
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous catalytic oxidation of glycerol over a UiO-66-derived ZrO2@C supported Au catalyst at room temperature†
Yi-Hu Ke *ab,
Chun-Mei Zhuab,
Huan-Huan Xuab,
Xue Wangab,
Hai Liuab and
Hong Yuanab
*ab,
Chun-Mei Zhuab,
Huan-Huan Xuab,
Xue Wangab,
Hai Liuab and
Hong Yuanab
aKey Laboratory for Chemical Engineering and Technology, State Ethnic Affairs Commission, North Minzu University, Yinchuan, 750021, P. R. China
bNingxia Key Laboratory of Solar Chemical Conversion Technology, North Minzu University, Yinchuan 750021, P. R. China
First published on 8th September 2023
Abstract
The catalytic conversion of biomass-derived glycerol into high-value-added products, such as glyceric acid (GLYA), using catalyst-supported Au nanoparticles (Au NPs) at room temperature presents a significant challenge. In this study, we constructed a series of supported Au catalysts, including Au/ZrO2@C, Au/C, Au/ZrO2, and Au/ZrO2-C, and investigated their effectiveness in selectively catalytic oxidizing glycerol to GLYA at room temperature. Among these catalysts, the Au/ZrO2@C catalyst exhibited the best catalytic performance, achieving a glycerol conversion rate of 73% and a GLYA selectivity of 79% under the optimized reaction conditions (reaction conditions: 30 mL 0.1 M glycerol, glycerol/Au = 750 mol mol−1, T = 25 °C, p(O2) = 10 bar, stirring speed = 600 rpm, time = 6 h). Physical adsorption, X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), and other characterization methods were employed to analyze the texture properties of the catalyst. The findings indicated that the support structure, the strong metal–support interactions between Au NPs and the support, and the presence of small metallic Au NPs were the primary factors contributing to the catalyst's high activity and selectivity. Moreover, the reusability of the Au/ZrO2@C catalyst was investigated, and a probable reaction mechanism for the oxidation of glycerol was proposed.
1 Introduction
As the main product produced by the transesterification of renewable bioenergy, biodiesel is known as an important green energy because of its renewability, excellent combustibility, and extensive sources of raw materials and has gradually attracted the attention of researchers.1 Furthermore, the generation of glycerol, an unavoidable byproduct of biodiesel production, has increased rapidly with the increasing production volume of biodiesel. Therefore, catalytic conversion of glycerol into high value added chemicals has attracted much interest from both academia and industry.2–9 From this glycerol, a sequence of valuable products, such as 1,3-dihydroxyacetone (DHA), glyceraldehyde (GLYHD), glyceric acid (GLYA), tartaric acid (TA), lactic acid (LA), formic acid (FA), and oxalic acid (OA), were obtained via a selective oxidation reaction.10–15 Among them, GLYA can be used in many applications because of its various biological activities, such as a liver stimulant with cholesterol activity, a base material for functional surfactants and a monomer for oligo-esters or polymers.16,17 Therefore, it is crucial to convert glycerol into GLYA via an economical, green and efficient approach.In recent decades, the catalytic conversion of glycerol to GLYA in aqueous solution over noble metal (Au, Pt, Pd) supported catalysts has been extensively investigated.12,18–20 It has been widely proved that supported Au catalysts exhibit some better catalytic performance than Pt and Pd catalysts. However, the higher activity of Au-based catalysts in the oxidation of glycerol depends on the presence of a strong base (NaOH). Many works have been published in the recent years, it was popularly accepted that Au based catalysts are active for this reaction in a strong base solution and Pt-based catalysts are active in base-free solution. However, adding base is generally necessary for carbon-supported Au catalyst to effectively oxidize the glycerol. Demirel-Gülen and co-workers21 found that Au catalyst had no glycerol oxidation activity under neutral condition, but could effectively catalyze the oxidation of glycerol under alkaline conditions. As the content of alkali increases, the glycerol oxidation is kinetically favored, demonstrating that Au catalyst requires the assistance of alkali to achieve an effective glycerol oxidation reaction. In this case, the drawbacks of Au-based catalyst is NaOH can make the C–C cleavage and the formed product should be acidified in order to get a Na-free product.22 And the drawbacks of Pt catalysts is that it deactivated easily because of the overoxidation of Pt, the leaching of Pt and the strong adsorption of formed acids.23,24 Furthermore, the selective catalytic oxidation of glycerol system is a structurally sensitive reaction. Catalyst properties, such as the composition and structure of Au NPs, the reaction conditions and the nature of the support, may greatly influence the catalyst activity, product selectivity and catalyst stability.25–28 It is challenging to achieve high catalytic performance at low temperatures with the currently available catalysts. At the same time, the effect of the support is also a very important factor. The support promotes Au NP distribution and prevents coalescence and agglomeration.
In supported Au catalysts, the main supports are carbon-based materials. For example, Hutchings et al.20,29,30 studied oxidation of glycerol over Au/charcoal and 1% Au/graphite catalysts. The yield of GLYA was 56% under mild reaction conditions. Gil et al.31 prepared a series of catalysts with Au NPs supported on different carbon materials (activated carbon, graphite and ribbon-type carbon nanofibres). They found that the activity and GLYA selectivity were significantly affected by the crystallinity of the carbon support. Ketchie et al.32 prepared different average Au nanoparticle sizes on carbon support and found that the catalytic performance was related to the particle size of Au nanoparticle and the nature of the support. Rodrigues et al.33,34 pointed out that the oxygen content of carbon-based material surfaces plays a key role in the oxidation of glycerol. When Au was supported on activated carbon (AC), which has surface oxygenated acid groups, it was detrimental to the catalytic activity in glycerol oxidation reaction. However, Au supported on multiwall carbon nanotubes with oxygen-free had a higher GLYA selectivity (60%) than oxygen-rich support (40%) at the same glycerol conversion of 20%. Many research groups have reported that carbon-based materials can affect the size, stability, dispersion, and electronic structure of the noble metal NPs and further affect the catalytic activity and selectivity of product.
Recently, MOF-derived porous carbon materials have received considerable attention as catalysts, particularly owing to their clear crystal structures, adjustable pore topology, ultrahigh surface areas, and excellent tail fin properties, and these new-style carbon materials have been well developed in the field of oxidation reactions, for instance, oxidative coupling of amines, oxidation of alcohols and aerobic epoxidation of styrene.35–39 To date, much effort has been focused on exploring ideal MOF-derived nanocomposites. UiO-66 is a type of MOF material that is composed of different organic ligands and Zr metal ions and generally possesses a regular dodecahedral secondary structural unit. Because of its excellent porosity, it has been used as a support for a variety of catalysts. Cabello et al.40 reported that porous carbon prepared with UiO-66 as the precursor has excellent adsorption capacity for the dye rhodamine Cao et al.41 prepared Ru/ZrO2@C and Ru/C catalysts for comparison. Ru/ZrO2@C was found to have a better catalytic effect and stability because ZrO2@C was obtained by pyrolysis of UiO-66; it possesses a nano t-ZrO2 (3.3 nm) structure embedded in amorphous carbon, so the stability is the result of the strong interaction between Ru and nano t-ZrO2. Li et al.42 successfully prepared ZrO2@CN from direct pyrolysis of the Pd@NH2-UiO-66 (Zr) precursor, and it exhibited excellent activity and reusability for the catalytic hydrogenation of TMBQ to TMHQ. Wang et al.43 fabricated a 0.5 wt% Pt/ZrO2@C catalyst and tested its catalytic performance in H2-SCR, achieving nearly 100% NOx conversion at 90 °C and verifying that the residual carbon formed by the pyrolysis treatment was encased in octagonal ZrO2, which effectively prevented Pt particles from coagulating.
In the present study, a ZrO2@C support was prepared by the pyrolysis of UiO-66 precursor, and then Au NPs were evenly dispersed on the ZrO2@C by colloidal deposition method (Scheme 1). By correlating the catalytic performance of the catalyst with its surface configuration, we found that the support had a significant and positive impact on stability in the selective oxidation of glycerol to GLYA. Compared with the Au/C, Au/ZrO2, and Au/ZrO2-C catalysts, the Au/ZrO2@C catalyst exhibited the best catalytic performance at 25 °C.
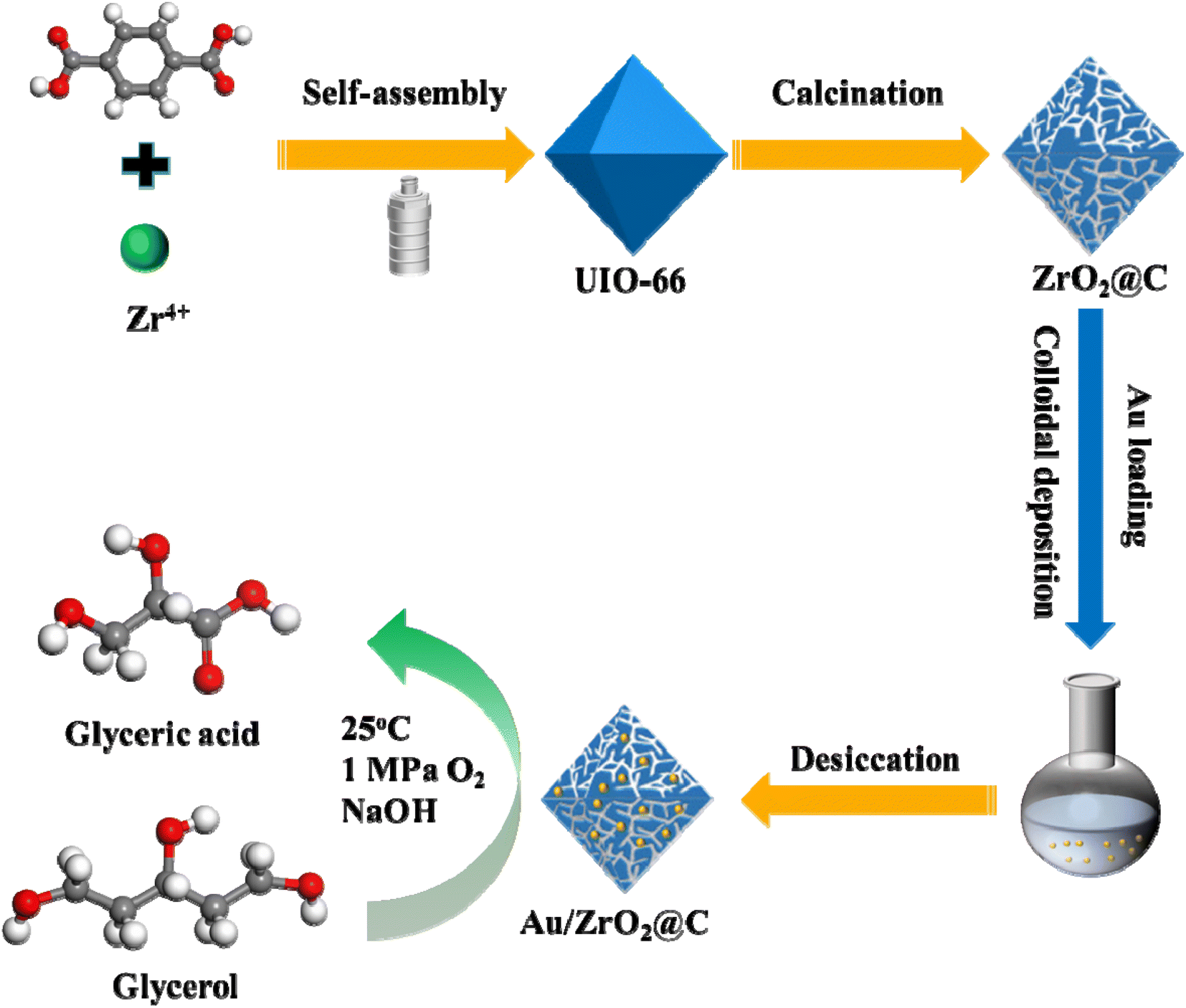 | ||
| Scheme 1 Schematic illustration of the preparation process of the Au/ZrO2@C catalyst. | ||
2 Results and discussion
2.1 Influence of the nature of the support
The selective oxidation of glycerol to GLYA was performed at 25 °C and 10 bar O2 under alkaline conditions. To contrast the catalytic performance of Au/ZrO2@C, Au NPs with the same weight loading supported on C, ZrO2 and ZrO2-C were also prepared as control catalysts. The catalytic performance of the supported Au catalysts for the selective aerobic oxidation of glycerol is shown in Fig. 1. The highest glycerol conversion (73%) was achieved over the Au/ZrO2@C catalyst after 6 h, together with the highest selectivity towards GLYA (79%). In contrast, the Au/ZrO2 catalyst exhibited the lowest catalytic activity for glycerol. Furthermore, the Au/ZrO2 catalyst exhibited the highest selectivity towards GLYA (72%), followed by Au/C (64%) and Au/ZrO2-C (55%). Moreover, the main byproduct of the reaction over the Au/ZrO2@C catalyst was DHA. For the Au/ZrO2-C catalyst, TA was also produced in addition to DHA and GLYCA.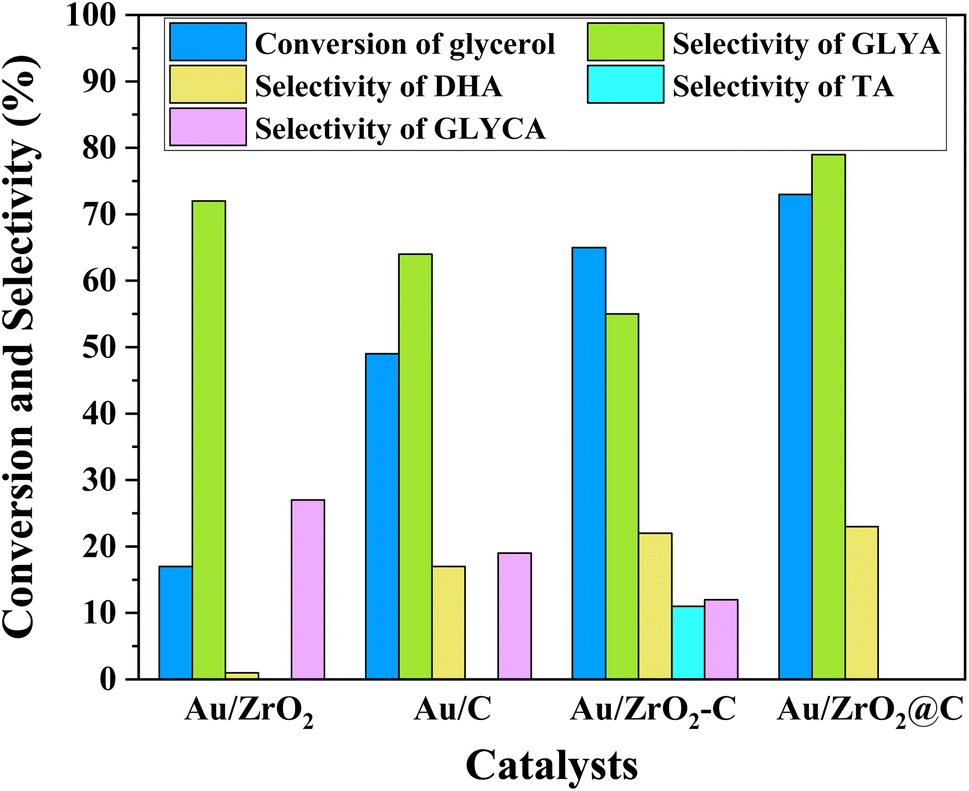 | ||
| Fig. 1 Catalytic performance of the Au/C, Au/ZrO2, Au/ZrO2-C and Au/ZrO2@C catalysts in glycerol oxidation (reaction conditions: 30 mL 0.1 M glycerol, glycerol/Au = 750 mol mol−1, T = 25 °C, p(O2) = 10 bar, stirring speed = 600 rpm, time = 6 h. GLYA: glyceric acid, DHA: dihydroxyacetone, GLYCA: glycolic acid, TA: tartronic acid). | ||
The physical–chemical properties of the Au/C, Au/ZrO2, Au/ZrO2-C and Au/ZrO2@C catalysts were analyzed to explain the distinct catalytic performance. The corresponding XRD patterns of pure UiO-66 are shown in Fig. S1,† and the diffraction peaks of UiO-66 with good crystallinity were very consistent with previously reported XRD patterns.44 The calcination temperature of UiO-66 was determined to be 600 °C through thermogravimetric (TG) analysis. This analysis revealed a mass loss of approximately 35%, as depicted in Fig S2.† The observed mass loss can be attributed to water evaporation and the decomposition of organic ligands. After reaching 600 °C, there were no significant changes in the mass loss, suggesting that the UiO-66 structure started to collapse and disintegrate, while simultaneously forming the stable ZrO2@C structure. Fig. 2a and b show XRD patterns of the support and the supported Au catalysts, respectively. After thermal decomposition of UiO-66 at 600 °C under an argon gas atmosphere, some new diffraction peaks appeared at 2θ = 30.2°, 35.3°, 50.2°, 59.3° and 60.2°, which corresponded to the (101), (110), (112), (103) and (211) lattice planes of tetragonal ZrO2, respectively (PDF card no. 79-1746). In addition, the characteristic peaks of carbon were not observed. This indicates that amorphous carbon was formed during the thermal decomposition process, which is in agreement with previous reports.45 For ZrO2 and ZrO2-C, diffraction peaks appeared at 2θ = 30.1°, 34.9°, 50.2°, and 59.7°, which corresponded to the (111), (200), (220) and (311) lattice planes of cubic ZrO2, respectively (PDF card no. 89-9069). Other diffraction peaks were attributed to monoclinic ZrO2 (PDF card no. 83-0936). These results indicated that ZrO2 was present in the monoclinic phase and tetragonal phase. For C, a broad peak centred at 24.6° was observed, which was ascribed to the formation of amorphous carbon regions and restacking due to attractive van der Waals interactions.46 The above results indicated that the different structures of the support affected the catalytic properties of the supported Au catalysts. In addition, two diffraction peaks were observed at 38.2° and 44.3° for all the supported Au catalysts, which were assigned to the (111) and (200) crystal planes of Au, respectively.
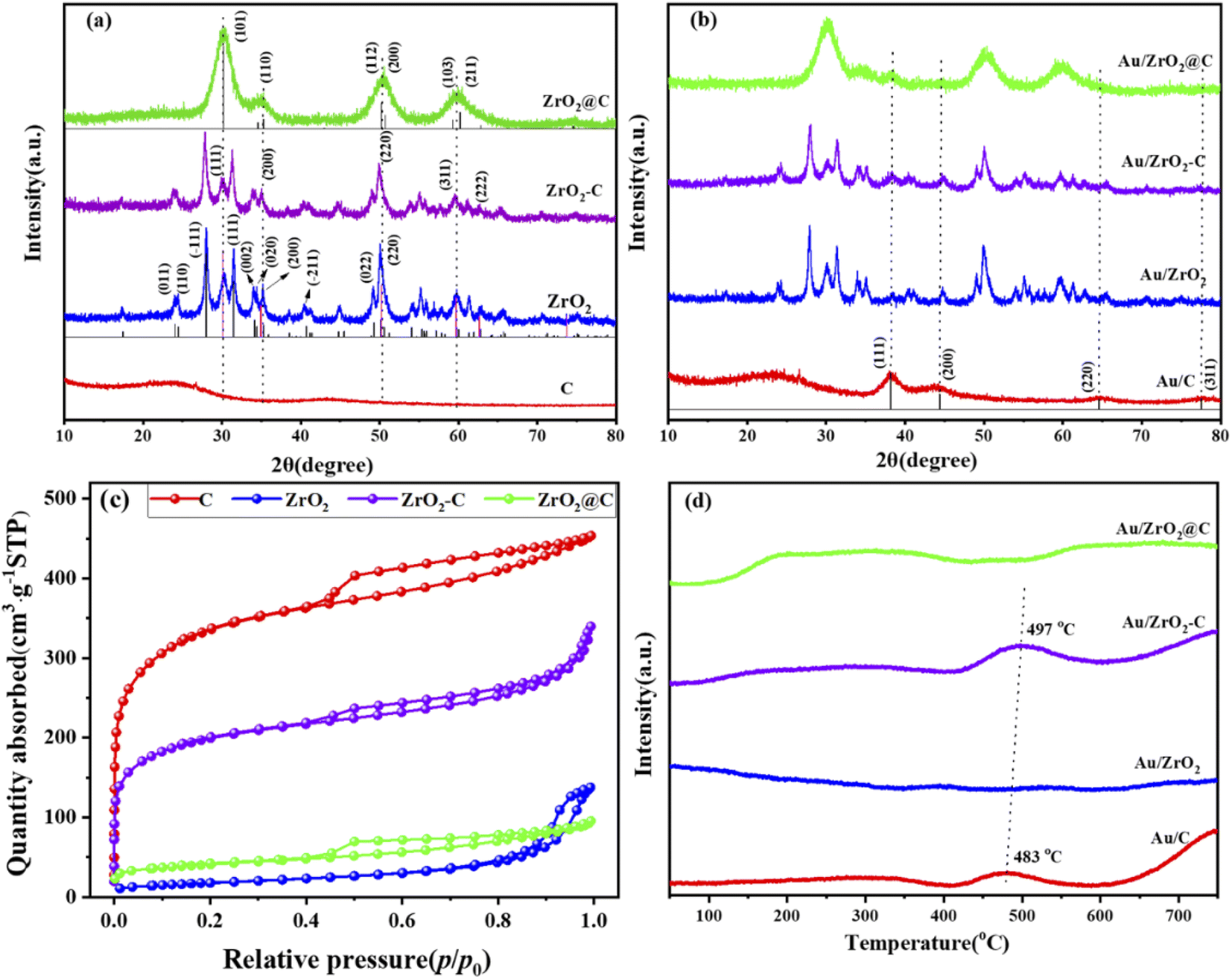 | ||
| Fig. 2 XRD patterns of the support (a) and supported Au catalysts (b). The nitrogen adsorption–desorption isotherms of the support (c). The H2-TPR profile of the supported Au catalysts (d). | ||
Fig. 2c and S4† show the N2 sorption isotherms of the support and supported Au catalysts, respectively. All samples except for ZrO2 gave rise to type I/IV isotherms with H4-shaped hysteresis loops, signifying the presence of both micropores and mesopores in these materials. Table 1 lists the texture parameters of the supports and corresponding supported Au catalysts. UiO-66 had a high specific surface area of 1320 m2 g−1. After calcination at a higher temperature, most micropores were lost, and ZrO2@C materials were formed. The specific surface area decreased considerably by 205 m2 g−1. The corresponding total pore volumes decreased from 0.57 cm3 g−1 to 0.15 cm3 g−1. After loading Au, slight decreases in the specific surface area and total pore volumes were observed for all catalysts (Table 1).
| Samples | BET surface area (m2 g−1) | Micropore area (m2 g−1) | External surface Area (m2 g−1) | Total pore volume (cm3 g−1) | Pore size distribution (nm) | Au loading (wt%) |
|---|---|---|---|---|---|---|
| UiO-66 | 1320 | 1138 | 182 | 0.57 | 2.28 | — |
| ZrO2@C | 205 | 77 | 128 | 0.23 | 4.40 | — |
| Au/ZrO2@C | 146 | 40 | 106 | 0.13 | 3.64 | 2.95 |
| C | 1120 | 532 | 588 | 0.70 | 2.50 | — |
| Au/C | 940 | 445 | 495 | 0.59 | 2.50 | 1.99 |
| ZrO2 | 64.5 | 0.7 | 63.8 | 0.21 | 13.19 | — |
| Au/ZrO2 | 61.5 | 0 | 61.5 | 0.17 | 11.12 | 2.41 |
| ZrO2-C | 663 | 322 | 341 | 0.53 | 3.17 | — |
| Au/ZrO2-C | 466 | 215 | 251 | 0.38 | 3.27 | 2.46 |
The H2-TPR profiles for the supported Au catalysts are shown in Fig. 2d. No H2 consumption was observed at less than 400 °C for all catalysts, suggesting that Au was present in the metallic state. For Au/ZrO2@C and Au/ZrO2 catalysts, because ZrO2 and C were irreducible, there were no peaks in the test temperature range. There was a small reduction peak at approximately 490 °C for the Au/ZrO2-C and Au/C catalysts, which was attributed to the reduction of oxygen-containing functional groups on the surface of activated carbon.47
Typical TEM micrographs, the average particle size distribution of Au NPs and the corresponding HRTEM images of the supported Au catalysts are shown in Fig. 3. It can be clearly seen that the Au NPs were well dispersed on the surface of all the supports. For the Au/ZrO2@C catalyst, the mean size of the Au NPs is smallest, which is approximately 2.65 ± 0.59 nm, followed by Au/ZrO2 > Au/C > Au/ZrO2-C. Furthermore, it was also found that the Au NPs were successfully supported onto the supports; for example, a crystalline structure of Au with an interplanar distance of 0.235 nm corresponding to the (111) plane was found.48 Moreover, the lattice fringes of ZrO2 can be clearly observed in Fig. 3(b-2) and (d-2). For the Au/ZrO2@C catalyst, the lattice spacing was 0.30 nm, corresponding to the (101) crystal plane of t-ZrO2. However, the lattice fringe of graphitic carbon was not observed. The results indicated that the t-ZrO2 were uniformly distributed in amorphous carbon.49
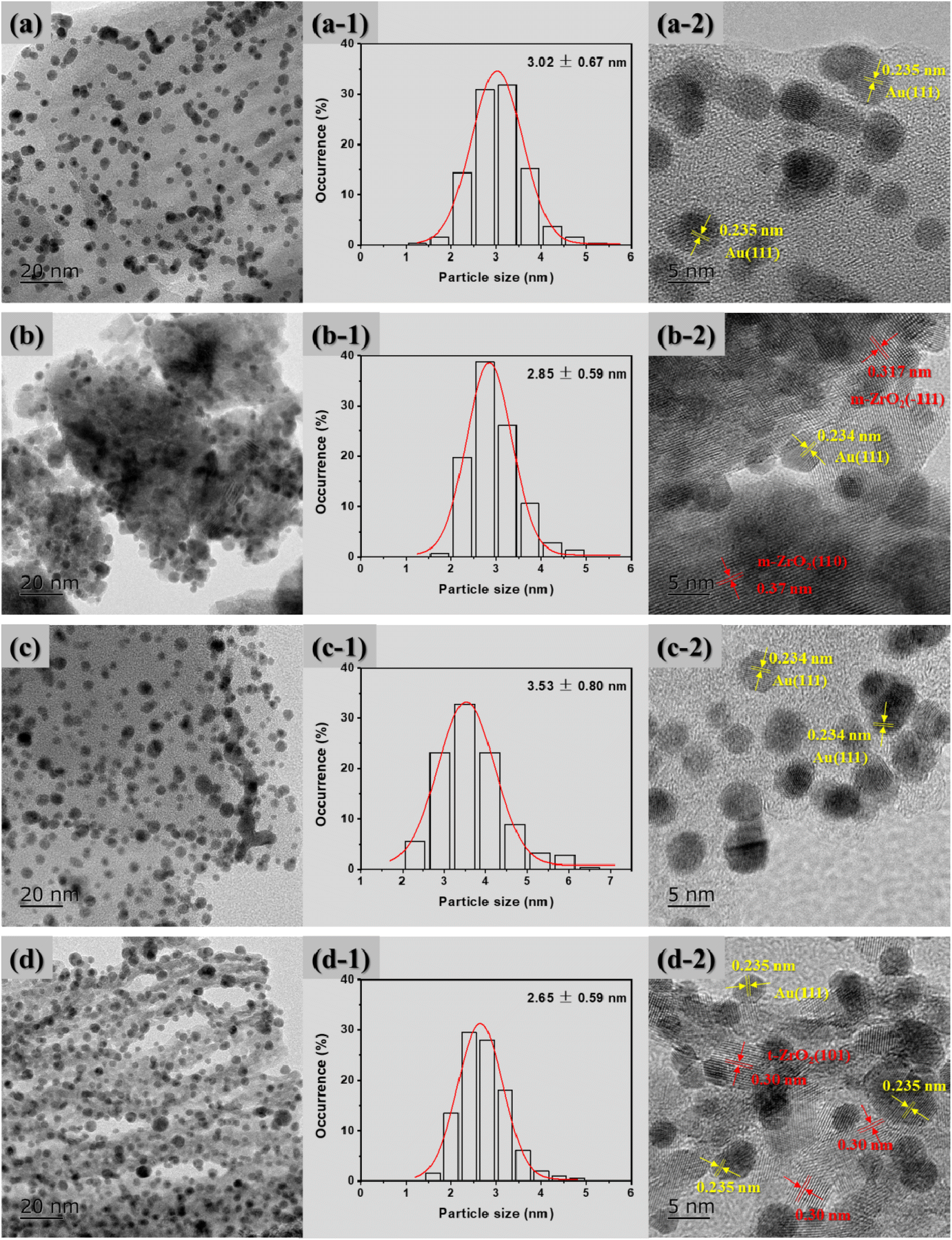 | ||
| Fig. 3 TEM micrographs, average particle size distribution of Au NPs and corresponding HRTEM images for the supported Au catalysts: (a) Au/C, (b) Au/ZrO2, (c) Au/ZrO2-C, and (d) Au/ZrO2@C. | ||
EDX mapping was used to investigate the element distribution in the Au/ZrO2@C catalyst. As shown in Fig. 4 and S3,† Au, Zr, C and O were well dispersed in the Au/ZrO2@C catalyst. These results also confirmed the successful incorporation of the ZrO2 species into the C framework.
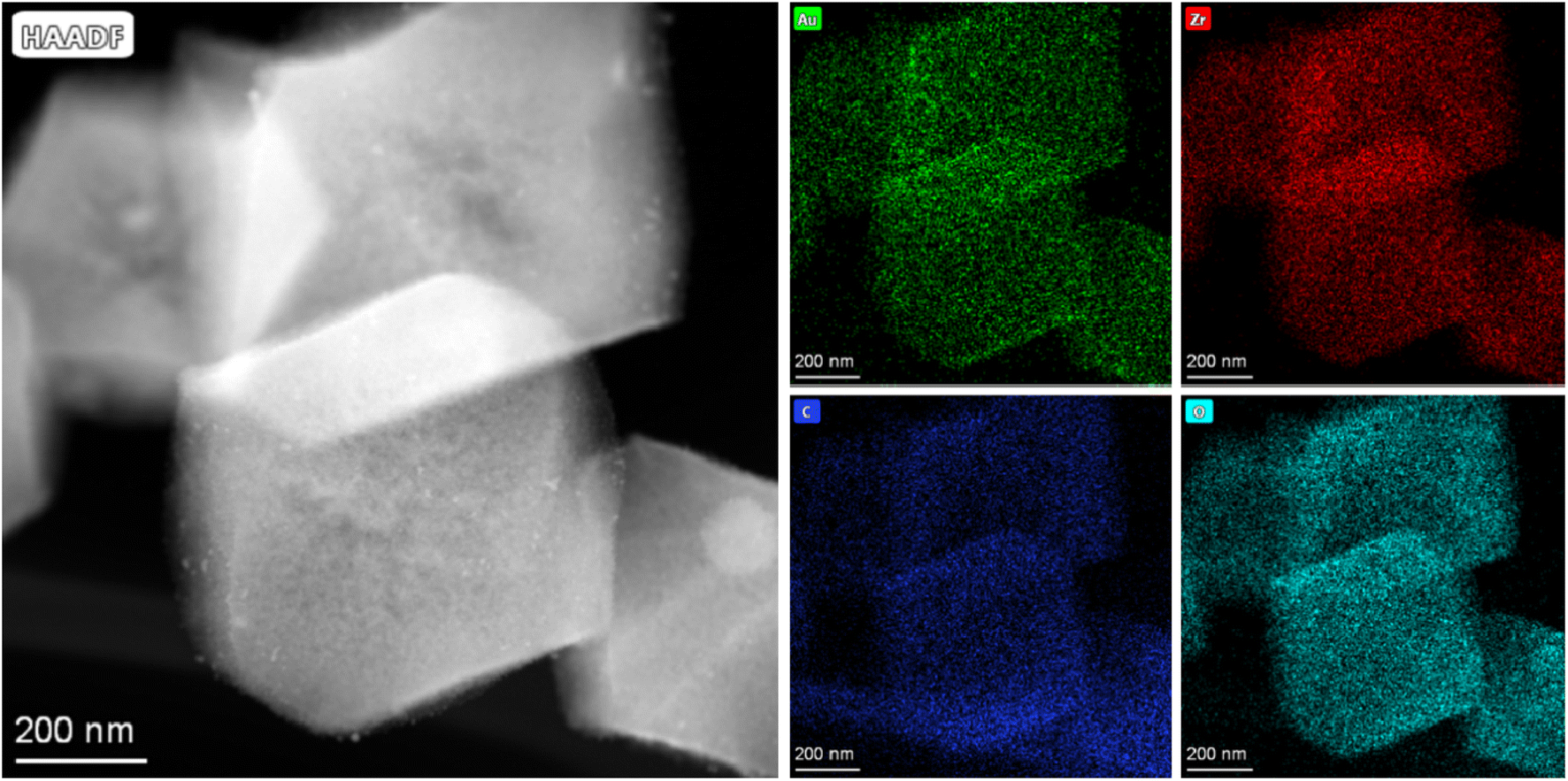 | ||
| Fig. 4 TEM images and corresponding EDX-mapping analysis of the Au/ZrO2@C catalyst. | ||
To further investigate the surface element composition and chemical state of all the catalysts, XPS was performed, and the results are shown in Fig. 5. Au, Zr, C and O were present in all catalysts (Fig. 5a). The Au 4f7/2 binding energies were 84.05 eV for all catalysts (Fig. 5b). These values are close to that of bulk Au, i.e., 84.0 eV, indicating that the Au particles were in the metallic state, which was also in good agreement with the H2-TPR characterization. Furthermore, the Au 4f peak of all catalysts was uniformly shifted to higher binding energies by 0.05 eV. Fitting of the O 1s data obtained for Au/ZrO2, Au/ZrO2-C, and Au/ZrO2@C catalysts resulted in peaks at approximately 529–530 eV, 531.5–532 eV and 533 eV, respectively, as shown in Fig. 3c, corresponding to O–C, O–C, and O–Zr bonds.50,51 The Zr 3d of zirconia usually shows two spin–orbit double peaks, Zr 3d5/2 and Zr 3d3/2, at binding energies of 182.1 eV and 184.5 eV, respectively. In Au/ZrO2 and Au/ZrO2-C catalysts, the binding energy of Zr 3d5/2 is lower than 182.1 eV, in contrast, there is a significant positive deviation of Zr 3d5/2 for Au/ZrO2@C, indicating electron transfer from Zr to Au. More specifically, the unique structure in Au/ZrO2@C can prevent oxidation of Au by providing access to electronic Au. In addition, no other peaks were found in the catalysts Au/ZrO2-C and Au/ZrO2@C, which demonstrates that this material does not contain ZrC, which is consistent with the XRD results.
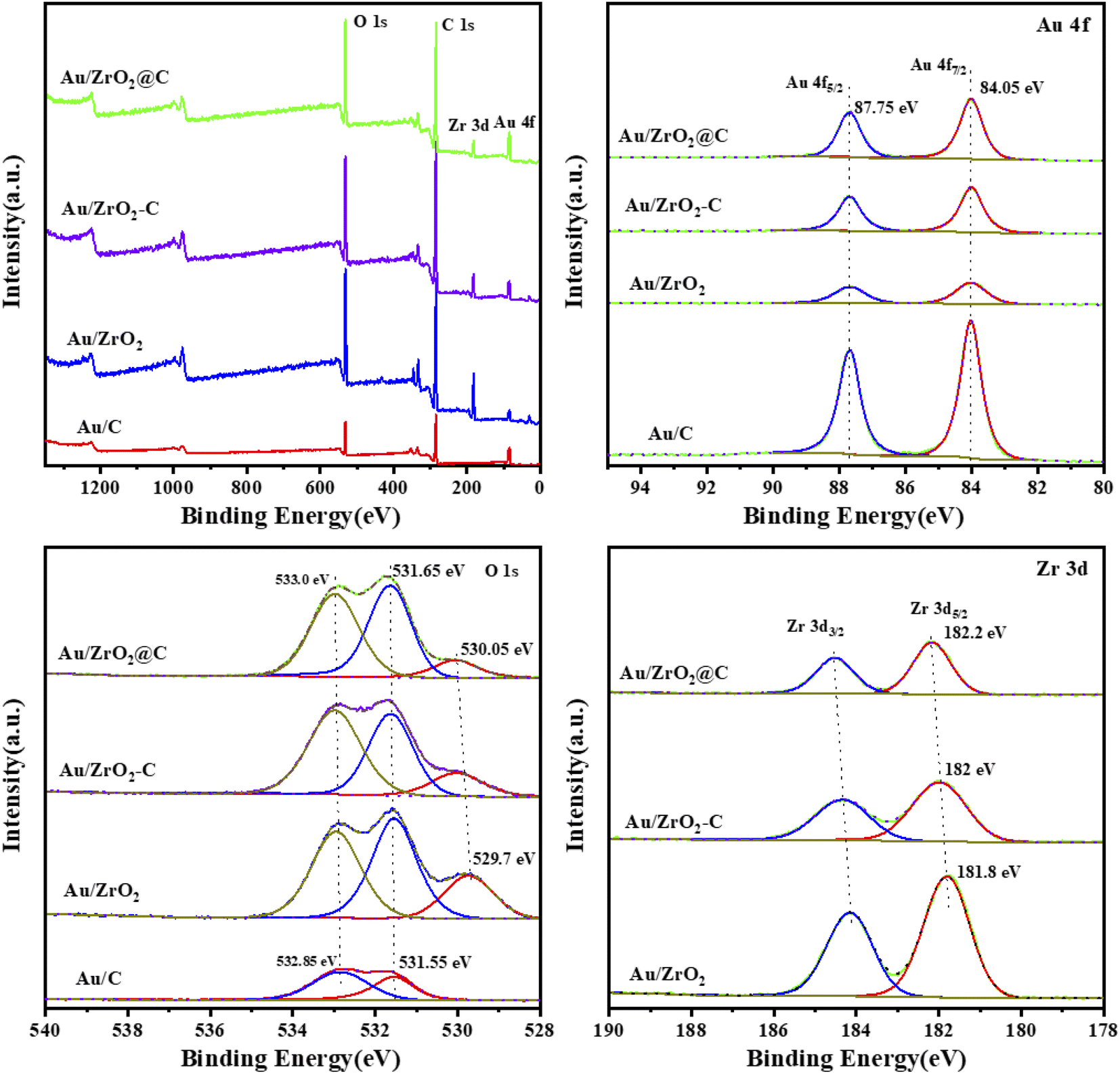 | ||
| Fig. 5 XPS profiles of various Au catalysts: (a) XPS survey spectrum, (b) Au 4f region, (c) O 1s region, and (d) Zr 3d region. | ||
The high activity of the Au/ZrO2@C catalyst can be explained based on the characterization of the catalysts. XRD showed that the structure of ZrO2@C was different from those of the other supports. TEM showed that the Au/ZrO2@C catalyst had the smallest metallic Au NPs, which were more active in the cleavage of the secondary C–H bond in glycerol molecules. EDX mapping showed that the high-density mesoporous structure of the Au/ZrO2@C catalyst effectively led to highly dispersed Au NPs. XPS showed that the highest electron transfer occurred from Zr to Au in the Au/ZrO2@C catalyst. This excellent catalytic performance was mainly ascribed to the strong metal–support interactions between Au NPs and the support on the interface.
2.2 Effect of reaction conditions
The influence of the reaction temperature was researched using the Au/ZrO2@C catalyst, and the results are shown in Fig. 6a. As expected, the conversion of glycerol increased with increasing reaction temperature. When the temperature was raised from 15 °C to 40 °C, the conversion of glycerol increased from 67% to 100%. Upon further increasing the temperature, no change was observed in the conversion of glycerol. The selectivity for GLYA first increased and then decreased with increasing temperature, and the highest selectivity for GLYA (79%) was achieved at 25 °C. Although high reaction temperatures ensure thermodynamic desorption of these molecules from the active site, the accompanying side reactions often result in reduced selectivity, especially for reactants with multiple functional groups.52,53 Fig. 6a shows that the yield of GLYA at 40 °C was much higher than that at other temperatures (67%); however, the amount of byproducts increased significantly. In general, a given catalyst with a strong adsorption energy for molecules at low temperatures has poor catalytic activity because strong adsorption of intermediates and products blocks the interfacial active site.54 From the point of view of energy conservation and environmental protection, the optimum temperature was 25 °C.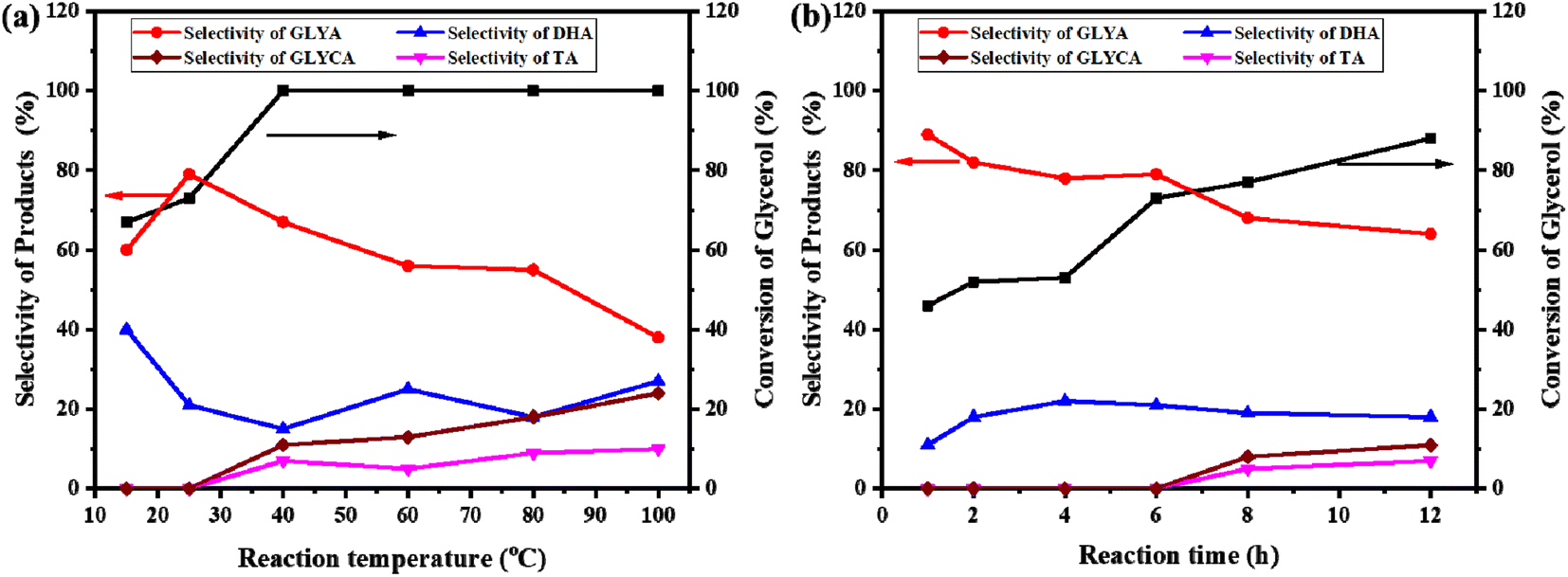 | ||
| Fig. 6 Influence of reaction conditions on the selective oxidation of glycerol to GLYA. (a) Effect of the reaction temperature; reaction conditions: 30 mL 0.1 M glycerol, glycerol/Au = 750 mol mol−1, 6 h, P(O2) = 10 bar. (b) Effect of the reaction time, reaction conditions: 30 mL 0.1 M glycerol, glycerol/Au = 750 mol mol−1, 25 °C, P(O2) = 10 bar. | ||
Fig. 6b shows the conversion of glycerol and the selectivity for products with different reaction times over the Au/ZrO2@C catalyst at 25 °C while keeping the other conditions constant. By prolonging the reaction time from 1 to 12 h, the conversion of glycerol increased from 46% to 88%. However, the selectivity towards GLYA decreased significantly with time. In addition to GLYA and DHA, other byproducts, such as GLYCA and TA, were also detected after 6 h, indicating that the reaction time has an effect on the synthesis of GLYA from glycerol.
2.3 Catalytic mechanism
For Au-based catalysts, alkaline conditions rather than acidic conditions are favourable for catalysis because under alkaline conditions, hydroxide anions can induce the removal of H on the primary hydroxyl group of glycerol, thus initiating the oxidation process of glycerol.20 Based on the results obtained in this work, a hypothetical reaction pathway of the selective oxidation of glycerol to GLYA over the Au/ZrO2@C catalyst was proposed, as shown in Scheme 2. The different surface Au and Zr species are involved in the proposed reaction cycle. The role of the hydroxyl groups from the reaction medium is pointed out.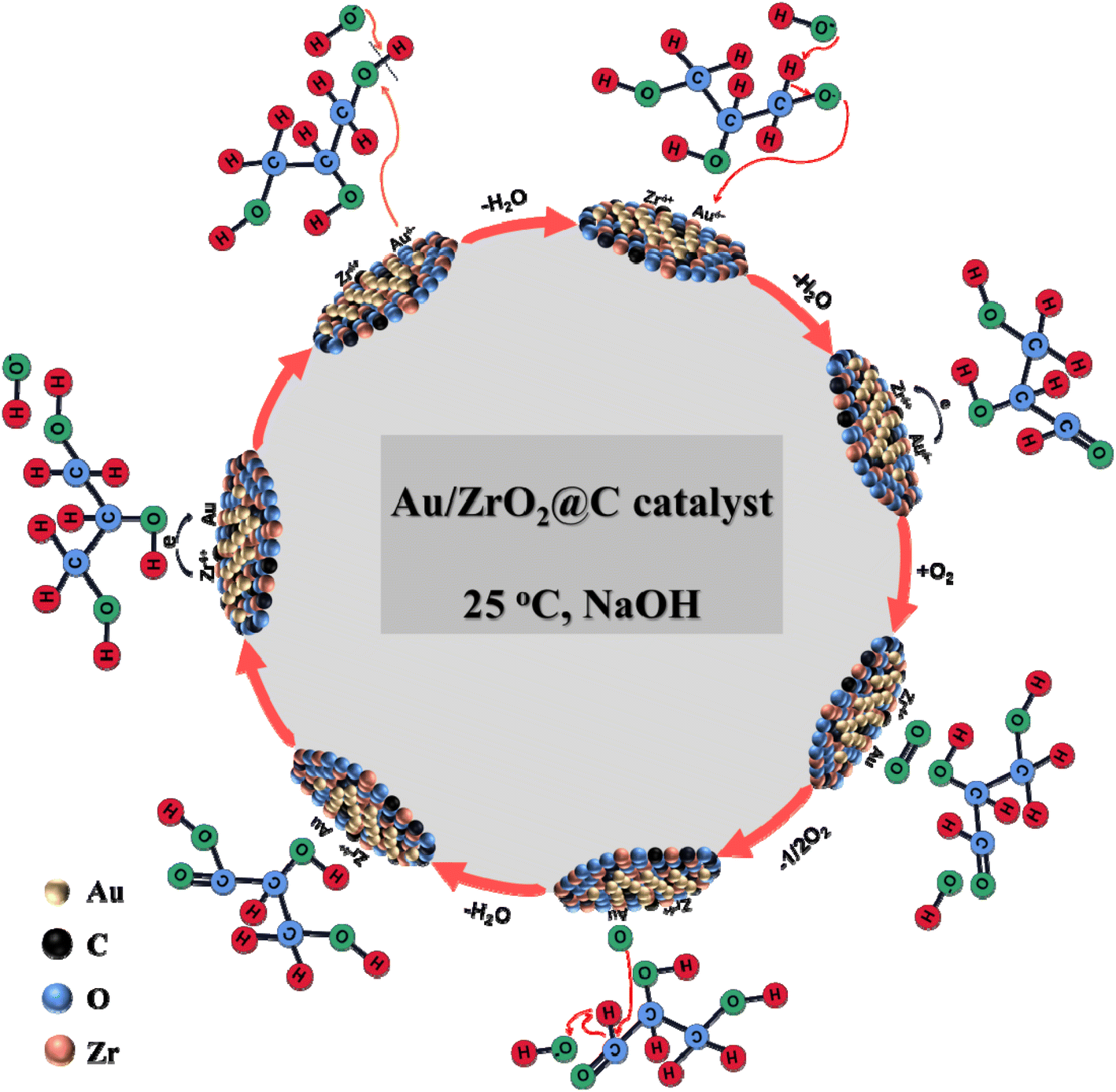 | ||
| Scheme 2 The reaction pathway of the oxidation glycerol to GLYA over the Au/ZrO2@C catalyst. | ||
First, it is commonly accepted that metal alkoxide formation by adsorption of an alcohol on the Au surface could be the initial step during alcohol oxidation over Au catalysts.55 In the presence of hydroxide anions, the O–H bond of the terminal hydroxyl group is activated and initiated dehydrogenation by H extraction. Furthermore, the interaction of negative charges on the Au formed by electron transfer of the Zr4+ species on the carbon-based material enhances the cleavage of the O–H bond. In the second step, aldehydes are produced by β-H elimination at Au NPs, which can occur via the transfer of either a hydrogen atom or hydride.56 Subsequently, the lattice oxygens are replenished by adsorbed oxygen on the support surface. Finally, the adsorbed oxygen is dissociatively chemisorbed on metallic Au NPs, hydrogen from the aldehyde group is oxidized to the carboxyl group under the synergistic action of the hydroxide anion, and GLYA is formed.57,58
2.4 Catalyst reusability
To evaluate the recyclability of the catalyst, the Au/ZrO2@C catalyst was tested over three runs. After each run, the catalyst was filtered, washed with deionized water and dried at 80 °C for 12 h. Then, the recycled catalyst was used again under the same reaction conditions. As shown in Fig. 7a. The fresh Au/ZrO2@C catalyst exhibited 73% glycerol conversion and 79% GLYA selectivity. The conversion of glycerol and the selectivity of GLYA obviously decreased with an increasing number of cycles. After the third cycle, the conversion of glycerol decreased to 43%, and the selectivity of GLYA decreased to 52%. To explain the deactivation of the Au/ZrO2@C catalyst, the used catalyst and the liquid-phase were characterized by XPS and ICP-AES, respectively. The results of the ICP-AES analysis are shown in Table S1.† It was observed that Au and Zr were dissolved in the reaction mixture after every cycle. These results indicated that the structure of the catalyst changed after the reaction, which may be a reason for the decrease in catalytic activity. The results of the XPS analysis (Fig. 7b–d) confirmed that the binding energy of Au 4f7/2 decreased after three runs (from 84.05 eV to 83.96 eV), indicating that the Au–support interaction changed after the reaction. In addition, the content of Au on the surface clearly decreased, which may be caused by Au dissolution during the reaction process. After three runs, the O 1s spectrum showed a significant decrease in the O–C peak intensities. These results indicated that the chemical state of O changed after recycling.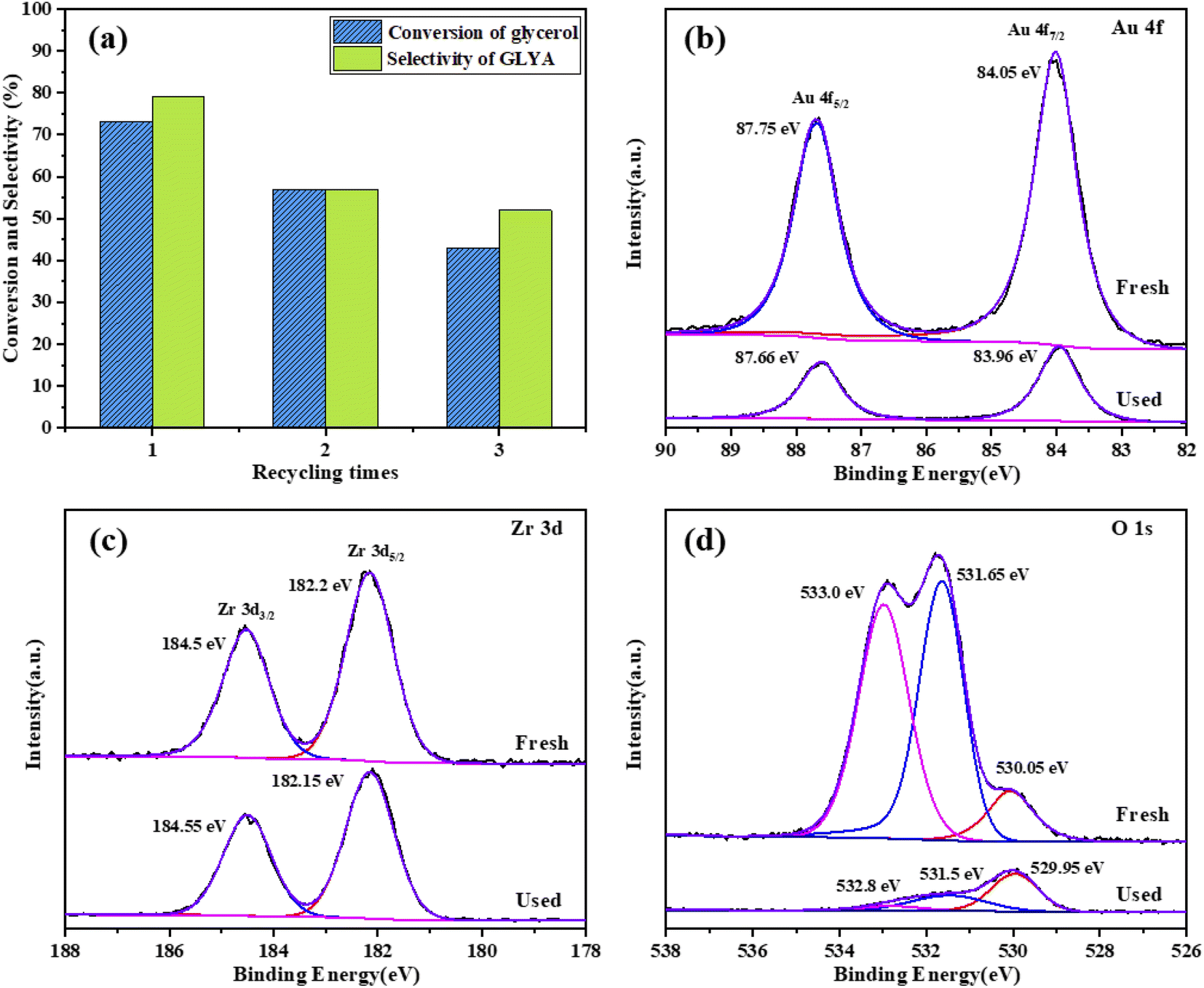 | ||
| Fig. 7 (a) Recycling of the Au/ZrO2@C catalyst. (b–d) XPS spectra of the Au/ZrO2@C catalyst: fresh and after being used three times. | ||
3 Conclusion
In this work, the Au/ZrO2@C catalyst exhibits excellent catalytic performance for the oxidation of glycerol to GLYA at room temperature (25 °C), which provides a prospective process for the catalytic conversion of biomass sources at room temperature. The catalytic activity of the Au/ZrO2@C catalyst is superior to that of the Au/C, Au/ZrO2, and Au/ZrO2-C catalysts. A combination study including XRD, TEM, and XPS confirms that the high activity of the Au/ZrO2@C catalyst is ascribed to the support structure, the strong metal–support interactions between Au NPs and the support, and the highly dispersed Au NPs. arising from the preparation method. Therefore, this work provides a facile strategy for glycerol oxidation at room temperature. Unfortunately, a few insufficiencies, such as catalyst stability, still remain to be solved.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number 21862001), the Foundation of Academic Backbone Talent Support Program of the North Minzu University (grant number 2019BGGZ12), the Innovative Team for Transforming Waste Cooking Oil into Clean Energy and High Value-Added Chemicals (grant number 2022QCXTD03) and the Ningxia Low-Grade Resource High Value Utilization and Environmental Chemical Integration Technology Innovation Team Project, China.References
- A. Demirbas, Progress and recent trends in biodiesel fuels, Energy Convers. Manage., 2009, 50(1), 14–34, DOI:10.1016/j.enconman.2008.09.001.
- J. Silva, M. Soria and L. Madeira, Challenges and Strategies for Optimization of Glycerol Steam Reforming Process, Renewable Sustainable Energy Rev., 2015, 42, 1187–1213, DOI:10.1016/j.rser.2014.10.084.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Glycerol hydrogenolysis into useful C3 chemicals, Appl. Catal., B, 2016, 193, 75–92, DOI:10.1016/j.apcatb.2016.04.013.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Glycerol as a potential renewable raw material for acrylic acid production, Green Chem., 2017, 19, 3186–3213, 10.1039/C7GC00358G.
- Y. Gu, A. Azzouzi, Y. Pouilloux and F. Jerome, J. Barrault. Heterogeneously catalyzed etherification of glycerol: new pathways for transformation of glycerol to more valuable chemicals, Green Chem., 2008, 10, 164–167, 10.1039/b715802e.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Lactic acid as a platform chemical in the biobased economy: the role of chemocatalysis, Energy Environ. Sci., 2013, 6, 1415–1442, 10.1039/c3ee00069a.
- V. O. Samoilov, D. N. Ramazanov, A. I. Nekhaev, A. L. Maximov and L. N. Bagdasarov, Heterogeneous catalytic conversion of glycerol to oxygenated fuel additives, Fuel, 2016, 172, 310–319, DOI:10.1016/j.fuel.2016.01.024.
- M. A. Ayude, L. I. Doumic, M. C. Cassanello and K. D. P. Nigam, Clean catalytic oxidation for derivatization of key biobased platform chemicals: ethanol, glycerol, and hydroxymethyl furfural, Ind. Eng. Chem. Res., 2019, 58(35), 16077–16095, DOI:10.1021/acs.iecr.9b00977.
- B. Katryniok, S. Paul and F. Dumeignil, Recent developments in the field of catalytic dehydration of glycerol to acrolein, ACS Catal., 2013, 3(8), 1819–1834, DOI:10.1021/cs400354p.
- X. M. Ning, Y. H. Li, H. Yu, F. Peng, H. J. Wang and Y. H. Yang, Promoting role of bismuth and antimony on Pt catalysts for the selective oxidation of glycerol to dihydroxyacetone, J. Catal., 2016, 335, 95–104, DOI:10.1016/j.jcat.2015.12.020.
- L. Yang, X. Li, P. Chen and Z. Hou, Selective oxidation of glycerol in a base-free aqueous solution: A short review, Chin. J. Catal., 2019, 40(7), 1020–1034, DOI:10.1016/S1872-2067(19)63301-2.
- Z. He, X. Ning, G. Yang, H. Wang, Y. Cao, F. Peng and H. Yu, Selective oxidation of glycerol over supported noble metal catalysts, Catal. Today, 2021, 365, 162–171, DOI:10.1016/j.cattod.2020.04.019.
- G. Dodekatos, S. Schünemann and H. Tüysüz, Recent advances in Thermo-, Photo-, and Electrocatalytic glycerol oxidation, ACS Catal., 2018, 8, 6301–6333, DOI:10.1021/acscatal.8b01317.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Glycerol oxidation using gold-containing catalysts, Acc. Chem. Res., 2015, 48, 1403–1412, DOI:10.1021/ar500426g.
- S. E. Davis, M. S. Ide and R. J. Davis, Selective oxidation of alcohols and aldehydes over supported metal nanoparticles, Green Chem., 2013, 15, 17–45, 10.1039/c2gc36441g.
- J. Zhang, X. Li, M. Xu, Y. Yang, Y. Li, N. Liu, X. Meng, L. Chen, S. Shi and M. Wei, Glycerol aerobic oxidation to glyceric acid over Pt/hydrotalcite catalysts at room temperature, Sci. Bull., 2019, 64(23), 1764–1772, DOI:10.1016/j.scib.2019.10.003.
- H. Habe, T. Fukuoka, D. Kitamoto and K. Sakaki, Biotechnological production of D-glyceric acid and its application, Appl. Microbiol. Biotechnol., 2009, 84(3), 445–452, DOI:10.1007/s00253-009-2124-3.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Reactivity of the gold–water interface during selective oxidation catalysis, Science, 2010, 330, 74–78, DOI:10.1126/science.1195055.
- G. Dodekatos, L. Abis, S. J. Freakley, H. Tueysuez and G. J. Hutchings, Glycerol oxidation using MgO- and Al2O3-supported gold and gold-palladium nanoparticles prepared in the absence of polymer stabilizers, ChemCatChem, 2018, 10(6), 1351–1359, DOI:10.1002/cctc.201800074.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffifin, C. J. Kiely and G. J. Hutchings, Oxidation of glycerol using supported Pt, Pd and Au catalysts, Phys. Chem. Chem. Phys., 2003, 5(6), 1329–1336, 10.1039/B212047J.
- S. Demirel-Gülen, M. Lucas and P. Claus, Liquid phase oxidation of glycerol over carbon supported gold catalysts, Catal. Today, 2005, 102, 166–172, DOI:10.1016/j.cattod.2005.02.033.
- D. Liang, J. Gao, H. Sun, P. Chen, Z. Y. Hou and X. M. Zheng, Selective oxidation of glycerol with oxygen in a base-free aqueous solution over MWNTs supported Pt catalysts, Appl. Catal., B, 2011, 106(3), 423–432, DOI:10.1016/j.apcatb.2011.05.050.
- Y. Sun, X. Li, J. G. Wang, W. S. Ning, J. Fu, X. Y. Lu and Z. Y. Hou, Carbon film encapsulated Pt NPs for selective oxidation of alcohols in acidic aqueous solution, Appl. Catal., B, 2017, 218, 538–544, DOI:10.1016/j.apcatb.2017.06.086.
- L. Yang, X. Li, Y. Y. Sun, L. H. Yue, J. Fu, X. Y. Lu and Z. Y. Hou, Selective oxidation of glycerol in base-free conditions over N-doped carbon film coated carbon supported Pt catalysts, Catal. Commun., 2017, 101, 107–110, DOI:10.1016/j.catcom.2017.08.008.
- Y. Zhou, K. Neyerlin, T. S. Olson, S. Pylypenko, J. Bult, H. N. Dinh, T. Gennett, Z. Shao and R. O′Hayre, Enhancement of Pt and Pt-alloy fuelcellcatalyst activity and durability vianitrogen-modified carbon supports, Energy Environ. Sci., 2010, 3(10), 1437–1446, 10.1039/C003710A.
- X. Ning, Y. Li, B. Dong, H. Wang, H. Yu, F. Peng and Y. Yang, Electron transfer dependent catalysis of Pt on N-doped carbon nanotubes: Effects of synthesis method on metal–support interaction, J. Catal., 2017, 348, 100–109, DOI:10.1016/j.jcat.2017.02.011.
- X. Wang, N. Li, J. A. Webb, L. D. Pfefferle and G. L. Haller, Effect of surface oxygen containing groups on the catalytic activity of multi-walled carbon nanotube supported Pt catalyst, Appl. Catal., B, 2010, 101(1–2), 21–30, DOI:10.1016/j.apcatb.2010.08.028.
- M. Sankar, Q. He, R. V. Engel, M. A. Sainna, A. J. Logsdail, A. Roldan, D. J. Willock, N. Agarwal, C. J. Kiely and G. J. Hutchings, Role of the support in gold-containing nanoparticles as heterogeneous catalysts, Chem. Rev., 2020, 120(8), 3890–3938, DOI:10.1021/acs.chemrev.9b00662.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin and G. J. Hutchings, Selective oxidation of glycerol to glyceric acid using a gold catalyst in aqueous sodium hydroxide, Chem. Commun., 2002, 7, 696–697, 10.1039/B201112N.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Solvent-free oxidation of primary alcohols to aldehydes using Au-Pd/TiO2 catalysts, Science, 2006, 311(5759), 362–365, DOI:10.1126/science.1120560.
- S. Gil, M. Marchena, L. Sánchez-Silva, A. Romero, P. Sánchez and J. L. Valverde, Effect of the operation conditions on the selective oxidation of glycerol with catalysts based on Au supported on carbonaceous materials, Chem. Eng. J., 2011, 178, 423–435, DOI:10.1016/j.cej.2011.10.048.
- W. C. Ketchie, Y. Fang, M. S. Wong, M. Murayama and R. J. Davis, Influence of gold particle size on the aqueous-phase oxidation of carbon monoxide and glycerol, J. Catal., 2007, 250(1), 94–101, DOI:10.1016/j.jcat.2007.06.011.
- E. G. Rodrigues, M. F. R. Pereira, X. Chen, J. J. Delgado and J. J. M. Órfão, Influence of activated carbon surface chemistry on the activity of Au/AC catalysts in glycerol oxidation, J. Catal., 2011, 281(1), 119–127, DOI:10.1016/j.jcat.2011.04.008.
- E. G. Rodrigues, J. J. Delgado, X. Chen, M. F. R. Pereira and J. J. M. Órfão, Selective oxidation of glycerol catalyzed by gold supported on multiwalled carbon nanotubes with different surface chemistries, Ind. Eng. Chem. Res., 2012, 51(49), 15884–15894, DOI:10.1021/ie302159m.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Su, Applications of metal–organic frameworks in heterogeneous supramolecular catalysis, Chem. Soc. Rev., 2014, 43(16), 6011–6061, 10.1039/C4CS00094C.
- K. Shen, X. Chen, J. Chen and Y. Li, Development of MOF-derived carbon-based nanomaterials for efficient catalysis, ACS Catal., 2016, 6, 5887–5903, DOI:10.1021/acscatal.6b01222.
- Q. Yang, Q. Xu and H. Jiang, Metal–organic frameworks meet metal nanoparticles: synergistic effect for enhanced catalysis, Chem. Soc. Rev., 2017, 46(15), 4774–4808, 10.1039/C6CS00724D.
- Y. Chen, R. Zhang, L. Jiao and H. Jiang, Metal–organic framework–derived porous materials for catalysis, Coord. Chem. Rev., 2018, 362, 1–23, DOI:10.1016/j.ccr.2018.02.008.
- R. Zhang, L. Jiao, W. Yang, G. Wan and H. Jiang, Single-atom catalysts templated by metal–organic frameworks for electrochemical nitrogen reduction, J. Mater. Chem. A, 2019, 7(46), 26371–26377, 10.1039/C9TA10206J.
- C. P. Cabello, M. F. F. Picó, F. Maya, M. D. Rio and G. T. Palomino, UiO-66 derived etched carbon/polymer membranes: High-performance supports for the extraction of organic pollutants from water, Chem. Eng. J., 2018, 346, 85–93, DOI:10.1016/j.cej.2018.04.019.
- W. Cao, W. Luo, H. Ge, Y. Su, A. Wang and T. Zhang, UiO-66 derived Ru/ZrO2@ C as a highly stable catalyst for hydrogenation of levulinic acid to γ-valerolactone, Green Chem., 2017, 19(9), 2201–2211, 10.1039/C7GC00512A.
- S. Li, J. Pan, X. Wu, Y. Fu, F. Zhang and W. Zhu, Metal–organic framework derived Pd/ZrO2@ CN as a stable catalyst for the catalytic hydrogenation of 2, 3, 5-trimethylbenzoquinone, Appl. Organomet. Chem., 2019, 33(12), e5233, DOI:10.1002/aoc.5233.
- Q. Wang, W. Sun, T. Xie, L. Cao and J. Yang, Metal–organic framework (MOF) template based efficient Pt/ZrO2@ C catalysts for selective catalytic reduction of H2 below 90 °C, Chem.–Asian J., 2019, 14(3), 416–421, DOI:10.1002/asia.201801680.
- L. Shi, T. Wang, H. Zhang, K. Chang and J. Ye, Electrostatic self–assembly of nanosized carbon nitride nanosheet onto a zirconium metal–organic framework for enhanced photocatalytic CO2 reduction, Adv. Funct. Mater., 2015, 25(33), 5360–5367, DOI:10.1002/adfm.201502253.
- S. Liu, Z. Yue and Y. Liu, Mesoporous carbon–ZrO2 composites prepared using thermolysis of zirconium based metal–organic frameworks and their adsorption properties, J. Porous Mater., 2015, 22(2), 465–471, DOI:10.1007/s10934-015-9915-y.
- R. Nie, J. Wang, L. Wang, Y. Qin, P. Chen and Z. Hou, Platinum supported on reduced graphene oxide as a catalyst for hydrogenation of nitroarenes, Carbon, 2012, 50(2), 586–596, DOI:10.1016/j.carbon.2011.09.017.
- L. Huang, X. Li and Y. Chen, The effect of the oxidation method of an activated carbon on the selective catalytic reduction of NOx with NH3 over CeO2/activated carbon catalysts, New Carbon Mater., 2018, 33(3), 237–244 CAS.
- T. Tian, Y. Liu and X. Zhang, Bimetallic synergistic Au/CuO-hydroxyapatite catalyst for aerobic oxidation of alcohols, Chin. J. Catal., 2015, 36(8), 1358–1364, DOI:10.1016/S1872-2067(15)60854-3.
- Y. Jiang, G. Cheng, Y. Li, Z. He, J. Zhu, W. Meng, L. Dai and L. Wang, Promoting vanadium redox flow battery performance by ultra-uniform ZrO2@C from metal–organic framework, Chem. Eng. J., 2021, 415, 129014, DOI:10.1016/j.cej.2021.129014.
- J. Qiao, X. Zhang, D. Xu, L. Kong, L. Lv, F. Yang, F. Wang, W. Liu and J. Liu, Design and synthesis of TiO2/Co/carbon nanofibers with tunable and efficient electromagnetic absorption, Chem. Eng. J., 2020, 380, 122591, DOI:10.1016/j.cej.2019.122591.
- M. A. Rahman, S. Rout, J. P. Thomas, D. McGillivray and K. T. Leung, Defect-rich dopant-free ZrO2 nanostructures with superior dilute ferromagnetic semiconductor properties, J. Am. Chem. Soc., 2016, 138(36), 11896–11906, DOI:10.1021/jacs.6b06949.
- M. S. Ide, B. Hao, M. Neurock and R. J. Davis, Mechanistic insights on the hydrogenation of α,β-Unsaturated ketones and aldehydes to unsaturated alcohols over metal catalysts, ACS Catal., 2012, 2(4), 671–683, DOI:10.1021/cs200567z.
- Y. Yang, D. Rao, Y. Chen, S. Dong, B. Wang, X. Zhang and M. Wei, Selective hydrogenation of cinnamaldehyde over Co-based intermetallic compounds derived from layered double hydroxides, ACS Catal., 2018, 8(12), 11749–11760, DOI:10.1021/acscatal.8b02755.
- S. Zhang, Z. Xia, M. Zhang, Y. Zou, H. Shen, J. Li, X. Chen and Y. Qu, Boosting selective hydrogenation through hydrogen spillover on supported-metal catalysts at room temperature, Appl. Catal., B, 2021, 297, 120418, DOI:10.1016/j.apcatb.2021.120418.
- J. Gong, D. W. Flaherty, T. Yan and C. B. Mullins, Selective oxidation of propanol on Au(111): mechanistic insights into aerobic oxidation of alcohols, ChemPhysChem, 2008, 9(17), 2461–2466, DOI:10.1002/cphc.200800680.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, Spin trapping of Au-H intermediate in the alcohol oxidation by supported and unsupported gold catalysts, J. Am. Chem. Soc., 2009, 131(20), 7189–7196, DOI:10.1021/ja809883c.
- H. Wang, W. Fan, Y. He, J. Wang, J. N. Kondo and T. Tatsumi, Selective oxidation of alcohols to aldehydes/ketones over copper oxide-supported gold catalysts, J. Catal., 2013, 299, 10–19, DOI:10.1016/j.jcat.2012.11.018.
- P. Kaminski, M. Ziolek and J. A. van Bokhoven, Mesoporous cerium–zirconium oxides modified with gold and copper–synthesis, characterization and performance in selective oxidation of glycerol, RSC Adv., 2017, 7(13), 7801–7819, 10.1039/C6RA27671G.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04300b |
| This journal is © The Royal Society of Chemistry 2023 |