DOI:
10.1039/D3RA04102F
(Paper)
RSC Adv., 2023,
13, 21021-21035
Solvatomorphism and first-time observation of acid–acid catemer in 4-phenylamino-benzoic acids†
Received
18th June 2023
, Accepted 30th June 2023
First published on 20th July 2023
Abstract
To investigate the polymorphism in 4-phenylamino-benzoic acids (4-PABAs) in general, and the effect on the polymorphism of these compounds exerted by substitution in particular, a series of 4-PABAs (1–8) varying in the substitution position and pattern were synthesized, and their polymorphic behavior was investigated for the first time. A relatively comprehensive polymorph screening led to the discovery of two forms, one solvent-free and the other solvate, for compounds 1, 3 and 8, and one form for the other compounds. The crystal structures were determined by single-crystal XRD. All the 4-PABAs in the crystal structures are highly twisted, and all the solvent-free crystals are based on the conventional acid–acid dimer motif, except for 2, which has a rarely observed acid–acid catemer motif. Two of the solvates (1-S and 8-S) have pyridine in the lattice while the other (3-S) has dichloromethane. The observation indicates that neither conformational flexibility or substitution alone nor the combination of both leads to polymorphism in these compounds, which is in dramatic contrast to the polymorphism of fenamic acids. The thermal properties of each system were investigated by differential scanning calorimetry and desolvation of the solvates was studied by thermogravimetric analysis. Hirshfeld surface analysis and molecular dynamics simulation were performed to study the mechanism of polymorphism and the intermolecular interactions contributing to the formation and stability of each crystal form.
1. Introduction
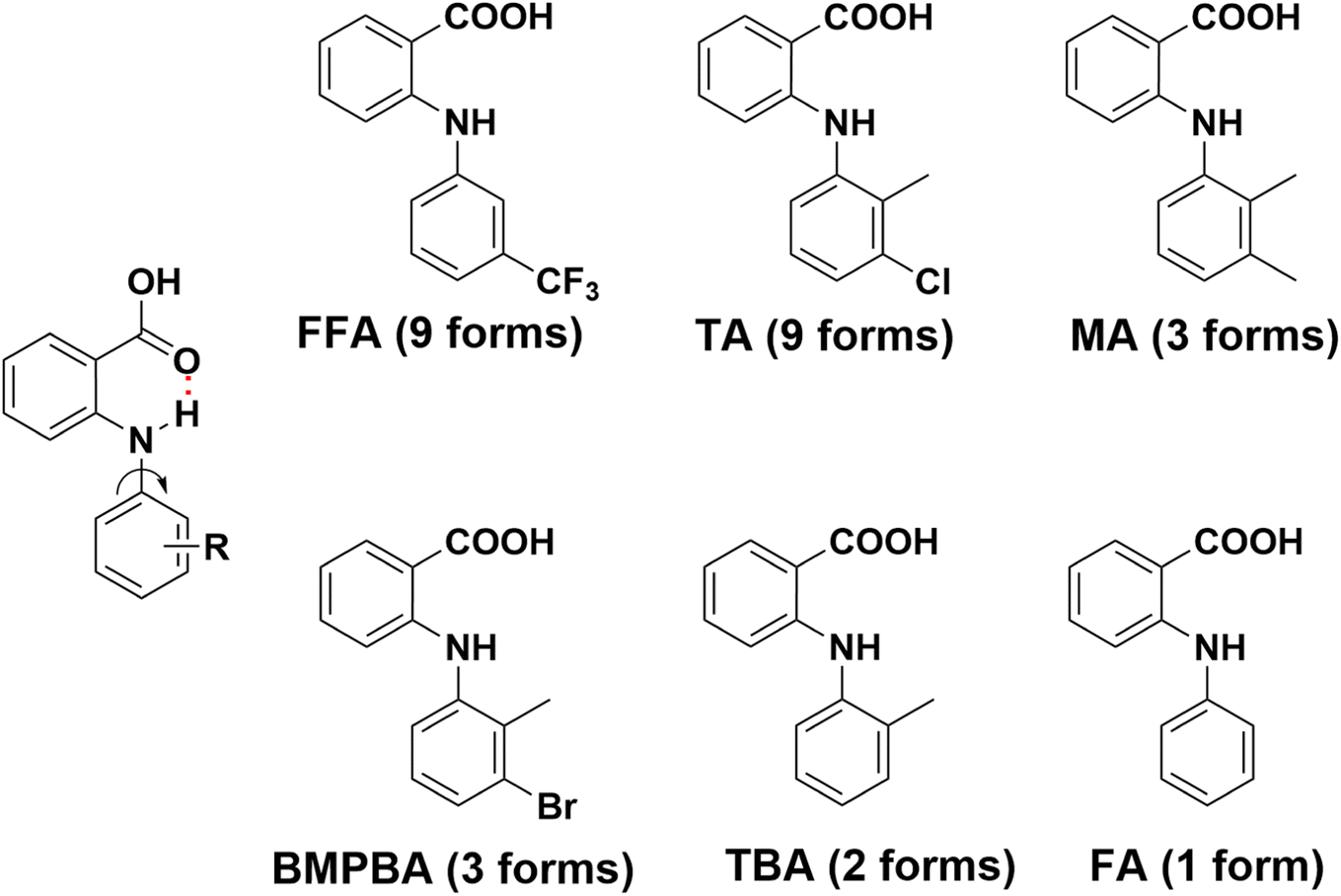
Anthranilic acids are diarylamines with medical applications in a variety of contexts, such as NSAIDs,1–3 analgesics4,5 and antirheumatics,6,7 antibacterial,8,9 antiviral,10 antitubercular11,12 etc., and recently they have been investigated as therapeutics for amyloid diseases,13 Alzheimer's disease,14,15 and cancer.16–18 These compounds are also fascinating as far as solid-state structures are concerned, due to their functional group diversity and conformational flexibility. Extensive solid-state studies have been focused on these compounds and many of them are polymorphic, i.e. the existence of multiple solid forms, and some are highly polymorphic (Fig. 1). For example, nine polymorphs have been discovered for both flufenamic acid (FFA)19 and tolfenamic acid (TA)20 so far. And mefenamic acid (MA),21 2-(3-bromo-2-methylphenyl)aminobenzoic acid (BMPBA) and 2-(o-tolylamino)benzoic acid (TBA)22 each has three, three and two forms reported, respectively. But not all the anthranilic acids are polymorphic. For example, the parent molecule, i.e. fenamic acid (FA) has only one crystal form discovered, despite an exhaustive polymorph screening.23,24 In nearly all the crystal forms of anthranilic acids, acid–acid dimer is the only observed structural motif, and an intramolecular hydrogen bond between NH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is persistent. The rise of polymorphism is mainly ascribed to the rotation about the N–Caniline bond (Fig. 1), since nearly all anthranilic acids are conformationally flexible to certain degrees. But other factors cannot be ruled out, for example, substitution obviously plays an important role. To account for the polymorphism of some anthranilic acids, Price and Matzger proposed that polymorphophore,25,26 a collective ensemble of conformational, steric, and electronic features, is what leads to the polymorphic behavior of these compounds. Polymorphophore was also identified for other systems.27–29
O is persistent. The rise of polymorphism is mainly ascribed to the rotation about the N–Caniline bond (Fig. 1), since nearly all anthranilic acids are conformationally flexible to certain degrees. But other factors cannot be ruled out, for example, substitution obviously plays an important role. To account for the polymorphism of some anthranilic acids, Price and Matzger proposed that polymorphophore,25,26 a collective ensemble of conformational, steric, and electronic features, is what leads to the polymorphic behavior of these compounds. Polymorphophore was also identified for other systems.27–29
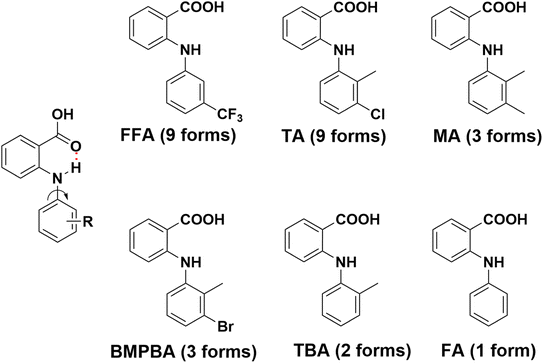 |
| | Fig. 1 Molecular structure and number of polymorphs identified for some representative anthranilic acids. | |
Fascinated by the polymorphism of anthranilic acids, we wondered what polymorphic behavior 4-phenylamino-benzoic acids (4-PABAs), structural isomers of anthranilic acids, would exhibit. By moving the anilino group to the para position of the carboxylic acid, not only its conformational flexibility is retained, but also the NH is freed as it is no longer locked in an intramolecular hydrogen bond with the carboxylic acid. Intuitively, the compounds would be more prone to be polymorphic both in a sense of conformational flexibility and synthon diversity. We were curious if the polymorphic behavior of 4-PABAs is similar to or dramatically different from that of anthranilic acids.
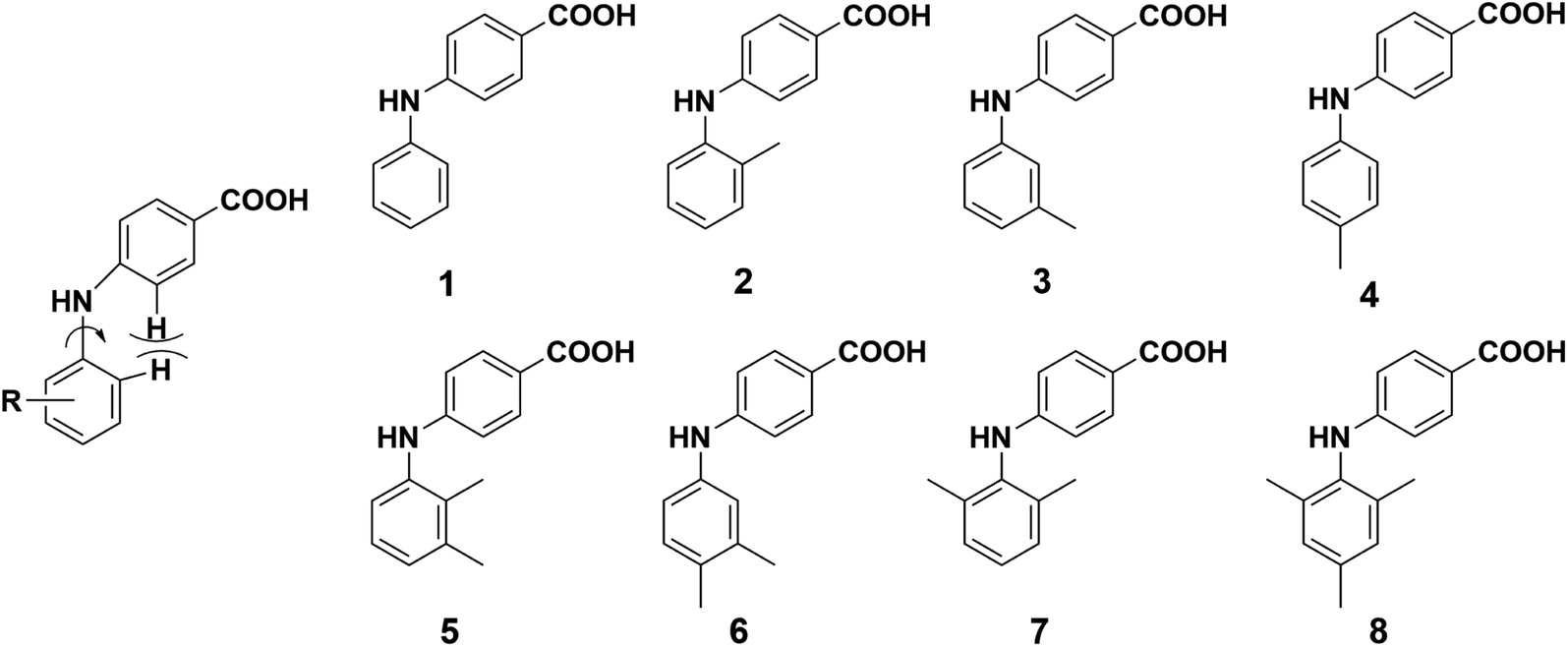
In this study, we designed and synthesized eight 4-PABAs (1–8, Fig. 2) by varying the substitution position and pattern to investigate the effect substitution would exert on the solid-state behavior of these compounds. To start, we limited the substituents to methyl group(s). Out of the eight compounds, compounds 2, 5, 6, 7, 8 are synthesized for the first time. The other compounds are known but their solid behavior has not been studied.30,31 Compounds 1, 3, and 4 are potent and selective aldo–keto reductase 1C3 (AKR1C3) inhibitors which are efficacious in a prostate cancer model and are potential therapeutic agents for the treatment of castration-resistant prostate cancer (CRPC).32,33
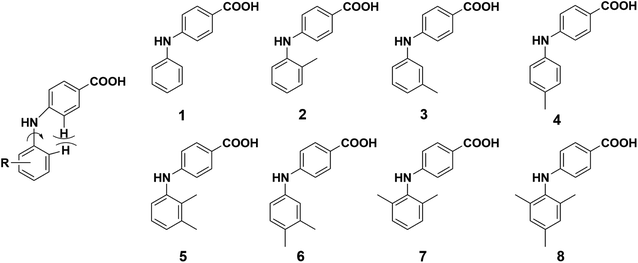 |
| | Fig. 2 Structure of compounds 1–8. | |
Crystallization of these compounds under the same conditions led to the discovery of varying numbers of crystal forms for each compound. All crystal forms were fully characterized by single-crystal XRD (SCXRD), powder XRD (PXRD), and FT-IR, except for 3-S, crystals of which were of poor quality. Their phase behavior was investigated with differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). To shed light on the role played by substitution and solvent(s) in the polymorphism of these compounds, theoretical studies were also performed.
2. Experimental section
2.1. Materials
Reagents for the synthesis of compounds 1–8 and solvents for crystallization were purchased from commercial sources: 4-chlorobenzoic acid, Cs2CO3, BINAP, and 2,4,6-trimethylaniline were from Bidepharm (Shanghai, China); aniline, 2-methylaniline, 3-methylaniline, 4-methylaniline, and 2,6-dimethylaniline were from Energy Chemical (Shanghai, China); 2,3-dimethylaniline and 3,4-dimethylaniline were from Meryer Chemical (Shanghai, China); Pd(OAc)2 was from Rock New Materials (Shaanxi, China); the solvents used for crystallization were from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
2.2. Synthesis and characterization
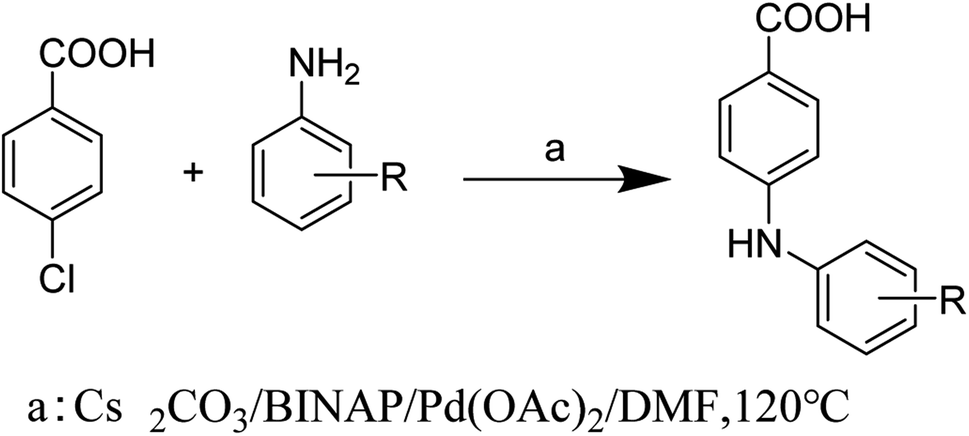
Compounds 1–8 were synthesized by the Buchwald–Hartwig reaction34,35 (Scheme 1) and purified by column chromatography and recrystallization. Each compound was characterized by 1H NMR, 13C NMR, IR, and melting point measurement, and for the new compounds, mass spectrometry (MS) was also performed (for details, see ESI†).
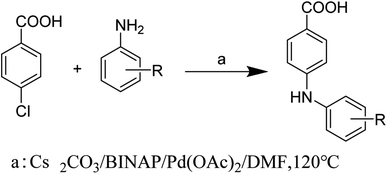 |
| | Scheme 1 Synthesis of compounds 1–8. | |
2.3. Crystallization, structure determination, and thermal studies
Slow evaporation in a variety of commonly used solvents (ranging from apolar to polar aprotic and polar protic) was employed for polymorph screening for all eight compounds,36 and high-quality single crystals were used for structure determination by SCXRD. The identity of individual batches of crystals was determined by PXRD.
The crystallographic data of all crystal forms were collected on a Rigaku Oxford diffractometer at ambient temperatures, except for 1-I, for which data collection was carried out on a Bruker APEX-II diffractometer with a CuKα radiation (λ = 1.54184 Å). Cell refinement and data reduction were performed using CrysAlisPro. Structure solution and refinement were carried out using the SHELXT37 and SHELXL38 programs, respectively. PXRD data for the crystal forms were collected on a Rigaku X-ray diffractometer with CuKα radiation (40 kV, 15 mA, λ = 1.5406 Å) between 5.0 and 50.0° (2θ) at ambient temperatures.
DSC experiments were performed on SII instruments DSC6220 (Seiko Instruments Inc., Japan). TGA experiments were run on SDT Q600 (TA Instruments, USA). Tzero® pans and aluminum hermetic lids were used for measuring a few milligrams of a finely ground sample, at a heating rate of 10 °C min−1.
2.4. Computational details
2.4.1. Hirshfeld surface analysis. Hirshfeld surface analyses39 were performed with CrystalExplorer (Version 3.1)40 to further understand the relative contributions to crystal stability by various intermolecular interactions in the crystals of these compounds.
2.4.2. Molecular dynamics simulation. API (4-PABAs herein) molecules and solvent molecules were described with the second generation generalized Amber force field (GAFF2),41 and charges of atoms in the molecules were calculated using the AM1-BCC method.42,43 Size of simulation box was 50 Å × 50 Å × 50 Å, and 35–38 API molecules (solubilities of 90 mg mL−1 in solvents) and a proper number of solvent molecules were put in the simulation box. Molecule simulations were performed with GROMACS 2022.3.44 van der Waals and electrostatic interactions were calculated using the cutoff method and particle mesh Ewald (PME) method with the same cutoff of 1.2 nm, respectively. NPT simulation of a total of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 steps was conducted at 300 K, and the time step was 2 fs. During the simulation, temperature and pressure were maintained by the Berendsen method. Four hundred structures were extracted from the trajectory of the last 4000000 steps with one structure every 10000 steps. Hydrogen bonds between the molecules in these structures were analyzed. The distance between hydrogen bond donor and acceptor was set to 3.5 Å.
000000 steps was conducted at 300 K, and the time step was 2 fs. During the simulation, temperature and pressure were maintained by the Berendsen method. Four hundred structures were extracted from the trajectory of the last 4000000 steps with one structure every 10000 steps. Hydrogen bonds between the molecules in these structures were analyzed. The distance between hydrogen bond donor and acceptor was set to 3.5 Å.
3. Results and discussion
3.1. Crystallization results
The condition and solvents used for crystallization, and the form(s) obtained in individual solvents are summarized in Table S1.† Two forms were obtained for compounds 1, 3, and 8, and only one form was discovered for the other compounds. Representative crystals of each crystal form are shown in Fig. 3.
 |
| | Fig. 3 Representative crystals of different crystal forms of 1–8. Scale bar 0.2 mm (EA: ethyl acetate, Py: pyridine; MeOH: methanol; CH3CN: acetonitrile; DCM: dichloromethane; CHCl3: chloroform; EtOH: ethanol). | |
The crystallographic data of all the crystal forms are listed in Table 1, and complete CIF files are provided in the ESI.† For 3-S, its crystal structure was not solved due to poor crystal quality and ease of losing solvent (dichloromethane), but its existence was confirmed by 1H NMR, PXRD, DSC and TGA.
Table 1 Crystallographic data of all crystal forms of compounds 1–8
| |
1-I |
1-S |
2 |
3-I |
4 |
| Formula |
C13H11NO2 |
C18H16N2O2 |
C14H13NO2 |
C14H13NO2 |
C14H13NO2 |
| Formula weight |
213.23 |
292.33 |
227.25 |
227.25 |
227.25 |
| Crystal size |
0.18 × 0.15 × 0.13 |
0.15 × 0.14 × 0.08 |
0.15 × 0.09 × 0.07 |
0.13 × 0.11 × 0.07 |
0.11 × 0.08 × 0.05 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21/n |
P21/n |
P21 |
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å |
5.8122(6) |
8.2172(2) |
8.71260(10) |
13.5689(6) |
5.8469(2) |
| b/Å |
29.731(3) |
11.5108(3) |
6.04470(10) |
5.8946(2) |
6.4881(3) |
| c/Å |
6.4123(7) |
16.6802(5) |
10.84520(10) |
14.9692(8) |
15.9753(7) |
| α/° |
90 |
90 |
90 |
90 |
80.106(4) |
| β/° |
106.167(11) |
95.056(3) |
91.9720(10) |
98.919(4) |
85.810(3) |
| γ/° |
90 |
90 |
90 |
90 |
73.682(4) |
| Z, Z′ |
4, 1 |
4, 1 |
2, 1 |
4, 1 |
2, 1 |
| V/Å3 |
1064.2(2) |
1571.59(8) |
570.825(13) |
1182.81(9) |
572.77(4) |
| Dcal/g × cm−3 |
1.331 |
1.235 |
1.322 |
1.276 |
1.318 |
| T/K |
294.9(5) |
282(20) |
302.98(10) |
302.98(10) |
302.99(10) |
| Abscoeff (mm−1) |
0.736 |
0.657 |
0.718 |
0.693 |
0.715 |
| F(000) |
448 |
616 |
240 |
480 |
240 |
| Flack parameter |
N/A |
N/A |
0.04(5) |
N/A |
N/A |
| θ range (deg) |
2.973–77.767 |
4.673–77.495 |
4.079–75.818 |
4.093–7.409 |
5.624–76.268 |
| Limiting indices |
−6 ≤ h ≤ 7 |
−10 ≤ h ≤ 9 |
−10 ≤ h ≤ 10 |
−13 ≤ h ≤ 17 |
−7 ≤ h ≤ 7 |
| −34 ≤ k ≤ 36 |
−14 ≤ k ≤ 8 |
−7 ≤ k ≤ 6 |
−7 ≤ k ≤ 6 |
−7 ≤ k ≤ 8 |
| −7 ≤ l ≤ 8 |
−20 ≤ l ≤ 20 |
−13 ≤ l ≤ 12 |
−18 ≤ l ≤ 18 |
−17 ≤ l ≤ 19 |
| Completeness to 2θ |
92.2% |
94.0% |
96.2% |
93.8% |
93.6% |
| Unique reflections |
2102 |
2563 |
2097 |
1719 |
1950 |
| R1 [I > 2σ(I)] |
0.0480 |
0.0429 |
0.0301 |
0.0519 |
0.0432 |
| wR2 (all data) |
0.1494 |
0.1283 |
0.0775 |
0.1678 |
0.1350 |
| CSD accession code |
2260090 |
2260091 |
2260092 |
2260093 |
2260094 |
| |
5 |
6 |
7 |
8-I |
8-S |
| Formula |
C15H15NO2 |
C15H15NO2 |
C15H15NO2 |
C16H17NO2 |
C21H22N2O2 |
| Formula weight |
482.56 |
241.28 |
241.28 |
255.30 |
334.40 |
| Crystal size |
0.12 × 0.09 × 0.06 |
0.18 × 0.15 × 0.13 |
0.21 × 0.15 × 0.12 |
0.14 × 0.11 × 0.07 |
0.15 × 0.12 × 0.11 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P |
P21/n |
P21/c |
P21/c |
P |
| a/Å |
6.2842(2) |
14.6295(14) |
5.8609(4) |
15.8309(2) |
8.0697(5) |
| b/Å |
13.1283(4) |
6.0707(6) |
21.0733(11) |
16.98433(19) |
8.4048(4) |
| c/Å |
15.8787(3) |
15.0512(17) |
10.4926(6) |
11.30860(15) |
14.8801(6) |
| α/° |
88.249(2) |
90 |
90 |
90 |
85.239(4) |
| β/° |
83.997(2) |
112.042(12) |
94.188(5) |
108.8907(15) |
74.568(5) |
| γ/° |
79.406(3) |
90 |
90 |
90 |
73.318(5) |
| Z, Z′ |
2, 2 |
4, 1 |
2, 1 |
8, 2 |
2, 1 |
| V/Å3 |
1280.52(6) |
1239.0(2) |
1292.46(13) |
2876.85(7) |
931.88(9) |
| Dcal/g × cm−3 |
1.252 |
1.293 |
1.240 |
1.179 |
1.192 |
| T/K |
293(2) |
303.88(10) |
301.97(10) |
287.3(3) |
303.99(10) |
| Abscoeff (mm−1) |
0.668 |
0.691 |
0.082 |
0.620 |
0.613 |
| F(000) |
512 |
512 |
512 |
1088 |
356 |
| Flack parameter |
N/A |
N/A |
N/A |
N/A |
N/A |
| θ range (deg) |
2.798–77.590 |
3.593–76.003 |
1.933–30.736 |
2.950–77.506 |
3.081–76.453 |
| Limiting indices |
−7 ≤ h ≤ 7 |
−17 ≤ h ≤ 18 |
−6 ≤ h ≤ 7 |
−19 ≤ h ≤ 19 |
−10 ≤ h ≤ 10 |
| −16 ≤ k ≤ 11 |
−5 ≤ k ≤ 7 |
−22 ≤ k ≤ 28 |
−19 ≤ k ≤ 21 |
−10 ≤ k ≤ 10 |
| −19 ≤ l ≤ 19 |
−18 ≤ l ≤ 18 |
−15 ≤ l ≤ 12 |
−14 ≤ l ≤ 14 |
−14 ≤ l ≤ 18 |
| Completeness to 2θ |
94.2% |
92.8% |
80.1% |
94.6% |
95.5% |
| Unique reflections |
4064 |
1766 |
2102 |
4782 |
2753 |
| R1 [I > 2σ(I)] |
0.0575 |
0.0552 |
0.0469 |
0.0501 |
0.0460 |
| wR2 (all data) |
0.1782 |
0.1638 |
0.1395 |
0.1639 |
0.1417 |
| CSD accession code |
2260096 |
2260099 |
2260100 |
2260101 |
2260102 |
All the crystal forms of compounds 1–8 can be grouped into two categories: three solvatomorphic systems, and five singletons. The solvatomorphic systems are of compounds 1, 3, and 8; compounds 2, 4, 5, 6, and 7 have only one form obtained.
3.2. Crystal structural properties
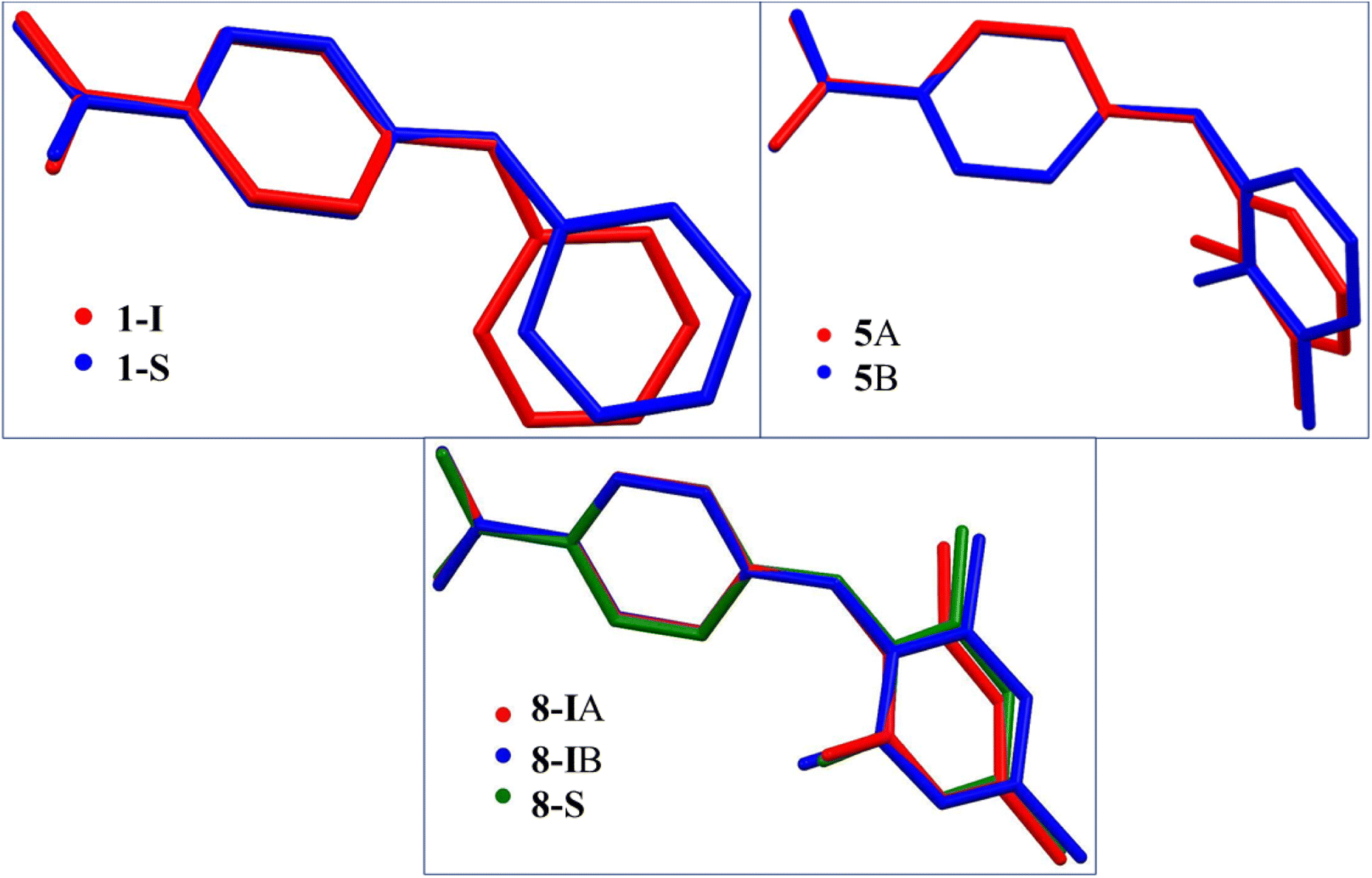
3.2.1. General aspects. There is only one molecule in the asymmetric units of 1-I, 2, 3-I, 4, 6 and 7. There are two molecules in the asymmetric units of the two solvates 1-S and 8-S, one is the host molecule and the other is pyridine, and there are two molecules in the asymmetric units of 5 and 8-I. Due to the repulsion between the CHs ortho to NH on both aromatic rings, the crystallographically independent molecules in all crystal structures are twisted to different degrees as evidenced by the dihedral angle between the two aromatic rings: 52.38(6)° for 1-I, 46.95(5)° for 1-S; 66.82(6)° for 2, 48.60(6)° for 3-I, 50.34(4)° for 4, 78.02(6)° for 5A, 67.42(6)° for 5B, 51.16(6)° for 6, 70.49(5)° for 7, 79.43(5)° for 8-IA, 65.59(5)° for 8-IB, and 70.96(5)° for 8-S. The superposition of the different conformations in compounds 1, 5, and 8 is provided in Fig. 4.
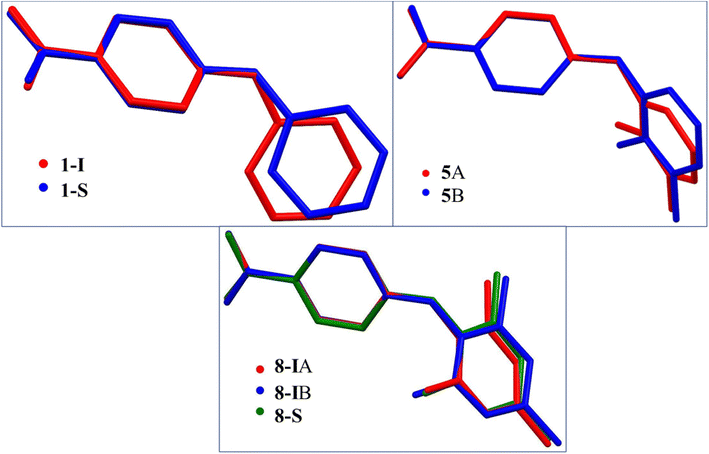 |
| | Fig. 4 Superposition of the crystallographically independent molecules in compounds 1, 5, and 8. | |
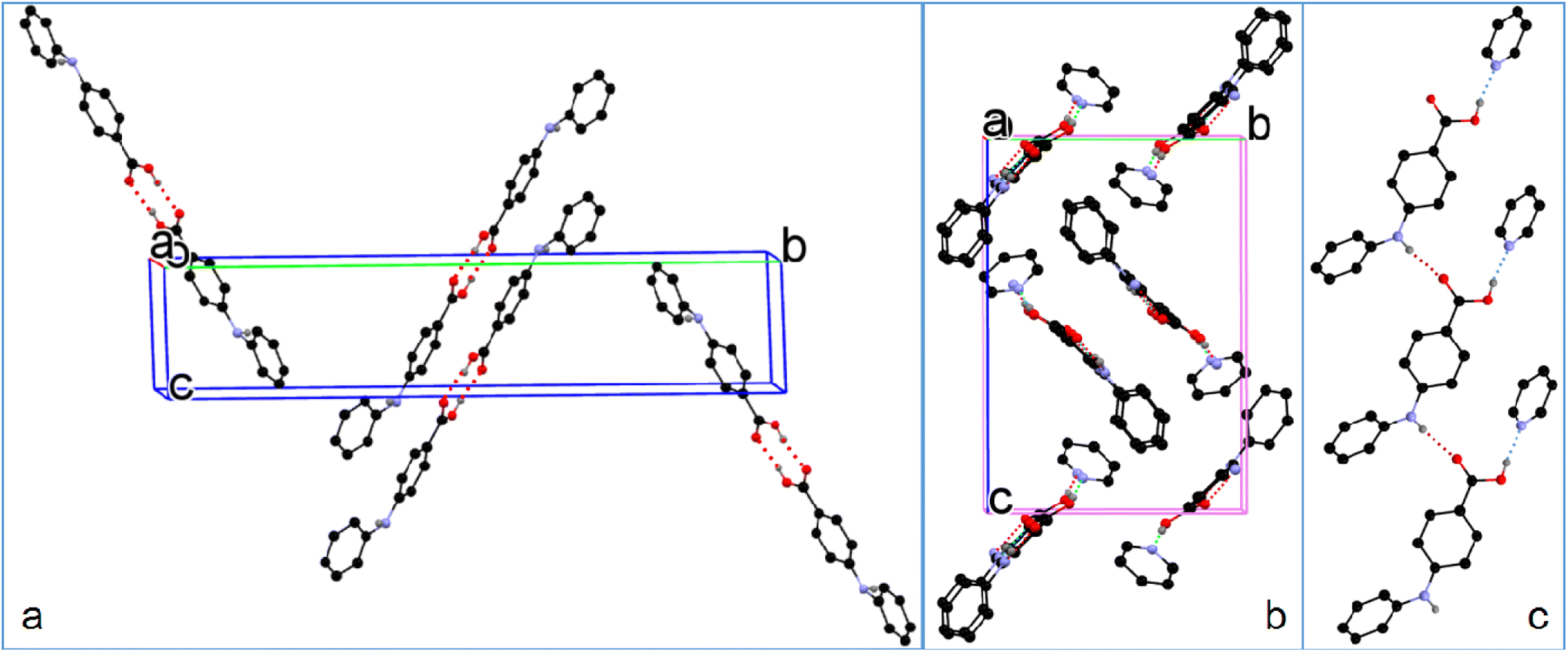
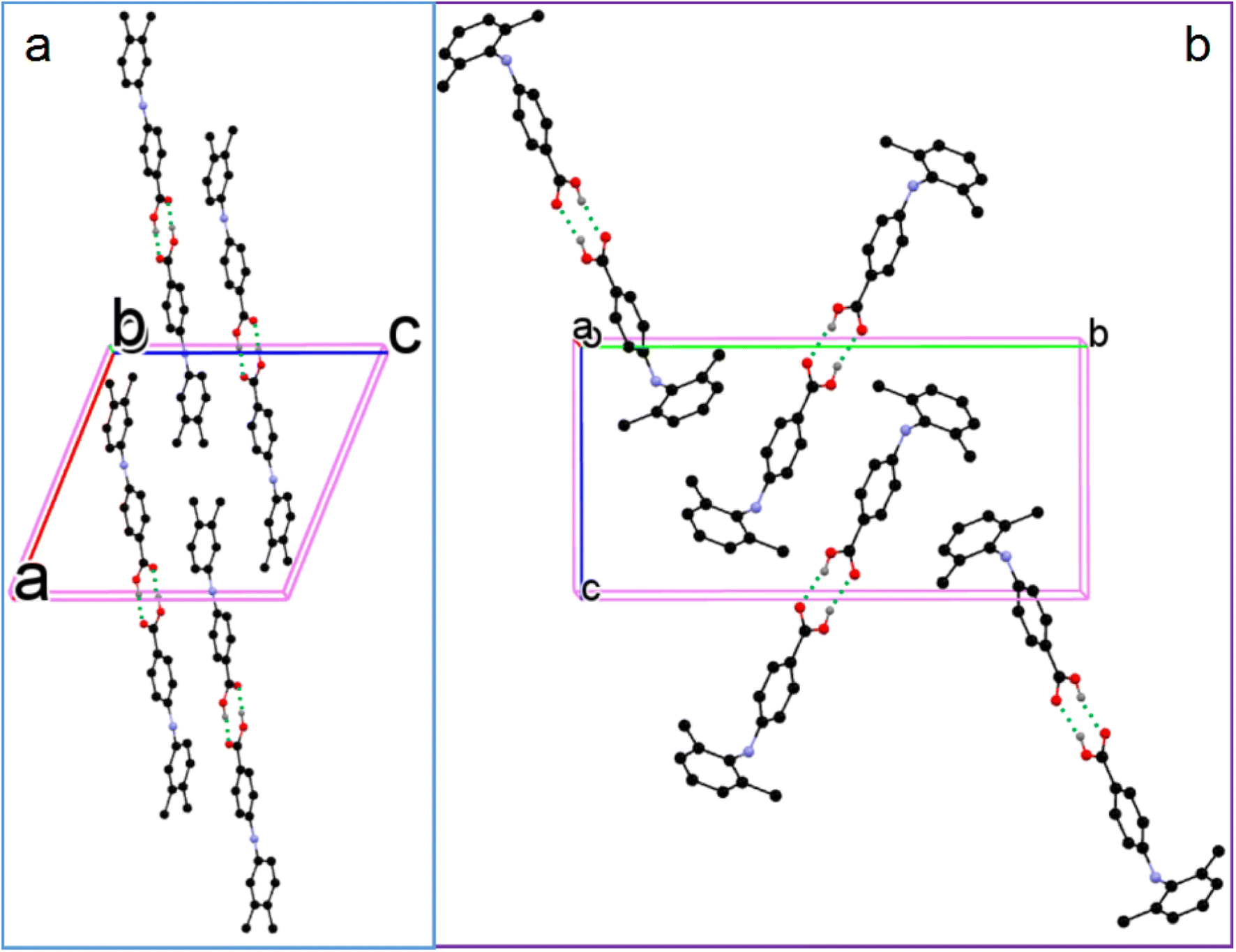
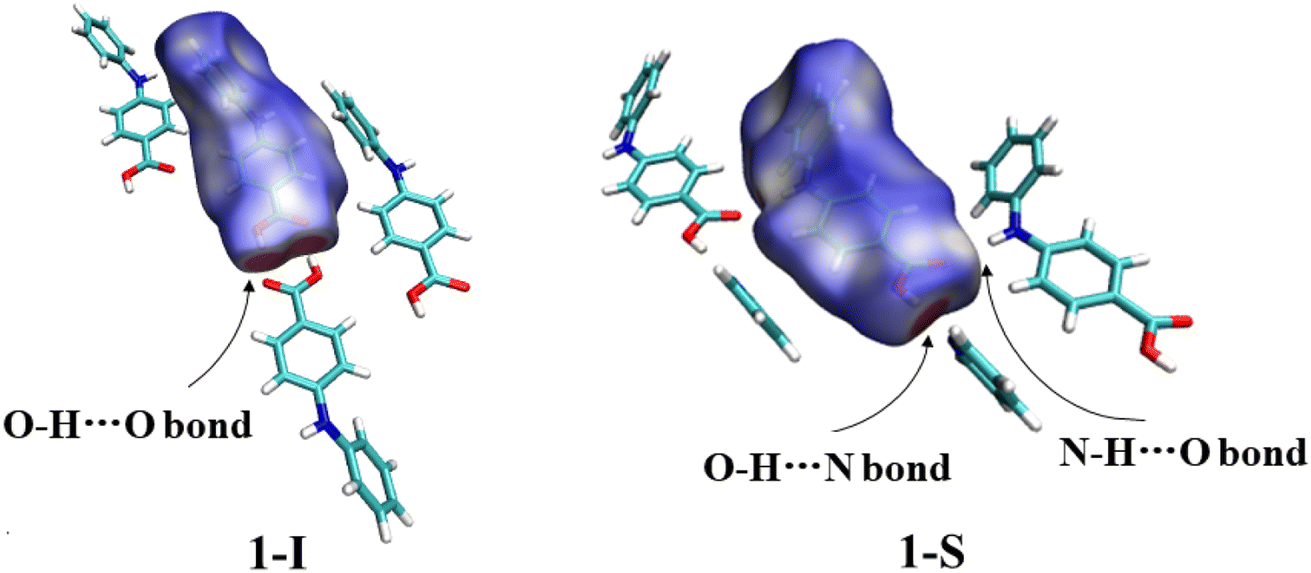
3.2.2. Solvatomorphic systems. The molecule in the asymmetric unit of 1-I and the corresponding molecule in 1-S are slightly different in conformation. Because the anilino group is at the para position of the carboxylic acid, the bond lengths of CBA–N (BA stands for benzoic acid) and N–Cbenzene are 1.401(3) Å and 1.395(3) Å for 1-I, and 1.3800(17) Å and 1.3996(16) Å for the molecule in 1-S, in contrast to 1.364 Å and 1.404 Å, and 1.359 Å and 1.419 Å for the corresponding bonds in the two molecules in FA. This difference should be due to the relatively weaker electron-withdrawing effect exerted by the para carboxylic acid compared with the ortho carboxylic acid. The molecules in 1-I form acid–acid dimers with hydrogen bond parameters of 1.60(4) Å and 174(4)°. The hydrogen bonding patterns in 1-S are starkly different from that of 1-I: no acid–acid hydrogen bond is observed; instead, the carboxylic acid OH forms a hydrogen bond with the N of the solvent molecule pyridine with hydrogen bond parameters of 1.85 Å and 174°. In addition, the aniline NH hydrogen bonds with the carbonyl O of carboxylic acid with parameters of 2.12 Å and 168° (Fig. 5).
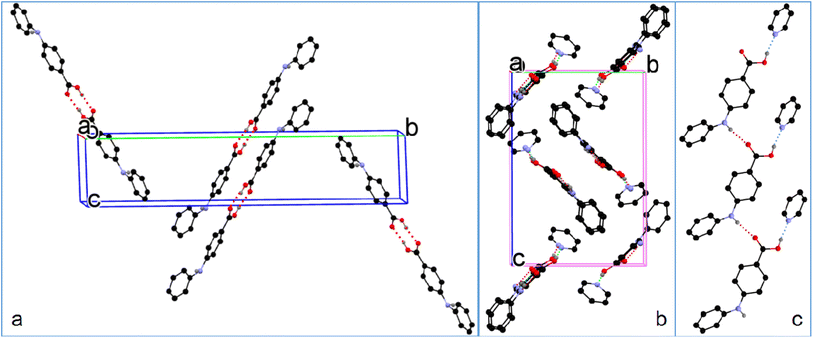 |
| | Fig. 5 (a) Crystal packing of 1-I, (b) 1-S, and (c) hydrogen bonding in 1-S (for clarity, H atoms not involved in hydrogen bonding are omitted). | |
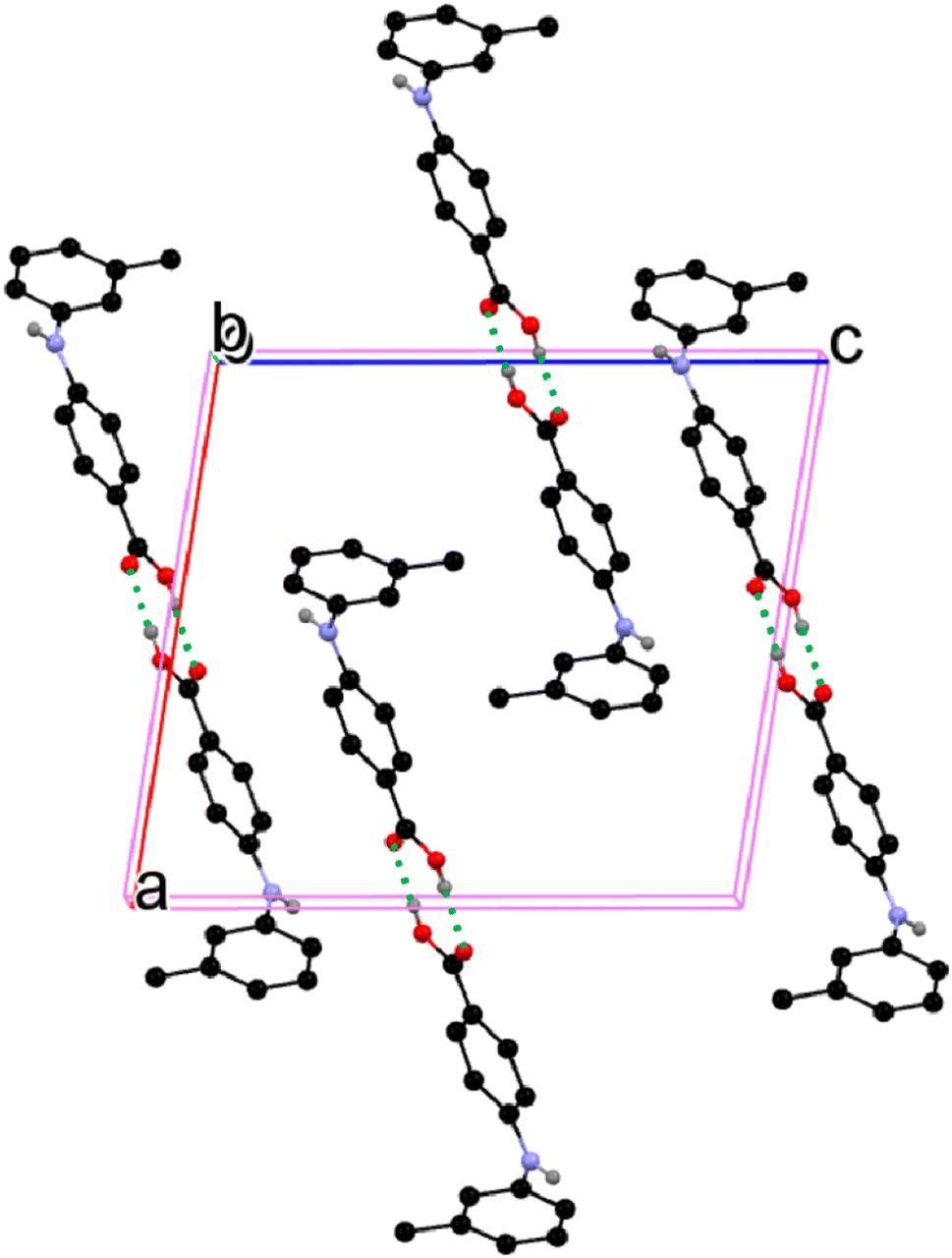
The CBA–N and N–Cbenzene bonds in 3-I have bond lengths of 1.368(3) Å and 1.417(2) Å, likely due to the electron-donating effect of the methyl group ortho to NH on the benzene ring. The molecules in 3-I pair up to form acid–acid dimers with hydrogen bond parameters of 1.82 Å and 174° (Fig. 6).
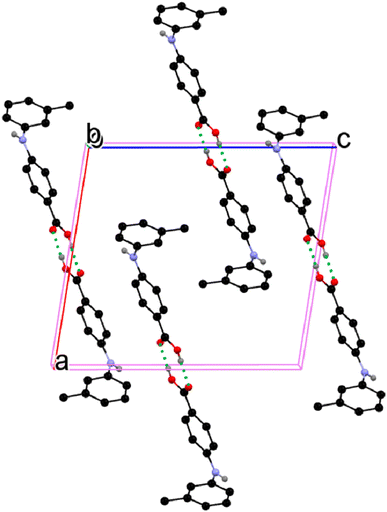 |
| | Fig. 6 Crystal packing of 3-I (for clarity, H atoms not involved in hydrogen bonding are omitted). | |
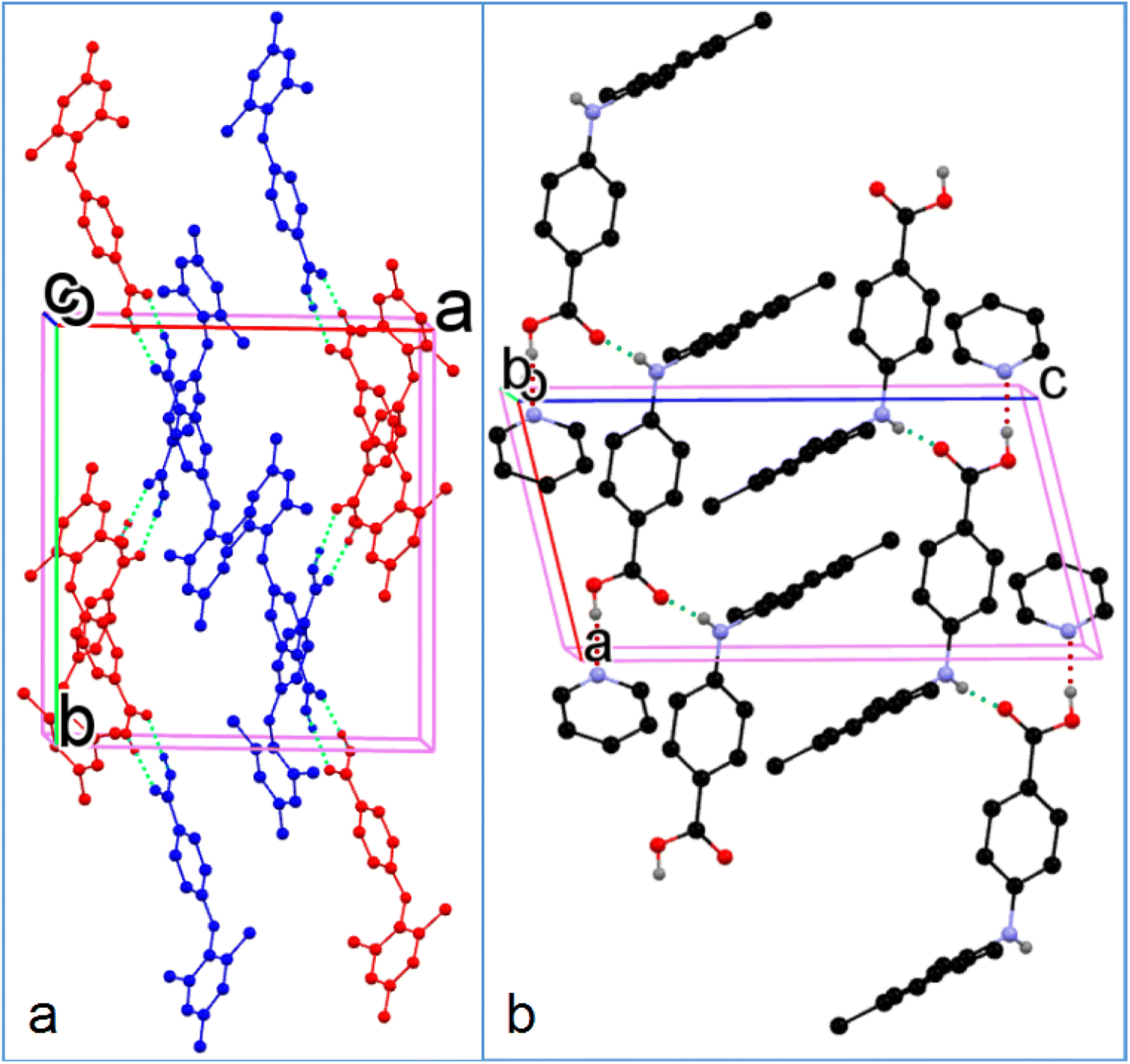
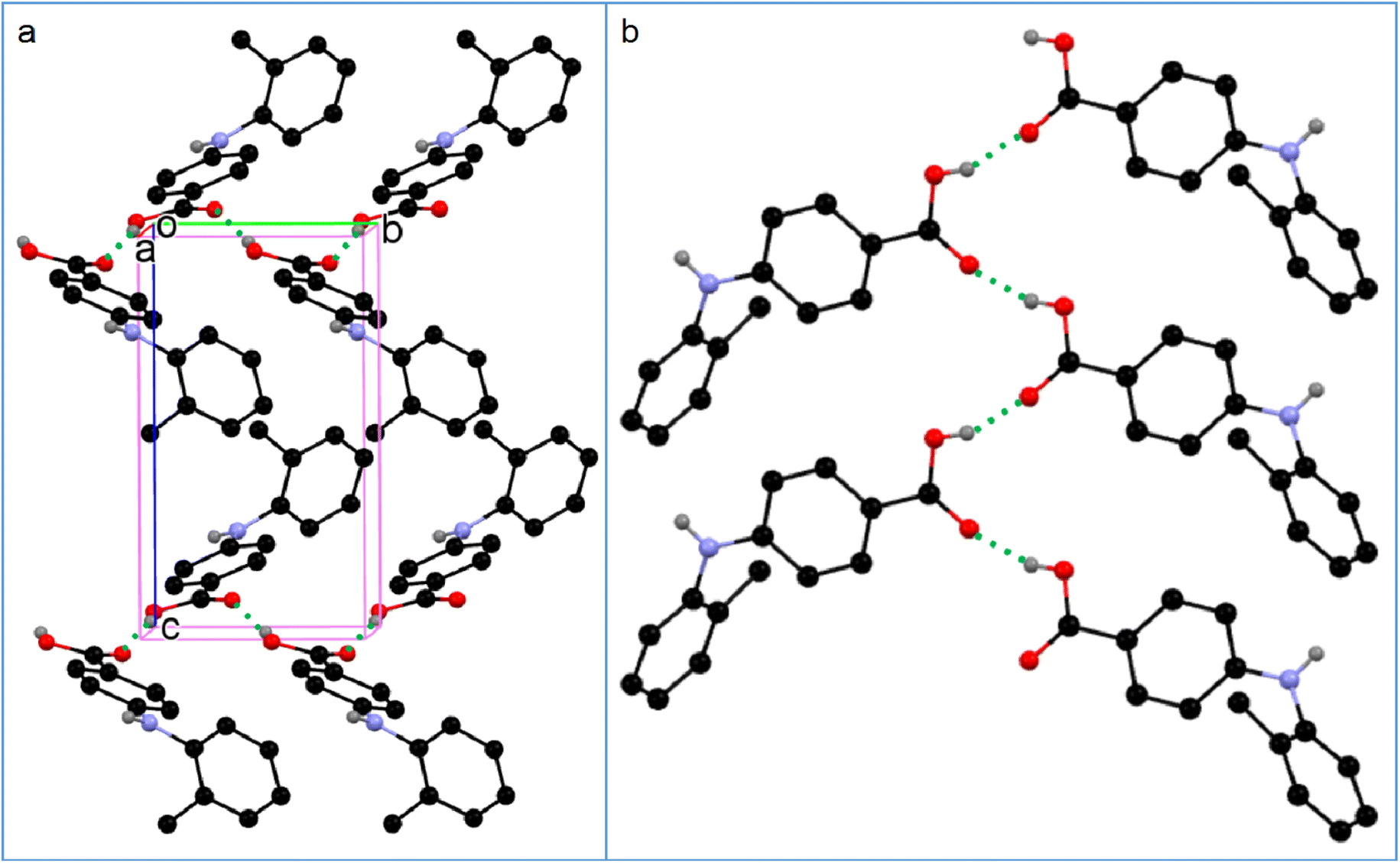
The two molecules (A, red; B, blue) in the asymmetric unit of 8-I and the host molecule in 8-S are highly twisted. The bond lengths of CBA–N and N–Cbenzene are 1.3677(18) Å and 1.4264(19) Å for A; 1.3754(18) Å and 1.422(2) Å for B; 1.380(2) Å and 1.4280(19) Å for the molecule in 8-S. Again, the obvious bond length difference between the two bonds in all three molecules are ascribed to the electron-withdrawing effect of the carboxylic acid and the electron-donating effect of the methyl groups. The two molecules in 8-I associate to form an A–B acid–acid dimer with hydrogen bonding parameters of 1.79 Å, 165° and 1.84 Å, 167°. For 8-S, the hydrogen bonding patterns are the same to those of 1-S. The hydrogen bond between NH and CO has parameters of 1.88 Å and 164°. The hydrogen bond between the COOH of the host molecule and pyridine-N has parameters of 2.10(2) Å and 171 (2)° (Fig. 7).
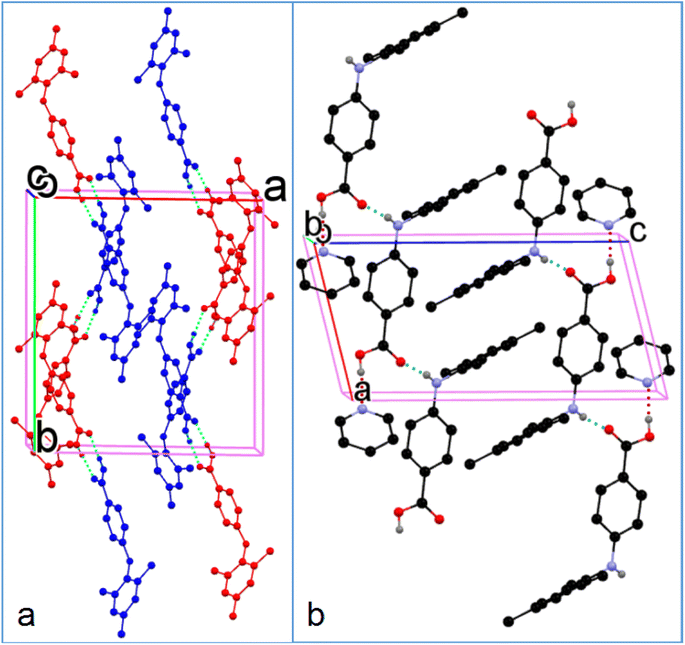 |
| | Fig. 7 Crystal packing of (a) 8-I and (b) 8-S (for clarity, H atoms not involved in hydrogen bonding are omitted). | |
3.2.3. Monomorphic systems. The molecule in 4 has CBA–N and N–Cbenzene bond lengths similar to those of molecule 1: 1.3957(17) Å and 1.3996(17) Å, likely because the methyl group is at the para position of the aniline. The molecules pair up to form acid–acid dimers with hydrogen bond parameters of 1.75(2) Å and 172(4)° (Fig. 8). The two molecules (A, red; B, blue) in the asymmetric unit of 5 are highly twisted with different dihedral angles. The CBA–N and N–Cbenzene have bond lengths of 1.377(2) Å and 1.428(2) Å for A, and 1.379(2) Å and 1.425(2) Å for B. Two identical molecules (AA and BB) associate to form an acid–acid dimer (Fig. 8). For the A–A dimer, the hydrogen bond parameters are 1.82(4) Å and 166(5)°; for the B–B dimer, the corresponding parameters are 1.80(4) Å and 170(5)°. The distance between the centroid of the benzoic acid ring of A and B is 3.676 Å with an angle of 8.10 (8)°, indicating they are nearly parallel. So it is inferred that π–π stacking exists in the benzoic acid rings of these two molecules.
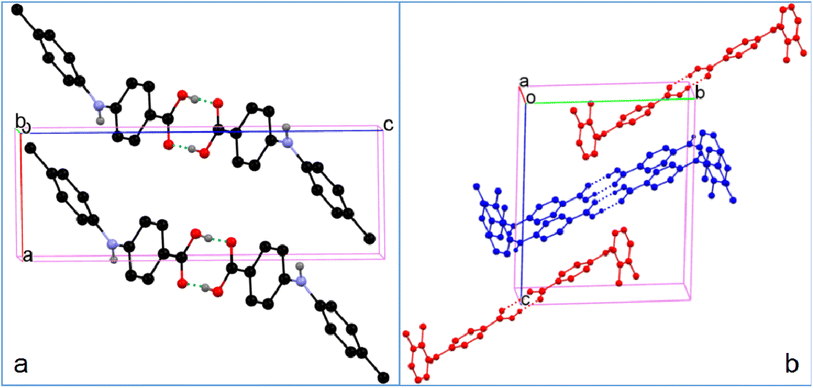 |
| | Fig. 8 Crystal packing of (a) 4 and (b) 5 (for clarity, H atoms not involved in hydrogen bonding are omitted). | |
Although molecules 6 and 7 are twisted to different degrees, the CBA–N and N–Cbenzene bond lengths of 6 and 7 are similar: 1.376(3) Å and 1.414(3) Å for 6; and 1.3742(18) Å and 1.4204(17) Å for 7. The molecules in both 6 and 7 pair up to form acid–acid homodimers with hydrogen bonding parameters of 1.75(2) Å and 177(3)° in 6, and 1.83 Å and 164° in 7 (Fig. 9). These parameters are listed in Table 2.
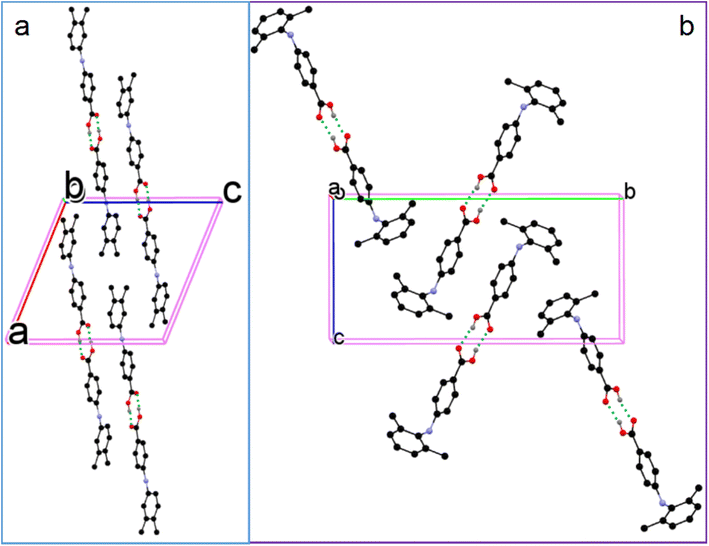 |
| | Fig. 9 Crystal packing of (a) 6 and (b) 7 (for clarity, H atoms not involved in hydrogen bonding are omitted). | |
Table 2 Bond lengths and bond angles of compounds 4–7
| Compound |
CBA–N (Å) |
N–Cbenzene (Å) |
Hydrogen bond length (Å) |
Hydrogen bond angle (°) |
| 4 |
1.3957(17) |
1.3996(17) |
1.75(2) |
172(4) |
| 5 (A–A dimer) |
1.377(2) |
1.428(2) |
1.82(4) |
166(5) |
| 5 (B–B dimer) |
1.379(2) |
1.425(2) |
1.80(4) |
170(5) |
| 6 |
1.376(3) |
1.414(3) |
1.75(2) |
177(3) |
| 7 |
1.3742(18) |
1.4204(17) |
1.83 |
164 |
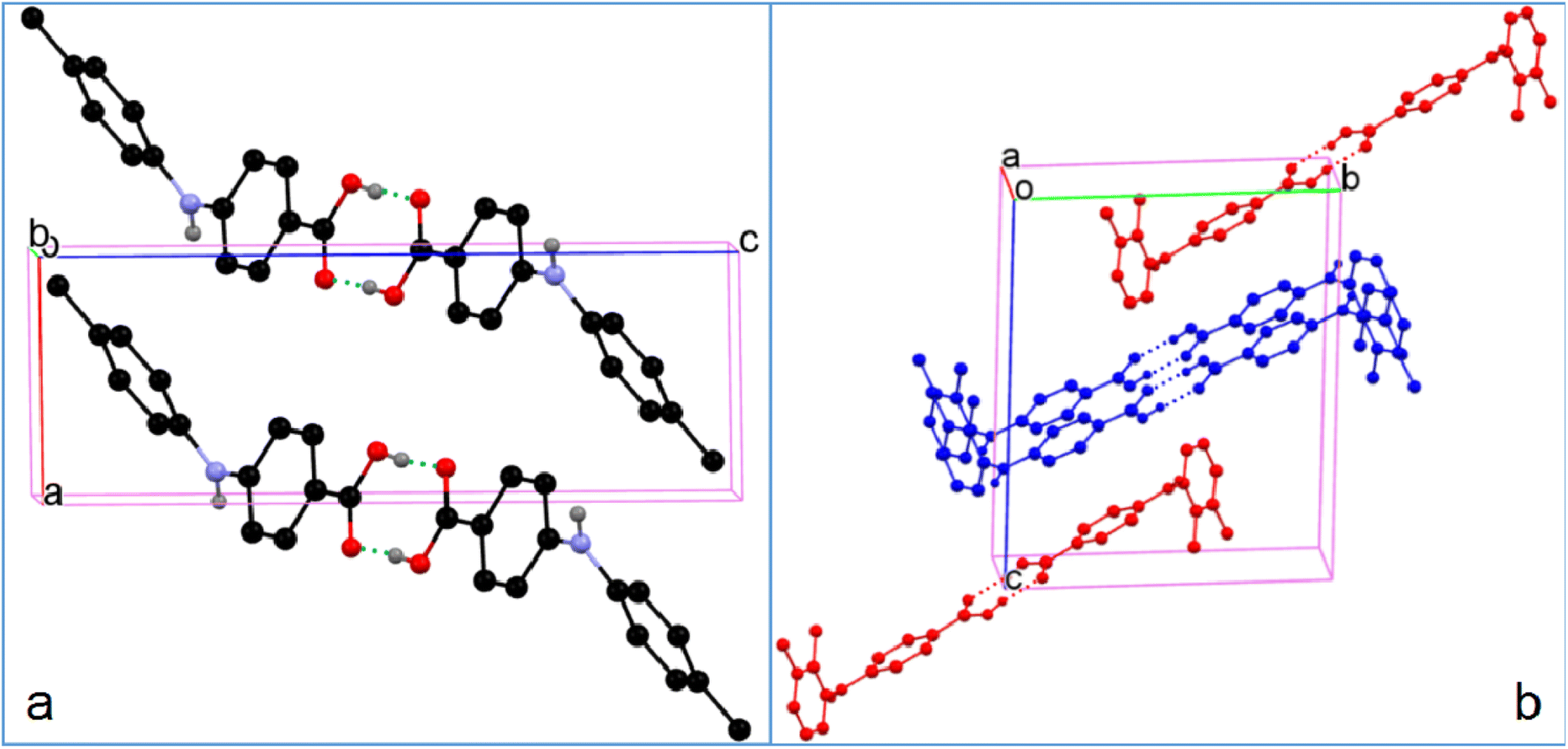
3.2.4. Catemer in compound 2. Although the molecule in 2 is also highly twisted, and the CBA–N and N–Cbenzene bonds (1.373(2) Å and 1.418(2) Å) are similar to those with ortho methyl group(s), the molecules in 2 form acid–acid catemers,45 instead of acid–acid homodimers, with hydrogen bond parameters of 1.92 Å and 160° (Fig. 10). Acid–acid dimer and catemer are two possible motifs in monocarboxylic acids. Yet, the acid–acid dimer motif is predominant in the crystal structures of monocarboxylic acids.46 The preponderance of the acid–acid dimer over catemer is ascribed to the prevention of catemer formation by the bulk of the R group in R–COOH.47 For example, formic acid and acetic acid with sufficiently small R groups have catemer motif in their crystals,45,48 tetrolic acid has two polymorphs, with α-form sustained on the acid–acid dimer, and β-form on catemer,49 benzoic acid forms an acid–acid dimer,50 and no catemer motif has been observed in FAs, until now. Thus the dogma that bulky R group prevents the formation of acid catemer in R–COOH doesn't hold for compound 2.
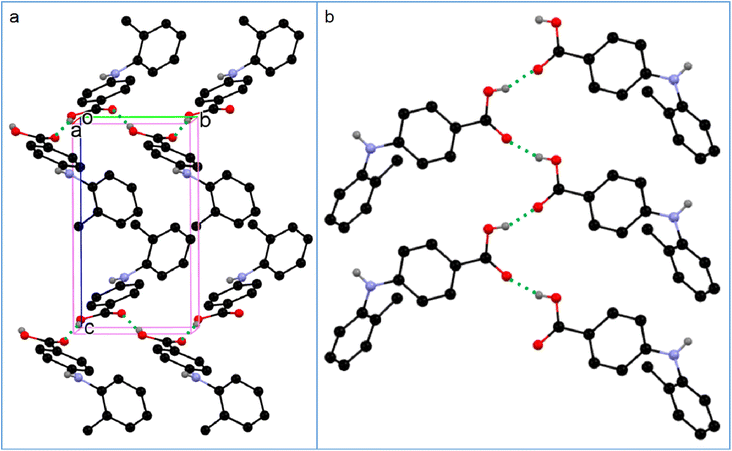 |
| | Fig. 10 (a) Crystal packing of 2, and (b) acid–acid catemer motif (for clarity, H atoms not involved in hydrogen bonding are omitted). | |
3.3. Thermal properties
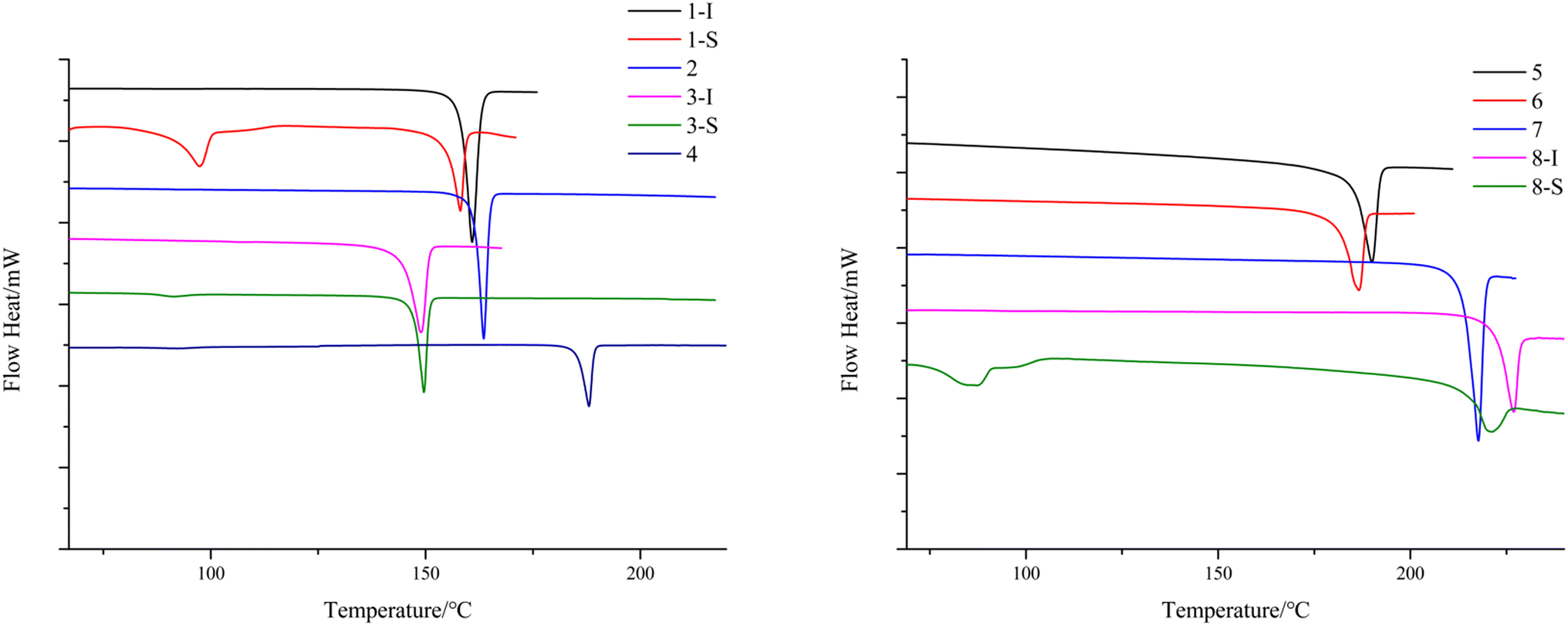
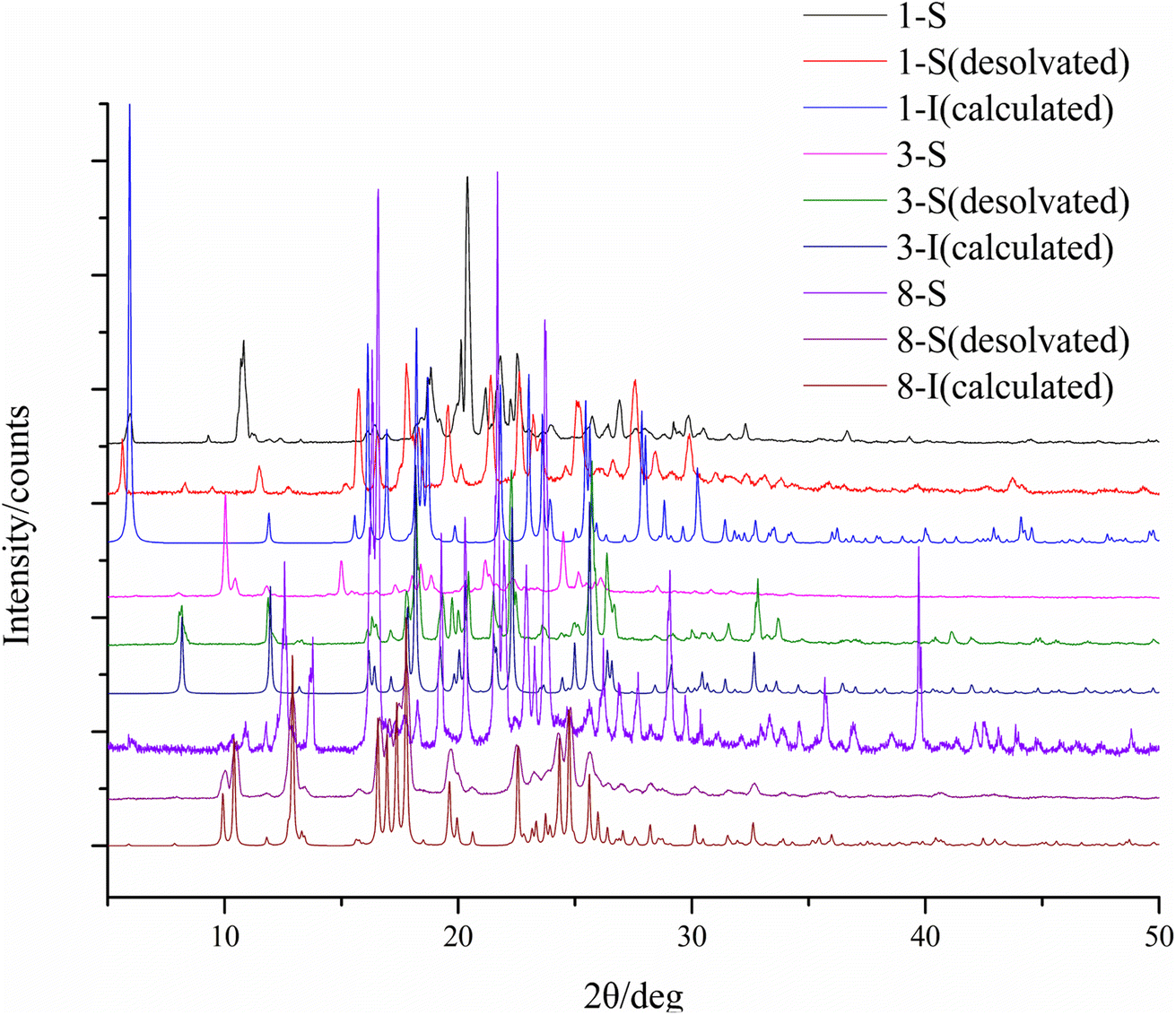
To study the thermal properties of the crystal forms of 1–8, pure samples of each form were obtained and confirmed by PXRD (Fig. S9†). The thermal properties of the crystal forms of 1–8 were studied by DSC. For compounds 2–7, each DSC trace shows one thermal event corresponding to the melting of each compound, with different onset temperatures: 163.9 °C for 2, 149.6 °C for 3-I, 187.6 °C for 4, 192.3 °C for 5, 186.6 °C for 6, and 217.7 °C for 7. For the solvent-free forms of 1 and 8, the DSC traces show one thermal event with an onset temperature of 158.1 °C and 226.9 °C respectively, which corresponds to the melting of each form. 1-S and 8-S display two thermal events with the first one with an onset temperature of 97.4 °C and 87.2 °C respectively, indicating the loss of pyridine, and the second one with an onset temperature of 158.1 °C and 221.1 °C, which suggests desolvation leads to the solvent-free form of 1 and 8 (Fig. 11). The phase transition caused by desolvation was also confirmed by PXRD, as the PXRD patterns of 1-S, 3-S and 8-S after thermal treatment match well with those of 1-I, 3-I and 8-I respectively (Fig. 12).
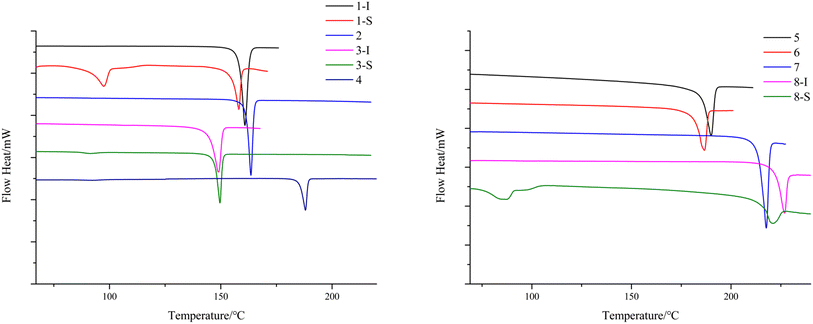 |
| | Fig. 11 DSC thermograms of the crystal form(s) of 1–8. | |
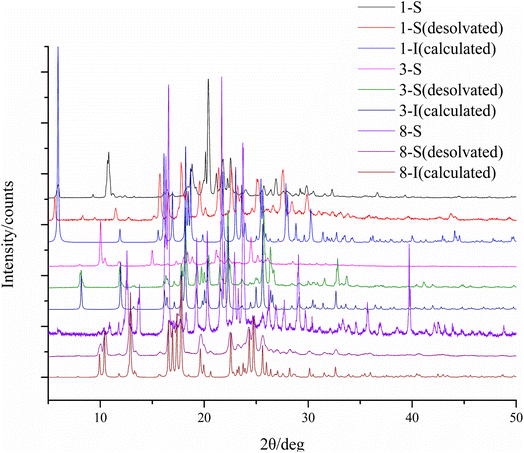 |
| | Fig. 12 PXRD patterns of 1-S, 3-S and 8-S before and after thermal treatment. | |
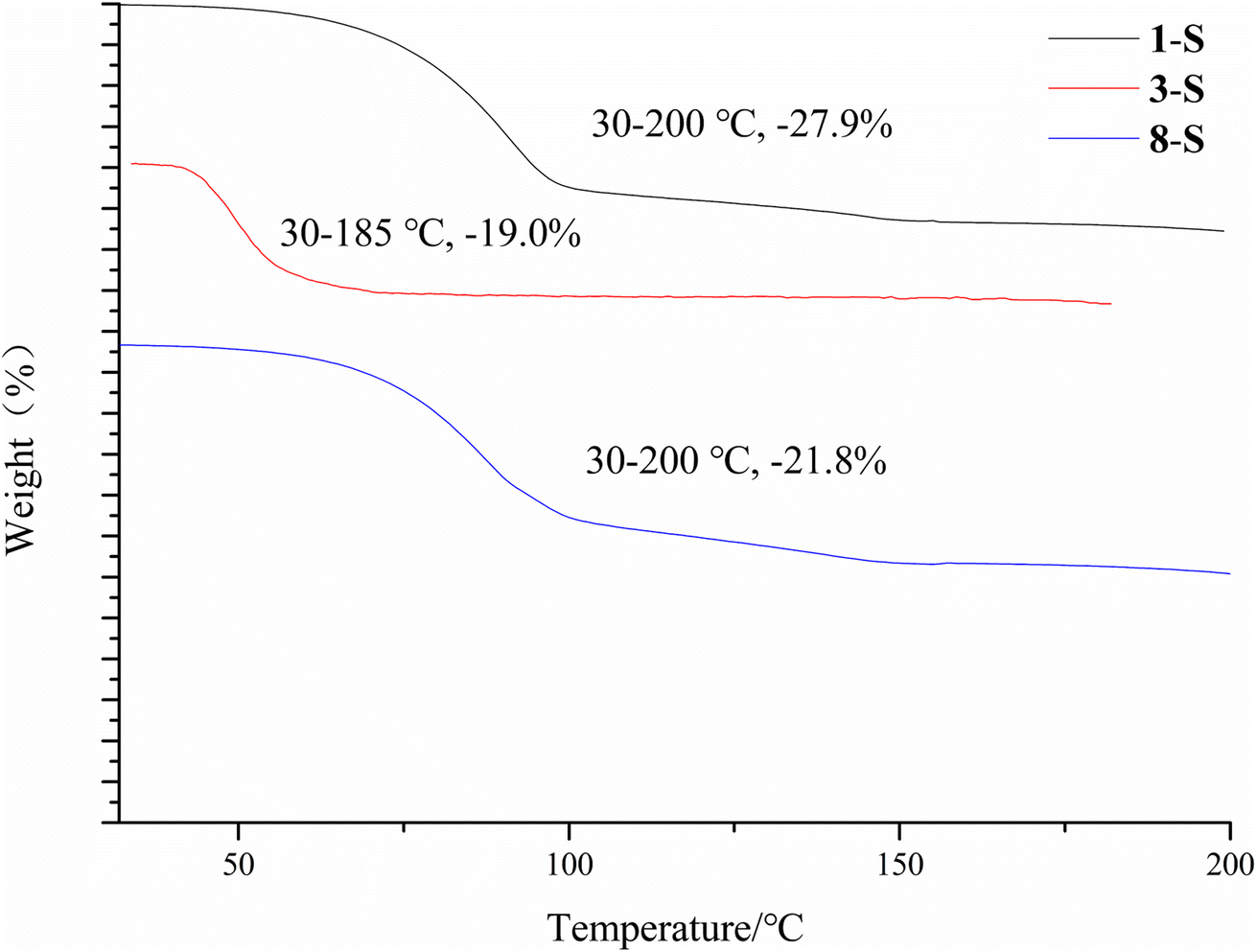
The desolvation was also confirmed by the TGA study. 1-S, 3-S and 8-S showed a major mass loss of 27.9%, 19.0%, 21.8% (Fig. 13) (theoretical value is 27.1%, 27.2%, and 23.6% for each form). The large discrepancy between the theoretical and experimental weight loss of 3-S may be due to the ease of escape of dichloromethane from the crystal lattice.
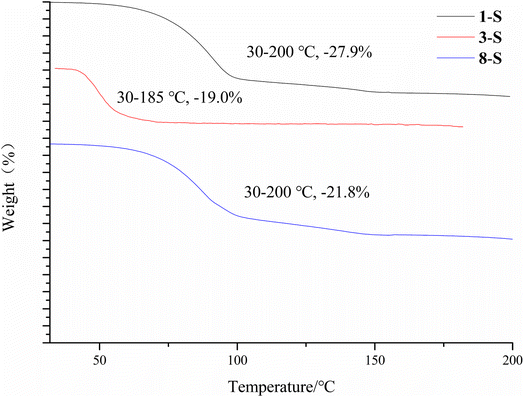 |
| | Fig. 13 TGA of 1-S, 3-S and 8-S. | |
3.4. Computational results
3.4.1. Hirshfeld analysis. Hirshfeld analysis was performed for all the crystal structures. The red areas in the Hirshfeld surface analysis indicate regions of significant intermolecular interactions between the central monomer and the surrounding molecules. The Hirshfeld surface analysis of compound 1 crystals (1-I and 1-S) is provided as an example (Fig. 14). As shown, the most dominant intermolecular interaction is the acid–acid hydrogen bond homodimer in 1-I, and the acid–pyridine and NH⋯OC hydrogen bond in 1-S. The white areas in the Hirshfeld surface analysis are regions of insignificant interactions, which are not comparable to hydrogen bonding in strength. The Hirshfeld analysis of compounds 2–8 are provided in the ESI.†
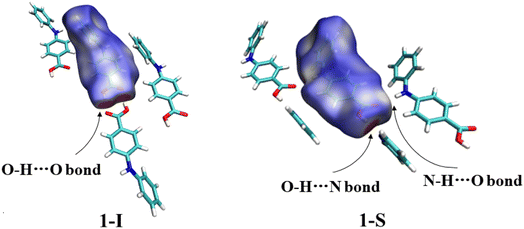 |
| | Fig. 14 Hirshfeld surface analysis of the two forms of compound 1. | |
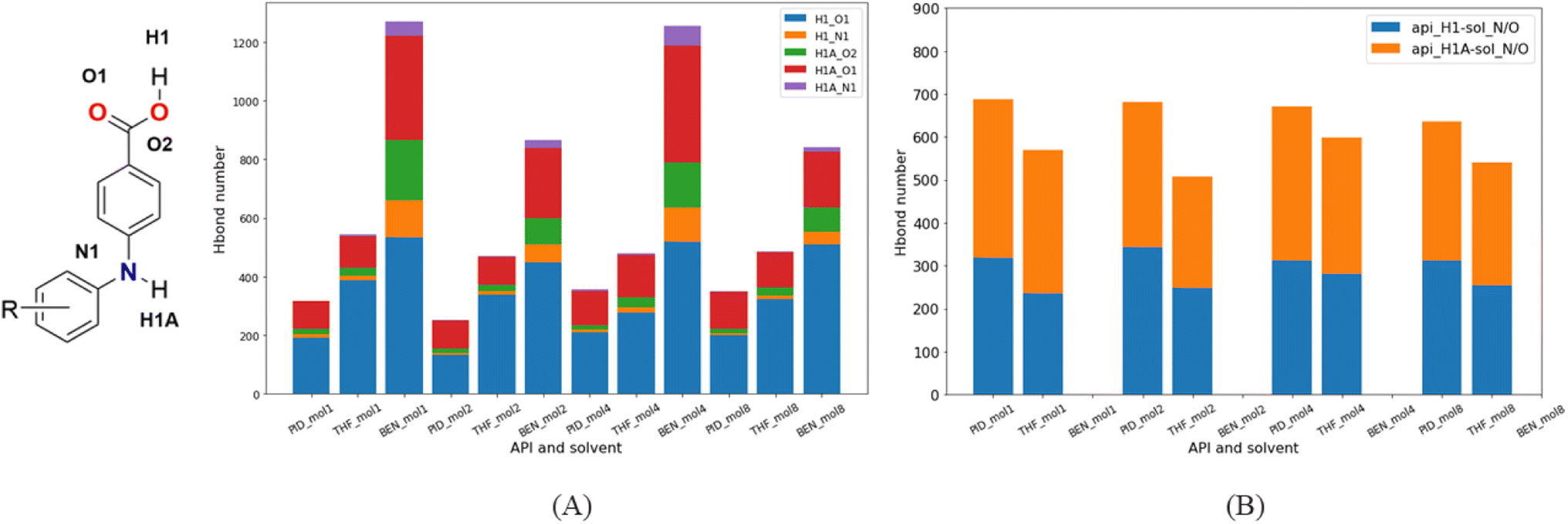
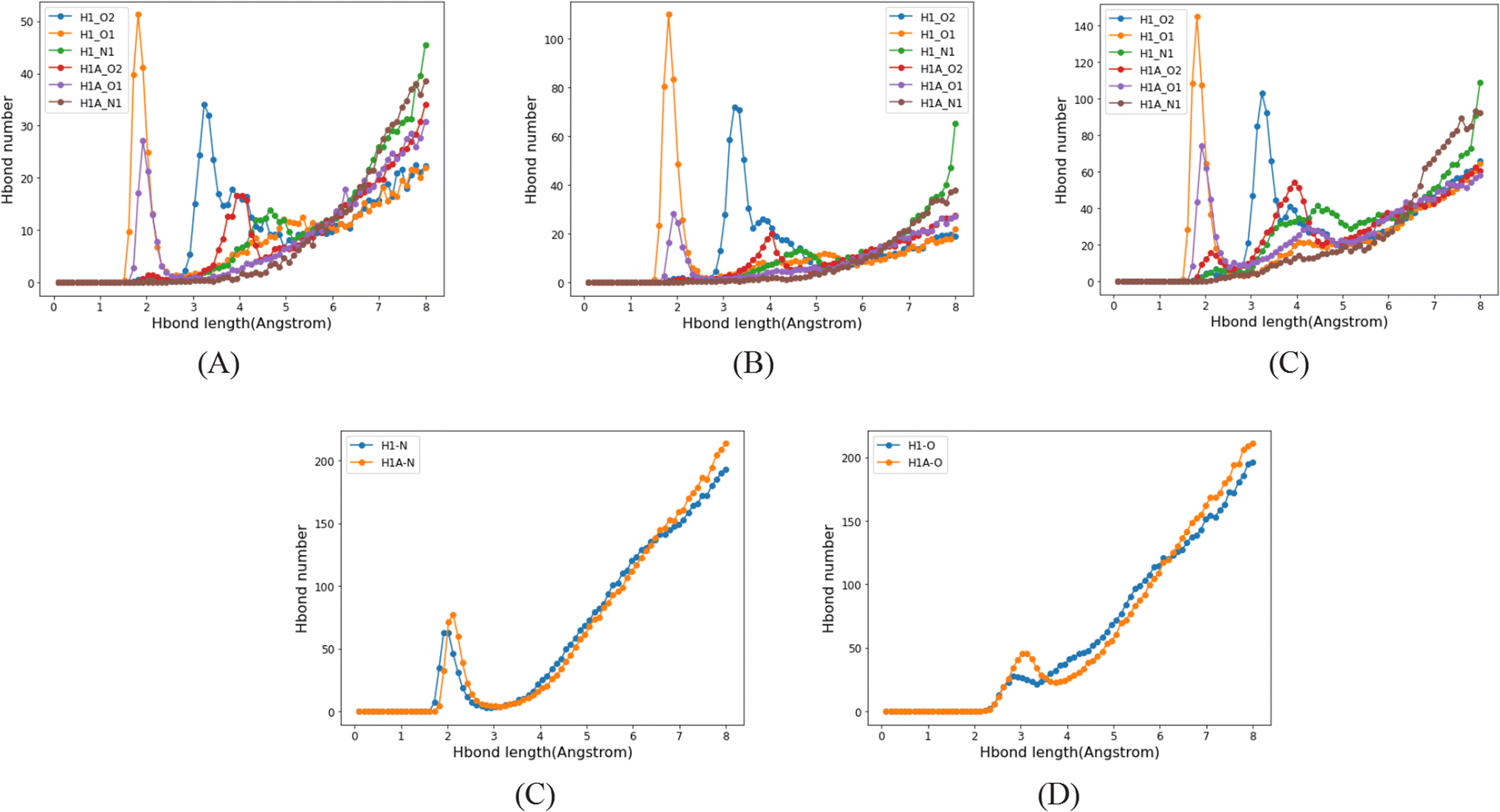
3.4.2. Molecular dynamics simulation. Crystallization in solvent systems of pyridine (PID), tetrahydrofuran (THF) and benzene (BEN) was simulated. The three solvents have similar molecule structures but different polarity, which can aid us to understand how the solvents affect the crystallization regardless of the other unwanted conformational factors. Hydrogen bond acceptors and donors exist in the API molecules, so hydrogen bonds between API molecules usually dominate the interactions between them. Furthermore, the –COOH in API molecules lead to a considerable number of API acid–acid dimers in solutions, due to a much stronger strength of double hydrogen bonds. In the crystallization of API molecules, these API dimers, instead of independent API molecules, become the basic units of nucleation. They stack together and grow in solution, and eventually form the crystals. Thus, most of the time, the API dimers in the API crystal structures originate from the API dimers in solutions.Since there is neither a hydrogen bond acceptor nor a donor in BEN, hydrogen bonds form only between API molecules. But as there is a hydrogen bond acceptor in PID and THF, the hydrogen bond acceptor of solvent molecules will compete with hydrogen bond acceptors of API molecules, that has a significant effect on the formation of API dimers in solutions. In contrast to the benzene system, the number of hydrogen-bonded API dimers decreases greatly in PID and THF (Fig. 15A), and besides, the number of API–solvent dimers is larger than that of the API dimers. Apart from the API dimers, API–solvent dimers become another important unit in the nucleation of API crystals. Especially in PID, the length of hydrogen bond between solvent molecule and API molecule and the length of hydrogen bond between API molecules is of the same magnitude. The hydrogen bond types and lengths between molecules in the solutions of molecule 1 are provided as an example (Fig. 16), and those of compounds 2, 4, 8 are provided in ESI.† Moreover, the number of API–solvent dimers is more than that of the API dimers (Fig. 15), so the original dominant basic units of API dimers in nucleation are replaced by API–solvent dimers, which gives rise to the solvates 1-S and 8-S. Because of the weaker hydrogen bonds between THF and API molecules (Fig. 16 and S11–S13†), the API–solvent dimers may form the initial crystal nuclei at the beginning of nucleation, but these metastable solvate nuclei may transform to thermodynamically favorable solvent-free crystal nuclei as they grow, or the metastable solvate crystal transforms to the thermodynamically more stable solvent-free crystal form during the crystallization, for the solvent-free API crystals have a lower energy.
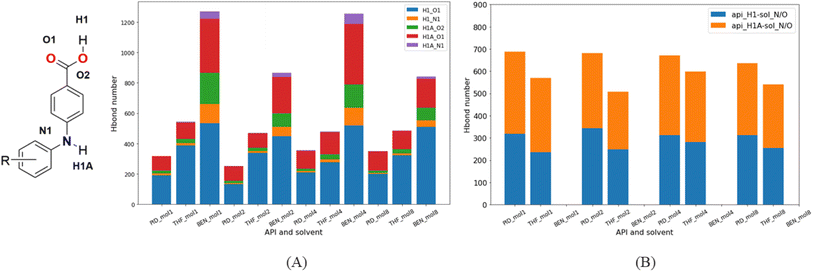 |
| | Fig. 15 Hydrogen bond types and numbers between molecules in different solutions. (A) Hydrogen bonds in API dimers; (B) hydrogen bonds in dimers of API and solvent. | |
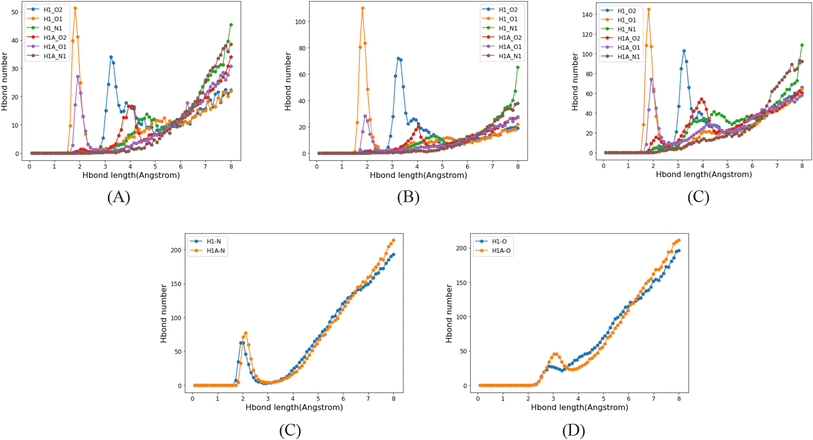 |
| | Fig. 16 Hydrogen bond types and lengths between molecules in solutions of molecule 1. (A–C), hydrogen bonds in API dimers in PID, THF and BEN; (D–E), hydrogen bonds in API–solvent dimers in PID and THF. | |
We hoped the simulation could provide an explanation for the formation of acid–acid catemer in compound 2, yet there is little difference between molecule 2 and other compounds regarding the number and strength of hydrogen bonds, and dimers of different molecules in different solutions. A more powerful approach, such as the comparison between the experimental structure and a predicted crystal with acid–acid dimer motif, should be sought to shed light on this phenomenon. Yet, the clue of the acid–acid catemer formation may be found in the close packing of the crystal, as it has the highest density among all the solvent-free crystals, which may be due to the catemer synthon in the crystal. Also analysis of a larger sample size with more analogues may provide insight.
4. Conclusions
Eight 4-phenylamino-benzoic acids differing from each other in the substitution position and pattern were synthesized by a Buchwald–Hartwig reaction. The compounds are isomers of FA analogues. Polymorph screening in the commonly used solvents generated one solvent-free form for compounds 2, 4, 5, 6, and 7, and one solvent-free form and one solvate for 1, 3, and 8. The acid–acid dimer is the synthon observed in the crystal structures of 4–7, and the solvent-free form of 1, 3, and 8. The acid–acid catemer motif is found in the crystal of 2, and this is a rarity in anthranilic acids and their isomers. The solvates of 1, 3, and 8 convert to the solvent-free form of each compound after thermal treatment. Hirshfeld analysis revealed the main intermolecular interaction and other less significant interactions contributing to the overall stability of each crystal form. Molecular dynamics simulations revealed the underlying mechanism that molecules 1 and 8 formed solvates with pyridine, but there is little difference between molecule 2 and other compounds considering the number, type, and strength of hydrogen bonds in different solvents. Nevertheless, the close packing of compound 2 may explain the rise of acid–acid catemer in the structure. In this study, we only investigated the substitution on the aniline ring with methyl group(s). The effects of changing the substitution pattern/substituents on the solid–state properties of 4-PABAs are currently under investigation.
Author contributions
Conceptualization, S. Long; methodology and investigation: synthetic experiments, X. Liu, J. Cui; characterization analysis, X. Liu, J. Cui; resources: S. Parkin, T. Li, P.-P. Zhou; formal analysis: computational analysis of experimental data, Q. Zeng, L. Fang, P.-Y. Liang, P.-P. Zhou; writing – original draft: S. Long, X. Liu, J. Cui; writing – review & editing: S. Long, J. Cui.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
SL thanks Natural Science Foundation of Hubei Province for financial support (2014CFB787).
References
- R. N. Brogden, Non-steroidal anti-inflammatory analgesics other than salicylates, Drugs, 1986, 32, 27–45 CrossRef.
- Y. Masubuchi, H. Saito and T. Horie, Structural requirements for the hepatotoxicity of nosteroidal anti-inflammatory drugs in isolated rat hepatocytes, J. Pharmacol. Exp. Ther., 1998, 287, 208–213 CAS.
- K. V. Belov, D. C. Batista, A. A. Dyshin, M. G. Kiselev, V. V. Sobornova and I. A. Khodov, Conformational analysis of mefenamic acid in scCO2-DMSO by the 2D NOESY method, Russ. J. Phys. Chem. B, 2022, 16, 1191–1199 CrossRef CAS.
- O. O. Fadeyi, C. A. Obafemi, C. O. Adewunmi and E. O. Iwalewa, Full length research paper – antipyretic, analgesic, anti-inflammatory and cytotoxic effects of four derivatives of salicylic acid and anthranilic acid in mice and rats, Afr. J. Biotechnol., 2004, 3, 198–207 Search PubMed.
- G. P. Stableforth, Mefenamic acid and dextropropoxyphene with paracetamol as analgesics in the accident department, Curr. Med. Res. Opin., 1977, 5, 189–191 CrossRef PubMed.
- M. E. Mavrikakis, M. Macleod, W. Watson Buchanan, L. A. Hernandez and J. A. Norris Rennie, Mefenamic acid: an under-rated antirheumatic?, Curr. Med. Res. Opin., 1977, 4, 535–539 CrossRef CAS PubMed.
- L. Nyfos, A comparative clinical study of a new antirheumatic agent, tolfenamic acid (Clotam), and phenylbutazone in rheumatoid arthritis, Scand. J. Rheumatol., 1979, 8, 5–7 CrossRef.
- A. Thorarensen, J. Li, B. D. Wakefield, D. L. Romero, K. R. Marotti, M. T. Sweeney, G. E. Zurenko and R. W. Sarver, Preparation of novel anthranilic acids as antibacterial agents: Extensive evaluation of structural and physical properties on antibacterial activity and human serum albumin affinity, Bioorg. Med. Chem. Lett., 2007, 17, 3113–3116 CrossRef CAS.
- L. K. Abdulkarem and S. H. Mahdi, Spectroscopic, structural and antibacterial activity of mixed ligand complexes from schiff base with anthranilic acid, J. Phys.: Conf. Ser., 2019, 1234, 012089–012103 CrossRef CAS.
- D. A. Inglot, Comparison of the antiviral activity in vitro of some non-steroidal anti-inflammatory drugs, J. Gen. Virol., 1969, 4, 203–211 CrossRef PubMed.
- H. Iqbal, S. Ali and S. Shahzadi, Anti-inflammatory and acute toxicity study of organotin(IV) complexes: A Review, Chem. J., 2016, 6, 59–73 CAS.
- D. Kovala-Demertzi, V. Dokorou, A. Primikiri, R. Vargas, C. Silvestru, U. Russo and M. A. Demertzis, Organotin meclofenamic complexes: Synthesis, crystal structures and antiproliferative activity of the first complexes of meclofenamic acid – novel anti-tuberculosis agents, J. Inorg. Biochem., 2009, 103, 738–744 CrossRef CAS PubMed.
- L. J. Simons, B. W. Caprathe, M. Callahan, J. M. Graham, T. Kimura, Y. Lai, L. V. Iii, W. Lipinski, A. T. Sakkab and Y. Tasaki, The synthesis and structure-activity relationship of substituted N-phenyl anthranilic acid analogs as amyloid aggregation inhibitors, Bioorg. Med. Chem. Lett., 2009, 19, 654–657 CrossRef CAS PubMed.
- A. Paul, K. C. Nadimpally, T. Mondal, K. Thalluri and B. Mandal, Inhibition of Alzheimer's amyloid-β peptide aggregation and its disruption by a conformationally restricted α/β hybrid peptide, Chem. Commun., 2015, 51, 2245–2248 RSC.
- P. Chatterjee, H. Zetterberg, K. Goozee, C. K. Lim, K. R. Jacobs, N. J. Ashton, A. Hye, S. Pedrini, H. R. Sohrabi and T. Shah, Plasma neurofilament light chain and amyloid-β are associated with the kynurenine pathway metabolites in preclinical Alzheimer's disease, J. Neuroinflammation, 2019, 16, 579–586 Search PubMed.
- M. Hiroi, Anti-tumor effect of N-[3,4-dimethoxycinnamoyl]-anthranilic acid (tranilast) on experimental pancreatic cancer, J. Nippon Med. Sch., 2002, 69, 224 CrossRef CAS.
- A. S. Culf, H. Yin, S. Monro, A. Ghosh, D. A. Barnett, R. J. Ouellette and S. A. Mcfarland, A spectroscopic study
of substituted anthranilic acids as sensitive environmental probes for detecting cancer cells, Bioorg. Med. Chem., 2016, 8, 929–937 CrossRef PubMed.
- F. Maestrelli, P. Rossi, P. Paoli, E. D. Luca and P. Mura, The role of solid state properties on the dissolution performance of flufenamic acid, J. Pharm. Biomed. Anal., 2019, 180, 113058–113071 CrossRef PubMed.
- V. Lopez-Mejias, J. W. Kampf and A. J. Matzger, Nonamorphism in flufenamic acid and a new record for a polymorphic compound with solved structures, J. Am. Chem. Soc., 2012, 134, 9872–9889 CrossRef CAS.
- P. Sacchi, S. M. Reutzel-Edens and A. J. Cruz-Cabeza, The unexpected discovery of the ninth polymorph of tolfenamic acid, CrystEngComm, 2021, 3, 168–179 Search PubMed.
- S. Seethalekshmi and T. Row, Conformational polymorphism in a non-steroidal anti-inflammatory drug, mefenamic acid, Cryst. Growth Des., 2012, 12, 4283–4289 CrossRef CAS.
- V. López-Mejías and J. A. Matzger, Structure–polymorphism study of fenamates: toward developing an understanding of the polymorphophore, Cryst. Growth Des., 2015, 15, 3955–3962 CrossRef.
- Y. Liu, M. Zhang, D. Xu, S. Parkin, T. Li, C. Li, Z. Yang, F. Yu and S. Long, Effect of substituent size and isomerization on the polymorphism of 2-(naphthalenylamino)-benzoic acids, Cryst. Growth Des., 2019, 19, 3694–3703 CrossRef CAS.
- R. Li, F. Yang, C. Yan, X. Yang, D. Zhou and P. Zhou, Fenamic acid crystal with two asymmetric units (Z′ = 2): why Z′ = 2 rather than Z′ = 1, CrystEngComm, 2017, 19, 1762–1770 RSC.
- K. M. Lutker, Z. P. Tolstyka and A. J. Matzger, Investigation of a privileged polymorphic motif: A dimeric ROY derivative, Cryst. Growth Des., 2008, 8, 136–139 CrossRef CAS PubMed.
- K. M. Lutker and A. J. Matzger, Crystal polymorphism in a carbamazepine derivative: Oxcarbazepine, J. Pharm. Sci., 2010, 99, 794–803 CrossRef CAS PubMed.
- O. G. Uzoh, A. J. Cruz-Cabeza and S. L. Price, Is the fenamate group a polymorphophore? Contrasting the crystal energy landscapes of fenamic and tolfenamic acids, Cryst. Growth Des., 2012, 12, 4230–4239 CrossRef CAS.
- C. F. Zipp, H. W. Dirr, M. A. Fernandes, H. M. Marques and J. P. Michael, Polymorphic diversity: N-phenylbenzamide as a possible polymorphophore, Cryst. Growth Des., 2013, 13, 3463–3474 CrossRef CAS.
- S. Bhandary, S. Gonde and D. Chopra, Dissecting the conformational and interaction topological landscape of N-ethynylphenylbenzamide by the device of polymorphic diversity, Cryst. Growth Des., 2019, 19, 129–141 CrossRef.
- W. Christian, S. Liu, X. Mei, T. Adam and M. August, Regioselective copper-catalyzed amination of bromobenzoic acids using aliphatic and aromatic amines, J. Chem. Res., 2006, 10, 232–245 Search PubMed.
- T. Zhou, L. Feng, F. Yan, W. Song, X. Mu, H. Zhang and W. Yue, Hydrogen-bonded dimer stacking induced emission of aminobenzoic acid compounds, Chem. Commun., 2009, 22, 3199–3201 RSC.
- A. O. Adeniji, B. M. Twenter, M. C. Byrns, J. Yi, J. D. Winkler and T. M. Penning, Discovery of substituted 3-(phenylamino)benzoic acids as potent and selective inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3), Bioorg. Med. Chem. Lett., 2011, 21, 1464–1468 CrossRef CAS PubMed.
- A. O. Adeniji, B. M. Twenter, M. C. Byrns, Y. Jin and T. M. Penning, Development of potent and selective inhibitors of aldo-Keto reductase 1C3 (type 5 17β-hydroxysteroid dehydrogenase) based on N-phenyl-aminobenzoates and their structure activity relationships, J. Med. Chem., 2012, 55, 2311–2326 CrossRef CAS PubMed.
- T. Lu, C. Xue and F. Luo, Palladium-catalyzed cross-coupling reaction of aryldioxaborolane with 2-bromo-N,N-dimethylacetamide, Tetrahedron Lett., 2003, 34, 20–32 Search PubMed.
- J. Hou, W. Zhao, Z. Huang, S. Yang, L. Wang, Y. Jiang, Z. Zhou, M. Zheng, J. Jiang and S. Li, Evaluation of novel N-(piperidine-4-yl)benzamide derivatives as potential cell cycle inhibitors in HepG2 cells, Chem. Biol. Drug Des., 2015, 86, 223–231 CrossRef CAS PubMed.
- S. Long, T. Mao, P. Chen, M. Liu, S. Parkin, M. Zhang, T. Li, P. Zhou and F. Yu, Strong hydrogen bond leads to a fifth crystalline form and polymorphism of clonixin, ChemistrySelect, 2017, 2, 4942–4950 CrossRef CAS.
- H. Imoto, Program for structure refinements, J. Crystallogr. Soc. Jpn., 1999, 41, 364–367 CrossRef.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. A. Spackman and D. Jayatilaka, Hirshfeld surface analysis, CrystEngComm, 2009, 11, 19–32 RSC.
- D. Jayatilaka, S. K. Wolff and D. J. Grimwoodx, CrystalExplorer: a tool for displaying Hirshfeld surfaces and visualising intermolecular interactions in molecular crystals, Acta Crystallogr., Sect. A: Found. Crystallogr., 2006, 62, 90–99 Search PubMed.
- X. He, V. H. Man, W. Yang, T. S. Lee and J. Wang, A fast and high-quality charge model for the next generation general AMBER force field, J. Chem. Phys., 2020, 153, 114502–114513 CrossRef CAS.
- A. Jakalian, B. L. Bush, D. And and C. I. Bayly, Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method, J. Comput. Chem., 2000, 10, 132–145 CrossRef.
- A. Jakalian, D. B. Jack and C. I. Bayly, Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation, J. Comput. Chem., 2010, 23, 1623–1641 CrossRef.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 23, 171–187 Search PubMed.
- Z. Berkovitch-Yellin and L. Leiserowitz, Atom-atom potential analysis of the packing characteristics of carboxylic acids. A study based on experimental electron-density distributions, J. Am. Chem. Soc., 1982, 104, 4052–4064 CrossRef CAS.
- A. Gavezzotti and G. Filippini, Geometry of the intermolecular X–H⋯Y (X, Y = N, O) hydrogen bond and the calibration of empirical hydrogen-bond potentials, J. Phys. Chem., 1994, 98, 4831–4837 CrossRef CAS.
- L. Leiserowitz, Molecular packing modes. Carboxylic acids, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 775–802 CrossRef.
- S. Lifson, A. T. Hagler and P. Dauber, Consistent force field studies of intermolecular forces in hydrogen-bonded crystals. 1. Carboxylic acids, amides, and the C:O⋯H-hydrogen bonds, J. Am. Chem. Soc., 1979, 101, 5111–5121 CrossRef CAS.
- R. Feld, M. S. Lehmann, K. W. Muir and J. C. Speakman, The crystal structure of benzoic acid: a redetermination with X-rays at room temperature; a summary of neutron-diffraction work at temperatures down to 5 K, Z. Kristallogr., 1981, 157, 215–231 CAS.
- G. M. Frankenbach and M. C. Etter, Relationship between symmetry in hydrogen-bonded benzoic acids and the formation of acentric crystal structures, Chem. Mater., 1992, 23, 272–278 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of the eight compounds, crystallization results in a series of solvents, and crystal structures of all the forms in the form of crystallographic information file (CIF). CCDC 2260090–2260094, 2260096, and 2260099–2260102. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra04102f |
| ‡ These two authors contributed to this work equally. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Sean Parkin
c,
Sean Parkin