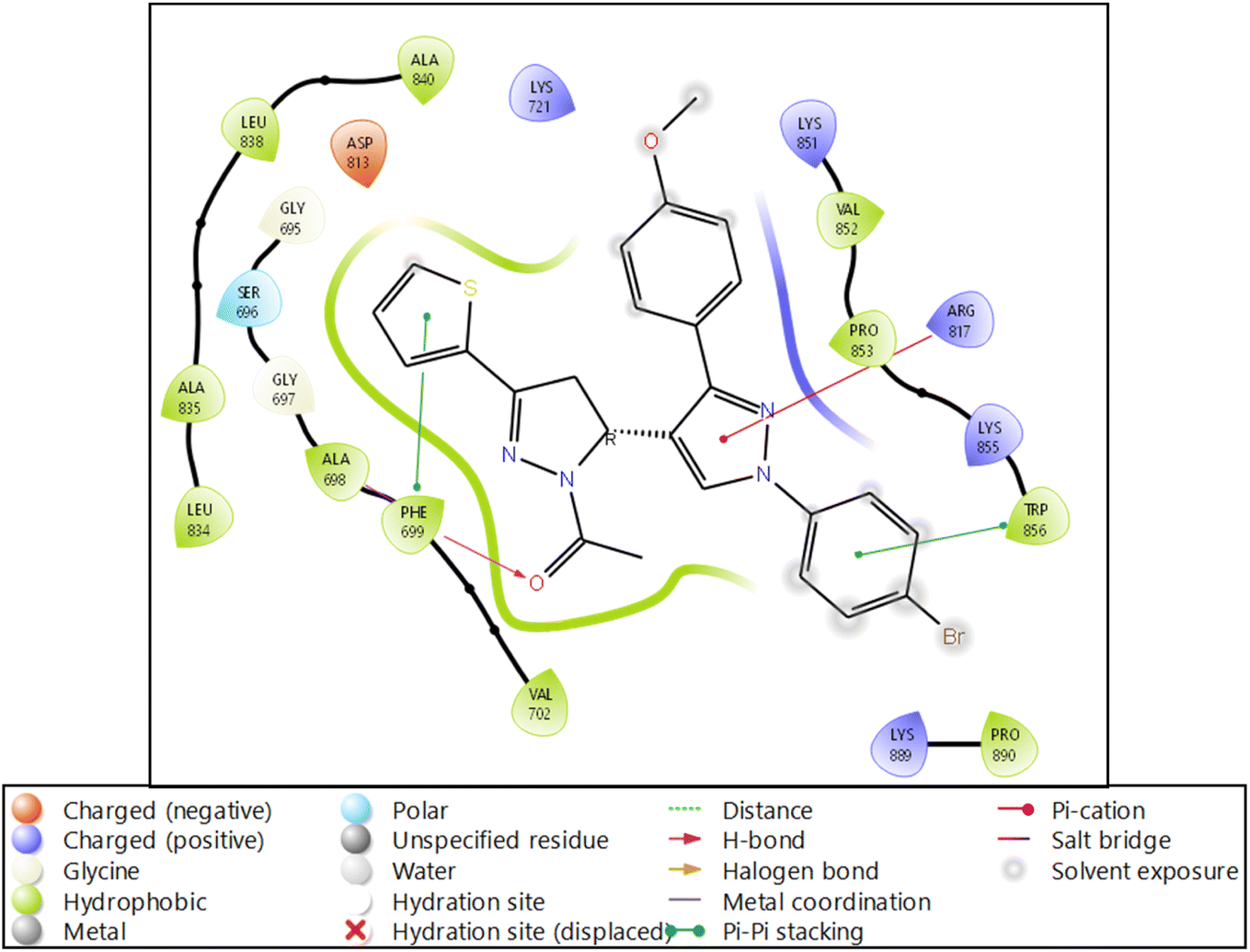 | ||
| Fig. 4 Ligand interaction diagram of EGFR tyrosine kinase with 9d and the legend is shown to understand the residues and interaction types. | ||
| Descriptors | Standard values | 9d | Descriptors | Standard values | 9d |
|---|---|---|---|---|---|
| #stars | 0–5 | 2 | QPlogS | −6.5–0.5 | −8.967 |
| #amine | 0–1 | 0 | CIQPlogS | −6.5–0.5 | −9.2 |
| #amidine | 0 | 0 | QPlogHERG | Concern below −5 | −7.252 |
| #acid | 0–1 | 0 | QPPCaco | <25 poor, >500 great | 3109.509 |
| #amide | 0–1 | 0 | QPlogBB | −3.0–1.2 | 0.203 |
| #rotor | 0–15 | 1 | QPPMDCK | <25 poor, >500 great | 7799.249 |
| #rtvFG | 0–2 | 0 | QPlogKp | −8.0 to −1.0 | −0.938 |
| CNS | −2 (inactive), +2 (active) | 1 | IP(eV) | 7.9–10.5 | 8.744 |
| mol MW | 130.0–725.0 | 521.43 | EA(eV) | −0.9–1.7 | 1.123 |
| Dipole | 1.0–12.5 | 7.054 | #metab | 1–8 | 4 |
| SASA | 300.0–1000.0 | 798.515 | QPlogKhsa | −1.5–1.5 | 1.324 |
| FOSA | 0.0–750.0 | 208.69 | HumanOralAbsorption | N/A | 1 |
| FISA | 7.0–330.0 | 53.064 | PercentHumanOralAbsorption | >80% is high, <25% is poor | 100 |
| PISA | 0.0–450.0 | 415.337 | SAfluorine | 0.0–100.0 | 0 |
| WPSA | 0.0–175.0 | 121.424 | SAamideO | 0.0–35.0 | 0 |
| Volume | 500.0–2000.0 | 1430.482 | PSA | 7.0–200.0 | 64.693 |
| donorHB | 0.0–6.0 | 0 | #NandO | 2–15 | 6 |
| accptHB | 2.0–20.0 | 5.25 | RuleOfFive | Maximum is 4 | 2 |
| dip^2/V | 0.0–0.13 | 0.0347858 | RuleOfThree | Maximum is 3 | 1 |
| ACxDN^.5/SA | 0.0–0.05 | 0 | #ringatoms | N/A | 27 |
| glob | 0.75–0.95 | 0.7689006 | #in34 | N/A | 0 |
| QPpolrz | 13.0–70.0 | 54.418 | #in56 | N/A | 27 |
| QPlogPC16 | 4.0–18.0 | 15.866 | #noncon | N/A | 2 |
| QPlogPoct | 8.0–35.0 | 21.98 | #nonHatm | N/A | 33 |
| QPlogPw | 4.0–45.0 | 9.82 | Jm | N/A | 0 |
| QPlogPo/w | −2.0–6.5 | 6.653 |
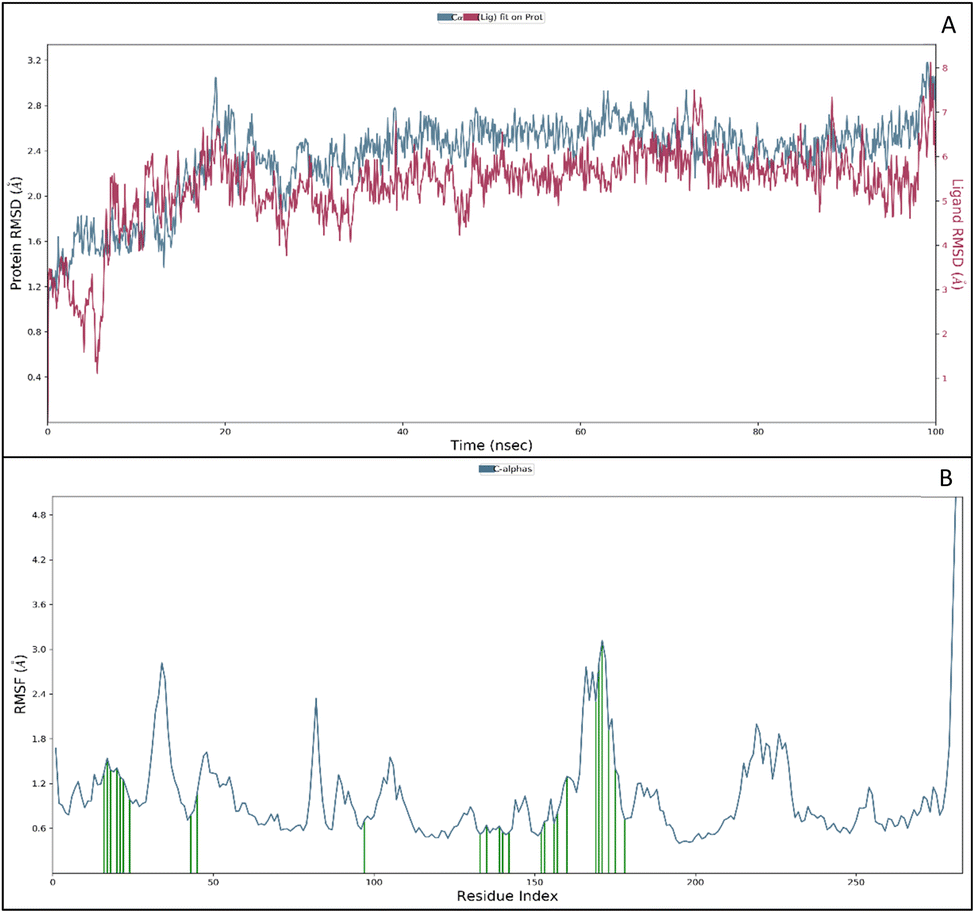 | ||
| Fig. 5 (A) Root Mean Square Deviation (RMSD) of protein (blue) and ligand (red) in Angstroms (Å) against time and (B) Root Mean Square Fluctuations (RMSF) of protein (Å) (blue) and ligand contact with the amino acids (green) against residue index. | ||
The RMSF measures the deviation of the positions of atoms or residues in a protein or other biological molecules from their average positions.31 It is often used in molecular dynamics (MD) simulations, where it can provide insight into the flexibility or stability of a protein or other biomolecules. The RMSF can be used to identify regions of a protein that are more flexible or less flexible, which can provide insight into the protein's function. It can also be used to compare the flexibility of different proteins or the results of different MD simulations. The RMSF is a valuable tool for understanding the structural and dynamic properties of proteins and other biological molecules at the atomic level. The protein did not show many fluctuations; however, a few residues, such as GLU710-LYS713, SER760, LYS843-GLY850, and GLN958-ASP960, show more fluctuation beyond 2 (Å). The fluctuated residues were not part of the interactions, so they do not bother much with the overall stability of the protein–ligand complex. There are many residues to which the ligand 9d formed the interactions. The interacting residues are LUE694, GLY695, SER696, ALA698PHE699, GLY700, VAL702, LYS721, LEU723, LEU775, HIS811, ASP813, ARG817, ASN818, LEU820, THR830, ASP831, LEU834, ALA835, LEU838, ALA847, GLU848, GLY849, LYS851, PRO853, and TRP856 (Fig. 5B).
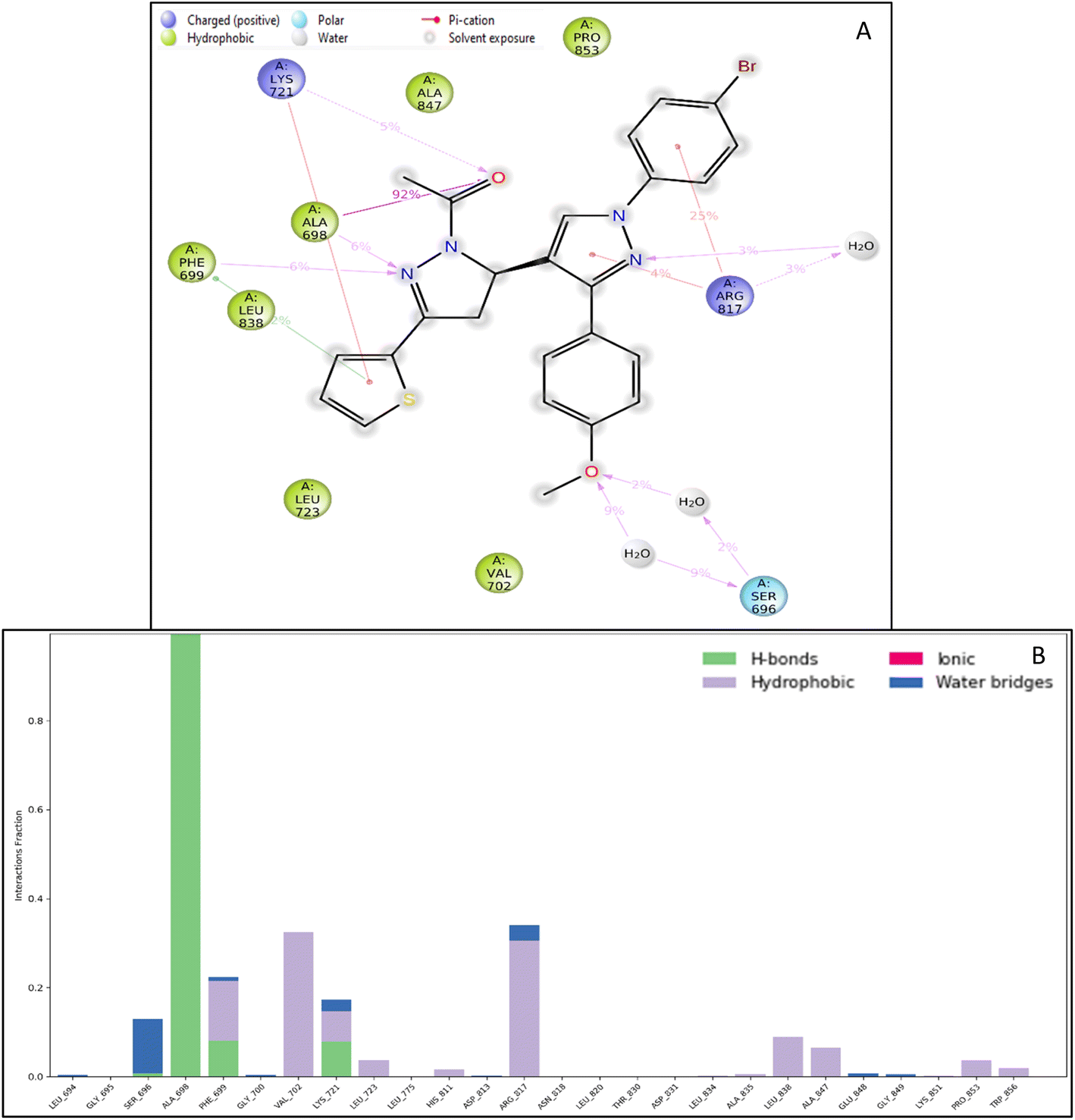 | ||
| Fig. 6 Intermolecular interactions formed during the molecular dynamics simulation (A) simulation interaction diagram and (B) histogram representation of the count of interactions and the legend is provided to understand each residue and bond type. | ||
2.5 DNA binding study
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) |
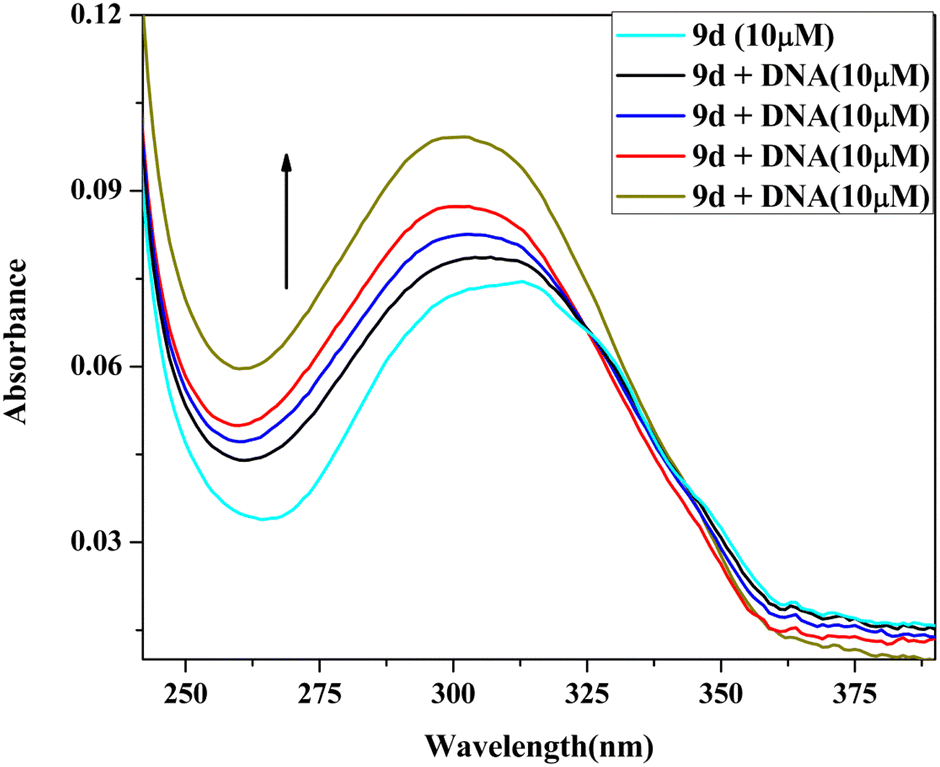 | ||
| Fig. 7 UV absorption spectra of 9d (10 μM) with increasing amounts of Ct-DNA (10–40 μM). | ||
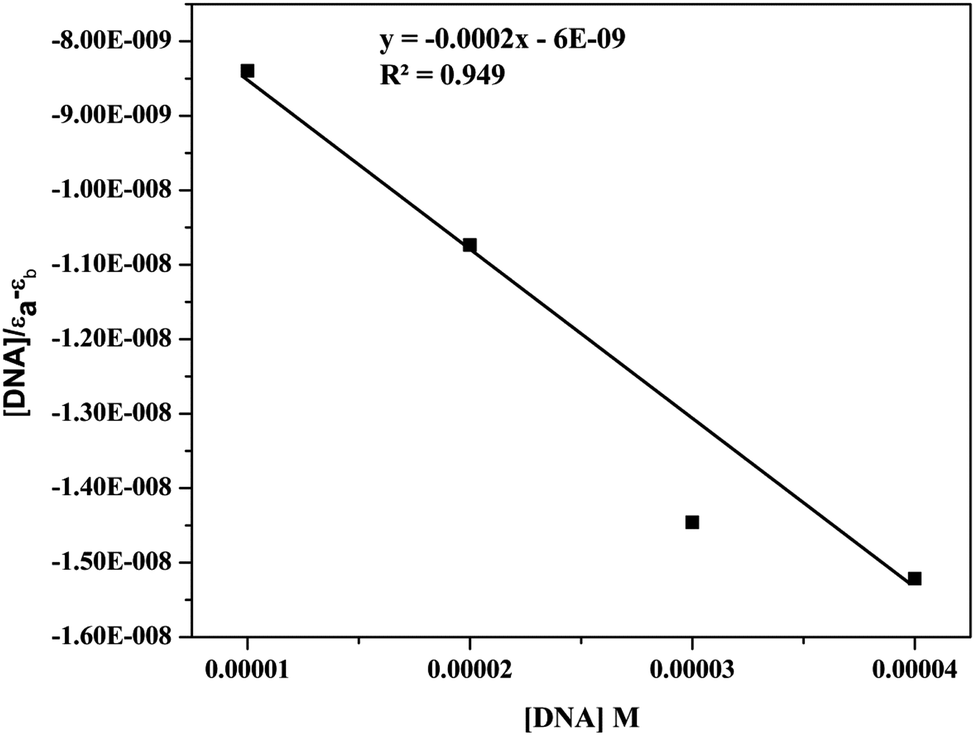 | ||
| Fig. 8 UV spectra and inset plot between [DNA]/[εa − εf] vs. [DNA] of 9d (10 μM) with increasing amounts of Ct-DNA (10–40 μM). | ||
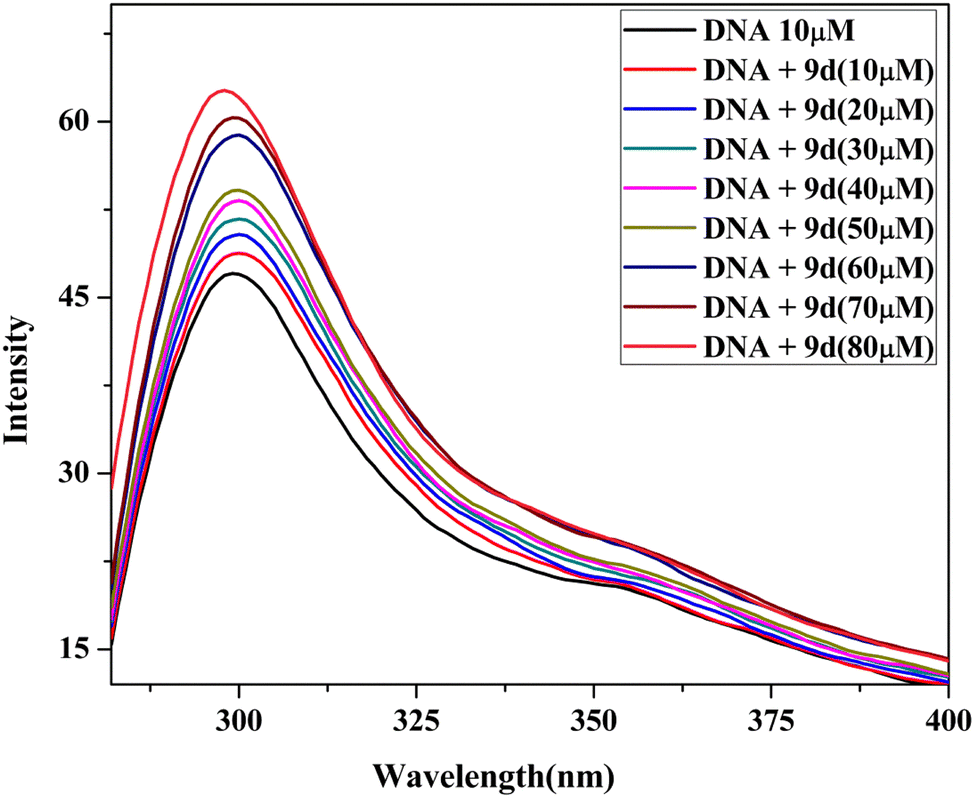 | ||
| Fig. 9 Emission titration of the lead compound 9d. | ||
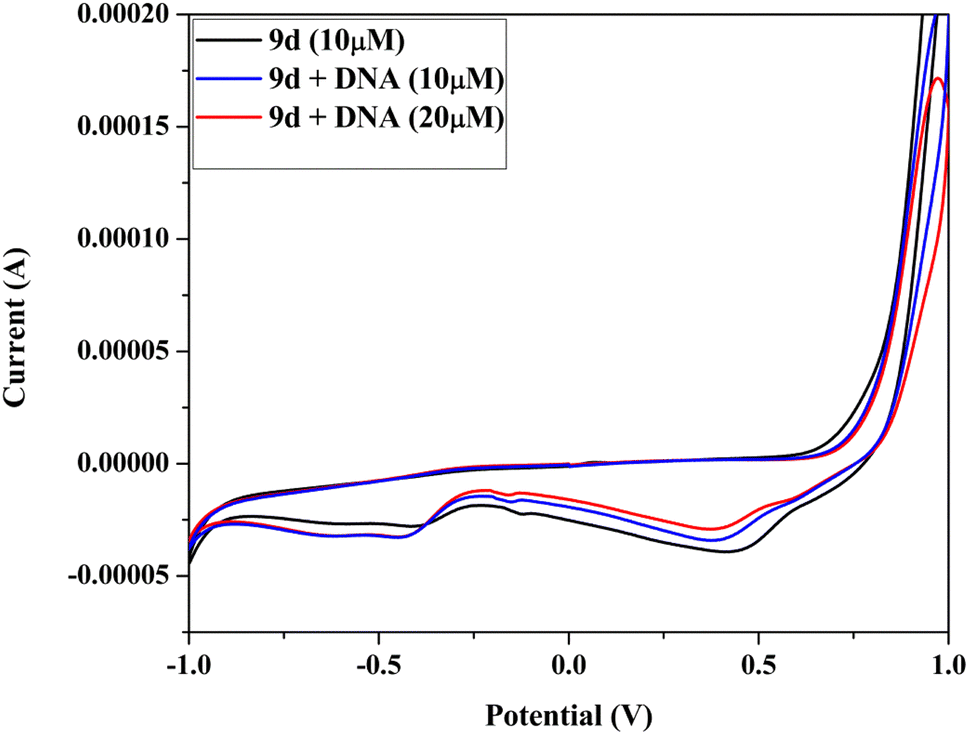 | ||
| Fig. 10 Cyclic voltammogram of analog 9d in Tris-buffer, at 100 mV s−1 scan rate with DNA (10–20 μM). | ||
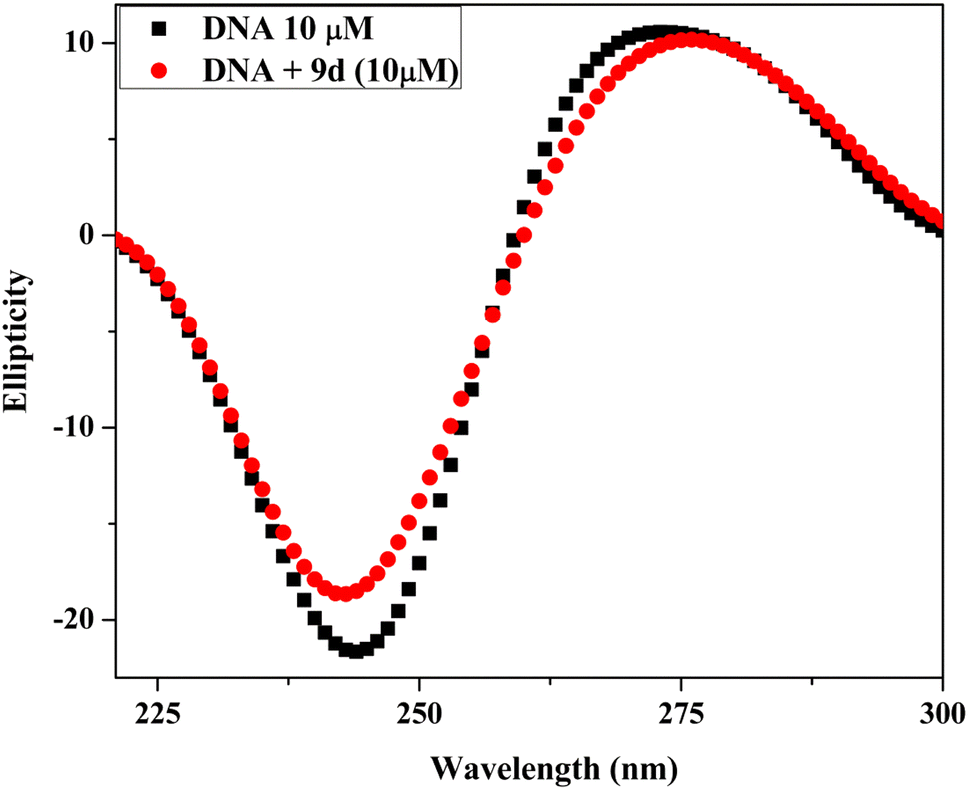 | ||
| Fig. 11 Circular dichroism spectra of Ct-DNA (10 μM) in the absence and presence of compound 9d (10 μM). | ||
2.6 Antioxidant assay
DPPH (2,2 diphenyl-1-picrylhydrazyl hydrate) is a free radical species that is stable and tends to accept an electron or a hydrogen and is therefore reduced by an electron or hydrogen provided by an oxidant. The presence of the antioxidant moiety was confirmed after 1 h of incubation when the colour changed from purple to yellow, and the absorbance dropped around 516 nm. Calculating the IC50 value of the compound has been done by monitoring the antioxidant activity.41,42 Ascorbic acid has been taken as standard. The calculated IC50 of the compound was 1.37 ± 0.015 mg mL−1. The obtained results demonstrate that the antioxidant activity of the test compound rises with concentration, as seen in Fig. 12.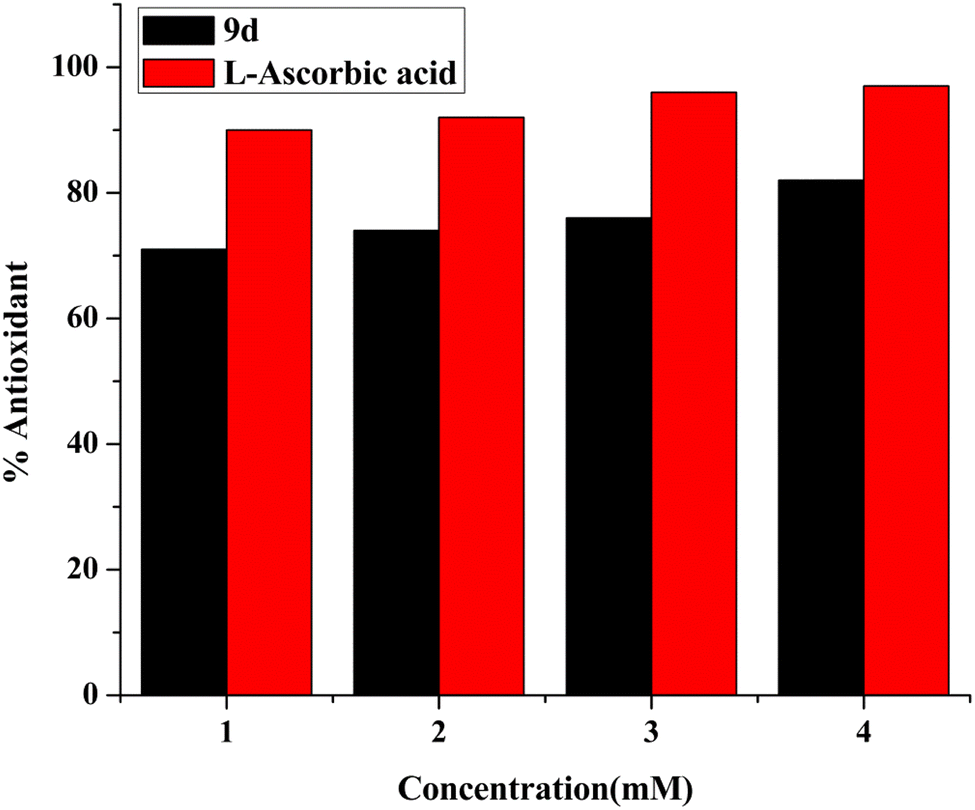 | ||
| Fig. 12 Concentration based bar graph of antioxidant ability of lead compound 9d. | ||
3. Experimental section
3.1 Material and methods
:1:1 (H2O:THF:Toluene) at N2 as inert atmosphere in the presence of catalyst Pd(PPh3)4 (5 mol%). The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture is extracted with EtOAc (5 × 30 mL), the organic layer was dried with anhydrous Na2SO4 and concentrated under reduced pressure. The precipitates obtained were filtered, washed, dried and recrystallized in ethanol.:ethanol (1:4) using 50% NaOH as a base. The precipitates obtained during the reaction were filtered, dried in a vacuum desiccator, and crystallized in ethanol.:3) as mobile phase was used to monitor the reaction progress. After the consume of chalcones, the hot reaction mixture was poured into ice-cold water. Precipitates were then filtered, washed with water, dried, and recrystallized in chloroform, and the affordable pyrazoline compounds (7a–7d) obtained are shown in Scheme 1.
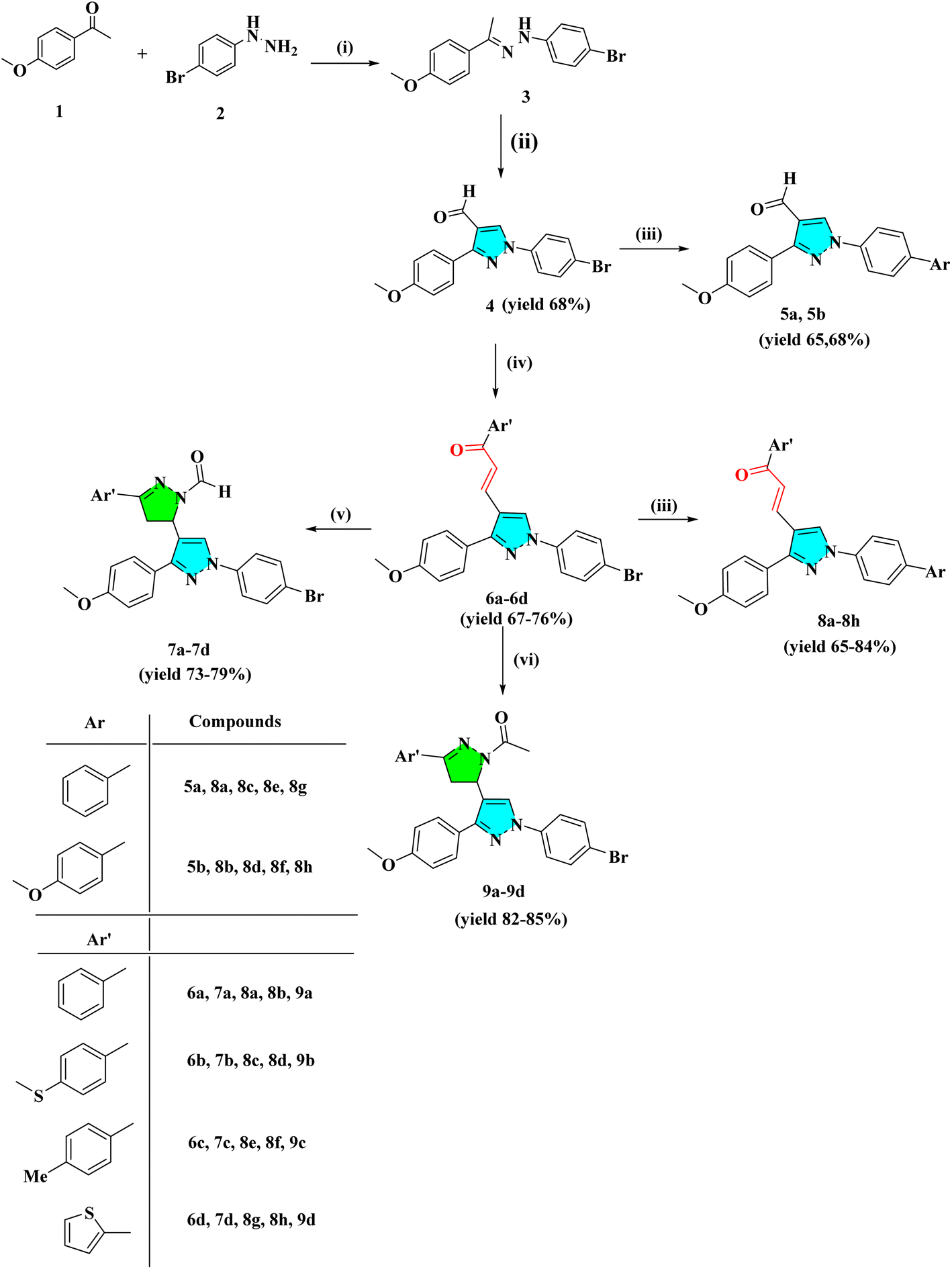 | ||
| Scheme 1 General procedure for the synthesis of heterocyclic derivatives. (i) Methanol, reflux for 4 h; (ii) DMF/POCl3 0–80 °C, NaHCO3 (iii) Pd(PPh3)4 (5 mol%), Ar-B(OH)2, H2O:THF:Toluene (1:1:1) reflux for 4–12 h (iv) Ar′COCH3, 50% NaOH, dioxane:ethanol (1:4), r.t (v) NH2NH2·H2O/HCOOH, reflux for 4–6 h (vi) NH2NH2·H2O/CH3COOH, reflux for 8–12 h. | ||
:1:1 (H2O:THF:Toluene) at N2 as inert atmosphere in the presence of catalyst Pd(PPh3)4 (5 mol%). The progress of the reaction was monitored by TLC. After the completion of the reaction, the reaction mixture is extracted with EtOAc (5 × 30 mL). The organic layer was dried with anhydrous Na2SO4 and concentrated under reduced pressure. The precipitates obtained were filtered, washed, dried and recrystallized in ethanol.:3) as mobile phase was used to monitor reaction progress. After the consume of chalcones, the hot reaction mixture was poured into ice-cold water. The precipitate was then filtered, washed with water, dried, and recrystallized in chloroform to obtain the affordable pyrazoline compounds. The synthetic reactions are shown in Scheme 1.
3.1.7.1 1-(4-Bromophenyl)-3-(4-methoxyphenyl)-1H-pyrazole-4-carbaldehyde (4). White solid; yield 68%; mp 168–170 °C; IR (neat, νmax): (–C
O str.) 3130, (–CN str.) 1678 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.52 (s, 1H), 7.78–7.80 (d, 2H), 7.69–7.72 (d, 2H), 7.64–7.66 (m, 3H), 7.04–7.06 (d, 2H), 3.90 (s, 3H); 13C NMR (CDCl3, δ): 185.12, 160.66, 154.73, 138.04, 132.77, 131.03, 130.26, 123.58, 122.62, 121.36, 121.07, 114.26, 55.41, ESI-MS. Calcd for C17H13BrN2O2, [M + H]+: m/z 356.02. Found: m/z 356.95.
3.1.7.2 1-([1,1′-Biphenyl]-4-yl)-3-(4-methoxyphenyl)-1H-pyrazole-4-carbaldehyde (5a). White solid; yield 65%; mp 220–225 °C; IR (neat, νmax): (–C
O str.) 2925, (–CN str.) 1674 cm−1; 1H NMR (Chloroform-D, δ ppm): δ 8.58 (s, 1H), 7.87–7.90 (m, 4H), 7.82–7.84 (d, 2H), 7.74–7.76 (d, 2H), 7.64–7.66 (d, 2H), 7.05–7.08 (m, 2H), 3.91 (s, 3H); 13C NMR (CDCl3, δ): 185.09, 160.67, 154.74, 140.82, 139.87, 138.09, 131.07, 130.30, 128.98, 128.26, 127.84, 127.04, 123.67, 119.97, 114.34, 55.26, ESI-MS. Calcd for C23H18N2O2, [M + H]+: m/z 354.14. Found: m/z 355.21.
3.1.7.3 1-(4′Methoxy-([1,1′-biphenyl]-4-yl))-3-(4-methoxyphenyl)-1H-pyrazole-4-carbaldehyde (5b). White solid; yield 68%; mp 210–212 °C; IR (neat, νmax): (–C
O str.) 2921, (–CN str.) 1607 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.56 (s, 1H), 7.82–7.84 (d, 2H), 7.69–7.71 (d, 2H), 7.57–7.59 (d, 2H), 7.49–7.51 (d, 2H), 6.97–7.07 (m, 4H), 3.86 (s, 3H); 13C NMR (CDCl3, δ): 185.27, 160.58, 159.58, 158.69, 154.58, 140.47, 137.63, 132.06, 131.04, 130.30, 128.10, 127.74, 123.85, 122.34, 119.99, 114.42, 114.23, 114.18, 55.36, ESI-MS. Calcd for C24H20N2O3, [M + H]+: m/z 384.15. Found: m/z 385.26.
3.1.7.4 (E)-3-((1-Bromophenyl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-phenylprop-2-en-1-one (6a). Yellow solid; yield 67%; mp 180–185 °C; IR (neat, νmax): (–C
O str.) 1596, (–CN str.) 1663 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.27 (s, 1H), 7.86–7.88 (d, 2H), 7.81–7.85 (d, 2H), 7.56–7.68 (m, 6H), 7.32–7.37 (d, 2H), 7.24–7.27 (d, 3H), 6.99–7.03 (m, 3H), 3.83 (s, 3H); 13C NMR (CDCl3, δ): 189.63, 160.38, 154.03, 144.66, 143.74, 138.65, 135.78, 134.93, 132.43, 130.20, 129.94, 128.71, 128.48, 126.61, 124.77, 121.82, 120.77, 120.51, 120.35, 118.77, 114.48, 114.25, 55.57, 21.85, ESI-MS. Calcd for C25H19BrN2O2, [M + H]+: m/z 459.97. Found: m/z 458.06.
3.1.7.5 (E)-3-(1-(4-Bromophenyl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-(4-(methylthio)phenyl)prop-2-en-1-one (6b). Yellow solid; yield 71%; mp 225–228 °C; IR (neat, νmax): (–C
O str.) 1592, (–CN str.) 1648 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.29 (s, 1H), 7.94–7.96 (d, 2H), 7.83–7.88 (d, 2H), 7.75–7.78 (d, 2H), 7.48–7.75 (m, 3H), 7.44–7.47 (d, 2H), 7.25–7.38 (d, 2H), 7.00–7.04 (d, 2H), 3.86 (s, 3H); 13C NMR (CDCl3, δ): 190.21, 160.42, 154.13, 138.66, 138.40, 135.44, 132.95, 132.91, 132.80, 131.24, 130.44, 130.22, 128.80, 128.58, 126.67, 124.74, 121.83, 121.25, 120.83, 120.61, 118.73, 114.51, 55.60, ESI-MS. Calcd for C26H21N2O2S, [M + H]+: m/z 505.43. Found: m/z 506.05.
3.1.7.6 (E)-3-(1-(4-bromophenyl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-(p-tolyl)prop-2-en-1-one (6c). Yellow solid; yield 71%; mp 218–222 °C; IR (neat, νmax): (–C
O str.) 1603, (–CN str.) 1663 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.29 (s, 1H), 7.85–7.89 (d, 2H), 7.75–7.78 (d, 2H), 7.52–7.74 (m, 4H), 7.20–7.25 (d, 2H), 7.13–7.16 (d, 2H), 6.99–7.07 (d, 3H), 3.87 (s, 3H); 13C NMR (CDCl3, δ): 181.93, 160.44, 154.12, 145.76, 138.65, 134.69, 134.14, 133.86, 132.81, 132.59, 132.49, 131.67, 130.26, 129.95, 128.42, 126.86, 124.74, 121.50, 120.84, 120.62, 120.45, 118.52, 114.52, 114.37, 55.61, 44.80 ESI-MS. Calcd for C26H21BrN2O2, [M + H]+: m/z 472.08. Found: m/z 473.06.
3.1.7.7 (E)-3-(1-(4-Bromophenyl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-(thiophen-2-yl)prop-2-en-1-one (6d). Yellow solid; yield 76%; mp 190–195 °C; IR (neat, νmax): (–C
O str.) 1592, (–CN str.) 1663 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.28 (s, 1H), 7.94–7.98 (d, 2H), 7.80–7.93 (d, 2H), 7.75–7.78 (d, 2H), 7.55–7.68 (m, 4H), 7.26–7.37 (m, 4H), 6.91–7.02 (m, 4H), 3.84 (s, 3H). 13C NMR (CDCl3, δ): 188.38, 163.56, 160.35, 153.96, 138.66, 134.51, 132.91, 132.74, 131.24, 130.85, 130.42, 130.19, 126.58, 124.81, 121.65, 121.21, 120.75, 120.47, 118.82, 114.46, 114.01, 55.60, ESI-MS. Calcd for C23H17BrN2O2S, [M + H]+: m/z 466.02. Found: m/z 466.93.
3.1.7.8 1′-(4-Bromophenyl)-3′-(4-methoxyphenyl)-5-phenyl-3,4-dihydro-1′H,2H-[3,4′-bipyrazole]-2-carbaldehyde (7a). White solid; yield 77%; mp 202–205 °C; IR (neat, νmax): (–C
O str.) 1596, (–CN str.) 1674 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.99 (s, 1H), 7.78 (s, 1H), 7.66–7.63 (d, 2H), 7.58–7.56 (d, 2H), 7.53–7.50 (d, 2H), 7.41–7.39 (m, 4H), 6.96–6.94 (d, 2H), 5.82–5.78 (dd, 1H), 3.81 (s, 3H), 3.71–3.64 (dd, 1H), 3.16–3.10 (dd, 1H); 13C NMR (CDCl3, δ): 160.44, 160.03, 154.98, 150.74, 138.5, 133.72, 133.01, 132.58, 130.47, 129.47, 129.75, 126.04, 125.40, 125.28, 123.15, 121.44, 120.58, 119.89, 114.40, 55.52, 51.78, 42.13, ESI-MS. Calcd for C26H21BrN4O2, [M + H]+: m/z 501.18. Found: m/z 503.08.
3.1.7.9 1′-(4-Bromophenyl)-3′-(4-methoxyphenyl)-5-(p-tolyl)-3,4-dihydro-1′H,2H-[3,4′-bipyrazole]-2-carbaldehyde (7b). White solid; yield 78%; mp 221–224 °C; IR (neat, νmax): (–C
O str.) 1592, (–CN str.) 1674 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.97 (s, 1H), 7.77 (s, 1H), 7.63–7.65 (d, 2H), 7.50–7.57 (m, 7H), 7.20–7.18 (d, 2H), 6.96–6.94 (d, 2H), 5.80–5.76 (dd, 1H), 3.81 (s, 3H), 3.66–3.61 (dd, 1H), 3.14–3.08 (dd, 1H), 2.37 (s, 3H). 13C NMR (CDCl3, δ): 160.33, 159.96, 156.52, 150.68, 141.41, 139.00, 132.95, 132.79, 132.54, 130.01, 129.75, 129.69, 128.18, 126.85, 125.92, 125.41, 121.80, 120.81, 120.54, 119.79, 114.39, 55.50, 51.51, 42.47, 21.70, ESI-MS. Calcd for C27H23BrN4O2, [M + H]+: m/z 515.00. Found: m/z 514.10.
3.1.7.10 1′-(4-Bromophenyl)-3′-(4-methoxyphenyl)-5-(4-(methylthio)phenyl)-3,4-dihydro-1′H,2H-[3,4′-bipyrazole]-2-carbaldehyde (7c). White solid; yield 79%; mp 240–244 °C; IR (neat, νmax): (–C
O str.) 1592, (–CN str.) 1670 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.97 (s, 1H), 7.78 (s, 1H), 7.68–7.49 (m, 4H), 7.30–7.27 (d, 2H), 7.25–7.21 (d, 2H), 6.96–6.93 (dd, 1H), 5.81–5.75 (dd, 1H), 3.81 (s, 3H), 3.68–3.58 (dd, 1H), 3.13–3.05 (dd, 1H), 2.49 (s, 3H). 13C NMR (CDCl3, δ): 160.31, 159.99, 156.02, 150.71, 142.78, 138.99, 138.63, 132.81, 132.56, 130.20, 130.24, 129.77, 129.03, 127.33, 127.16, 126.70, 125.97, 125.38, 125.32, 121.70, 120.83, 120.56, 119.83, 114.39, 55.51, 51.58, 42.29, 15.32, ESI-MS. Calcd for C27H23BrN4O2S, [M + H]+: m/z 548.12. Found: m/z 548.07.
3.1.7.11 1′-(4-Bromophenyl)-3′-(4-methoxyphenyl)-5-(thiophen-2-yl)-3,4-dihydro-1′H,2H-[3,4′-bipyrazole]-2-carbaldehyde (7d). White solid; yield 73%; mp 207–210 °C; IR (neat, νmax): (–C
O str.) 1503, (–CN str.) 1666 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.94 (s, 1H), 7.79 (s, 1H), 7.64–7.61 (d, 2H), 7.58–7.56 (d, 2H), 7.53–7.51 (d, 2H), 7.44–7.43 (d, 2H), 7.13–7.12 (d, 2H), 7.04–7.02 (d, 2H), 6.96–6.94 (d, 2H) 5.82–5.78 (dd, 1H), 3.82 (s, 3H), 3.71–3.64 (dd, 1H), 3.13–3.08 (dd, 1H). 13C NMR (CDCl3, δ): 160.42, 160.01, 155.33, 150.73, 138.96, 136.98, 132.58, 129.79, 129.48, 129.26, 128.10, 126.09, 125.33, 121.48, 120.58, 119.89, 114.39, 55.50, 51.78, 42.17, ESI-MS. Calcd for C24H19BrN4O2S, [M + H]+: m/z 508.15. Found: m/z 507.41.
3.1.7.12 (E)-3-(1-([1,1′-Biphenyl]-4-yl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-phenylprop-2-en-1-one (8a). Yellow solid; yield 78%; mp 250–252 °C; IR (neat, νmax): (–C
O str.) 1529, (–CN str.) 1663 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.33 (s, 1H), 8.28–8.27 (s, 1H), 8.00–7.90 (d, 2H), 7.80–7.88 (d, 2H), 7.78–7.58 (m, 5H), 7.56–7.48 (d, 2H), 7.44–7.29 (m, 4H), 7.06–7.04 (d, 2H), 3.90 (s, 3H). 13C NMR (CDCl3, δ): 190.19, 160.24, 153.99, 138.45, 138.17, 135.65, 135.38, 133.57, 132.78, 132.71, 132.63, 131.14, 130.10, 130.05, 128.94, 128.63, 128.42, 128.17, 128.00, 127.01, 126.53, 124.51, 121.62, 121.34, 120.67, 120.46, 119.58, 118.53, 114.33, 55.41, ESI-MS. Calcd for C32H26N2O2, [M + H]+: m/z 456.55. Found: m/z 458.98.
3.1.7.13 (E)-3-(1-(4′-Methoxy-[1,1′-biphenyl]-4-yl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-phenylprop-2-en-1-one (8b). Yellow solid; yield 72%; mp 209–211 °C; IR (neat, νmax): (–C
O str.) 1529, (–CN str.) 1637 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.39 (s, 1H), 8.01–7.99 (s, 1H), 7.87–7.85 (d, 2H), 7.70–6.68 (d, 2H), 7.60–7.58 (d, 2H), 7.53–7.50 (d, 2H), 7.43–7.39 (m, 4H), 7.29–7.28 (d, 2H), 7.07–7.02 (d, 2H), 3.91 (s, 3H). 13C NMR (CDCl3, δ): 189.90, 160.23, 159.34, 158.34, 153.61, 138.49, 138.43, 138.31, 138.07, 135.69, 132.66, 132.42, 132.06, 130.09, 128.60, 128.40, 128.05, 127.65, 126.56, 124.86, 124.62, 121.25, 119.60, 118.18, 114.39, 55.40, ESI-MS. Calcd for C33H28N2O3, [M + H]+: m/z 486.19. Found: m/z 487.28.
3.1.7.14 (E)-3-(1-([1,1′-Biphenyl]-4-yl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-(4(methylthio) phenyl)prop-2-en-1-one (8c). Yellow solid; yield 80%; mp 240–242 °C; IR (neat, νmax): (–C
O str.) 1599, (–CN str.) 1659 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.40 (s, 1H), 8.28–8.27 (s, 1H), 7.94–7.89 (d, 2H), 7.75–7.74 (d, 2H), 7.70–7.65 (d, 2H), 7.56–7.54 (d, 2H), 7.53–7.49 (d, 2H), 7.44–7.41 (d, 2H), 7.33–7.31 (d, 2H), 7.07–7.05, (d, 2H), 3.91 (s, 3H), 2.46 (s, 3H). 13C NMR (CDCl3, δ): 188.82, 160.18, 153.87, 145.58, 140.07, 139.98, 138.64, 135.32, 134.53, 130.17, 129.03, 128.94, 128.23, 127.08, 125.12, 120.99, 119.63, 119.60, 119.57, 118.36, 114.40, 114.38, 114.35, 114.32, 55.49, 29.81 ESI-MS. Calcd for C23H17N3O, [M + H]+: m/z 502.17. Found: m/z 503.08.
3.1.7.15 (E)-3-(1-(4′-Methoxy-[1,1′-biphenyl]-4-yl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-(4-(methylthio)phenyl)prop-2-en-1-one (8d). Yellow solid; yield 76%; mp 248–250 °C; IR (neat, νmax): (–C
O str.) 1544, (–CN str.) 1659 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.38 (s, 1H), 7.93–7.90 (d, 2H), 7.87–7.85 (d, 2H), 7.70–7.68 (d, 2H), 7.60–7.58 (d, 2H), 7.51–7.49 (d, 2H), 7.43–7.40 (m, 4H), 7.32–7.28 (d, 2H), 7.07–7.02 (d, 2H), 3.89 (s, 3H), 2.46 (s, 3H). 13C NMR (CDCl3, δ): 188.99, 159.53, 153.60, 137.48, 135.56, 135.48, 135.35, 132.15, 130.10, 128.86, 128.56, 128.11, 127.64, 126.60, 126.44, 125.11, 120.98, 120.86, 119.60, 116.07, 114.36, 113.44, 55.41, 29.71 ESI-MS. Calcd for C30H24N2O3S, [M + H]+: m/z 532.18. Found: m/z 533.39.
3.1.7.16 (E)-3-(1-([1,1′-Biphenyl]-4-yl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-(p-tolyl)prop-2-en-1-one (8e). Yellow solid; yield 68%; mp 260–262 °C; IR (neat, νmax): (–C
O str.) 1611, (–CN str.) 1633 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.39 (s, 1H), 7.94–7.88 (d, 2H), 7.75–7.65 (d, 2H), 7.52–7.48 (d, 2H), 7.44–7.39 (d, 2H), 7.32–7.30 (d, 2H), 7.07–7.05 (d, 2H), 3.90 (s, 3H), 2.45 (s, 3H). 13C NMR (CDCl3, δ): 189.67, 160.12, 153.77, 143.55, 140.01, 138.61, 135.67, 135.16, 132.15, 132.05, 130.09, 129.32, 128.94, 128.56, 128.15, 127.68, 127.00, 126.57, 124.80, 121.37, 119.55, 118.32, 114.29, 55.41, 29.72 ESI-MS. Calcd for C32H36N2O2, [M + H]+: m/z 470.20. Found: m/z 471.34.
3.1.7.17 (E)-3-(1-(4′-Methoxy-[1,1′-biphenyl]-4-yl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-(p-tolyl)prop-2-en-1-one (8f). Yellow solid; yield 65%; mp 280–282 °C; IR (neat, νmax): (–C
O str.) 1611, (–CN str.) 1633 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.37 (s, 1H), 7.93–7.89 (d, 2H), 7.86–7.84 (d, 2H), 7.70–7.67 (d, 2H), 7.60–7.58 (d, 2H), 7.51–7.49 (d, 2H), 7.43–7.39 (m, 4H), 7.32–7.28 (d, 2H), 7.07–7.01 (d, 2H), 3.89 (s, 3H), 2.45 (s, 3H). 13C NMR (CDCl3, δ): 189.65, 160.10, 159.45, 153.70, 143.52, 138.10, 135.69, 135.19, 130.09, 129.31, 128.55, 128.04, 127.62, 126.53, 124.84, 121.29, 119.57, 118.23, 114.38, 114.28, 55.41, 29.72 ESI-MS. Calcd for C33H28N2O3, [M + H]+: m/z 500.21. Found: m/z 501.32.
3.1.7.18 (E)-3-(1-([1,1′-Biphenyl]-4-yl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-(thiophen-2-yl)prop-2-en-1-one (8g). Yellow solid; yield 78%; mp 275–277 °C; IR (neat, νmax): (–C
O str.) 1596, (–CN str.) 1640 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.32 (s, 1H), 7.93–7.88 (d, 2H), 7.82–7.77 (d, 2H), 7.70–7.64 (d, 2H), 7.54–7.48 (d, 2H), 7.43–7.41 (d, 2H), 7.19–7.17 (m, 4H), 7.07–7.04 (d, 2H), 3.90 (s, 3H). 13C NMR (CDCl3, δ): 191.14, 160.24, 153.97, 145.44, 134.62, 133.70, 132.62, 132.14, 132.04, 128.73, 128.61, 127.00, 121.25, 120.67, 120.57, 119.59, 115.37, 114.33, 55.33 ESI-MS. Calcd for C29H22N2O2S, [M + H]+: m/z 462.14. Found: m/z 463.25.
3.1.7.19 (E)-3-(1-(4′-Methoxy-[1,1′-biphenyl]-4-yl)-3-(4-methoxyphenyl)-1H-pyrazol-4-yl)-1-(thiophen-2-yl)prop-2-en-1-one (8h). Yellow solid; yield 84%; mp 268–270 °C; IR (neat, νmax): (–C
O str.) 1599, (–CN str.) 1655 cm−1; 1H NMR (Chloroform-D, δ ppm): 8.32 (s, 1H), 8.20–8.18 (d, 2H), 7.94–7.77 (m, 2H), 7.72–7.57 (d, 2H), 7.51–7.49 (d, 2H), 7.24–7.17 (d, 2H), 7.10–6.92 (d, 2H), 3.90 (s, 3H). 13C NMR (CDCl3, δ): 181.40, 160.25, 145.44, 132.65, 132.15, 132.05, 130.08, 128.68, 128.56, 128.25, 127.24, 121.27, 120.67, 119.61, 118.32, 116.07, 114.78, 114.38, 114.33, 114.18, 113.46, 55.36 ESI-MS. Calcd for C30H24N2O3S, [M + H]+: m/z 492.15. Found: m/z 493.28.
3.1.7.20 1-(1′-(4-Bromophenyl)-3′-(4-methoxyphenyl)-5-phenyl-3,4-dihydro-1′H,2H-[3,4′-bipyrazol]-2-yl)ethan-1-one (9a). White solid; yield 82%; mp 185–187 °C; IR (neat, νmax): (–C
O str.) 1596, (–CN str.) 1663 cm−1; 1H NMR (Chloroform-D, δ ppm): 7.80 (s, 1H), 7.67–7.57 (d, 2H), 7.42–7.40 (d, 2H), 7.39–7.35 (d, 2H), 7.33–7.25 (d, 2H), 6.97–6.94 (d, 2H), 5.85–5.79 (dd, 1H), 3.81 (s, 3H), 3.83–3.73 (dd, 1H), 3.31–3.23 (dd, 1H), 2.43 (s, 3H). 13C NMR (CDCl3, δ): 169.65, 159.93, 154.10, 150.93, 139.08, 133.18, 132.53, 131.17, 131.11, 130.54, 130.50, 129.90, 127.11, 125.89, 125.58, 122.67, 120.59, 119.71, 114.32, 55.50, 52.72, 44.90, 22.28. ESI-MS. Calcd for C27H23BrN4O2, [M + H]+: m/z 514.10. Found: m/z 515.00.
3.1.7.21 1-(1′-(4-Bromophenyl)-3′-(4-methoxyphenyl)-5-(4-(methylthio)phenyl)-3,4-dihydro-1′H,2H-[3,4′-bipyrazol]-2-yl)ethan-1-one (9b). White solid; yield 85%; mp 214–217 °C; IR (neat, νmax): (–C
O str.) 1592, (–CN str.) 1655, cm−1; 1H NMR (Chloroform-D, δ ppm): 7.72 (s, 1H), 7.66–7.64 (d, 2H), 7.63–7.49 (d, 2H), 7.42–7.40 (d, 2H), 7.11–7.10 (d, 2H) 7.09–6.92 (d, 2H), 5.87–5.82 (dd, 1H), 3.82 (s, 3H), 3.66–3.57 (dd, 1H), 3.09–3.01 (dd, 1H), 2.40 (s, 3H). 13C NMR (CDCl3, δ): 169.09, 167.79, 159.28, 150.71, 150.28, 139.04, 134.95, 132.51, 129.80, 129.10, 128.99, 127.80, 125.86, 125.51, 122.67, 120.55, 119.70, 114.31, 55.51, 52.61, 42.76, 22.21, 20.86. ESI-MS. Calcd for C28H25BrN4O2S, [M + H]+: m/z 560.09. Found: m/z 561.16.
3.1.7.22 1-(1′-(4-Bromophenyl)-3′-(4-methoxyphenyl)-5-(p-tolyl)-3,4-dihydro-1′H,2H-[3,4′-bipyrazol]-2-yl)ethan-1-one (9c). White solid; yield 82%; mp 197–199 °C; IR (neat, νmax): (–C
O str.) 1592, (–CN str.) 1663 cm−1; 1H NMR (Chloroform-D, δ ppm): 7.74 (s, 1H), 7.67–7.63 (d, 2H), 7.58–7.49 (d, 2H), 7.42–7.36 (d, 2H), 6.96–6.91 (d, 2H), 5.88–5.83 (dd, 1H), 3.80 (s, 3H), 3.65–3.56 (dd, 1H), 3.11–3.04 (dd, 2H), 2.46 (s, 3H), 2.08 (s, 3H). 13C NMR (CDCl3, δ): 169.45, 159.87, 154.83, 150.74, 139.04, 132.49, 131.37, 130.66, 129.81, 128.89, 126.79, 125.86, 125.55, 122.80, 120.57, 119.67, 114.30, 55.48, 52.52, 42.06, 22.24, 20.91. ESI-MS. Calcd for C28H25BrN4O2, [M + H]+: m/z 529.10. Found: m/z 528.12.
3.1.7.23 1-(1′-(4-Bromophenyl)-3′-(4-methoxyphenyl)-5-(thiophen-2-yl)-3,4-dihydro-1′H,2H-[3,4′-bipyrazol]-2-yl)ethan-1-one (9d). White solid; yield 85%; mp 191–193 °C; IR (neat, νmax): (–C
O str.) 1568, (–CN str.) 1666 cm−1; 1H NMR (Chloroform-D, δ ppm): δ 7.72 (s, 1H), 7.69–7.64 (d, 2H), 7.55–7.47 (d, 2H), 7.25–7.17 (d, 2H), 6.97–6.92 (d, 2H), 5.85–5.80 (dd, 1H), 3.81 (s, 3H), 3.63–3.54 (dd, 1H), 3.09–3.02 (dd, 1H), 2.44 (s, 3H). 13C NMR (CDCl3, δ): 169.12, 159.85, 154.65, 150.70, 140.98, 139.06, 132.47, 129.79, 129.59, 128.65, 126.73, 125.69, 125.61, 123.02, 120.51, 119.60, 114.29, 55.47, 52.39, 42.17, 21.68. ESI-MS. Calcd for C25H21BrN4O2S, [M + H]+: m/z 522.05. Found: m/z 523.00.
3.2 In vitro cytotoxicity
The cytotoxicity evaluation of compounds 4, 5a–5b, 6a–6d, 7a–7d, 8a–8h, and 9a–9d was performed using MTT assay on human adenocarcinoma (A549) and cervical cancer (HeLa) cell lines purchased from ATCC. The chemical compounds were tested at 24 h and 48 h with various concentrations ranging from 0 to 200 μM. The following day, cells were treated with various concentrations of chemical compounds and incubated for 24 h and 48 h at 37 °C with 5% CO2. After that, 0.5 mg mL−1 MTT was added to each well and incubated for 4 h. Next, the solution was removed, and 150 μl of dimethyl sulfoxide (DMSO) was added to the wells. Then, the plate was read at 575 nm by Infinite M200 pro. The results were analyzed as the percentage of viable cells to the concentration of the compounds.433.3 DNA binding study and antioxidant activity
The Calf-thymus DNA interactions and antioxidant activity of the active compound have been studied by a previously reported method.44,453.4 MD simulation
665 atoms for the complete protein, added ions, and water as a solute medium. The production run was kept for 100 ns to record the trajectory at 100 ps at an energy level of 1.2 that has produced 1000 frames. The NPT ensemble class was used at 300 K temperature and 1.01325 pressure (bar) and then relaxed the model system before simulation. The trajectories were analyzed with the help of a simulation interaction diagram tool to understand the deviation, fluctuation, and intermolecular interactions.4. Conclusions
In this study, a series of novel pyrazole–pyrazoline derivatives have been synthesized. Structures of all synthesized compounds were fully characterized by detailed FT-IR, 1H NMR, 13C NMR, and mass spectrometry. Compound 9d exhibited significant anti-proliferative activity against A549 (lung cancer) and HeLa (human cervix) cancer cell lines. By utilizing UV-visible, fluorescence, circular dichroism, and cyclic voltammetry, the binding affinity of the active analog 9d with Ct-DNA was investigated. The molecular dynamics analysis of derivative 9d was also carried out with the KGFR receptor.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for financial assistance through a large group Research Project under grant number RGP-2/104/44.References
- K. P. Rakesh, H. K. Kumara, H. M. Manukumar and D. Channe Gowda, Bioorg. Chem., 2019, 87, 252–264 CrossRef CAS PubMed.
- B. Moku, L. Ravindar, K. P. Rakesh and H.-L. Qin, Bioorg. Chem., 2019, 86, 513–537 CrossRef CAS PubMed.
- G. C. Arya, K. Kaur and V. Jaitak, Eur. J. Med. Chem., 2021, 221, 113511 CrossRef CAS PubMed.
- P. Dandawate, K. Ahmed, S. Padhye, A. Ahmad and B. Biersack, Adv. Anticancer Agents Med. Chem., 2021, 21, 558–566 CrossRef CAS PubMed.
- L.-C. Chou, L.-J. Huang, M.-H. Hsu, M.-C. Fang, J.-S. Yang, S.-H. Zhuang, H.-Y. Lin, F.-Y. Lee, C.-M. Teng and S.-C. Kuo, Eur. J. Med. Chem., 2010, 45, 1395–1402 CrossRef CAS PubMed.
- A. A. WalyEldeen, S. Sabet, H. M. El-Shorbagy, I. A. Abdelhamid and S. A. Ibrahim, Chem.-Biol. Interact., 2023, 369, 110297 CrossRef CAS PubMed.
- Y. N. Nayak, S. L. Gaonkar and M. Sabu, J. Heterocycl. Chem., 2022, 60, 1301–1325 CrossRef.
- L. C. C. Vieira, M. W. Paixão and A. G. Corrêa, Tetrahedron Lett., 2012, 53, 2715–2718 CrossRef CAS.
- K. Dahlén, E. A. A. Wallén, M. Grøtli and K. Luthman, J. Org. Chem., 2006, 71, 6863–6871 CrossRef PubMed.
- M. Selepe and F. Van Heerden, Molecules, 2013, 18, 4739–4765 CrossRef CAS PubMed.
- K. S. Girisha, B. Kalluraya, V. Narayana and Padmashree, Eur. J. Med. Chem., 2010, 45, 4640–4644 CrossRef CAS PubMed.
- N. C. Desai, D. V. Vaja, J. D. Monapara, V. Manga and T. Vani, J. Heterocycl. Chem., 2021, 58, 737–750 CrossRef CAS.
- M. Asad, M. N. Arshad, M. Oves, M. Khalid, S. A. Khan, A. M. Asiri, M. Rehan and H. Dzudzevic-Cancar, Bioorg. Chem., 2020, 99, 103842 CrossRef CAS PubMed.
- X. Lu, J. Tang, S. Cui, B. Wan, S. G. Franzblauc, T. Zhang, X. Zhang and K. Ding, Eur. J. Med. Chem., 2017, 125, 41–48 CrossRef CAS PubMed.
- Z. Yang, P. Li and X. Gan, Molecules, 2018, 23, 1798 CrossRef PubMed.
- G. Kumar, O. Tanwar, J. Kumar, M. Akhter, S. Sharma, C. R. Pillai, M. M. Alam and M. S. Zama, Eur. J. Med. Chem., 2018, 149, 139–147 CrossRef CAS PubMed.
- S. Kuthyala, G. K. Nagaraja, S. Sheik, M. Hanumanthappa and M. Kumar S, J. Mol. Struct., 2019, 1177, 381–390 CrossRef CAS.
- S. Ravula, R. R. Bobbala and B. Kolli, J. Heterocycl. Chem., 2020, 57, 2535–2538 CrossRef CAS.
- A. T. Taher, M. T. Mostafa Sarg, N. R. El-Sayed Ali and N. Hilmy Elnagdi, Bioorg. Chem., 2019, 89, 103023 CrossRef CAS PubMed.
- N. C. Desai, D. V. Vaja, K. A. Jadeja, S. B. Joshi and V. M. Khedkar, Anti-Infect. Agents, 2020, 18, 306–314 CAS.
- X. Cai, S. Zhao, D. Cai, J. Zheng, Z. Zhu, D. Wei, Z. Zheng, H. Zhu and Y. Chen, Steroids, 2019, 146, 70–78 CrossRef CAS PubMed.
- A. R. Sayed, S. M. Gomha, F. M. Abdelrazek, M. S. Farghaly, S. A. Hassan and P. Metz, BMC Chem., 2019, 13, 116 CrossRef PubMed.
- D. Havrylyuk, O. Roman and R. Lesyk, Eur. J. Med. Chem., 2016, 113, 145–166 CrossRef CAS PubMed.
- M. Asad, S. A. Khan, M. N. Arshad, A. M. Asiri and M. Rehan, J. Mol. Struct., 2021, 1234, 130131 CrossRef CAS.
- M. Rana, R. Arif, F. I. Khan, V. Maurya, R. Singh, M. I. Faizan, S. Yasmeen, S. H. Dar, R. Alam, A. Sahu, T. Ahmad and Rahisuddin, Bioorg. Chem., 2021, 108, 104665 CrossRef CAS PubMed.
- T. S. Reddy, H. Kulhari, V. G. Reddy, V. Bansal, A. Kamal and R. Shukla, Eur. J. Med. Chem., 2015, 101, 790–805 CrossRef CAS PubMed.
- k. Venu, B. Saritha and B. B. V. Sailaja, Tetrahedron, 2022, 124, 132991 CrossRef CAS.
- M. J. Lavecchia, R. Puig de la Bellacasa, J. I. Borrell and C. N. Cavasotto, Bioorg. Med. Chem., 2016, 24, 768–778 CrossRef CAS PubMed.
- N. Fayyazi, A. Fassihi, S. Esmaeili, S. Taheri, J. B. Ghasemi and L. Saghaie, Int. J. Biol. Macromol., 2020, 142, 94–113 CrossRef CAS PubMed.
- Y. Zhang and Q.-C. Zheng, J. Theor. Biol., 2018, 447, 118–125 CrossRef CAS PubMed.
- N. Songtawee, D. R. Bevan and K. Choowongkomon, J. Mol. Graphics Modell., 2015, 58, 16–29 CrossRef CAS PubMed.
- A. Shanmugapriya, R. Jain, D. Sabarinathan, G. Kalaiarasi, F. Dallemer and R. Prabhakaran, New J. Chem., 2017, 41, 10324–10338 RSC.
- K. Jeyalakshmi, J. Haribabu, C. Balachandran, N. S. P. Bhuvanesh, N. Emi and R. Karvembu, New J. Chem., 2017, 41, 2672–2686 RSC.
- J. Li, D. Li, Y. Han, S. Shuang and C. Dong, Spectrochim. Acta, Part A, 2009, 73, 221–225 CrossRef PubMed.
- P. Kundu and N. Chattopadhyay, J. Photochem. Photobiol., B, 2017, 173, 485–492 CrossRef CAS PubMed.
- R. Gul, M. K. Rauf, A. Badshah, S. S. Azam, M. N. Tahir and A. Khan, Eur. J. Med. Chem., 2014, 85, 438–449 CrossRef CAS PubMed.
- M. S. Muneera and J. Joseph, J. Photochem. Photobiol., B, 2016, 163, 57–68 CrossRef CAS PubMed.
- H. Muslu, A. Golcu, M. Tumer and M. Ozsoz, J. Coord. Chem., 2011, 64, 3393–3407 CrossRef.
- R. Vijayalakshmi, Biochim. Biophys. Acta, Gen. Subj., 2000, 1475, 157–162 CrossRef CAS PubMed.
- S. Mahadevan and M. Palaniandavar, Inorg. Chem., 1998, 37, 693–700 CrossRef CAS.
- B. P. Bandgar, L. K. Adsul, H. V. Chavan, S. S. Jalde, S. N. Shringare, R. Shaikh, R. J. Meshram, R. N. Gacche and V. Masand, Bioorg. Med. Chem. Lett., 2012, 22, 5839–5844 CrossRef CAS PubMed.
- T. Taj, R. R. Kamble, T. M. Gireesh, R. K. Hunnur and S. B. Margankop, Eur. J. Med. Chem., 2011, 46, 4366–4373 CrossRef CAS PubMed.
- P. Singla, V. Luxami, R. Singh, V. Tandon and K. Paul, Eur. J. Med. Chem., 2017, 126, 24–35 CrossRef CAS PubMed.
- S. Hussain Dar, N. Hasan, M. Rana, A. Fatima, S. Noorul Sabah Andrabi, S. Ahmedi, N. Manzoor, S. Javed and Rahisuddin, Inorg. Chim. Acta, 2023, 545, 121271 CrossRef CAS.
- M. Rana, A. Fatima, N. Siddiqui, S. Ahmedi, S. H. Dar, N. Manzoor, S. Javed and Rahisuddin, Polycyclic Aromat. Compd., 2022, 43, 6181–6201 CrossRef.
- G. Madhavi Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef CAS PubMed.
- M. P. Jacobson, R. A. Friesner, Z. Xiang and B. Honig, J. Mol. Biol., 2002, 320, 597–608 CrossRef CAS PubMed.
- T. T. H. Hajalsiddig, A. B. M. Osman and A. E. M. Saeed, ACS Omega, 2020, 5, 18662–18674 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2196195 and 2235766. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra04873j |
| This journal is © The Royal Society of Chemistry 2023 |