
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances and prospects of porphyrin derivatives in the energy field
Mingfa Xie,
Jinyuan Liu,
Lianghong Dai,
Hongjian Peng * and
Youqing Xie
* and
Youqing Xie
College of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, China. E-mail: Hongjianpeng@126.com
First published on 18th August 2023
Abstract
At present, porphyrin is developing rapidly in the fields of medicine, energy, catalysts, etc. More and more reports on its application are being published. This paper mainly takes the ingenious utilization of porphyrin derivatives in perovskite solar cells, dye-sensitized solar cells, and lithium batteries as the background to review the design idea of functional materials based on the porphyrin structural unit in the energy sector. In addition, the modification and improvement strategies of porphyrin are presented by visually showing the molecular structures or the design synthesis routes of its functional materials. Finally, we provide some insights into the development of novel energy storage materials based on porphyrin frameworks.
1. Introduction
Against the current social background of rapid economic development, the global greenhouse effect, the fossil energy crisis, environmental pollution, and other problems are in the foreground. To reduce carbon dioxide emissions and achieve the goal of carbon neutrality, the trend towards low-carbon energy and industrial development has become inevitable, and green development is the only solution. Developing solar, wind, hydro, geothermal, and other renewable energies by modern scientific and technological means is an effective measure. With the advent of third-generation solar cells, such as perovskite solar cells and dye-sensitized solar cells, the focus of our scientific research is on exploiting photovoltaic power generation technologies that enable efficient conversion and storage of solar energy. And porphyrins, as protagonists in our review, play a crucial role in solar cells, as well as in lithium-ion batteries, which are widely applied to current energy devices.Porphyrin with a macrocyclic conjugated structure is a class of aromatic organic compounds containing substituents in the porphin ring. As the framework of porphyrins, porphins usually coordinate with metal ions, introduce electron withdrawing or donating groups in the meso-site, or introduce large steric hindrance groups to adjust the electron cloud density, the ability of charge storage and transport, molecular orbital energy, π–π stacking degree, UV-visible light absorption, solubility, and self-assembly to varying degrees. In this way, some adaptation performance in different application fields can be enhanced or weakened. The electrostatic potential, HOMO and LUMO energies, absorption spectra, and self-assembly principles of porphins are presented in Fig. 1.
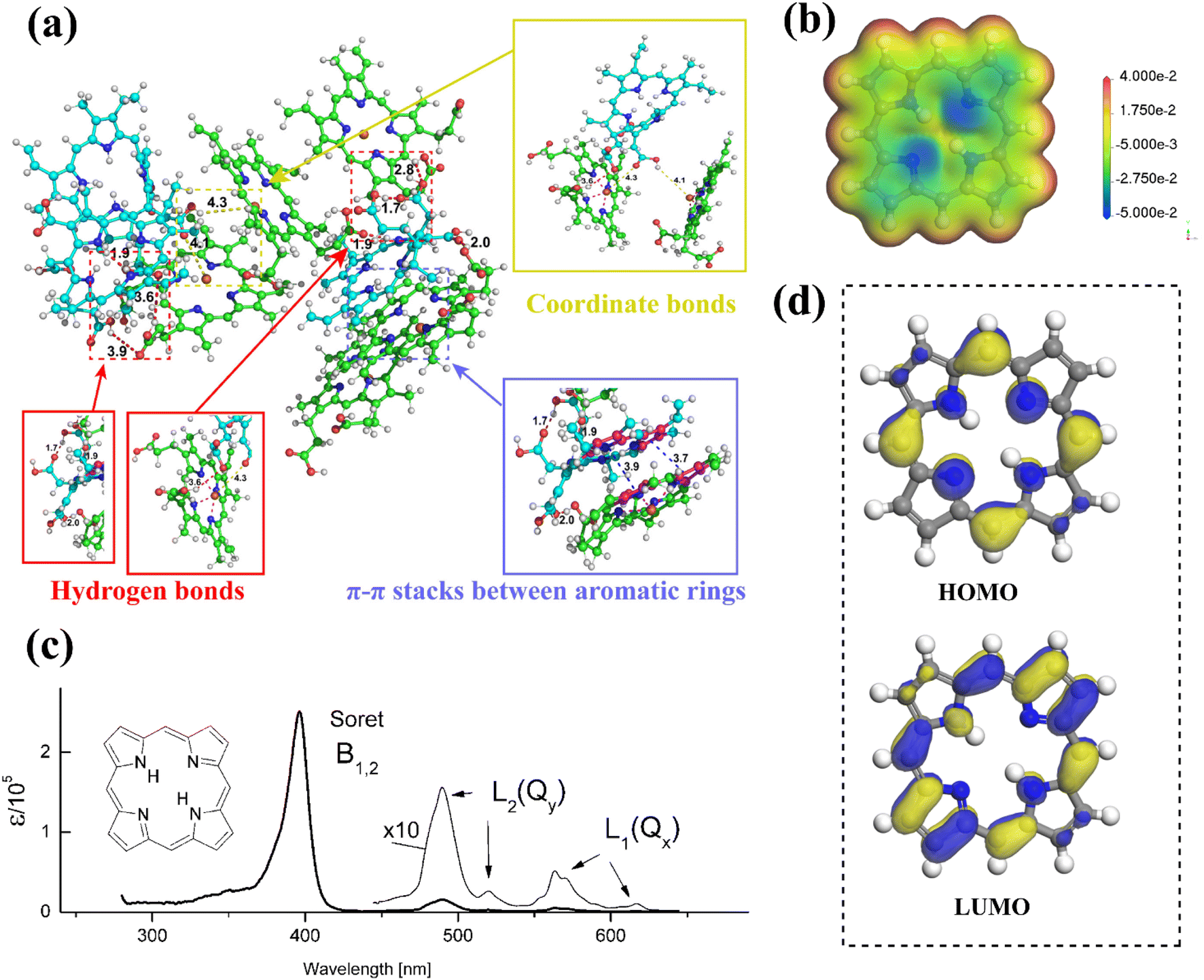 | ||
| Fig. 1 The self-assembled MD simulation of porphyrin derivatives Ce6 and hemin molecules.1 Copyright 2021, ACS. (b) The electrostatic potential of porphin molecule. (c) The absorption spectrum of porphin molecule.,2 Copyright 2016, ACS. (d) HOMO and LUMO energy levels of porphin molecule obtained from DFT calculation. | ||
Along with the study of porphyria in the 19th century, scientists generated great cognitive interest in porphyrins. Fischer first reported the synthesis of porphyrin in 1926, which initiated the progressive transition from biosynthesis to artificial synthesis.3 Subsequently, scientists such as Rothemund, Macdonald, Adler, and Lindsey conducted continuous explorations and optimizations, which opened up new methods for the artificial preparation of porphyrins.4–8 Modern methods for the fabricating of porphyrins and their derivatives, thoroughly described in reviews by Sabine Horn et al. in 2008, provided ideas for its structural modifications.9 Due to the shortcomings of traditional synthesis methods in terms of yield and purity, a novel green synthetic route of porphyrins was explored by Mondal et al. in 2021.10 Common synthetic routes for porphyrin compounds are shown in Fig. 2. The increasing sophistication of artificial synthesis methods for porphyrins has laid the foundation for their structural design and application. Originally, porphyrins were rapidly developed in medicine thanks to their specific structure and physiological functions, which led to porphyrin complexes receiving much attention. With constant research into the properties of porphyrins, their applications are expanding in materials, batteries, catalysts, etc. Porphyrins benefit from their excellent photochemical performance, which allows them to serve as a vital character in the energy sector, for example in batteries and capacitors.
 | ||
| Fig. 2 Common synthetic routes of porphyrin compounds. | ||
2. Porphyrins for perovskite solar cells
As a new type of photovoltaic cell, perovskite solar cells (PSCs) have the characteristics of a high absorption coefficient, controllable band gap, long carrier diffusion length, and high charge carrier mobility. Due to its low cost, easy preparation, and high photoelectric conversion efficiency, it is considered an alternative to silicon-based solar cells.11–13 The power conversion efficiency (PCE) of organic–inorganic hybrid perovskite solar cells (PSCs) has increased from an initial 3.8% to over 25% by 2023,14,15 representing great promise for growth. However, the defects formed by the perovskite film severely limit the photovoltaic conversion efficiency and long-term operational stability of PSCs.16 Therefore, the development of functional materials, such as porphyrins and their derivatives, to enhance the PCE and the hydrothermal stability of PSCs is an effective measure.For the sake of subsequent description, we first present the structure and principle of PSCs. The core structure of a PSC consists of an electron transport layer (ETL), a perovskite absorber layer (PAL), and a hole transport layer (HTL). First, the perovskite photoactive material generates electron–hole pairs by absorbing photons when irradiated by sunlight. Before compounding, electrons and holes are transported via electron transport materials (ETMs) and hole transport materials (HTMs) respectively, then collected by the relevant functional materials. Finally, a complete circuit is formed by connecting loads (Fig. 3). Therefore, it's necessary to improve the performance and quality of interfaces, perovskite films, and ETMs/HTMs. The main applications of porphyrins in PSCs are doped in perovskite films or deposited on the surface of perovskite films to modulate its quality and promote the transport of charge as well as enhance the stability of battery devices, or designing them as HTMs.
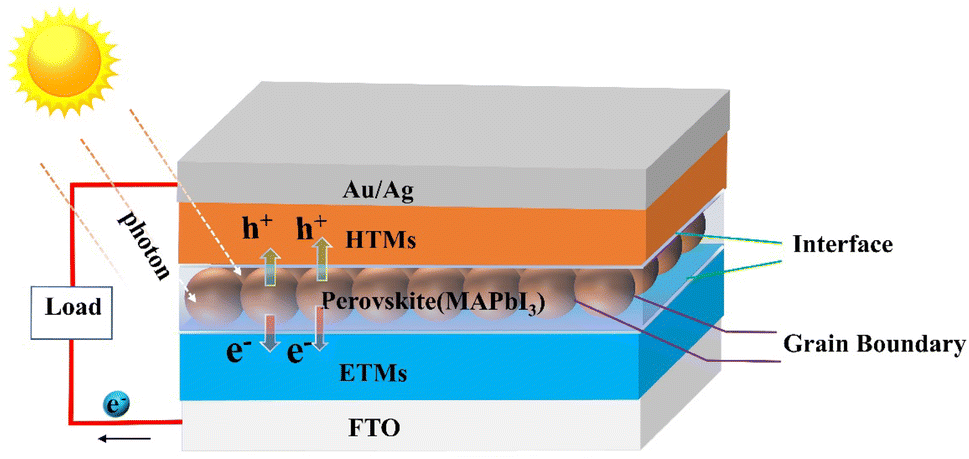 | ||
| Fig. 3 Mechanism and structure (n-i-p) of planar heterojunction perovskite solar cells. | ||
2.1. Porphyrins are doped in the perovskite film as additives
In recent years, some valuable porphyrin compounds have been developed and produced to improve the quality of perovskite films by doping, to achieve a high PCE.17–25 As we know, the structural formula of a perovskite light-absorbing material can be expressed as ABX3, where A is an organic cation, usually, the HC(NH2)2+ (referred to as FA+) or the CH3NH3+ (referred to as MA+), B is a metal cation, usually a lead ion with good stability, and X is a halogen group, usually the iodide ion, which is less electronegative and has a strong covalent interaction with the lead ion. During the preparation of the perovskite films in the solution, the undercoordinated Pb2+ and free iodide ion can induce the formation of defects that existed in the surface of the film and grain boundaries, which further affects the quality of the perovskite film by providing sites for nonradiative recombination.16,26–28From this point of view, it is a reasonable strategy to passivate the defects by employing functional groups such as pyridine,23,29 amine,17,20 carbonyl,18,22 and sulfonate30 to react with free Pb2+ in perovskite film. For example, Zhang et al. manufactured a novel D–π–A-type porphyrin derivative (coded as ZnPP, Fig. 4a) by grafting pyridine functional groups onto a porphyrin and used it as a passivation molecule to modify the perovskite bulk thin film.23 The triarylamine persuade in the porphyrin molecule can not only act as an electron donor to enhance the coordination ability of the pyridine group with Pb2+ but also augment the steric hindrance to reducing the aggregation of porphyrin molecules. Therefore, the crystallinity of perovskite film adjusted with ZnPP was improved compared to a control group without additives. As listed in Table 1, a, this contributed to the upgrade of battery performance.
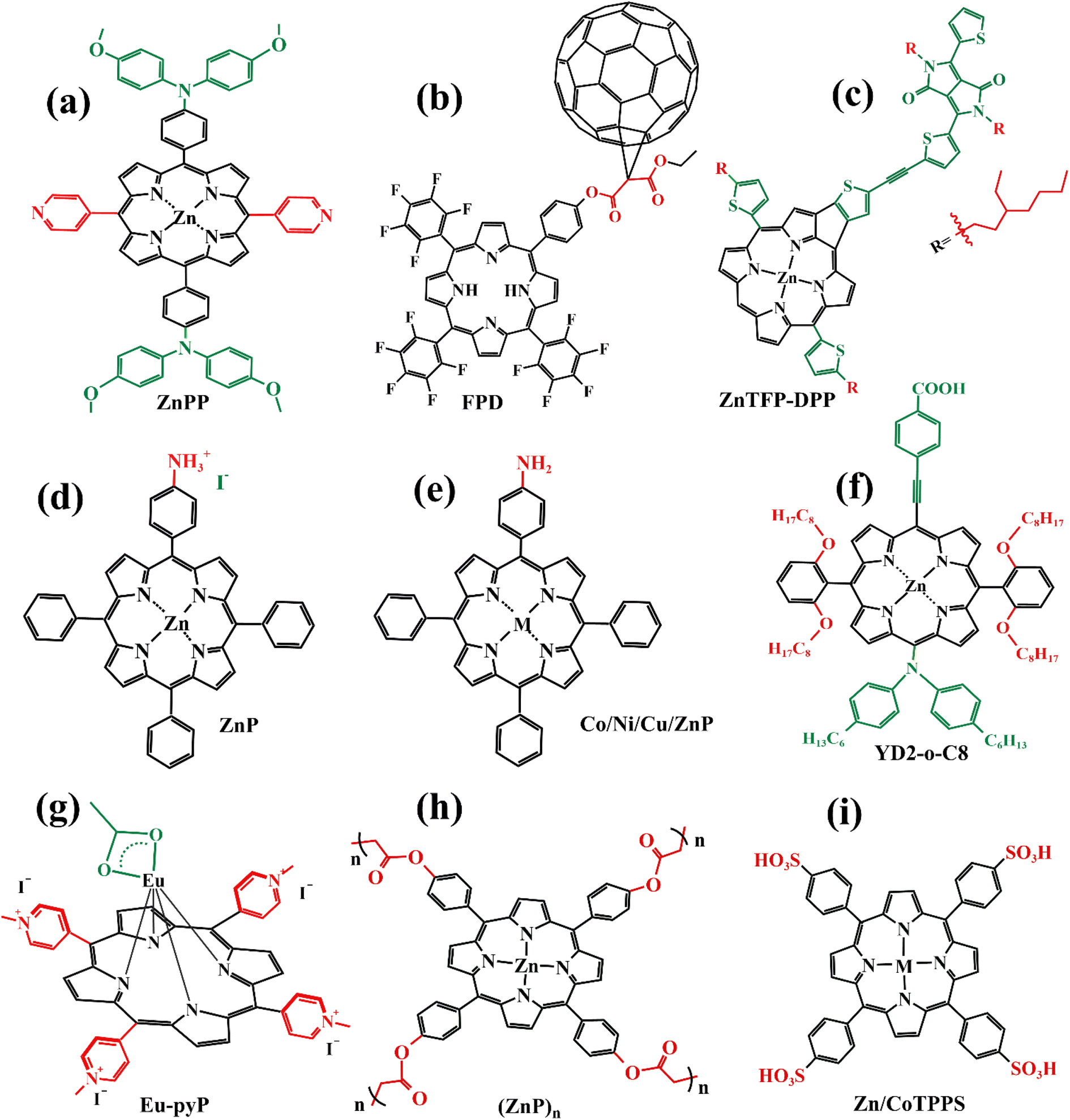 | ||
| Fig. 4 (a)–(i) The structures of porphyrins doped into perovskite film mentioned in 2.1. | ||
| Materials | Code | PCE′b (%) | PCE (%) | FF′b (%) | FF (%) |
|
VOC (V) |
|
JSC (mA cm−2) | Ref., year |
|---|---|---|---|---|---|---|---|---|---|---|
a Measured under AM 1.5G illumination (100 mW cm−2).b PCE′, FF′, 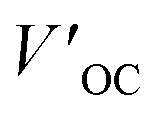 , and , and  represent the battery performance parameters of the control group. represent the battery performance parameters of the control group. |
||||||||||
| ZnPP | a | 20.18 | 21.08 | 82.17 | 82.91 | 1.097 | 1.126 | 22.37 | 22.57 | Ref. 23, 2022 |
| FPD | b | 20.99 | 23.00 | 79.20 | 80.86 | 1.082 | 1.129 | 24.49 | 25.19 | Ref. 22, 2022 |
| ZnTFP-DPP | c | 18.4 | 19.3 | 76 | 75 | 1.06 | 1.08 | 22.7 | 24.3 | Ref. 24, 2021 |
| ZnP | d | 18.81 | 20.46 | 74.58 | 77.63 | 1.09 | 1.12 | 22.9 | 23.27 | Ref. 20, 2019 |
| NiP | e | 22.84 | 24.16 | 79.01 | 81.01 | 1.14 | 1.17 | 25.31 | 25.49 | Ref. 17, 2021 |
| YD2-o-C8 | f | 18.5 | 20.5 | 77 | 80 | 1.03 | 1.05 | 23.3 | 24.4 | Ref. 25, 2019 |
| Eu-pyP | g | 18.96 | 18.35 | 75.64 | 74.92 | 1.09 | 1.08 | 22.99 | 22.68 | Ref. 18, 2019 |
| (ZnP)n | h | 19.77 | 20.53 | 77.89 | 79.29 | 1.07 | 1.11 | 23.12 | 23.19 | Ref. 21, 2022 |
| CoTPPS | i | 19.89 | 21.00 | 78.40 | 78.13 | 1.097 | 1.134 | 23.13 | 23.71 | Ref. 19, 2022 |
| ZnTPPS | 19.89 | 20.74 | 78.40 | 78.40 | 1.097 | 1.132 | 23.13 | 23.37 | ||

For another instance, fullerene cages can interact with iodides to inhibit the formation of deep traps in perovskite films,31 but common fullerenes such as C60 have a weak chemical bond with perovskite lattice, resulting in a weak ability to trap free Pb2+.32 However, porphyrins can introduce functional groups to enhance the ability to combine Pb2+ besides themselves. Based on this theory, Wei et al. synthesized a novel bi-functional complex (coded as FPD, Fig. 4b).22 On the one hand, the electron-deficient C60 combines with the electron-rich I− to achieve binding, reducing defects due to iodine ion shifts. On the other hand, the ester group introduced on the porphyrin ring is coordinated with lead ions, reducing defects caused by free lead ions. Both work together to improve the passivation effect of PDF on defects. In addition, the fluorine on the benzene ring forms a hydrogen bond with the nearby ammonium salt, preventing its decomposition and improving its stability. Even if the perovskite material is damaged by water, the porphyrin on its surface can chelate with the dissociated lead ions to form a stable and water-insoluble metalloporphyrin complex, thus preventing the pollution caused by the leakage of lead ions. When using FPD as a passivation molecule, we can see a significant increase in PCE of the battery device from 20.99% to 23.00% in Table 1, b.
In addition to suppressing carrier recombination through passivating defects in films, porphyrins used as a dopant can also promote hole transfer. For example, Cao et al. designed and synthesized a series of monoamine metalloporphyrin compounds (Fig. 4e) to fill the grain-to-grain boundaries in perovskite thin films by self-assembling into supramolecules.17 Amino groups on porphyrins firmly attached to the grain surface in perovskite crystals through the protonation effect passivated the defects of perovskite films and promote the transport of extracted holes across boundaries, reducing nonradiative carrier recombination. Especially, the best efficiency of the PSCs with 0.5% NiP doping was up to 24.16%, much higher than 22.84% of the control device under the active area of 0.1 cm2 (Table 1, e). Admirably, Cao et al. further study revealed that a supra-molecule (CuP–S1) self-assembled by CuPs in perovskite grain boundaries can change into a new supramolecule (CuP–S2) via simple post-annealing treatment, and CuP–S2 can generate homogeneously large polarons in a periodic lattice to promote holes transport between the perovskite grains and reduce the non-radiative carrier recombination.33
Like that studies, adding functional porphyrin materials into the perovskite layer can passivate defects in grain boundaries, thus reducing the non-radiative recombination of carriers, and certain special functional porphyrin compounds can also promote carrier transfer. Moreover, these additives can provide a stable environment for perovskite films, especially Eu-pyP (Fig. 4g) designed by Cao et al. deserves to be mentioned. Although the PCE of the battery device whose perovskite material (MAPbI3) doped with Eu-pyP complex was slightly less than the reference device, it presented outstanding stability.18 This was attributed to the porphyrin addictive filled between grain boundaries for defect passivation and improving the resistibility of the perovskite to moisture, heat, and solar light including UV.
Besides the aromatic porphyrins applied to PSCs, Yutaka Matsuo's team designed an anti-aromatic donor–acceptor (D–A) type porphyrin (coded as ZnTFP-DPP, Fig. 4c) for the first time and used it as a light-absorbing dopant to enhance the light absorption capability of PSC devices.24 A diketopyrrolopyrrole (DPP) moiety introduced on the porphyrin ring is an excellent chromophore widely used in D–A molecules. It increases the HOMO level of ZnTFP-DPP to −5.36 eV, which is close to the HOMO level of perovskite absorption material (MAPbI3), thus achieving the level matching. ZnTFP-DPP had stronger absorption than perovskite materials at wavelengths less than 400 nm and longer than 450 nm. Additionally, the presence of the DPP unit significantly enhanced the light absorption of both the Soret band and Q band. The PCE of PSCs doped with ZnTFP-DPP in perovskite absorbent material increased to 19.3% compared with the control group. This study provides a reference for the design of anti-aromatic porphyrins as dopants of perovskite absorbent materials.
In this subsection, the chemical structures of the porphyrins doped in the perovskite film are presented in Fig. 4, and their effects on the performance parameters of PSCs compared to the control group are correspondingly summarized in Table 1.
2.2. Porphyrins as interface materials for perovskite solar cell
Interfacial engineering is also an effective strategy to ameliorate the performance of PSCs.34–36 Utilizing porphyrin compounds as interfacial materials, spinning coated or deposited on the perovskite surface, thus passivating the surface defects of perovskite film, promoting charge transfer, and generating a hydrophobic environment to enhance the stability of battery devices. For instance, Gkini et al. fabricated a novel Mn–porphyrin compound (Fig. 5b) and filled them in the form of a thin film between the TiO2 electron transport layer (ETL) and the perovskite absorb layer (PAL)(Fig. 5a).37 The introduction of Mn–porphyrins not only brought the ETL and PAL closer together but also created a waterproof environment for perovskite crystals to grow better. Moreover, the high paramagnetic character of manganese porphyrin facilitated the transport of electrons. So that the PCE of the battery device increased from 16.94% to 18.70% compared to the control group without the porphyrin film (Table 2, a).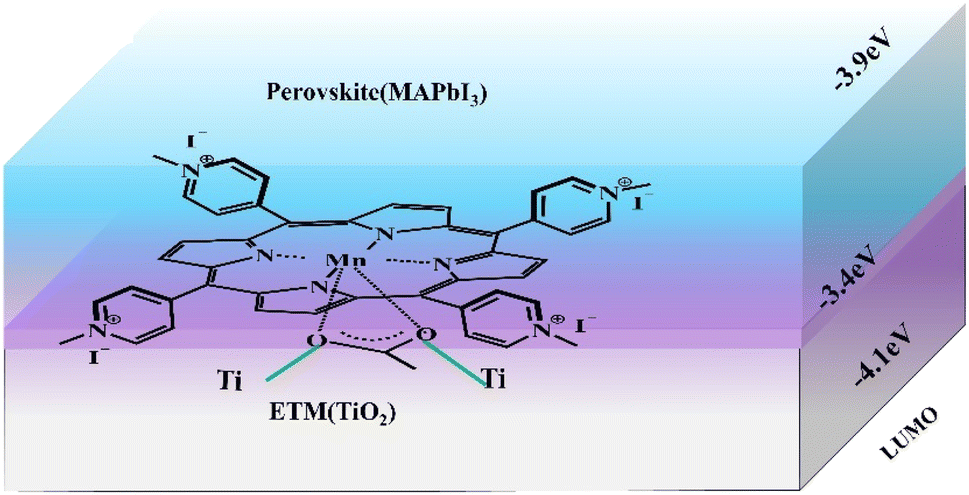 | ||
| Fig. 5 The schematic diagram of Mn–porphyrins at perovskite and ETM anchored on TiO2 through C–O–Ti bond. | ||
| Interface materials | Code | Acting site | PCE′b (%) | PCE (%) | FF′b (%) | FF (%) |
|
VOC (V) |
|
JSC (mA cm−2) | Ref., year |
|---|---|---|---|---|---|---|---|---|---|---|---|
a Measured under AM 1.5G illumination (100 mW cm−2).b PCE′, FF′,  and and  represents the battery performance parameters of the control group. represents the battery performance parameters of the control group. |
|||||||||||
| (TMePyP)-I4Mn(AcO) | a | ETL-PAL | 16.94 | 18.70 | 71 | 75 | 1.05 | 1.04 | 22.62 | 23.52 | Ref. 37, 2020 |
| BZnTPP | b | ETL-PAL | 16.63 | 17.34 | 72 | 74 | 1.12 | 1.11 | 20.62 | 21.11 | Ref. 38, 2020 |
| Por | c | HTL-PAL | 19.92 | 20.34 | 76.95 | 77.57 | 1.10 | 1.11 | 23.53 | 23.62 | Ref. 42, 2020 |
| Zn-Por | 20.76 | 78.48 | 1.12 | 23.60 | |||||||
| Cu-Por | 21.24 | 79.11 | 1.13 | 23.76 | |||||||
| CuP | d | HTL-PAL | 20.01 | 21.76 | 76.83 | 79.32 | 1.11 | 1.14 | 23.44 | 23.91 | Ref. 44, 2021 |
| ZnP | 21.12 | 78.51 | 1.13 | 23.79 | |||||||
| Co(II)P | e | HTL-PAL | 19.58 | 21.28 | 76.91 | 78.97 | 1.10 | 1.14 | 23.12 | 23.45 | Ref. 45, 2020 |
| CS0 | f | HTL-PAL | 21.04 | 22.14 | 81.7 | 84.3 | 1.07 | 1.096 | 24.07 | 23.96 | Ref. 43, 2021 |
| CS1 | 22.37 | 84.2 | 1.100 | 23.14 | |||||||
| CS2 | 22.17 | 83.6 | 1.117 | 23.74 | |||||||
| H2P | g | HTL-PAL | 19.86 | 20.11 | 76.83 | 78.05 | 1.10 | 1.10 | 23.51 | 23.43 | Ref. 46, 2021 |
| ZnP | 20.41 | 78.31 | 1.11 | 23.45 | |||||||
| CuP | 20.71 | 78.87 | 1.11 | 23.64 | |||||||
| Por-BTA | h | HTL-PAL | 21.30 | 22.30 | 77.3 | 80.4 | 1.10 | 1.10 | 24.95 | 25.22 | Ref. 39, 2021 |
| ZnTPPS4 | i | HTL-PAL | 11.65 | 14.37 | 74.43 | 74.69 | 0.90 | 0.93 | 17.34 | 20.65 | Ref. 41, 2020 |
| CuTPPS4 | 14.61 | 75.91 | 0.94 | 20.42 | |||||||


Later, Gkini et al. reported another porphyrin complex (coded as BZnTPP, Fig. 6b), which introduced a triazine unit as the π-bridge to improve charge transfer from the electron–donor (5,10,15,20-tetraphenyl-porphyrin zinc) to the electron–acceptor (bodipy) segment through the polarizable electron structure of the π-bridge, to further explain the interface engineering using suitable materials to act as a universal electron transfer mediator.38 Thanks to the porphyrin mediator with a “push–pull” character filled between ETL and PAL, the PCE and stability of perovskite solar cell devices are significantly improved.
 | ||
| Fig. 6 (a)–(i) The structures of porphyrins as an interfacial material mentioned in 2.2. | ||
To enhance the hole extraction ability between the interface of the perovskite layer and the hole transport layer while passivating defects, Zhang et al. designed a new porphyrin complex (coded as Por-BTA, Fig. 6h) as a multifunctional interface material.39 The carbonyl unit and basic sites of the pyrrole unit in Por-BTA can coordinate with the exposed lead ions, thereby passivating the perovskite surface defects. Moreover, the shortening of the distance between interfaces and the strong intermolecular π–π stacking of Por-BTA are good for charge transfer. The battery device treated by Por-BTA achieved a PCE of 22.30%.
Extending the photoresponse of perovskite light-absorbing material to sunlight by coating a layer of bulk-heterojunction (BHJ) organic semiconductors on the top of the perovskite layer is an approach worth considering. Gao et al. designed and synthesized a new porphyrin compound (coded as DPPZn-TSEH, Fig. 7a) and blended it with PCBM (Fig. 7b) to create an efficient BHJ for organic photovoltaics/perovskite hybrid solar cells. The highest occupied molecular orbital energy level of DPPZnP-TSEH, adjusted by the alkyl-sulfide thienyl electron-donating groups, matches the energy level of perovskite materials. Moreover, profiting from the porphyrin special structure, it has wide light absorption and high hole mobility. The optimized cell device achieved a high PCE of 19.02%.40
 | ||
| Fig. 7 (a) The chemical structure of DPPZn-TSEH; (b) the chemical structure of PCBM. | ||
As we listed in Fig. 6 and Table 2, there are many reports on various functional porphyrin molecules as interface materials to passivate defects, regulate the quality of perovskite films, and promote charge transfer.37–39,41–46 To avoid the repetition of the work, we just describe certain typical examples of porphyrin compounds at different acting sites and diverse functions.
2.3. Porphyrins as hole-transport materials in perovskite solar cells
As an essential constituent element of PSCs, hole transport materials (HTMs) can extract carriers and transfer them to the corresponding electrode, simultaneously, they also protect the perovskite absorption layer from corrosion by the external water, heat, and ultraviolet. The ideal HTMs should have a high carrier mobility, energy level matching perovskite materials, low production cost, good stability, and film-formation.47,48 The most widely used organic semiconductor materials in n-i-p solar cell devices, such as spiro-OMeTAD (Fig. 9a), are difficult to synthesize, with a high cost and poor crystallinity as well as stability.49 In contrast, porphyrin has a large π–electron conjugated plane and relative stability to light and heat. Moreover, a porphyrin large ring can be deposited on the perovskite layer in the way of π–π stacking, to promote vertical charge transmission, and can achieve a high power conversion efficiency. In principle, the HTM layer should preferably be colorless to avoid loss of light absorption from the perovskite layer. However, porphyrins have strong light absorption in the Soret zone (400–450 nm). Undoubtedly, it is a challenge to design porphyrins as hole transport materials.In 2016, Yeh's team designed and synthesized two novel types of zinc porphyrins (coded as Y2 and Y2A2, Fig. 8b), which have different lengths of carbon chains, and used them as HTM for PSCs for the first time.50 The HOMO energy of Y2 and Y2A2 is close to the spiro-OMeTAD energy level. UV-Visible Spectroscopy Testing suggests that Y2 and Y2A2 are limited in their effectiveness as HTMs in competing with perovskite for light absorption. It should be noted that the alkyl chain of Y2A2 is much longer than that of Y2, resulting in a large steric hindrance that blocks the close stacking between molecules. Therefore, the cavity extraction capacity of Y2A2 is much weaker compared to Y2. Afterward, Yeh et al. continued to develop two porphyrin dimers (coded as WT3, YR3, Fig. 8c and d) with better hole-transporting ability owing to their dimeric conjugate structure, which can enhance the effect of intermolecular π–π stacking.51 Based on their experience, Yeh's team designed another four porphyrin derivatives (Fig. 8e) with simpler structures and easier preparation.52 However, the PCE of batteries device used YZT-series as HTMs doped with tBP and FK209 is much lower than spiro-OMeTAD under the same condition (Table 3).
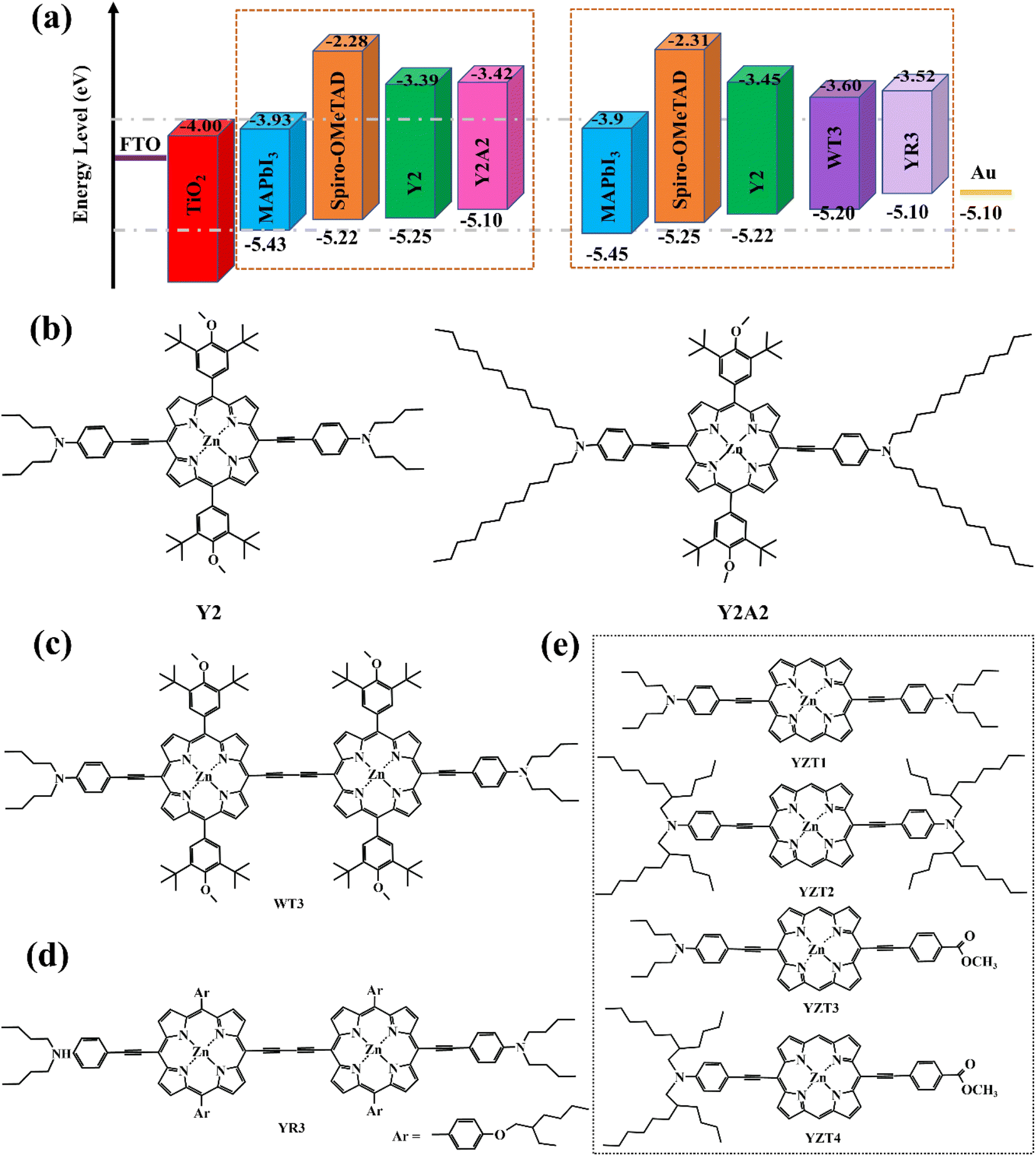 | ||
| Fig. 8 (a) Energy level diagram for device components including varied HTMs; (b–e) the structure of porphyrins designed by Yeh's team. | ||
| HTM material | Hole mobility (cm2 V−1 s−1) | VOC (V) | JSC (mA cm−2) | FF (%) | PCE (%) | Ref., year |
|---|---|---|---|---|---|---|
| Spiro-OMeTAD | 9.46 × 10−4 | 1.06 | 22.82 | 74.39 | 18.03 | Ref. 50, 2016 |
| Y2 | 2.04 × 10−4 | 0.99 | 17.80 | 73.34 | 16.60 | |
| Y2A2 | 1.53 × 10−5 | 1.01 | 22.79 | 58.69 | 10.55 | |
| Spiro-OMeTAD | 1.4 × 10−4 | 1.086 | 22.64 | 75.74 | 18.62 | Ref. 51, 2018 |
| Y2 | 3.2 × 10−5 | 1.086 | 22.62 | 73.02 | 17.93 | |
| WT3 | 4.2 × 10−4 | 1.096 | 22.60 | 78.52 | 19.44 | |
| YR3 | 9.3 × 10−5 | 1.039 | 22.97 | 74.75 | 17.84 | |
| Spiro-OMeTAD | — | 1.07 | 21.58 | 76.74 | 17.77 | Ref. 52, 2020 |
| YZT1 | 7.61 × 10−5 | 0.97 | 21.19 | 70.21 | 14.46 | |
| YZT2 | 2.69 × 10−6 | 0.93 | 11.86 | 52.77 | 5.81 | |
| YZT3 | 4.48 × 10−5 | 0.96 | 21.32 | 70.90 | 14.50 | |
| YZT4 | 5.48 × 10−5 | 0.95 | 21.40 | 73.51 | 14.95 |
Although WT3 and Y2 were used as HTMs achieving a high PCE, the fabricating process of the hole transfer layer is complicated because of the introduction of additives. The tedious preparation step of porphyrin compounds used as HTMs is also a limitation. For example, Co(II)P and Co(III)P (Fig. 9d–e) synthesized via poly-step reaction were mixed as HTMs, even though the best PCE of successfully fabricated PSCs was 20.5%, reported by Cao et al.53 On the contrary, in a one-pot, Yong Hua's team synthesized a new porphyrin molecule as well as complexes with the metals zinc and copper (coded as CuP, ZnP, Fig. 9b).54
 | ||
| Fig. 9 (a)–(i) The molecular structures of organic compounds as HTMs for PSCs mentioned above. | ||
More and more compounds of the porphyrin family are being designed and synthesized as HTMs for batteries, either directly or by doping (Table 4).55–60 The relevant experimental conclusions provide a constructive reference for the subsequent design of better and more valuable porphyrin compounds as HTMs. For example, Chen Hong's team designed and synthesized two organic compounds coded as and Y4 (Fig. 9f). By comparing their performance as HTMs of PSCs, it was concluded that the ligand coupling induced by planar porphyrin nuclei and intermolecular π–π stacking led to higher hole mobility and PSC efficiency in the obtained molecular materials. This highlights the central importance of considering molecular structural geometry, the potential to exploit intermolecular interactions, and possible molecular fillers when designing novel charge transport materials for future applications.59 Hwan's team designed and synthesized three porphyrin derivatives with triarylamine donor units, coded as SGT-061, SGT-062, and SGT-063 (Fig. 9i), and compared their performance as HTMs in battery devices. It is concluded that SGT-061 has a smaller volume of the donor so that the π–π stacking degree between molecules is large. This gives SGT-061 higher pore mobility than SGT-062 and SGT-063, suggesting that smaller electron donor units are the preferred peripheral group for the development of porphyrin-based HTM for efficient perovskite solar cells.55 The above porphyrin compounds are all single or in the form of dopants as HTM of PSCs. It is worth mentioning that in 2022, the Faezeh team reported for the first time that ZnPor/rGO/CuO bio-nano composite was prepared by self-assembly of zinc porphyrin (Fig. 9h), graphene oxide, and nano-copper oxide through π–π stacking and hydrogen bonding.61 This novel green synthesis method has the advantage of low cost.
| HTMs | Code | HOMO (eV) | LUMO (eV) | PCE (%) | VOC (V) | JSC (mA cm−2) | FF | Reference/year |
|---|---|---|---|---|---|---|---|---|
| Spiro-OMeTAD | a | −5.22 | −2.28 | Ref. 50/2016 | ||||
| MAPbI3 | −5.43 | −3.93 | Ref. 50/2016 | |||||
| ZnP | b | −5.13 | −3.23 | 17.78 | 1.10 | 22.68 | 0.71 | Ref. 54/2017 |
| CuP | −5.37 | −3.40 | 15.36 | 1.07 | 21.61 | 0.66 | ||
| CoP | b | −5.3 | — | 16.51 | 1.03 | 21.13 | 0.76 | Ref. 57/2019 |
| CuPro | c | −5.12 | — | 18.21 | 1.10 | 22.57 | 0.73 | Ref. 58/2020 |
| Co(II)P/Co(III)P | d | −5.30 | −3.30 | 19.61 | 1.12 | 23.61 | 0.745 | Ref. 53/2018 |
| e | −5.28 | −3.27 | ||||||
| Y3 | f | −5.38 | −2.94 | 0.01 | 0.87 | 0.06 | 0.16 | Ref. 59/2019 |
| Y4 | −5.28 | −3.23 | 16.05 | 1.01 | 20.13 | 0.79 | ||
| HPPHT | g | −5.24 | — | 9.85 | 0.95 | 15.8 | 0.65 | Ref. 56/2020 |
| ZPPHT | −5.16 | — | 11.0 | 0.98 | 18.5 | 0.60 | ||
| CPPHT | −5.27 | — | 12.8 | 0.98 | 21.3 | 0.60 | ||
| ZnPor/rGO/CuO | h | −5.25 | −2.055 | 9.8 | 0.776 | 20.41 | 0.62 | Ref. 61/2022 |
| SGT-061 | i | −5.03 | −3.22 | 12.6 | 1.072 | 17.2 | 0.682 | Ref. 55/2019 |
| SGT-062 | −5.27 | −3.47 | 11.5 | 1.051 | 17.3 | 0.629 | ||
| SGT-063 | −5.05 | −3.28 | 9.0 | 1.012 | 14.5 | 0.606 |
Above, we discussed the influences of the volume of the introduced group, the length of the alkyl group, the coordination metal, and the degree of π–π stacking on the performance of porphyrin molecules as the hole transport materials for perovskite solar cells, which provides a reference for the modification design of porphyrin compounds into HTMs with better performance.
2.4. Summary and outlook of porphyrins applied to PSCs
In this section, it is summarized that whether porphyrins are designed as functional molecules to passivate defects or as hole transport materials, based on the stability and regulatory ability of porphyrins derivatives, their battery devices also generally show high stability to water, heat, light, and oxygen, and the PCE of PSCs using functional porphyrin compounds has been improved at a different level compared with those control groups under the same conditions.Although PSCs have a broad prospect for industrialization, they also face some challenges, such as the poor water stability of the perovskite layer and easy decomposition in humid environments. The methylamine salt decomposition temperature is low, so the battery device is not suitable for a high-temperature working environment. Spiro-OMeTAD, as a hole transport material, is expensive to prepare in a large area. Lead leakage from battery devices can bring an adverse impact on the environment. Porphyrin compounds show great promise in solving these troubles, as the successful examples we inserted in this section.
The common modification strategies of porphyrins are coordinating with metal ions or modifying the framework. We can design and synthesize some molecular fragments or functional materials that can bind to iodine ions or lead ions to couple porphyrins, and then doping the porphyrin derivatives in the perovskite film to passivate the defects in situ to regulate the quality of the perovskite film. Or synthesize hydrophobic porphyrins with strong conductivity to encapsulate the perovskite layer to improve its stability and prevent lead leakage. In addition, it is also a good choice to modify the porphyrin by introducing a conjugated electron donor to improve its π–π stacking effect as a hole transport material.
3. Porphyrins for dye-sensitized solar cells
Dye-sensitized solar cells (DSSCs) are the third generation of solar cell products by imitating the principle of photosynthesis and have the advantages of a simple preparation procedure, easy access to raw materials, long service life, unique flexible structure, and better deformation ability. It not only meets the needs of general electronic devices but also provides good energy for specific electronic devices. Therefore, dye-sensitized solar cells have basic conditions for industrial application.62–64Firstly, we take the common iodine electrolyte-based dye-sensitized solar cell as an example to describe its battery structure and operating principle (Fig. 10). Under sunlight, the dye molecule absorbs photons from the ground state into the excited state, that is to say, an electron on HOMO of the dye molecule absorbs energy into the LUMO. The HOMO level of semiconductor materials (such as TiO2) is lower than the LUMO level of the dye molecule, so the LUMO electron of the excited dye molecule will be injected into the conductor band of the semiconductor material. And gathered on the semiconductor glass (FTO), through the external circuit to the counter electrode; The oxidized dye molecules that lose electrons are reduced to the ground state by the reducing electrolyte I− and achieve regeneration in this process. And the oxidizing electrolyte I3− gets electrons at the opposite electrode, transforming into the reducing state. This cycle is then repeated.
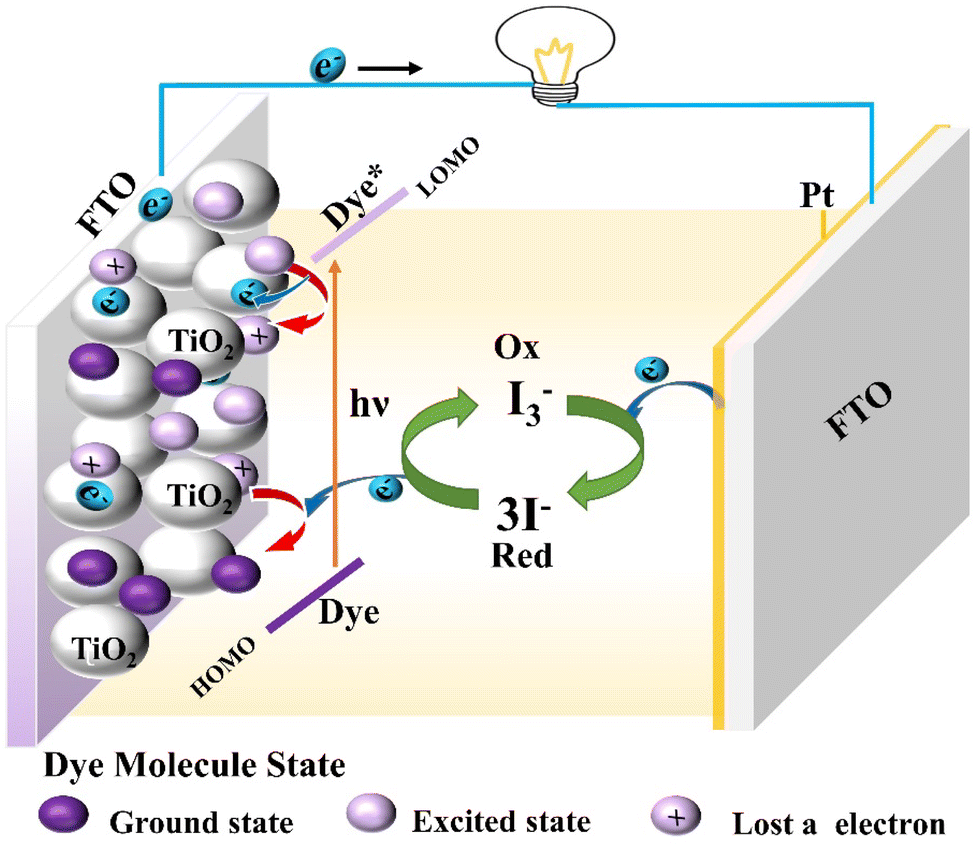 | ||
| Fig. 10 Schematic representation of the principle of iodine electrolyte-based DSSC. | ||
As an essential component of dye-sensitized solar cells, the function of dye sensitizer is to absorb visible light and contribute electrons. Efficient dye sensitizers must meet the following requirements: first, they must have a strong absorption band in the visible region, and the energy level between HOMO and LUMO should be as narrow as possible to facilitate the electron transition from the ground state to the excited state. Second, dye sensitizers need to have enough adsorption capacity, so that it is not easy to fall off from the semiconductor material with a large surface area (such as TiO2) film, and the LUMO energy level should be higher than the TiO2 conduction band energy level so that excited electrons can be smoothly injected into the semiconductor material. Third, for the sake of promoting the separation of electrons and holes and preventing recombination, the oxidation state of the dye should have a certain stability. Fourth, dye molecules should have an appropriate steric hindrance to prevent mass aggregation due to π–π stacking.
In 1991, professor Gratzel initiated the research of DSSCs by using three ruthenium complexes as dye sensitizers (Fig. 11a).65 Besides polypyridine–ruthenium complexes (Fig. 11c and d), phthalocyanine complexes (Fig. 11e), porphyrin complexes (Fig. 11f), and pure sensitized dyes, such as ZL003 and NKX-2311 (Fig. 11b and 9g), porphyrin complexes are also commonly used as organic dye sensitizers.66 Among them, porphyrins, as the main role of this paper, are characterized by a wide absorption spectrum, large absorption coefficient, easy adjustment of molecular structure, long electron injection time, strong chemical stability, etc. Compared to ruthenium-organic dyes containing expensive rare earth metals, and phthalocyanine dyes with pure synthetic and poor solubility, porphyrins are greener, environmentally friendly, cheap, and have excellent performance.
 | ||
| Fig. 11 (a)–(g) The molecular structure of common dyes for dye-sensitized solar cell. | ||
3.1. Molecular design engineering of porphyrins as sensitizers for DSSCs
The molecular design strategies of porphyrins as sensitizers for DSSCs have been reported in detail in recent years.67,68 Therefore, our work mainly selects the achievements of some teams with outstanding contributions in this field and takes the increase of PCE as the development line to intuitively present the design idea of porphyrins applied to DSSCs in the form of modifying molecular structure.Porphyrin molecules with D–π–A (donor–π–acceptor) type structure can be used as high-performance dye sensitizers. Therefore, it is a common strategy to improve the performance of DSSCs by changing the structure of electron donor, electron acceptor, and π-conjugated bridge in porphyrin dye sensitizer molecules to achieve the redshift of spectral absorption range, enhance the adsorption anchoring ability of dyes to TiO2, and improve the stability of the molecule.
In 2010, Prof. Gratzel et al. designed and synthesized a D–π–A zinc porphyrin dye molecule coded as YD-2 (Fig. 12a), which attached a diarylamino donor group to the porphyrin ring as an electron donor, introduced benzoic acid through an acetylene bridge, as electron acceptors and an anchoring group on the surface of the semiconductor film. In addition, the introduction of tert-butyl groups (t-Bu) can increase steric hindrance, thus reducing the aggregation of porphyrin molecules. When I−/I3− was used as the redox electrolysis pair, the PCE of the battery device reached an astonishing 11%.69 This work has laid the foundation for the development of high-performance D–π–A porphyrin dyes. Then Prof. Gratzel's team replaced tert-butyl with alkoxy with stronger electronic ability, and more steric hindrance and produced a new porphyrin compound (coded as YD2-o-C8, Fig. 12b). When used Co3+/Co2+as redox electrolysis pair, the PCE of the battery device based on YD2-o-C8 dye-sensitized molecule was as high as 11.9%. In addition, the PCE of the battery device co-sensitized with dye Y123 (Fig. 11a) can reach 12.3%.70 In 2013, Yeh and Gratzel et al. further ameliorated the molecular structure of YD2-O-C8 by inserting 2,1,3-benzothiadiazole (BTD), an electron acceptor, between the acetylene bridge and benzene ring, and created a new functional porphyrin compound (coded as GY50, Fig. 12c). The introduction of BTD as a π-conjugated linker enhanced the electron-pulling effect, broadens the absorption spectrum of porphyrin molecules. Thus the light absorption capacity of porphyrin is improved.71 In 2014, Gratzel continued to improve the structure of GY50 and developed a new porphyrin compound (coded as SM315, Fig. 12d). Compared to the performance of the battery based on the GY50 sensitizer, the open-circuit voltage of the battery device based on the SM315 sensitizer is large, while the short-circuit current density is small, suggesting that the introduction of long-chain alkoxy groups does not significantly increase the capacity of electron-donating ability, but rather better inhibits the recombination of the electrolyte with the photogenerated electrons, so this greatly reduces the generation of dark currents and leads to higher photoelectric conversion efficiency.72 The conclusion provides an important reference for future porphyrin dye designs. In 2016, Jong et al. introduced a larger volume of the electron-donate group on the amino base on the structure of SM315, increasing the short-circuit current and open-circuit voltage. This new compound (coded as SGT-021, Fig. 12e) is more capable of capturing light and effectively prevents charge compounding, and its batteries have an average PCE of 12.0%, which is higher than the 11.6% of the battery assembly device with SM315 as the sensitizer under the same conditions.73
 | ||
| Fig. 12 (a)–(e) Comparison of the molecular structure of porphyrin dye sensitizers and performance parameters of DSSCs devices mentioned in 3.1. | ||
Besides the above porphyrin compounds we described, there are also other D–π–A porphyrin dye molecules, all of which have been designed along the same idea and for the same purpose, so we will not elaborate on them.74–77
3.2. Co-sensitization and co-adsorption engineering for DSSCs based on porphyrins
Porphyrins inherently lack absorption in the 450–550 nm wavelength range, while some metal-free organic dyes (Fig. 13) exhibit a broad absorption band below 500 nm. Therefore, it is an appropriate intermolecular project to employ co-sensitizers to compensate for the light absorption deficiencies of porphyrin dyes and to achieve a wide range of responses to sunlight, thus enhancing the photovoltaic performance of DSSCs. In addition, electrolyte ions in the oxidation state, such as Co3+ or I3−, can cross the gaps between the dye molecules and burst the electrons on the semiconductor materials (TiO2). To prevent current carriers from compounding with electrolyte ions, we introduced a co-adsorbent (Fig. 14) to achieve that the TiO2 is completely encapsulated. Admittedly many scholars have made numerous contributions to the co-sensitization and co-adsorption of DSSCs, but to prevent length and redundancy, we focus on representative research results and design strategies for porphyrins as sensitizers. | ||
| Fig. 13 (a)–(h) Metal-free organic dyes as co-sensitizer for DSSCs mentioned in the review. | ||
 | ||
| Fig. 14 The structure of the co-adsorbent mentioned in this paper. | ||
In 2015, Xie's team designed and manufactured a new series of functional porphyrin dyes, coded as XW9, XW10, and XW11 (Fig. 15a–c), all of which used the phenothiazine framework and long-chain alkoxy groups as electron donators. Even more, long-chain alkoxy groups can further prevent the aggregation of porphyrin dye molecules. The PCE of the cell devices was measured using I−/I3− as the redox pair and it was found that especially for XW11 when the CDCA adsorbent was added, the PCE of its battery device increased from 7.8% to 9.3%, and when co-sensitized with dyes C1 and WS-5 respectively, the PCE correspondingly increased to 10.6% and 11.5% (Table 5), which is great progress in iodine-based solar cells.78 In 2019, Xie et al. modified the molecular structure of XW10 and XW11 by replacing four long alkoxy chains with two circular chains and obtained two corresponding porphyrin complexes (coded as XW40, Fig. 15d, XW41, Fig. 15e). The double-tied circular chains on the XW40 matrix weakened the charge recombination while inhibiting the dyes aggregation, resulting in an improvement of the dye open-circuit voltage and loading. Under the same conditions, the PCE of the battery device based on XW40 as dye sensitizer increased to 9.3% compared to 8.6% for the device with XW10 as dye sensitizer. When the dye Z1 (Fig. 13f) was added as a co-sensitizer, the PCE of the battery devices all increased to varying levels, especially when Z1 was co-sensitized with XW41, the PCE increased significantly from 8.16% to 9.71%. In addition, in the presence of a co-sensitizer, CDCA (Fig. 14) was added as a co-adsorbent, and it was found that the combined strategy of co-adsorption and co-sensitization further improved the PCE.79 In the same year, Xie's team obtained two new porphyrin dye compounds (coded as XW50, Fig. 15f, XW51, Fig. 15g) based on the XW40 molecular structure by expanding the volume of the electron donor group. When using I−/I3− as the redox pair, the PCE of the battery devices, which used respectively XW50 and XW51 alone as dye sensitizers and were without co-adsorbent and co-sensitizer, was significantly improved over those batteries that used XW40 as dye sensitizer. XW51 contained a larger electron donator compared to XW50, resulting in a high PCE of 11.1%, but the PCE of the battery used XW50 as a sensitizer just is 10.1%.80
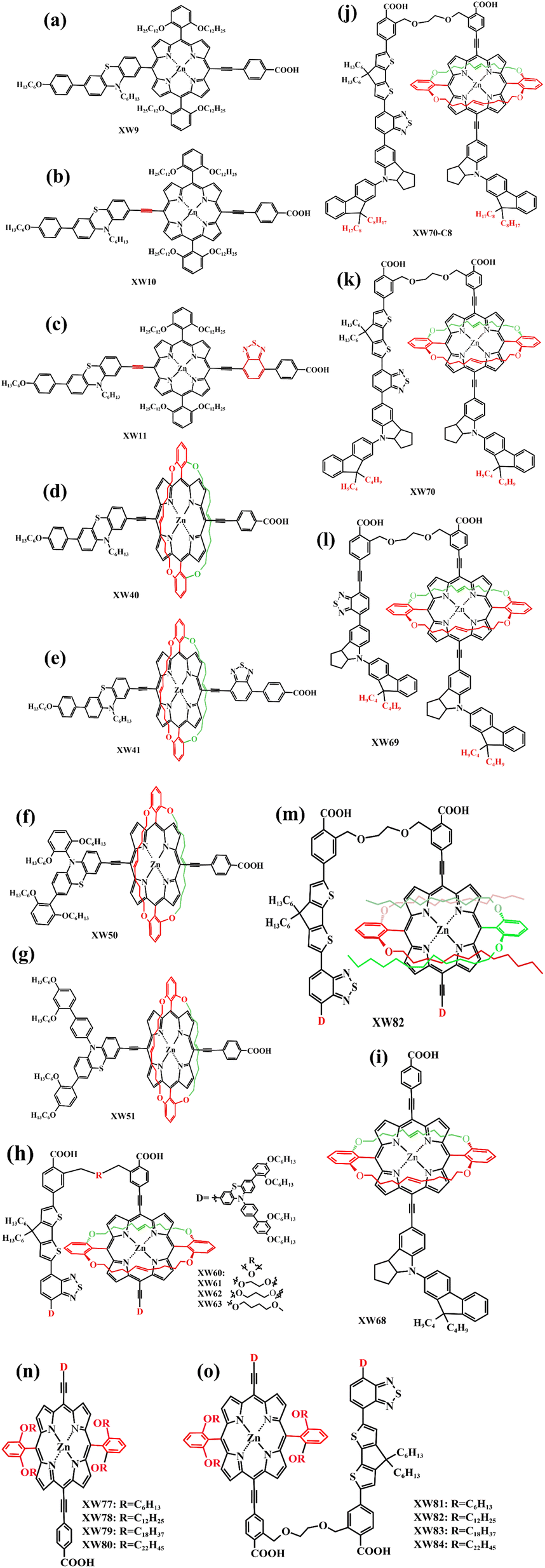 | ||
| Fig. 15 (a)–(o) The molecular structure of porphyrin dyes for DSSC mentioned in 3.2. | ||
| Dye | JSC (mA cm−2) | VOC (mV) | FF | Redox couple | PCE | Reference |
|---|---|---|---|---|---|---|
| XW9 | 16.27 | 728 | 0.698 | I−/I3− | 8.2 | Ref. 78, 2015 |
| XW10 | 17.90 | 711 | 0.684 | 8.6 | ||
| XW11 | 18.83 | 645 | 0.642 | 7.8 | ||
| XW11 + CDCA | 18.26 | 727 | 0.701 | 9.3 | ||
| XW11 + C1 | 19.52 | 746 | 0.740 | 10.6 | ||
| XW11 + WS-5 | 20.33 | 760 | 0.744 | 11.5 | ||
| XW40 | 18.67 | 730 | 0.683 | I−/I3− | 9.31 | Ref. 79, 2019 |
| XW41 | 16.77 | 695 | 0.701 | 8.16 | ||
| XW40 + Z1 | 19.36 | 742 | 0.694 | 9.97 | ||
| XW41 + Z1 | 18.32 | 728 | 0.728 | 9.71 | ||
| XW40 (CDCA) + Z1 | 19.59 | 748 | 0.719 | 10.55 | ||
| XW41 (CDCA) + Z1 | 19.63 | 726 | 0.715 | 10.19 | ||
| XW50 | 18.96 | 761 | 0.702 | I−/I3− | 10.1 | Ref. 80, 2019 |
| XW51 | 20.07 | 781 | 0.702 | 11.1 | ||
| Z2 | 17.81 | 740 | 0.741 | I−/I3− | 9.8 | Ref. 81, 2020 |
| XW51 (CDCA) + Z2 | 13.99 | 822 | 0.799 | 9.6 | ||
| XW60 | 16.77 | 715 | 0.731 | 8.8 | ||
| XW61 | 20.75 | 763 | 0.739 | 11.7 | ||
| XW62 | 20.70 | 762 | 0.732 | 11.6 | ||
| XW63 | 20.63 | 763 | 0.737 | 11.6 | ||
| XW61 (CDCA) | 21.41 | 775 | 0.747 | 12.4 | ||
| XW68 | 20.48 | 723 | 0.620 | I−/I3− | 9.2 | Ref. 82, 2021 |
| XW69 | 22.03 | 720 | 0.623 | 9.9 | ||
| XW70 | 22.19 | 730 | 0.625 | 10.1 | ||
| XW70-C8 | 22.00 | 733 | 0.649 | 10.5 | ||
| XW70-C8 (CDCA) | 22.25 | 750 | 0.665 | 11.1 | ||
| XW77 | 19.26 | 755 | 0.727 | I−/I3− | 10.5 | Ref. 83, 2022 |
| XW78 | 19.00 | 744 | 0.725 | 10.3 | ||
| XW79 | 18.58 | 735 | 0.722 | 9.9 | ||
| XW80 | 17.93 | 725 | 0.716 | 9.3 | ||
| XW81 | 20.58 | 745 | 0.714 | 10.9 | ||
| XW82 | 21.44 | 773 | 0.724 | 12.0 | ||
| XW83 | 21.34 | 784 | 0.728 | 12.2 | ||
| XW84 | 20.47 | 762 | 0.713 | 11.1 |
Although the performance of DSSC is significantly improved by adopting the co-sensitization strategy, the adsorption of the two dyes on the TiO2 thin film competes, and it is difficult to control the ratio and distribution of the two dyes. Therefore, it is hard to achieve the best PCE by complementary light absorption.
In 2020, Xie et al. reported a novel strategy for combining complementary co-sensitive dyes by covalent bonding with porphyrin dye molecules to obtain “concerted companion dyes” (CC dyes). A series of CC dyes (coded as XW60-XW63, Fig. 15h) were obtained by linking Z2 (Fig. 13c) as a co-sensitizer to XW51 through flexible chains with different lengths. The performance of the battery devices was measured using I−/I3− as the redox electrolyte pair and adding CDCA as the co-adsorbent, and it was surprisingly found that the battery devices used XW61 CC dye as sensitizer achieved a high PCE of 12.4%. By comparing the parameters of the battery based on XW60-XW63 as a dye sensitizer, we can conclude that the suitability length of the bridge chain linking Z2 and XW51 brought a positive effect on the battery performance.81 It is of great interest and promises for exploiting CC dyes such as XW61. In 2021, Xie et al. used a stronger fluorenyl indoline-based electron donor to construct the porphyrin dye (coded as XW68, Fig. 15i) and the co-sensitizer (coded as Y1, Y2, Fig. 13b and d), then obtained a series of CC dyes (coded as XW69, XW70, XW70-C8, Fig. 15l, 13k and j) by linking the porphyrin and co-sensitizer. CC dyes benefited from the excellent electron-donating capability of the fluorenyl indoline moiety on its molecule, resulting in extended panchromatic absorption and excellent light-trapping ability. Comparing the electrochemical parameters of cell devices (Table 5) based on different sensitized dyes indicates that the introduction of octyl groups on the fluorenyl indoline unit can further reduce dye aggregation and suppress charge recombination.82
Admittedly, CC dyes can only afford moderate open circuit voltages (VOC) and are generally relatively weak against aggregation. To address this issue, in 2022, Xie and his team wrapped porphyrin cores in four different long alkoxy chains, and obtained another four porphyrin dyes (XW77-XW80, Fig. 15m). Corresponding CC dyes XW81–XW84 (Fig. 15n) were produced via using the chemical bond respectively link them and co-sensitizer Z2 (Fig. 13c). XW83 with the alkoxy chains of eighteen carbons had the largest VOC of 784 mV. As a result, when using CDCA as a co-sorbent for anti-aggregation and adopting XW83 as dye sensitizer, the battery device afforded the highest PCE of 12.2%, setting a new record for the iodine electrolyte-based solar cells sensitized with a single dye.83 This suggests that using long chains to bind porphyrins to improve molecule anti-aggregation is a reasonable and feasible approach.
In addition to co-sensitization, co-adsorption, and designing CC dyes, other strategies for achieving the maximum efficiency of dye-sensitized solar cells include tandem cells73 and using ideal electrolytes.
3.3. Summary and outlook of porphyrins applied to DSSCs
To briefly summarize, porphyrins are used as sensitizers in DSSCs mainly through: firstly, molecular engineering to design the ideal dyes with excellent performance for the DSSCs. The common strategies are to enhance the light capture ability of porphyrin compounds and promote charge transfer by altering the electron donor and electron acceptor (anchoring group), and to inhibit the porphyrin molecular own aggregation by introducing large site resistance groups. Secondly, intermolecular engineering, including the addition of co-adsorbents to the electrolyte to prevent the inopportune combination of the carriers and electrolyte, or employing co-insensitive dyes to compensate for the light absorption defects of the porphyrins. Third, designing and manufacturing CC dyes through the chemical bond to combine two dye molecules that have complementary effects on sunlight absorption.As we know, the PCE of dye-sensitized solar cells is much lower than other solar cells. For dye sensitizers, if they are easy to desorb from the anode material such as TiO2, or are not tightly bound, it will prevent the electrons from being injected smoothly, or cause the extinction of electrons by the electrolyte ions. In addition, if the stability of the dye is poor, it will easily decompose, resulting in a short battery life. More importantly, if the dye photo-responsive ability is not prominent, it will directly limit the photogenerated charge carrier yield.
The porphyrin molecule structure is easy to modify for improving its photochemical properties and is relatively mature in terms of synthesis and design, so specific porphyrin dyes are promising for DSSCs. For example, to enhance the adsorption ability of porphyrin to TiO2, we can introduce some molecular fragments containing carboxyl groups to the porphyrin ring. In order to improve the photo-response range of porphyrin dyes, we can co-sensitize them with other photo-complementary dyes by coupling or combination. Usually, we also introduce large hindrance molecular fragments to inhibit porphyrin self-assembly. Besides, chlorophyll, as a natural porphyrin derivative, can be degraded and modified to make it suitable for a sensitizer.
4. Porphyrins for lithium batteries
“When an iron block dropped on the batteries, the metal–lithium battery ignited violently and a roaring fire spread across all the battery, while the lithium-ion battery did not trigger a spark and showed no signs of burning”, at this moment, the lithium-ion battery was truly born.84 The successful passing of the lithium-ion battery safety test by Professor Akira Yoshino in 1986 provided a security guarantee for its commercialization. Over the past three decades, lithium-ion batteries have been widely used as power sources for computers, mobile phones, electric vehicles, and so on because of their advantages such as high working voltage, high energy storage density, long cycle life, good safety, and environmental protection. They also make it possible to store renewable energy sources such as wind and solar energy, so it can be said that they play a pivotal role in the sector of power supply and energy storage. The 2019 Nobel Prize in Chemistry was awarded to Professors Akira Yoshino, John Goodenough, and Stanley Whittingham for their contributions to lithium-ion battery research. As lithium-ion batteries are continuously commercialized, higher demands are being placed on their performance. Therefore, better range, long cycling life, and greater safety have become a top priority in the development of lithium-ion batteries.A lithium-ion battery consists of four core components: cathode, anode, electrolyte, and separator. We use carbon material as the anode and a specific porphyrin as the cathode material to explain the operating principle of lithium-ion batteries. As a rechargeable battery, during the discharge process, the negative material loses electrons, meanwhile, lithium ions are released and migrate to the positive electrode, where they are embedded. Finally, electrons from the external circuit flow through the load back to the positive terminal, converting chemical energy into electrical energy. When charging, lithium ions are forced by an external voltage to unembed from the cathode material and migrate to the anode to complete lithium storage. At the same time, the electrons provided by the external circuit arrive at the negative electrode, ensuring the anode material is electrically neutral. In this process, electrical energy is converted into chemical energy and stored (Fig. 16). Undoubtedly, the development of functional electrode materials to achieve efficient and reversible storage of lithium ions is a vital step to improve the capacity, cycle retention, and charge–discharge rates of organic lithium batteries.
 | ||
| Fig. 16 Schematic representation of the principle of organic lithium-ion battery based on the specific porphyrin. | ||
4.1. Functionalized small molecule porphyrins as an electrode for organic lithium battery
The aromatic porphyrins with 18π electrons and their complexes can be oxidized to the oxidation state of 16π electrons or reduced to the reduced state of 20π electrons, presenting a bipolar nature (Fig. 17). Therefore, it can act as the cathode or anode in the electrochemical energy storage system. The HOMO–LUMO gap of small molecule porphyrins meets the condition of fast redox kinetics and can be enhanced by the introduction of functional groups and coordination to metals, thus forming functionalized porphyrin molecules to improve their electrode performance. | ||
| Fig. 17 Diagram of porphyrin redox. | ||
In 2017, Gao et al. synthesized a small molecule of copper porphyrin complex (coded as CuDEPP, Fig. 18a) and found the rapid redox activity of the porphyrin molecule based on its relevant mesomeric structures. CuDEPP electrode exhibited excellent capacity retention and fast charge–discharge rates owing to its self-conditioned capability. When used as a cathode material paired with anode Li in the LiPF6 electrolyte or as a negative electrode paired with graphite in PP14+/TFSI− ionic liquid electrolyte.85 Later the overall electrochemical reaction mechanism of this organic full rechargeable battery was studied by Gao et al. via Operando Raman Spectroscopy and complementary Density Functional Theory (DFT).86 The effect of different anion-based electrolytes on the charge storage performance of CuDEPP as cathode material in an organic lithium battery was also explored by Bo Ren et al.87 The conclusion of the above reports reveals that CuDEPP can be used as both a positive or negative electrode, and it presents excellent stability in cycling and high energy density, which lays a foundation for the molecular modification and improvement of the porphyrin with functional acetylene group to be as the electrode material of organic lithium batteries (OLiBs).
 | ||
| Fig. 18 (a–f) The structure of small molecule porphyrins; (g–h) the mesomeric transformations of CuDEPT. | ||
Based on the CuDEPP molecular structure, Gao et al. synthesized CuTEPP (Fig. 18d) by replacing two phenyl groups with two ethynyl groups. And compared the electrochemical performance of CuTEPP and ZnTEPP as cathodes in OLIBs (Table 6). As a bipolar molecule, M-TEPP has an excellent storage capacity for Li+ as well as for Na+ and K+, and even larger PF6−, TFSI− anions.88 To increase the redox-active site of the porphyrin molecule, Gao's team replaced the two phenyl groups in the CuDEPP structure with two thiophene groups while retaining the acetylene group, obtained a new porphyrin molecule (coded as CuDEPT, Fig. 18b) with a bipolar thiophene functionalization. A porphyrin compound (coded as CuTTP, Fig. 18f) with four thiophene groups also has been synthesized. The electrochemical properties of CuDEPT and CuTTP as a novel cathode material for organic lithium batteries were studied (Table 6), as well as the relationship between the reaction mechanism and structural changes of the CuDEPT molecule during the charging and discharging process (Fig. 18g and h). As a result, CuTTP without acetylene groups as an electrode dissolved in the electrolyte and lead to poor cycle stability, whereas porphyrins with acetylene and thiophene groups is no significant dissolution, so batteries based on CuDEPT as an electrode exhibited remarkable cycling stability up to 9000 cycles.89 Similarly, furan and thiophene have similar aromatic properties and good electrophilicity; when the thiophene group in CuDETP is replaced by a furan group, the newly obtained porphyrin compound (code as CuDEOP, Fig. 18e), can still be used as a new cathode material for rechargeable organic lithium-based batteries. Both CuTEPP and CuDETP can provide high specific capacity and admirable cycling stability as anode materials for organic lithium batteries.90
| Material | Role | Specific capacity | Capacity retention | Reference |
|---|---|---|---|---|
| CuDEPP | Cathode | 132 mA h g−1 | 85% | Ref. 85, 2017 |
| 4 A g−1 | 4 A g−1 | |||
| Cycle: 2000 | ||||
| CuDEPP | Cathode | PF6-PC: 176 mA h g−1 | 85.84% | Ref. 87, 2022 |
| PF6-EPD: 165 mA h g−1 | 75.33% | |||
| BOB-PC: 142 mA h g−1 | 78.2% | |||
| BF4-PC: 117 mA h g−1 | 56.43% | |||
| 3M-TFSI-PC: 115 mA h g−1 | 98% | |||
| 1 A g−1, cycle: 40th | 1 A g−1 | |||
| Cycle: 1000 | ||||
| CuTEPP | Cathode | 118 mA h g−1 | 73.1% | Ref. 88, 2021 |
| 129 mA h g−1 | 83.6% | |||
| ZnTEPP | 1 A g−1 | 1 A g−1 | ||
| Cycle: 1000 | ||||
| CuDETP | Cathode | 300 mA h g−1 | 137 mA h g−1 | Ref. 89, 2022 |
| 150 mA h g−1 | — | |||
| CuTTP | 0.2 A g−1 | 1 A g−1 | ||
| Cycle: 9000 | ||||
| CuDEOP | Cathode | 223 mA h g−1 | 76% | Ref. 90, 2022 |
| 0.2 A g−1 | 5 A g−1 | |||
| Cycle: 6000 | ||||
| TCPP | Anode | 548.4 mA h g−1 | 89% | Ref. 91, 2019 |
| 8 A g−1 | 6 A g−1 | |||
| Cycle: 2500 |
Organic lithium batteries require electrode materials with good conductivity and high insolubility in the electrolyte. To this end, Xu et al. introduced carboxyl groups on the benzene ring of tetraphenyl-porphyrin to increase the molecular polarity, which combined with the large π-conjugation system of the porphyrin reduced the solubility of this porphyrin compound (coded as TCPP, Fig. 19) in conventional lithium-ion battery electrolytes (LiDFOB/PC). When used TCPP as an anode for lithium-ion batteries, the anode material was found to exhibit excellent rate capability and superior cycling performance (Table 6).91
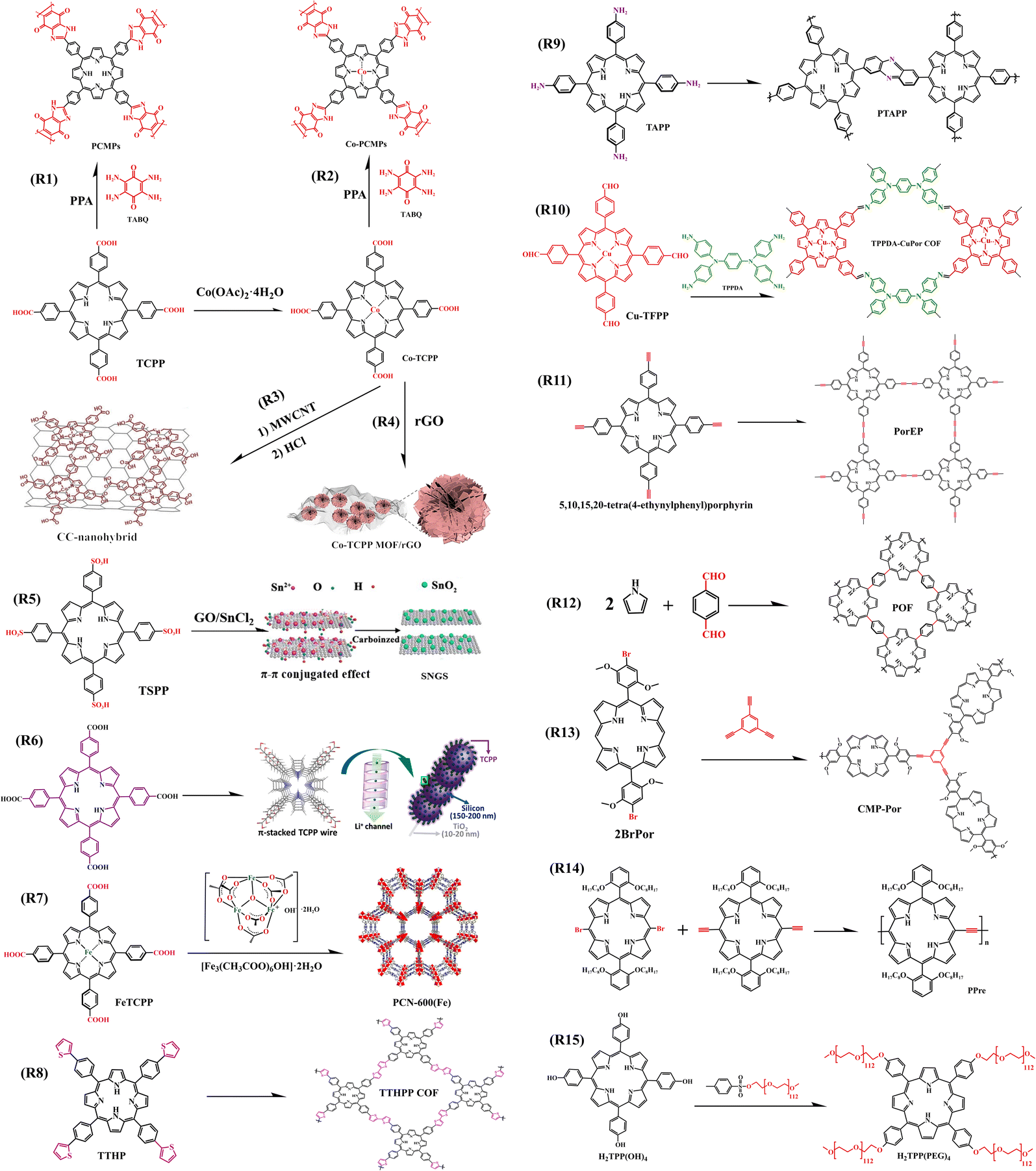 | ||
| Fig. 19 Synthesis route of COF, MOF, and CMP based on porphyrin unit. CC-nanohybrid,92 Copyright 2019, Wiley. Co-TCPP MOF/rGO,93 Copyright 2021, Elsevier. SNGS,96 Copyright 2018, Elsevier. TCPP/Si/TBNW,97 Copyright 2020, American Chemical Society. PCN-600 (Fe),99 Copyright 2018, The Royal Society of Chemistry. | ||
As we presented above, some functionalized small-molecule porphyrins can be used as electrode materials for lithium-ion batteries. In addition, micro-polymers and organic framework materials based on porphyrins are also promising electrode materials.
4.2. CMPs, MOFs, and COFs based on porphyrin unit in organic lithium battery
It has been reported that CoTCPP (Fig. 19) exhibits outstanding performance as an anode in organic lithium batteries,91 Lee combined it with a new multi-walled carbon nanotube (MWCNT) through strong non-covalent interactions (π–π stacking and van der Waals forces) to form a CC-nanohybrid as an anode in LIBs (Fig. 19, R3). The unique chemical structure of the CC-nanohybrids can promote electron transfer, thus significantly improving the redox kinetics of the electrode compared to that of a single CoTCPP electrode. Furthermore, the nanomaterial anode sheet presents excellent electrochemical properties and mechanical flexibility, far beyond what can be achieved with conventional LIB anode materials.92Similarly, Han et al. synthesized Co-TCPP MOF/rGO composites by adding rGO to Co-TCPP MOF (Fig. 19, R4). The introduction of rGO not only effectively reduced the accumulation of MOF but also formed a continuous conductive network consisting of graphene and MOF entanglement, which facilitated fast electron transport. As a result, the ultra-thin nanosheet structure provided cavities for the diffusion and storage of lithium ions, reducing the diffusion distance to the internal active site and exposing more active sites, thus enhancing the electrochemical performance of the MOF composite. When used as anode materials for lithium-ion batteries, the composite electrode exhibits excellent lithium storage performance, and these results suggest that porphyrin MOFs-based composites are worthy candidates to be considered as anode.93
The cellular structure of conjugated microporous polymers (CMPs), besides increasing the specific surface area, it can allow the material to have more redox active sites and faster ion conduction rates. Yuan's team combined CoTCPP and TCPP with the redox-active unit TABQ respectively to form new porphyrin-based Co-PCMP and PCMP with a large conjugated backbone (Fig. 19, R2, R1). Both the porphyrin unit and TABQ unit can contribute to capacity, so the microporous complexes formed with these two units can possess a high capacity and excellent multiplication capabilities when used as anode materials for lithium-ion batteries.94 Similarly, Yang et al. designed and synthesized a porphyrin-conjugated microporous polymer (coded as CMP-Por, Fig. 19, R13), when used as the anode material for lithium-ion batteries, it exhibited excellent performance for lithium-ion storage.95
Wang et al. anchored the porphyrin (TSPP) to a graphene oxide surface by π–π stacking and van der Waals force, and sulfur/nitrogen co-doped graphene complex (SNGS) formed by doping SnO2. The heteroatom-containing organic composite can be used as the anode material for lithium-ion batteries or sodium-ion batteries (Fig. 19, R5).96
Silicon nanowire (SiNW) electrodes have a capacity of up to 4200 mA h g−1, which is 12 times as much as conventional graphite electrodes, and are commonly used as an anode material for lithium-ion micro-batteries. However, silicon suffers from problems such as volume expansion, which makes it difficult to embed lithium ions. For this reason, Yin et al. used TCPP via covering SiNW to prepare TCPP/Si/TBNW chain nanowires as an anode material for Li-ion battery (Fig. 19, R6) and found it was able to deliver a capacity of 4752.2 mA h g−1, which was 40% higher than SiNW anode without TCPP coating. This is attributed to TCPP as an additive that not only relieved the volume expansion of silicon but also enhanced the transmission ability of the electrode material to lithium ions.97
PCN-600 (Fe) has been reported to be a mesoporous MOF material with strong chemical stability and extraordinary porosity.98 Based on the characteristics of PCN-600 (Fe), Li et al. fabricated a metal–organic framework (MOF) of iron-porphyrins, when used as an anode material for LIB (Fig. 19, R7) indicated that the introduction of Fe clusters enriched the electrode active sites and increased the capacity, while TCPP, as a carrier for electron and lithium ion transport, enhanced the rate capacity of the PCN-600 anode.99
In addition to porphyrin-based MOFs that can be used as electrode materials for organic lithium batteries, two-dimensional covalent organic frameworks (2D COFs) with large overlapping of π-orbitals, which are well suited for charge transport, are also promising electrode materials. Yang et al. synthesized a 2D COF(coded as TTHPP, Fig. 19, R8). When using TTHPP film as an anode for LIBs found that it had lithium-ion adsorption and diffusion to such an extent that the battery device exhibited a high specific capacity as well as excellent rate performance and long cycle stability. This provided a successful example of porphyrin-based 2D COFs as novel anode materials in Li-ion batteries and capacitors.100 It's also been reported the use of nanocube materials (TAPP-NC) formed by the polymerization of TAPP as electrodes for LIBs (Fig. 19, R9), which has excellent rate performance and long cycle stability (Table 7). High specific capacities of nearly 650 mAhg−1 can be achieved at a current density of 100 mA g−1.101
| Active Material | Role | Specific reversible capacity | Capacity retention | Ref., year |
|---|---|---|---|---|
| Co-TCPP | Anode | 144 mA h g−1 | — | Ref. 92, 2019 |
| CC-nanohybrid | 377 mA h g−1 | 226 mA h g−1 | ||
| 20C | 5.0C, cycle: 1500 | |||
| Co-TCPP MOF | Anode | 854 mA h g−1 | 152 mA h g−1 | Ref. 93, 2021 |
| Co-TCPP MOF/rGO | 1050 mA h g−1 | 616.6 mA h g−1 | ||
| 0.1 A g−1 | 1 A g−1, cycle: 300 | |||
| PCMP | Anode | 276.4 mA h g−1 | 445 mA h g−1 | Ref. 94, 2022 |
| 331.3 mA h g−1 | 545 mA h g−1 | |||
| Co-PCMPs | 1.0 A g−1 | 0.05 A g−1 | ||
| Cycle: 50 | ||||
| CMP-Por | Anode | 1200 mA h g−1 | 546 m Ah g−1 | Ref. 95, 2022 |
| 1.0 A g−1 | 3.0 A g−1 | |||
| Cycle: 1000th | Cycle: 5000 | |||
| SNGS | Anode | 711.1 mA h g−1 | 80% | Ref. 96, 2018 |
| 0.2 A g−1 | 0.2 A g−1 | |||
| Cycle: 200 | ||||
| Si/TBNW | Anode | 3396.2 mA h g−1 | 37.7% | Ref. 97, 2020 |
| 4752.2 mA h g−1 | 45% | |||
| TCPP/Si/TBNW | 0.1C | 0.1C | ||
| 1C = 0.15 mA cm−2 | Cycle: 70 | |||
| PCN-600 (Fe) | Anode | 1300 mA h g−1 | 1000 mA h g−1 | Ref. 99, 2018 |
| 0.4 A g−1 | 0.4 A g−1 | |||
| Cycle: 300 | ||||
| TTHPP | Anode | 666 mA h g−1 | 61.6% | Ref. 100, 2016 |
| 0.2 A g−1 | 1.0 A g−1 | |||
| Cycle: 200 | ||||
| PTAPP-NC | Anode | ≈650 m Ah g−1 | 75% | Ref. 101, 2020 |
| 0.1 A g−1 | 2.0 A g−1 | |||
| Cycle: 2000 | ||||
| TPPDA-CuPor-COF | Cathode | 142 mA h g−1 | 102 mA h g−1 | Ref. 102, 2022 |
| 60 mA g−1 | 1.0 A g−1 | |||
| Cycle: 3000 | ||||
| PPre | Cathode | 79 mA h g−1 | 75.4% | Ref. 105, 2020 |
| PPre9 | 113 mA h g−1 | 80.5% | ||
| PPre15 | 122 mA h g−1 | 98% | ||
| PPre25 | 110 mA h g−1 | 90.1% | ||
| 200 mA g−1 | 0.5 A g−1 | |||
| Cycle: 10th | Cycle: 500 |
Lei et al. prepared a bipolar two-dimensional covalent organic framework material (TPPDA-CuPor-COF) by condensing TPPDA with Cu-TFP (Fig. 19, R10). The 2D COF has a porous structure that can provide a pathway for ion/electron diffusion. Because TPPDA is capable of p-type doping reactions and the Cu-TFPP segment can act as a bipolar active center, TPPDA-CuPor-COF has superior rate performance and decent cycling stability when used as the positive electrode in organic lithium batteries.102
Besides COF materials based on porphyrins as electrodes for lithium-ion batteries, Li et al. also reported that two-dimensional porous conjugated porphyrin polymers (PorEP) synthesized via situ coupling reaction on the copper foil can be employed as the lithiophilic host (Fig. 19, R11). When using PorEP@Cu as the electrode for lithium batteries, it turned out that the intervention of porphyrin COF can inhibit the formation of lithium dendrites and facilitate the uniform lithium deposition on the electrode, and this discovery promotes the development of lithium metal anode.103 Similarly, Zhang et al. designed and fabricated a framework porphyrin (POF) material by the one-pot method (Fig. 19, R12). G@POF composite material, formed through using graphene as a substrate to hybridize with POF, can extensively attract lithium ions to its surface, so that the dendrite growth of Li deposition is uniform, resulting in subsequent smooth lithium plating and dendrite-free lithium metal anode.104
The small molecule porphyrin containing acetylene group as organic lithium battery electrode materials presented good electrode performance in this review reported previously. Different from low molecular alkynyl porphyrin, Gao et al. designed and synthesized a highly conjugated porphyrin polymer (coded as PPre, Fig. 19, R14) with acetylene group as a new positive electrode material of lithium organic battery, and measured the influence of different polymerization degrees on the electrochemical performance (Table 7).105
As reported in the literature we reviewed, the performance of porphyrins as organic lithium battery electrodes is obvious to all. In addition, Zhang's team applied a polyether chain to modify porphyrins to composite solid polymer electrolytes (CSPE) (Fig. 19, R15). The introduction of modified porphyrins into polymer electrolytes of all-solid-state lithium-ion batteries (ASSLIB) has improved the electrochemical properties of the electrolytes, and this work has broadened the application range of porphyrins.106
4.3. Porphyrins for other lithium batteries
The theoretical density of Li–CO2 batteries is as high as 1876 W h kg−1, but the slow kinetics of carbon dioxide reduction reaction and carbon dioxide precipitation reaction leads to a series of problems such as large overpotential of the battery and poor cycling performance. Therefore, exploiting the cathode catalyst to improve the electrochemical reaction kinetics, and then improve the overall performance of the battery is very sensible. Recently, it has been reported that H2TCPP and Cu-TCPP (Fig. 20c) were used as cathode catalysts of Li–CO2 batteries, which had a strong ability to capture CO2. Moreover, the introduction of Cu2+ through coordination can synergistically catalyze the reduction reaction of CO2.107 Besides small molecule porphyrin, Zhang et al. fabricated a porphyrin-based covalent organic framework material (TTCOF-Mn, Fig. 20, R2), and TTCOF-Mn presented high efficiency as the cathode catalyst for Li–CO2 battery owing to its microporous channels and porphyrin units with active sites.108 | ||
| Fig. 20 (a)–(c) The molecular structure of porphyrins and the synthetic route of porphyrin polymers mentioned in 4.3. (R1)–(R3) The route diagram for R3 comes directly from the ref. 114, Copyright 2022, Willey. | ||
Lithium–sulfur (Li–S) battery is a kind of lithium battery with sulfur-based materials as the positive electrode and lithium metal as the negative electrode. Li–S batteries have an ultrahigh theoretical energy density (2600 W h kg−1), but the multiphase sulfur redox reactions with sophisticated homogeneous and heterogeneous electrochemical processes are sluggish in kinetics, resulting in a large degree of actual capacity degradation. Nakamura et al. introduced the reaction principle of Li–S batteries and reviewed the challenges these batteries faced in commercializing.109 At present, the most intensively studied carbon-supported single-atom catalysts in Li–S batteries are porphyrins with metal–nitrogen structures.110 Quan et al. used the H2TCPP molecule unit as an active site and grafted it onto conductive and flexible polypyrrole linkers via an amidation reaction to form a semi-immobilized electrocatalyst (G@ppy-Por) for accelerating the reaction kinetics of sulfur and regulating the multiple opposite reduction reactions (Fig. 20, R1). Li–S batteries using the semi-immobilized catalytic strategy exhibited excellent electrochemical performance and achieved a practical energy density of 343 W h kg−1.111 Zhang et al. also used G@POF as electrocatalysts for Li–S batteries, and the battery device delivered a capacity of up to 1611 mA h g−1 and outstanding cycling stability (Table 8).112
| Material | Role/battery | Specific capacity | Capacity retention | Ref., year |
|---|---|---|---|---|
| H2TPP | Cathode | 101.2 mA h g−1 | 98% | Ref. 133, 2021 |
| H2TCPP | AIBs | 21.3 mA h g−1 | — | |
| 0.1 A g−1 | 0.2 A g−1, cycle: 5000 | |||
| H2TCPP | Cathode catalyst | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 979 mA h g−1 979 mA h g−1 |
— | Ref. 107, 2022 |
| Cu-TCPP | Li–CO2 | 20393 mA h g−1 |
— | |
| 0.1 A g−1 | ||||
| TTCOF-Mn | Cathode catalyst | 13018 mA h g−1 |
1000 mA h g−1 | Ref. 108, 2021 |
| Li–CO2 | 0.1 A g−1 | 0.3 A g−1 | ||
| Cycle: 180 | ||||
| G@ppy-Por | Electrocatalyst | 649 mA h g−1 | 904 mA h g−1 | Ref. 111, 2021 |
| Li–S | 4.0C | 2.0C | ||
| 1C = 1672 mA g−1 | Cycle: 50 | |||
| G@POF | Electrocatalyst | 1611 mA h g−1 | 936 mA h g−1 | Ref. 112, 2019 |
| Li–S | 0.1C | 0.5C | ||
| 1C = 1672 mA g−1 | Cycle: 400 | |||
| CMT@MXene | Electrocatalyst | 6850 mA h g−1 | 94.1% | Ref. 114, 2022 |
| Li–O2 | 0.2 A g−1 | 0.2 A g−1, cycle: 312 | ||
| CuDEPP | Cathode | 104 mA h g−1 | 87% | Ref. 137, 2020 |
| KIBs | 1.0 A g−1 | 0.3 A g−1 | ||
| Cycle: 300 | ||||
| SNGS | Anode | 286.6 mA h g−1 | 143 mA h g−1 | Ref. 96, 2018 |
| SIBs | 0.1 A g−1 | 0.1 A g−1 | ||
| Cycle: 300 | ||||
| Sb2S3/CZM | Anode | 550 mA h g−1 | 88.9% | Ref. 136, 2022 |
| SIBs | 0.1 A g−1 | 3.0 A g−1 | ||
| Cycle: 1000 |
2D transition metal carbides and/or nitrides (MXenes), have excellent electrocatalytic activity. Liu et al. profoundly reviewed the potential of MXene-based heterostructures for energy conversion and storage.113 However, the formation of its metal oxides brings about performance degradation. Yury et al. employed the bimetallic cobalt-manganese organic framework (CMT) formed by coordination of H2TCPP with Co2+ and Mn2+ grown directly on MXene sheet via solvothermal treatment to form CMT@MXene material (Fig. 20, R3), which can be used as a bifunctional electrocatalyst on the positive electrode of Li–O2 batteries, and the ligand chemistry using TCPP organic junction can enhance the antioxidant capacity of MXene.114 Additionally, there are some reports on the application of porphyrins as electrocatalysts to Li–SOCl2 batteries.115–117
4.4. Summary and outlook of porphyrins applied to lithium batteries
As for the application of porphyrin in lithium batteries, we can summarize as follows: functional porphyrin derivatives are usually obtained through molecular modification strategies, or by designing and synthesizing porphyrin-based MOFs, COFs, and CMPs. These materials with appropriate performance can be used as positive or negative electrode materials for lithium batteries, or by regulating the electrochemical performance of electrodes or electrolytes to improve the storage and transportation of lithium ions.Although lithium-ion batteries have been commercialized due to their excellent performance, the recovery of rare metals such as cobalt, nickel, and lithium from the cathode materials is costly.118 Compared with inorganic electrode materials, organic electrode materials are rich in resources, easy to modify their structures, and can also form functional lithium storage materials such as MOFs and COFs.119–121 However, organic electrode materials also have serious problems such as dissolution in electrolytes.122
As we have reviewed, small-molecule porphyrins containing carboxyl or alkynyl groups are less soluble in organic electrolytes and can enhance conductivity by coordinating with transition metal ions such as copper ions or increase lithium storage active sites by linking aromatic groups such as thiophene. Therefore, the newly developed small molecule porphyrin compounds as electrode materials based on theoretical computational chemistry need to consider their solubility, conductivity, and whether they can form conjugated structures with more active sites after electron gain and loss.
From the references we have cited in this section, porphyrin-based framework materials and their composites with MWCNT or rGO present decent prospects as electrode materials for LIBs, SIBs, and KIBs or as electrocatalysts for Li–S batteries, Li–O2 batteries, Li–CO2 batteries, and Li–SOCl2 batteries due to their large specific surface area, charge channels and more active sites. Many reviews have summarized the classification of organic electrode materials as well as the redox mechanism and discussed the challenges and prospects for their development.123–132 Recently, Li et al. further reviewed the influences of different electrolytes on organic electrode materials and their applicability to different organic batteries.122 These reports have laid a theoretical foundation for the rational design of porphyrin-based organic electrode materials.
5. Porphyrins for other energy storage
In addition to H2TCPP (Fig. 20b) being used as the positive electrode of the lithium-ion battery, it was also reported that Jiao et al. employed H2TCPP and tetraphenyl porphyrins (coded as H2TPP, Fig. 20a) as the cathode electrode materials for organic aluminum ion battery (AIBs). It was found that the introduction of the carboxyl group weakened the complexing ability of porphyrin and Al3+, resulting in a higher charge–discharge capacity of H2TPP than H2TCPP.133 Another research suggested that H2TPP deposited on the surface of graphite electrodes can perturb and inhibit its blister formation induced by anion intercalation in sulfuric acid electrolyte solution.134 Moreover, ZnO modified by the H2TPP molecule with a protective function can exhibit higher cycle stability as an anode material for rechargeable Ni–Zn batteries in an alkaline medium.135As we mentioned above in this review SNGS can be used as anode materials for lithium-ion batteries but also used for sodium-ion batteries (SIBs).96 SIBs have superior cost performance, but the Na+ radius is relatively large compared to Li+, so the construction of microporous sodium ion channels can enhance the performance of SIBs. Sun et al. fabricated zirconium-based MOF material (coded as PCN-222) by ligating TCPP as a ligand with metallic zirconium ions (Zr4+), adsorbed trivalent antimony and the n vulcanized at high temperature to obtain Sb2S3/CZM composites. When the composites were used as SIB-negative electrodes, they facilitated the cyclic storage of Na+ and exhibited good electrochemical properties (Table 8).136
Rechargeable potassium-ion batteries (KIBs) are promising alternatives to lithium-ion batteries for large-scale electrochemical energy-storage applications because of the abundance and low cost of potassium. As already mentioned in our review, Gao's team reported excellent performance of CuDEPP as a cathode material for organic lithium batteries.85 Moreover, owing to the functionalized effect of the acetylene group, the CuDEPP molecule also can be used as a cathode material for KIBs (Table 8).137
We have reviewed in the text that porphyrin derivatives can be served as electrocatalysts for Li-ion batteries, and similarly, Zhang et al. reported that a variety of porphyrin@MOF hybrids were synthesized by grafting metal–porphyrins onto MOF materials through the exchange of metal ion ligands, which could act as oxygen reduction catalysts for Zn-air batteries.138
The microporous structures of CMPs, MOFs, and COFs, constructed based on porphyrin units, are capable of adsorbing anions and cations. Moreover, the research indicated that nitrogen-rich boron nitride anode materials can enhance the adsorption of cations,139 this is good news for porphyrins, which are rich in nitrogen. And these results provide theoretical support for porphyrin polymers as supercapacitor materials. A series of novel porous coordination polymers (MCP-PCP) have been reported using a catechol-substituted porphyrin (CP) as the basic unit (Fig. 21, R1).140 These hybrid nanomaterials exhibit specific capacitance of up to 380 F g−1 at 1 A g−1 and possess good cycling properties, which are of great utility in supercapacitor energy storage materials.
 | ||
| Fig. 21 Synthesis route of porphyrin micro-polymers for capacitors. | ||
Coupling porphyrins with functional small molecule organics to form porous polymers is a common strategy. Guo et al. used tetra(4-aminophenyl)porphyrin (TAPP) and 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTCA) as the basic building blocks, synthesized a porous organic polymer (POP-TAPP-NTCA, Fig. 21, R2) microsphere (1.5–3 μm in diameter) as a zinc ion storage anodes with an energy density of 48 W h kg−1, and assembled aqueous zinc ion hybrid capacitors (ZIHCs) based on POP-TAPP-NTCA anodes can provide a specific capacitance of 172 F g−1.141 It has also been reported that a composite material formed by graphene oxide (GO), TAPP, and nylon 66 was utilized as a supercapacitor electrode to achieve energy storage (Fig. 21, R3).142
As we have introduced in this section, porphyrin-based functional materials can be used not only to store lithium ions but also as electrode materials of SIBs, AIBs, KIBs, and capacitors. In addition, they can also be exploited to modify the electrode (ZnO) of Ni–Zn batteries and as a redox catalyst for Zn-air batteries. In Section 4.4, we have discussed the development direction and ideas of porphyrin-based electrode materials and electrocatalysts in detail, so we will not repeat and over-describe them in this subsection.
6. Summary and perspective
In the past decade, porphyrins and their derivatives have been actively applied in the fields of new energy,68,143–145 biomedicine,146–149 catalysts,150–152 and analytical chemistry153–155 et al. due to their excellent photoelectrochemical properties. Porphyrins can not only coordinate with metal ions and be modified by functional groups but also can form supramolecular polymers,156 MOFs,157,158 COFs,159 porous organic polymers160 and organic/inorganic composites.161 Therefore, porphyrin-based functional materials are still the focus of scientific research.As we have reviewed, porphyrin-based functional materials in the energy field are mainly designed and explored as charge transport materials, modifiers, battery interface materials, electrode materials, dye sensitizers, and electrocatalysts (Fig. 22). They play a significant role in energy storage. In particular, porphyrin-based MOFs, COFs, and CMPs formed through intermolecular engineering have been exploited as lithium storage materials for organic lithium batteries. Moreover, small molecular porphyrin compounds improved by introducing specific groups, functional organic small molecules or their fragments on the porphyrin ring as electrode materials and modifiers for perovskite solar cells and lithium batteries, or as organic dyes for dye-sensitized solar cells. The project of twinning porphyrins with newly developed organic small molecules or functional materials to complement or enhance their performance undoubtedly has great prospects. Therefore, it can inspire the creativity and enthusiasm of researchers.
 | ||
| Fig. 22 The application schematic illustration of porphyrin and its derivatives in the energy field. | ||
Based on the latest research literature, this review timely highlighted the advanced progress and design idea of porphyrin materials in the energy field and made a comparative summary and objective evaluation. Admittedly, there is much literature on the application of porphyrin in batteries, and we cannot list them all. To provide readers with the convenience of access, we classified and sorted out some review articles, such as the application of porphyrin in solar cells76,162–168 and the application of porphyrin in alkali metal ion batteries.169–171
Finally, we offer some personal insights into the design of porphyrin-based materials. Practice needs to be guided by theory, so we should rely on Density Functional Theory (DFT), Molecular Dynamics (MD) simulation, and other methods in the construction and development of functional porphyrin-based materials. The preparation of these specific materials can be initiated from the following three precursors.
The porphyrin and metalloporphyrin frameworks specially designed by the chemical synthesis method are used directly or further modified. To regulate the energy level distribution of the conjugated structure and expand the absorption spectrum, we usually introduce electron donors, such as the arylamine group, at the meso-site in porphyrin ring, and introduce electron acceptors with the acetylene group as the connecting chain on the opposite meso-site to construct D–π–A (donor–π–acceptor) type porphyrins. Regarding the modification of the benzene ring at the meso-site, we can link specific functional groups on the benzene ring as reaction sites for the preparation of polymers such as COFs and MOFs. Some functional small molecules can also be connected to the benzene ring, such as organic electrode materials, organic photovoltaic materials, etc. In addition, the effect of π–π stacking of porphyrin molecules can be reduced by introducing alkoxy and alkyl groups into the benzene ring (Fig. 23a).
 | ||
| Fig. 23 (a) Design ideas for modification of porphyrin-based materials, the meso-site marked with numbers 5, 10, 15, 20. (b) Reaction sites and modification strategies of pyropheophorbide (a). (c) Selected carbaporphyrinoid systems. | ||
Although the cost of chlorophyll a derivation is relatively high in purification and separation, which limits its large-scale use, it is also an alternative path to develop porphyrin derivatives as functional materials for energy applications using them as starting materials. For example, we can employ pyropheophorbide a, an acid degradation product of chlorophyll a, as the initial materials, through a series of addition, substitution, and ring–opening reactions to obtain some specific porphyrin derivatives (Fig. 23b). In particular, the carboxyl group they contained can be used as the active site of the reaction to couple with other organic molecules such as dye sensitizers. In addition, the carbaporphyrinoid systems formed by replacing the nitrogen atom in the porphyrin ring with one or more carbon atoms can still exhibit aromatic properties, which has great application prospects in materials science and catalysis.172–174 Therefore, these carbaporphyrins and confused porphyrinoids such as N-confused porphyrins (Fig. 23c) can be modified to regulate their conjugate structure and charge distribution, to enhance their applicability in the energy field.
As we have reviewed, porphyrins and their derivatives remain promising in the energy field. Based on some relevant scientific research results introduced in this article, we put forward some of our personal views on the design ideas of porphyrin-based functional materials applied in the energy field, and further research needs to be practiced. Finally, we hope this review could inspire readers and researchers working in related fields.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (U19A2019), The Hunan Provincial Science and Technology Plan (2020GK2100).References
- J. Chen, F. Chen, L. Zhang, Z. Y. Yang, T. Deng, Y. F. Zhao, T. Y. Zheng, X. L. Gan, H. T. Zhong, Y. Q. Geng, X. W. Fu, Y. Q. Wang and C. Yu, ACS Appl. Mater. Interfaces, 2021, 13, 27856–27867, DOI:10.1021/acsami.1c04868.
- J. Waluk, Chem. Rev., 2017, 117, 2447–2480, DOI:10.1021/acs.chemrev.6b00328.
- H. Fischer and W. Gleim, Justus Liebigs Ann. Chem., 1936, 521, 157–160, DOI:10.1002/jlac.19365210110.
- P. Rothemund, J. Am. Chem. Soc., 1935, 57, 2010–2011, DOI:10.1021/ja01313a510.
- P. Rothemund, J. Am. Chem. Soc., 1936, 58, 625–627, DOI:10.1021/ja01295a027.
- G. P. Arsenault, E. Bullock and S. F. Macdonald, J. Am. Chem. Soc., 1960, 82, 4384–4389, DOI:10.1021/ja01501a066.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476, DOI:10.1021/jo01288a053.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827–836, DOI:10.1021/jo00381a022.
- S. Horn, K. Dahms and M. O. Senge, J. Porphyrins Phthalocyanines, 2008, 12, 1053–1077, DOI:10.1142/s108842460800042x.
- S. Mondal, T. Pain, K. Sahu and S. Kar, ACS Omega, 2021, 6, 22922–22936, DOI:10.1021/acsomega.1c03534.
- R. Sharma, A. Sharma, S. Agarwal and M. S. Dhaka, Sol. Energy, 2022, 244, 516–535, DOI:10.1016/j.solener.2022.08.001.
- T. H. Wu, Z. Z. Qin, Y. B. Wang, Y. Z. Wu, W. Chen, S. F. Zhang, M. L. Cai, S. Y. Dai, J. Zhang, J. Liu, Z. M. Zhou, X. Liu, H. Segawa, H. R. Tan, Q. W. Tang, J. F. Fang, Y. W. Li, L. M. Ding, Z. J. Ning, Y. B. Qi, Y. Q. Zhang and L. Y. Han, Nano-Micro Lett., 2021, 13, 152, DOI:10.1007/s40820-021-00672-w.
- C. G. Huang, E. Gajewiak, A. Wright, W. Rodriguez-Kazeem, D. Heift and J. C. Bear, Z. Anorg. Allg. Chem., 2023, 649(13), e202300045, DOI:10.1002/zaac.202300045.
- https://www.nrel.gov/pv/cell-efficiency.html.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051, DOI:10.1021/ja809598r.
- Z. D. Chu, M. J. Yang, P. Schulz, D. Wu, X. Ma, E. Seifert, L. Y. Sun, X. Q. Li, K. Zhu and K. J. Lai, Nat. Commun., 2017, 8, 8, DOI:10.1038/s41467-017-02331-4.
- Z. Fang, L. Wang, X. Mu, B. Chen, Q. Xiong, W. D. Wang, J. Ding, P. Gao, Y. Wu and J. Cao, J. Am. Chem. Soc., 2021, 143, 18989–18996, DOI:10.1021/jacs.1c07518.
- X. X. Feng, R. H. Chen, Z. A. Nan, X. D. Lv, R. Q. Meng, J. Cao and Y. Tang, Adv. Sci., 2019, 6, 9, DOI:10.1002/advs.201802040.
- X. X. Feng, X. D. Lv, J. Cao and Y. Tang, Chin. J. Chem., 2022, 41(11), 43–49, DOI:10.1002/cjoc.202200468.
- C. P. Li, J. Yin, R. H. Chen, X. D. Lv, X. X. Feng, Y. Y. Wu and J. Cao, J. Am. Chem. Soc., 2019, 141, 6345–6351, DOI:10.1021/jacs.9b01305.
- H. X. Liang, W. D. Wang, S. B. Mai, X. D. Lv, J. Fang and J. Cao, Chem. Eng. J., 2022, 429, 6, DOI:10.1016/j.cej.2021.132405.
- Y. M. Liang, P. Q. Song, H. R. Tian, C. B. Tian, W. J. Tian, Z. A. Nan, Y. T. Cai, P. P. Yang, C. Sun, J. F. Chen, L. Q. Xie, Q. Y. Zhang and Z. H. Wei, Adv. Funct. Mater., 2022, 32, 8, DOI:10.1002/adfm.202110139.
- K. Su, W. T. Chen, Y. Q. Huang, G. Yang, K. G. Brooks, B. Zhang, Y. Q. Feng, M. K. Nazeeruddin and Y. Zhang, Sol. RRL, 2022, 6, 8, DOI:10.1002/solr.202100964.
- X. L. Zheng, H. S. Lin, B. W. Zhang, S. Maruyama and Y. Matsuo, J. Org. Chem., 2022, 87, 5457–5463, DOI:10.1021/acs.joc.1c01947.
- Y. Zhou, H. Zhong, J. H. Han, M. Q. Tai, X. W. Yin, M. H. Zhang, Z. Y. Wu and H. Lin, J. Mater. Chem. A, 2019, 7, 26334–26341, 10.1039/c9ta09369a.
- X. J. Gu, W. C. Xiang, Q. W. Tian and S. Z. Liu, Angew. Chem., Int. Ed., 2021, 60, 23164–23170, DOI:10.1002/anie.202109724.
- S. Singh, Laxmi and D. Kabra, J. Phys. D: Appl. Phys., 2020, 53, 24, DOI:10.1088/1361-6463/abb487.
- Y. Z. Zhang, Y. J. Wang, L. C. Zhao, X. Y. Yang, C. H. Hou, J. Wu, R. Su, S. Jia, J. J. Shyue, D. Y. Luo, P. Chen, M. T. Yu, Q. Y. Li, L. Li, Q. H. Gong and R. Zhu, Energy Environ. Sci., 2021, 14, 6526–6535, 10.1039/d1ee02287c.
- H. Zhang, Y. Z. Wu, C. Shen, E. P. Li, C. X. Yan, W. W. Zhang, H. Tian, L. Y. Han and W. H. Zhu, Adv. Energy Mater., 2019, 9, 9, DOI:10.1002/aenm.201803573.
- R. H. Chen, Y. K. Wang, S. Q. Nie, H. Shen, Y. Hui, J. Peng, B. H. Wu, J. Yin, J. Li and N. F. Zheng, J. Am. Chem. Soc., 2021, 143, 10624–10632, DOI:10.1021/jacs.1c03419.
- E. Castro, J. Murillo, O. Fernandez-Delgado and L. Echegoyen, J. Mater. Chem. C, 2018, 6, 2635–2651, 10.1039/c7tc04302c.
- B. Chen, P. N. Rudd, S. Yang, Y. B. Yuan and J. S. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867, 10.1039/c8cs00853a.
- J. H. Zhao, X. Mu, L. Wang, Z. Fang, X. Zou and J. Cao, Angew Chem. Int. Ed. Engl., 2022, 61, e202116308, DOI:10.1002/anie.202116308.
- A. Fakharuddin, L. Schmidt-Mende, G. Garcia-Belmonte, R. Jose and I. Mora-Sero, Adv. Energy Mater., 2017, 7(22), 1700623, DOI:10.1002/aenm.201700623.
- J. J. Shi, X. Xu, D. M. Li and Q. B. Meng, Small, 2015, 11, 2472–2486, DOI:10.1002/smll.201403534.
- L. Zhang, W. G. Hu and S. F. Shao, Org. Electron., 2022, 107, 15, DOI:10.1016/j.orgel.2022.106545.
- K. Gkini, N. Balis, M. Papadakis, A. Verykios, M. C. Skoulikidou, C. Drivas, S. Kennou, M. Golomb, A. Walsh, A. G. Coutsolelos, M. Vasilopoulou and P. Falaras, ACS Appl. Energy Mater., 2020, 3, 7353–7363, DOI:10.1021/acsaem.0c00710.
- K. Gkini, A. Verykios, N. Balis, A. Kaltzoglou, M. Papadakis, K. S. Adamis, K. K. Armadorou, A. Soultati, C. Drivas, S. Gardelis, I. D. Petsalakis, L. C. Palilis, A. Fakharuddin, M. I. Haider, X. C. Bao, S. Kennou, P. Argitis, L. Schmidt-Mende, A. G. Coutsolelos, P. Falaras and M. Vasilopoulou, ACS Appl. Mater. Interfaces, 2020, 12, 1120–1131, DOI:10.1021/acsami.9b17580.
- K. Su, P. Zhao, Y. Ren, Y. Zhang, G. Yang, Y. Q. Huang, Y. Q. Feng and B. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 14248–14257, DOI:10.1021/acsami.1c00146.
- K. Gao, Z. L. Zhu, B. Xu, S. B. Jo, Y. Y. Kan, X. B. Peng and A. K. Y. Jen, Adv. Mater., 2017, 29, 8, DOI:10.1002/adma.201703980.
- X. C. Feng, Y. H. Huan, C. Y. Zheng, C. Tan, H. Meng, B. Liu, D. Q. Gao and W. Huang, Org. Electron., 2020, 77, 7, DOI:10.1016/j.orgel.2019.105522.
- C. Li, L. Wang, P.-J. Yan, H. Liu, J. Cao, C.-C. Chen and Y. Tang, Chem. Eng. J., 2021, 409, 128167, DOI:10.1016/j.cej.2020.128167.
- C. L. Mai, Q. Zhou, Q. Xiong, C. C. Chen, J. B. Xu, Z. Z. Zhang, H. W. Lee, C. Y. Yeh and P. Gao, Adv. Funct. Mater., 2021, 31, 8, DOI:10.1002/adfm.202007762.
- G. B. Xiao, L. Y. Wang, X. J. Mu, X. X. Zou, Y. Y. Wu and J. Cao, CCS Chem., 2021, 3, 25–36, DOI:10.31635/ccschem.020.202000516.
- G. B. Xiao, Z. F. Yu, J. Cao and Y. Tang, CCS Chem., 2020, 2, 488–494, DOI:10.31635/ccschem.020.202000163.
- P. J. Yan, J. Cao, J. Pang, Z. R. Yang, X. L. Wang and X. Q. Yao, Org. Electron., 2021, 93, 7, DOI:10.1016/j.orgel.2021.106158.
- X. J. Zhao and M. K. Wang, Mater. Today Energy, 2018, 7, 208–220, DOI:10.1016/j.mtener.2017.09.011.
- M. Urbani, G. De La Torre, M. K. Nazeeruddin and T. Torres, Chem. Soc. Rev., 2019, 48, 2738–2766, 10.1039/c9cs00059c.
- H. D. Pham, T. C. J. Yang, S. M. Jain, G. J. Wilson and P. Sonar, Adv. Energy Mater., 2020, 10, 23, DOI:10.1002/aenm.201903326.
- H. H. Chou, Y. H. Chiang, M. H. Li, P. S. Shen, H. J. Wei, C. L. Mai, P. Chen and C. Y. Yeh, ACS Energy Lett., 2016, 1, 956–962, DOI:10.1021/acsenergylett.6b00432.
- Y. H. Chiang, H. H. Chou, W. T. Cheng, Y. R. Li, C. Y. Yeh and P. Chen, ACS Energy Lett., 2018, 3, 1620–1626, DOI:10.1021/acsenergylett.8b00607.
- H. H. Chou, Y. H. Chiang, Y. H. Chen, C. J. Guo, H. Y. Zuo, W. T. Cheng, P. Y. Lin, Y. Y. Chiu, P. Chen and C. Y. Yeh, Sol. RRL, 2020, 4, 8, DOI:10.1002/solr.202000119.
- J. Cao, X. D. Lv, P. Zhang, T. T. Chuong, B. H. Wu, X. X. Feng, C. F. Shan, J. C. Liu and Y. Tang, Adv. Mater., 2018, 30, 9, DOI:10.1002/adma.201800568.
- S. Chen, P. Liu, Y. Hua, Y. Y. Li, L. Kloo, X. Z. Wang, B. Ong, W. K. Wong and X. J. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 13231–13239, DOI:10.1021/acsami.7b01904.
- S. H. Kang, C. Y. Lu, H. R. Zhou, S. Choi, J. Kim and H. K. Kim, Dyes Pigm., 2019, 163, 734–739, DOI:10.1016/j.dyepig.2018.12.065.
- G. Reddy, P. Basak, L. A. Jones, E. Della Gaspera, N. Islavath and L. Giribabu, Sol. Energy, 2020, 206, 539–547, DOI:10.1016/j.solener.2020.06.040.
- M. N. Shah, S. R. Pathipati and N. Ahmed, J. Mater. Sci.: Mater. Electron., 2019, 30, 7866–7872, DOI:10.1007/s10854-019-01106-5.
- C. D. Si, X. D. Lv and S. J. Long, Inorg. Chem. Commun., 2020, 112, 5, DOI:10.1016/j.inoche.2019.107701.
- W. Zhang, Y. Hua, L. Q. Wang, B. B. Zhang, Y. Y. Li, P. Liu, V. Leandri, Y. Guo, H. Chen, J. M. Gardner, L. C. Sun and L. Kloo, ACS Appl. Energy Mater., 2019, 2, 6768–6779, DOI:10.1021/acsaem.9b01223.
- U. H. Lee, R. Azmi, S. Sinaga, S. Hwang, S. H. Eom, T. W. Kim, S. C. Yoon, S. Y. Jang and I. H. Jung, ChemSusChem, 2017, 10, 3780–3787, DOI:10.1002/cssc.201701526.
- F. Arjmand, S. J. Fatemi, S. Maghsoudi and A. Naeimi, J. Mater. Res. Technol., 2022, 16, 1008–1020, DOI:10.1016/j.jmrt.2021.12.012.
- A. B. Munoz-Garcia, I. Benesperi, G. Boschloo, J. J. Concepcion, J. H. Delcamp, E. A. Gibson, G. J. Meyer, M. Pavone, H. Pettersson, A. Hagfeldt and M. Freitag, Chem. Soc. Rev., 2021, 50, 12450–12550, 10.1039/d0cs01336f.
- J. W. Gong, K. Sumathy, Q. Q. Qiao and Z. P. Zhou, Renewable Sustainable Energy Rev., 2017, 68, 234–246, DOI:10.1016/j.rser.2016.09.097.
- M. Kokkonen, P. Talebi, J. Zhou, S. Asgari, S. A. Soomro, F. Elsehrawy, J. Halme, S. Ahmad, A. Hagfeldt and S. G. Hashmi, J. Mater. Chem. A, 2021, 9, 10527–10545, 10.1039/d1ta00690h.
- B. O’regan and M. Gratzel, Nature, 1991, 353, 737–740, DOI:10.1038/353737a0.
- A. Carella, F. Borbone and R. Centore, Front. Chem., 2018, 6, 24, DOI:10.3389/fchem.2018.00481.
- S. Ramasamy, M. Bhagavathiachari, S. A. Suthanthiraraj and M. Pichai, Dyes Pigm., 2022, 203, 19, DOI:10.1016/j.dyepig.2022.110380.
- K. W. Zeng, Z. F. Tong, L. Ma, W. H. Zhu, W. J. Wu and Y. S. Xie, Energy Environ. Sci., 2020, 13, 1617–1657, 10.1039/c9ee04200h.
- T. Bessho, S. M. Zakeeruddin, C. Y. Yeh, E. W. G. Diau and M. Gratzel, Angew. Chem., Int. Ed., 2010, 49, 6646–6649, DOI:10.1002/anie.201002118.
- A. Yella, H. W. Lee, H. N. Tsao, C. Y. Yi and A. K. Chandiran, Science, 2011, 334, 1203 CrossRef CAS PubMed.
- A. Yella, C. L. Mai, S. M. Zakeeruddin, S. N. Chang, C. H. Hsieh, C. Y. Yeh and M. Gratzel, Angew. Chem., Int. Ed., 2014, 53, 2973–2977, DOI:10.1002/anie.201309343.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Gratzel, Nat. Chem., 2014, 6, 242–247, DOI:10.1038/nchem.1861.
- S. H. Kang, M. J. Jeong, Y. K. Eom, I. T. Choi, S. M. Kwon, Y. Yoo, J. Kim, J. Kwon, J. H. Park and H. K. Kim, Adv. Energy Mater., 2017, 7, 10, DOI:10.1002/aenm.201602117.
- J. F. Lu, S. S. Liu and M. K. Wang, Front. Chem., 2018, 6, 19, DOI:10.3389/fchem.2018.00541.
- O. Birel, S. Nadeem and H. Duman, J. Fluoresc., 2017, 27, 1075–1085, DOI:10.1007/s10895-017-2041-2.
- V. Piradi, F. Yan, X. J. Zhu and W. Y. Wong, Mater. Chem. Front., 2021, 5, 7119–7133, 10.1039/d1qm00952d.
- C. C. Chen, Y. H. Chen, V. S. Nguyen, S. Y. Chen, M. C. Tsai, J. S. Chen, S. Y. Lin, T. C. Wei and C. Y. Yeh, Adv. Energy Mater., 2023, 13(20), 2300353, DOI:10.1002/aenm.202300353.
- Y. S. Xie, Y. Y. Tang, W. J. Wu, Y. Q. Wang, J. C. Liu, X. Li, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2015, 137, 14055–14058, DOI:10.1021/jacs.5b09665.
- K. W. Zeng, Y. Y. Lu, W. Q. Tang, S. L. Zhao, Q. Y. Liu, W. H. Zhu, H. Tian and Y. S. Xie, Chem. Sci., 2019, 10, 2186–2192, 10.1039/c8sc04969f.
- K. W. Zeng, W. Q. Tang, C. J. Li, Y. Y. Chen, S. L. Zhao, Q. Y. Liu and Y. S. Xie, J. Mater. Chem. A, 2019, 7, 20854–20860, 10.1039/c9ta06911a.
- K. W. Zeng, Y. Y. Chen, W. H. Zhu, H. Tian and Y. S. Xie, J. Am. Chem. Soc., 2020, 142, 5154–5161, DOI:10.1021/jacs.9b12675.
- Y. Y. Chen, Y. Y. Tang, J. Z. Zou, K. W. Zeng, G. Baryshnikov, C. J. Li and Y. S. Xie, ACS Appl. Mater. Interfaces, 2021, 13, 49828–49839, DOI:10.1021/acsami.1c12448.
- J. Z. Zou, Y. Q. Wang, G. Baryshnikov, J. X. Luo, X. Y. Wang, H. Agren, C. J. Li and Y. S. Xie, ACS Appl. Mater. Interfaces, 2022, 14, 33274–33284, DOI:10.1021/acsami.2c07950.
- A. Yoshino, Angew. Chem., Int. Ed., 2012, 51, 5798–5800, DOI:10.1002/anie.201105006.
- P. Gao, Z. Chen, Z. Zhao-Karger, J. E. Mueller, C. Jung, S. Klyatskaya, T. Diemant, O. Fuhr, T. Jacob, R. J. Behm, M. Ruben and M. Fichtner, Angew. Chem., Int. Ed., 2017, 56, 10341–10346, DOI:10.1002/anie.201702805.
- X. M. Lin, D. Y. Wu, P. Gao, Z. Chen, M. Ruben and M. Fichtner, Chem. Mater., 2019, 31, 3239–3247, DOI:10.1021/acs.chemmater.9b00077.
- B. Ren, Y. Sun, X. Feng, J. F. Peng, R. Ding, X. J. Sun, E. H. Liu and P. Gao, Sustainable Energy Fuels, 2022, 6, 361–370, 10.1039/d1se01649k.
- X. Feng, X. Wu, X. Chen, J. J. Yuan, S. S. Lv, B. Ren, X. J. Sun, E. H. Liu, S. T. Tan and P. Gao, Energy Storage Mater., 2021, 42, 454–463, DOI:10.1016/j.ensm.2021.08.003.
- X. Wu, X. Feng, J. J. Yuan, X. K. Yang, H. B. Shu, C. K. Yang, Z. Y. Liu, J. F. Peng, E. H. Liu, S. T. Tan and P. Gao, Energy Storage Mater., 2022, 46, 252–258, DOI:10.1016/j.ensm.2022.01.020.
- Y. Zhou, X. Huang, X. Chen, F. He, D. Chen, X. Sun, S. Tan and P. Gao, ACS Appl. Mater. Interfaces, 2022, 14, 40862–40870, DOI:10.1021/acsami.2c09649.
- H. Wu, J. J. Zhang, X. F. Du, M. Zhang, J. F. Yang, J. N. Zhang, T. Luo, H. Liu, H. Xu and G. L. Cui, Chem. Commun., 2019, 55, 11370–11373, 10.1039/c9cc05474j.
- K. Jeong, J. M. Kim, S. H. Kim, G. Y. Jung, J. Yoo, S. H. Kim, S. K. Kwak and S. Y. Lee, Adv. Funct. Mater., 2019, 29, 10, DOI:10.1002/adfm.201806937.
- Y. Han, Z. J. Liu, F. F. Zheng, Y. X. Bai, Z. Q. Zhang, X. G. Li, W. W. Xiong, J. H. Zhang and A. H. Yuan, J. Alloys Compd., 2021, 881, 8, DOI:10.1016/j.jallcom.2021.160531.
- L. L. Shu, J. Yu, Y. Cui, Y. Q. Ma, Y. H. Li, B. Gao and H. G. Wang, Int. J. Hydrogen Energy, 2022, 47, 10902–10910, DOI:10.1016/j.ijhydene.2022.01.146.
- Y. Yang, J. X. Yuan, S. H. Huang, Z. Y. Chen, C. B. Lu, C. Q. Yang, G. Q. Zhai, J. H. Zhu and X. D. Zhuang, J. Power Sources, 2022, 531, 7, DOI:10.1016/j.jpowsour.2022.231340.
- H. G. Wang, C. Jiang, C. P. Yuan, Q. Wu, Q. Li and Q. Duan, Chem. Eng. J., 2018, 332, 237–244, DOI:10.1016/j.cej.2017.09.081.
- W. Y. Ko, M. J. Fang, M. S. Li, W. C. Hsu, Y. W. Huang, C. W. Cheng, B. J. Hwang, S. Gwo and K. J. Lin, ACS Appl. Energy Mater., 2020, 3, 6098–6106, DOI:10.1021/acsaem.0c01090.
- K. C. Wang, D. W. Feng, T. F. Liu, J. Su, S. Yuan, Y. P. Chen, M. Bosch, X. D. Zou and H. C. Zhou, J. Am. Chem. Soc., 2014, 136, 13983–13986, DOI:10.1021/ja507269n.
- L. Sun, J. Xie, Z. D. Chen, J. Wu and L. Li, Dalton Trans., 2018, 47, 9989–9993, 10.1039/c8dt02161a.
- H. Yang, S. L. Zhang, L. H. Han, Z. Zhang, Z. Xue, J. Gao, Y. J. Li, C. S. Huang, Y. P. Yi, H. B. Liu and Y. L. Li, ACS Appl. Mater. Interfaces, 2016, 8, 5366–5375, DOI:10.1021/acsami.5b12370.
- Z. L. Han, Y. Ai, X. L. Jiang, Y. X. You, F. C. Wei, H. Luo, J. Cui, Q. Y. Bao, J. W. Fu, Q. G. He, S. H. Liu and J. G. Cheng, Chem.–Eur. J., 2020, 26, 10433–10438, DOI:10.1002/chem.202001943.
- L. Gong, X. Y. Yang, Y. Gao, G. X. Yang, Z. H. Yu, X. Z. Fu, Y. H. Wang, D. D. Qi, Y. Z. Bian, K. Wang and J. Z. Jiang, J. Mater. Chem. A, 2022, 10, 16595–16601, 10.1039/d2ta03579k.
- C. H. Li, Y. Gu, Y. B. Wang, B. Sun and H. Shang, Dalton Trans., 2021, 50, 15849–15854, 10.1039/d1dt02923a.
- B. Q. Li, X. R. Chen, X. Chen, C. X. Zhao, R. Zhang, X. B. Cheng and Q. Zhang, Research, 2019, 2019, 11, DOI:10.34133/2019/4608940.
- J. J. Yuan, B. Ren, X. Feng, P. Gao, E. H. Liu and S. T. Tan, Chem. Commun., 2020, 56, 5437–5440, 10.1039/c9cc09846a.
- Q. H. Zeng, P. P. Chen, Z. F. Li, X. Wen, W. Wen, Y. Liu, H. L. Zhao, S. P. Zhang, H. H. Zhou and L. Y. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 48569–48581, DOI:10.1021/acsami.1c12086.
- Y. Y. Xu, H. Gong, H. Ren, X. L. Fan, P. Li, T. F. Zhang, K. Chang, T. Wang and J. P. He, Small, 2022, 18, 8, DOI:10.1002/smll.202203917.
- Y. Zhang, R. L. Zhong, M. Lu, J. H. Wang, C. Jiang, G. K. Gao, L. Z. Dong, Y. F. Chen, S. L. Li and Y. Q. Lan, ACS Cent. Sci., 2021, 7, 175–182, DOI:10.1021/acscentsci.0c01390.
- N. Nakamura, S. Ahn, T. Momma and T. Osaka, J. Power Sources, 2023, 558, 232566, DOI:10.1016/j.jpowsour.2022.232566.
- S. Li, J. D. Lin, B. Chang, D. W. Yang, D. Y. Wu, J. H. Wang, W. J. Zhou, H. Liu, S. H. Sun and L. Zhang, Energy Storage Mater., 2023, 55, 94–104, DOI:10.1016/j.ensm.2022.11.045.
- C. X. Zhao, X. Y. Li, M. Zhao, Z. X. Chen, Y. W. Song, W. J. Chen, J. N. Liu, B. Wang, X. Q. Zhang, C. M. Chen, B. Q. Li, J. Q. Huang and Q. Zhang, J. Am. Chem. Soc., 2021, 143, 19865–19872, DOI:10.1021/jacs.1c09107.
- B. Q. Li, H. J. Peng, X. Chen, S. Y. Zhang, J. Xie, C. X. Zhao and Q. Zhang, CCS Chem., 2019, 1, 128–137, DOI:10.31635/ccschem.019.20180016.
- J. B. Pang, B. Chang, H. Liu and W. J. Zhou, ACS Energy Lett., 2022, 7, 78–96, DOI:10.1021/acsenergylett.1c02132.
- S. Nam, M. Mahato, K. Matthews, R. W. Lord, Y. Lee, P. Thangasamy, C. W. Ahn, Y. Gogotsi and I. K. Oh, Adv. Funct. Mater., 2023, 33, 10, DOI:10.1002/adfm.202210702.
- X. Q. Su, J. Li, G. P. Yao, J. L. Wang, J. S. Zhao and F. X. Zhang, Catal. Commun., 2013, 37, 23–26, DOI:10.1016/j.catcom.2013.03.024.
- X. Q. Su, W. J. Sun, J. Li, Z. Q. Zhang, M. Jiang and J. S. Zhao, ECS Electrochem. Lett., 2014, 3, A39–A40, DOI:10.1149/2.010405eel.
- Z. Q. Zhang, L. Y. Kong, Y. Xiong, Y. Luo and J. Li, J. Solid State Electrochem., 2014, 18, 3471–3477, DOI:10.1007/s10008-014-2571-3.
- Y. L. Zhao, X. Z. Yuan, L. B. Jiang, J. Wen, H. Wang, R. P. Guan, J. J. Zhang and G. M. Zeng, Chem. Eng. J., 2020, 383, 123089, DOI:10.1016/j.cej.2019.123089.
- T. Sun, J. Xie, W. Guo, D. S. Li and Q. C. Zhang, Adv. Energy Mater., 2020, 10(19), 1904199, DOI:10.1002/aenm.201904199.
- Z. Zhao-Karger, P. Gao, T. Ebert, S. Klyatskaya, Z. Chen, M. Ruben and M. Fichtner, Adv. Mater., 2019, 31(26), 1806599, DOI:10.1002/adma.201806599.
- X. Y. Li, C. Y. Tang, L. Zhang, M. Y. Song, Y. J. Zhang and S. J. Wang, Biomimetics, 2023, 8(2), 171, DOI:10.3390/biomimetics8020171.
- M. J. Li, R. P. Hicks, Z. F. Chen, C. Luo, J. C. Guo, C. S. Wang and Y. H. Xu, Chem. Rev., 2023, 123(4), 1712–1773, DOI:10.1021/acs.chemrev.2c00374.
- M. E. Bhosale, S. Chae, J. M. Kim and J. Y. Choi, J. Mater. Chem. A, 2018, 6, 19885–19911, 10.1039/c8ta04906h.
- C. P. Han, H. F. Li, R. Y. Shi, T. F. Zhang, J. Tong, J. Q. Li and B. H. Li, J. Mater. Chem. A, 2019, 7, 23378–23415, 10.1039/c9ta05252f.
- S. Lee, G. Kwon, K. Ku, K. Yoon, S. K. Jung, H. D. Lim and K. Kang, Adv. Mater., 2018, 30(42), 1704682, DOI:10.1002/adma.201704682.
- Y. L. Liang and Y. Yao, Joule, 2018, 2, 1690–1706, DOI:10.1016/j.joule.2018.07.008.
- Y. Lu, Y. Cai, Q. Zhang and J. Chen, Adv. Mater., 2022, 34(22), 2104150, DOI:10.1002/adma.202104150.
- Y. Lu and J. Chen, Nat. Rev. Chem, 2020, 4, 127–142, DOI:10.1038/s41570-020-0160-9.
- Y. Lu, Q. Zhang, L. Li, Z. Q. Niu and J. Chen, Chem, 2018, 4, 2786–2813, DOI:10.1016/j.chempr.2018.09.005.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. L. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557, DOI:10.1021/acs.chemrev.9b00482.
- R. Rajagopalan, Y. G. Tang, C. K. Jia, X. B. Ji and H. Y. Wang, Energy Environ. Sci., 2020, 13, 1568–1592, 10.1039/c9ee03637g.
- J. J. Shea and C. Luo, ACS Appl. Mater. Interfaces, 2020, 12, 5361–5380, DOI:10.1021/acsami.9b20384.
- X. Han, S. J. Li, W. L. Song, N. Chen, H. S. Chen, S. Y. Huang and S. Q. Jiao, Adv. Energy Mater., 2021, 11(32), 2101446, DOI:10.1002/aenm.202101446.
- R. Yivlialin, G. Bussetti, M. Penconi, A. Bossi, F. Ciccacci, M. Finazzi and L. Duo, ACS Appl. Mater. Interfaces, 2017, 9, 4100–4105, DOI:10.1021/acsami.6b12359.
- J. H. Huang, Z. H. Yang and T. T. Wang, Electrochim. Acta, 2014, 123, 278–284, DOI:10.1016/j.electacta.2014.01.033.
- S. Y. Zhao, H. N. Jia, Y. Wang, N. Ju, X. Y. Zhang, Y. Guo, Y. M. Wang, H. P. Wang, S. Y. Niu, Y. M. Lu, L. Zhu and H. B. Sun, Dalton Trans., 2022, 51, 12524–12531, 10.1039/d2dt01898e.
- S. S. Lv, J. J. Yuan, Z. Chen, P. Gao, H. B. Shu, X. K. Yang, E. H. Liu, S. T. Tan, M. Ruben, Z. Zhao-Karger and M. Fichtner, ChemSusChem, 2020, 13, 2286–2294, DOI:10.1002/cssc.202000425.
- Z. Z. Liang, H. B. Guo, G. J. Zhou, K. Guo, B. Wang, H. T. Lei, W. Zhang, H. Q. Zheng, U. P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 8472–8476, DOI:10.1002/anie.202016024.
- H. H. Jiang, D. Shi, X. C. Sun, S. Z. Wang, Y. L. Li, B. Chang, B. G. Zhang, Y. L. Shao, Y. Z. Wu and X. P. Hao, ACS Appl. Mater. Interfaces, 2020, 12, 47425–47434, DOI:10.1021/acsami.0c12163.
- S. B. Jin, J. P. Hill, Q. M. Ji, L. K. Shrestha and K. Ariga, J. Mater. Chem. A, 2016, 4, 5737–5744, 10.1039/c6ta00516k.
- F. Z. Cui, Z. C. Liu, D. L. Ma, L. L. Liu, T. Huang, P. P. Zhang, D. M. Tan, F. X. Wang, G. F. Jiang and Y. P. Wu, Chem. Eng. J., 2021, 405, 8, DOI:10.1016/j.cej.2020.127038.
- C. A. Garcia-Perez, C. Menchaca-Campos, M. A. Garcia-Sanchez, Y. I. Vega-Cantu, O. Rodriguez-Perez and J. Uruchurtu, Diamond Relat. Mater., 2019, 96, 44–51, DOI:10.1016/j.diamond.2019.04.023.
- M. Urbani, M. Gratzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396, DOI:10.1021/cr5001964.
- Y. H. Zhang, K. Ren, L. Wang, L. Wang and Z. J. Fan, Chin. Chem. Lett., 2022, 33, 33–60, DOI:10.1016/j.cclet.2021.06.013.
- L. L. Li and E. W. G. Diau, Chem. Soc. Rev., 2013, 42, 291–304, 10.1039/c2cs35257e.
- J. J. Chen, Y. F. Zhu and S. Kaskel, Angew. Chem., Int. Ed., 2021, 60, 5010–5035, DOI:10.1002/anie.201909880.
- J. Y. Kou, D. Dou and L. M. Yang, Oncotarget, 2017, 8, 81591–81603, DOI:10.18632/oncotarget.20189.
- M. A. Rajora, J. W. H. Lou and G. Zheng, Chem. Soc. Rev., 2017, 46, 6433–6469, 10.1039/c7cs00525c.
- J. Tian, B. X. Huang, M. H. Nawaz and W. A. Zhang, Coord. Chem. Rev., 2020, 420, 213410, DOI:10.1016/j.ccr.2020.213410.
- E. Nikoloudakis, I. Lopez-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045, 10.1039/d2cs00183g.
- X. L. Li, H. T. Lei, L. S. Xie, N. Wang, W. Zhang and R. Cao, Acc. Chem. Res., 2022, 55, 878–892, DOI:10.1021/acs.accounts.1c00753.
- W. Zhang, W. Z. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797, DOI:10.1021/acs.chemrev.6b00299.
- Y. B. Ding, W. H. Zhu and Y. S. Xie, Chem. Rev., 2017, 117, 2203–2256, DOI:10.1021/acs.chemrev.6b00021.
- Y. B. Ding, Y. Y. Tang, W. H. Zhu and Y. S. Xie, Chem. Soc. Rev., 2015, 44, 1101–1112, 10.1039/c4cs00436a.
- C. Di Natale, C. P. Gros and R. Paolesse, Chem. Soc. Rev., 2022, 51, 1277–1335, 10.1039/d1cs00662b.
- H. S. W. Lee, H. Park, D. Ryu and W. D. Jang, Chem. Soc. Rev., 2023, 52, 1947–1974, 10.1039/d2cs01066f.
- X. Zhang, M. C. Wasson, M. Shayan, E. K. Berdichevsky, J. Ricardo-Noordberg, Z. Singh, E. K. Papazyan, A. J. Castro, P. Marino, Z. Ajoyan, Z. J. Chen, T. Islamoglu, A. J. Howarth, Y. Y. Liu, M. B. Majewski, M. J. Katz, J. E. Mondloch and O. K. Farha, Coord. Chem. Rev., 2021, 429, 213615, DOI:10.1016/j.ccr.2020.213615.
- M. Zhao, S. Ou and C. D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207, DOI:10.1021/ar400265x.
- S. S. Huang, K. Chen and T. T. Li, Coord. Chem. Rev., 2022, 464, 214563, DOI:10.1016/j.ccr.2022.214563.
- W. Y. Ji, T. X. Wang, X. S. Ding, S. B. Lei and B. H. Han, Coord. Chem. Rev., 2021, 439, 213875, DOI:10.1016/j.ccr.2021.213875.
- G. Bottari, M. A. Herranz, L. Wibmer, M. Volland, L. Rodriguez-Perez, D. M. Guldi, A. Hirsch, N. Martin, F. D'souza and T. Torres, Chem. Soc. Rev., 2017, 46, 4464–4500, 10.1039/c7cs00229g.
- J. Chen, C. J. Zhu, Y. Xu, P. W. Zhang and T. X. Liang, Curr. Org. Chem., 2018, 22, 485–504, DOI:10.2174/1385272821666171002122055.
- Y. Matsuo, K. Ogumi, I. Jeon, H. Wang and T. Nakagawa, RSC Adv., 2020, 10, 32678–32689, 10.1039/d0ra03234d.
- S. T. Wu and J. Cao, J. Coord. Chem., 2022, 75, 1494–1519, DOI:10.1080/00958972.2022.2079410.
- Z. Yu, A. Hagfeldt and L. C. Sun, Coord. Chem. Rev., 2020, 406, 16, DOI:10.1016/j.ccr.2019.213143.
- J. M. Park, J. H. Lee and W. D. Jang, Coord. Chem. Rev., 2020, 407, 29, DOI:10.1016/j.ccr.2019.213157.
- E. W.-G. Diau, L.-L. Li, G. C. Ferreira, K. M. Kadish, K. M. Smith and R. Guilard, Chem. Soc. Rev., 2014, 28, 279–317 CAS.
- Y. Z. Zhang, T. Higashino and H. Imahori, J. Mater. Chem. A, 2023, 11, 12659–12680, 10.1039/d2ta09264f.
- H. G. Wang, Q. Wu, L. Q. Cheng, L. Chen, M. F. Li and G. S. Zhu, Energy Storage Mater., 2022, 52, 495–513, DOI:10.1016/j.ensm.2022.08.022.
- D. Y. Wang, R. L. Liu, W. Guo, G. Li and Y. Z. Fu, Coord. Chem. Rev., 2021, 429, 213650, DOI:10.1016/j.ccr.2020.213650.
- Z. Zhao-Karger, P. Gao, T. Ebert, S. Klyatskaya, Z. Chen, M. Ruben and M. Fichtner, Adv. Mater., 2019, 31, e1806599, DOI:10.1016/j.ccr.2020.213650.
- M. J. Bialek, K. Hurej, H. Furuta and L. Latos-Grazynski, Chem. Soc. Rev., 2023, 52, 2082–2144, 10.1039/d2cs00784c.
- T. D. Lash, Chem. Rev., 2017, 117, 2313–2446, DOI:10.1021/acs.chemrev.6b00326.
- M. Toganoh and H. Furuta, Chem. Rev., 2022, 122, 8313–8437, DOI:10.1021/acs.chemrev.1c00065.
| This journal is © The Royal Society of Chemistry 2023 |