
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReinvestigation of Passerini and Ugi scaffolds as multistep apoptotic inducers via dual modulation of caspase 3/7 and P53-MDM2 signaling for halting breast cancer†
Mohammed Salah Ayoup*a,
Yasmin Wahbya,
Hamida Abdel-Hamida,
Marwa M. Abu-Serieb,
Sherif Ramadan cd,
Assem Barakat*e,
Mohamed Telebf and
Magda M. F. Ismailg
cd,
Assem Barakat*e,
Mohamed Telebf and
Magda M. F. Ismailg
aChemistry Department, Faculty of Science, Alexandria University, P. O. Box 426, Alexandria, 21321, Egypt. E-mail: mohammedsalahayoup@gmail.com; Mohamed.salah@alexu.edu.eg
bMedical Biotechnology Department, Genetic Engineering and Biotechnology Research Institute, City of Scientific Research and Technological Applications (SRTA-City), Egypt
cChemistry Department, Michigan State University, East Lansing, MI 48824, USA
dDepartment of Chemistry, Benha University, Benha, Egypt
eDepartment of Chemistry, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia. E-mail: ambarakat@ksu.edu.sa
fDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Alexandria University, Alexandria, 21521, Egypt
gDepartment of Pharmaceutical Medicinal Chemistry, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo, 11754, Egypt
First published on 20th September 2023
Abstract
Selective induction of breast cancer apoptosis is viewed as the mainstay of various ongoing oncology drug discovery programs. Passerini scaffolds have been recently exploited as selective apoptosis inducers via a caspase 3/7 dependent pathway. Herein, the optimized Passerini caspase activators were manipulated to synergistically induce P53-dependent apoptosis via modulating the closely related P53-MDM2 signaling axis. The adopted design rationale and synthetic routes relied on mimicking the general thematic features of lead MDM2 inhibitors incorporating multiple aromatic rings. Accordingly, the cyclization of representative Passerini derivatives and related Ugi compounds into the corresponding diphenylimidazolidine and spiro derivative was performed, resembling the nutlin-based and spiro MDM-2 inhibitors, respectively. The study was also extended to explore the apoptotic induction capacity of the scaffold after simplification and modifications. MTT assay on MCF-7 and MDA-MB231 breast cancer cells compared to normal fibroblasts (WI-38) revealed their promising cytotoxic activities. The flexible Ugi derivatives 3 and 4, cyclic analog 8, Passerini adduct 12, and the thiosemicarbazide derivative 17 were identified as the study hits regarding cytotoxic potency and selectivity, being over 10-folds more potent (IC50 = 0.065–0.096 μM) and safer (SI = 4.4–18.7) than doxorubicin (IC50 = 0.478 μM, SI = 0.569) on MCF-7 cells. They promoted apoptosis induction via caspase 3/7 activation (3.1–4.1 folds) and P53 induction (up to 4 folds). Further apoptosis studies revealed that these compounds enhanced gene expression of BAX by 2 folds and suppressed Bcl-2 expression by 4.29–7.75 folds in the treated MCF-7 cells. Docking simulations displayed their plausible binding modes with the molecular targets and highlighted their structural determinants of activities for further optimization studies. Finally, in silico prediction of the entire library was computationally performed, showing that most of them could be envisioned as drug-like candidates.
1. Introduction
Breast cancer is the most recognized cancer among women as the universal cause of death. The high incidence and mortality rate of breast cancer have promptly increased, requiring effective treatment protocols.1 One of the fundamental paths to breast cancer treatment is chemotherapeutic agents; nonetheless, their efficacy for therapeutic control of cancer is extremely restricted by multidrug resistance (MDR), where cancer cells gain resistance, and several cases deteriorate in the first five years.2–7 Deregulations of apoptosis is regarded as a central aspect that empowers cancer cells to develop MDR; thus, apoptosis induction is a critical approach for breast cancer control and therapy.Apoptosis evidently requires solid control via two proteins: the family of caspase enzymes and inhibitors of apoptosis proteins (IAPs). Concerning the caspase family, there are two main strategies for apoptosis induction by caspases; the first one is by activation of the mitochondrial apoptotic pathway as an intrinsic pathway and the second strategy commits the activation of an extrinsic pathway (transmembrane-apoptotic pathway).8,9 Afterwards, both tracks assemble at the effective level of caspases; in the extrinsic pathway, caspases 8 and 10 are engaged, whereas caspases 9 and 2 are involved in the intrinsic pathway. Eventually, apoptosis is initiated by executioner caspases 3 and 7 as feedback to stimuli from both paths.10 Bcl-2 family proteins and their regulator, the tumor suppressor P53, are adopted in the intrinsic pathway of apoptosis. P53, the “guardian of the genome”, is a transcription factor regulating several pivotal genes controlling apoptosis.11,12 It primarily uses pro-apoptotic proteins to initiate the apoptotic cascade activation13 and consequent inhibition of the proteins of the mitochondrial anti-apoptotic family, such as Bcl-2.14,15 Altough P53 activation induces caspase-independent apoptosis,16 cell senescence, and associated opsonization signal,17 P53 signals can also promote caspase activation through mitochondrial cytochrome c release.18 The transcriptional activity of P53 and its levels are tightly controlled by the E3 ubiquitin protein ligase MDM2 that can directly conceal P53 N-terminal transactivation domain,19 inducing its proteasomal degradation.20–22 Consistent with its role, MDM2 is viewed as an oncogen when overexpressed.23 These findings have been mirrored by continuous medicinal chemistry research for drugging these signalling pathways towards efficient anticancer protocols24–30 Based on the fact that the activity of mature caspases is abrogated by IAPs that are intrinsically inactivated by the secondary mitochondria-derived activator of caspases (SMAC).31 A tetrapeptide motif of SMAC was viewed as the early lead for designing peptidomimetic apoptotic inducers. Successive optimization studies led to a series of capped tripeptides32 and small-molecule caspase activators,25 of which some reached preclinical evaluation.26–30 Further fragment-based design protocols identified fragment-derived nonpeptidic clinical-stage apoptotic inducers based on amide core as an essential motif.33 Recently, our group has developed series of tumor-selective amide-based caspase activators via Passerini multicomponent reaction mimicking the structural features of lead caspase-dependent apoptotic inducers.34
On the other hand, substantial investment in direct MDM2 inhibition35 led to the discovery of nearly twenty different classes of highly potent small-molecule MDM2 inhibitors, of which several efficacious inhibitors have been raised to clinical trials on various human cancers36–44 including spirooxindoles developed by Sanofi-Aventis (SAR405838),45 Hoffmann-La Roche (RO2468),46 and Daiichi Sankyo (DS-3032b)47 in addition to dozens of leads and active molecules.48 Nutlins based on imidazoline scaffold have also been investigated by both academic and industrial groups.48 Clinical studies revealed that combinations of P53 inducers and other agents acting on related signalling pathways, such as Bcl-2 functions, can synergistically promote apoptosis and sensitize the tumor cells.49,50 Inspired by these outcomes, we speculated that the next frontier towards efficient apoptotic inducers is to combine the apoptotic induction potential of direct caspase activation and P53-MDM2 axis inhibition for efficient tumor sensitization.
2. Rationale design
The privileged Passerini and related Ugi scaffolds sparked our interest, given their facile synthesis, ease of structure diversification, chemical stability, and efficiency as caspase-dependent apoptotic inducers.34,51 Accordingly, in continuation of our previous optimization studies of tumor-selective Passerini-based caspase activators,34,51 we set our design protocol to reinvestigate the potential of these scaffolds to modulate the P53-MDM2 axis. This rationale was considered based on mimicking the general thematic features of MDM2 inhibitors incorporating multiple aromatic rings. Utilizing the optimized tumor-selective Passerini scaffold (Fig. 1), a library of new Passerini and Ugi derivatives with versatile substituents influencing the scaffolds' electronic environment and spatial arrangement was designed and synthesized to allow us to perform structure–activity relationship study. As part of our proposed mimicry design approach, we introduced various functionalities that conferred promising caspase-mediated antitumor activities to lead compounds, especially sulfonamide,52 nitro,53 and azido54 groups. Moreover, these substituents imparted high potency to our previously reported Passerini51 and Ugi55 caspase activators. On the other hand, these functionalities were also represented in several efficient P53 activators, e.g., nonpeptidic sulfonamide P53-MdM2 interaction inhibitors,56 and nitro-substituted spirooxindoles.21 A special focus was placed on the designed scaffold flexibility versus the relatively constrained nutlins (I) ((4S,5R)-2,4,5-triphenyl-4,5-dihydro-1H-imidazole) and spirooxindoles ((2′S,3R,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-N-((1S,3S)-3-hydroxy-3-methylcyclobutyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide) (II); thus, the adopted synthetic route involved cyclization of a representative Ugi derivative into the corresponding diphenylimidazolidine derivative (ethyl-4-(2,5-dioxo-3,4-diphenylimidazolidin-1-yl)benzoate) resembling the nutlins (I) family. Similarly, a representative spiro derivative was successfully synthesized from the appropriate Passerini adduct. The study was also extended to probe the effect of scaffold simplification and modifications to enrich the SAR study. The cytotoxicity of our library against the breast cell lines MCF-7 and MDA-MB 231 in comparison to normal human fibroblasts (Wi-38) was examined. Flow cytometric analysis of apoptosis followed by succesive mechanistic apoptosis studies on the promising cytotoxic hits were performed to evaluate their potential to activate caspase 3/7 and modulate P53-MDM2 axis. Other related signalling nodes were also investigated, e.g., Bcl-2 and BAX. Molecular modelling studies were employed to simulate the binding modes of the active compounds within their molecular targets. Finally, brief in silico studies were performed to predict their drug-like candidate properties. | ||
| Fig. 1 Target adducts and their derivatives. | ||
3. Materials and methods
3.1. Chemistry
The materials and equipment are described in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The stirred mixture was refluxed for 2 days and was continuously monitored by TLC using n-hexane/ethyl acetate (2:1) as an eluent. Then, the reaction mixture was neutralized with a saturated solution of NaHCO3, followed by extraction with EtOAc (3 × 20 ml). The organic layer was separated, dried over anhydrous Na2SO4, and loaded to column chromatography (silica gel, n-hexane/EtOAc (3:1)).55
:1). The stirred mixture was refluxed for 2 days and was continuously monitored by TLC using n-hexane/ethyl acetate (2:1) as an eluent. Then, the reaction mixture was neutralized with a saturated solution of NaHCO3, followed by extraction with EtOAc (3 × 20 ml). The organic layer was separated, dried over anhydrous Na2SO4, and loaded to column chromatography (silica gel, n-hexane/EtOAc (3:1)).55
3.1.1.1. Ethyl-4-(-phenyl-2-(2,2,2-trifluoro-N-phenylacetamido)acetamido)benzoate (2). Yellow crystals: yield 59%; m.p. = 102–104 °C; Rf 0.4 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3314 (–NH), 1690, 1662 (–CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.74 (s, 1H, –NH), 7.89 (d, J = 9.0 Hz, 2H, aromatic–H), 7.72 (d, J = 8.5 Hz, 3H, aromatic–H), 7.32–7.27 (m, 1H, aromatic–H), 7.16–7.10 (m, 6H, aromatic–H), 6.98 (t, J = 8.0 Hz, 1H, aromatic–H), 6.79 (d, J = 8.0 Hz, 1H, aromatic–H), 6.14 (s, 1H, N–CH), 4.24 (q, J = 7.5 Hz, 2H, CH3–CH2O), 1.26 (t, J = 7.0 Hz, 3H, CH3–CH2O); 13C-NMR (125 MHz, DMSO-d6) δC: 168.4 (NH–CO), 165.8 (EtO–CO), 156.5, 156.3 (d, J = 33.3 Hz, N–CO–CF3), 143.5, 135.9, 132.2, 131.7, 131.4, 131.3, 130.9, 130.3, 129.5, 129.4, 129.2, 128.7, 128.4, 128.3, 127.6, 125.1, 122.1, 119.1 (aromatic–C), 67.0 (N–CH), 61.0 (OCH2), 14.7 (CH3). Anal. calc. for C25H21F3N2O4 (470.45); C, 63.83; H, 4.50; N, 5.95; found: C, 63.73; H, 4.72; N, 6.03.
3.1.1.2. Ethyl-4-(2-(2-methyl-N-phenylbenzamido)-2-phenylacetamido)benzoate (3). Pale brown powder: yield 56%; m.p. = 80–82 °C; Rf 0.20 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3118 (–NH), 1709 (br, –CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.71 (s, 1H, –NH), 7.91 (d, J = 9.0 Hz, 2H, aromatic–H), 7.77 (d, J = 9.0 Hz, 2H, aromatic–H), 7.13–6.97 (m, 10H, aromatic–H), 6.88–6.82 (m, 4H, aromatic–H), 6.37 (s, 1H, N–CH), 4.25 (q, J = 7.0 Hz, 2H, CH3–CH2O), 2.27 (s, 3H, aromatic–CH3), 1.27 (t, J = 7.0 Hz, 3H, CH3–CH2O); 13C-NMR (125 MHz, DMSO-d6) δC: 171.0 (N–CO), 170.0 (NH–CO), 165.8 (EtO–CO), 143.9, 139.6, 137.3, 134.6, 134.3, 131.3, 130.9, 130.2, 128.7, 128.2, 127.6, 127.4, 125.2, 124.8, 119.0 (aromatic–C), 65.3 (N–CH), 61.0 (OCH2), 19.6 (aromatic–CH3), 14.8 (OCH2–CH3). Anal. calc. for C31H28N2O4 (492.58); C, 75.59; H, 5.73; N, 5.69; found: C, 75.73; H, 5.59; N, 5.74.
3.1.1.3. Ethyl-4-(2-(2-iodo-N-phenylbenzamido)-2-phenylacetamido)benzoate (4). Buff powder: yield 67%; m.p. = 130–132 °C; Rf 0.27 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3394 (–NH), 1701 (br, –CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.77 (s, 1H, –NH), 7.94 (d, J = 9.0 Hz, 2H, aromatic–H), 7.81 (d, J = 8.5 Hz, 2H, aromatic–H), 7.65–7.61 (m, 2H, aromatic–H), 7.36–7.14 (m, 8H, aromatic–H), 7.04–6.86 (m, 4H, aromatic–H), 6.42 (s, 1H, N–CH), 4.28 (q, J = 7.0 Hz, 2H, CH3–CH2O), 1.30 (t, J = 7.5 Hz, 3H, CH3–CH2O); 13C-NMR (125 MHz, DMSO-d6) δC: 169.7 (N–CO), 169.0 (NH–CO), 165.3 (EtO–CO), 153.5, 143.3, 142.2, 138.5, 138.4, 133.6, 131.1, 130.4, 130.3, 129.8, 128.4, 128.3, 127.8, 127.6, 127.3, 124.4, 118.6, 112.6 (aromatic–C), 94.1 (C–I), 64.6 (N–CH), 60.5 (OCH2), 14.3 (CH3). Anal. calc. for C30H25IN2O4 (604.44); C, 59.61; H, 4.17; N, 4.63; found: C, 59.76; H, 4.05; N, 4.69.
3.1.1.4. Ethyl-4-(2-(2-nitro-N-phenylbenzamido)-2-phenylacetamido)benzoate (5). Green crystals: yield 70%; m.p. = 193–195 °C; Rf 0.29 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3327 (–NH), 1719 (br, –CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.79 (s, 1H, –NH), 7.92 (dd, J = 8.0, 1.5 Hz, 3H, aromatic–H), 7.78 (d, J = 8.5 Hz, 2H, aromatic–H), 7.58 (t, J = 8.5 Hz, 1H, aromatic–H), 7.43–7.39 (m, 2H, aromatic–H), 7.15 (t, J = 4.0 Hz, 6H, aromatic–H), 6.89 (bs, 3H, aromatic–H), 6.44 (s, 1H, N–CH), 4.25 (q, J = 7.0 Hz, 2H, CH3–CH2O), 1.27 (t, J = 7.0 Hz, 3H, CH3–CH2O); 13C-NMR (125 MHz, DMSO-d6) δC: 168.8 (N–CO), 166.9 (NH–CO), 165.3 (EtO–CO), 144.9, 143.2, 138.3, 134.2, 133.4, 132.5, 130.6, 130.4, 130.3, 130.1, 129.4, 128.4, 128.3, 128.1, 127.8, 124.4, 124.1, 118.6 (aromatic–C), 64.6 (N–CH), 60.5 (OCH2), 14.2 (CH3). Anal. calc. for C30H25N3O6 (523.55); C, 68.83; H, 4.81; N, 8.03; found: C, 68.95; H, 4.77; N, 8.10.
3.1.1.5. Ethyl-4-(2-(3,5-dinitro-N-phenylbenzamido)-2-phenylacetamido)benzoate (6). Orange powder: yield 60%; m.p. = 170–172 °C; Rf 0.19 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3328 (–NH), 1709 (br, –CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.80 (s, 1H, –NH), 8.63 (t, J = 2.0 Hz, 1H, aromatic–H), 8.36 (d, J = 1.5 Hz, 2H, aromatic–H), 7.91 (d, J = 9.0 Hz, 2H, aromatic–H), 7.78 (d, J = 8.5 Hz, 2H, aromatic–H), 7.21–7.14 (m, 6H, aromatic–H), 7.01–6.93 (m, 4H, aromatic–H), 6.42 (s, 1H, N–CH), 4.25 (q, J = 7.5 Hz, 2H, CH3–CH2O), 1.27 (t, J = 7.0 Hz, 3H, CH3–CH2O); 13C-NMR (125 MHz, DMSO-d6) δC: 169.4 (N–CO), 166.2 (NH–CO), 165.8 (EtO–CO), 147.9, 143.7, 139.5, 139.3, 133.7, 131.6, 131.0, 130.9, 129.0, 128.9, 128.8, 128.7, 128.4, 125.0, 119.8, 119.2 (aromatic–C), 65.9 (N–CH), 61.0 (OCH2), 14.7 (CH3). Anal. calc. for C30H24N4O8 (568.16); C, 63.38; H, 4.26; N, 9.85; found: C, 63.16; H, 4.18; N, 9.91.
3.1.1.6. Ethyl-4-(2,5-dioxo-3,4-diphenylimidazolidin-1-yl)benzoate (8). Off-white powder: yield 58%; m.p. = 62–64 °C; Rf 0.48 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 1779 (–CO), 1724 (CO–N–CO); 1H-NMR (500 MHz, DMSO-d6) δH: 8.07 (d, J = 8.5, 2H, aromatic–H), 7.64 (d, J = 8.5 Hz, 2H, aromatic–H), 7.54 (d, J = 8.0 Hz, 2H, aromatic–H), 7.49 (d, J = 7.5 Hz, 2H, aromatic–H), 7.34–7.28 (m, 5H, aromatic–H), 7.07 (t, J = 7.0 Hz, 1H, aromatic–H), 6.12 (s, 1H, N–CH), 4.30 (q, J = 7.5 Hz, 2H, CH3–CH2O), 1.30 (t, J = 7.5 Hz, 3H, CH3–CH2O); 13C-NMR (125 MHz, DMSO-d6) δC: 169.8 (N–CO–CH), 165.6 (EtO–CO), 153.4 (N–CO–N), 136.6, 136.4, 134.1, 130.3, 129.8, 129.5, 129.4, 128.3, 127.6, 125.4, 122.1, 64.0 (N–CH), 61.6 (OCH2), 14.7 (CH3). Anal. calc. for C24H20N2O4 (400.43); C, 71.99; H, 5.03; N, 7.00; found: C, 72.13; H, 5.11; N, 7.08.
:1) as an eluent. Then, the reaction was quenched by adding 5 ml of dichloromethane and neutralized by NaHCO3 saturated solution. The organic layer was separated, dried over anhydrous Na2SO4, and loaded to column chromatography for purification.34
3.1.2.1. 1-((4-(Ethoxycarbonyl)phenyl)carbamoyl)cyclohexyl-2-methylbenzoate (9). Off-white powder: yield 50%; m.p. = 121–124 °C; Rf 0.54 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3337 (–NH), 1720, 1697 (–CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.98 (s, 1H, –NH), 7.95 (d, J = 7.5 Hz, 1H, aromatic–H), 7.88 (d, J = 8.0 Hz, 2H, aromatic–H), 7.78 (d, J = 9.0 Hz, 2H, aromatic–H), 7.50 (t, J = 6.0 Hz, 1H, aromatic–H), 7.38–7.32 (m, 2H, aromatic–H), 4.26 (q, J = 7.0 Hz, 2H, CH3–CH2O), 2.41 (s, 3H, CH3–aromatic), 2.29 (d, J = 13.5 Hz, 2H, cyclohexylidene–H), 1.91–1.85 (td, J = 13.5, 3.5 Hz, 2H, cyclohexylidene–H), 1.69–1.55 (m, 5H, cyclohexylidene–H), 1.34–1.32 (m, 1H, cyclohexylidene–H), 1.29 (t, J = 7.5 Hz, 3H, CH3–CH2O); 13C-NMR (125 MHz, DMSO-d6) δC: 171.4 (NH–CO), 165.7 (C–O–CO), 165.4 (EtO–CO), 143.5, 139.3, 132.3, 131.6, 130.2, 130.0, 126.1, 124.4, 119.5 (aromatic–C), 81.9 (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CO), 60.5 (OCH2), 31.4, 24.6, 21.2, 20.9 (cyclohexylidene–C), 14.3 (CH3). Anal. calc. for C24H27NO5 (409.48); C, 70.40; H, 6.65; N, 3.42; found: C, 70.34; H, 6.57; N, 3.49.
C–CO), 60.5 (OCH2), 31.4, 24.6, 21.2, 20.9 (cyclohexylidene–C), 14.3 (CH3). Anal. calc. for C24H27NO5 (409.48); C, 70.40; H, 6.65; N, 3.42; found: C, 70.34; H, 6.57; N, 3.49.
3.1.2.2. 1-((4-(Ethoxycarbonyl)phenyl)carbamoyl)cyclohexyl-4-azidobenzoate (10). Off-white crystals: yield 56%; m.p. = 182–184 °C; Rf 0.74 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3319 (–NH), 2125 (–N3), 1711 (br, –CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.94 (s, 1H, –NH), 8.02 (d, J = 8.5 Hz, 2H, aromatic–H), 7.84 (d, J = 8.5, 2H, aromatic–H), 7.72 (d, J = 8.5 Hz, 2H, aromatic–H), 7.25 (d, J = 8.5 Hz, 2H, aromatic–H), 4.23 (q, J = 7.0 Hz, 2H, CH3–CH2O), 2.27 (d, J = 13.5 Hz, 2H, cyclohexylidene–H), 1.84 (td, J = 13.5, 3.0 Hz, 2H, cyclohexylidene–H), 1.64 (d, J = 10.0 Hz, 3H, cyclohexylidene–H), 1.55–1.48 (m, 2H, cyclohexylidene–H), 1.33–1.30 (m, 1H, cyclohexylidene–H), 1.26 (t, J = 7.0 Hz, 3H, CH3–CH2O); 13C-NMR (125 MHz, DMSO-d6) δC: 171.8 (NH–CO), 165.9 (C–O–CO), 164.2 (EtO–CO), 145.2, 143.9, 132.1, 130.4, 126.8, 124.9, 120.1, 119.9 (aromatic–C), 82.2 (OC–CO), 61.0 (OCH2), 31.9, 25.1, 21.6 (cyclohexylidene–C), 14.7 (CH3). Anal. calc. for C23H24N4O5 (436.47); C, 63.29; H, 5.54; N, 12.84; found: C, 63.17; H, 5.59; N, 12.77.
3.1.2.3. 1-((4-(Ethoxycarbonyl)phenyl)carbamoyl)cyclohexyl 4-nitrobenzoate (11). Off-white crystals: yield 51%; m.p. = 239–241 °C; Rf 0.58 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3328 (–NH), 1714 (br, –CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.99 (s, 1H, –NH), 8.36 (d, J = 7.5 Hz, 2H, aromatic–H), 8.25 (d, J = 7.5 Hz, 2H, aromatic–H), 7.85 (d, J = 8.0 Hz, 2H, aromatic–H), 7.72 (d, J = 7.5 Hz, 2H, aromatic–H), 4.23 (q, J = 6.0 Hz, 2H, CH3–CH2O), 2.29 (d, J = 14.0 Hz, 2H, cyclohexylidene–H), 1.90 (t, J = 12.5 Hz, 2H, cyclohexylidene–H), 1.65 (bs, 3H, cyclohexylidene–H), 1.58–1.51 (m, 2H, cyclohexylidene–H), 1.40–1.31 (m, 1H, cyclohexylidene–H), 1.26 (t, J = 7.0 Hz, 3H, CH3–CH2O); 13C-NMR (125 MHz, DMSO-d6) δC: 171.3 (NH–CO), 165.8 (C–O–CO), 163.5 (EtO–CO), 150.9, 143.7, 135.7, 131.6, 130.5, 125.0, 124.5, 120.2 (aromatic–C), 83.3 (OC–CO), 61.0 (OCH2), 31.9, 25.0, 21.6 (cyclohexylidene–C), 14.7 (CH3). Anal. calc. for C23H24N2O7 (440.45); C, 62.72; H, 5.49; N, 6.36; found: C, 62.85; H, 5.42; N, 6.22.
3.1.2.4. 1-((4-(Ethoxycarbonyl)phenyl)carbamoyl)cyclohexyl-4-(phenylsulfonamido)benzoate (12). Yellow powder: yield 45%; m.p. = 220–222 °C; Rf 0.56 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3404 (–NH), 1717, 1669 (–CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.93, 9.86 (s, 2H, 2NH's), 7.87–7.81 (m, 6H, aromatic–H), 7.69 (d, J = 8.0 Hz, 2H, aromatic–H), 7.62–7.53 (m, 3H, aromatic–H), 7.22 (d, J = 8.5 Hz, 2H, aromatic–H), 4.22 (q, J = 7.0 Hz, 2H, CH3–CH2O), 2.20 (d, J = 13.0 Hz, 2H, cyclohexylidene–H), 1.79 (t, J = 11.5 Hz, 2H, cyclohexylidene–H), 1.61–1.48 (m, 5H, cyclohexylidene–H), 1.27 (t, J = 7.0 Hz, 3H, CH3–CH2O), 1.19 (s, 1H, cyclohexylidene–H); 13C-NMR (125 MHz, DMSO-d6) δC: 171.9 (NH–CO), 165.9 (C–O–CO), 164.4 (EtO–CO), 143.9, 143.4, 139.8, 133.8, 131.6, 130.4, 130.0, 127.2, 124.9, 124.8, 120.1, 118.5 (aromatic–C), 81.9 (OC–CO), 61.0 (OCH2), 31.9, 25.1, 21.6 (cyclohexylidene–C), 14.7 (CH3). Anal. calc. for C29H30N2O7S (550.63); C, 63.26; H, 5.49; N, 5.09; S, 5.82; found: C, 63.14; H, 5.17; N, 5.03; S, 5.75.
3.1.2.5. Ethyl 4-(1-(2-(phenylsulfonamido)acetoxy)cyclohexanecarboxamido)benzoate (13). White powder: yield 49%; m.p. = 198–199 °C; Rf 0.23 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3228 (–NH), 1757, 1702 (–CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.76 (s, 1H, –NH), 8.22 (t, J = 6.5 Hz, 1H, CH2–NH), 7.85 (d, J = 9.0 Hz, 2H, aromatic–H), 7.73–7.68 (m, 4H, aromatic–H), 7.55–7.52 (m, 1H, aromatic–H), 7.44 (t, J = 8.0 Hz, 2H, aromatic–H), 4.24 (q, J = 7.5 Hz, 2H, CH3–CH2O), 3.87 (d, J = 6.5 Hz, 2H, OCH2–NH), 1.98 (d, J = 13.0 Hz, 2H, cyclohexylidene–H), 1.68 (td, J = 14.5, 4.0 Hz, 2H, cyclohexylidene–H), 1.56–1.50 (m, 3H, cyclohexylidene–H), 1.44–1.37 (m, 2H, cyclohexylidene–H), 1.26 (t, J = 7.0 Hz, 3H, CH3–CH2O); 1.24 (m, 1H, cyclohexylidene–H); 13C-NMR (125 MHz, DMSO-d6) δC: 171.4 (NH–CO), 168.3 (C–O–CO), 165.9 (EtO–CO), 143.7, 141.1, 133.0, 130.4, 129.6, 127.0, 125.0, 120.0 (aromatic–C), 82.6 (OC–CO), 61.0 (OCH2), 44.4 (CH2–NH), 31.8, 25.0, 21.2 (cyclohexylidene–C), 14.7 (CH3). Anal. calc. for C24H28N2O7S (488.56); C, 59.00; H, 5.78; N, 5.73; S, 6.56; found: C, 59.09; H, 5.81; N, 5.69; S, 6.50.
3.1.2.6. 4-(1-(Carboxyoxy)cyclohexane-1-carboxamido)benzoic acid (15). Ethyl-4-(2,4-dioxo-1-oxa-3-azaspiro[4.5]decan-3-yl)benzoate 14 (0.315 mmol, 1 eq.) and NaOH (0.946 mmol, 3 eq.) were placed in EtOH/H2O (1
:1) mixture. The stirred mixture was refluxed for 1 h and was monitored continuously by TLC till reaction completion. After that, the reaction mixture was cooled, acidified till pH = 2 using HCl (2M), the crude product was precipitated, and collected by filtration. Off-white crystals: yield 51%; m.p. = 220–222 °C; Rf 0.54 (2:1, n-hexane/EtOAc); IR vmax/cm−1: 3291 (–NH), 1700 (br, –CO, –NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 12.66 (s, 2H, 2OH's), 10.04 (s, 1H, –NH), 7.82 (d, J = 9.0 Hz, 2H, aromatic–H), 7.52 (d, J = 8.5 Hz, 2H, aromatic–H), 2.07 (d, J = 13.5 Hz, 2H, cyclohexylidene–H), 1.73 (dist.td, J = 14.0, 3.0 Hz, 2H, cyclohexylidene–H), 1.57–1.46 (m, 5H, cyclohexylidene–H), 1.30–1.22 (m, 1H, cyclohexylidene–H); 13C-NMR (125 MHz, DMSO-d6) δC: 173.9 (NH–CO), 167.0 (aromatic–CO–OH), 152.1 (O–CO–OH), 143.3, 130.5, 124.4, 117.4 (aromatic–C), 79.2 (OC–CO), 32.0, 24.7, 21.0 (cyclohexylidene–C). Anal. calc. for C15H17NO6 (307.30); C, 58.63; H, 5.58; N, 4.56; found: C, 58.77; H, 5.65; N, 4.48.
3.1.2.7. N-(4-(Hydrazinecarbonyl)phenyl)-1-hydroxycyclohexane-1-carboxamide (16). 1-((4-(Ethoxycarbonyl)phenyl)carbamoyl)cyclohexyl-4-nitrobenzoate (11) (70 mg, 0.16 mmole) in 2 ml of hydrazine (99%) was refluxed for 2 days and monitored continuously by TLC. The product precipitate is formed after cooling the mixture. The product was filtered and collected without any further purification. Off-white powder: yield 46%; m.p. = 201–204 °C; Rf 0.48 (1
:1:1, n-hexane/EtOAc/methanol); IR vmax/cm−1: 3549 (–OH), 3411, 3332, 3270 (–NH's), 1651 (–NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 9.76, 9.61 (2s, 2H, 2NH's), 7.82–7.71 (m, 4H, aromatic–H), 7.22 (s, 1H, –NH), 5.49 (s, 1H, –OH), 4.42 (s, 1H, –NH), 1.70 (m, 2H, cyclohexylidene–H), 1.57–1.47 (m, 7H, cyclohexylidene–H), 1.21–1.16 (m, 1H, cyclohexylidene–H); 13C-NMR (125 MHz, DMSO-d6) δC: 176.9, 167.9 (2NH–CO), 142.0, 141.9, 129.2, 128.7, 128.2, 128.1, 119.3, 119.2 (aromatic–C), 74.5 (OC–CO), 34.2, 25.5, 21.3 (cyclohexylidene–C). Anal. calcd for C14H19N3O3 (277.32): C, 60.63; H, 6.91; N, 15.15; found C, 60.77; H, 6.99; N, 15.08.
3.1.2.8. 1-Hydroxy-N-(4-(2-(phenylcarbamothioyl)hydrazine-1-carbonyl)phenyl)cyclohexane-1-carboxamide (17). A solution of N-(4-(hydrazinecarbonyl)phenyl)-1-hydroxycyclohexane-1-carboxamide 16 (300 mg, 1.08 mmol) and phenyl isothiocyanate (160 mg, 1.19 mmol) in 5 mL absolute ethanol was refluxed for 8 h. The reaction mixture was continuously monitored by TLC till reaction completion. After that, the mixture was cooled at room temperature. The formed solid was collected by filtration and purified using column chromatography. Orange powder: yield 69%; m.p. = 165–168 °C; Rf 0.5 (2
:1, n-hexane/EtOAc); IR vmax/cm−1: 3346, 3259 (br, –NH's), 1671 (–NCO); 1H-NMR (500 MHz, DMSO-d6) δH: 10.39, 9.86, 9.65 (3s, 3H, 3NH's), 9.76 (s, 1H, –NH), 7.85 (q, J = 9 Hz, 4H, aromatic–H), 7.39 (bs, 2H, aromatic–H), 7.29 (t, J = 7.5 Hz, 2H, aromatic–H), 7.12 (t, J = 6.5 Hz, 1H, aromatic–H), 5.51 (s, 1H, –OH), 1.71 (td, J = 13.5, 4.5 Hz, 2H, cyclohexylidene–H), 1.58–1.48 (m, 7H, cyclohexylidene–H), 1.28–1.14 (m, 1H, cyclohexylidene–H); 13C-NMR (125 MHz, DMSO-d6) δC: 177.0, 166.0 (2NH–CO), 142.6, 139.8, 129.2, 128.4, 127.4, 126.6, 119.1 (aromatic–C), 74.5 (OC–CO), 34.2, 25.5, 21.3 (cyclohexylidene–C). Anal. calcd for C21H24N4O3S (412.51): C, 61.14; H, 5.86; N, 13.58; S, 7.77; found C, 61.22; H, 5.93; N, 13.49; S, 7.83.
3.2. Biological evaluation
4. Results and discussion
4.1. Chemistry
Multicomponent reactions (MCRs) are straightforward chemical reactions that include a group of chemical transformations in a single step without the isolation of intermediates.Ugi-MCR is a one-pot condensation reaction of amines, carboxylic acid derivatives, and isonitriles for the generation of classical bisamide products. The application of the Ugi reaction generates a variety of biologically active molecules, which has attracted attention in recent years.57–61 Herein, we utilized ethyl-4-isocyanobenzoate (1) as an entry to Passerini and Ugi products; it was prepared via esterification of 4-aminobenzoic acid to afford the ester; then, formylation using HCOOH/toluene protocol yielded the formamide, which underwent dehydration by Wang's method62,63 to give the corresponding isonitrile, 1 in good yield.
α-Aminoacyl amides (2–6) were prepared via Ugi reaction where the interaction occurs between the isonitrile 1, aniline, benzaldehyde, and various carboxylic acids (trifluoroacetic acid, o-toluic acid, o-iodobenzoic acid, o-nitrobenzoic acid, and 3,5-dinitrobenzoic acid) using trifluoroethanol/ethanol mixture (1:1) under reflux condition (Scheme 1). Ugi structures were confirmed based on IR and NMR spectral data. Their 1H-NMR spectra clearly recorded –NH singlet signals between δH 10.81 and 10.71 ppm, and the aromatic proton signals appeared at δH 6.94–6.79 ppm. Methine protons (N–CH) of all products were recorded as singlet signals between δH 6.44 and 6.14 ppm. Also, protons of CH2 and CH3 of the ethyl group appeared in the range δH 4.28–4.24 and 1.30–1.26 ppm, respectively. Moreover, the 13C-NMR spectrum showed the carbonyl carbons of bis-amide in all Ugi products resonating from δc 171.0 to 156.3 ppm. The aromatic carbon signals were recorded between δC 147.9 and 118.6 ppm. Methine carbons (N–CH–CO) appeared between δC 67.5 and 64.6 ppm. Carbon signals of the carbonyl group of COCF3 for compound 2 appeared as a doublet at 156.5 and 156.3 ppm with J coupling 33.3 Hz. The methyl group (Ar–CH3) in product 3 showed signals at δC 19.6 ppm. For product 4, carbon of C–I showed a signal recorded at δC 94.1 ppm.
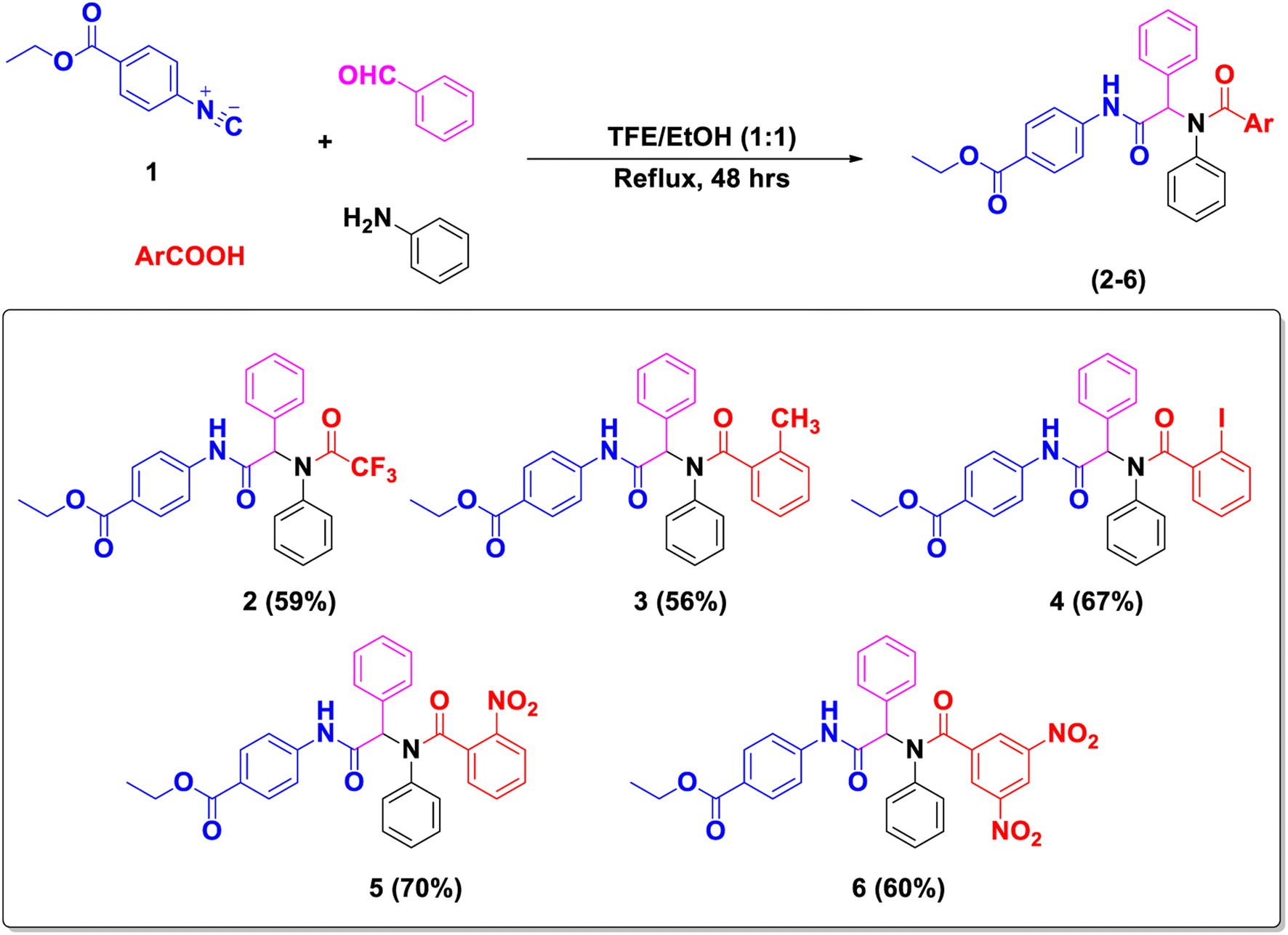 | ||
| Scheme 1 Synthesis of α-acylamino amide derivatives 2–6 via Ugi reaction. | ||
Moreover, the synthesis of ethyl 4-(2,5-dioxo-3,4-diphenylimidazolidin-1-yl)benzoate (8) was performed using ethyl-4-isocyanobenzoate 1, aniline, benzaldehyde and trichloroacetic acid under reflux in EtOH/TFE to yield the intermediate 7. The latter was cyclized immediately via intramolecular attack of the nucleophile NH to the electrophilic center of the carbonyl of COCCl3 to afford the unexpected compound 8 with the elimination of one molecule of CHCl3 (Scheme 2). 1H-NMR confirmed the structure of 8, revealing the lack of –NH signal; in addition, methine proton (Ph–CH) appeared as a singlet signal at δH 6.12 ppm. 13C-NMR showed a methine carbon (Ph–CH) signal at δC 64.0 ppm and 2 CO signals of oxazolinedione at δC 169.8 and 153.4 ppm.
 | ||
| Scheme 2 Synthesis of ethyl 4-(2,5-dioxo-3,4-diphenylimidazolidin-1-yl)benzoate (8). | ||
Furthermore, Mario Passerini explored the first multicomponent reaction nearly a century ago. Passerini reactions, in a one-pot fashion,34 offer great advantages over classical bimolecular reactions. Novel α-acyloxy carboxamides (9–13) were synthesized according to the Passerini method by using ethyl-4-isocyanobenzoate 1, cyclohexanone with miscellaneous carboxylic acids, namely 4-(phenylsulfonamido)benzoic acid, 2-(phenylsulfonamido)acetic acid, o-toluic acid, p-azidobenzoic acid or p-nitrobenzoic acid, at room temperature (Scheme 3). The obtained compounds (9–13) were analyzed by FT-IR and NMR spectroscopy. 1H-NMR spectra showed signals from δH 9.99–9.76 ppm for –NH in amido groups. Protons of aromatic moiety showed signals at δH 7.25–8.36 ppm and c-hexylidene protons (10 Hs) at δH 2.29–1.19 ppm. Aliphatic protons of –OCH2 and –CH3 protons were shielded and detected clearly at δH 4.26–4.22 ppm and δH 1.29–1.26 ppm, respectively; for instance, methyl protons (CH3–Ar) in 9 showed signal resonating at δH 2.41 ppm. 13C-NMR spectrum confirmed the existence of amido and ester CO groups by showing signals at δC 171.9–163.5 ppm. The aromatic carbons resonated between δC 150.9–118.5 ppm. c-Hexylidene (O–C–CO) carbon signals ranged from δc 83.3–79.2 ppm. Also, cyclohexylidene carbon signals at δc 31.9–20.9 ppm. Signals were detected resonating at δC 61.0–60.5 ppm and δC 14.7–14.3 ppm for –OCH2 and –CH3 carbons, respectively. Carbons of the methyl group (CH3–Ar) in product 9 showed signal resonating at δC 20.9 ppm. Also, product 13 showed a carbon signal at δC 44.4 ppm for methylene carbon (CH2–NH).
 | ||
| Scheme 3 Synthesis of α-acyloxy carboxamide derivatives 9–13 via Passerini reaction. | ||
Alternatively, Passerini reaction using 1 with trichloroacetic acid and cyclohexanone proceeded to afford the unexpected Passerini adduct ethyl 4-(2,4-dioxo-1-oxa-3-azaspiro[4.5]decan-3-yl)benzoate 14.62 Saponification of 14 in NaOH followed by acidification led to the formation of 4-(1-(carboxyoxy)cyclohexane-1-carboxamido)benzoic acid 15 (Scheme 4). The obtained monocarbonate ester 15 was confirmed by 1H-NMR spectroscopy where two carboxylic protons showed a signal at δH 12.66 ppm and one proton of –NH at δH 10.04 ppm. 13C-NMR spectrum recorded signals at δC 173.9 and 167.0 ppm, corresponding to NH–CO and Ar–CO–OH carbons, respectively. The carbon signal of O–CO–OH resonated at δC 152.1 ppm, while the carbon signals of OC–CO appeared at δc 79.2 ppm.
 | ||
| Scheme 4 Synthesis of 4-(1-(carboxyoxy)cyclohexane-1-carboxamido)benzoic acid 15. | ||
Moreover, hydrazinolysis of 11 by reflux with hydrazine afforded N-(4-(hydrazinecarbonyl)phenyl)-1-hydroxycyclohexane-1-carboxamide (16), which upon treatment with phenylisothiocyanate (PhNCS) afforded 1-hydroxy-N-(4-(2-(phenylcarbamothioyl)hydrazine-1-carbonyl)phenyl)cyclohexane-1-carboxamide (17) (Scheme 5). The products 16 and 17 were confirmed by 1H-NMR spectroscopy. With respect to 16, its 1H-NMR recorded three isolated signals at δH 9.76, 9.61, and 7.22 ppm, representing 3-NH protons and another signal at δH 5.49 ppm for the OH group. 13C-NMR spectrum recorded signals resonating at δC 176.9 and 167.9 ppm, corresponding to carbons of two NH–CO groups. Also, the signal was detected for carbon in the OC–C–OH group at δc 74.4 ppm. On the other hand, the disubstituted semicarbazide 17 showed four signals at δH: 10.39, 9.86, 9.76, and 9.65 ppm, corresponding to 4 NH. Also, another singlet appeared at δH 5.51 ppm, equivalent to the quaternary OH group. 13C-NMR spectrum of 17 recorded two carbon signals at δC 177.0 and 166.0 ppm, corresponding to two carbons of 2 NH–CO. Also, the signal was detected for carbon in the OC–C–OH group at δC 74.5 ppm.
 | ||
| Scheme 5 Synthesis of the hydrazide 16 and thiosemicarbazide derivative 17. | ||
4.2. Biological evaluation
| Compound no. | Wi-38 | MCF-7 | MDA-MB 231 | |||
|---|---|---|---|---|---|---|
| IC50 (μM) | EC100 (μM) | IC50 (μM) | SI | IC50 (μM) | SI | |
| a Values are demonstrated as mean ± SEM. | ||||||
| 2 | 0.147 ± 0.020 | 0.024 ± 0.004 | 0.369 ± 0.014 | 0.398 | 0.472 ± 0.013 | 0.311 |
| 3 | 1.329 ± 0.084 | 0.302 ± 0.064 | 0.086 ± 0.016 | 15.453 | 0.306 ± 0.014 | 4.343 |
| 4 | 0.418 ± 0.033 | 0.141 ± 0.018 | 0.096 ± 0.007 | 4.354 | 0.188 ± 0.044 | 2.223 |
| 5 | 0.570 ± 0.072 | 0.197 ± 0.006 | 0.229 ± 0.042 | 2.489 | 0.302 ± 0.024 | 1.887 |
| 6 | 0.538 ± 0.025 | 0.172 ± 0.009 | 0.143 ± 0.016 | 3.762 | 0.370 ± 0.094 | 1.454 |
| 8 | 0.953 ± 0.037 | 0.376 ± 0.004 | 0.078 ± 0.072 | 12.217 | 0.146 ± 0.034 | 6.527 |
| 9 | 0.608 ± 0.012 | 0.133 ± 0.013 | 0.440 ± 0.006 | 1.382 | 0.445 ± 0.004 | 1.366 |
| 10 | 0.505 ± 0.001 | 0.242 ± 0.031 | 0.439 ± 0.005 | 1.150 | 0.470 ± 0.012 | 1.074 |
| 11 | 0.516 ± 0.011 | 0.110 ± 0.013 | 0.591 ± 0.035 | 0.873 | 0.756 ± 0.175 | 0.683 |
| 12 | 0.633 ± 0.001 | 0.208 ± 0.003 | 0.065 ± 0.050 | 9.738 | 0.135 ± 0.000 | 4.689 |
| 13 | 0.590 ± 0.026 | 0.147 ± 0.028 | 0.142 ± 0.027 | 4.155 | 0.513 ± 0.066 | 1.150 |
| 15 | 0.827 ± 0.004 | 0.200 ± 0.028 | 0.264 ± 0.159 | 3.133 | 0.261 ± 0.025 | 3.169 |
| 16 | 3.327 ± 0.032 | 1.194 ± 0.020 | 0.968 ± 0.036 | 3.437 | 1.028 ± 0.036 | 3.236 |
| 17 | 1.535 ± 0.122 | 0.466 ± 0.070 | 0.082 ± 0.002 | 18.720 | 0.518 ± 0.081 | 2.963 |
| Dox. | 0.272 ± 0.053 | 0.074 ± 0.008 | 0.478 ± 0.076 | 0.569 | 2.828 ± 0.173 | 0.096 |
| Compound no. | MCF-7 | MDA-MB 231 |
|---|---|---|
| a Values are demonstrated as mean ± SEM. | ||
| 3 | 4.110 ± 0.1014 | ND |
| 4 | 4.137 ± 0.0407 | 3.309 ± 0.110 |
| 8 | 4.810 ± 0.1014 | 3.733 ± 0.023 |
| 12 | 5.168 ± 0.0676 | 4.312 ± 0.075 |
| 17 | 4.377 ± 0.144 | ND |
| Doxorubicin | 2.148 ± 0.174 | 1.623 ± 0.122 |
| Compound no. | MCF-7 | MDA-MB 231 | ||
|---|---|---|---|---|
| BAX | Bcl-2 | BAX | Bcl-2 | |
| a Values are demonstrated as mean ± SEM. | ||||
| 3 | 2.448 ± 0.057 | 0.217 ± 0.006 | ND | ND |
| 4 | 2.747 ± 0.1285 | 0.233 ± 0.0017 | 2.338 ± 0.021 | 0.496 ± 0.005 |
| 8 | 3.195 ± 0.082 | 0.163 ± 0.0035 | 2.479 ± 0.030 | 0.454 ± 0.010 |
| 12 | 3.337 ± 0.1485 | 0.129 ± 0.0027 | 2.683 ± 0.065 | 0.376 ± 0.037 |
| 17 | 3.042 ± 0.0365 | 0.201 ± 0.0075 | ND | ND |
| Doxorubicin | 1.556 ± 0.054 | 0.592 ± 0.031 | 1.232 ± 0.019 | 0.798 ± 0.044 |
| Compound no. | MCF-7 | MDA-MB 231 |
|---|---|---|
| a ND: not done; values are demonstrated as mean ± SEM. | ||
| 3 | 3.470 ± 0.0485 | ND |
| 4 | 3.113 ± 0.10115 | 2.618 ± 0.1615 |
| 8 | 3.763 ± 0.011 | 2.945 ± 0.0885 |
| 12 | 4.105 ± 0.0795 | 3.304 ± 0.0745 |
| 17 | 3.677 ± 0.0145 | ND |
| Doxorubicin | 1.881 ± 0.028 | 1.272 ± 0.074 |
 | ||
| Fig. 2 Morphological changes of breast cancer cells after 72 h treatment with the promising hits. | ||
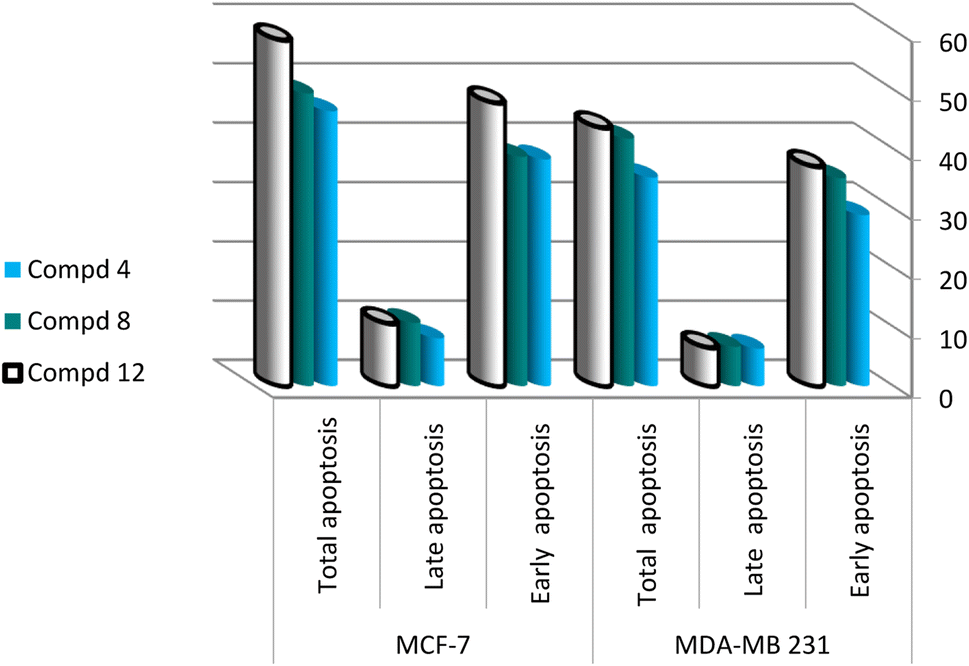 | ||
| Fig. 3 Apoptotic cell population (%) in the treated breast cancer lines. | ||
 | ||
| Fig. 4 Flow charts of annexin-PI analysis of 4, 8 and 12-treated breast cancer cell lines. | ||
4.3. Molecular modeling studies
4.3.1.1. Binding mode analysis of the caspase 3/7 activator hits into XIAP BIR2 domain. Based on our design strategy, the binding mode of the hit compounds into XIAP, the SMAC mimetics molecular target (Fig. 1; I & II), was explored. XIAP functions as an apoptosis inhibitor through direct binding to BIR domains. It is known that the BIR2 domain inhibits caspase 3 and 7,10,70–74 whereas the BIR3 domain selectively targets caspase 9. Various structure-based studies reported that SMAC mimetics that can bind into the XIAP BIR domains directly antagonize the inhibitory potential of XIAP against caspase, thus inducing apoptosis75–78 Herein, docking of the hit caspase activators was simulated within the XIAP BIR2 domain that selectively binds caspase 3/7 employing MOE 2019.102.79 The crystal structure of the XIAP BIR2 domain complexed with a benzodiazepinone-based inhibitor was retrieved from the protein data bank (PDB ID: 4KJU)80 (Fig. 5). After default structure preparation, the studied compounds were built in silico, energy minimized, and docked into the ligand binding site of XIAP BIR2 domain. The docking protocol was preliminarily validated by redocking the reference ligand (ΔG = −8.66 kcal mol−1) with restoration of nearly all the experimental interactions (Fig. 5, panels K and L). The most potent caspase activator 12 fitted well into the ligand's site within the BIR2 domain, recording the most favored free binding energy (ΔG = −7.16 kcal mol−1) among the group (Table 5). The aromatic rings encompassing the sulphonamide group interacted with Leu207 and Asp214 via π–H and hydrogen bonds resembling the reference inhibitor in addition to H-bonds linking the amide and terminal ester carbonyl oxygens of the scaffold with Lys206 and Arg156 (Fig. 5, panels G and H). The cyclized derivative 8 displayed π–H and hydrogen bond interactions with the ligand essential residues Leu207, Ly208, and His223, besides the nearby Lys206 (Fig. 5, panels E and F). The thiosemicarbazide motif in 17 interacted with Asp214 and Arg222 through hydrogen bonds, whereas the backbone phenyl ring exhibited H–π interactions with Lys208 (Fig. 5, panels I and J). The Ugi adduct 3 also shared interactions with Lys208, Arg222, and His223 (Fig. 5, panels A and B). The iodophenyl analog 4 displayed multiple H–π interactions with Lys208, Trp210, and Phe224 (Fig. 5, panels C and D).
 | ||
| Fig. 5 (A) Overlay of the 3D binding mode of the co-crystallized ligand (green sticks) and 3 (yellow sticks), (B) 2D binding mode of 3, (C) overlay of the 3D binding mode of the co-crystallized ligand (green sticks) and 4 (cyan sticks), (D) 2D binding mode of 4, (E) overlay of the 3D binding mode of the co-crystallized ligand (green sticks) and 8 (pink sticks), (F) 2D binding mode of 8, (G) overlay of the 3D binding mode of the co-crystallized ligand (green sticks) and 12 (magenta sticks), (H) 2D binding mode of 12 (I) overlay of the 3D binding mode of the co-crystallized ligand (green sticks) and 17 (red sticks), (J) 2D binding mode of 17 into the XIAP BIR2 domain (PDB ID: 4KJU80,81), (K) superposition of the modeled (blue sticks) and the co-crystallized benzodiazepinone inhibitor (light pink sticks), and (L) 2D binding mode of the co-crystallized inhibitor in the XIAP BIR2 domain (PDB ID: 4KJU80,81). | ||
| Compound no. | ΔG (kcal mol−1) |
|---|---|
| 3 | −6.30 |
| 4 | −6.53 |
| 8 | −6.19 |
| 12 | −7.16 |
| 17 | −6.60 |
| Co-crystallized inhibitor | −8.66 |
Herein, it could be assumed that the compounds could bind to the XIAP BIR2 domain, the molecular target of SMAC mimetics caspase 3/7 activators. Interestingly, these results could be correlated to the in vitro caspase 3/7 activation assay results, highlighting the main pharmacophoric features of the studied compounds. The cyclization of Ugi scaffold in future studies, given their expected key interactions with the BIR2 domain, should be specially considered.
4.3.1.2. Binding mode analysis of the P53 activator hits into MDM2. The E3 ubiquitin-protein ligase MDM2 crystal structure was retrieved from the protein databank co-crystallized with the reference inhibitor spiro[3H-indole-3,2′-pyrrolidin]-2(1H)-one 6SJ (PBD ID: 5LAW82). The protein was prepared, employing MOE “QuickPrep” module after eliminating unwanted residues. The studied derivatives (Table 6) were built in silico and energy minimized, then docked into the co-crystallized ligand's binding site after validating the adopted protocol (Fig. 6). The best simulated binding modes (Fig. 6) showed that the hit compounds were able to reside well in the ligand's site with free binding energies (Table 6) comparable to the redocked reference inhibitor 6SJ (ΔG = −8.40 kcal mol−1) while sharing some essential interactions (Fig. 6, panels K and L). Interestingly, the most active Passerini-derived P53 inducer 12 recorded the most favorable free binding energies among the group (ΔG = −6.93 kcal mol−1), displaying π–π interactions between the terminal phenyl group and His96(MDM2) in the Leu26(P53) pocket (Fig. 6, panels G and H). The less active tolyl 3 and iodo 4 Ugi derivatives showed similar π–π interactions at less favored free binding energies (ΔG = −6.91 and −6.73 kcal mol−1) (Fig. 6, panels A–D). The cyclic analog 8 (ΔG = −6.80 kcal mol−1) was buried into the Trp23(P53) and Phe19(P53) pockets, forming H–π and hydrogen bond interactions with Leu54(MDM2) and Lys94(MDM2), respectively, as the reference 6SJ (Fig. 6, panels E and F). Similarly, the thiosemicarbazide derivative 17 (ΔG = −6.28 kcal mol−1) exhibited similar interactions with additional hydrogen bonding between the terminal alcoholic group and Glu69 (Fig. 6, panels I and J). Considering these observations, it could be postulated that the studied derivatives share some predicted key interactions with the reference inhibitor. The molecular modeling results could be nearly correlated to the P53 induction results (Table 6).
| Compound no. | ΔG (kcal mol−1) |
|---|---|
| 3 | −6.91 |
| 4 | −6.72 |
| 8 | −6.80 |
| 12 | −6.93 |
| 17 | −6.28 |
| 6SJ | −8.40 |
 | ||
| Fig. 6 (A) Overlay of the 3D binding mode of the co-crystallized ligand (green sticks) and 3 (yellow sticks), (B) 2D binding mode of 3, (C) overlay of the 3D binding mode of the co-crystallized ligand (green sticks) and 4 (cyan sticks), (D) 2D binding mode of 4, (E) overlay of the 3D binding mode of the co-crystallized ligand (green sticks) and 8 (pink sticks), (F) 2D binding mode of 8, (G) overlay of the 3D binding mode of the co-crystallized ligand (green sticks) and 12 (magenta sticks), (H) 2D binding mode of 12 (I) overlay of the 3D binding mode of the co-crystallized ligand (green sticks) and 17 (red sticks), (J) 2D binding mode of 17 into the co-crystallized inhibitor 6SJ binding site (PBD ID: 5LAW).82 (K) Superposition of the modeled (blue sticks) and the co-crystallized benzodiazepinone inhibitor (light pink sticks), and (L) 2D binding mode of the co-crystallized inhibitor in the MDM2 (PBD ID: 5LAW).82 | ||
P, while compounds 4–6 and 12 demonstrated violations of Lipinski's rule concerning molecular weight (MWt). All compounds except 6 were in the reasonable range for TPSA values. As a consequence, the majority of our compounds elicited acceptable %ABS ranging from 51.26–85.91%, suggesting promising cell permeability and oral bioavailability.
| Compound no. | LogP |
MWt | HBAa | HBDb | Lipinski's violation | TPSA | ABS (%) | Volume (A3) | NROTB |
|---|---|---|---|---|---|---|---|---|---|
| a HBA: number of hydrogen bond acceptors.b HBD: number of hydrogen bond donors. | |||||||||
| 2 | 5.22 | 470.45 | 6 | 1 | 1 | 75.71 | 82.88 | 399.47 | 9 |
| 3 | 6.41 | 492.57 | 6 | 1 | 1 | 75.71 | 82.88 | 456.14 | 9 |
| 4 | 7.04 | 604.44 | 6 | 1 | 2 | 75.71 | 82.88 | 463.57 | 9 |
| 5 | 5.92 | 523.54 | 9 | 1 | 2 | 121.54 | 67.07 | 462.92 | 10 |
| 6 | 5.85 | 568.54 | 12 | 1 | 3 | 167.36 | 51.26 | 486.25 | 11 |
| 8 | 4.94 | 400.43 | 6 | 0 | 0 | 66.92 | 85.91 | 357.71 | 6 |
| 9 | 5.50 | 409.48 | 6 | 1 | 1 | 81.71 | 80.81 | 382.37 | 8 |
| 10 | 6.48 | 436.47 | 9 | 1 | 1 | 131.46 | 63.65 | 390.70 | 9 |
| 11 | 5.39 | 440.45 | 9 | 1 | 1 | 127.53 | 65.00 | 389.14 | 9 |
| 12 | 6.20 | 550.63 | 9 | 2 | 2 | 127.88 | 64.88 | 481.05 | 11 |
| 13 | 4.19 | 488.56 | 9 | 2 | 0 | 127.88 | 64.88 | 426.44 | 11 |
| 15 | 2.22 | 307.30 | 7 | 3 | 0 | 112.93 | 70.04 | 268.09 | 5 |
| 16 | 0.84 | 277.32 | 6 | 5 | 0 | 104.45 | 72.96 | 255.79 | 3 |
| 17 | 2.66 | 412.51 | 7 | 5 | 0 | 102.48 | 73.64 | 368.58 | 7 |
Moreover, Swiss ADME software88 was applied to predict drug-like and medical chemistry characteristics of the test compounds, as described in Table 8. All compounds showed high bioavailability scores (0.55) except compounds 4 and 6, with a bioavailability score of 0.17. Besides, no alerts are predicted for all compounds except 10 (containing the N3 group). Overall, it could be established that the promising compounds presented acceptable physicochemical values as well as drug-like characteristics, which may introduce them as promising drug-like compounds.
| Compound no. | Bioavailability score | PAINS | Synthetic accessibility |
|---|---|---|---|
| 2 | 0.55 | 0 | 3.29 |
| 3 | 0.55 | 0 | 3.63 |
| 4 | 0.17 | 0 | 3.69 |
| 5 | 0.55 | 0 | 3.91 |
| 6 | 0.17 | 0 | 4.07 |
| 8 | 0.55 | 0 | 3.23 |
| 9 | 0.55 | 0 | 2.95 |
| 10 | 0.55 | 1 | 3.12 |
| 11 | 0.55 | 0 | 3.04 |
| 12 | 0.55 | 0 | 3.64 |
| 13 | 0.55 | 0 | 3.55 |
| 15 | 0.56 | 0 | 2.24 |
| 16 | 0.55 | 0 | 1.86 |
| 17 | 0.55 | 0 | 3.03 |
5. Structure–activity relationship
Generally, the activity motif proposes that the bisamide-based scaffold preserved the intrinsic antitumor activity of the reported lead apoptotic inducers (Fig. 1). Concerning the cytotoxicity of the Ugi adducts (2–6, 8) towards the two cancer breast cell lines, the activity profile demonstrated higher potency against MCF-7 than MDA-MB 231. On examining the effect of substituents on the adduct activities, results showed that the derivative 4 bearing 2-iodophenyl was more potent, particularly against MDA-MB 231 (IC50 = 0.188 μM) than its analog, 3 bearing 2-tolyl group (IC50 = 0.306 μM). The influence of the substituent effect on the observed cytotoxic profile of the scaffold was also deduced by introducing 2-nitrophenyl or 3,5-dinitrophenyl groups to the Ugi adducts 5 and 6, respectively. Herein, the 2-nitrophenyl derivative 5 was less potent and selective than the 2-iodophenyl 4 and the 2-methylphenyl 3 derivatives. The 3,5-dinitrophenyl analog 6 showed a supplemental increase in potency and selectivity relative to the 2-nitrophenyl derivative, however, still less active than the 2-idophenyl derivative. The notable potency of the iodophenyl derivative 4 may be attributed to the possibility of halogen bonding with the target receptors. Based on previous studies, interactions between C–I and oxygen of carbonyl, hydroxyl, charged carboxylate, or phosphate group (OY) in biological receptors (C–I⋯OY) potentially stabilize inter- and intramolecular interactions that can affect ligand binding and molecular folding where the I⋯O distance is less than or equal to the sums of the respective Van der Waals radii.89,90Deletion of the phenyl group and introduction of a trifluoromethyl group in compound 2 was detrimental to the scaffold's selectivity. Obviously, rigidification of the intermediate 7 furnished the imidazolidindione 8 with remarkable cytotoxicities against both MCF-7 (IC50 = 0.078 μM) and MDA-MB 231 (IC50 = 0.146 μM). When comparing the cytotoxic results of Ugi adduct, 3 (IC50 0.086) and its isostere, Passerini adduct, 9 (IC50 = 0.44 μM) on MCF-7 cell line, remarkable cytotoxicity of 3 carrying phenyl can be attributed to its higher lipophilic character than 9, which bears a cyclohexylidene moiety. Notably, compound 12 bearing central phenyl exhibited superior activity on both cell lines than its counterpart 13, which bears methylene spacer instead; for instance, on the MCF-7 cell line, 12 elicited IC50 = 0.065 μM while 13 demonstrated IC50 = 0.142 μM. This emphasizes the importance of lipophilicity for the cytotoxic activity of our studied compounds. Moreover, extension strategy on the acid hydrazide, 16 (MCF-7, IC50 = 0.968 μM) and (MDA-MB 231, IC50 = 1.028 μM) gave compound 17 with escalated anticancer activity towards both MCF-7s (IC50 = 0.082 μM) and MDA-MB 231 (IC50 = 0.518 μM). Among all tested compounds, compound 12 elicited the highest cytotoxicity against both types of breast cancer.
6. Conclusions
The current study reports a quick mechanistic investigation of the apoptotic induction potential of rationally designed flexible and constrained Passerini and Ugi derivatives. The promising adducts 3, 4, and 12, the cyclized imidazolidine derivative 8, as well as the thiosemicarbazide derivative 17 exhibited selective cytotoxic activity against breast cancer cell line (MCF-7, MDA-MB231) activated caspase 3/7 by 3.1–4.1 folds and promoted increased P53 signaling by 4.1–5.1 fold changes in MCF-7 cells line. Among the group, only 4, 8 and 12 demonstrated dual activities in MDA-MB231 cells. Further mechanistic studies showed that the compounds induced overexpression of BAX and downregulated the antiapoptotic oncogene, Bcl-2. Docking studies aided by SAR studies and ADME calculations indicated their acceptable pharmacokinetics and drug-like properties.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to extend their sincere appreciation to the Researchers Supporting Project (RSP2023R64), King Saud University, Riyadh, Saudi Arabia.References
- L. Yuan, Y. Cai, L. Zhang, S. Liu, P. Li and X. Li, Front. Pharmacol, 2021, 12, 1–45 Search PubMed
.
- O. Yersal and S. Barutca, World J. Clin. Onco., 2014, 5, 412 CrossRef PubMed
- I. Tsaur, I. Heidegger, A. Kretschmer, H. Borgmann, G. Gandaglia, A. Briganti, P. De Visschere, R. Mathieu, M. Valerio and R. van den Bergh, Cancer Treat. Rev., 2019, 75, 20–26 CrossRef PubMed
- K.-S. H. Nguyen, J. W. Neal and H. Wakelee, World J. Clin. Onco., 2014, 5, 576 CrossRef PubMed
- C. M. Neophytou, I. P. Trougakos, N. Erin and P. Papageorgis, Cancers, 2021, 13, 4363 CrossRef CAS PubMed
- R. Siegel, K. Miller and A. Jemal, Am. Cancer Soc., 2020, 70, 7–30 Search PubMed
- Z. Kozovska, V. Gabrisova and L. Kucerova, Biomed. Pharmacother., 2014, 68, 911–916 CrossRef CAS PubMed
- R. Rathore, J. E. McCallum, E. Varghese, A.-M. Florea and D. Büsselberg, Apoptosis, 2017, 22, 898–919 CrossRef CAS PubMed
- S. Fulda, Clin. Cancer Res., 2014, 20, 3915–3920 CrossRef CAS PubMed
- Q. L. Deveraux and J. C. Reed, Genes Dev., 1999, 13, 239–252 CrossRef CAS PubMed
- A. Dey, V. Tergaonkar and D. P. Lane, Nat. Rev. Drug Discovery, 2008, 7, 1031–1040 CrossRef CAS PubMed
- P. Ak and A. J. Levine, Faseb. J., 2010, 24, 3643–3652 CrossRef CAS PubMed
- T. Liu, L. Zhang, D. Joo and S.-C. Sun, Signal Transduction Targeted Ther., 2017, 2, 1–9 Search PubMed
- T. Scholzen and J. Gerdes, J. Cell. Physiol., 2000, 182, 311–322 CrossRef CAS PubMed
- E. C. Kauffman, V. L. Robinson, W. M. Stadler, M. H. Sokoloff and C. W. Rinker-Schaeffer, J. Urol., 2003, 169, 1122–1133 CrossRef PubMed
- A. Beloglazkina, N. Zyk, A. Majouga and E. Beloglazkina, Molecules, 2020, 25, 1211 CrossRef CAS PubMed
- C. Riedinger and J. M. McDonnell, Future Med. Chem., 2009, 1, 1075–1094 CrossRef CAS PubMed
- M. Millard, D. Pathania, F. Grande, S. Xu and N. Neamati, Curr. Pharm. Des., 2011, 17, 536–559 CrossRef CAS PubMed
- A. Barakat, M. S. Islam, H. M. Ghawas, A. M. Al-Majid, F. F. El-Senduny, F. A. Badria, Y. A. M. M. Elshaier and H. A. Ghabbour, Bioorg. Chem., 2019, 86, 598–608 CrossRef CAS PubMed
- M. S. Islam, H. M. Ghawas, F. F. El-Senduny, A. M. Al-Majid, Y. A. M. M. Elshaier, F. A. Badria and A. Barakat, Bioorg. Chem., 2019, 82, 423–430 CrossRef CAS PubMed
- G. Lotfy, Y. M. A. Aziz, M. M. Said, H. El Sayed, H. El Sayed, M. M. Abu-Serie, M. Teleb, A. Dömling and A. Barakat, Bioorg. Chem., 2021, 117, 105427 CrossRef CAS PubMed
- Y. M. A. Aziz, G. Lotfy, M. M. Said, E. S. H. El Ashry, E. S. H. El Tamany, S. M. Soliman, M. M. Abu-Serie, M. Teleb, S. Yousuf and A. Dömling, Front. Chem., 2021, 9, 735236 CrossRef PubMed
- J. Li, J. Viallet and E. B. Haura, Cancer Chemother. Pharmacol., 2008, 61, 525–534 CrossRef CAS PubMed
- V. Tergaonkar and N. D. Perkins, Mol. Cell, 2007, 26, 158–159 CrossRef CAS PubMed
- S. X. Cai, B. Nguyen, S. Jia, J. Herich, J. Guastella, S. Reddy, B. Tseng, J. Drewe and S. Kasibhatla, J. Med. Chem., 2003, 46, 2474–2481 CrossRef CAS PubMed
- L. Bai, D. C. Smith and S. Wang, Pharmacol. Ther., 2014, 144, 82–95 CrossRef CAS PubMed
- H. Sun, Z. Nikolovska-Coleska, C.-Y. Yang, D. Qian, J. Lu, S. Qiu, L. Bai, Y. Peng, Q. Cai and S. Wang, Acc. Chem. Res., 2008, 41, 1264–1277 CrossRef CAS PubMed
- C. H. A. Cheung, Y.-C. Chang, T.-Y. Lin, S. M. Cheng and E. Leung, J. Biomed. Sci., 2020, 27, 1–10 CrossRef PubMed
- H. Cong, L. Xu, Y. Wu, Z. Qu, T. Bian, W. Zhang, C. Xing and C. Zhuang, J. Med. Chem., 2019, 62, 5750–5772 CrossRef CAS PubMed
- P. Cetraro, J. Plaza-Diaz, A. MacKenzie and F. Abadía-Molina, Cancers, 2022, 14, 1671 CrossRef CAS PubMed
- M. Olsson and B. Zhivotovsky, Cell Death Differ., 2011, 18, 1441–1449 CrossRef CAS PubMed
- T. K. Oost, C. Sun, R. C. Armstrong, A.-S. Al-Assaad, S. F. Betz, T. L. Deckwerth, H. Ding, S. W. Elmore, R. P. Meadows and E. T. Olejniczak, J. Med. Chem., 2004, 47, 4417–4426 CrossRef CAS PubMed
- C. N. Johnson, J. S. Ahn, I. M. Buck, E. Chiarparin, J. E. H. Day, A. Hopkins, S. Howard, E. J. Lewis, V. Martins and A. Millemaggi, J. Med. Chem., 2018, 61, 7314–7329 CrossRef CAS PubMed
- M. S. Ayoup, Y. Wahby, H. Abdel-Hamid, M. Teleb, M. M. Abu-Serie and A. Noby, Eur. J. Med. Chem., 2019, 168, 340–356 CrossRef PubMed
- Z. Mrkvová, S. Uldrijan, A. Pombinho, P. Bartůněk and I. Slaninová, Molecules, 2019, 24, 2152 CrossRef PubMed
- L.-t. Wu, Z. Jiang, J.-j. Shen, H. Yi, Y.-c. Zhan, M.-q. Sha, Z. Wang, S.-t. Xue and Z.-r. Li, Eur. J. Med. Chem., 2016, 114, 328–336 CrossRef CAS PubMed
- K. Błaszczak-Świątkiewicz, Molecules, 2019, 24, 3902 CrossRef PubMed
- P. Boggu, E. Venkateswararao, M. Manickam, D. Kwak, Y. Kim and S.-H. Jung, Bioorg. Med. Chem., 2016, 24, 1872–1878 CrossRef CAS PubMed
- P. Boggu, E. Venkateswararao, M. Manickam, Y. Kim and S.-H. Jung, Arch. Pharmacal Res., 2017, 40, 469–479 CrossRef CAS PubMed
- K. J. Okolotowicz, R. Shi, X. Zheng, M. MacDonald, J. C. Reed and J. R. Cashman, Bioorg. Med. Chem., 2010, 18, 1918–1924 CrossRef CAS PubMed
- G. Dunphy, S. M. Flannery, J. F. Almine, D. J. Connolly, C. Paulus, K. L. Jønsson, M. R. Jakobsen, M. M. Nevels, A. G. Bowie and L. Unterholzner, Mol. Cell, 2018, 71, 745–760 CrossRef CAS PubMed
- R. Poltz and M. Naumann, BMC Syst. Biol., 2012, 6, 1–19 CrossRef PubMed
- K. Jonak, M. Kurpas, K. Szoltysek, P. Janus, A. Abramowicz and K. Puszynski, BMC Syst. Biol., 2016, 10, 1–12 CrossRef PubMed
- M. Barani, M. Mirzaei, M. Torkzadeh-Mahani and M. Adeli-Sardou, Sci. Rep., 2019, 9, 7139 CrossRef PubMed
- M. Ríos-Gutiérrez, A. Barakat and L. R. Domingo, Organics, 2022, 3, 122–136 CrossRef
- J. Araujo, E. Gonzalez-Mira, M. A. Egea, M. L. Garcia and E. B. Souto, Int. J. Pharm., 2010, 393, 168–176 CrossRef PubMed
- A. M. Al-Mahallawi, O. M. Khowessah and R. A. Shoukri, Int. J. Pharm., 2017, 522, 157–164 CrossRef CAS PubMed
- H. Zhu, H. Gao, Y. Ji, Q. Zhou, Z. Du, L. Tian, Y. Jiang, K. Yao and Z. Zhou, J. Hematol. Oncol., 2022, 15, 91 CrossRef CAS PubMed
- E. A. Mazyed and A. E. Abdelaziz, Pharmaceutics, 2020, 12, 465 CrossRef CAS PubMed
- S. T. Sp, M. Mothilal, N. Damodharan and D. Jaison, Int. J. Appl. Pharm., 2018, 79–85 CrossRef
- M. S. Ayoup, A. F. Mansour, H. Abdel-Hamid, M. M. Abu-Serie, S. M. Mohyeldin and M. Teleb, Eur. J. Med. Chem., 2023, 245, 114865 CrossRef CAS PubMed
- H. Liang, R. A. Salinas, B. Z. Leal, T. Kosakowska-Cholody, C. J. Michejda, S. J. Waters, T. S. Herman, J. M. Woynarowski and B. A. Woynarowska, Mol. Cancer Ther., 2004, 3, 1385–1396 CrossRef CAS PubMed
- K. O. Mohamed, Y. M. Nissan, A. A. El-Malah, W. A. Ahmed, D. M. Ibrahim, T. M. Sakr and M. A. Motaleb, Eur. J. Med. Chem., 2017, 135, 424–433 CrossRef CAS PubMed
- R. Ponce-Cusi and G. M. Calaf, Int. J. Oncol., 2016, 48, 774–782 CrossRef CAS PubMed
- M. S. Ayoup, M. A. Fouad, H. Abdel-Hamid, M. M. Abu-Serie, A. Noby and M. Teleb, Eur. J. Med. Chem., 2020, 186, 111875 CrossRef CAS PubMed
- P. S. Galatin and D. J. Abraham, J. Med. Chem., 2004, 47, 4163–4165 CrossRef CAS PubMed
- Y. Wen, X. Chen, Z. Liu, Q. Zhu, Z. Li, G. He, H. Yan and Q. Lin, ChemistrySelect, 2021, 6, 10965–10973 CrossRef CAS
- L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Chem. Heterocycl. Compd, 2017, 53, 382–408 CrossRef CAS
- P. S. G. Nunes, H. D. A. Vidal and A. G. Corrêa, Org. Biomol. Chem., 2020, 18, 7751–7773 RSC
- J. G. Rudick, S. Shaabani and A. Dömling, Front. Chem., 2020, 7(918), 1–3 Search PubMed
- M. Tandi and S. Sundriyal, J. Indian Chem. Soc., 2021, 98, 100106 CrossRef CAS
- M. S. Ayoup, Y. Wahby, H. Abdel-Hamid, M. M. Abu-Serie and M. Teleb, Sci. Rep., 2022, 12, 22390 CrossRef CAS PubMed
- X. Wang, Q.-G. Wang and Q.-L. Luo, Synthesis, 2015, 47, 49–54 CAS
- T. Mosdam, J. Immunol. Methods, 1983, 65, 55–63 CrossRef PubMed
- J. T. Zilfou and S. W. Lowe, Cold Spring Harbor Perspect. Biol., 2009, 1, a001883 Search PubMed
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed
- S. Salakou, D. Kardamakis, A. C. Tsamandas, V. Zolota, E. Apostolakis, V. Tzelepi, P. Papathanasopoulos, D. S. Bonikos, T. Papapetropoulos and T. Petsas, In Vivo, 2007, 21, 123–132 CAS
- P. J. Lakshmi, B. S. Kumar, R. S. Nayana, M. S. Mohan, R. Bolligarla, S. K. Das, M. U. Bhanu, A. K. Kondapi and M. Ravikumar, Bioorg. Med. Chem., 2009, 17, 6040–6047 CrossRef PubMed
- Y. A. Ammar, A. M. S. El-Sharief, A. Belal, S. Y. Abbas, Y. A. Mohamed, A. B. Mehany and A. Ragab, Eur. J. Med. Chem., 2018, 156, 918–932 CrossRef CAS PubMed
- G. S. Salvesen and C. S. Duckett, Nat. Rev. Mol. Cell Biol., 2002, 3, 401–410 CrossRef CAS PubMed
- S. J. Riedl and Y. Shi, Nat. Rev. Mol. Cell Biol., 2004, 5, 897–907 CrossRef CAS PubMed
- E. C. LaCasse, S. Baird, R. G. Korneluk and A. E. MacKenzie, Oncogene, 1998, 17, 3247–3259 CrossRef PubMed
- M. Holcik, H. Gibson and R. G. Korneluk, Apoptosis, 2001, 6, 253–261 CrossRef CAS PubMed
- Y. Huang, Y. C. Park, R. L. Rich, D. Segal, D. G. Myszka and H. Wu, Cell, 2001, 104, 781–790 CAS
- H. Sun, Z. Nikolovska-Coleska, J. Lu, S. Qiu, C.-Y. Yang, W. Gao, J. Meagher, J. Stuckey and S. Wang, J. Med. Chem., 2006, 49, 7916–7920 CrossRef CAS PubMed
- H. Sun, J. A. Stuckey, Z. Nikolovska-Coleska, D. Qin, J. L. Meagher, S. Qiu, J. Lu, C.-Y. Yang, N. G. Saito and S. Wang, J. Med. Chem., 2008, 51, 7169–7180 CrossRef CAS PubMed
- Y.-C. Chang and C. H. A. Cheung, Appl. Sci., 2020, 11, 335 CrossRef
- E. Morrish, G. Brumatti and J. Silke, Cells, 2020, 9(406), 1–24 Search PubMed
- Molecular Operating Environment (MOE), 102 Chemical Computing Group ULC, Suite #910, 1010 Sherbooke St. West, 2R7, Canada, H3A, Montreal, QC, 2019, p. 2022 Search PubMed
- R. F. Kester, A. F. Donnell, Y. Lou, S. W. Remiszewski, L. J. Lombardo, S. Chen, N. T. Le, J. Lo, J. A. Moliterni and X. Han, J. Med. Chem., 2013, 56, 7788–7803 CrossRef CAS PubMed
- P. Andrews, D. Craik and J. Martin, J. Med. Chem., 1984, 27, 1648–1657 CrossRef CAS PubMed
- M. H. Nematollahi, A. Pardakhty, M. Torkzadeh-Mahanai, M. Mehrabani and G. Asadikaram, RSC Adv., 2017, 7, 49463–49472 RSC
- Molinspiration
Cheminformatics, August, 2023, https://www.molinspiration.com Search PubMed
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed
- P. Ertl, B. Rohde and P. Selzer, J. Med. Chem., 2000, 43, 3714–3717 CrossRef CAS PubMed
- A. Polinsky and G. Shaw, Pract. Med. Chem., 2003, 147–157 CAS
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci., 2004, 101, 16789–16794 CrossRef CAS PubMed
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04029a |
| This journal is © The Royal Society of Chemistry 2023 |