
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGap-enhanced gold nanodumbbells with single-particle surface-enhanced Raman scattering sensitivity†
Ran Cheng a,
Danchen Jiab,
Zhiyi Dua,
Ji-Xin Chengbc and
Chen Yang*ab
a,
Danchen Jiab,
Zhiyi Dua,
Ji-Xin Chengbc and
Chen Yang*ab
aDepartment of Chemistry, Boston University, Boston, MA 02215, USA. E-mail: cheyang@bu.edu
bDepartment of Electrical & Computer Engineering, Boston University, Boston, MA 02215, USA
cDepartment of Biomedical Engineering, Boston University, Boston, MA 02215, USA
First published on 13th September 2023
Abstract
Gap-enhanced Raman tags (GERTs) have been widely used for surface-enhanced Raman scattering (SERS) imaging due to their excellent SERS performances. Here, we reported a synthetic strategy for novel gap-enhanced dumbbell-like nanoparticles with anisotropic shell coatings. Controlled shell growth at the tips of gold nanorods was achieved by using cetyltrimethylammonium bromide (CTAB) as a capping agent. A mechanism related to the shape-directing effects of CTAB was proposed to explain the findings. Optimized gap-enhanced gold dumbbells exhibited highly enhanced SERS responses compared to rod cores, with an enhancement ratio of 101.5. We further demonstrated that gap-enhanced AuNDs exhibited single-particle SERS sensitivity with an acquisition time as fast as 0.1 s per spectrum, showing great potential for high-speed SERS imaging.
Introduction
Surface-enhanced Raman scattering (SERS) spectroscopy is a powerful tool that gains sensitivity from plasmonic enhancement. It also offers advantages inherited from Raman spectroscopy, including direct readouts of enriched molecular fingerprint information, superior multiplexity due to narrow Raman bandwidths, high photostability, and low background from biological tissues.1 Therefore, SERS imaging has been extensively used for bioimaging to visualize biocomponents spanning from biomolecules to entire organisms,2 enabling broad biomedical applications with the capability of observing subcellular or cellular structures, mapping intercellular connections, probing tissue complexities, and in vivo imaging of organs or detection of tumors.3,4SERS probes are the labeling agents that empower SERS bioimaging.3–6 Typically, a SERS probe consists of a plasmonic nanostructure to enhance the Raman signals from Raman indicators coated on or embedded in the plasmonic structure. Designing strong plasmonic enhancers with “hot spots” where the local electromagnetic field is highly amplified is a key for developing bright SERS probes. Hot spots could be created in multiple ways.7 Nanoparticles with sharp tips or corners, such as nanorods, nanocubes, or nanostars, present intraparticle hot spots located at the protruding ends.8–10 However, such a local “hot spot” area only counts for a small portion of the overall surface of the probe. Another strategy for generating hot spots is to introduce nanogaps. Electromagnetic amplification is usually more effective within an ultrasmall gap than that from rough surfaces or sharp tips.7,11 Nanogaps could be introduced in an inter-particle way through aggregations of plasmonic nanoparticles. SERS responses of the aggregates are strong but typically not uniform, due to randomized hot spot formation caused by limited controllability over the aggregation process.12,13 SERS uniformity could be improved but it requires either well-controlled step-by-step synthesis14,15 or tedious purifications,13,16,17 bringing difficulties for efficient fabrication of uniform nanoprobes (such as dimers, trimers or beyond) with high yields.
Gap-enhanced nanoparticles provide a new design for SERS probes by creating intraparticle gaps between the core and the shell and embedding indicator molecules inside these nanogaps.18 The internal gaps could be finely controlled by choosing desired supporting molecules or materials, such as DNA strands, polymers, or small molecules, leading to bright and uniform SERS performances. Specifically, some gap-enhanced Raman tags (GERTs) reported have an internal gap supported by a monolayer of thiolated aromatic molecules with a size around or under 1 nm. The thiolated molecules act as both the supporting layer and Raman indicator molecules at the same time. These GERTs demonstrate superior SERS performances with an outstanding enhancement factor up to 1011 as reported,19 opening up the broad opportunities for bioanalysis, such as immunoassay development,20,21 multiplex bio-detection,20,22–24 in vitro cellular imaging,19,23,24 in vivo tumor diagnosis25–27 and lymph node imaging.23,28 Till now, GERTs with various core designs, such as spheres, rods, triangles, stars, cubes, covered by smooth or rough external shells with one or multiple in-built gaps have been developed.18 However, among these only a few structures have been reported to reach single-particle SERS sensitivity.19,23,29,30 For example, bilayered Raman-intense gold nanostructures with hidden tags (BRIGHTs)19 and petal-like GERTs fabricated from spherical cores23,29,30 have demonstrated excellent SERS responses at the single-particle level, enabling high-speed or high-resolution bioimaging. To further empower SERS imaging with superior sensitivity for high-speed detection, further developing GERTs with novel morphologies that exhibit single-particle brightness are still strongly needed.
Gold nanorod (AuNR)-core GERTs are bright SERS probes with improved SERS performances of 1 to 2 orders stronger than AuNRs21,31 and at least a few times stronger than regular GERTs with spherical cores.21,32 It has been showed that just a few single AuNR-core GERTs presented in the test zone of lateral flow immunoassay (LFIA) strips could enable sensitive detection with an average intensity of 40 counts with 0.5 s acquisition time,21 showing the potential of reaching single-particle SERS sensitivity by AuNR-core GERTs.
AuNR-core GERT reported so far contains an isotropic gold shell that fully encapsulates a AuNR core.21,31 Notably, a AuNR presents inhomogeneous electromagnetic field distribution due to its anisotropy, with hot spots located at the AuNR tips contributing to at least 65% of the overall enhancement from the AuNR.33 Therefore, we hypothesize that AuNR-core GERTs with anisotropic coatings only at the tips, i.e. a dumbbell nanostructure, could demonstrate the SERS performances at the same level as the isotropic ones and have a potential reaching single-particle SERS sensitivity.
To test this hypothesis, in this work we developed a new synthetic method for gap-enhanced gold nanodumbbells (AuNDs). The structure is composed of a AuNR core, gold shells specifically coated on two tips of the AuNR, and internal nanogaps embedded between AuNR tips and shells that are supported by a monolayer of thiolated aromatic indicator molecules, 4-nitrobenzenethiol (NBT) (Fig. 1a). NBT molecules were chosen due to their largest Raman cross sections compared to other popularly used thiolated aromatic indicator molecules.34 Notably, the gap-enhanced AuND is a new type of gap-enhanced nanostructure with anisotropic morphology due to tip-selective Au shells deposited on the AuNR tips while with the AuNR side wall exposed. It is different from previously reported GERTs with isotropic morphologies, where AuNR cores were fully covered by nearly spherical shells.21,31 It was found that the key to drive the anisotropic shell growth on the ends of the AuNR cores was shape-directing effects of cetyltrimethylammonium bromide (CTAB) surfactants introduced during synthesis. We have systematically examined the morphologies of grown shells at different reaction stages in CTAB. We also compared the reaction to the one using cetyltrimethylammonium chloride (CTAC), a capping agent regularly used for synthesis of AuNR-core GERTs with isotropic shells. Based on our results, we suggested a potential mechanism related to the strong facet-dependent absorption of CTAB molecules on the surfaces of AuNRs. Gap-enhanced AuNDs presented substantially improved SERS performances compared to AuNRs. An enhancement ratio, defined by SERS intensities of gap-enhanced AuNDs over the SERS intensities of AuNRs, of 101.5 has been achieved by optimizing the shell thicknesses, comparable to the previously reported enhancement ratio of 102 for fully encapsulated AuNR-core GERTs.21 We further demonstrated single-particle SERS response from single gap-enhanced AuND with a detection speed as fast as 0.1 s per pixel through SERS followed by scanning electron microscopy (SEM) study of the same area. Our work on developing gap-enhanced AuNDs may benefit high-speed SERS bioimaging and provide new possibilities for designing gap-enhanced nanoparticles with different geometries and morphologies.
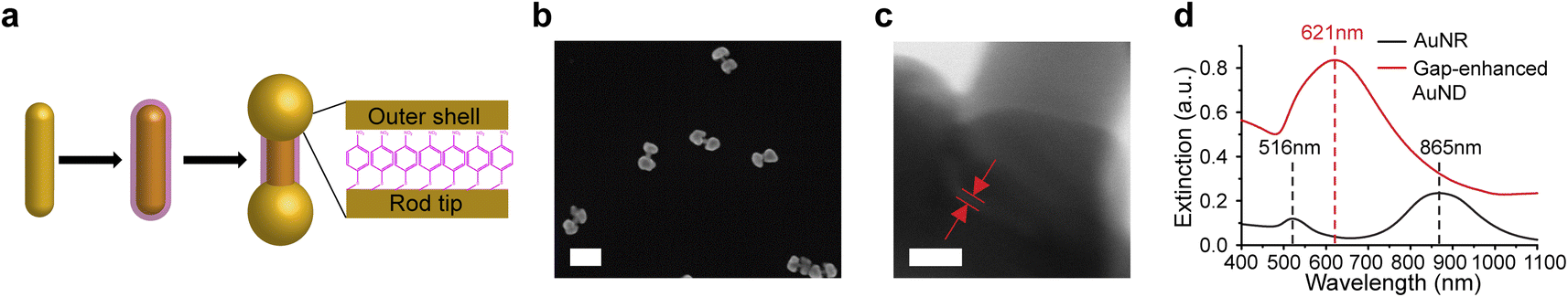 | ||
| Fig. 1 Synthesis of gap-enhanced AuNDs. (a) Schematic showing three main steps of gap-enhanced AuND synthesis. The monolayered NBT molecules are represented by the pink layer. (b) Representative SEM image of gap-enhanced AuNDs. Scale bar: 200 nm. (c) TEM image zooming in on one end of a gap-enhanced AuND. The embedded nanogap is labeled by the red lines between the two red arrows. Scale bar: 10 nm. (d) Extinction spectra of AuNRs and gap-enhanced AuNDs. Gap-enhanced AuNDs were synthesized from the same concentration of the AuNRs. | ||
Results and discussion
Synthesis and characterization of gap-enhanced AuNDs
As shown in Fig. 1a, three main steps were used to synthesize AuNDs, including synthesis of AuNR cores, functionalization of AuNRs by thiolated indicator molecules NBT, and preferential growth of gold shells on the ends of AuNRs. Firstly, AuNRs were synthesized using a protocol reported previously and described briefly in the Experimental section.35 As-synthesized AuNRs showed high morphological uniformity (Fig. S1a†). Through a SEM analysis of over 100 AuNRs, the average size of AuNRs was 103 ± 8 nm in length and 24 ± 3 nm in diameter with an aspect ratio around 4.3. Secondly, AuNRs were functionalized by NBT molecules in CTAC solution by one-hour vigorous sonication followed by overnight incubation. After functionalization, a layer of NBT molecules was covalently bound to the AuNR surface via Au–S bond, serving as the embedded Raman tags as well as the supporting layer to define the gap between the core and the shells in the following synthetic step. NBT molecules absorb on the AuNR surface in a monolayered manner through the interaction of the thiol group in NBT with the gold surface, since the nitro group presents low affinity towards the gold surface and surface-absorbed NBTs lack the ability to form intermolecular cross-linkage with another NBT molecules. It is noticed that the AuNR longitudinal resonance shifted from 865 nm to 868 nm and the transverse mode shifted from 516 nm to 521 nm after binding of NBT molecules, due to the change of refractive index between surfactants and NBT molecules absorbed on the surface (Fig. S1b†). Finally, outer shells were formed through the reduction of gold precursor, Au3+ ions, by ascorbic acid in CTAB solution. Au shells preferably grew at both ends of NBT-functionalized AuNRs in the presence of CTAB molecules and resulted in dumbbell shapes. To clearly visualize the morphologies of AuNDs under SEM, we picked the areas where the AuNDs were well separated (Fig. 1b). In addition, statistical analysis was performed over 100 nanoparticles in 10 SEM images, and a yield over 77% was estimated for AuND synthesis. TEM imaging was performed to identify the nanogap embedded between the AuNR core and outer shells, supported by a monolayer of NBT molecules. Specifically, TEM was performed at high magnification at 4 MX. Fig. 1c shows a representative TEM image zooming in on one end of AuND. The shell of the AuND was presented by the darker contrast observed at the left bottom corner of the TEM image and the uncovered AuNR body of the AuND was at the upper right corner. Moreover, it could also be observed that the outer shell and the AuNR core were separated by a nanoscale gap with a gap width of 0.9 nm, indicated by red lines between two red arrows (Fig. 1c). The gaps in the TEM images presented a gap size ranging from 0.7 to 1.2 nm based on 10 gaps from 9 AuNDs examined, with a size distribution centered around 1.0 nm (Fig. S2†), consistent with the reported thickness of a monolayer of NBT molecules as molecular spacers.21,29,36Extinction spectra were measured from AuNRs and gap-enhanced AuNDs. AuNRs present a transverse resonance of 516 nm and a longitudinal resonance of 865 nm, corresponding to an aspect ratio slightly above 4.37 Interestingly, gap-enhanced AuNDs only showed one single resonance peak at 621 nm in the detection range of 400 to 1100 nm (Fig. 1d). Previously, nanodumbbells without an embedded gap have been reported to have two resonance peaks in the same detection range of the original AuNR cores, typically from 400 to 1100 nm.38–43 These two peaks were the transversal resonance peak slightly red-shifted and the longitudinal resonance peak blue-shifted compared to AuNR cores, due to the increased diameters and decreased aspect ratio, the length over the diameter, of the nanoparticles. However, for our gap-enhanced AuNDs, only one resonance peak was observed in the same spectrum range, different from two peaks of nanodumbbells without the internal gap, suggesting that the existence of the gap played an important role in tuning the optical response.
Growth mechanism of gap-enhanced AuNDs
Unlike previously reported isotropic shell growth in the CTAC system,21,31 our shell growth in the presence of CTAB showed an anisotropic preference at the ends, indicating the key role of the capping agents in controlling the shell growth in the reaction. To understand the role of these capping agents better, shell growth processes in both CTAB (Fig. 2a–f) and CTAC solution (Fig. 2g–l) were monitored more systematically. For better comparison, all reaction conditions were kept the same except for the surfactants (CTAB or CTAC) that existed in the system. To understand differences at different reaction stages, the reactions were paused by dilution and immediately followed by centrifugation at specific reaction stages. The nanostructure morphologies were then examined by SEM and corresponding extinction spectra were taken at each stage.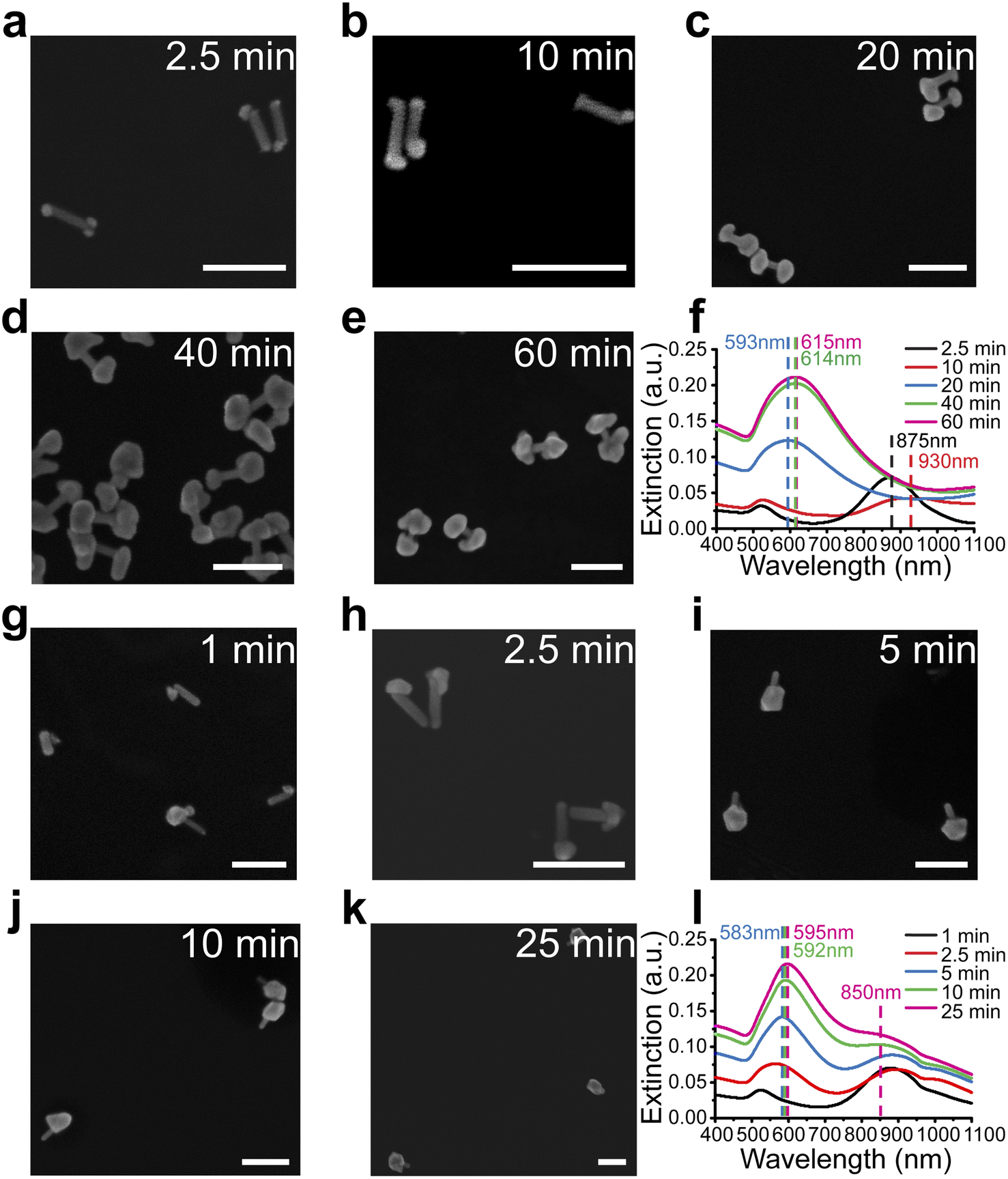 | ||
| Fig. 2 Different shell growth mechanisms in the presence of CTAB or CTAC as capping agents. (a–e) Shell growth of gap-enhanced AuNDs in the CTAB system at different reaction stages. SEM images showing the shell morphologies at 2.5 min (a), 10 min (b), 20 min (c), 40 min (d), and 60 min (e) after the initiation of the reaction. Scale bars: 200 nm. (f) Extinction spectra at corresponding reaction stages in (a–e). (g–k) Shell growth of gap-enhanced AuNLs in CTAC system at different reaction stages. SEM images showing the shell morphologies at 1 min (g), 2.5 min (h), 5 min (i), 10 min (j), and 25 min (k) after the initiation of the reaction. Scale bars: 200 nm. (l) Extinction spectra at corresponding reaction stages in (g–k). | ||
For the CTAB system, the shells formed at both tips of AuNR cores, leading to the formation of gap-enhanced AuNDs. At the initial stage at 2.5 min, Au precursors were reduced to form multiple “seeds” at the tips of the AuNRs (Fig. 2a). Then, reduction of Au3+ by ascorbic acid was catalyzed at the surface of pre-formed Au seeds44 to form patchy protrusions partially covering the tips (Fig. 2b). As the reaction continued, the protrusions grew into complete shells covering the tips (Fig. 2c). Shells continued growing thicker over time till Au ions were depleted (Fig. 2d and e). The whole reaction took about 60 min to complete. The extinction spectra of the reaction at the corresponding reaction stages are plotted in Fig. 2f. At 2.5 minutes (black curve, Fig. 2f), the longitudinal peak shifted from 868 nm to 875 nm due to the formation of the seeds. As the seed grew into irregular protrusions at 10 min, the longitudinal peak broadened and further redshifted (red curve, Fig. 2f). When the protrusions further grew into complete shells covering the AuNR tips at 20 min, the single resonance peak occurred at 593 nm (blue curve, Fig. 2f). As the shell thickness increased during the reaction, the extinction peak further redshifted to around 615 nm, and the extinction coefficient increased. Collectively, the spectrum results were consistent with what we observed in SEM images.
For the CTAC system, the trend of growing shells at both ends of AuNR no longer existed. In the initial stage, the “seed” was formed at one relatively random location on the surface of the AuNR, meaning at either one of the tips or occasionally on side walls (Fig. 2g). During the growth process, the seed continued to grow into a larger area (Fig. 2h). Some then grew into a complete shell covering one of the AuNR tips (Fig. 2i–k). Notably, we ended up with gold nanolollipops (AuNLs) as the final product for the CTAC group. The spectrum evolution of the reaction showed somewhat different features. AuNL sample showed a shoulder peak around 850 nm in addition to the peak around 590 nm (Fig. 2l). Such lollipop-shaped nanoparticles were previously reported when an insufficient amount of Au3+ precursors was added21 or as an intermediate product toward the synthesis of AuNR-core GERTs with isotropic shells.31 Therefore, AuNLs observed are expected to grow into AuNR-core GERTs with complete isotropic shell if sufficient Au3+ precursors were added.
We further investigated whether the formation of gap-enhanced AuNDs in the CTAB system was also due to insufficient Au3+ precursors. We used a fixed amount of AuNR cores containing 162 nmol gold in total. The amounts of Au3+ precursors in the shell growth step were 324 nmol, 648 nmol and 1296 nmol, respectively, corresponding to a molar ratio between the gold added for the shell growth and the gold in AuNR cores, nAu(shell)/nAu(NR) of 2, 4, 8, respectively. In all groups, we observed AuND growth on both tips of the AuNR cores (Fig. 3a–c). The successful synthesis of AuNDs with different amounts of precursors not only proved the high reproducibility of AuND synthesis, but also showed that solely increasing the amount of Au3+ precursors didn't change the tip-growing preference of the shells. With increased Au3+ precursors, the shells on the two ends grew thicker, leading to smaller distances between two shells and less exposed AuNR side walls in between. This observation was confirmed by an analysis of over 50 nanoparticles. When nAu(shell)/nAu(NR) = 2, the average diameter of the shells was around 65 nm with 43 nm AuNR side wall exposed (Fig. 3a). The average diameter increased to 78 nm when nAu(shell)/nAu(NR) = 4, leaving around 29 nm uncovered AuNR in between (Fig. 3b). Further increasing Au3+ precursors to reach nAu(shell)/nAu(NR) of 8 led to an even larger average shell diameter of 99 nm and the AuNR cores were almost fully covered by the shell for most of the nanoparticles (Fig. 3c). The thickened shells also led to an increased extinction coefficient and a redshift of the resonance peak of gap-enhanced AuNDs, which suggested that the plasmonic peak observed originated from the transverse resonance of AuNDs. As shown in Fig. 3d, the extinction peak shifted from 621 nm to 633 nm and 651 nm, respectively. We speculate that the longitudinal resonance of gap-enhanced AuNDs was in the far red, thus falling out of the current detection range (400–1100 nm) and leading to the single resonance observed in the extinction spectra. In conclusion, we showed that, unlike AuNLs, the formation of AuNDs was not due to insufficient Au3+ precursors but a direct outcome of the usage of CTAB surfactants during the reaction.
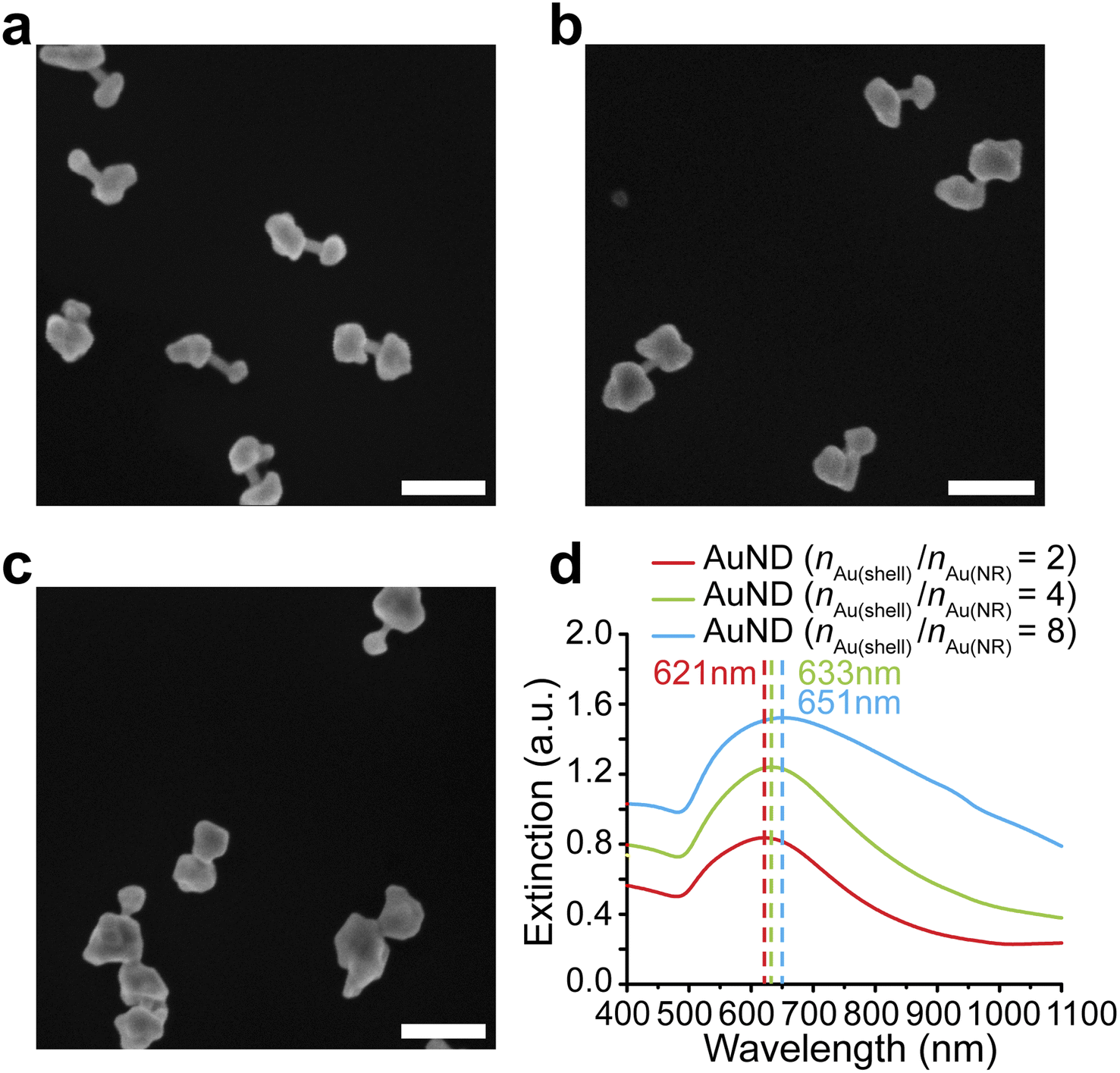 | ||
| Fig. 3 Synthesis of gap-enhanced AuNDs with increased amounts of Au3+ precursors. (a) SEM image of gap-enhanced AuNDs (nAu(shell)/nAu(NR) = 2). Scale bar: 200 nm. (b) SEM image of gap-enhanced AuNDs (nAu(shell)/nAu(NR) = 4). Scale bar: 200 nm. (c) SEM image of gap-enhanced AuNDs (nAu(shell)/nAu(NR) = 8). Scale bar: 200 nm. (d) Extinction spectra of gap-enhanced AuNDs from (a), (b), (c). The concentrations and amounts of AuNR cores added during shell synthesis were kept the same for each group. | ||
To explain our observations, we firstly considered the binding affinities of CTAB and CTAC on the gold surface. Previous studies suggested that different halide ions show different absorption affinities towards the gold surface.45 Due to a higher solvation energy of chloride (Cl−) than that of bromide (Br−), the Gibbs free energy for Br− absorption to the gold surface is lower, meaning Br− ions tend to bind to the gold surface more than Cl−.45 In specific context of AuNRs dispersed in CTA-halide system (CTAB/CTAC), simulation results have revealed that the surface occupation density of Br− anions on the AuNR surface was around 10 times higher than that of Cl− anions.46 Surface-absorbed Br− anions with high binding density functioned as the driving force to attract the co-absorption of CTA+ cations.46 As a result, the AuNR surface is tightly coated by CTAB surfactants in a bilayered manner due to the hydrophobicity of the CTA+ tail chain.47 In comparison, the low affinity of Cl− to AuNRs leads to a lower surface coverage of CTAC, compared to CTAB. Such conclusion has been experimentally confirmed by dark-field spectroscopy.46 A high surface coverage by a thick surfactant layer acts as a blockage that slows down the diffusion rate of reactive gold species to the AuNR surface. As a result, the gold reduction rate on the AuNR surface was slowed down and the reaction time elongated. In our case, we observed that it took 2.5 minutes or less to observe small seeds in CTAB system (Fig. 2a) while only 1 minute was enough to see even larger seeds in CTAC system (Fig. 2g), which is consistent with the thicker coverage of CTAB expected.
Different facet selectivity of CTAB and CTAC influence the surface coverage of the surfactants on AuNRs, dependent on the locations of the AuNRs. Br− ions show a higher packing density on {100} than on {111}.46 Since AuNR contains {111} at the tips and {100} at the side walls,48 it leads to higher Br− coverage on the side walls but less at the tips (Fig. S3a†). The uneven distribution of Br− will further lead to differentiated cation (CTA+) absorption and thus anisotropic distribution of CTAB with less coverage on the ends (Fig. S3b†). It has been experimentally observed that the coverage of CTAB bilayer on the ends is 30% less than that on the side walls of the AuNR body.49 The shell deposition is expected to occur preferably where the surfactant coverage was less, since these areas were relatively more accessible for gold ions, therefore resulting AuNDs. In comparison, the facet-selective absorption of CTAC was overall not as sensitive due to the low surface coverage of CTAC discussed above, meaning AuNR surface is nearly “unprotected” by CTAC and isotropically exposed (Fig. S3c and d†).
Secondly, the binding area-selectivity of surfactants controlled the seed deposition sites. When the surfactant layer presented anisotropic distribution with less CTAB presented at the rod ends, the seed formation occurred at the ends of the AuNRs in the CTAB system. For the CTAC system, seeds formed at relatively random locations due to weak absorption and low selectivity of CTAC surfactants on the surface. For example, in Fig. 2g, three seeds formed at one end of the AuNR while one formed near the AuNR body. Interestingly, we observed the formation of multiple seeds at both ends of the AuNR cores in the CTAB system (Fig. 2a), which might be due to the synergistic effect of multiple channels formed at the less covered ends for gold diffusion by overall densely coated CTAB46 and also a slow reaction rate led to sufficient time allowing for depositions at different sites located at both of the tips simultaneously (Fig. S4a–c†). However, for the CTAC system, only a single seed was typically observed for a AuNR core (Fig. S4d–f†), which we think was mainly related to the rapid seed deposition process controlled by the high reaction rate.
The mechanism we proposed above is a simplified model. There might be many factors synergistically controlling the shell growth process, such as surfactant packing density change on the AuNR surface after NBT functionalization, AuNR surface energy change induced by chemisorption of thiolated molecules,50 influences of the chemical structures and intrinsic properties (such as pKa) of thiolated indicator molecules used,29 etc. Future quantitative studies exploring molecular absorption (surfactants, indicator molecules with thiol groups, etc.) on Au facets are needed to better explain our observations and to systematically understand the shell growth mechanisms of gap-enhanced nanoparticles.
SERS performance of gap-enhanced AuNDs in colloids
We tested the SERS performances of gap-enhanced AuNDs and AuNR cores in colloids. The SERS signals from AuNR samples with the same concentration as the gap-enhanced AuNDs were very low and only the major peak at 1338 cm−1 could be slightly distinguished from the baseline (data not shown). To clearly resolve the SERS spectrum of NBTs absorbed on AuNR cores, we therefore measured SERS directly from the AuNR stock solution that was used for synthesis of gap-enhanced AuNDs. 0.25 mL of NBT-modified AuNR stocks were added to the reaction to reach a total volume of 1.94 mL for synthesis of AuNDs, resulting in that the concentration of AuNR solution measured was 7.76 times that of gap-enhanced AuNDs. SERS spectra after background removal were shown in Fig. 4a. Characteristic Raman bands of the indicator molecules, NBT, were presented at 1081, 1108, 1338, and 1570 cm−1, among which the mode of ν(NO2) at 1338 cm−1 was the strongest. Even though the concentration of the AuNR sample was higher, a higher SERS signal intensity was still observed for gap-enhanced AuNDs.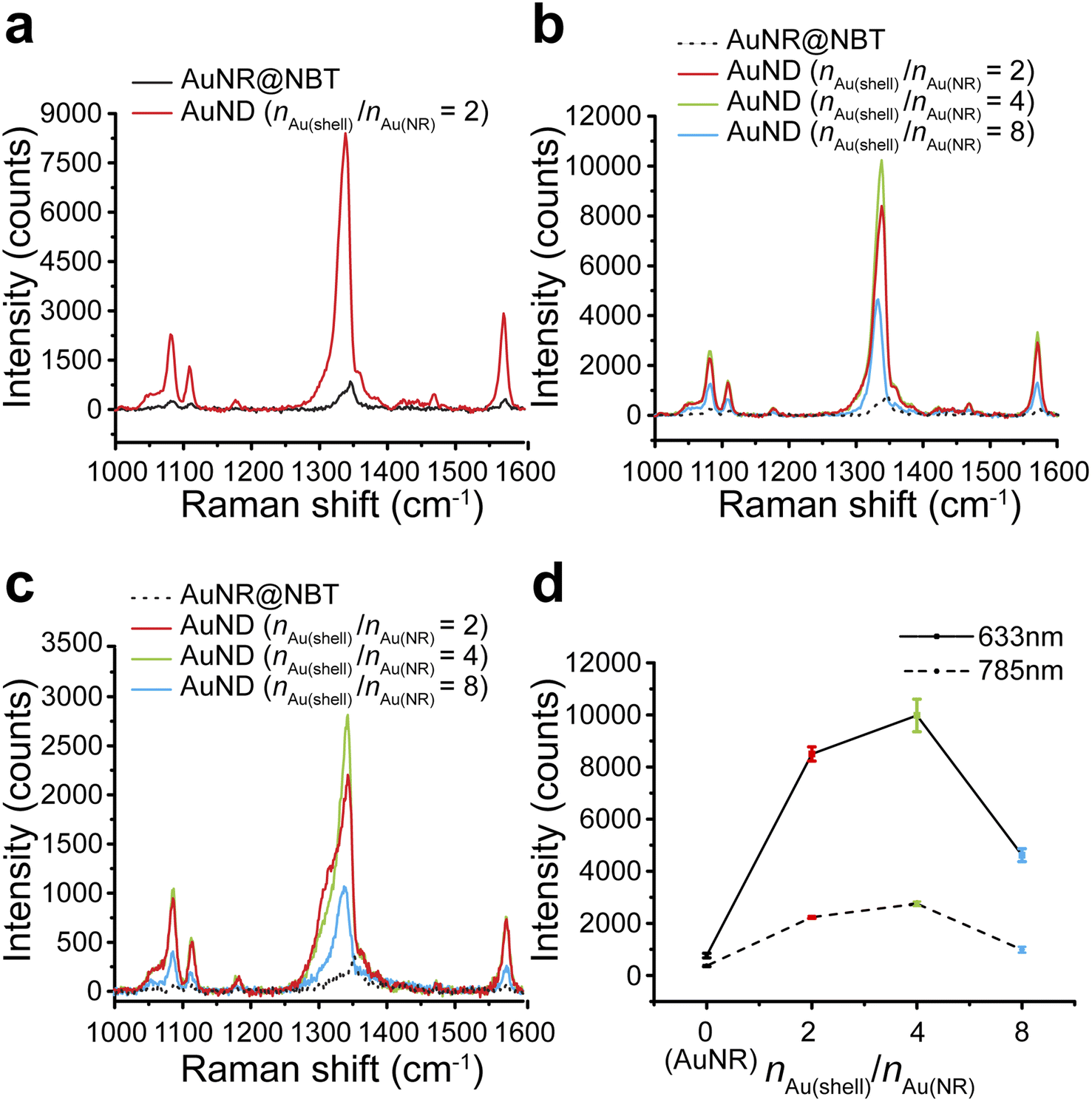 | ||
| Fig. 4 SERS performance of AuNDs in colloids. (a) SERS spectra of NBT-functionalized AuNRs and gap-enhanced AuNDs (nAu(shell)/nAu(NR) = 2) under excitation of 633 nm laser. The concentration of AuNR solution was 7.76 times that of gap-enhanced AuNDs. (b) SERS spectra of AuNRs and gap-enhanced AuNDs with different shell thicknesses (nAu(shell)/nAu(NR) = 2, 4, 8) under excitation of 633 nm laser. The concentrations of all gap-enhanced AuNDs were approximately the same. The concentration of AuNRs was 7.76 times those of gap-enhanced AuNDs. (c) SERS spectra of AuNRs and gap-enhanced AuNDs with different shell thicknesses (nAu(shell)/nAu(NR) = 2, 4, 8) under excitation of 785 nm laser. (d) SERS intensities of the strongest ν(NO2) mode in AuNR (i.e., nAu(shell)/nAu(NR) = 0) and gap-enhanced AuND colloids with different shell thicknesses (nAu(shell)/nAu(NR) = 2, 4, 8) under 633 nm and 785 nm laser excitation. The error bars represented the standard deviations of 3 measurements of the same sample at 633 nm and 2 measurements at 785 nm. The concentration of AuNR solution measured was 7.76 times that of gap-enhanced AuNDs. | ||
We further explored how Au shell thickness would affect the SERS performance of AuNDs using the samples from Fig. 3. SERS intensity firstly increased for the AuNDs prepared with nAu(shell)/nAu(NR) = 4, due to increased shell thicknesses. However, when nAu(shell)/nAu(NR) increased to 8, SERS intensity decreased. We attribute this to the possible shading of NBT as previously observed by Khlebtsov et al.21 meaning the internal field was not effectively excited due to the thick shells and therefore leading to a drop in SERS intensity.
Same trends were observed when gap-enhanced AuNDs were excited by both 633 nm laser (Fig. 4b) and 785 nm laser (Fig. 4c), respectively. The averaged signal intensities of the strongest ν(NO2) mode for each group were plotted in Fig. 4d. Under 633 nm excitation, an averaged SERS intensity out of 3 individual measurements of the same sample was 763 ± 89 counts for AuNR cores (nAu(shell)/nAu(NR) = 0). The SERS intensities reached 8450 ± 273, 9979 ± 625, and 4617 ± 247 counts, respectively, indicating enhanced performances by 86, 101.5, and 47 times for AuNDs with shell thickness of 65 nm, 78 nm, and 99 nm, compared to AuNRs. We further calculated the corresponding enhancement factors (EFs) based on experimental results. According to our calculations (Section S1†), under 633 nm laser excitation, EF was found to be 1.6 × 104 for AuNRs, which is on the same order as the previously reported experimental EF for NBT-functionalized AuNRs with similar size and aspect ratio.42 For AuNDs, the EFs ranged from 0.77 × 106 to 1.7 × 106, depending on the shell thicknesses of gap-enhanced AuNDs. When under 785 nm excitation, the SERS intensities for AuNRs (nAu(shell)/nAu(NR) = 0) and AuNDs (nAu(shell)/nAu(NR) = 2, 4, 8) with different shell thicknesses were 371 ± 30, 2235 ± 47, 2753 ± 87, and 988 ± 114 counts, separately as shown in Fig. 4d. The SERS performances were enhanced by 46, 58, and 21 times compared to AuNR cores. A higher enhancement ratio between gap-enhanced AuNDs and AuNRs was observed when excited by a 633 nm laser, due to a better match between AuND localized surface plasmonic resonances (LSPRs) and laser wavelength. Collectively, under both conditions, gap-enhanced AuNDs with different shell thicknesses all showed improved SERS performances compared to AuNRs, among which gap-enhanced AuNDs (nAu(shell)/nAu(NR) = 4) with 78 nm shell thickness (Fig. 3b) presented the highest SERS intensities. AuNR-core GERTs with isotropic shells have been previously reported to present about 20 times stronger SERS performance than AuNR cores by Wang et al.31 Later on, Khlebtsov et al. reported AuNR-core GERTs presenting an enhancement ratio of 102.34, compared to AuNR cores.21 Our gap-enhanced AuNDs show the enhancement ratio of 101.5, suggesting the essential enhancement could be enabled by the gapped shells at the tips. Note that nanodumbbells without the gap showed a much smaller enhancement ratio around 5.42 Collectively, the significantly improved SERS performances of gap-enhanced AuNDs over AuNR cores could be attributed to the creation of the nanometer gap and the coupling of the shells and AuNR cores. The coated shells also stabilized the AuND structures and retained their SERS performances by entrapping the NBT molecules inside the nanometer gaps. After 3 weeks of storage at room temperature, the colloidal gap-enhanced AuNDs (nAu(shell)/nAu(NR) = 2) still presented a peak intensity of 7237 counts at 1338 cm−1 (Fig. S5†). Compared to the freshly prepared gap-enhanced AuNDs with 8450 counts detected at 1338 cm−1, 86% of the original SERS performances were maintained after 3 weeks, showing the good stability of the gap-enhanced AuNDs to present prolonged SERS brightness.
Single-particle SERS of gap-enhanced AuNDs
Based on the highly enhanced SERS performances observed in colloids, we further explored the possibility of single-particle SERS detection of AuNDs using a confocal Raman microscope. Here, gap-enhanced AuNDs synthesized with nAu(shell)/nAu(NR) = 2 were used. Gap-enhanced AuNDs were cast on a piece of the pre-marked silicon substrate (Fig. S6a†). For co-localization of the same nanostructures in Raman imaging and SEM, we used specific features of the markers to identify the area of interest (Fig. S6a–c†). Raman spectroscopy and SEM imaging were then performed to examine the same sample area sequentially. Raman spectroscopy with pixel acquisition times of 0.1 s and 1 s, at a step size of 1 μm was firstly used to collect the SERS signals. SEM imaging was performed afterwards on the same area guided by the markers, to confirm the single particles and to correlate the SERS spectra with the morphologies of the individual nanostructures.SEM image and Raman mapping images at 1338 cm−1 (Fig. S6e–h†) recorded in two regions (Fig. S6d†) were selected for single-particle analysis. These two regions were specifically chosen due to the sparse nanoparticle distributions with inter-particle distances over 3 μm. First, we confirmed that no obvious background peak was observed from the bare substrate in the detected spectral window ranging from 1000 to 1600 cm−1 with detection speeds of 1 s or 0.1 s per point, respectively (Fig. 5a). When nanoparticles were present, SERS signals could be clearly observed. Fig. 5b–d presented the single-particle SERS responses from three individual AuNDs, shown by SEM images presented below. With 1 s acquisition time per spectrum, Raman peaks from NBT embedded could be clearly resolved at 1081, 1108, 1338, and 1570 cm−1, matching our previous colloidal results (Fig. 4b and c). More importantly, even with only 0.1 s acquisition time, the strongest peak of NBT molecules could still be clearly seen at 1338 cm−1. We observed consistent SERS intensities of around 30 counts from all three of the gap-enhanced AuNDs when 0.1 s was used for collection. With acquisition time increased to 1 s, signal intensities started to show some variations. For instance, the intensities ranged from 226, 65, and 135 counts for the major peak at 1338 cm−1 and 67, 38, and 47 counts for the peak at 1570 cm−1 respectively. We think morphological differences of the nanoparticles, laser intensity fluctuations and slight photodamage during 1 s irradiation for the second particle might be the main contributors to the variations we observed here.
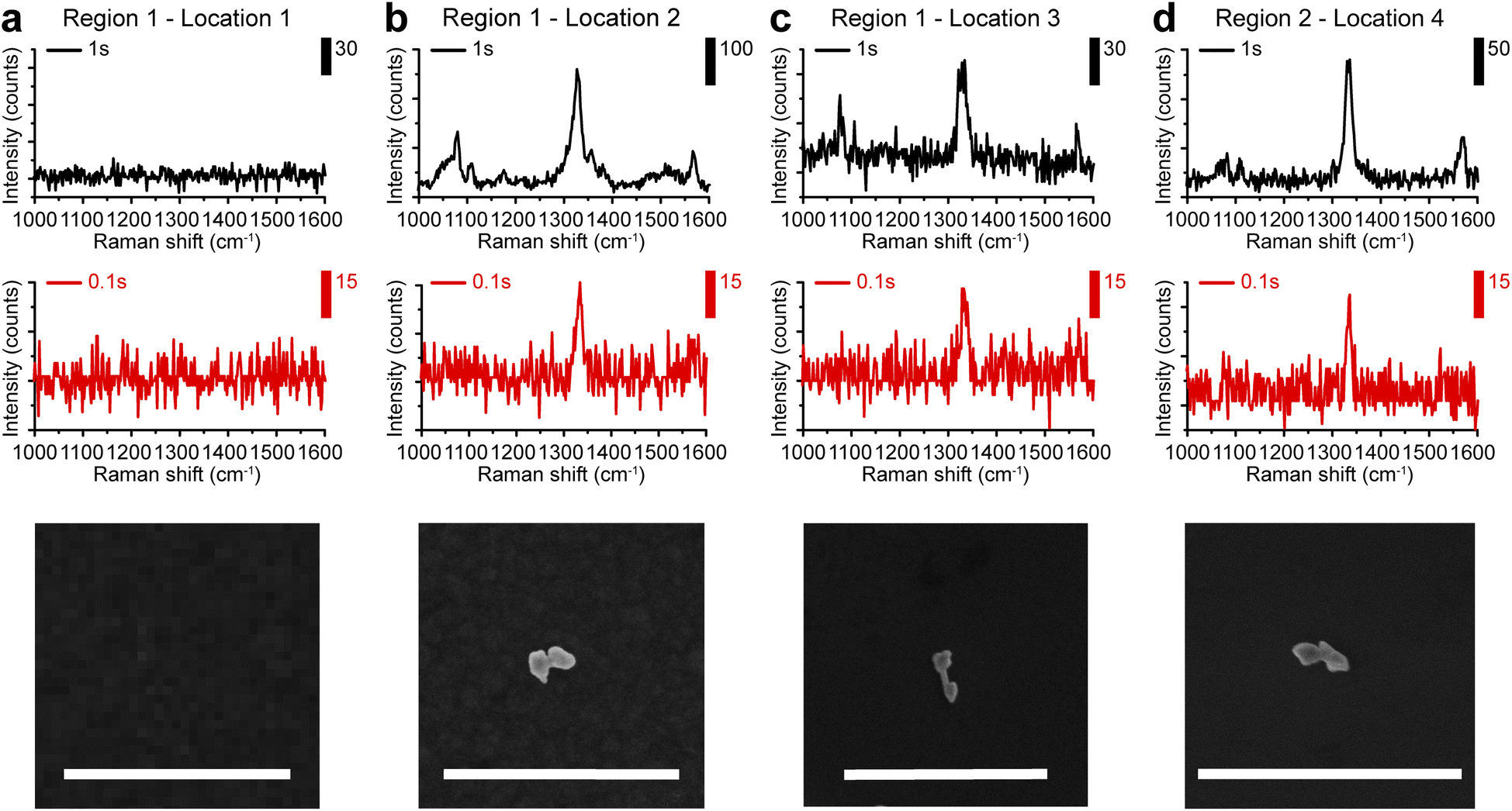 | ||
| Fig. 5 Single-particle SERS measurements. (a) SERS spectra of the bare silicon substrate without nanoparticles presented. (b–d) SERS spectra were recorded at three different locations with single gap-enhanced AuNDs presented. Corresponding SEM images were shown below. Scale bars: 1 μm. | ||
Conclusions
In summary, we reported a novel synthetic methodology of gap-enhanced AuNDs with embedded nanometer gaps supported by monolayered NBT molecules. By choosing the capping agent from CTAB over CTAC, we found that the shell growth preference can be altered from isotropy to anisotropy. We proposed the preferential tip-coating Au shell growth was due to stronger and more facet-dependent binding affinities of CTAB molecules to Au surfaces, compared to CTAC molecules.Colloidal studies suggested that compared to their AuNR cores, gap-enhanced AuNDs highly enhanced the SERS intensities by up to 101.5 times. By adjusting the shell thicknesses, SERS performances of AuNDs could be further optimized. We further demonstrated that a single-particle SERS sensitivity could be reached with an acquisition time of 0.1 s by a co-localized study by SEM and Raman microscopy.
Notably, Jiang et al. have fabricated core–shell dumbbell-like nanoparticles, in which two spatially separate gold shells were specifically deposited at the tips of AuNR with a 6 nm-thick polystyrene-block-poly(acrylic acid) (PSPAA) layer as the gap-supporting layer.51 This nanodumbbell, with the thick gap of 6 nm, exhibited SERS signals with an enhancement ratio of 2 compared to PSPAA-encapsulated AuNR cores. The much-improved SERS performance of our gap-enhanced AuNDs is attributed to the well-controlled thin gap around 1 nm supported by a monolayer of NBT to also act as the indicator molecules at the same time. Our findings open the opportunities for controllable design and synthesis of gap-enhanced nanoparticles with variable and tunable morphologies. With uniform single-particle SERS performances detectable with a detection speed as fast as 0.1 s per spectrum, gap-enhanced AuNDs could serve as novel ultrabright SERS probes for rapid bioimaging in the future.
Experimental section
Materials
All chemicals were obtained from commercial suppliers and used without further purification. Cetyltrimethylammonium bromide (CTAB, >99%), cetyltrimethylammonium chloride (CTAC, 25% water solution), L-ascorbic acid (AA, >99%), sodium borohydride (NaBH4, >98%), silver nitrate (AgNO3, >99%), gold(III) chloride trihydrate (HAuCl4·3H2O, >99.9%), hydrochloric acid (HCl, 37 wt% in water), and sulfuric acid (H2SO4, 95–98%) were purchased from Sigma-Aldrich. 4-Nitrobenzenethiol (NBT, >95%) and sodium oleate (NaOL, >97%) were purchased from TCI America. Deionized water (18.2 MΩ) was provided by a synergy UV water purification system. All reactions were carried out in glassware cleaned by aqua regia and piranha solution.Synthesis of AuNRs
AuNRs were synthesized by following a protocol developed by Ye et al. with modifications.35 In brief, the seed solution was synthesized as follows: 5 mL of HAuCl4 solution (0.54 mM) was mixed with 5 mL of CTAB solution (0.2 M) in a 20 mL glass vial. 1 mL of fresh NaBH4 solution (6 mM) was added to the above mixture solution under vigorous stirring (2000 rpm). The color of the solution changed from yellow to brownish yellow and the stirring was stopped after 2 min. The seed solution was aged at room temperature for 45 min before use. The growth solution was prepared by dissolving 1.8 g of CTAB and 246.8 mg of NaOL in 50 mL of warm water (∼50 °C) in a 250 mL flask. The solution was cooled down to 30 °C and 4.8 mL of AgNO3 solution (4 mM) was added. The mixture was kept undisturbed at 30 °C for 15 min after which 50 mL of HAuCl4 solution (1.08 mM) was added. The solution became colorless after 90 min of stirring (700 rpm) and 0.72 mL of HCl solution (37 wt% in water) was added to adjust the pH value. After another 15 min of slow stirring at 400 rpm, 0.25 mL of ascorbic acid (AA, 0.064 M) was added, and the solution was vigorously stirred at 1500 rpm for 30 s. Finally, 0.02 mL of seed solution was added to the growth solution. The mixture was stirred at 1500 rpm for another 30 s and left undisturbed at 30 °C for 12 h for AuNR growth.Functionalization of AuNRs with NBT
The obtained CTAB-capped AuNRs (peak OD = 1.525) were centrifuged at 1500g for 20 min. To remove CTAB, the pellet of AuNRs was resuspended in CTAC solution (0.1 M) and then washed twice with CTAC solution (0.1 M) at 1500g for 10 min. AuNRs were then resuspended in CTAC solution (0.1 M). The final concentration of Au in the colloid was ∼0.2 mM Au (peak OD ∼0.6). 2 mL of NBT solution in ethanol (5 mM) was added to 20 mL of the above AuNR solution. The mixture was vigorously sonicated for 1 h and then left undisturbed overnight for NBT molecules to absorb on the AuNR surface. To remove excess unbound NBT molecules and CTAC, AuNRs were washed with 0.1 M, 0.05 M, and 0.01 M CTAC solution sequentially and were then resuspended in DI water to reach a final concentration of 0.648 mM Au in the colloid (peak OD = 1.83).Synthesis of gap-enhanced AuNDs
To synthesize gap-enhanced AuNDs, 0.25 mL of NBT-modified AuNRs (AuNRs@NBT, 0.648 mM Au) was added to the growth solution consisting of 1.6 mL of CTAB solution (0.1 M), 0.06 mL of HAuCl4 solution (5.4 mM) and 0.03 mL of AA (40 mM) under vigorous sonication. The mixture was sonicated for 1 min and then kept undisturbed at 30 °C for 1 h. To ensure the reaction was fully completed, the system was kept undisturbed for another 1 h. For the synthesis of gap-enhanced AuNDs with different shell thicknesses, the amount of added HAuCl4 was varied from 0.03 mL, 0.06 mL, 0.12 mL and up to 0.24 mL, which corresponded to an Au molar ratio between the shells and AuNR cores (nAu(shell)/nAu(NR)) of 1, 2, 4 and 8 separately. For each group, the molar ratio between Au3+ precursors and reductants was kept constant.Synthesis of gap-enhanced AuNLs
For a comparison with gap-enhanced AuNDs, gap-enhanced AuNLs were synthesized using the same reaction parameters except for different surfactants. Briefly, 0.25 mL of NBT-modified AuNRs (AuNRs@NBT, 0.648 mM Au) was added to the growth solution consisting of 1.6 mL of CTAC solution (0.1 M), 0.06 mL of HAuCl4 solution (5.4 mM) and 0.03 mL of AA (40 mM) under vigorous sonication. The mixture was kept undisturbed at room temperature for 1 h to ensure a complete reaction.Sample preparation for growth mechanism study
A small portion (0.25 mL) of the reaction mixtures of gap-enhanced AuNDs or gap-enhanced AuNLs was extracted from the system at various times after the initiation of the reaction. The reaction at different stages was temporarily paused by immediately diluting the extracted reaction solution to 1 mL. Centrifugation (3000g, 2 min) was conducted to remove the reactants from the system right after the extinction spectra were quickly measured. The nanoparticles were then washed two times by centrifugation with DI water for SEM measurement.Nanoparticle characterizations
Extinction spectra were measured by UV-1900i spectrometer (Shimadzu). SEM images were obtained by Supra55-VP scanning electron microscope (Zeiss). For SEM measurements, nanoparticles were washed 2–3 times by centrifugation with DI water and then 5 μL of washed nanoparticles were dried on a silicon wafer under vacuum at room temperature. SEM imaging was conducted at 5 KV with a working distance of around 6 mm and an aperture size of 10 μm. The diameters of nanoparticles were measured by ImageJ. To calculate the quantities of nanoparticles presented in the field of view, we only considered the nanoparticles that were not tightly packed with other nanoparticles and showed complete structures in the SEM images, with sharp and clearly definable boundaries of individual particles. Nanostructures in the SEM showing the following three features are defined as AuNDs and numbers of AuNDs were counted correspondingly. First, the AuNDs should present a dumbbell structure with two shells coated at both ends of the AuNRs. Second, the diameters of shells on both ends should be no less than 1.5 times the diameters of the exposed AuNR portion of the structures. Third, if the shells were uneven, the diameters of the smaller shells should be no less than 1/4 of the diameters of the larger shells. The yield is calculated by total quantities of the AuNDs/total quantities of all the nanoparticles in the 10 SEM images from the measurements of one batch of AuND synthesis.To better visualize the ultrathin gap in TEM imaging, AuNDs with the thinnest shells, where 0.03 mL gold precursor was added during synthesis, were used for TEM examination. For TEM imaging, the gap-enhanced AuNDs were washed 3 times by centrifugation with DI water to remove CTAB surfactants. Then, we recovered the nanoparticles to 10 times of the original volume with DI water. A TEM grid was placed on a piece of clean tissue and a drop of diluted nanoparticles (5 to 7 μL) was deposited to the grid. The TEM sample was dried at the room temperature before TEM imaging. TEM imaging was conducted by atomic resolution electron microscope (JEM-ARM 200F) in STEM mode with an accelerating voltage of 200 kV. The gap size was analyzed using high-magnification (1.5–4 MX) TEM images. We only measured the size of a gap if the gap feature revealed in the TEM image had a length over 5 nm along the surface of the AuNR core. For each gap feature, one measurement was taken. We measured a total of 10 gaps in 9 different AuNDs from 9 TEM images in the batch prepared as described in the previous TEM sample preparation section. The size distribution of the internal gaps is shown in Fig. S2.†
Raman and SERS measurements
NBT powder was sandwiched between two pieces of cover glass with a gap of 1 mm, spaced by a piece of rubber film. Raman measurements of NBT powder were conducted by a confocal Renishaw InVia Raman microscope (objective, 20×, NA 0.4; laser condition, 633 nm, 0.504 mW; integration time, 10 s; grating, 1200 lines per mm). The laser beam was focused on the sample's top surface.To measure the SERS performances of nanoparticles in colloids, a 20 μL drop of each sample was sandwiched between two pieces of cover glass with a gap of 1 mm, supported by a piece of cut rubber film spacer. All samples were sonicated right before SERS spectra were measured by a confocal Renishaw InVia Raman microscope (objective, 20×, NA 0.4; laser condition, 633 nm, 2.4 mW or 785 nm, 39.9 mW; integration time, 10 s; grating, 1200 lines per mm). The laser beam was focused on the sample's top. In this case, the collecting area is around 2 μm on the sample and has a z depth of around 10 μm.
For single-particle SERS measurement, 150 μL of AuNDs was washed three times by centrifugation with DI water and then resuspended in 1.5 mL of water. A 10 μL drop was cast on a labeled silicon substrate and blow-dried with nitrogen gas. Raman mapping was obtained by a Renishaw InVia Raman microscope (objective, 100×, NA 0.85; laser condition, 633 nm, 504 μW; integration time, 0.1 s, 1 s; grating, 1200 lines per mm). In this case, the laser spot size is less than 1 μm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Brain Initiative R01 NS109794 to J-XC and CY by National Institute of Health, United States.References
- C. Zong, M. Xu, L.-J. Xu, T. Wei, X. Ma, X.-S. Zheng, R. Hu and B. Ren, Surface-enhanced Raman spectroscopy for bioanalysis: reliability and challenges, Chem. Rev., 2018, 118(10), 4946–4980 CrossRef CAS PubMed.
- D. Cialla-May, X.-S. Zheng, K. Weber and J. Popp, Recent progress in surface-enhanced Raman spectroscopy for biological and biomedical applications: from cells to clinics, Chem. Soc. Rev., 2017, 46(13), 3945–3961 RSC.
- Y. Wang, B. Yan and L. Chen, SERS tags: novel optical nanoprobes for bioanalysis, Chem. Rev., 2013, 113(3), 1391–1428 CrossRef CAS PubMed.
- H. Liu, X. Gao, C. Xu and D. Liu, SERS tags for biomedical detection and bioimaging, Theranostics, 2022, 12(4), 1870 CrossRef CAS PubMed.
- A. Kumar, S. Kim and J.-M. Nam, Plasmonically engineered nanoprobes for biomedical applications, J. Am. Chem. Soc., 2016, 138(44), 14509–14525 CrossRef CAS PubMed.
- E. Lenzi, D. Jimenez de Aberasturi and L. M. Liz-Marzan, Surface-enhanced Raman scattering tags for three-dimensional bioimaging and biomarker detection, ACS Sens., 2019, 4(5), 1126–1137 CrossRef CAS PubMed.
- J.-M. Nam, J.-W. Oh, H. Lee and Y. D. Suh, Plasmonic nanogap-enhanced Raman scattering with nanoparticles, Acc Chem. Res., 2016, 49(12), 2746–2755 CrossRef CAS PubMed.
- S.-Y. Ding, J. Yi, J.-F. Li, B. Ren, D.-Y. Wu, R. Panneerselvam and Z.-Q. Tian, Nanostructure-based plasmon-enhanced Raman spectroscopy for surface analysis of materials, Nat. Rev. Mater., 2016, 1(6), 1–16 Search PubMed.
- J. Zheng, X. Cheng, H. Zhang, X. Bai, R. Ai, L. Shao and J. Wang, Gold nanorods: the most versatile plasmonic nanoparticles, Chem. Rev., 2021, 121(21), 13342–13453 CrossRef CAS PubMed.
- N. M. Ngo, H.-V. Tran and T. R. Lee, Plasmonic nanostars: systematic review of their synthesis and applications, ACS Appl. Nano Mater., 2022, 5(10), 14051–14091 CrossRef CAS.
- D.-K. Lim, K.-S. Jeon, J.-H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J.-M. Nam, Highly uniform and reproducible surface-enhanced Raman scattering from DNA-tailorable nanoparticles with 1-nm interior gap, Nat. Nanotechnol., 2011, 6(7), 452–460 CrossRef CAS PubMed.
- K. L. Wustholz, A.-I. Henry, J. M. McMahon, R. G. Freeman, N. Valley, M. E. Piotti, M. J. Natan, G. C. Schatz and R. P. Van Duyne, Structure–activity relationships in gold nanoparticle dimers and trimers for surface-enhanced Raman spectroscopy, J. Am. Chem. Soc., 2010, 132(31), 10903–10910 CrossRef CAS PubMed.
- N. Pazos-Perez, C. S. Wagner, J. M. Romo-Herrera, L. M. Liz-Marzán, F. J. García de Abajo, A. Wittemann, A. Fery and R. A. Alvarez-Puebla, Organized plasmonic clusters with high coordination number and extraordinary enhancement in surface-enhanced Raman scattering (SERS), Angew. Chem., Int. Ed., 2012, 51(51), 12688–12693 CrossRef CAS PubMed.
- H. Cha, J. H. Yoon and S. Yoon, Probing quantum plasmon coupling using gold nanoparticle dimers with tunable interparticle distances down to the subnanometer range, ACS Nano, 2014, 8(8), 8554–8563 CrossRef CAS PubMed.
- J. H. Yoon, F. Selbach, L. Langolf and S. Schlücker, Ideal dimers of gold nanospheres for precision plasmonics: synthesis and characterization at the single-particle level for identification of higher order modes, Small, 2018, 14(4), 1702754 CrossRef.
- G. Chen, Y. Wang, L. H. Tan, M. Yang, L. S. Tan, Y. Chen and H. Chen, High-purity separation of gold nanoparticle dimers and trimers, J. Am. Chem. Soc., 2009, 131(12), 4218–4219 CrossRef CAS PubMed.
- K. Esashika, R. Ishii, S. Tokihiro and T. Saiki, Simple and rapid method for homogeneous dimer formation of gold nanoparticles in a bulk suspension based on van der Waals interactions between alkyl chains, Opt. Mater. Express, 2019, 9(4), 1667–1677 CrossRef CAS.
- N. G. Khlebtsov, L. Lin, B. N. Khlebtsov and J. Ye, Gap-enhanced Raman tags: fabrication, optical properties, and theranostic applications, Theranostics, 2020, 10(5), 2067 CrossRef CAS PubMed.
- N. Gandra and S. Singamaneni, Bilayered Raman-intense gold nanostructures with hidden tags (BRIGHTs) for high-resolution bioimaging, Adv. Mater., 2013, 25(7), 1022–1027 CrossRef CAS PubMed.
- B. Khlebtsov, T. Pylaev, V. Khanadeev, D. Bratashov and N. Khlebtsov, Quantitative and multiplex dot-immunoassay using gap-enhanced Raman tags, RSC Adv., 2017, 7(65), 40834–40841 RSC.
- B. N. Khlebtsov, D. N. Bratashov, N. A. Byzova, B. B. Dzantiev and N. G. Khlebtsov, SERS-based lateral flow immunoassay of troponin I by using gap-enhanced Raman tags, Nano Res., 2019, 12, 413–420 CrossRef CAS.
- Y. Gu, C. He, Y. Zhang, L. Lin, B. D. Thackray and J. Ye, Gap-enhanced Raman tags for physically unclonable anticounterfeiting labels, Nat. Commun., 2020, 11(1), 516 CrossRef CAS PubMed.
- Y. Zhang, Y. Gu, J. He, B. D. Thackray and J. Ye, Ultrabright gap-enhanced Raman tags for high-speed bioimaging, Nat. Commun., 2019, 10(1), 3905 CrossRef PubMed.
- J. H. Lee, J. W. Oh, S. H. Nam, Y. S. Cha, G. H. Kim, W. K. Rhim, N. H. Kim, J. Kim, S. W. Han and Y. D. Suh, Synthesis, optical properties, and multiplexed Raman bio-imaging of surface roughness-controlled nanobridged nanogap particles, Small, 2016, 12(34), 4726–4734 CrossRef CAS PubMed.
- Y. Qiu, Y. Zhang, M. Li, G. Chen, C. Fan, K. Cui, J.-B. Wan, A. Han, J. Ye and Z. Xiao, Intraoperative detection and eradication of residual microtumors with gap-enhanced Raman tags, ACS Nano, 2018, 12(8), 7974–7985 CrossRef CAS PubMed.
- C. Jiang, Y. Wang, W. Song and L. Lu, Delineating the tumor margin with intraoperative surface-enhanced Raman spectroscopy, Anal. Bioanal. Chem., 2019, 411, 3993–4006 CrossRef CAS.
- Y. Zhang, Y. Qiu, L. Lin, H. Gu, Z. Xiao and J. Ye, Ultraphotostable mesoporous silica-coated gap-enhanced Raman tags (GERTs) for high-speed bioimaging, ACS Appl. Mater. Interfaces, 2017, 9(4), 3995–4005 CrossRef CAS.
- Z. Bao, Y. Zhang, Z. Tan, X. Yin, W. Di and J. Ye, Gap-enhanced Raman tags for high-contrast sentinel lymph node imaging, Biomaterials, 2018, 163, 105–115 CrossRef CAS PubMed.
- B. N. Khlebtsov, A. M. Burov, D. N. Bratashov, R. S. Tumskiy and N. G. Khlebtsov, Petal-like gap-enhanced Raman tags with controllable structures for high-speed Raman imaging, Langmuir, 2020, 36(20), 5546–5553 CrossRef CAS PubMed.
- Y. Gu, X. Bi and J. Ye, Gap-enhanced resonance Raman tags for live-cell imaging, J. Mater. Chem. B, 2020, 8(31), 6944–6955 RSC.
- S. Wang, Z. Liu, C. Bartic, H. Xu and J. Ye, Improving SERS uniformity by isolating hot spots in gold rod-in-shell nanoparticles, J. Nanopart. Res., 2016, 18, 1–11 CrossRef.
- J. H. Hwang, N. K. Singhal, D. K. Lim and J. M. Nam, Au nanocucumbers with interior nanogap for multiple laser wavelength-compatible surface-enhanced Raman scattering, Bull. Korean Chem. Soc., 2015, 36(3), 882–886 CAS.
- T. Chen, C. Du, L. H. Tan, Z. Shen and H. Chen, Site-selective localization of analytes on gold nanorod surface for investigating field enhancement distribution in surface-enhanced Raman scattering, Nanoscale, 2011, 3(4), 1575–1581 RSC.
- B. Khlebtsov, V. Khanadeev, A. Burov and N. Khlebtsov, A new type of SERS tags: Au@Ag core/shell nanorods with embedded aromatic molecules, Nanotechnol. Russ., 2017, 12, 495–507 CrossRef CAS.
- X. Ye, C. Zheng, J. Chen, Y. Gao and C. B. Murray, Using binary surfactant mixtures to simultaneously improve the dimensional tunability and monodispersity in the seeded growth of gold nanorods, Nano Lett., 2013, 13(2), 765–771 CrossRef CAS PubMed.
- S. Chen, L. Meng, L. Wang, X. Huang, S. Ali, X. Chen, M. Yu, M. Yi, L. Li, X. Chen, L. Yuan, W. Shi and G. Huang, SERS-based lateral flow immunoassay for sensitive and simultaneous detection of anti-SARS-CoV-2 IgM and IgG antibodies by using gap-enhanced Raman nanotags, Sens. Actuators, B, 2021, 348, 130706 CrossRef CAS PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, Cancer cell imaging and photothermal therapy in the near-infrared region by using gold nanorods, J. Am. Chem. Soc., 2006, 128(6), 2115–2120 CrossRef CAS PubMed.
- P. Wang, M. Liu, G. Gao, S. Zhang, H. Shi, Z. Li, L. Zhang and Y. Fang, From gold nanorods to nanodumbbells: a different way to tailor surface plasmon resonances by a chemical route, J. Mater. Chem., 2012, 22(45), 24006–24011 RSC.
- K. Sohn, F. Kim, K. C. Pradel, J. Wu, Y. Peng, F. Zhou and J. Huang, Construction of evolutionary tree for morphological engineering of nanoparticles, ACS Nano, 2009, 3(8), 2191–2198 CrossRef CAS PubMed.
- M. Grzelczak, A. Sánchez-Iglesias, B. Rodríguez-González, R. Alvarez-Puebla, J. Pérez-Juste and L. M. Liz-Marzán, Influence of iodide ions on the growth of gold nanorods: tuning tip curvature and surface plasmon resonance, Adv. Funct. Mater., 2008, 18(23), 3780–3786 CrossRef CAS.
- T. Song, L. Tang, L. H. Tan, X. Wang, N. S. R. Satyavolu, H. Xing, Z. Wang, J. Li, H. Liang and Y. Lu, DNA-encoded tuning of geometric and plasmonic properties of nanoparticles growing from gold nanorod seeds, Angew. Chem., Int. Ed., 2015, 54(28), 8114–8118 CrossRef CAS PubMed.
- B. N. Khlebtsov, V. A. Khanadeev, A. M. Burov, E. C. Le Ru and N. G. Khlebtsov, Reexamination of surface-enhanced Raman scattering from gold nanorods as a function of aspect ratio and shape, J. Phys. Chem. C, 2020, 124(19), 10647–10658 CrossRef CAS.
- B. N. Khlebtsov, V. A. Khanadeev, J. Ye, G. B. Sukhorukov and N. G. Khlebtsov, Overgrowth of gold nanorods by using a binary surfactant mixture, Langmuir, 2014, 30(6), 1696–1703 CrossRef CAS PubMed.
- A. Gole and C. J. Murphy, Seed-mediated synthesis of gold nanorods: role of the size and nature of the seed, Chem. Mater., 2004, 16(19), 3633–3640 CrossRef CAS.
- O. M. Magnussen, Ordered anion adlayers on metal electrode surfaces, Chem. Rev., 2002, 102(3), 679–726 CrossRef CAS PubMed.
- S. K. Meena, S. Celiksoy, P. Schäfer, A. Henkel, C. Sönnichsen and M. Sulpizi, The role of halide ions in the anisotropic growth of gold nanoparticles: a microscopic, atomistic perspective, Phys. Chem. Chem. Phys., 2016, 18(19), 13246–13254 RSC.
- S. Gomez-Grana, F. Hubert, F. Testard, A. Guerrero-Martínez, I. Grillo, L. M. Liz-Marzán and O. Spalla, Surfactant (bi) layers on gold nanorods, Langmuir, 2012, 28(2), 1453–1459 CrossRef CAS PubMed.
- J.-Y. Kim, M.-G. Han, M.-B. Lien, S. Magonov, Y. Zhu, H. George, T. B. Norris and N. A. Kotov, Dipole-like electrostatic asymmetry of gold nanorods, Sci. Adv., 2018, 4(2), e1700682 CrossRef PubMed.
- B. E. Janicek, J. G. Hinman, J. J. Hinman, S. H. Bae, M. Wu, J. Turner, H.-H. Chang, E. Park, R. Lawless and K. S. Suslick, Quantitative imaging of organic ligand density on anisotropic inorganic nanocrystals, Nano Lett., 2019, 19(9), 6308–6314 CrossRef CAS PubMed.
- J. Huang, Y. Zhu, C. Liu, Z. Shi, A. Fratalocchi and Y. Han, Unravelling thiol's role in directing asymmetric growth of Au nanorod–Au nanoparticle dimers, Nano Lett., 2016, 16(1), 617–623 CrossRef CAS PubMed.
- T. Jiang, X. Wang, J. Tang, J. Zhou, C. Gu and S. Tang, The seeded-synthesis of core–shell Au dumbbells with inbuilt Raman molecules and their SERS performance, Anal. Methods, 2017, 9(30), 4394–4399 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04365g |
| This journal is © The Royal Society of Chemistry 2023 |