
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimized synthesis of polyacrylic acid-coated magnetic nanoparticles for high-efficiency DNA isolation and size selection†
Nesrine Bali a,
Svein J. Brennhauga,
Magnar Bjørås*bcd,
Sulalit Bandyopadhyay‡
*a and
Adeel Manaf‡*bcd
a,
Svein J. Brennhauga,
Magnar Bjørås*bcd,
Sulalit Bandyopadhyay‡
*a and
Adeel Manaf‡*bcd
aParticle Engineering Centre, Department of Chemical Engineering, NTNU, Trondheim, Norway. E-mail: sulalit.bandyopadhyay@ntnu.no
bDepartment of Clinical and Molecular Medicine, NTNU, Trondheim, Norway
cDepartment of Microbiology, Oslo University Hospital, Oslo, Norway
dCentre of Embryology, University of Oslo, Oslo, Norway
First published on 4th October 2023
Abstract
Solid-phase reversible immobilization (SPRI) bead technology is widely used in molecular biology for convenient DNA manipulation. However, commercial SPRI bead kits lack cost advantages and flexibility. It is, therefore, necessary to develop new and alternative cost-effective methods of on-par or better quality. Herein, an easy and cost-effective method is proposed for synthesizing polyacrylic acid-coated iron oxide nanoparticles (PAA-IONPs) through in situ polymerization at lab scale for high-efficiency nucleic acid extraction and size selection. A design of experiment (DoE) approach was used to investigate the influence of iron oxide nanoparticles (IONPs), acrylic acid (AA) monomer, and sodium dodecyl sulfate (SDS) surfactant amounts on the sizes and carboxyl group densities of PAA-IONPs. Thorough characterization by thermogravimetric analysis (TGA), attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) and vibrating sample magnetometry (VSM) highlights the importance of a low starting pH achieved by a high ratio of AA/IONPs, to yield the largest sizes (554 nm) and highest carboxyl group densities (2.13 mmol g−1) obtained in this study. An efficient DNA purification strategy is then presented using homemade beads-suspension buffer and optimized bead concentrations (17% PEG 8000, 2.5 M NaCl, and 3 mg mL−1 PAA-IONPs). This method shows comparable performance to the control (AMPure XP beads) for DNA recovery. An adjustable PAA-IONPs DNA purification system was also developed to be used for DNA-size selection at low DNA amounts (50–100 ng) with a high degree of resolution and recovery. In conclusion, this work offers an optimized PAA-IONPs synthesis protocol and a flexible DNA purification approach that will enable researchers to manipulate DNA under various conditions, holding the significant potential to benefit future molecular biology research and diagnostics.
1. Introduction
In the field of nucleic acid extraction and manipulation, magnetic bead-based commercial kits, particularly solid-phase reversible immobilization (SPRI) paramagnetic beads, have been widely employed. These beads play a fundamental role in various genomic and epigenomic analyses, such as DNA extraction and size selection of next-generation sequencing (NGS) libraries,1–3 recovery of longer DNA fragments (>6 kbp) for nanopore sequencing,4 and manipulation of target DNA fragments in 3D chromosome conformation capture5 among others. Compared to traditional DNA extraction methods,6,7 these beads offer several advantages, including simplicity, efficiency, high-throughput capability, and the avoidance of tedious steps and toxic chemicals.Several factors come into play to optimize the SPRI bead DNA purification method. The surface chemistry, diameter, shape, and magnetic properties of the beads, along with the binding threshold, all affect DNA binding and recovery. Additionally, the concentration of polyethylene glycol (PEG) and NaCl, which induce the conformational transition of DNA and facilitate its binding to the bead surface through ‘salty ion bridging,’ also influence DNA binding and recovery. Adjusting these variables allows the DNA purification method to be fine-tuned to meet specific experimental requirements.
Numerous methods have been reported in the literature to synthesize iron oxide nanoparticles (IONPs) of varying shapes, sizes, magnetic strengths and dispersion properties.8–11 An organic coating is usually necessary to increase their colloidal stability and biocompatibility, while allowing specific surface functionalization. Different techniques such as nanoprecipitation,12 flash nanoprecipitation,13 dendronization,14 and emulsion-based methods15,16 can be employed for this purpose. Herein, free radical polymerization of polyacrylic acid (PAA) is conducted in the presence of IONPs. This method presents the advantages of being a simple and straight-forward strategy to functionalize IONPs with carboxyl groups while providing control over the size and carboxyl group density of PAA-IONPs. Although the mechanism of free PAA polymerization has been extensively studied,17–20 limited research is available on polymerization mechanism for coating nanomaterials with a monolayer of carboxyl groups that does not compromise the magnetic properties of IONPs.21,22 Hence, this work aims to investigate the influence of the amounts of acrylic acid (AA), IONPs and sodium dodecyl sulfate (SDS) on the physical properties of PAA-IONPs. The findings are then correlated to the performance of the beads for DNA extraction and size selection under various conditions. By determining the optimal concentrations of PEG 8000, NaCl, PAA-IONPs' amount, and reaction volume ratio, variable-volume samples could be accommodated, thus enabling flexible DNA manipulation. The recovery of DNA fragments using the adjustable PAA-IONPs DNA purification system was studied with a control method employing AMPure XP beads. Fragment selection was also assessed under different conditions, including different types of PAA-IONPs, varying amounts of NPs, and buffer conditions. The results demonstrate that the PAA-IONPs DNA purification system is a cost-effective and high-performance method for DNA extraction and manipulation, offering a flexible option for DNA purification for a wide range of genomic applications. The following sections provide detailed synthesis and characterization methods, along with results and discussions that shed light on the polymerization mechanism and highlight the performance and advantages of PAA magnetic nanoparticles in high-efficiency DNA extraction and size selection.
2. Experimental section
2.1. Materials
Iron(III) chloride hexahydrate (FeCl3·6H2O, ≥99%), ammonia solution (NH4OH 25%), sodium dodecyl sulfate (SDS, ≥99%), acrylic acid (AA, 99%), potassium persulfate (KPS, ≥99%), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, ≥99.5%), pyrocatechol violet (PV) and dialysis tubing (12 kDa cut-off) were purchased from Sigma-Aldrich (Schnelldorf, Germany). Iron(II) chloride tetrahydrate (FeCl2·4H2O, ≥99%) and nickel(II) chloride hexahydrate (NiCl2·6H2O, ≥98%) were obtained from VWR. All the chemicals were used as received. Milli-Q water having a resistivity of ca. 18.2 MΩ cm−1 at 25 °C was produced by a Sartorius Arium mini Water System from distilled water.The following reagents were used in the size selection experiments: home-made polyacrylic acid coated IONPs (PAA-IONPs); UltraPure™ Nuclease free Water (Thermo Fisher Scientific, cat. #10977035); UltraPure™ 1 M Tris–HCl buffer, pH-8 (Thermo Fisher Scientific, cat. #15568-025); polyethylene glycol (PEG) 8000 (Merk, cat. # 89510-250G-F); 5 M sodium chloride (Merck, cat. # 71386); diamine tetra acetic acid (EDTA) (EMELCA Biosciences, cat. #40520000-1); Tween-20 (Merk, cat. #11332465001); DNA ladder (Thermo Fisher Scientific, cat. #50473). All test experiments were performed with 50 ng of DNA ladder. Commercial extraction kit Agencourt® AMPure® XP (Beckman Coulter, cat. # A63880) was used as a positive control.
2.2. Preparation of PAA-IONPs and bare PAA
Bare IONPs were synthesized through co-precipitation as previously reported by our group.23,24 The in situ polymerization of PAA on bare IONPs was adapted from earlier work.25 Bare IONPs were added in the amounts mentioned in Table 1 to a stirred solution of SDS surfactant in 20 mL of MQ water under nitrogen atmosphere. The NP solution was heated to 73 °C before the addition of acrylic acid monomers. After 45 min, 400 mg of KPS initiator dissolved in 10 mL of MQ water were added and the reaction was stirred for 2 h at 73 °C under inert atmosphere. Upon completion, the NP solution was cooled down to room temperature, washed several times with MQ water by magnetic separation and redispersed in MQ water.| Condition | IONPs amount (mg) | SDS amount (mg) | AA amount (μL) |
|---|---|---|---|
| A1 | 166 | 230 | 77 |
| A2 | 498 | 0 | 77 |
| A3 | 498 | 115 | 231 |
| A4 | 166 | 0 | 77 |
| A5 | 166 | 230 | 231 |
| A6 | 332 | 115 | 154 |
| A7 | 332 | 115 | 154 |
| A8 | 498 | 230 | 77 |
| A9 | 166 | 0 | 231 |
Bare PAA was also synthesized following the same method in the presence of 1.54 mL of AA and 100 mg of KPS in 20 mL of MQ water. The viscous solution was then dialysed against MQ water for 24 h.
2.3. Design of experiment
A custom design was created using JMP® Pro software version 16.1.0. SAS Institute Inc., Cary, North Carolina, USA, to build the experimental design and analyse the data. The levels of the components and responses studied are shown in Table 2. The hydrodynamic size (Hd size) and COOH density were assumed to be the most relevant outcomes to influence the interaction between PAA-IONPs and DNA. It was hypothesized that such interaction would be maximized when the hydrodynamic size and COOH density of PAA-IONPs would be respectively the lowest and the highest, in order to provide a large surface area functionalized with carboxyl groups to bind DNA. The design consisted of 9 runs, including two center points (A6 and A7) as listed in Table 1. The center points were placed midway between the highest and lowest values of the design space to assess the linearity and stability of the model, as well as to increase its statistical power. Standard least squares method was used later to run the fitted model and evaluate the interaction between components with a statistical significance set at 0.05.| Factors | Low level | High level |
|---|---|---|
| IONPs amount (mg) | 166 | 498 |
| SDS amount (mg) | 0 | 230 |
| AA amount (μL) | 77 | 231 |
| Responses | Goal |
|---|---|
| Hd size (nm) | Minimize |
| COOH density (mmol g−1) | Maximize |
2.4. Characterization
2.5. DNA isolation and size selection
Different buffer compositions (the critical concentration of PEG 8000 and beads) and working conditions, such as incubation time for DNA binding, magnetic separation, and incubation time for elution tested, are reported in ESI Table 1.† The beads preparation was performed as follows: the desired amount of PAA-IONPs (6–1 mg) was transferred to a 1.5 mL tube and placed on the magnet to allow the beads to be drawn to the magnet. The supernatant was carefully removed, NPs were washed twice with 1 mL of TE buffer (10 mM Tris–HCl, pH-8, 1 mM EDTA), and were fully resuspended in 1 mL of binding buffer (10 mM Tris–HCl, pH 8, 1 mM EDTA, 24% or 17% PEG-8000, 2.5 M or 1 M NaCl, Tween-20 0.044%) by gentle mixing, resulting in a final desired concentration. The resuspended PAA-IONPs were wrapped in tinfoil or placed in a dark container and stored at 4 °C.For the total DNA isolation or size-selection test, 50 ng of DNA ladder was diluted to 50 μL. An appropriate volume of bare IONPs, PAA-IONPs, or AMPure XP (the specific volume depends on the desired fractionation) was added to each sample. Samples were mixed thoroughly. The mixture was incubated at room temperature for 8 minutes, followed by placement on a magnetic stand for 3 minutes. The supernatant was then removed, and each sample was washed twice with 180 μL of freshly prepared 80% ethanol without removing from the magnet. The pellet was allowed to dry, ensuring not to over-dry to avoid cracking. The pellet was rehydrated with 11 μL of Qiagen Elution Buffer and incubated for 5 minutes at room temperature. Afterward, samples were placed on the magnetic stand for 3 minutes. The supernatant was then transferred to a new tube. All eluted DNA was quantified with the Qubit® Fluorometer dsDNA HS Assay Kit (Invitrogen), and the fragment distribution of samples was assessed using the TapeStation HS DNA kit (Agilent Technologies).
3. Results and discussion
Magnetite IONPs were synthesized via co-precipitation method and subsequently coated with PAA to enable the binding of DNA fragments of varying sizes. The following sections will cover the characterization of bare IONPs and PAA-IONPs by several techniques to gain a deeper understanding of the influence of the synthesis parameters over the sizes and carboxyl densities, the interaction between PAA and IONPs, and the amount of PAA formed in different experimental conditions. Later, selected PAA-IONPs are tested for DNA extraction and size selection.3.1. Bare IONPs
The XRD pattern (Fig. 1c) displays characteristic peaks corresponding to spinel iron oxide of magnetite. The 2θ values of 30°, 36°, 43°, 54°, 57° and 63° are attributed respectively to the planes (220), (311), (400), (422), (511) and (440) as previously reported.24,28,29 The crystal size of the NPs derived from Scherrer equation was calculated to be 9.6 nm which closely aligns with the TEM size (Fig. 1b) estimated at 9.3 ± 2 nm for the spheroid IONPs.30 | ||
| Fig. 1 (a) Schematic of the synthesis process. (b) TEM image of bare IONPs with 50 nm scale bar and inset of particle size distribution. (c) XRD spectrum of magnetite IONPs. (d) VSM magnetization curve for bare IONPs. (e) Study of zeta potential as a function of pH. | ||
Fig. 1d depicts the superparamagnetic properties of IONPs where no remanent magnetization was observed in the absence of a magnetic field. The saturation magnetization of these NPs, measured at 54 emu g−1, falls within the typical range reported in the literature, indicating substantial magnetic strength.31 After conducting Langevin fitting of the experimental data, the magnetic core diameter was determined to be 11.3 nm. The size estimations of IONPs in dry state obtained from TEM, XRD and VSM data exhibit good agreement with minimal discrepancies attributed to batch-to-batch variations. Considering the requirement of biomedical applications for NPs to be suspended in aqueous solutions, the hydrodynamic size also needs to be considered. Dynamic light scattering (DLS) was employed to measure the hydrodynamic diameter which accounts for the corona of water molecules diffusing at the same speed as the NPs. For bare IONPs, it increased from 93 ± 7 nm at pH 3 to 175 ± 5 nm at pH 8. As shown in Fig. 1e, the NPs exhibit positive surface charges above +20 mV at pH < 6 and negative charges below −20 mV at pH > 8.5. Standard deviations of triplicates measurements are too small to be distinguished from marker points. According to the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory, IONPs demonstrate good colloidal stability in these pH ranges due to high electrostatic repulsive forces.32 It is well documented that these interactions are prevented close to the isoelectric point (IEP) of IONPs at pH = 7.5, resulting in NP aggregation and larger DLS sizes (Fig. 3a).33 At pH 7.5, NPs possess a neutral global charge, leading to the predominance of van der Waals forces.34,35 Furthermore, interparticle magnetic dipole–dipole interactions also contribute to aggregate formation, particularly at high concentrations.36 This underscores the need to improve the colloidal stability of IONPs at neutral pH through functionalization, such as with PAA, especially for biomedical applications. Oxidation and loss of magnetic properties are also common challenges that require a protective layer over bare IONPs.37
3.2. Carboxyl group quantification assay
A colorimetric assay was used to quantify the number of surface carboxyl groups after in situ polymerization of AA on the above IONPs. This method relies on the affinity of nickel ions to bind carboxyl groups with a stoichiometry of 2.65 carboxyl groups per nickel ion.26 NP solutions were incubated shortly with 0.2 mM Ni2+ solution. After centrifugation, the supernatant containing unbound Ni2+ was collected and reacted with pyrocatechol violet (PV) for UV-VIS measurement. The absorption spectrum of 0.04 mM PV in 10 mM HEPES pH 7.5 displays two peaks at ca. 450 and 600 nm (Fig. 2a). In the presence of increasing concentrations of Ni2+, higher absorbance values are measured at 600 nm due to complexation with the indicator dye. A calibration curve was constructed with known concentrations of Ni2+ ranging from 0 to 0.2 mM with R2 > 0.99 (Fig. 2b) and the previously established equation was employed to determine the number of carboxyl groups.27
 | (1) |
 | ||
| Fig. 2 (a) UV-VIS spectra at different concentrations of Ni2+ in the presence of 40 μM of PV in 10 mM HEPES pH 7.5. (b) Calibration curve of the carboxyl group quantification assay. (c) Carboxyl group titration of DOE conditions. (d) Carboxyl group density prediction model. (e) Hydrodynamic sizes measured for DOE batches. (f) Hydrodynamic size prediction model. | ||
This quantification assay was previously shown to yield comparable results to other titration methods such as conductimetry, with the advantages of being faster and more sensitive.38 Hence, it was chosen to characterize all PAA-IONPs. The results of the assay reported in Fig. 2c varied between 0.82 and 1.85 mmol g−1, with the highest densities observed for A3, A5 and A9. Notably, these coating conditions involved a twofold increase of AA compared to the previous study,25 suggesting that this parameter exerted the most significant influence on carboxyl group densities. To validate this hypothesis, a comprehensive statistical analysis was performed using JMP software. Fig. 2d displays a remarkable fit of the model with an R2 of 0.99348 and a p-value of 0.0194. The p-values of the parameters assessed in the model presented in Table 3 indicate a strong influence of the monomer and IONPs amounts on the number of carboxyl groups. Higher concentrations of both IONPs and AA provide a larger surface area available for adsorption of potentially longer PAA chains. Bare IONPs were also tested to confirm that the interaction between hydroxyl groups and nickel ions was too weak to significantly affect the results for PAA-IONPs with a maximum background signal of 0.38 mmol g−1.
| Model terms | P-Value for COOH density | P-Value for Hd size |
|---|---|---|
| IONPs amount (mg) | 0.02768* | 0.08904 |
| SDS amount (mg) | 0.13178 | 0.05443 |
| AA amount (μL) | 0.00458* | 0.00427* |
| IONPs amount (mg) × SDS amount (mg) | 0.87146 | 0.10349 |
| IONPs amount (mg) × AA amount (μL) | 0.31306 | 0.02261* |
| SDS amount (mg) × AA amount (μL) | 0.22374 | 0.05443 |
To gain deeper insights into the underlying polymerization process, a new set of parameters was explored (Table 4). In the absence of SDS surfactant, which was shown to have no impact on the outcomes, the same concentrations of IONPs were mixed with 1.56 mL (A10) and 156 μL (A11) of AA. The results validate the dependence of carboxyl group density on the AA/IONPs ratio. Interestingly, a ten-fold increase of AA led to a carboxyl group density of 2.13 mmol g−1 for A10, although an even higher value could have been expected. Another study on in situ polymerization of AA demonstrated that AA monomers adsorb onto hydroxyl surfaces prior to polymerization initiation.21 This finding suggests that PAA coating of IONPs can only occur up to a certain extent and is limited by the available surface area of IONPs estimated to be 300 nm2 per NP in our case.
| Cond. | IONPs amount (mg) | SDS amount (mg) | AA amount (μL) | COOH density (mmol g−1) | Hd size (nm) |
|---|---|---|---|---|---|
| A10 | 166 | 0 | 1560 | 2.13 (±0.03) | 554 (±93) |
| A11 | 166 | 0 | 156 | 1.58 (±0.03) | 332 (±12) |
3.3. Size control
The hydrodynamic sizes measured for PAA-IONPs, as presented in Fig. 2e, were larger than those of bare IONPs in MQ water. The diameters ranged from 275 to 453 nm (Fig. 2e) with a similar trend to the carboxyl quantification results (Fig. 2c). These results confirm the successful coating of IONPs with PAA. The prediction model constructed based on these data exhibited good accuracy, with an R2 of 0.9928 and a p-value of 0.0214. The outcomes of the statistical analysis summarized in Table 3 highlight the significant role of the investigated AA amounts with a p-value = 0.00427. The smallest diameters were obtained for conditions having the lowest AA amount (77 μL): A1, A2, A4 and A8. On the other hand, A3 displayed the largest size, corresponding to the highest concentrations of both AA and IONPs. This strong dependence on AA concentration was further confirmed when the concentration was increased by a factor of 10 for A10 (Table 4) resulting in a diameter of 535 nm. These data suggest that IONPs were either coated with a thick layer of PAA or encapsulated within PAA networks, forming clusters of IONPs. TEM imaging of PAA-IONPs stained by uranyl acetate as a contrast agent did not reveal any discernible difference compared to bare IONPs. It is thus unlikely that multiple layers of PAA were formed on IONPs, as previously reported.25SDS can form negatively charged micelles in aqueous solutions above its critical micelle concentration (cmc) of 8.2 mM.39 Previous studies have shown that SDS could partially adsorb onto hydroxyl coated NPs and form supercharged systems with even higher electrostatic repulsion forces.39 This finding by Ahualli et al. was unexpected since other studies conducted at different SDS and NPs concentrations demonstrated that such interactions could not occur.40,41 In the case of partial adsorption of SDS on IONPs, the interaction between AA and IONPs as well as the PAA coating could be affected. The DOE included experimental conditions without SDS and at concentrations slightly below cmc (8 mM) and above cmc (16 mM). The SDS term scored a p-value of 0.0544, making it challenging to infer the interaction between SDS and IONPs.
3.4. PAA coating of IONPs
Further investigations were conducted to gain more insights into the coating process by examining zeta potential as a function of pH for A2 and A9 (Fig. 1e). A9, which had the lowest IONPs/AA ratio produced PAA-IONPs with a size of 370 nm and one of the highest carboxyl group densities (1.85 mmol g−1). Conversely, A2 was synthesized with the highest IONPs/AA ratio and yielded smaller sizes and much lower carboxyl group values (300 nm and 0.88 mmol g−1). It was expected that A2 would be partially coated with PAA, exhibiting an intermediary trend between A9 and bare IONPs. At pH > 9, all three NPs displayed negative zeta potentials. The zeta potentials of both A2 and A9 remained negative as the pH decreased between −22 and −40 mV owing to efficient carboxyl group functionalization. The PAA coating clearly improved the colloidal stability of IONPs across the pH range studied as previously reported.34,42Additional evidence of carboxyl groups on A5 was obtained through FT-IR data, which showed a distinctive –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration peak from –COOH at 1719 cm−1 (Fig. 3a) that is also found in bare PAA prominently at 1697 cm−1. Asymmetric and symmetric –CO stretching vibrations from –COO− also appeared at 1595 cm−1 and 1420 cm−1 respectively, along with –CH2 scissoring vibration from PAA at 1455 cm−1.43,44 Similar peaks were observed in bare PAA, although shifted respectively to a shoulder ca. 1630 cm−1 and two distinct peaks at 1406 cm−1 and 1450 cm−1 in the absence of IONPs. Both IONPs and PAA-IONPs show Fe–O bond from magnetite at 547 cm−1 and 558 cm−1 respectively due to a change of surface chemistry upon PAA adsorption.
O stretching vibration peak from –COOH at 1719 cm−1 (Fig. 3a) that is also found in bare PAA prominently at 1697 cm−1. Asymmetric and symmetric –CO stretching vibrations from –COO− also appeared at 1595 cm−1 and 1420 cm−1 respectively, along with –CH2 scissoring vibration from PAA at 1455 cm−1.43,44 Similar peaks were observed in bare PAA, although shifted respectively to a shoulder ca. 1630 cm−1 and two distinct peaks at 1406 cm−1 and 1450 cm−1 in the absence of IONPs. Both IONPs and PAA-IONPs show Fe–O bond from magnetite at 547 cm−1 and 558 cm−1 respectively due to a change of surface chemistry upon PAA adsorption.
 | ||
| Fig. 3 (a–c) FT-IR spectra of different PAA-IONPs and controls. (d) Proposed binding mechanism of PAA containing of uncoordinated carboxyl groups and carboxylates adsorbed on IONPs through bidentate bridging. (e) Thermogravimetric analysis of IONPs and PAA-IONPs in inert atmosphere. (f) Magnetic saturation curves of PAA-IONPs exhibiting clear correlation with TGA data. | ||
A control experiment was conducted without the initiator nor the surfactant, and with three washing steps to discard unbound AA (AA-IONPs). This was done to rule out the possibility of unpolymerized AA monomers being adsorbed on IONPs instead of PAA. The carboxyl and carboxylate peaks observed previously shifted respectively to 1639, 1524 and 1439 cm−1, suggesting different binding interactions between IONPs and AA vs. PAA. This confirms the in situ polymerization of PAA on IONPs surface, as well as the strong interaction between AA and IONPs.21,45 Spectra of PAA-IONPs synthesized with and without SDS, respectively A5 and A2, were also compared to pure SDS in Fig. 3b. Both spectra were similar and did not show any characteristic peaks reported elsewhere for SDS.46 This result indicates that no SDS molecules assisted in PAA-IONPs stabilization after the washing steps.
The complexation mechanism of surface Fe atoms on IONPs with carboxyl groups from PAA can be investigated using eqn (2):
| Δν = (νasym − νsym) | (2) |
O vs. asymmetric –COO− provides information about the protonation state of PAA.45,47 Consistent trends were observed across most PAA-IONPs conditions run with different IONPs/AA ratios. Addition of high IONPs amounts dispersed in basic solution neutralized the acidic pH of AA solution, thereby determining the initial pH of the solution. FT-IR spectra in Fig. 3c revealed that as the pH of the initial solution containing AA and IONPs decreased, the peak attributed to –COOH was more prominent than the one for –COO−. This indicates the presence of different proportions of free carboxyl groups vs. adsorbed carboxylates in PAA-IONPs (Fig. 3d).48 Low starting pH is also one of the factors that increases the polymerization rate of bulk PAA.17–20 Carboxylic groups possess higher reactivity than carboxylates due to the electrostatic repulsion forces of their negative charges that trigger the unfolding of PAA chains. Growing polymers become more accessible to further monomer addition unless cationic species are present in solution. This competition phenomenon arises from the shielding effect of cations that interact with carboxylates and prevent monomer addition.18 This effect is even more pronounced at low AA concentrations. Scott et al. demonstrated such influence of the pH for AA concentrations ranging from 1.45 to 10.9 M, which are much higher than the ones used in this work (0.02–0.07 M). While the exact concentration of cationic species in solution is unknown, it is reasonable to assume that it exceeds 0.03 M, the concentration of K+ added through KPS. In comparison, most of the works previously cited were conducted with initiator concentrations lower or equal to 0.01 M. Higher AA concentrations are also linked with faster polymerization kinetics due to faster decomposition of KPS. Therefore, it can be inferred that the polymerization rate in A10 was much higher than in the other conditions owing to its lowest IONPs/AA ratio which lowered the pH to 2.5. On the other hand, A2 exhibited the slowest kinetics with a starting pH of 8. Although the kinetics of PAA polymerization in the presence of IONPs are unknown, it is assumed that all conditions reached completion after 2 h due to such a high concentration of KPS and a clear colour change of NPs from black to brown.
TGA was used to quantify the mass of PAA coating IONPs. The results presented in Fig. 3e show a mass loss of 5% for bare IONPs, likely originating from the evaporation of surface hydroxyl groups. For PAA-IONPs, two different profiles were recorded: one mass loss for A2 and two consecutive losses for A9 and A10. The first one occurring between 200–500 °C is commonly associated with the degradation of organic compounds.49,50 The other one between 500 and 800 °C results from the oxidation of Fe3O4 to FeO, as FeO is thermodynamically stable above 570 °C.49,51,52 The amount of PAA in A2, A9 and A10 represents respectively 12, 17.7 and 16.2% of the total mass of NPs. The data for A2 confirms the previous findings and the fact that AA amount used was not sufficient to completely functionalize IONPs. A10, with the highest amount of AA and the fastest kinetics, was expected to contain a higher fraction of PAA than A9. The results for A9 and A10 can be explained by the faster kinetics in A10 which favoured a mono-layer coating of larger aggregates of IONPs than in A9, where AA concentration was much lower and the kinetics slower.53 This hypothesis is substantiated by the results obtained from hydrodynamic size analysis, VSM measurements and TEM where no distinct polymer layer could be imaged with and without staining agent in Fig. S1 and S2,† most likely due to the thinness of the shell. The magnetization data presented in Fig. 3f confirm this trend with recorded values of 50, 46 and 43 emu g−1 for A2, A10 and A9 respectively. It is worth noting that the slight decrease in magnetic saturation compared to bare IONPs (54 emu g−1) is usually attributed to PAA acting as a magnetically dead layer.51 These values underscore the great potential for PAA-IONPs to be magnetically extracted from solutions and used for diagnostic purposes.
3.5. Determining the optimal conditions for DNA isolation with PAA-IONPs
A comprehensive analysis of factors contributing to the effectiveness of the beads-based purification system was conducted, considering the type and concentration of beads and the conditions of the PEG/NaCl binding buffer. Based on previous studies demonstrating its superior performance, PEG 8000 was chosen as the binding buffer. Different concentrations of PEG-8000 and NaCl were tested with three PAA-IONPs types, including bare IONPs as control, as shown in Fig. 4. These results indicate that a combination of 17% PEG and 2.5 M NaCl provided ease of handling due to the reduced viscosity of PEG without compromising the DNA recovery. Additionally, we tested the relationship between bead concentration (ranging from 6 mg mL−1 to 1 mg mL−1) and recovery efficiency and found that 3 mg mL−1 was an optimal working concentration. Through a side-by-side comparison of A2, A9, and A10, A10 was shown to perform better. As expected, control IONPs did not precipitate DNA on the beads. Consequently, we recommend utilizing the PAA-IONPs DNA purification method, employing 3 mg mL−1 PAA-IONPs and a binding buffer composed of 17% PEG and 2.5 M NaCl, as an efficient and effective approach for DNA extraction. | ||
| Fig. 4 The determining factors of the PAA-IONPs-based DNA recovery. (a) The percentage DNA recovery with different particle and PEG/NaCl concentrations for control IONPs at 2× beads to DNA sample volume. (b) The percentage DNA recovery with different particle and PEG/NaCl concentrations for A2 at 2× beads to DNA sample volume. (c) The percentage DNA recovery with different particle and PEG/NaCl concentrations for A9 at 2× beads to DNA sample volume. (d) The percentage DNA recovery with different particle and PEG/NaCl concentrations for A10 at 2× beads to DNA sample volume. | ||
3.6. Performance and size-selection comparison
To thoroughly evaluate the suitability of PAA beads as a standard for size selection in DNA analysis, a meticulous comparison was conducted against the widely accepted AMPure XP beads, which served as a control. Additionally, bare IONPs were included as a negative control. With a specific focus on the input range spanning from 100 bp to 1500 bp, the comprehensive analysis produced insightful results. Both the AMPure XP beads and the PAA-IONPs, designated as A2, A9, and A10, respectively, exhibited remarkable efficacy in recovering DNA fragments larger than 100 bp. Nevertheless, some differences were observed. For example, we found that A2 had relatively lower magnetic strength as compared to A9 and A10. This difference is hypothesized to stem from the smaller size of A2 in the viscous solution, which impeded their magnetic behaviour. A2 still exhibited enough carboxyl groups to bind to DNA coils in the presence of PEG, and Na+ ions which shield the negative phosphate charges.54 Importantly, upon closer examination of the data presented in Fig. 5, a slightly superior cut-off performance was displayed by A9 compared to A10. On the other hand, A10 had a better recovery than A9. This observation underscores the exceptional capabilities of the PAA-IONPs in extracting and precisely selecting DNA fragments for a broad size range. The recovery rates achieved by PAA-IONPs were found to be highly comparable to those obtained with the AMPure XP beads, affirming their excellent performance (ESI Table 2†). These findings solidify the position of PAA-IONPs as a highly efficient and reliable tool for size selection in DNA analysis, offering researchers a valuable alternative to achieve accurate and consistent results in their experiments at a fraction of the cost.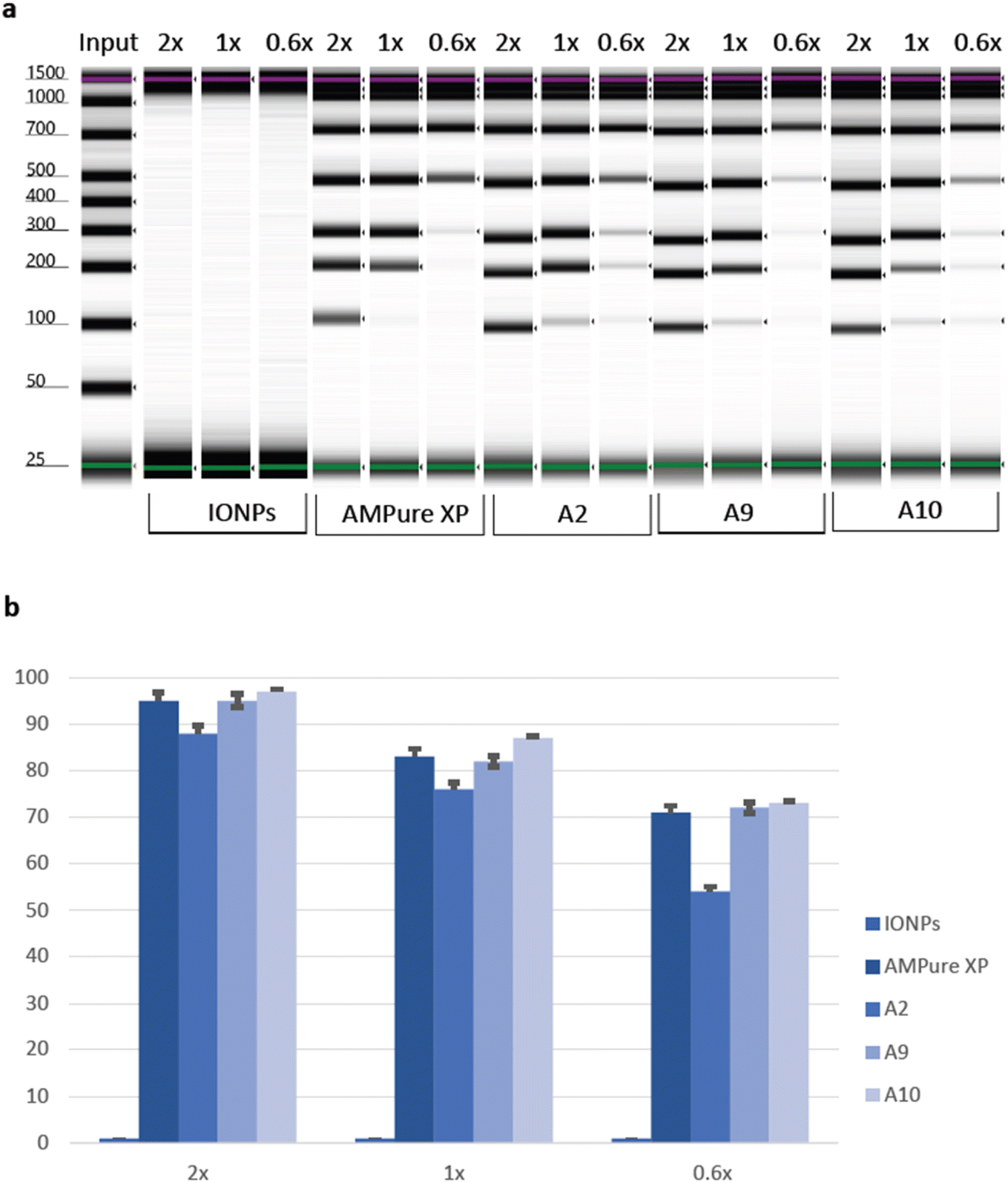 | ||
| Fig. 5 Performance evaluation of the PAA beads against bare IONPs and AMPure XP controls. (a) The TapeStation visualization of DNA extracted using the bare IONPs and AMPure XP controls, A2, A9, and A10 at different beads-to-sample ratios. (b) The average percentage of DNA recovery of the bare IONPs and AMPure XP controls, A2, A9, and A10 at different beads-to-sample ratios. Each sample had three replicates. | ||
4. Conclusion
This study highlighted the role of the pH in the size and carboxyl density fine-tuning of PAA-IONPs. The use of high initiator concentration has shown to produce a mono-layer coverage of PAA, regardless of the amount of monomer, thus preserving the magnetic strength of IONPs and enhancing their stability. This strategy can be easily used to functionalize other kinds of nanomaterial with carboxyl groups, for subsequent binding to proteins or nucleic acid sequences for example. In this work, a DNA extraction and size-selection protocol was tailored by capitalizing on the relationship between polyethylene glycol (PEG) and NaCl concentrations and the precipitation size of DNA molecules. These NPs demonstrated exceptional performance in DNA isolation and size selection, offering high resolution and recovery rates even at low DNA concentrations. Compared to existing solutions, PAA-IONPs present a superior alternative for efficiently fractionating DNA with high capacity. The synthesis process is cost-effective and scalable, making it highly advantageous for large-scale applications. By utilizing these NPs, researchers and clinicians can achieve optimal size selection of nucleic acid fragments for next-generation sequencing (NGS) without the financial burden associated with traditional magnetic bead-based kits. Moreover, this method simplifies the workflow, saving valuable time and resources.Author contributions
A. M., M. B. conceived the study. N. B. and A. M. designed the experiments and developed the method with critical inputs from M. B. and S. B. N.B. carried out all the experiments for beads synthesis with assistance from S. J. B. A. M. carried out all the experiments for DNA extraction and size-selection. All authors discussed the data and provided critical inputs. A. M. and N. B. prepared the manuscript with guidance from M. B. and S. B. All authors read and commented on the manuscript.Conflicts of interest
The authors declare no competing interests for the current study.Acknowledgements
We thank the members of the Bjørås and Bandyopadhyay labs for their support and critical suggestions throughout this work. We also thank Solon Oikonomopoulos for TGA measurements. The Research Council of Norway is acknowledged for the support to the Norwegian Micro- and Nano-Fabrication Facility, NorFab, project number 295864. Access to TEM was provided by the Cellular and Molecular Imaging Core Facility (CMIC), Norwegian University of Science and Technology (NTNU). CMIC is funded by the Faculty of Medicine at NTNU and Central Norway Regional Health Authority.Notes and references
- N. Rohland and D. Reich, Genome Res., 2012, 22, 939–946 CrossRef CAS PubMed.
- S. Behjati and P. S. Tarpey, Arch. Dis. Child., 2013, 98, 236–238 CrossRef PubMed.
- I. F. Bronner, M. A. Quail, D. J. Turner and H. Swerdlow, Curr. Protoc. Hum. Genet., 2013, 79, 18 Search PubMed.
- A. Stortchevoi, N. Kamelamela and S. S. Levine, J. Biomol. Tech., 2020, 31, 7 CrossRef PubMed.
- N. L. Van Berkum, E. Lieberman-Aiden, L. Williams, M. Imakaev, A. Gnirke, L. A. Mirny, J. Dekker and E. S. Lander, J. Visualized Exp., 2010, e1869 Search PubMed.
- J. Sambrook and D. W. Russell, Cold Spring Harb. Protoc., 2006, 2006, pdb.prot4455 CrossRef PubMed.
- M. R. Green and J. Sambrook, Cold Spring Harb. Protoc., 2016, 2016, pdb.prot093377 CrossRef PubMed.
- E. Amstad, M. Textor and E. Reimhult, Nanoscale, 2011, 3, 2819–2843 RSC.
- B. H. McDonagh, C. Staudinger, P. S. Normile, J. A. De Toro, S. Bandyopadhyay, W. R. Glomm and G. Singh, Appl. Mater. Today, 2021, 24, 101084 CrossRef.
- A. Kusior, K. Michalec, P. Jelen and M. Radecka, Appl. Surf. Sci., 2019, 476, 342–352 CrossRef CAS.
- F. N. Sayed and V. Polshettiwar, Sci. Rep., 2015, 5, 9733 CrossRef CAS PubMed.
- A. L. V. Zumaya, D. Martynek, T. Bautkinová, M. Šoóš, P. Ulbrich, J.-M. Raquez, M. Dendisová, J. Merna, J. Vilčáková, D. Kopecký and F. Hassouna, Eur. Polym. J., 2020, 133, 109795 CrossRef CAS.
- E. G. Fuller, H. Sun, R. D. Dhavalikar, M. Unni, G. M. Scheutz, B. S. Sumerlin and C. Rinaldi, ACS Appl. Polym. Mater., 2019, 1, 211–220 CrossRef CAS.
- G. Cotin, C. Blanco-Andujar, F. Perton, L. Asín, J. M. de la Fuente, W. Reichardt, D. Schaffner, D.-V. Ngyen, D. Mertz, C. Kiefer, F. Meyer, S. Spassov, O. Ersen, M. Chatzidakis, G. A. Botton, C. Hénoumont, S. Laurent, J.-M. Greneche, F. J. Teran, D. Ortega, D. Felder-Flesch and S. Begin-Colin, Nanoscale, 2021, 13, 14552–14571 RSC.
- K. Li, P. Y. Dugas and E. Bourgeat-Lami, Macromolecules, 2016, 49, 7609–7624 CrossRef CAS.
- N. Ahmed, M. Michelin-Jamois, H. Fessi and A. Elaissari, Soft Matter, 2012, 8, 2554–2564 RSC.
- J. Gao and A. Penlidis, J. Macromol. Sci. Polym. Rev., 1996, 36, 199–404 CrossRef.
- R. A. Scott and N. A. Peppas, AIChE J., 1997, 43, 135–144 CrossRef CAS.
- S. P. Manickam, K. Venkatarao and N. R. Subbaratnam, Eur. Polym. J., 1979, 15, 483–487 CrossRef CAS.
- T. Klement, N. Kockmann, C. Schwede and T. Röder, Ind. Eng. Chem. Res., 2021, 60, 4240–4250 CrossRef CAS.
- Y.-P. Wang, P. Zhou, S.-Z. Luo, X.-P. Liao, B. Wang, Q. Shao, X. Guo and Z. Guo, Langmuir, 2018, 34, 7859–7868 CrossRef CAS PubMed.
- B. Farkaš, U. Terranova and N. H. de Leeuw, J. Mater. Chem. B, 2021, 9, 4915–4928 RSC.
- A. Sharma, J. W. Foppen, A. Banerjee, S. Sawssen, N. Bachhar, D. Peddis and S. Bandyopadhyay, Nanoscale Res. Lett., 2021, 16, 24 CrossRef CAS PubMed.
- Z. Ali, J.-P. Andreassen and S. Bandyopadhyay, Ind. Eng. Chem. Res., 2023, 62, 4831–4839 CrossRef CAS.
- S. Yu and G. M. Chow, J. Mater. Chem., 2004, 14, 2781–2786 RSC.
- A. Hennig, A. Hoffmann, H. Borcherding, T. Thiele, U. Schedler and U. Resch-Genger, Anal. Chem., 2011, 83, 4970–4974 CrossRef CAS PubMed.
- M. Li, F. Cheng, C. Xue, H. Wang, C. Chen, Q. Du, D. Ge and B. Sun, Langmuir, 2019, 35, 14688–14695 CrossRef CAS PubMed.
- M. Puddu, D. Paunescu, W. J. Stark and R. N. Grass, ACS Nano, 2014, 8, 2677–2685 CrossRef CAS PubMed.
- N. Mahmed, O. Heczko, A. Lancok and S. P. Hannula, J. Magn. Magn. Mater., 2014, 353, 15–22 CrossRef CAS.
- T. Köhler, A. Feoktystov, O. Petracic, E. Kentzinger, T. Bhatnagar-Schöffmann, M. Feygenson, N. Nandakumaran, J. Landers, H. Wende, A. Cervellino, U. Rücker, A. Kovács, R. E. Dunin-Borkowski and T. Brückel, Nanoscale, 2021, 13, 6965–6976 RSC.
- S. Khashan, S. Dagher, N. Tit, A. Alazzam and I. Obaidat, Surf. Coat. Technol., 2017, 322, 92–98 CrossRef CAS.
- M. Baalousha, Sci. Total Environ., 2009, 407, 2093–2101 CrossRef CAS PubMed.
- J. J. Carlson and S. K. Kawatra, Miner. Process. Extr. Metall. Rev., 2013, 34, 269–303 CrossRef CAS.
- K. Hayashi, H. Tomonaga, T. Matsuyama and J. Ida, J. Appl. Polym. Sci., 2022, 139, e51581 CrossRef.
- L. Chekli, S. Phuntsho, M. Roy, E. Lombi, E. Donner and H. K. Shon, Water Res., 2013, 47, 4585–4599 CrossRef CAS PubMed.
- J. Lim, S. P. Yeap, H. X. Che and S. C. Low, Nanoscale Res. Lett., 2013, 8, 381 CrossRef PubMed.
- G. C. Lavorato, A. A. de Almeida, C. Vericat and M. H. Fonticelli, Nanotechnology, 2023, 34, 192001 CrossRef PubMed.
- A. Hennig, H. Borcherding, C. Jaeger, S. Hatami, C. Würth, A. Hoffmann, K. Hoffmann, T. Thiele, U. Schedler and U. Resch-Genger, J. Am. Chem. Soc., 2012, 134, 8268–8276 CrossRef CAS PubMed.
- S. Ahualli, G. R. Iglesias, W. Wachter, M. Dulle, D. Minami and O. Glatter, Langmuir, 2011, 27, 9182–9192 CrossRef CAS PubMed.
- S. Kumar and V. K. Aswal, J. Phys.: Condens. Matter, 2011, 23, 035101 CrossRef PubMed.
- H. Singh and V. K. Aswal, J. Appl. Phys., 2021, 129, 234703 CrossRef CAS.
- J. Liu, J. Zhao, S. M. Louie, X. Gao, P. Zhang, D. Liang and Y. Hu, Chemosphere, 2023, 319, 137992 CrossRef CAS PubMed.
- A. Hajdú, M. Szekeres, I. Y. Tóth, R. A. Bauer, J. Mihály, I. Zupkó and E. Tombácz, Colloids Surf., B, 2012, 94, 242–249 CrossRef PubMed.
- L. M. Sanchez, D. A. Martin, V. A. Alvarez and J. S. Gonzalez, Colloids Surf., A, 2018, 543, 28–37 CrossRef CAS.
- Y. Xu, L. Zhuang, H. Lin, H. Shen and J. W. Li, Thin Solid Films, 2013, 544, 368–373 CrossRef CAS.
- M. K. Singh, A. Agarwal, R. Gopal, R. K. Swarnkar and R. K. Kotnala, J. Mater. Chem., 2011, 21, 11074–11079 RSC.
- S. Khanlari and M. A. Dubé, J. Macromol. Sci., Part A: Pure Appl.Chem., 2015, 52, 587–592 CrossRef CAS.
- L. J. Kirwan, P. D. Fawell and W. van Bronswijk, Langmuir, 2003, 19, 5802–5807 CrossRef CAS.
- C.-L. Lin, C.-F. Lee and W.-Y. Chiu, J. Colloid Interface Sci., 2005, 291, 411–420 CrossRef CAS PubMed.
- N. Jain, Y. Wang, S. K. Jones, B. S. Hawkett and G. G. Warr, Langmuir, 2010, 26, 4465–4472 CrossRef CAS PubMed.
- E. T. Tenório-Neto, T. Jamshaid, M. Eissa, M. H. Kunita, N. Zine, G. Agusti, H. Fessi, A. E. El-Salhi and A. Elaissari, Polym. Adv. Technol., 2015, 26, 1199–1208 CrossRef.
- E. T. Tenorio-Neto, A. Baraket, D. Kabbaj, N. Zine, A. Errachid, H. Fessi, M. H. Kunita and A. Elaissari, Mater. Sci. Eng., C, 2016, 61, 688–694 CrossRef CAS PubMed.
- Q. Yue, M. Wang, Z. Sun, C. Wang, C. Wang, Y. Deng and D. Zhao, J. Mater. Chem. B, 2013, 1, 6085–6093 RSC.
- X. Wang, L. Zhao, X. Wu, H. Luo, D. Wu, M. Zhang, J. Zhang, M. Pakvasa, W. Wagstaff, F. He, Y. Mao, Y. Zhang, C. Niu, M. Wu, X. Zhao, H. Wang, L. Huang, D. Shi, Q. Liu, N. Ni, K. Fu, K. Hynes, J. Strelzow, M. El Dafrawy, T.-C. He, H. Qi and Z. Zeng, Genes Dis., 2021, 8, 298–306 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04687g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |