
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUsing phenolic polymers to control the size and morphology of calcium carbonate microparticles†
Yurie Nakanishia,
Bohan Cheng a,
Joseph J. Richardsonab and
Hirotaka Ejima*a
a,
Joseph J. Richardsonab and
Hirotaka Ejima*a
aDepartment of Materials Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ejima@g.ecc.u-tokyo.ac.jp
bSchool of Engineering, RMIT University, Melbourne, VIC 3000, Australia
First published on 18th October 2023
Abstract
Calcium carbonate (CaCO3) is a naturally occurring mineral that occurs in biology and is used industrially. Due to its benign nature, CaCO3 microparticles have found use in the food and medical fields, where the specific size of the microparticles determine their functionality and potential applications. We demonstrate that phenolic polymers with different numbers of hydroxy groups can be used to control the diameter of CaCO3 microparticles in a range of 2–9 μm, and obtained particles were relatively uniform. The largest particles (∼9 μm in diameter) were obtained using poly(2,3,4,5-tetrahydroxystyrene) (P4HS), which showed the highest water solubility among the tested phenolic polymers. The polymer concentration and stirring speed influenced the size of microparticles, where the size of the obtained particles became smaller as the concentrations of phenolic polymers increased and as the stirring speed increased, both likely due to promoting the formation of a large number of individual crystal seeds by shielding seed–seed fusion and increasing the chances for precursor contact, respectively. The preparation time and temperature had a great influence on the morphology of the CaCO3 particles, where vaterite transforms into calcite over time. Specifically, aragonite crystals were observed at preparation temperature of 80 °C and vaterite particles with rough surfaces were obtained at 40 °C. Molecular weight and scale of reaction were also factors which affect the size and morphologies of CaCO3 particles. This research represents a facile method for producing relatively monodisperse CaCO3 microparticles with diameters that have previously proven difficult to access.
Introduction
Calcium carbonate (CaCO3) is one of the most abundant inorganic minerals naturally found in the exoskeletons of algae, eggshells, mussel shells, and sea urchin spines.1 It is also an important compound actively used in biomedical and industrial applications, such as drug delivery, paper manufacturing, cosmetics, fillers, and rubber.2 CaCO3 has six different polymorphs:3 amorphous calcium carbonate (ACC), calcium carbonate hexahydrate, calcium carbonate monohydrate, vaterite, aragonite, and calcite. Among these six polymorphs, ACC is the least stable phase and transforms into one of the crystalline polymorphs unless stabilized with specific additives.4,5 Of the crystalline phases, calcite is the most stable at room temperature and pressure, and vaterite is the least stable that rarely occurs in nature.6 Recently, the controlled synthesis and crystallization of CaCO3 have been investigated because controlling the size and morphology of CaCO3 particles is of particular importance for applications such as drug delivery.7The simplest synthesis method of CaCO3 spheres is via the vigorous mixing of solutions of CaCl2 and Na2CO3,8 where formation and crystallization generally follow Ostwald's rules of states.3,9 After mixing, CaCO3 temporarily exists as amorphous calcium carbonate (ACC), which rapidly transforms into more stable crystals. As this precipitation process is very rapid, a mixture of polymorphs and polydisperse particles form, and therefore controlling the size and morphology of these CaCO3 particles are generally challenging. To control the crystal growth and particle size of CaCO3, certain stabilizing polymers can be added to the precipitation solution to control precipitation in two main ways.10 First, the hydroxy, carboxy, or sulfonic groups on the polymer chains can complex with Ca2+ in the mixed solution due to electrostatic interactions, thereby delaying nucleation. This allows for the nano-sized amorphous intermediates to aggregate and gradually crystallize. The second effect of the polymer is to bind to the surface of these aggregated amorphous intermediates and prevent particles from growing along a particular crystal plane. As a result, the particles aggregate spherically and grow into microscale spherical particles, where recrystallization into more stable structures is inhibited.
Many anionic polymer additives such as polystyrene sulfonate (PSS),11–16 poly(acrylic acid) (PAA),4,17–22 poly(vinylsulfonic acid) (PVSA),23 and double hydrophilic block copolymers (DHBC)24–27 have shown a profound effect on crystallization. Furthermore, precipitation conditions such as pH,3,25 temperature,28,29 polymer concentration,2,30 incubation time,31,32 and stirring speed33 impact the size, dispersity, and polymorphs of CaCO3. Despite the numerous efforts devoted to the controlled synthesis of CaCO3 particles, a systematic study of monodisperse particles bigger than 4.0 μm has yet to be achieved and remains challenging. The possible reason for the difficulty in preparing particles with a large particle size is that it is difficult to achieve both delaying nucleation and effectively stimulating nucleation growth at the same time. For example, one effective way to delay nucleation is increasing the concentrations of polymers. Higher polymer concentration means providing the more chelation sites for Ca2+. However, those higher amounts of polymer would prevent particles from growing or provide more nucleation sites, and it results in the particles with a small diameter. We could obtain bigger particles by decreasing the polymer concentration because a lower amount of polymer doesn't prevent particles from growing, but the resulting particles become very heterogeneous. This is because the Ca2+ is not chelated well, so the nucleation is not delayed, and it results in the uncontrolled heterogeneous nucleation growth. Most of the studies that have already been reported to obtain large fine particles by polymer addition method are not uniform in particle size or contain various crystal polymorphs. Based on these results, new polymers that can simultaneously delay nucleation and promote nucleation growth are required to obtain bigger and uniform CaCO3 particles.
We recently reported the synthesis of phenolic polymers with three, four, and five hydroxy groups on one styrene monomer unit, and demonstrated their ultrastrong underwater adhesion.34,35 The number of hydroxy groups determined the underwater adhesion strength and it is known that catechol (2 hydroxy groups) and gallol (3 hydroxy groups) have an affinity for metal ions. We hypothesized that the number of hydroxy groups of phenolic polymers could also play an important role in the chelation of Ca2+ ions, and therefore might be able to control the crystallization of CaCO3. Herein, five kinds of phenolic polymers, poly(4-hydroxystyrene) (P1HS), poly(3,4-dihydroxystyrene) (P2HS), poly(3,4,5-trihydroxystyrene) (P3HS), poly(2,3,4,5-tetrahydroxystyrene) (P4HS), and poly(2,3,4,5,6-pentahydroxystyrene) (P5HS) were used as polymer additives for CaCO3 crystallization (Fig. 1a), and CaCO3 particles with diameters ranging from 2 to 9 μm were successfully synthesized. The largest particles were obtained when using poly(2,3,4,5-tetrahydroxystyrene) (P4HS), which showed the highest water solubility among the tested phenolic polymers. Moreover, the stirring speed, concentration of phenolic polymers, molecular weight of phenolic polymers, and the scale of reaction also influenced the crystal size, while the temperature and preparation time influenced the morphology. Based on these experimental results, the mechanism of CaCO3 crystallization in the presence of phenolic polymers was discussed. The current research presents a facile method to produce relatively uniform CaCO3 microparticles and has a potential in applications that require larger microparticles, such as porous scaffold materials36 or orally administered drug delivery vehicles.37
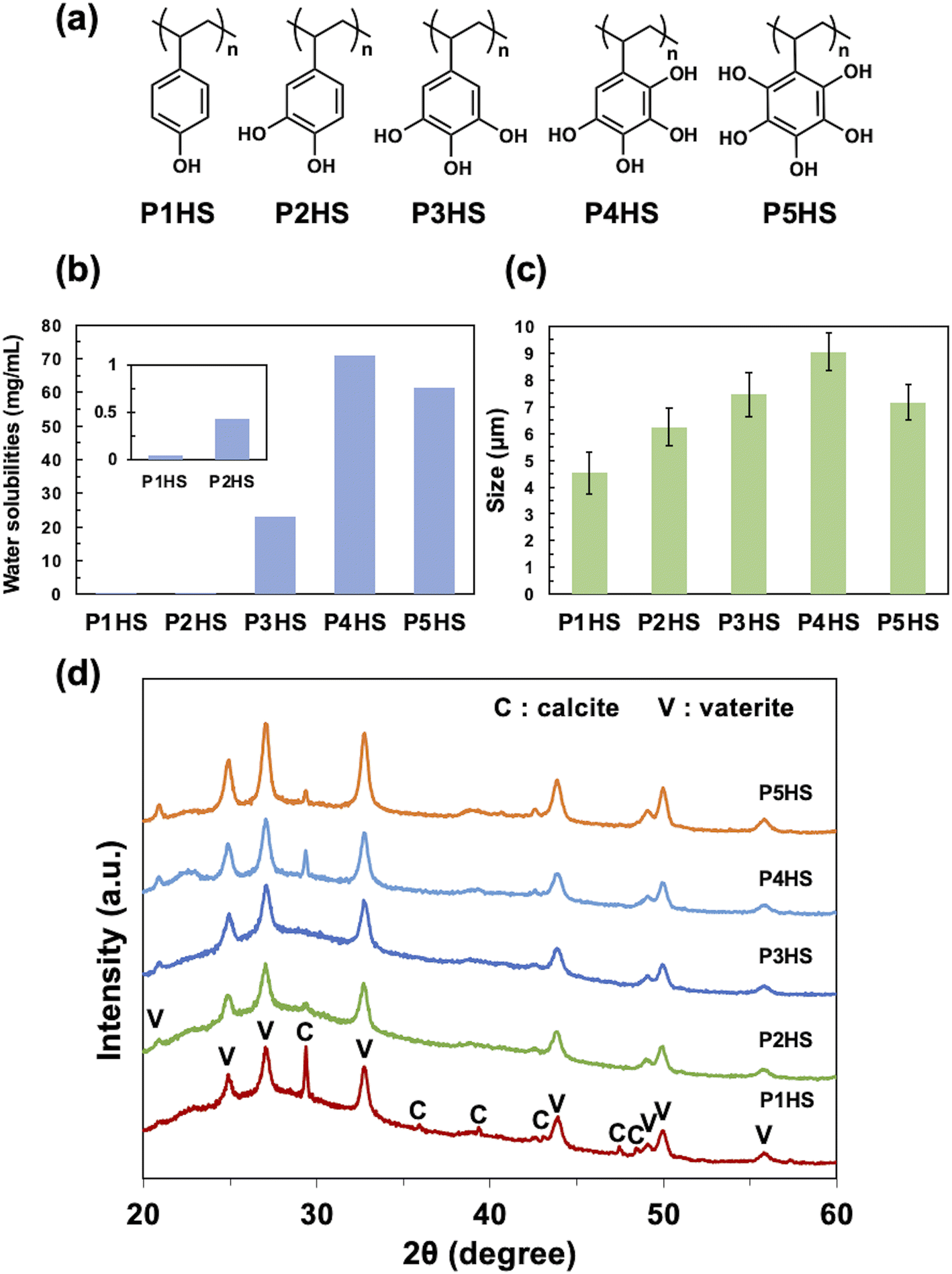 | ||
| Fig. 1 (a) Chemical structures of poly(4-hydroxystyrene) (P1HS), poly(3,4-dihydroxystyrene) (P2HS), poly(3,4,5-trihydroxystyrene) (P3HS), poly(2,3,4,5-tetrahydroxystyrene) (P4HS), and poly(2,3,4,5,6-pentahydroxystyrene) (P5HS). (b) Water solubilities of the different phenolic polymers. (c) The average size of CaCO3 particles obtained in the presence of the phenolic polymers (0.1 mg mL−1). (d) XRD patterns of CaCO3 particles obtained in the presence of the phenolic polymers (0.1 mg mL−1). | ||
Experimental details
Chemicals and materials
The syntheses of phenolic polymers were carried out as reported by Cheng et al.34 Calcium chloride (CaCl2), sodium carbonate (Na2CO3) and methanol were purchased from FUJIFILM Wako Pure Chemical, and used without further purification.Preparation of CaCO3 particles
We prepared CaCO3 particles using a simple mixing method of two solutions. Typically, 240 μL of 1 M CaCl2 solution was added to 19 mL of 12 mM Na2CO3 ([Ca2+] ∼ 24 mM, [CO32−] ∼ 12 mM) solution containing phenolic polymers (P1HS–P5HS) at room temperature. As for P1HS–P3HS, equal volumes of polymer/methanol solution prepared at 10 mg mL−1 were added, and then pure water was added to the solution because the water solubilities of these phenolic polymers are lower than P4HS and P5HS. Although the solvent plays a key role in controlling the growth, polymorphism, and the shape of CaCO3 particles,38,39 the methanol added in our experiment accounted for only 1 vol% of the total solvent, and therefore the solubility of P1HS, P2HS, and P3HS in the mixed solutions was still low. The addition of methanol had a negligible effect on the size of CaCO3 particles. The polymer concentration was varied in the range of 0–0.5 mg mL−1 and the reaction temperature was varied in the range of 25–80 °C. After mixing, the solution was stirred for 60 s. Stirring speed differs in the range of 500–1500 rpm. The precipitated CaCO3 was soon collected by centrifugation (5000g, 60 s) and washed twice with DI water. Typically, the precipitates were collected soon after the stirring, but the precipitates were kept without any treatment for 0 min to 7 days in order to examine the effect of preparation time. After centrifugation, the resulting particles were calcined at 500 °C for 2 h to remove the remaining phenolic polymers.Characterization
The number-average molecular weight (Mn) and polydispersity index (PDI) of phenolic polymers were both measured by gel permeation chromatography (Prominence-i GPC System, SHIMADZU, Japan) using THF as the eluent. The calibration curve was prepared using polystyrene standards. Water solubilities of phenolic polymers were measured by dissolving excess polymer in 10 mL DI water at room temperature. The mixture was sonicated for 30 min and aqueous solutions of the polymers were filtered. The filtrate was collected and weighed after freeze-drying using a FDS-2000 freeze-drier (EYELA, Tokyo, Japan). Water solubility was then calculated by the weight of polymer remaining. The size of the CaCO3 particles was measured using an inverted optical microscope (Eclipse TE2000, Nikon, Japan). CaCO3 particles were observed under the microscope with a 40× objective lens immediately after centrifugation. 100 particles were analysed for each sample to calculate mean values and standard deviations. A coefficient of variation (CV) was calculated by dividing standard deviation by the mean value. The calcined CaCO3 samples were dispersed in DI water and dropped on silicon substrates. They were dried at room temperature, then platinum was sputtered on the surface by a sputter coater (JEC-3000FC, JEOL, Japan). The samples were characterized using a scanning electron microscope (JCM-7000, JEOL, Japan) and a field-emission scanning electron microscope (JSM-7000F, JEOL, Japan) at an accelerating voltage of 5 kV. X-ray diffraction (XRD) patterns were recorded on a Rigaku Mini Flex 600/PTN with Cu-Kα radiation at tube parameters of 40 kV and 15 mA over a scanning range of 2θ from 20° to 60° and a step size of 0.02°. The analysis was conducted soon after the centrifugation in a typical experiment. The polymorphic ratio of calcite and vaterite in the CaCO3 particles was calculated with Rao's equation.40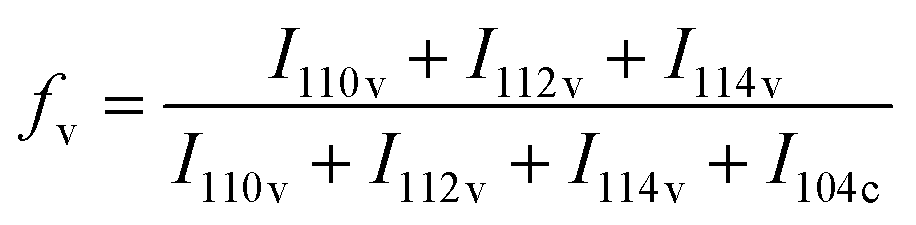
The surface area of the CaCO3 particles was determined using the BET method with nitrogen adsorption and desorption (MicrotacBEL Corp., Japan). To perform microgravimetric measurements, a quartz crystal microbalance (QCM-992A, SEIKO EG&G) was used. The QCM Au chips (9 MHz, Seiko EG&G) were employed as working electrodes. Specifically, 3 μL of a suspension of CaCO3 particles with a certain particle concentration was dropped on the QCM chips and dried for 20 min in an oven at 65 °C before measurement of the frequency change. Thermogravimetric analysis (TGA) curves of the samples were collected using a DTG-60H (Shimadzu, Japan) within a temperature range of 25–800 °C and with the temperature increase rate set as 10 °C min−1.
Results and discussion
Synthesis and water solubility of phenolic polymers
Phenolic polymers (Fig. 1a) were synthesized according to our previous report,34 and the molecular weights of the synthesized polymers are summarized in Table S1.† The total number of hydroxy groups on a single aromatic ring greatly influenced the water solubility of the polymers (Fig. 1b), where generally increasing the number of hydroxy groups improve the solubilities of polymers (P1HS < P2HS < P3HS < P4HS). Although P5HS had the highest numbers of hydroxy groups, P4HS showed the highest solubility (71 mg mL−1), possibly due to both the high symmetry of P5HS and the intramolecular hydrogen bonds hindering interactions with water molecules.The effect of phenolic polymers on the crystallization of CaCO3
Without any polymer additive, rhombic crystalline CaCO3 particles with a diameter of 2.0–5.0 μm formed (Fig. S1a†) that were a mixture of calcite and vaterite (Fig. S1b†). The addition of P1HS, P2HS, P3HS, P4HS, or P5HS yielded spherical CaCO3 particles with diameters of 4.53 ± 0.79 μm, 6.24 ± 0.70 μm, 7.45 ± 0.81 μm, 9.04 ± 0.71 μm, and 7.16 ± 0.65 μm (mean ± S.D.), respectively (Fig. 1c). The optical microscope image of CaCO3 particles synthesized in the presence of P4HS was shown in Fig. S2.† It is likely that the coordination of the hydroxy groups of the polymers to Ca2+ delayed crystallization and shielded the seeds from growing along crystal planes, and therefore produced spherical crystal polymorphs. The coefficients of variation (CV) were about 10% for P2HS–P5HS and about 17% for P1HS. These results suggested that relatively monodisperse CaCO3 particles were obtained. The corresponding XRD results showed both vaterite and calcite peaks although the calcite peak is much lower than that of vaterite (Fig. 1d), as the relative mass percentage of vaterite is over 85% regardless of the type of added polymer. Overall, these results indicated that phenolic polymers have a significant effect on the morphology of CaCO3, even at concentrations as low as 0.1 mg mL−1.Among the phenolic polymers tested, the largest CaCO3 particles were obtained when using P4HS, which has more hydroxy groups per monomer than P1HS–P3HS. We note that higher numbers of hydroxy groups promoted aggregation of the nanoscale particles that make up CaCO3 particles, because P4HS has more chelation sites for Ca2+, which therefore resulted in larger particles. On the other hand, the average particle size in the presence of P5HS was smaller than that of P4HS, although P5HS has one more hydroxy group than P4HS. Trushina et al.3 reported that when the viscosity of the precipitation solution is high, the restricted diffusion of Ca2+ promotes nucleation and the resulting CaCO3 particles become smaller. The P5HS used in this experiment has a larger molecular weight than P1HS–P4HS (Table S1†) due to limitations in controlling the molecular weight of such a hydroxylated polymer. This is assumed to be a possible reason for the high viscosity of the P5HS solution and the inhibition of crystal growth. The solubilities of the polymers correlate with the particle size (Fig. 1b and c), suggesting that the strength of nanoparticle aggregation depends on the solubility of the capping polymers, which in turn can relate to interact with dissolved species, like metal ions, and water molecules.
BET analysis of the CaCO3 particles precipitated in the presence of P4HS revealed a surface area of 8.6 m2 g−1, which is consistent with previously reported values for CaCO3 particles (6.18 and 8.8 m2 g−1).41,42 Furthermore, the average pore size was determined to be 37 nm. Those nano-sized pores were all throughout the particles (Fig. S3†). The apparent density of the CaCO3 particles was estimated as 1.4 g cm−3 by QCM according to the previously reported method.41 Considering the density of vaterite (2.7 g cm−3),43 ∼48% of the total volume of the particle is void (i.e., empty). After calcination at 500 °C for 2 h, the density of the CaCO3 particles decreased to 1.2 g cm−3 as the remaining P4HS was removed and therefore, the void volume increased to ∼56%.
The effect of polymer concentrations on the crystallization of CaCO3
To further examine the effects of phenolic polymers on the controlled crystallization of CaCO3 particles, a series of experiments with various polymer concentrations (0.3 mg mL−1 and 0.5 mg mL−1, compared with the standard of 0.1 mg mL−1) were carried out. Overall, particles with average diameters ranging from 2.0 to 9.0 μm were obtained at the stirring speed of 500 rpm with varying concentrations (Fig. 2a). The average particle size decreased with increasing concentration for all phenolic polymers tested (Fig. 2b, S4, and S5†), likely due to the higher number of nucleation sites at higher concentrations and the increased polymer shielding hindering fusion and growth along crystal planes. Moreover, the higher the polymer concentration, the more hydroxy groups are available to bind to Ca2+, which results in the formation of more seeds of CaCO3 nanoparticles. As a result, the crystal growth was inhibited and CaCO3 particles with smaller diameters were obtained at higher concentrations, which is consistent with previous studies.3 The CV values of obtained particles were about 20% or less, which showed that polymer concentrations didn't affect the particle uniformity so much.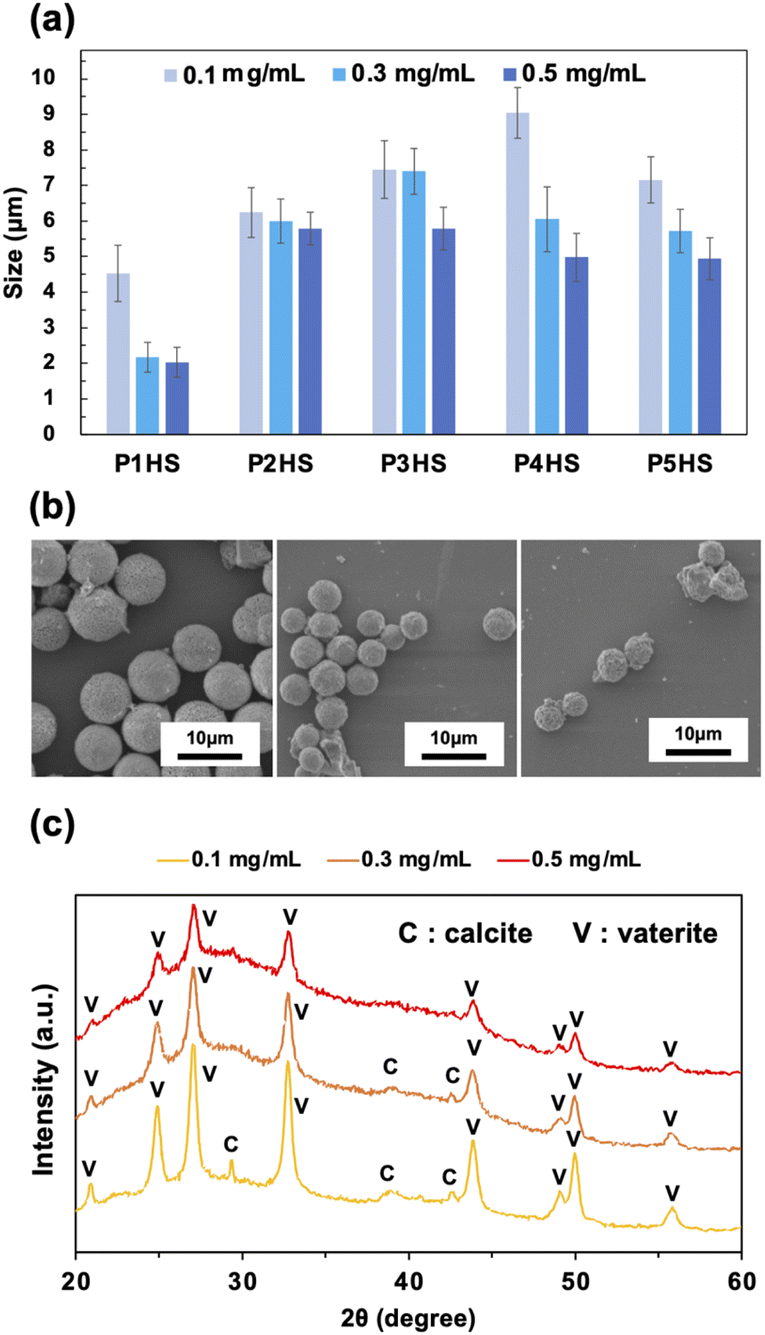 | ||
| Fig. 2 (a) The relationship between the concentration of phenolic polymers and the final particle sizes. The data for 0.1 mg mL−1 are the same as Fig. 1c but also shown here for comparison. (b) SEM images of CaCO3 particles in the presence of P4HS. (c) XRD patterns of CaCO3 particles obtained in the presence of P5HS with different concentrations. | ||
Only vaterite peaks were present in the XRD patterns of the CaCO3 particles when the concentration of P5HS was 0.5 mg mL−1 (Fig. 2c). However, when decreasing the concentration to 0.3 mg mL−1, calcite peaks appeared along with the vaterite peaks, and even larger calcite peaks appeared at 0.1 mg mL−1. Their relative mass percentage of vaterite is 100% (0.5 mg mL−1), 99% (0.3 mg mL−1), and 94% (0.1 mg mL−1), respectively. CaCO3 particles prepared in the presence of P3HS showed only vaterite peaks at all the concentrations, however P1HS, P2HS, and P4HS showed both calcite and vaterite peaks at 0.1 mg mL−1, but the calcite peaks became smaller as the concentration increased (Fig. S6†). These differences occurred because a large amount of polymer was bound around the CaCO3 particles, which prevented the crystal polymorphs from changing.
The compositions of CaCO3 particles in the presence of P4HS were estimated by TGA (Fig. 3). The weight loss between 200 °C and 650 °C were due to the decomposition of the phenolic polymers. At higher polymer concentrations, more polymer molecules were adsorbed on or occluded in the CaCO3 particles, meaning higher polymer concentrations showed a stronger effect on the size of the CaCO3 particles. Distinct thermal decomposition of the polymer and CaCO3 were clearly observed in the TGA curves of the CaCO3 particles prepared with P1HS (Fig. S7†). On the other hand, gradual weight losses were observed for P2HS–P5HS. This is because PS and P1HS decomposed at 400 °C while P2HS, P3HS, P4HS and P5HS were partially carbonized and remained in the CaCO3 particles (Fig. S8a†). A sharp peak of graphite (002) in XRD (Fig. S8b†) confirmed that the P4HS was partially graphitized during the carbonization process.
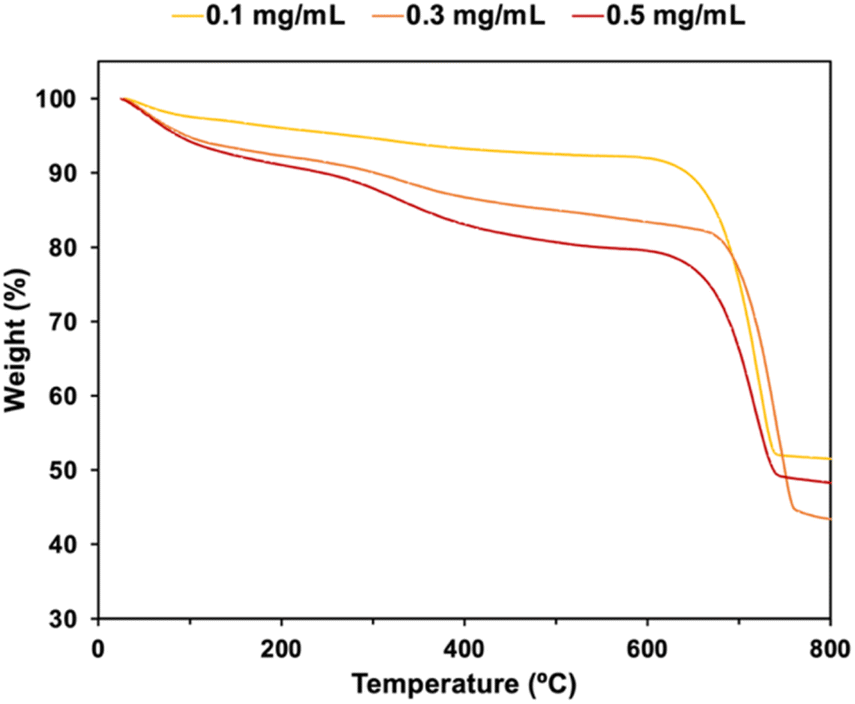 | ||
| Fig. 3 TGA curves of the CaCO3 particles prepared with P4HS at various concentrations. | ||
Diamond-shaped crystals as well as spherical particles could be observed by scanning electron microscopy (SEM, Fig. S4†), likely because the spherical particles recrystallized into rhombic crystals after precipitation. The obtained CaCO3 particles were aggregates of nanoscale particles as previously reported (Fig. S9a†). The hexagonal-shaped crystals made using 0.5 mg mL−1 of P2HS indicated that the oriented growth of the microspheres occurred along crystal planes (Fig. S9b†), which is consistent with the fact that CaCO3 particles have a hierarchical architecture composed of oriented nanometric crystals.11 After calcination at 500 °C, the surface of CaCO3 particles became porous because the phenolic polymers were partially removed (Fig. S9c and d†).
The effect of stirring speed on the crystallization of CaCO3
CaCO3 particles with average diameters ranging from 1.8 to 9.0 μm were obtained at different stirring speeds (1000 rpm and 1500 rpm, compared with the standard of 500 rpm) when using 0.1 mg mL−1 of phenolic polymers (Fig. 4). For all polymers, the average size of the CaCO3 particles became smaller as the stirring speed increased, because the coordination of the phenolic hydroxy groups to Ca2+ was inhibited when the stirring speed increased and there was higher contact between the precursors, resulting in a higher number of individual seeds, which collectively prevented the formation of larger particles. In the presence of P4HS, the average CaCO3 particle size was 9.04 ± 0.71 μm, 4.42 ± 0.65 μm, and 3.56 ± 0.71 μm at stirring speeds of 500 rpm, 1000 rpm, and 1500 rpm, respectively. At 1000 rpm and 1500 rpm, the particle size became smaller, and more rhombic crystals formed, as the interaction between the hydroxy groups of the phenolic polymers and Ca2+ was inhibited and mixing was promoted, which resulted in the formation of more stable crystals. The particle size distribution was narrow and did not significantly depend on the stirring speed or type of phenolic polymer added, as relatively uniform CaCO3 particles could be prepared at any stirring speed (Fig. S10 and S11†). The CV values were about 15% or less for P2HS–P5HS, and about 20% or less for P1HS. These CV values also confirm that the particle size distribution is narrow.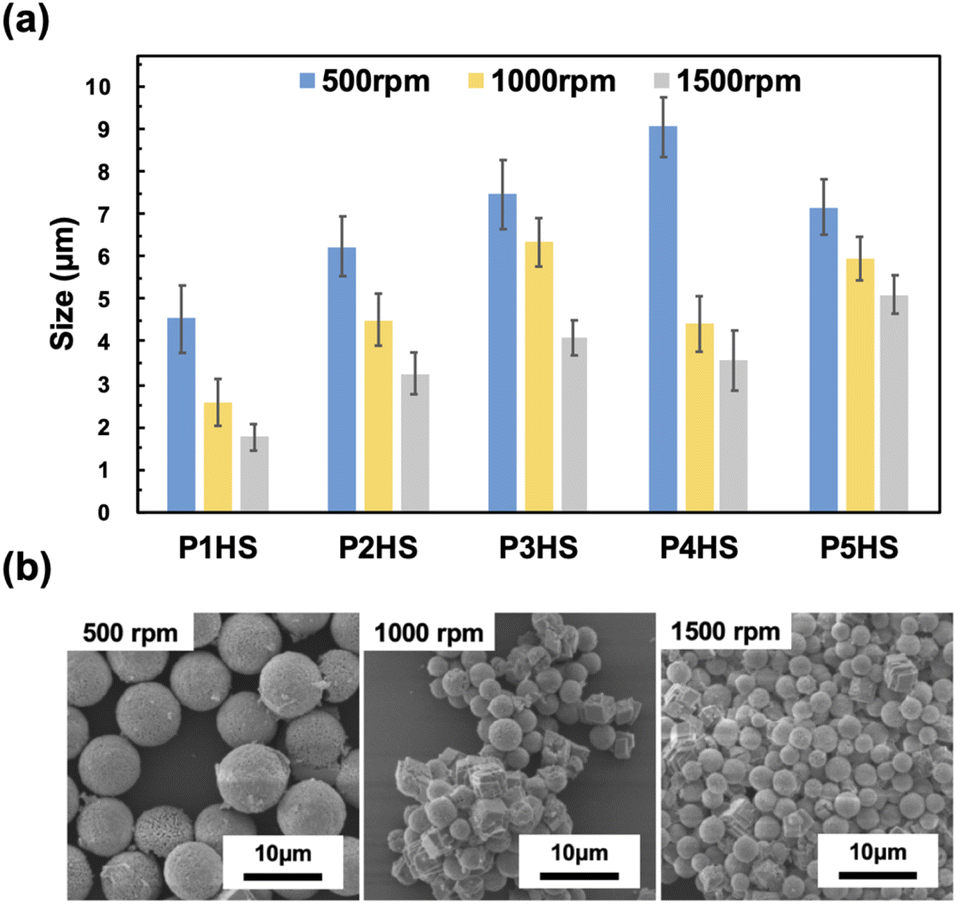 | ||
| Fig. 4 (a) The relationship between the stirring speed of phenolic polymers and the particle size. The data for 500 rpm are the same as Fig. 1c but also shown here for comparison. (b) SEM images of CaCO3 particles in the presence of P4HS (0.1 mg mL−1) at different stirring speed. | ||
The effect of temperature on the crystallization of CaCO3
To examine the effect of temperature on the size and morphology of the CaCO3 particles, P4HS was used to synthesize CaCO3 particles at different temperatures (40 °C and 80 °C, compared with room temperature). As shown in Fig. 5, significant differences were observed in the morphology of the CaCO3 particles prepared at different temperatures. At 40 °C, particles with rough surfaces were observed (Fig. 5a) and the corresponding XRD peaks (Fig. 5b) were mainly from vaterite. Meanwhile, at 80 °C, needle-like crystals (∼6.8 μm) were observed (Fig. 5a) and XRD patterns demonstrated that only aragonite peaks were present (Fig. 5b). This temperature dependence is consistent with previous reports, which discuss the kinetics of formation deeply.17,44 It is suggested that higher temperature is the favourable condition for the nucleation of aragonite crystals. SEM and XRD of CaCO3 particles prepared at 80 °C in the absence of phenolic polymers (Fig. S12†) demonstrated peaks of vaterite, aragonite, and calcite and a mixture of needle-like crystals and flower-like crystals with diameters much smaller than those prepared with P4HS (Fig. 5a). These results indicate that P4HS contributed to the growth and stability of needle-like aragonite crystals at 80 °C.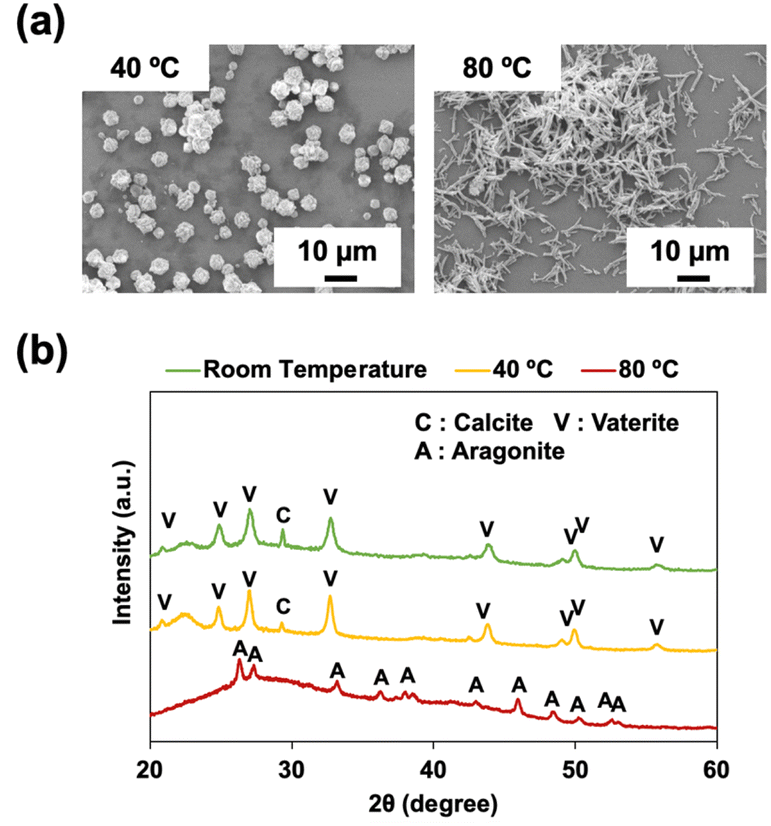 | ||
| Fig. 5 (a) SEM images of CaCO3 particles in the presence of P4HS (0.1 mg mL−1) at different temperatures. (b) XRD patterns of CaCO3 particles obtained in the presence of P4HS (0.1 mg mL−1) at different temperatures. The data for room temperature are the same as Fig. 1d but also shown here for comparison. | ||
The effect of preparation time on the crystallization of CaCO3
The morphology of the CaCO3 particles prepared with P4HS changed depending on the incubation time after stirring for 1 min, where 1 h, 6 h, 12 h, 1 d, 3 d, and 7 d were studied (Fig. 6). As shown in Fig. 6a, the transformation from vaterite to calcite occurred as the preparation time increased, and the calcite crystals became bigger and more complex over time, because the crystal growth proceeded in different directions. The XRD patterns in Fig. 6b clearly show that the calcite peaks became higher and the vaterite peaks became lower over time. The corresponding mass percentage of vaterite was 75% for 1 h, 57% for 12 h, 11% for 24 h, and 0% for 72 h. Taken together, this suggests that CaCO3 particles obtained using P4HS as an additive are not stable unless thermal treatment is applied after stirring. However, if the CaCO3 particles were once calcined at 500 °C soon after precipitation, the recrystallization was prevented, and spherical particles were obtained as shown in Fig. 2b. These calcined particles were stable in terms of morphology in DI water for more than 2 months after re-dispersion. Moreover, XRD patterns of the calcined particles showed that the spherical particles were all calcite (Fig. S13†), confirming that the transformation from vaterite to calcite took place without changing the spherical morphology.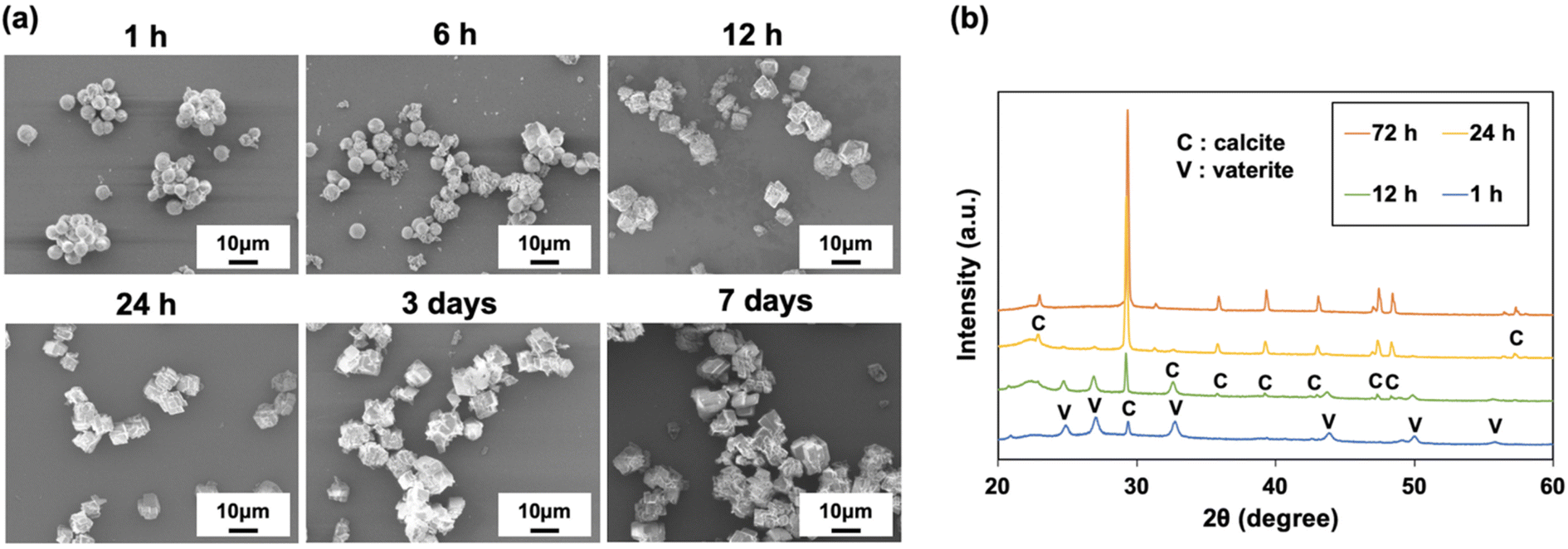 | ||
| Fig. 6 (a) SEM images and (b) XRD patterns of CaCO3 particles obtained in the presence of P4HS (0.1 mg mL−1) at different preparation times. | ||
The effect of molecular weight and scale of reaction on the crystallization of CaCO3
To examine the effect of the molecular weight and scale of reaction, P4HS was used to synthesize CaCO3 particles at a different molecular weight (Mn ∼ 12 kDa, compared with the standard of ∼7 kDa) and at different scales of reaction (0.5× scale, and 5× scale, compared with the standard of 1× scale). When CaCO3 particles were prepared with P4HS (Mn ∼ 12 kDa), particles with diameters of 4.47 ± 0.43 μm were obtained as shown in Fig. S14a.† Compared with those prepared with P4HS (Mn ∼ 7 kDa), precipitated particles became smaller. As already discussed, the viscosity has an important effect on the size of CaCO3 particles. P4HS solution with higher molecular weight has a higher viscosity. The inhibition of crystal growth occurred, and the particles became smaller. The corresponding XRD peaks (Fig. S14b†) demonstrated that the crystals obtained were mainly vaterite demonstrating that the length of polymer chain has a negligible effect on the polymorphs of CaCO3 crystals. The effect of scale of reaction was examined by varying the amount of solution added without changing the final concentrations for each of the solutions. The total amount of solution was 20 mL for 1×, and 10 mL for 0.5× scale, and 100 mL for 5× scale. When CaCO3 particles were obtained at 0.5× scale, smaller particles with a diameter of 2.57 ± 0.49 μm were precipitated (Fig. S15a†). The corresponding XRD results (Fig. S15b†) showed both vaterite and calcite peaks. Smaller vaterite particles were obtained because mixing was promoted at smaller scale of reaction, which resulted in the formation of more seeds of CaCO3 nanoparticles. On the other hand, when CaCO3 particles were obtained at 5× scale, heterogeneous particles with a diameter of 5.29 ± 0.69 μm were precipitated (Fig. S15a†). Rough surfaces were observed demonstrating that microparticle were composed of the aggregation of relatively larger grown nanoparticles. At the larger scale of reaction, weaker sheer forces were created, and the growth of nanoparticles was promoted before aggregation. The corresponding XRD results (Fig. S15b†) showed vaterite peaks mainly. From these results, it is suggested that both the molecular weight and scale of reaction had effects on the size of CaCO3 particles.Discussion
Overall, the effects of six experimental variables were investigated, where polymer concentration, stirring speed, molecular weight, and scale of reaction significantly influenced the particle size, and temperature and incubation time affected the crystal structure and morphology. Specifically, the effect of stirring speed is more prominent than polymer concentration in terms of final particle size. Increasing the concentration of phenolic polymers did not always lead to an increase in the number of nucleation sites because of the low solubilities of P1HS–P3HS. Regarding crystal structure and morphology, the effects of both temperature and preparation time are prominent. Three kinds of crystal structures were all observed by changing the reaction conditions. As shown in Fig. S1,† in the absence of polymer, a mixture of calcite and vaterite was found soon after, however, as time goes on the dissolution of vaterite and growth of calcite proceeds in water (Fig. 7a). In the presence of phenolic polymers, the hydroxy groups of the polymers bind to Ca2+ and slow the nucleation growth and shield seed fusion. Nano-sized amorphous particles form in solution, and then the amorphous precursors transform into nanosized crystals slowly. Finally, the nanoparticles aggregate and grow into microparticles (Fig. 7b). When increasing the polymer concentration, the number of nucleation sites increased, shielding increased, and the obtained microparticles were relatively small. When the stirring speed was higher, sheer forces were created that drastically increased the number of crystallization nuclei and therefore the obtained microparticles became smaller. The sizes and standard deviations of CaCO3 particles prepared under various conditions using different polymers were summarized in Table S2.† Collectively, the size range of CaCO3 particles obtained in this work was compared to previous reports in Fig. 7c, which highlights the usefulness of phenolic polymers as an additive for CaCO3 particle synthesis.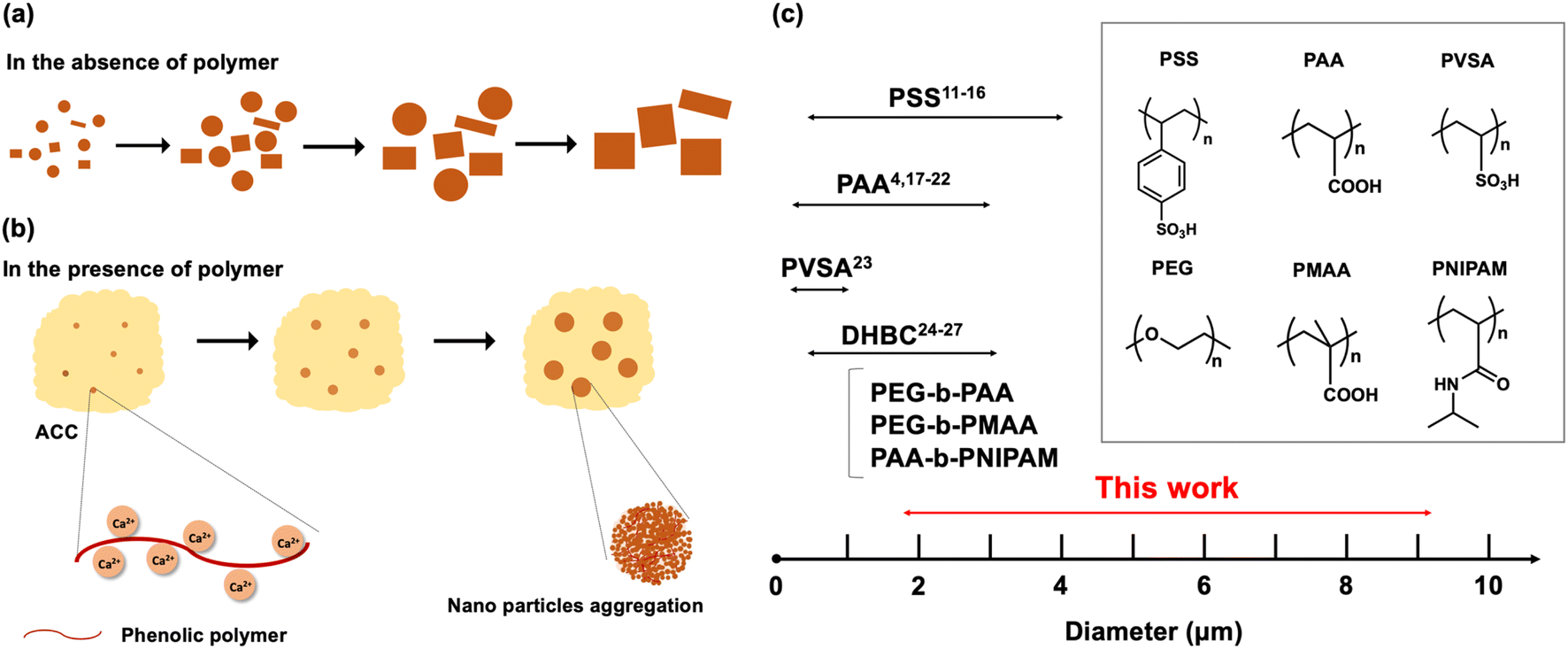 | ||
| Fig. 7 Proposed mechanisms of CaCO3 growth in the absence (a) and presence (b) of phenolic polymers. (c) The size range of CaCO3 particles obtained in the previous reports compared to the range obtained in this work. | ||
Conclusions
In summary, CaCO3 particles with relatively uniform particle diameters in the range of 2–9 μm were successfully synthesized by adding polymers with anywhere from one to five phenolic hydroxy groups, and by changing the polymer concentration, stirring speed, temperature, preparation time, molecular weight, and/or the scale of reaction. In particular, the largest monodisperse particles were precipitated in the presence of P4HS. The particle diameters of the obtained particles became smaller as the polymer concentration increased, and the particle diameters became smaller when the stirring speed was high. Analysis of XRD patterns showed mainly vaterite peaks at higher polymer concentrations though calcite peaks were also detected at low concentration. Temperature and preparation time affect the morphologies of the obtained crystals. At high temperature (80 °C), needle-like aragonite crystals were easily obtained. When incubated in water, spherical vaterite particles transformed to calcite over time if the particles were not calcined. However, once calcined at 500 °C, the particles kept the initial spherical shape in DI water for at least 2 months. Molecular weight and scale of reaction also affected the size of CaCO3 particles showing that the size of CaCO3 particles is determined by various factors. Our findings introduce a new strategy for producing relatively uniform CaCO3 microparticles with diameters that have previously proven difficult to access.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was partially supported by Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant No. 20H02581 and 20K20641), Japan Science and Technology Agency (JST) through the Precursory Research for Embryonic Science and Technology (PRESTO) grant number JPMJPR21N4 (H. E.) and “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Grant Number JPMXP1223UT0277. H. E. acknowledges Katsu Research Encouragement Award of the University of Tokyo. J. J. R. acknowledges JSPS for the postdoctoral fellowship for research in Japan (P20373). J. J. R. is the recipient of an Australian Research Council Future Fellowship (project number FT210100669) funded by the Australian Government. Biorender was used for graphical illustrations.References
- S. Kim and C. B. Park, Langmuir, 2010, 26, 14730–14736 CrossRef CAS PubMed.
- X. Yang, G. Xu, Y. Chen, F. Wang, H. Mao, W. Sui, Y. Bai and H. Gong, J. Cryst. Growth, 2009, 311, 4558–4569 CrossRef CAS.
- D. B. Trushina, T. V. Bukreeva and M. N. Antipina, Cryst. Growth Des., 2016, 16, 1311–1319 CrossRef CAS.
- S. C. Huang, K. Naka and Y. Chujo, Langmuir, 2007, 23, 12086–12095 CrossRef CAS PubMed.
- X. R. Xu, A. H. Cai, R. Liu, H. H. Pan, R. K. Tang and K. Cho, J. Cryst. Growth, 2008, 310, 3779–3787 CrossRef CAS.
- D. Konopacka-Łyskawa, Crystals, 2019, 9, 223 CrossRef.
- Y. Boyjoo, V. K. Pareek and J. Liu, J. Mater. Chem. A, 2014, 2, 14270–14288 RSC.
- P. Fadia, S. Tyagi, S. Bhagat, A. Nair, P. Panchal, H. Dave, S. Dang and S. Singh, 3 Biotech, 2021, 11, 1–30 CrossRef PubMed.
- A. Vikulina, J. Webster, D. Voronin, E. Ivanov, R. Fakhrullin, V. Vinokurov and D. Volodkin, Mater. Des., 2021, 197, 109220 CrossRef CAS.
- M. Abebe, N. Hedin and Z. Z. Bacsik, Cryst. Growth Des., 2015, 15, 3609–3616 CrossRef CAS.
- H. Imai, N. Tochimoto, Y. Nishino, Y. Takezawa and Y. Oaki, Cryst. Growth Des., 2012, 12, 876–882 CrossRef CAS.
- C. Wang, C. He, Z. Tong, X. Liu, B. Ren and F. Zeng, Int. J. Pharm., 2006, 308, 160–167 CrossRef CAS PubMed.
- M. Lei, W. H. Tang, L. Z. Cao, P. G. Li and J. G. Yu, J. Cryst. Growth, 2006, 294, 358–366 CrossRef CAS.
- Y. Wang, Y. X. Moo, C. Chen, P. Gunawan and R. Xu, J. Colloid Interface Sci., 2010, 352, 393–400 CrossRef CAS PubMed.
- A. Jada and A. Verraes, Colloids Surf., A, 2003, 219, 7–15 CrossRef CAS.
- H. Kawaguchi, H. Hirai, K. Sakai, S. Seral, T. Nakajima, Y. Ebisawa and K. Koyama, Colloid Polym. Sci., 1992, 270, 1176–1181 CrossRef CAS.
- S. Ouhenia, D. Chateigner, M. A. Belkhir, E. Guilmeau and C. Krauss, J. Cryst. Growth, 2008, 310, 2832–2841 CrossRef CAS.
- J. Yu, M. Lei, B. Cheng and X. Zhao, J. Solid State Chem., 2004, 177, 681–689 CrossRef CAS.
- J. Tark Han, X. Xu and K. Cho, J. Cryst. Growth, 2007, 308, 110–116 CrossRef.
- B. Cheng, M. Lei, J. Yu and X. Zhao, Mater. Lett., 2004, 58, 1565–1570 CrossRef CAS.
- H. Matahwa, V. Ramiah and R. D. Sanderson, J. Cryst. Growth, 2008, 310, 4561–4569 CrossRef CAS.
- K. Naka, S. C. Huang and Y. Chujo, Langmuir, 2006, 22, 7760–7767 CrossRef CAS PubMed.
- A. T. Nagaraja, S. Pradhan and M. J. McShane, J. Colloid Interface Sci., 2014, 418, 366–372 CrossRef CAS PubMed.
- I. M. Weiss, N. Tuross, L. Addadi, S. Weiner, E. Beniash and J. Aizenberg, Proc. R. Soc. London, Ser. B, 2001, 135, 1875–1879 Search PubMed.
- H. Cölfen and L. Qi, Chem.–Eur. J., 2001, 7, 106–116 CrossRef.
- S. Guragain, N. L. Torad, Y. G. Alghamdi, A. A. Alshehri, J. Kim, B. P. Bastakoti and Y. Yamauchi, Mater. Lett., 2018, 230, 143–147 CrossRef CAS.
- M. Lei, W. H. Tang and J. G. Yu, Mater. Res. Bull., 2005, 40, 656–664 CrossRef CAS.
- Ç. M. Oral and B. Ercan, Powder Technol., 2018, 339, 781–788 CrossRef.
- G. Yan, L. Wang and J. Huang, Powder Technol., 2009, 192, 58–64 CrossRef CAS.
- L. Wang, Z. Meng, Y. Yu, Q. Meng and D. Chen, Polymer, 2008, 49, 1199–1210 CrossRef CAS.
- Y. Tanaka and K. Naka, Polym. J., 2010, 42, 676–683 CrossRef CAS.
- Y. Tanaka and K. Naka, Polym. J., 2012, 44, 586–593 CrossRef CAS.
- R. Febrida, A. Cahyanto, E. Herda, V. Muthukanan, N. Djustiana, F. Faizal, C. Panatarani and I. M. Joni, Materials, 2021, 14, 4425 CrossRef CAS PubMed.
- B. Cheng, J. Yu, T. Arisawa, K. Hayashi, J. J. Richardson, Y. Shibuta and H. Ejima, Nat. Commun., 2022, 13, 1892 CrossRef CAS PubMed.
- J. Yu, B. Cheng and H. Ejima, J. Mater. Chem. B, 2020, 8, 6798–6801 RSC.
- A. Wulf, R. I. Mendgaziev, R. Fakhrullin, V. Vinokurov, D. Volodkin and A. S. Vikulina, Adv. Funct. Mater., 2022, 32, 2109824 CrossRef CAS.
- F. Tewes, O. L. Gobbo, C. Ehrhardt and A. M. Healy, ACS Appl. Mater. Interfaces, 2016, 8, 1164–1175 CrossRef CAS PubMed.
- X. H. Guo, S. H. Yu and G. B. Cai, Angew. Chem., Int. Ed., 2006, 45, 3977–3981 CrossRef CAS PubMed.
- X. Geng, L. Liu, J. Jiang and S. H. Yu, Cryst. Growth Des., 2010, 10, 3448–3453 CrossRef CAS.
- M. S. Rao, Bull. Chem. Soc. Jpn., 1973, 46, 1414–1417 CrossRef CAS.
- D. V. Volodkin, A. I. Petrov, M. Prevot and G. B. Sukhorukov, Langmuir, 2004, 20, 3398–3406 CrossRef CAS PubMed.
- C. B. Tovani, D. C. Zancanela, A. N. Faria, P. Ciancaglini and A. P. Ramos, J. Mater. Chem., 2011, 6, 90509–90515 Search PubMed.
- A. G. Christy, Cryst. Growth Des., 2017, 17, 3567–3578 CrossRef CAS.
- L. N. Zhao, J. K. Wang and Z. C. Wang, Chem. Res. Chin. Univ., 2013, 29, 969–973 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04791a |
| This journal is © The Royal Society of Chemistry 2023 |