
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrosion process of stainless steel in natural brine as a source of chromium and iron – the need for routine analysis†
Beata Krasnodębska-Ostręga ,
Krzysztof Drwal,
Monika Sadowska*,
Dominika Bluszcz and
Krzysztof Miecznikowski
,
Krzysztof Drwal,
Monika Sadowska*,
Dominika Bluszcz and
Krzysztof Miecznikowski
Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland. E-mail: msadowska@chem.uw.edu.pl
First published on 2nd October 2023
Abstract
The corrosion of the installations carrying high salinity water is particularly important in the case of utility and therapeutic waters, such as natural brine. The analysis of such complex systems is challenging in the context of routine analysis of Fe and Cr. Both elements can be determined by UV-Vis spectrophotometry after only 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 dilution and liquid–liquid extraction (LODFe = 0.03 mg L−1) or thermal chloride stripping (LODCr = 0.02 mg L−1). The corrosion process was characterized. Electrochemically accelerated corrosion showed uneven decomposition of the steel components – Cr is less easily released to the solution than Fe. Iron comprises 53% of the dissolved part, and Cr comprises 8.6%, while the steel originally contained 62% of Fe and 18% of Cr. During the short-term (one week) contact of the brine with stainless steel, the amounts of Fe and Cr released to the brine were insignificant from the perspective of its therapeutic value.
:100 dilution and liquid–liquid extraction (LODFe = 0.03 mg L−1) or thermal chloride stripping (LODCr = 0.02 mg L−1). The corrosion process was characterized. Electrochemically accelerated corrosion showed uneven decomposition of the steel components – Cr is less easily released to the solution than Fe. Iron comprises 53% of the dissolved part, and Cr comprises 8.6%, while the steel originally contained 62% of Fe and 18% of Cr. During the short-term (one week) contact of the brine with stainless steel, the amounts of Fe and Cr released to the brine were insignificant from the perspective of its therapeutic value.
1. Introduction
The corrosion of water system installations is one of the major problems connected with the exploitation of water with high salinity. Corrosion processes cause partial dissolution of installation components. As a result, various solid and dissolved chemical compounds are carried by the exploited solution. The corrosion of stainless steel installation components is an uncontrolled source of metal species, including some undesirable elements that may be harmful to human health. There are legal regulations from various countries, e.g., from Poland, Germany, Slovakia, and the Czech Republic, regarding the composition of therapeutic waters.1–4 In general, brines are defined as groundwater having mineralization (total dissolved mineral solids) not less than 35 g L−1. Brine is a specific case of a highly saline solution. In addition to large amounts of dissolved NaCl, it contains other components, such as bromides, boron, iodides, hydrogen sulfide, carbon dioxide, and radon. Most often, the chemical composition of brines is dominated by chloride, sodium, and calcium ions. Brines are officially recognized as therapeutic waters when they meet the quantitative requirements for beneficial components, and are not contaminated chemically or microbiologically.1 The brine is delivered from the intake to the medical treatment places by means of a direct installation or transported in a special tank.Steel used during brine exploitation has to be highly resistant to corrosion. The contact of the steel with brine occurs during mining, transport, and storage. The proportions of Fe and Cr determine the physicochemical properties and corrosion resistance of steel in the environment in which it operates.5 Pitting corrosion is facilitated by non-metallic inclusions, such as carbon, phosphorus, and sulphur, while chromium, molybdenum, and nitrogen prevent this from happening. The addition of chromium enables the formation of a passive layer consisting of Cr2O3, CrOOH, and Cr(OH)3.6 Stainless steels are resistant to atmospheric conditions, nitric acid, very dilute sulphuric acid, organic acids, and an alkaline environment (except for hot, concentrated NaOH in the presence of stresses).7 They corrode in an environment with acidic and reducing properties, such as dilute solutions of acids containing aggressive ions (HCl, HF, HBr, and their salts), solutions of oxidizing chlorides (e.g., FeCl3, MgCl2), sea waters, some organic acids (oxalic, formic, lactic), as well as in the water environment at a temperature above 80 °C in the presence of chlorides, which cause local destruction of the passive layers.8–10
In the case of stainless steels, pitting, crevice, intercrystalline and general corrosion deserve special attention.11 The environment containing halide ions contributes to the formation of pitting, i.e., local cavities that are formed by dissolving the passive metal through complexation with halides. Oxidation is mediated by oxygen or (if present in a corrosive environment) Fe3+ or Cu2+ ions. In an environment rich in chlorides, localized corrosion is strongly exacerbated when the cracks already exist. Water-dependent corrosion can lead to uniform pitting corrosion, erosion and stress cracking. The main cause of the corrosion of steels and iron alloys is the presence of dissolved CO2 and H2S.12 Metallic iron does not remain in a stable phase over the entire pH range. The production of hydrogen ions has the potential to remove Fe from the metal surface. In the presence of CO2 at the steel/liquid interface, an anode reaction takes place and the iron atoms are oxidized to cations (Fe(II)). At the same time, carbonate anions can participate in the formation of solid corrosion products such as FeCO3.13 Siderite (FeCO3) is poorly soluble in water at 25 °C and may limit the corrosion rate of steel.14 In solutions with a higher pH, the carbonate film dissolves and a soluble Fe(II) complex is formed.15 At the same time, a cathode reaction takes place in which the protons are reduced and form bicarbonate ions (HCO3−). In general, increasing the CO2 content increases the corrosion rate in aqueous solutions by increasing the rate of hydrogen evolution.16,17
Determination of iron and chromium in water samples can be performed using various methods but usually some sample pretreatment is required to simplify the sample matrix or preconcentrate the analytes. Direct analysis of brine is problematic for many advanced analytical techniques due to the presence of chlorides. For example, in techniques with a nebulizing step (e.g., ICP), there is a decrease in selectivity and/or an increase in the detection limits.18 In the case of ICP-MS, isobaric ions (interfering ions) cause positive errors.19 Thus, various strategies are applied to overcome the problem of spectral interferences during Fe and Cr detection.20 For example, Cr was separated from the highly saline water matrix using DETA-modified magnetic carbon composites (Fe3O4@C@DETA)21 and core–shell magnetic mesoporous carbon (Fe3O4@void@C),22 which served as sorbents for Cr(III)–EDTA. Both Fe and Cr were determined using ICP-OES in water from the Dead Sea, after a large dilution of the samples.23 Iron was determined in seawater with ICP-MS after preconcentration on a resin,24 and Cr was determined directly25 or after solid phase extraction.26–28 In the case of Fe, extending the procedure by an additional step of extraction to the solid phase (NTA resin column) lowered the LOD from 11 to 3 μg L−1, and the recovery of Fe from sea water was 101%.29 Various types of atomic absorption spectrometry also have been applied for the determination of both metals, usually after a proper sample pretreatment, including separation or preconcentration using liquid–liquid extraction.30–32 Electroanalytical methods seem to be useful as well. Adsorptive stripping voltammetry can be used for the simultaneous determination of Fe and Cr,33 whereas catalytic cathodic stripping voltammetry allows for the determination of Fe in seawater even at picomolar levels.34 However, for the voltammetric determination of Cr and Fe, the adsorptive preconcentration of their complex compounds is used. This step may be substantially impaired because of the presence of competing ions for the complexation of Cr by EDTA or DTPA35,36 and Fe by dihydroxynaphtalene.34 Also, in UV-Vis spectrophotometry, where complexation reactions play a significant role, the high content of inorganic ions interferes with the complexation of the analyte, especially when ions with complexing properties are present, such as Cl−, Br− or I−. Application of this technique to highly saline solutions also requires a sample pretreatment step to remove the excess of chlorides, but this step can also be used to improve the detection limit of the method. After preconcentration by SPE, the LOD for Fe determination in seawater using UV-Vis spectrophotometry was 0.6 nmol L−1.37
The aims of the study were to characterize the corrosion process of stainless steel in brine, and assess whether it can be a source of metals in amounts significant enough to affect the brines' therapeutic properties, in both short-term and prolonged perspectives. Possession of suitable analytical tools is necessary to study the problem of corrosion processes as a source of Fe and Cr during the exploitation of brines, such as mining, transport or application. Therefore, the study also included the development of analytical methods for Fe and Cr determination in samples with a high content of chlorides (natural brine). In the case of routine analysis, the procedure should be simple, precise, reproducible, and financially affordable, which makes UV-Vis spectrophotometry a favorable method of determination. Due to the excessively high chloride concentration, a significant dilution of the sample may result in the concentration of analytes being below the detection limit. Thus, the application of a step to reduce the chloride content is advised to enable the analysis of brine.
2. Materials and methods
2.1. Materials
Standard solutions used in this study were: 2.54 g L−1 Cr(III) as chromium(III) chloride (Merck), 1.00 g L−1 Cr(VI) as potassium dichromate (Merck), and 1.00 g L−1 Fe(III) as iron(III) chloride (Merck). Other utilized reagents were: HCl 30% ultranal (Ciech), 1,5-diphenylcarbazide (P.P.H Gliwice), pure acetone for analysis (POCH), potassium permanganate (POCH), sulfosalicylic acid (P.P.H. Gliwice), sodium azide (ReagentPlus), pure sulphuric acid for analysis (POCH), 4-methyl-2-pentanone (EMSURE® ACS Reag.), ammonia 25% (Chempur), and EDTA disodium salt (Merck). The 0.25% solution of 1,5-diphenylcarbazide was prepared by dissolving 0.0625 g of DPC in 25 mL of acetone, and the addition of 250 μL 10% sulphuric acid. All water solutions were prepared using deionized water from the Sartorius Arium® Mini Plus Ultrapure Water System.The examined steel was a widely used 316L stainless steel, containing 18% Cr, 14% Ni, 2% Mn, and 2.5% Mo. The remaining elements are mainly Fe and admixtures of C, Cu, P, S, Si and N.
The model brine was prepared by dissolving 1.428 g of non-iodized evaporated salt (commercially available salt for consumption purposes) in 2.000 L of deionized water. The residue was not separated.
The natural brine used in this research was a commercially available therapeutic brine from a Polish health resort. This brine is exploited in the borehole from a depth more than 1000 m, from sedimentary rocks (claystones, sandstones, with anhydrites) of the Upper Keuper (Upper Triassic) age,38 and it is a chloride-sodium, iodide-type brine with mineralization of about 79 g L−1. A characteristic feature of this brine is the elevated concentrations of bromides (220 mg L−1), boron (15.6 mg L−1) and iodides (2.32 mg L−1).39 The concentrations of ions declared by the producer are 43.3 g L−1 Cl−, 24.3 g L−1 Na+, 2.5 mg L−1 I−, and 3070 mg L−1 SO42−.
2.2. Apparatus
The UV-Vis spectrophotometer Cary 600 (Agilent Technologies) was used for the determination of Fe and Cr. The samples were heated on the heating plate Ceran 500. Examination of the polarization curve in brine solutions and electrochemical digestion was performed by Electrochemical Workstation CHI760E potentiostat (CH Instruments). For temperature stabilization, the aggregate circulator EAC-10 PLUS (LABOPLAY) was utilized.2.3. Methods
For assessment of the corrosion processes, the electrochemically accelerated corrosion of steel was performed. Here, two methods were utilized. In the first approach, the 316L steel electrode was placed in a model brine solution and a potential of 1600 mV was applied for 30 minutes, resulting in the partial dissolution of steel. The second was the monitoring of the corrosion products of 316L steel electrode at the open circuit potential (0.06 V) over one week. To determine the scale of corrosion, the difference in the mass of the electrode was analyzed, and it was also theoretically calculated on the basis of the total measured charge that flowed through it. The obtained suspension was collected and prepared for chemical analysis by dissolving the particulate matter in concentrated HCl. Before the analysis, the obtained solution was filtered through a 0.22 μm syringe filter.
3. Results and discussion
3.1. Corrosion process in brine – basic study
To determine the tendency to corrosion (general or pitting) of the examined 316L steel in a brine environment, experiments were carried out based on the measurement of the open circuit potential (OCP) transients and the electrode (the tested steel) polarization in therapeutic brine and model brine. The open circuit potential of the tested steel was evaluated after the steel was immersed in the therapeutic and model brine solutions (Fig. 1). The recorded OCP was unstable during the initial measurement period; the voltage–time curves for both brines are characterized by a fluctuation of the recorded potential. However, further extension of the measurement time led to a progressive stabilization of the recorded OCP in both brines. At the beginning, the OCP of 316L steel sample was ca. −0.30 V for the therapeutic brine and ca. −0.13 V for the model brine. During the 2 hours of the examination, the OCP of the 316L stainless steel reached the maximum value of −0.06 V and minimum value of −0.29 V for the therapeutic brine, and −0.045 V and −0.2 V for the model brine, respectively.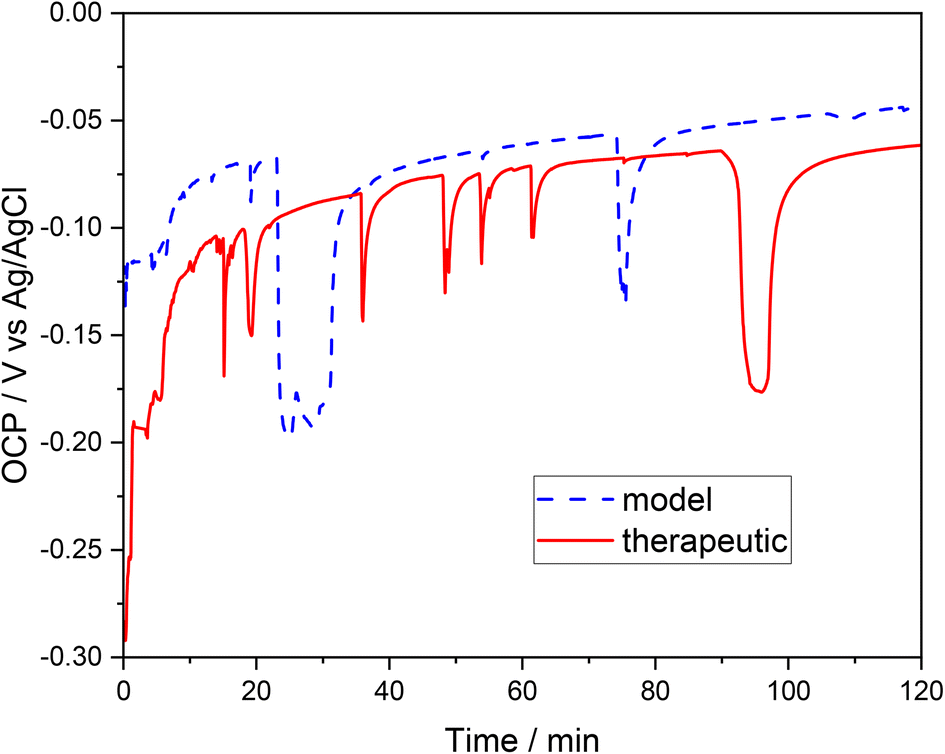 | ||
| Fig. 1 Monitoring of the open circuit potential (OCP) of stainless steel immersed in model brine (dashed blue line) and therapeutic brine (solid red line), as a function of the exposure time. | ||
Further examination of the passivation properties of the 316L stainless steel in the therapeutic brine and model brine were made utilizing the potentiodynamic polarization curves recorded at a 10 mV s−1 scan rate. Typical potentiodynamic polarization curves of the examined stainless steel submerged in therapeutic and model brines are exhibited in Fig. 2. The shapes of the polarization curves obtained for the model brine and therapeutic brine were comparable due to the passive area with the roughly constant current density. On the basis of the obtained polarization curves, the corrosion potential of the 316L stainless steel was determined as −0.951 ± 0.027 V for the model brine and −1.041 ± 0.005 V for the therapeutic brine. The corrosion potential of the 316L stainless steel recorded in the model brine is shifted by approximately 0.090 V towards positive values relative to the corrosion potential values recorded in the therapeutic brine. These results imply better corrosion resistance properties of the 316L stainless steel in model brine in comparison to therapeutic brine. The observed difference is probably a consequence of a more complex composition of the therapeutic brine (additional content of I−, SO42−, etc.) in comparison to the model brine. Furthermore, the pitting nucleation potential (Epit) amounts to 0.180 V for therapeutic brine and 0.105 V for model brine, demonstrating that the 316L stainless steel was more readily rusted in therapeutic brine than in model brine. At higher potential value (above the passive range), the current density on the polarization curves increases rapidly associated with the initiation of pitting in the steel, implying that the kinetics of the corrosion reaction was mainly the same.
 | ||
| Fig. 2 Polarization curves of the 316L stainless steel electrode measured in the therapeutic brine (solid line) and model brine (dashed line) at a scan rate of 10 mV s−1. | ||
In order to further verify the resistance properties of the 316L stainless steel against pitting corrosion in both types of corrosive solutions with high concentration of chlorides (model and therapeutic brine), the chronoamperometric method was employed. Based on the shape of the chronoamperometric curves, the nucleation of pits can be determined. In each case, the variations in the current intensity were monitored as a function of time after applying a constant potential (0.1 V) to the stainless steel working electrode until a rapid current increase was observed (Fig. 3). In both corrosive solutions, the initiation of pit formation on the steel occurs within several seconds, which is manifested by the dramatically increasing value of the recorded current. The recorded current increases referred to the passive layer failure (initiation of pitting).
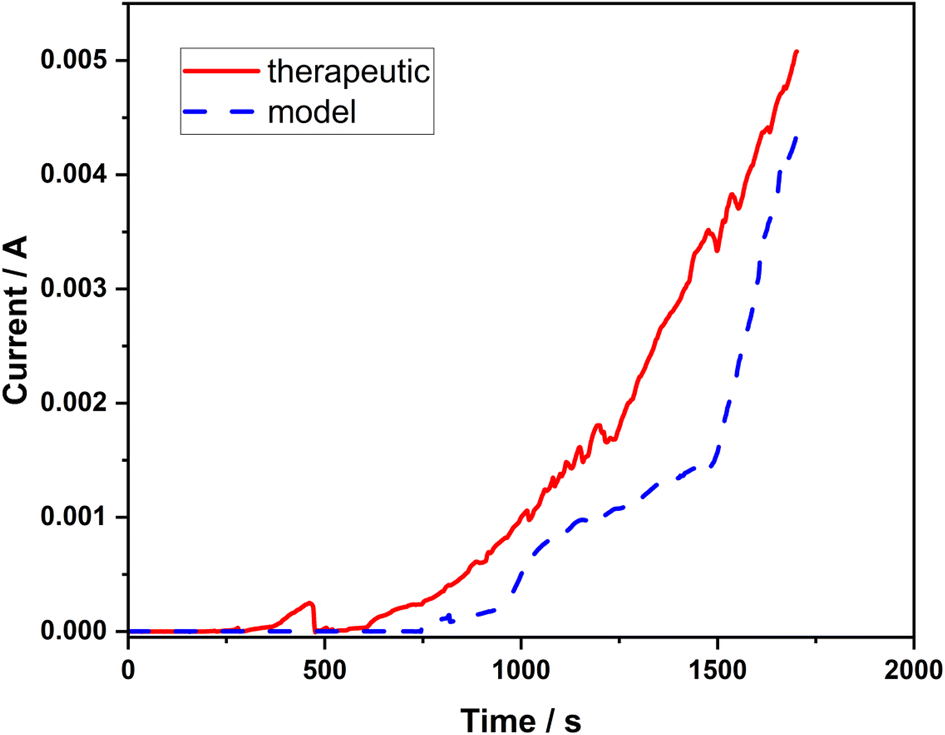 | ||
| Fig. 3 Chronoamperometric curves recorded at a potential of 0.1 V vs. Ag/AgCl in the model and therapeutic brines for 316L stainless steel. | ||
Additionally, the scanning electron microscopy images of 316L stainless steel were recorded before and after immersion for 30 minutes in model and therapeutic brine solutions at an applied potential of 0.1 V, which resulted in the partial dissolution of the stainless steel plate. Fig. 4A shows that the surface of the 316L stainless steel sample before immersion is smooth without any visual pits, which appear after contact with the model brine (Fig. 4B). The changes of the surface occurring in therapeutic brine were very similar to that in the model brine.
 | ||
| Fig. 4 SEM images of the 316L stainless steel surface before (A) and after (B) immersion for 30 minutes in a model brine solution at an applied potential of 0.1 V. | ||
3.2. Corrosion as a source of traces of Cr and Fe
For evaluation of the release of Cr and Fe as a result of corrosion, the electrochemically accelerated corrosion of 316L stainless steel in highly saline solutions was performed. Based on the polarization curve in model brine, the potential chosen for this experiment was +1600 mV. The 316L steel electrode was placed in the model brine solution and electrolysis was carried out for 30 minutes. After digestion of the residue, the solution was stored for further analysis of the total content of metals that were oxidized and released during this process. The purpose was to simulate the corrosion of stainless steel, and obtain a solution containing its products. The electrode was weighed before and after digestion, and the mass difference was 2.2 mg. The theoretical mass of the digested elements was also determined by integration of the current from the intensity versus time plot. The mean current was 3.52 mA and the obtained charge was 6.34 C. On the basis of the mass composition of the steel, molar fractions of individual components were calculated. The elements considered important in the electrochemical reaction were Fe, Cr, Ni, Mo, Mn and Cu, and C, P, S, Si and N were omitted. For the theoretical estimation of the mass difference, it was assumed that the electrochemical reactions occurred at a proportional rate for the mole fractions for the individual elements and in the systems: Fe/Fe(II), Cr/Cr(III), Ni/Ni(II), Mo/Mo(III), Mn/Mn(II), Cu/Cu(II). With such assumptions made, the mass loss was calculated as 1.71 mg.Firstly, an attempt was made to separate Cr from the brine matrix by liquid–liquid extraction. Model samples were prepared containing 0.5 μg mL−1 Cr(III) or Cr(VI) in model brine. To extract the total chromium, it was first oxidized to Cr(VI) by KMnO4 in the presence of 0.1 mol L−1 H2SO4. The solution was heated in a boiling water bath (T = 100 °C). The remaining KMnO4 was reduced by the dropwise addition of 2.5% NaN3 until the purple color disappeared. Then, the samples were acidified with HCl to obtain the final concentration of 2 or 3 mol L−1, and 15.0 mL of the sample was placed in a separating funnel. Cr(VI) was extracted with three portions (5.0 mL each) of 4-methyl-2-pentanone at a temperature below 10 °C, and next re-extracted with three portions of deionized water (5.0 mL) at room temperature. For spectrophotometric determination of Cr, DPCI was added to the obtained aqueous solutions, and the measurements were carried at a wavelength of 542 nm. The experiment showed that 2 mol L−1 HCl is a better extraction medium than 3 mol L−1 HCl because for the latter, a lower recovery was obtained (only 50%) and a strong turbidity of the aqueous phase was observed. However, the results obtained for 2 mol L−1 HCl were not reproducible. The recoveries for the model samples containing Cl− at concentrations 100 times lower than in the therapeutic brine were in the range of 68–84% (n = 6) for the samples initially containing Cr(VI) (the step of oxidation with KMnO4 was omitted), and 68–127% (n = 7) for samples initially containing Cr(III) (after oxidation with KMnO4 and extraction). The whole procedure is a multi-step process, which makes it not only time-consuming, but also results in additional uncertainties that are difficult to estimate. For this reason, it was decided to limit the simplification of the matrix to thermal chloride stripping in the presence of H2SO4.
Model samples were prepared, containing 0.5 μg mL−1 Cr(III) or Cr(VI) in model brine. Concentrated H2SO4 was added to each sample in an amount that allowed for obtaining the final concentration (after evaporation and dilution) within the range of 0.05–0.1 mol L−1. The sample was then heated to approximately 210 °C until characteristic white fumes were observed. After cooling and dilution with DI water, KMnO4 solution was added in order to oxidize chromium to Cr(VI), which was determined using spectrophotometry after the addition of DPCI. In the model samples, the recovery of Cr was 117 ± 3% (n = 4) for solutions initially containing Cr(VI), and 80 ± 10% (n = 4) for solutions containing Cr(III).
Apart from the determination of the total content of the elements, it is very important to analyze their speciation, i.e., the concentration of their various physicochemical forms. In the case of chromium, there are large differences in the toxicity of individual forms – Cr(III) shows low mobility, toxicity and bioavailability (additionally, it plays an important biological role as a component of many enzymes), while Cr(VI) is more mobile, toxic and carcinogenic. Iron, regardless of the speciation form, is not highly toxic. Fe(II) shows greater mobility and low stability, while Fe(III) is much more stable but not very mobile (it precipitates as oxides and hydroxides).
Considering that the determination of total Cr requires its oxidation to Cr(VI), it was checked whether the procedure of thermal chloride stripping would allow for the determination of only Cr(VI) if the oxidation step with KMnO4 was omitted. In this experiment, the preparation of the samples was limited to chloride stripping with sulphuric acid only. For samples containing Cr(III), the recovery was 0% (n = 3), and for samples containing Cr(VI), the recovery was in the range of 3–38% (n = 4). Unfortunately, such procedure of sample preparation is inadequate for speciation analysis.
According to literature data, a significant excess of Fe(III) ions over Cr(VI) may disrupt spectrophotometric determinations of Cr with DPC. To eliminate interferences from naturally present Fe and other metal cations, EDTA can be used as a masking agent. This approach was verified during determinations of Cr in samples of natural brine, before and after “short contact” with steel under OCP conditions. These samples were chosen for the experiment due to the expected low concentrations of Cr and strong interferences. The samples were diluted 100-times, subjected to thermal chloride stripping and oxidation with KMnO4, and spiked with 0.1 μg mL−1 Cr(VI). Then, Cr was determined using spectrophotometry, with and without the addition of EDTA (avoiding interference from Fe) (Table 1).
| Total Cr [μg mL−1] | Brine (fresh) | Brine (after one week) |
|---|---|---|
| Without EDTA | 0.107 ± 0.004 | 0.070 ± 0.006 |
| With EDTA | 0.118 ± 0.003 | 0.114 ± 0.003 |
The values obtained in the presence of EDTA were higher than that without the addition of a masking agent. The difference was especially significant for the brine that remained in contact with steel, and contained increased amounts of Fe compared to natural brine. After addition of EDTA, the results obtained for both samples (before and after contact with steel) did not differ statistically, which indicates that in the short time period, the corrosion of steel is not a source of significant amounts of Cr. In samples of natural brine (100-times diluted), the measured absorbance for chromium (A = 0.009) was equal to the absorbance limit of detection (LODabsorbance = 0.010). It was below the limit of Cr determination (20 μg L−1), and remained the same after the “short contact” period with steel under OCP conditions.
A sample preparation method based on thermal chloride stripping and oxidation with KMnO4 was also used to determine the total chromium in the solution obtained by the electrochemically accelerated corrosion of 316L stainless steel in a model brine. The weight loss of corroding steel was measured after 30 minutes of electrolysis, as well as calculated theoretically on the basis of the total charge and assuming that the electroactive components of the steel dissolve in a proportion consistent with their molar fractions in the steel. The measured weight loss was 2.2 mg, while the theoretical (calculated) weight loss was 1.7 mg. The mass of steel that was dissolved and the concentration of Cr in the obtained solution were used to calculate the share of chromium in the dissolved part of the steel, which was equal to 8.6% (Table 2). The content of Cr in the investigated stainless steel is 18%. Such discrepancies between theoretical and experimental values may be caused by the uneven rate of dissolution of various steel components.
| Sample no. | Concentration of Cr in the solution [μg mL−1] | Cr content in steel |
|---|---|---|
| 1 | 6.30 | 7.6% |
| 2 | 7.30 | 8.9% |
| 3 | 7.66 | 9.2% |
| 4 | 7.48 | 9.1% |
| 5 | 6.64 | 8.1% |
| Mean | 7.07 | 8.6% |
| RSD | 8.2% | 8.1% |
The use of a simple method of removing the excess chlorides (thermal chloride stripping) together with a widely available spectrophotometry results in an increase in the LOD compared to the method of hyphenating solid phase extraction (SPE) with Amberlite XAD-2000 as a sorbent (mostly used to absorb the organic matter) and atomic absorption spectrometry (GF-AAS) as a determination technique.41 It should also be emphasized that brine is a much more difficult matrix than wastewater, and the LOD would be much higher if the excess chloride ions were as high as in the case of the tested matrix. However, it should be noted that the precision of the proposed methodology is really good at around 2–3%.
This method of sample preparation was used before the determination of Fe in a solution obtained as a result of the electrochemically accelerated corrosion of 316L stainless steel in a model brine, and in natural brine after contact with steel for one week. The results obtained for the first sample (Table 3) allowed for the calculation that iron constitutes 53% of the mass that was dissolved electrochemically (with the mass content of Fe in steel at approx. 62%).
| Sample no. | Concentration of Fe in the solution [μg mL−1] | Fe content in steel |
|---|---|---|
| 1 | 1.04 | 52% |
| 2 | 1.06 | 53% |
| 3 | 1.10 | 55% |
| 4 | 1.02 | 51% |
| Mean | 1.06 | 53% |
| RSD | 2.9% | 3.2% |
The second experiment simulated a short-time contact time (mining and transport) of brine with devices made of steel. In samples of natural brine (100-times diluted), the measured absorbance for iron (A = 0.003) was equal to the limit of absorbance detection (LODabsorbance = 0.003). After the “short contact” time with steel under OCP conditions, it was equal to 0.005. It can be concluded that iron is slightly released into the brine, but it is still below the absorbance limit of quantification (LOQabsorbance = 0.010), and is thus below the limit of Fe determination (30 μg L−1). It should be noted that a similar LOQ was obtained using the spectrophotometry method in fresh and marine water.29 If the detected amount must be lower, then some additional pretreatment is needed using SPE as a separation technique. However, in the case of brine, the effectiveness of the sorbent will be disturbed because it has a much higher salinity. The experiments showed that the short-time contact of the brine with steel is not a source of significant amounts of Fe.
Results of Fe and Cr determinations in the suspension obtained as a product of electrolytic decomposition of steel show that chromium comprises 8.6% of the dissolved part, and iron comprises 53%. The content of Cr in steel is 18%, and Fe – 62%. This means that the steel is not evenly decomposed, and Fe is released more easily than Cr. In this case, iron and chromium are released in the ratio of 6:1. In the case of atmospheric corrosion, the literature data report that the release of chromium and iron per unit area of steel equals 0.2–0.7 mg per m2 per year and 10–200 mg per m2 per year, respectively.42 Considering the wide range, especially for Fe, the ratio of Fe to Cr release is between 50:1 and 1000:1. The numbers are significantly different for corrosion under atmospheric conditions and in brine, indicating that a highly saline medium enhances the release of Cr, mostly due to its complexation in the presence of chlorides.
4. Conclusions
Simulation of the corrosion process (i.e., electrochemically accelerated dissolution of steel immersed in model brine) showed that in the long-term perspective, the corrosion process is a source of significant amounts of Fe, Cr, and certainly also other elements (although in smaller amounts). Under the conditions of an open circuit potential, after one week of contact with the steel, the amounts of Fe and Cr released to the brine were insignificant from the perspective of its therapeutic value. Short-term storage in metal containers, or passing of the solution through steel parts of the installations, should not increase the content of metals in brine. Results of the Fe and Cr determinations in the suspension obtained as a product of electrolytic dissolution of steel show that Fe comprises 53% of the dissolved part, and Cr comprises 8.6%. The steel originally contained 62% of Fe and 18% of Cr, which shows that it is not evenly decomposed. Chromium seems to be less easily released to the solution than iron.Brine is a highly saline, complex matrix, and thus it is difficult to analyze. Determination of Fe and Cr can be performed by UV-Vis spectrophotometry but only after proper preparation of the samples, which should include simplification of the sample matrix (reduction of the chloride content). In the case of Fe, the sample preparation can be based on liquid–liquid extraction with hexone, and following re-extraction with diluted HCl. Iron is then determined as a complex of Fe(III) with sulfosalicylic acid. In the case of Cr, the extraction process is a source of significant errors, but it can be replaced with thermal chloride stripping in the presence of H2SO4. Next, Cr is oxidized to Cr(VI) with KMnO4, and determined as a complex with 1,5-diphenylcarbazide. Analytical procedures for both elements are characterized by good repeatability. They are also simple and do not require sophisticated equipment or reagents, which makes them reproducible and applicable in routine analysis.
Conflicts of interest
There are no conflicts of interest to declare.References
- Obwieszczenie Ministra Zdrowia z dnia 27 lutego 2018 r. w sprawie ogłoszenia jednolitego tekstu rozporządzenia Ministra Zdrowia w sprawie zakresu badań niezbędnych do ustalenia właściwości leczniczych naturalnych surowców leczniczych i właściwości leczniczych klimatu, kryteriów ich oceny oraz wzoru świadectwa potwierdzającego te właściwości Dz.U. 2018 poz. 605, https://isap.sejm.gov.pl/isap.nsf/DocDetails.xsp?id=WDU20180000605, accessed 29 August 2022 Search PubMed.
- Min/TafelWV - Verordnung über natürliches Mineralwasser, Quellwasser und Tafelwasser, https://www.gesetze-im-internet.de/min_tafelwv/BJNR010360984.html, accessed 13 March 2023 Search PubMed.
- Slov-lex, 100/2006 Z.z. - Vyhláška Ministerstva zdravotníctva, https://www.slov-lex.sk/pravne-predpisy/SK/ZZ/2006/100/20200901, accessed 15 March 2023 Search PubMed.
- A. C.- info@aion.cz, 423/2001 Sb. Vyhláška o zdrojích přírodních minerálních vod a lázních, https://www.zakonyprolidi.cz/cs/2001-423, accessed 15 March 2023 Search PubMed.
- Z. Tao, X.-Q. Wang, M. K. Hassan, T.-Y. Song and L.-A. Xie, Behaviour of three types of stainless steel after exposure to elevated temperatures, J. Constructional Steel Res., 2019, 152, 296–311 CrossRef.
- Z. H. Jin, H. H. Ge, W. W. Lin, Y. W. Zong, S. J. Liu and J. M. Shi, Corrosion behaviour of 316L stainless steel and anti-corrosion materials in a high acidified chloride solution, Appl. Surf. Sci., 2014, 322, 47–56 CrossRef CAS.
- J. Chen and B. Young, Stress–strain curves for stainless steel at elevated temperatures, Eng. Struct., 2006, 28, 229–239 CrossRef.
- J. E. Truman, The influence of chloride content, pH and temperature of test solution on the occurrence of stress corrosion cracking with austenitic stainless steel, Corros. Sci., 1977, 17, 737–746 CrossRef CAS.
- F. Sarıoğlu, The effect of tempering on susceptibility to stress corrosion cracking of AISI 4140 steel in 33% sodium hydroxide at 80 °C, Mater. Sci. Eng., A, 2001, 315, 98–102 CrossRef.
- N. Mundhenk, P. Huttenloch, R. Bäßler, T. Kohl, H. Steger and R. Zorn, Electrochemical study of the corrosion of different alloys exposed to deaerated 80 °C geothermal brines containing CO2, Corros. Sci., 2014, 84, 180–188 CrossRef CAS.
- M. P. Ryan, D. E. Williams, R. J. Chater, B. M. Hutton and D. S. McPhail, Why stainless steel corrodes, Nature, 2002, 415, 770–774 CrossRef PubMed.
- J. Banaś, U. Lelek-Borkowska, B. Mazurkiewicz and W. Solarski, Effect of CO2 and H2S on the composition and stability of passive film on iron alloys in geothermal water, Electrochim. Acta, 2007, 52, 5704–5714 CrossRef.
- B. R. Linter and G. T. Burstein, Reactions of pipeline steels in carbon dioxide solutions, Corros. Sci., 1999, 41, 117–139 CrossRef CAS.
- D. A. López, T. Pérez and S. N. Simison, The influence of microstructure and chemical composition of carbon and low alloy steels in CO2 corrosion. A state-of-the-art appraisal, Mater. Des., 2003, 24, 561–575 CrossRef.
- D. H. Davies and G. T. Burstein, The Effects of Bicarbonate on the Corrosion and Passivation of Iron, Corrosion, 1980, 36, 416–422 CrossRef CAS.
- S. Nešić, Key issues related to modelling of internal corrosion of oil and gas pipelines – A review, Corros. Sci., 2007, 49, 4308–4338 CrossRef.
- P. Alt-Epping, H. N. Waber, L. W. Diamond and L. Eichinger, Reactive transport modeling of the geothermal system at Bad Blumau, Austria: Implications of the combined extraction of heat and CO2, Geothermics, 2013, 45, 18–30 CrossRef CAS.
- F. Alshahri, Heavy metal contamination in sand and sediments near to disposal site of reject brine from desalination plant, Arabian Gulf: Assessment of environmental pollution, Environ. Sci. Pollut. Res., 2017, 24, 1821–1831 CrossRef CAS PubMed.
- V. K. Karandashev, A. Yu. Leikin, V. A. Khvostikov, N. K. Kutseva and S. V. Pirogova, Water analysis by inductively coupled plasma mass spectrometry, Inorg. Mater., 2016, 52, 1391–1404 CrossRef CAS.
- T.-S. Lum and K. S.-Y. Leung, Strategies to overcome spectral interference in ICP-MS detection, J. Anal. At. Spectrom., 2016, 31, 1078–1088 RSC.
- J. Wang, Y. Chen, T. Sun, A. Saleem and C. Wang, Enhanced removal of Cr(III)-EDTA chelates from high-salinity water by ternary complex formation on DETA functionalized magnetic carbon-based adsorbents, Ecotoxicol. Environ. Saf., 2021, 209, 111858 CrossRef CAS PubMed.
- L. Liang, J. Wang and Y. Zhang, Magnetic mesoporous carbon hollow microspheres adsorbents for the efficient removal of Cr(III) and Cr(III)-EDTA in high salinity water, Microporous Mesoporous Mater., 2023, 347, 112344 CrossRef CAS.
- M. El-khateeb, Determination of metals' contents in the Dead Sea's water, mud and sediments, Int. J. Energy Water Res., 2020, 4, 205–212 CrossRef.
- D. V. Biller and K. W. Bruland, Analysis of Mn, Fe, Co, Ni, Cu, Zn, Cd, and Pb in seawater using the Nobias-chelate PA1 resin and magnetic sector inductively coupled plasma mass spectrometry (ICP-MS), Mar. Chem., 2012, 130–131, 12–20 CrossRef CAS.
- H. Louie, M. Wu, P. Di, P. Snitch and G. Chapple, Direct determination of trace elements in sea-water using reaction cell inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2002, 17, 587–591 RSC.
- L. Abrankó, L. Yang, R. E. Sturgeon, P. Fodor and Z. Mester, Solid phase microextraction for the determination of chromium in sea-water, J. Anal. At. Spectrom., 2004, 19, 1098–1103 RSC.
- S. Hirata, K. Honda, O. Shikino, N. Maekawa and M. Aihara, Determination of chromium(III) and total chromium in seawater by on-line column preconcentration inductively coupled plasma mass spectrometry, Spectrochim. Acta, Part B, 2000, 55, 1089–1099 CrossRef.
- I. Sánchez Trujillo, E. Vereda Alonso, A. García de Torres and J. M. Cano Pavón, Development of a solid phase extraction method for the multielement determination of trace metals in natural waters including sea-water by FI-ICP-MS, Microchem. J., 2012, 101, 87–94 CrossRef.
- T. C. F. Ribas, R. B. R. Mesquita, T. Moniz, M. Rangel and A. O. S. S. Rangel, Greener and wide applicability range flow-based spectrophotometric method for iron determination in fresh and marine water, Talanta, 2020, 216, 120925 CrossRef CAS PubMed.
- A. Baysal, S. Akman, S. Demir and M. Kahraman, Slurry sampling electrothermal atomic absorption spectrometric determination of chromium after separation/enrichment by mercaptoundecanoic acid modified gold coated TiO2 nanoparticles, Microchem. J., 2011, 99, 421–424 CrossRef CAS.
- N. Kamakura, T. Inui, M. Kitano and T. Nakamura, Determination of Chromium(III), Chromium(VI), and Chromium(III) acetylacetonate in water by ion-exchange disk extraction/metal furnace atomic absorption spectrometry, Spectrochim. Acta, 2014, 93, 28–33 CrossRef CAS.
- W. M. Landing and K. W. Bruland, The contrasting biogeochemistry of iron and manganese in the Pacific Ocean, Geochim. Cosmochim. Acta, 1987, 51, 29–43 CrossRef CAS.
- D. Deswati, E. Munaf, H. Suyani, R. Zein and H. Pardi, Simultaneous Determination of Trace Amounts of Iron, Cobalt, Nickel and Chromium in Water Samples with Calcon as Complexing Agent by Adsorptive Stripping Voltammetry, Asian J. Chem., 2015, 27, 3978–3982 CrossRef.
- H. Obata and C. M. G. van den Berg, Determination of Picomolar Levels of Iron in Seawater Using Catalytic Cathodic Stripping Voltammetry, Anal. Chem., 2001, 73, 2522–2528 CrossRef CAS PubMed.
- M. Grabarczyk and M. Korolczuk, Modification of catalytic adsorptive stripping voltammetric method of hexavalent chromium determination in the presence of DTPA and nitrate, Anal. Bioanal. Chem., 2003, 376, 1115–1118 CrossRef CAS PubMed.
- Y. Li and H. Xue, Determination of Cr(III) and Cr(VI) species in natural waters by catalytic cathodic stripping voltammetry, Anal. Chim. Acta, 2001, 448, 121–134 CrossRef CAS.
- D. W. King, J. Lin and D. R. Kester, Spectrophotometric determination of iron(II) in seawater at nanomolar concentrations, Anal. Chim. Acta, 1991, 247, 125–132 CrossRef CAS.
- A. Krawiec and K. Dulski, Wody lecznicze Połczyna Zdroju, Przegl. Geol., 2004, 52, 147 Search PubMed.
- Świadectwo potwierdzające właściwości lecznicze wody z ujęcia IG-1 w miejscowości Połczyn-Zdrój, Narodowy Instytut Zdrowia Publicznego – Państwowy Zakład Higieny, Warszawa, Archiwum, 2018.
- A. A. Christie, J. R. W. Kerr, G. Knowles and G. F. Lowden, The colorimetric determination of cadmium, chromium, copper, iron, lead, manganese, nickel and zinc in sewage and industrial wastes, Analyst, 1957, 82, 336–342 RSC.
- L. Elci, A. A. Kartal and M. Soylak, Solid phase extraction method for the determination of iron, lead and chromium by atomic absorption spectrometry using Amberlite XAD-2000 column in various water samples, J. Hazard. Mater., 2008, 153, 454–461 CrossRef CAS PubMed.
- I. O. Wallinder, S. Bertling, D. B. Kleja and C. Leygraf, Corrosion-Induced Release and Environmental Interaction of Chromium, Nickel and Iron from Stainless Steel, Water, Air, Soil Pollut., 2006, 170, 17–35 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04801b |
| This journal is © The Royal Society of Chemistry 2023 |