
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtraction of rare earth elements from aqueous solutions using the ionic liquid trihexyltetradecylphosphonium 3-hydroxy-2-naphthoate†
Andreas Gradwohl a,
Jakob Windischa,
Matthias Weissensteinera,
Bernhard K. Kepplera,
Wolfgang Kandiollera and
Franz Jirsa*ab
a,
Jakob Windischa,
Matthias Weissensteinera,
Bernhard K. Kepplera,
Wolfgang Kandiollera and
Franz Jirsa*ab
aDepartment of Inorganic Chemistry, University of Vienna, Währinger Straße 42, Vienna 1090, Austria. E-mail: franz.jirsa@univie.ac.at
bDepartment of Zoology, University of Johannesburg, PO Box 524, Auckland Park, Johannesburg 2006, South Africa
First published on 21st August 2023
Abstract
The task-specific ionic liquid trihexyltetradecylphosphonium 3-hydroxy-2-naphthoate has been described as a suitable extraction agent for numerous metals from aqueous phases, while additionally providing reduced leaching into the used matrices. Here, we investigate the extraction properties of this extractant towards rare earth elements. Of these, La, Ce, Nd, Ho und Lu were chosen as a representative mix of light and heavy elements. Single- as well as double-element extractions were carried out under varying conditions regarding pH, temperature and extraction time. The highest extraction efficacies and minimalized precipitation of the respective metals were recorded at a pH of 2.5. Satisfactory extraction efficacies (>80%) were achieved already after 6 hours for the elements Ce, Nd and Lu in single-element extraction experiments at room temperature. Increased temperatures improved the extraction efficacy for Nd from 36% at 20 °C to 80% at 30 °C after only 2 hours. Surprisingly, this effect was not observed for Ce in single-element experiments. In double-element feed solutions containing both Ce and Nd, however, the time-dependant extraction efficacy of Ce mirrored that of Nd. The pH in the aqueous extraction matrix changed during the extraction, showing a positive correlation with the extraction efficacy and revealing the extraction mechanism to be via anion exchange. The leaching was in good agreement with literature values, showed a positive correlation with extraction efficacies, and ranged for all extractions between 0.8 and 1.2%. Remarkably, increasing the temperature from 20 °C to 30 °C had no significant influence on leaching.
Introduction
Ionic liquids as extraction agents
Ionic liquids (ILs) are generally defined as low-melting salts, consisting of large, non-symmetrical organic cations and organic or inorganic anions. The currently accepted definition of a melting point below 100 °C was originally proposed to distinguish ILs from traditional high-temperature molten salts. ILs usually display special properties such as a low vapor pressure, low flammability and a high chemical and thermal stability. The tuneable physical and chemical properties and the extremely high variability of substances within this class, along with their possible applications in extraction, synthesis, separation or polymer chemistry, led to a booming interest in ILs.1–6 In the literature, several specific descriptions of ILs, as molten salts or non-aqueous ILs are used to state their special behaviour or purpose. The most frequently used terms are room-temperature ILs (RTILs), describing ILs that are liquid at room temperature, and task-specific ILs (TSILs). The key feature of TSILs is that they have been specifically designed for a special purpose, e.g. as a selective extraction agent.6 The assumption is that approximately 1018 combinations of ILs may be possible,7 explaining the huge interest in ILs as, amongst others, extraction agents for metals from the aqueous phase.8–10 Besides the tremendous variety of cations and anions, there are several other reasons for the interest in and the necessity for research in the field of ionic liquids as extraction and separation agents for metals from the aquatic phase. The selectivity for elements (respectively ions), by choosing and anchoring specific functional groups on ionic liquids, can be tuned and selected in a design and try-out process considering previously published knowledge.8,9The TSIL trihexyltetradecylphosphonium 3-hydroxy-2-naphthoate (Fig. 1), [P66614][HNA], was synthesised and characterized by Pirkwieser et al., who described it as a suitable extraction agent for numerous metals from diverse aqueous phases.11 This TSIL was designed from similar predecessors to provide reduced dissolvability (leaching) into the used matrices in order to reduce toxicity towards aquatic biota. Good extraction efficacies were shown for Cu, Ag, Cd, and Pb not only in “pure water” but also in more complex matrices such as drinking water, hypersaline water and wastewater. Moreover, this TSIL showed reduced leaching and decreased toxicity towards the two freshwater green algae Raphidocelis subcapitata and Tetradesmus obliquus compared to similar compounds.12 These findings suggest investigating its extraction behaviour towards other metals of interest in regard of recovery and recycling, especially rare earth elements.
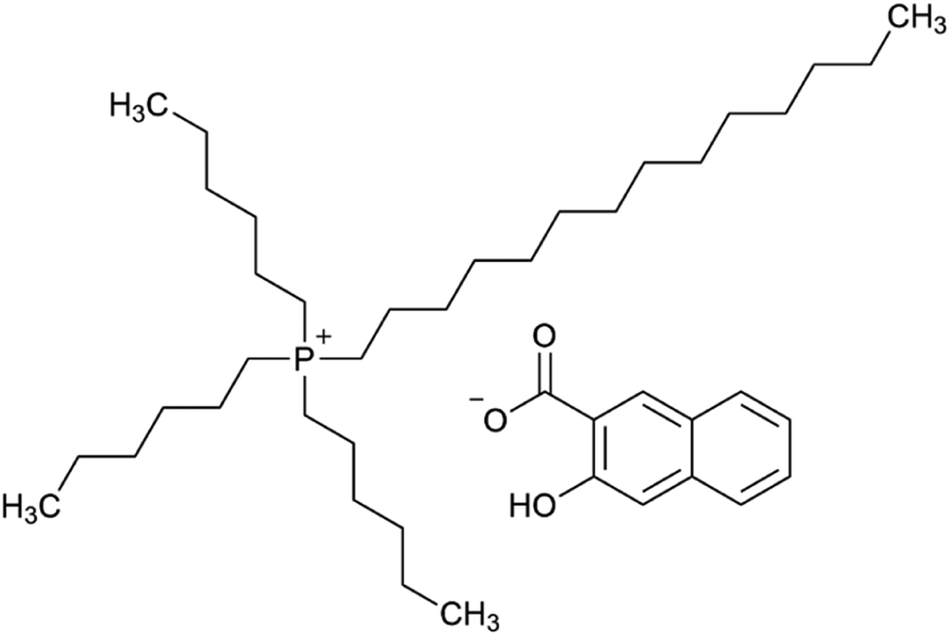 | ||
| Fig. 1 The task-specific ionic liquid trihexyltetradecylphosphonium 3-hydroxy-2-naphthoate, [P66614][HNA]. | ||
Rare earth elements
Rare earth elements (REE) include lanthanides (Ln), namely La, Ce, Pr, Nd, Pm, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, as well as the elements Sc and Y.13 The lanthanides themselves are known for their similar physical and chemical properties, the progressive filling of the 4f orbitals with increasing atomic number, causing the so-called ‘lanthanide contraction’ and the common occurrence in the same natural ores.14 Several classifications within the REE have been proposed in the past, discriminating the elements based on their chemical or technical properties. A common differentiation is between light (LREE) and heavy rare earth elements (HREE).15 The unique physical and chemical properties of REE – delivering specific magnetic, luminescent or strength characteristics to the high-tech end products in which they are used – make them indispensable in a broad range of modern technologies. This is especially the case in the production of modern computers and mobile phones (e.g. batteries and screens), magnets and lasers.16The extensive usage of REE raises issues regarding the recycling and reusability of products containing those metals. Amongst others, hydrometallurgical methods for recovering REE from e.g. magnets are described.16,17 These methods include the dissolution of the REE containing matrix in acids, so that the REE can be regained from the liquid phase. Optimally, this proceeds selectively. The speciation of REE in solution is therefore crucial for research on the applicability of ILs using liquid–liquid extraction for recovery and recycling.
In aqueous solutions, the dominant oxidation state of all REE is +3. The trivalent ions can be classified as hard ions and preferably complex with hard ligands, the strongest complexes of lanthanides (Ln) are formed with the ligands fluoride, sulphate, carbonate, phosphate and hydroxide.18 Complexes with chloride and nitrate play only a subordinate role, but might occur in the absence of the others listed above, e.g. as LnCl2+ in brine waters or as LnNO32+ under high nitrate concentrations.18
Extraction of rare earth elements using ionic liquids
The recovery and recycling of REE is often considered to be expensive, inefficient and facing limitations, e.g. the uneven distribution of small amounts of elements within a much bigger matrix, as in mobile phones. Also, the chemical similarities of the individual REE are a major obstacle regarding separation and purification. Nonetheless, the development of cost-effective and resource-saving technologies would bear a huge potential, both economically and environmentally.14 Fang et al., for example, proposed using redox-active ligands for electro-kinetic separation, with the additional potential of adding recycled REE into the supply chain.19 Beyond the idea of using tailored organic molecules to separate and recover REE, the usage of IL-based systems has gained increasing research interest in the last 20 years.20Davris et al. reported the application of the functionalized IL betainium bis(trifluoromethyl sulfonyl)imide (HbetTf2N) to directly leach REE from bauxite residue (red mud) and selectively separate all REE except for Sc from Fe.21 The yields were approx. 70–85%. With extraction efficacies <3% for Fe, the authors successfully developed a pathway for REE separation from Fe out of secondary REE resources. The IL Aliquat 336® and EHEHPA (2-ethylhexyl phosphonic acid) were used for the specific design of the novel TSIL [R4N][EHEHP] for REE extraction.22,23 The pH and the chloride content of the extraction solution influenced the TSIL. Due to an unsatisfactory loading capacity at the necessary high pH values, the authors did not recommend an application for LREE separation, but suggested potential in HREE separation. Zhou et al. reported a different, multi-step recovery process for REE.24 After removing Fe ions with the IL Cyphos® IL 101, the authors successfully used the novel IL [trihexyl(tetradecyl)phosphonium]2[4,4′-isopropylidene bis(phenoxyacetate)], [P66614]2[IOPAA], as the extraction agent in a solution of dissolved Nd-Fe-B scrap. The approach of using IL mixtures as synergist extractants was also published by Hidayah and Abidin.25 They co-utilized Aliquat336® and [C2mim][NTf2] (1-ethyl-3-methylimidazolium bis(trifluoro-methanesulfonyl)imide) and showed an increase in Pr, Gd and Dy extraction efficiency compared to using the conventional extractant Aliquat336® alone. With the newly synthesised IL [N8888][DEHOX] (tetraoctylammonium di(2-ethylhexyl)-oxamate), Maria et al. reported unique discrimination factors along the lanthanide series for the separation between LREE and HREE. The factors ranged from close to zero for Ce3+ to more than 90% for Yb3+.26
Following the promising results for other metals, we chose the REE La, Ce, Nd, Ho and Lu for the present study. They constitute a representative mix of light and heavy REE for possibly greener recovery and recycling strategies using the TSIL [P66614][HNA]. We elucidate the extraction mechanism and present optimal conditions for REE extraction, demonstrating the applicability of [P66614][HNA] for REE extraction from simple aqueous solutions.
Experimental
Solvents and reagents
For synthesis, the IL precursor trihexyltetradecylphosphonium chloride (95%) and 3-hydroxy-2-naphthoic acid (98%) were purchased from Sigma-Aldrich (USA) along with potassium hydroxide (p.a., ≥86%) and the solvents methanol (HPLC grade, ≥99.9%) and dichloromethane (p.a., ≥99.9%). Element standard solutions of La, Ce and Lu (1000 mg L−1 ± 4) in 2% (w/w) HNO3 for the preparation of the extraction solutions were purchased from Sigma-Aldrich (USA), Nd and Ho (999 μg mL−1 ± 5) in 5% (w/w) HNO3 from AlfaAesar. The Pt standard solution (1000 mg L−1 in 10% (w/w) HCl) used for sample preparations before TXRF measurements was purchased from VWR. A 3.0 g L−1 polyvinyl alcohol (PVA) solution was prepared from hydrolysed polyvinyl alcohol (86–89%) with low molecular weight (AlfaAesar). For pH adjustment and dilutions, we used sodium hydroxide (50% (w/w) in H2O, extra pure, Acros Organics) and nitric acid (>68%, PrimarPlus-trace analysis grade, Fisher Scientific). Ultra-pure water of resistivity >18.2 MΩ cm was obtained from a Millipore Milli-Q apparatus (Merck Millipore, USA).Synthesis of the ionic liquid
The IL was synthesised following the deprotonation metathesis route described by Pirkwieser et al.11 In short, 3-hydroxy-2-naphthoic acid was dissolved in methanol, deprotonated using potassium hydroxide and subsequently mixed with trihexyltetradecylphosphonium chloride.The pure product was achieved after preparative extraction in dichloromethane/water and removal of the solvent in vacuo. Subsequent 1H- and 31P-NMR measurements confirmed the sufficient purity of the synthesised compound and no further analysis was necessary.
Apparatus
The ionic liquid trihexyltetradecylphosphonium 3-hydroxy-2-naphthoate was characterized by 1H and 31P NMR analysis. The spectra were recorded on an Avance III™ 600 MHz spectrometer (Bruker, USA) in CDCl3 at 298.1 K using pulse programs at 600.25 MHz (1H) and 242.99 MHz (31P). The metal content was determined using the total-reflection X-ray fluorescence (TXRF) spectrometer S2 PICOFOX (Bruker, USA) and quartz glass sample carrier discs. Total organic carbon (TOC) was determined using a TOC-VCPH analyser (Shimadzu, Japan). pH values were determined using a ProLab 2000 pH meter (Schott Instruments, Germany). All masses were weighed using a METTLER AT200 analytical balance (Mettler-Toledo, USA). Samples were shaken for extraction on a Vibramax 100 orbital shaker (Heidolph, Germany). To ensure constant temperature during temperature-dependent extraction experiments, the orbital shaker was placed in either a 20 °C thermostat cabinet (TS 606/2, WTW) or a heating cabinet (TK/L 4250, Ehret).Extraction and back-extraction experiments
Metal feed solutions were prepared by diluting the respective metal standard solutions with 2% nitric acid. Each feed solution contained 10 mg L−1 La, Ce, Nd, Ho or Lu, respectively at the beginning of the experiment. The pH was set to 2.50 ± 0.03 using sodium hydroxide solutions (0.5%, 5.0% and 50% (w/w)). For double-element extraction experiments, 2% nitric acid was spiked with 10 mg L−1 of La and Lu, La and Nd, Ce and Lu or Ce and Nd. The pH was set the same way to 2.50 ± 0.03 using sodium hydroxide dilutions. All extraction experiments were conducted in triplicates, including reference triplicates of feed solution without IL to determine metal stability during the respective time (Fig. 2).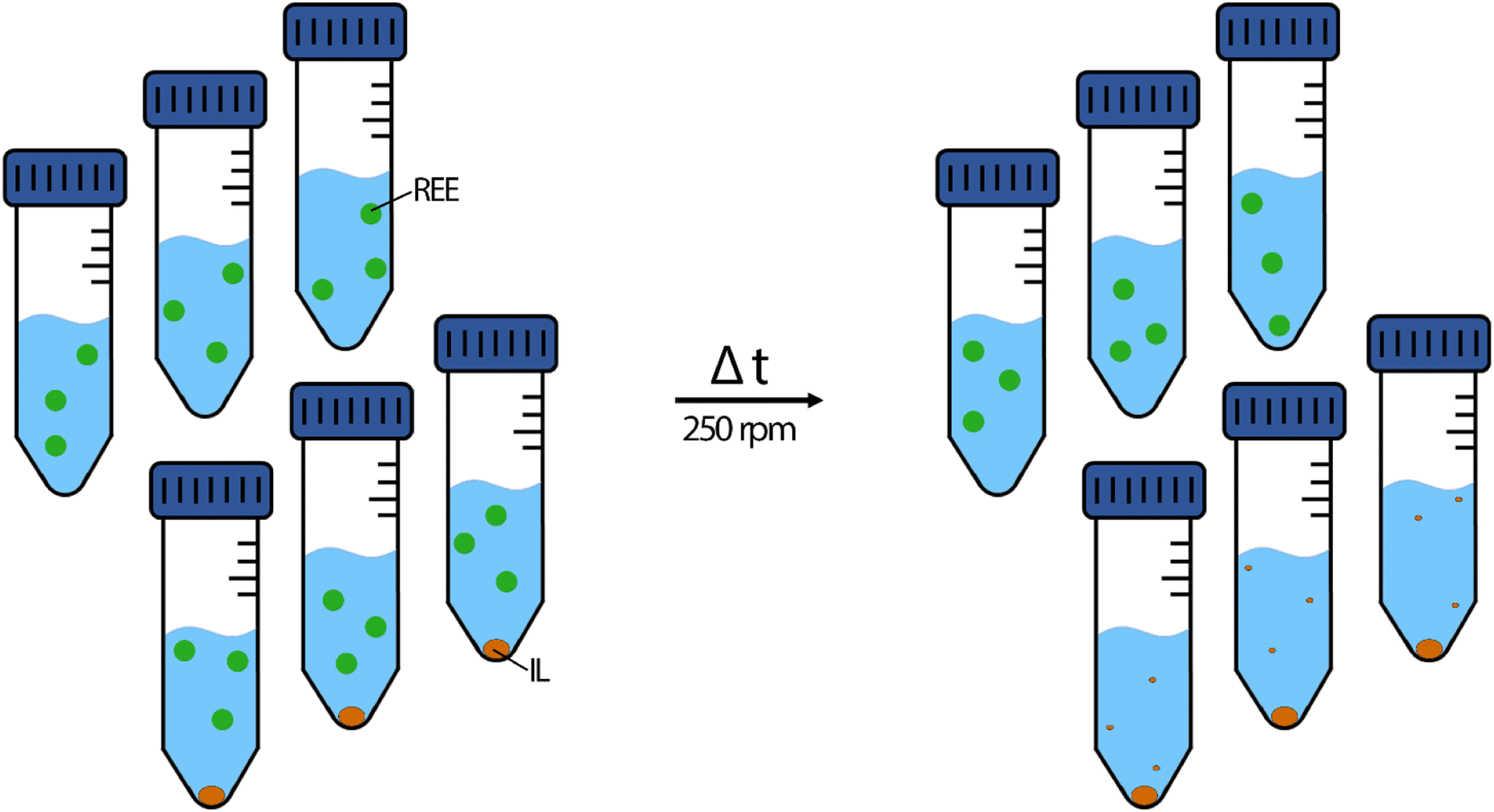 | ||
| Fig. 2 Schematic depiction of the extraction experiments. Left side: starting conditions; right side: the ideal result after the respective time. Green dot: dissolved REE metals. Orange dot: the IL. | ||
The IL trihexyltetradecylphosphonium 3-hydroxy-2-naphtoate was weighed into 50 mL polypropylene centrifuge tubes (100 ± 10 mg) using an analytical balance accurate to 0.1 mg. 20 mL of prepared metal feed solution was added onto the IL and agitated for either 1 hour, 2 hours, 4 hours, 6 hours or 24 hours using an orbital shaker (250 rpm). The experiments were conducted at room temperature (23 ± 1 °C), and thereafter either at 20 °C or 30 °C. The temperature was controlled by placing the orbital shaker either in a climate- (20 °C) or heating cabinet (30 °C). Feed solutions were brought to the respective temperature before the extraction experiments. After the respective extraction time, an aliquot of 10 mL of the aqueous phase was removed with a syringe and filtered through a 0.45 μm nylon syringe filter for further analysis.
For back-extraction experiments, we used 0.5 M nitric acid, following Platzer et al., who achieved best results using this agent.27 All back-extraction experiments were performed in triplicates. The feed solution was removed after the 24 hours extraction experiments and the metal containing IL was used for the back-extraction experiments. The determined concentration in the metal feed solution was used to calculate the metal content in the IL. 20 mL nitric acid (0.5 M) was pipetted onto the metal containing IL and agitated for 2 hours using an orbital shaker (250 rpm). Then, an aliquot of 10 mL of the aqueous phase was removed with a syringe, filtered through a 0.45 μm nylon syringe filter and stored for further analysis.
Analysis and quantification
For temperature-dependent extraction experiments and time-dependent extraction of Lu, the pH value after extraction was determined in the filtered sample. For analysis of the metal content, TXRF measurements (S2 PICOFOX, Bruker) were performed. 0.5 mL of the filtered sample were transferred into 1.5 mL Eppendorf tubes and mixed with 0.5 mL Pt standard solution (5 mg L−1 in 2% (w/w) HCl). 100 μL of PVA solution (3.0 g L−1) was added and the sample was mixed thoroughly using a laboratory shaker. An aliquot of 5 μL sample was pipetted on a siliconized quartz glass sample carrier plate and dried for 40 min under an IR lamp and under reduced pressure. After drying, TXRF spectra of the samples were recorded and evaluated using the software Spectra (Bruker, version 7.8.2.0). Recovery rates for REE using Pt as internal standard were determined by fivefold measurements. As the results were within a 95% confidence interval, the mean value of these measurements was used as the respective correction factor (Table 1) for all further measurements and calculations.| Element | Factor | Elements | Factor | |
|---|---|---|---|---|
| La | 0.763 | La & Lu | La | 0.792 |
| Lu | 1.111 | |||
| Ce | 0.863 | La & Nd | La | 0.798 |
| Nd | 0.882 | |||
| Nd | 0.834 | Ce & Lu | Ce | 0.852 |
| Lu | 1.105 | |||
| Ho | 1.101 | Ce & Nd | Ce | 0.799 |
| Nd | 0.908 | |||
| Lu | 1.123 | Ho & Lu | Ho | 1.040 |
| Lu | 0.890 | |||
The extraction efficacy for metals from feed solutions was calculated using eqn (1). It is defined as the percentage of removed metal from the feed solution after the respective extraction times in relation to the mean metal concentration in the reference samples after the respective time. 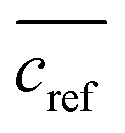 is the mean metal concentration of the reference samples, ct is the metal concentration in the respective sample after the extraction.
is the mean metal concentration of the reference samples, ct is the metal concentration in the respective sample after the extraction.
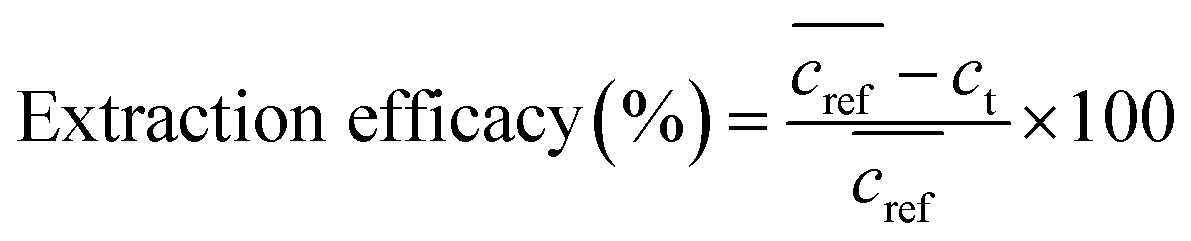 | (1) |
Leaching was determined based on total organic carbon (TOC) measurements. 5 mL of filtered sample were diluted with 5 mL Milli-Q water and measured in a TOC-VCPH analyser (Shimadzu). The measured TOC value was divided by the carbon content factor of the IL (CIL) to determine IL leaching using eqn (2). The carbon content factor equals 0.7697 and was derived from the carbon content of the used ionic liquid (76.97%). The leaching (%) was calculated using eqn (3), whereby Vs represents the volume (L) of the aqueous phase during extraction and mIL the mass (mg) of used IL. Cation and anion of the IL were assumed to leach equally. Leaching therefore represents the percentual loss of IL during the extraction.
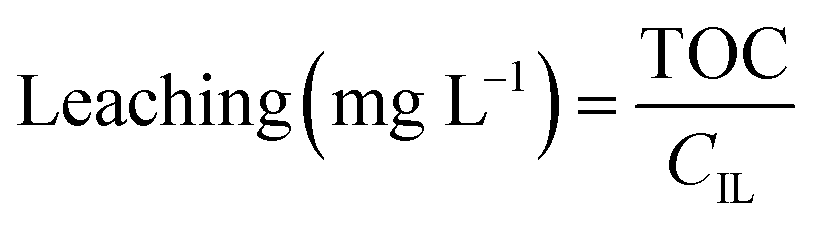 | (2) |
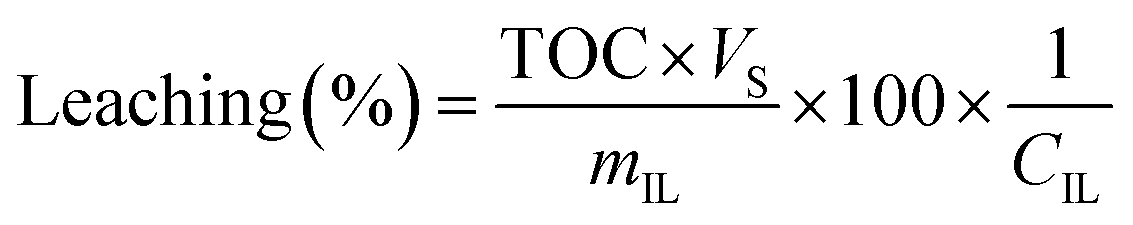 | (3) |
The back-extraction efficacy was calculated using eqn (4). It is defined as the percentage of metal in feed solution after the back extraction in relation to the metal content in the used IL. The metal concentration after back extraction, caq, is multiplied by the volume used for back extraction (Vbe). This equals the mass of REE in the aqueous phase, mREE,aq. The calculated value for the mass of REE is divided by the mass of REE in the IL (mREE,org) and multiplied by 100 to determine the efficacy of back-extracted metal. The REE mass in the organic phase (mREE,org) is calculated by using the measured concentration after the respective extraction, cext, which is subtracted from the initial concentration of 10 mg L−1 and subsequently multiplied with the volume used during extraction, Ve. The resulting value equals the mass of REE in the organic IL phase (mREE,org).
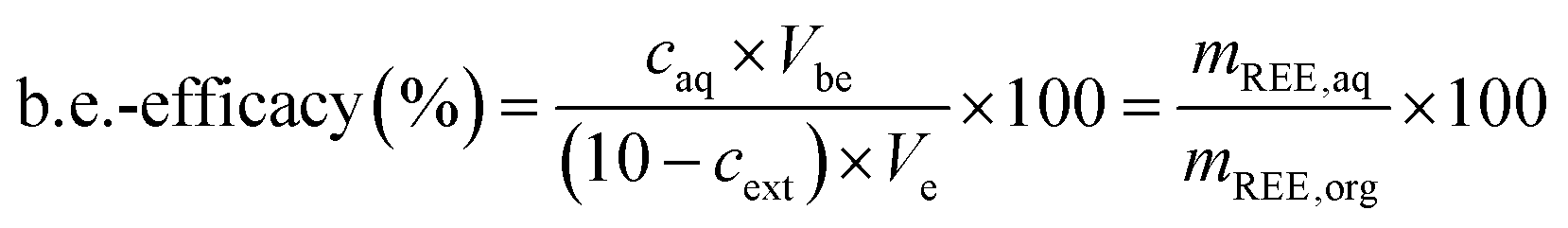 | (4) |
Results and discussion
Stability of REE under varying pH conditions
Preliminary experiments, performed at pH values ranging from 2.5 to 6 revealed (partial) precipitation of the metal during 3 hours. The determined stabilities for dissolved metal ranged from 76.76 ± 5.26% at pH 3, 74.23 ± 3.37% at pH 4, 70.40 ± 3.14% at pH 5 to only 48.51 ± 3.99% at pH 6. At pH 2.5, however, >95% of Nd were stable in solution even after 4 hours, and still were at 86.48 ± 1.03% after 24 hours. These findings are in good agreement with the aqueous chemistry of REE described in the literature.28,29 We used Nd2O3 (in nitric acid) to prepare the feed solution. Han suggested a cut-off pH of approximately pH 6 for the stability of this oxide in solution.29 This means precipitation of the respective oxides in all solutions with pH > 2.5 in increasing percentages, which our results confirmed. We used filtration throughout all experiments to ensure the elimination of potentially precipitated metal oxides or other anti-soluble salts.Extraction of La, Ce, Nd, Ho and Lu from 10 mg L−1 feed solutions
During the first experiments, feed solutions were prepared by spiking Milli-Q water with 10 mg L−1 Ce or 10 mg L−1 Nd and the pH value was set to 2.5 with NaOH. Hereby, extraction efficacies of only 13.53 ± 2.53% (Ce) and 2.84 ± 1.86% (Nd) were achieved. Consequently, feed solutions were prepared using 2% nitric acid (brought to pH 2.5 with NaOH), which yielded significantly higher efficacies. The achieved extraction efficacies over time for the elements Ce, Nd and Lu are shown in Fig. 3.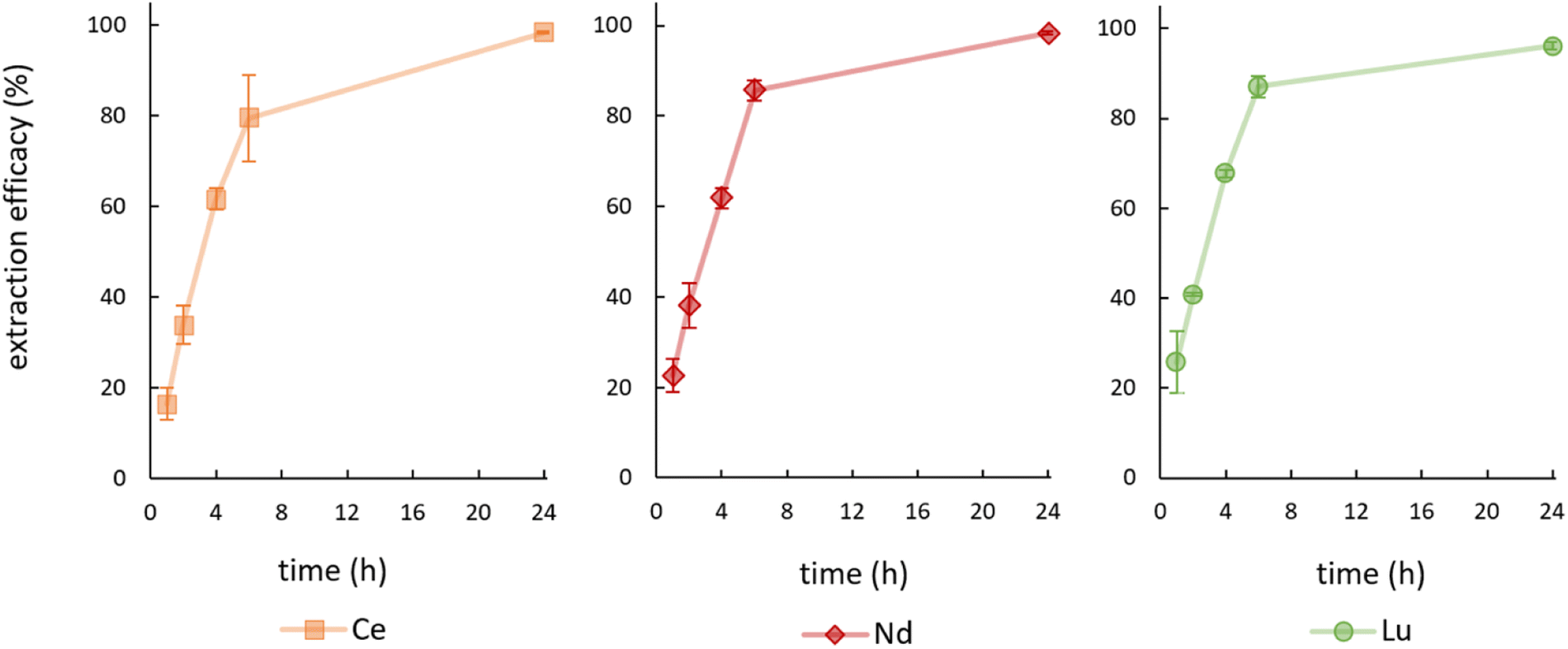 | ||
| Fig. 3 Mean (±SD) extraction efficacies over time for Ce, Nd and Lu at room temperature (23 ± 1 °C), initial pH = 2.5, initial metal concentration = 10 mg L−1 (n = 3). | ||
See Table S1† for detailed data on remaining metal concentration in the feed solution as well as the corresponding extraction efficacies including data for La and Ho. We achieved efficacies of ≥96% for Ce, Nd and Lu after 24 hours at room temperature. About 80 to 85% of the metals were thereby already removed from the aqueous phase after only 6 hours. Extraction efficacies for La (88.80 ± 0.54%) and for Ho (62.29 ± 4.76%) after 24 hours are significantly lower and therefore not presented graphically. The extraction efficacies and the necessary time for reaching the extraction equilibrium of [P66614][HNA] for Ce, Nd and Lu agree with various published extraction results, using conventional extractants.30–39 A table summarizing the results is given in S2.† One of the obvious advantages of using [P66614][HNA] is the abdication of volatile solvents during the extraction process, which were used in all cited publications. Especially for scientific applications the TSIL can compete with the traditionally used extractants and clearly represents an interesting alternative.
While the extraction of La using [P66614][HNA] can still be considered somewhat sufficient, the values for Ho are unsatisfactory. The equilibrium between aqueous and IL phase for Ho seems to be reached once about 60% of the metal is extracted. The full reason for this remains unclear. Maria et al. reported various discriminating factors of extraction along the lanthanide series, especially between light and heavy REE.26 These selectivities are not always progressive along the Ln series and depend on various factors such as pH and ionic strength. When these parameters are kept constant, differing maximum extraction efficacies (such as in our case for Ho) along the Ln series are not unusual.
In general, the metal extraction abilities of ILs rely on the mechanisms of ion exchange and neutral extraction, with a balance between the two or the dominance of one mechanism. This depends strongly on sample composition and speciation of the metal.40 Excess concentrations of nitrate ions in the feed solution are apparently crucial for successful extraction. Nitrate complexes of REE play only a subordinate role in natural waters due to competitive complexation with other anions.18 Nonetheless, nitrates typically form highly extractable complexes with REE.41 Those authors successfully extracted REE with a derivative of the ammonium-based IL Aliquat®336, [A336][NO3], from 2 M feed solutions containing high nitrate concentrations (up to 11 M). Their findings agree well with our results and reflect the advantage of high nitrate ion concentrations when extracting REE from aqueous solutions. Zhou et al. used three different yet, comparable ionic liquids for REE extraction featuring the same phosphonium-based cation: [trihexyltetradecylphosphonium]2[4,4′-isopropylidene bis(phenoxy acetate)], [P66614]2[IOPAA], [trihexyltetradecyl-phosphonium][sec-octylphenoxy acetate], [P66614][SOPAA], and trihexyltetradecyl-phosphonium chloride, [P66614]Cl.24 They reported extraction efficacies between 60 and 70% for most REE using [P66614]2[IOPAA] at pH 4.2 and sodium chloride concentrations of 0.5 mol L−1. [P66614][SOPAA] extracted about 50% Lu and less of the other REE under the same conditions. Using [P66614]Cl led to no extraction of REE. The presence of the functionalized anion [IOPAA]2−, compared to Cl−, seemed crucial for successful extraction. Van der Hoogerstraete et al. drew similar conclusions when successfully separating Fe, Co, Cu, Mn and Zn from mixtures with REE using [P66614][Cl] without a functionalized anion.42 This clearly points to an ion exchange mechanism in REE extraction, calling for using ILs containing exchangeable anions for such extractions. Onghena et al. extracted REE using the ionic liquids [P66614]NO3 and the Aliquat®336 derivative [A336]NO3 from nitrate-containing feed extraction media.43 This involved the formation of the pentanitrato complex [Ln(NO3)5]2−, which together with two cations (Q+) from the IL formed the complex [Q]2[Ln(NO3)5] in the organic phase. Due to comparable nitrate concentrations in our extraction media, our findings support the extraction of REE in the proposed experimental setup via anion exchange and confirm a high suitability of the 3-hydroxy-2-napthoate anion of [P66614][HNA] to be exchanged with nitrato complexes.
Temperature-dependent extractions of Ce and Nd
We investigated the influence of temperature on the extraction during time-dependent experiments. Fig. 4 presents the results for single element experiments from Ce and Nd solutions as well as the double-element experiment from a feed solution containing Ce and Nd. See Table S3† for detailed data on remaining metal concentrations in the feed solution as well as the corresponding extraction efficacies.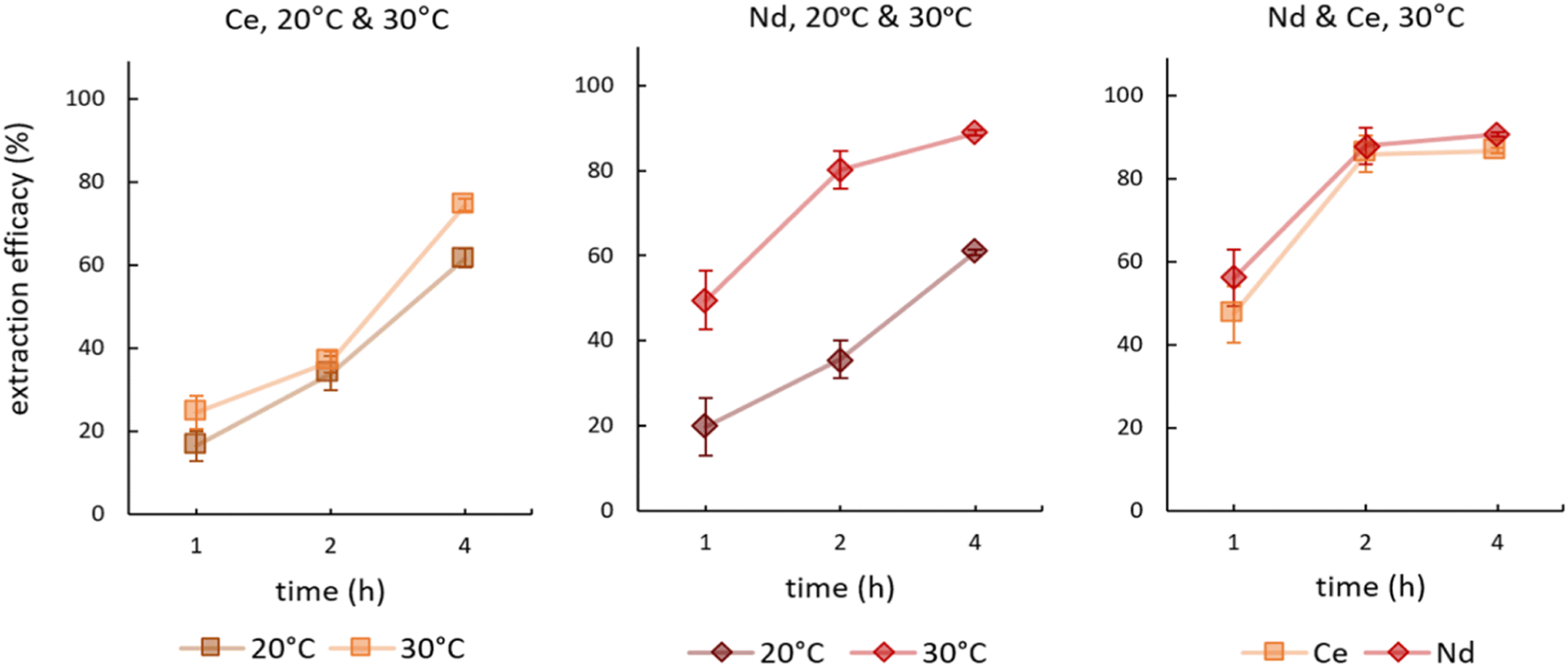 | ||
| Fig. 4 Mean (±SD) extraction efficacies over time for Ce (left), Nd (centre) at 20° and 30 °C, and a mixture of both (right) at 30 °C, initial pH 2.5, initial metal concentration = 10 mg L−1 (n = 3). | ||
In extracting Ce, increasing the temperature from 20 °C to 30 °C showed no significant effect on the time-dependent efficacy. The only minor differences were recorded after 4 hours, where at 20 °C 61.67 ± 2.32% and at 30 °C 74.47 ± 3.81% of Ce were extracted. In contrast, the impact of temperature was pronounced for Nd. An increase by 10 °C more than doubled the extraction efficacy after 1 hour from 19.87 ± 9.36% at 20 °C to 49.58 ± 6.81% at 30 °C. After 2 hours at 30 °C, the value jumped to 80.19 ± 4.39%. Interestingly, the double-element experiments at 30 °C showed that the time-dependent Ce extraction adapted to that of Nd. Nd extraction was not influenced by the presence of Ce, but Ce was extracted to a significantly higher degree when Nd was present (Fig. 4, right).
The minor Ce extraction differences at the two temperatures possibly reflects the oxidation state of Ce in solution. The elemental standard used provides a 5% nitric acid solution of CeO2, resulting in Ce4+ in the feed solution. In contrast to the long-held assumption that Ce4+ is present as a hexanitrato complex [Ce(NO3)6]2−, Demars et al. and Antonio et al. found oxo-bridged Ce4+ dimers as predominant species in the presence of excess nitrate concentrations.44,45 Nevertheless, we expect the hexanitrato complex [Ce(NO3)6]2− to be the extracted species.43 The expectation is that both species, dimers and nitrato complexes, are present and occur in equilibrium. Moreover, Buchanan et al. described a stronger stabilisation of Ce4+ versus trivalent cations in aqueous solutions.46 We hypothesise that the stronger stabilisation and the equilibrium of Ce4+ species explain the low influence of temperature on the extraction in single-element Ce solutions. Nd, on the other hand, is present as Nd3+, derived from Nd2O3, forming solely penta- and hexanitrato complexes.43 Both complexes are extractable with the IL via anionic exchange. The enhanced extraction at higher temperature agrees with the findings of Mishra and Devi, who described a temperature-dependency of Eu3+ extraction when using Cyphos® IL 104.47 The underlying assumption is that higher temperature boosts extraction efficacies due to decreased viscosity and therefore accelerated mass transfer. For the double-element experiment, our findings suggest that the formation of the stabilising oxo-bridged Ce4+ dimers is potentially hindered in the presence of an equimolar amount of Nd in solution. Furthermore, extraction conditions for double-element extractions are apparently enhanced at pH 2.5 at 30 °C and the described nitrate concentrations, leading to a satisfactory co-extraction of Ce and Nd (>85%) already after 2 hours. Overall, our findings highlight the importance of temperature constancy during extraction experiments, not only in regard of reproducibility but also the optional acceleration of the time-dependent extraction.
Co-extraction from other double-element feed solutions and back-extraction
The successful co-extraction of Ce and Nd triggered further experiments using double-element feed solutions. We used 10 mg L−1 La and Lu, La and Nd, Ce and Lu, as well as Ce and Nd, respectively. The co-extraction of REE is of interest for recycling and separation of REE if certain discriminating factors for one REE can be achieved. Also, back-extraction experiments were performed using 0.5 M nitric acid as stripping agent to investigate the options for REE recovery and reusability of the IL. Fig. 5 details the results for extraction from double-element feed solutions and back-extraction. Detailed data on remaining metal concentration in the feed solution after extraction, the corresponding extraction efficacies and back-extraction efficacies are presented in Table S4.†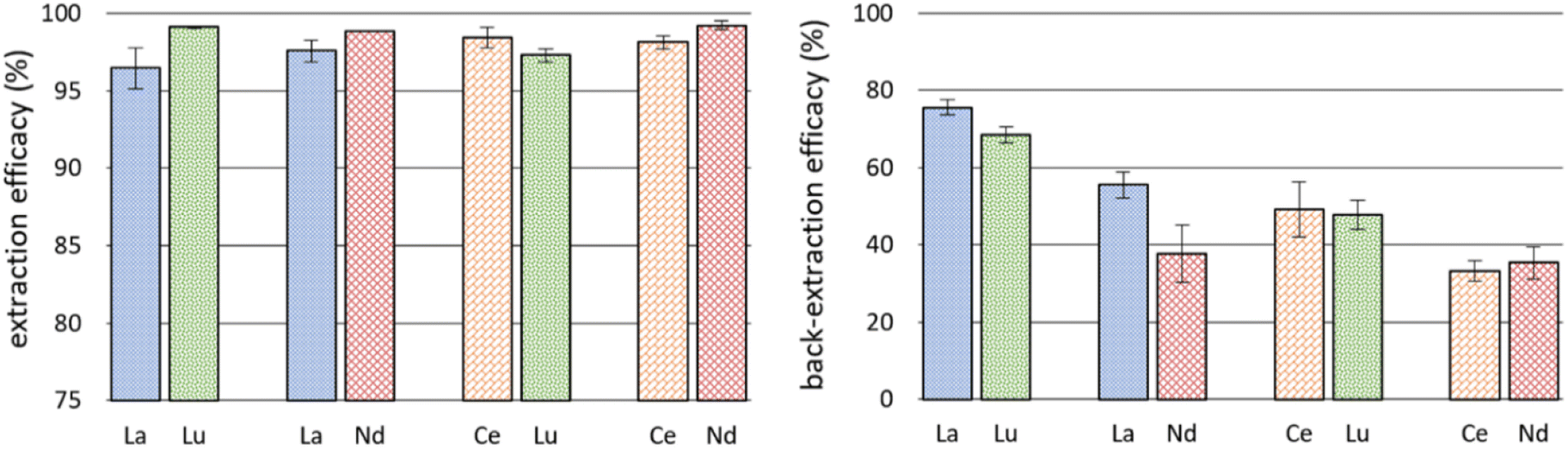 | ||
| Fig. 5 Left: mean extraction efficacies (%, ±SD) after 24 hours from double-element feed solutions at pH = 2.5 at 23 ± 1 °C (n = 3); right: mean back-extraction efficacies (%, ±SD) after 2 hours using 0.5 M HNO3 (n = 3). | ||
Remarkably, La was extracted with efficacies >95% when Lu or Nd were present as well. This moderately increased value, compared to single element extraction, is coherent with the observed enhanced extraction of Ce when Nd is present (see previous subchapter). Literature on the influence of metal concentration in feed solutions on extraction efficacies is scarce. Maria et al. used a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 IL to metal ratio and described decreased efficacies when the ratio was decreased to 2:1.26 Our findings do not confirm those results, whereby the experiments are poorly comparable because our IL-to-metal ratio was orders of magnitude higher and therefore doubling the experimental metal concentration seems irrelevant for the outcome. Nonetheless, increasing the concentration from 10 mg L−1 in single-element experiments to a total of 20 mg L−1 in double-element experiments appears to be advantageous for REE extraction using [P66614][HNA]. The explanation remains unknown and is being further investigated. Importantly, however, all efficacies from double-element feed solutions were ≥95%, indicating that the metal loading capacity was not fully exploited.
:1 IL to metal ratio and described decreased efficacies when the ratio was decreased to 2:1.26 Our findings do not confirm those results, whereby the experiments are poorly comparable because our IL-to-metal ratio was orders of magnitude higher and therefore doubling the experimental metal concentration seems irrelevant for the outcome. Nonetheless, increasing the concentration from 10 mg L−1 in single-element experiments to a total of 20 mg L−1 in double-element experiments appears to be advantageous for REE extraction using [P66614][HNA]. The explanation remains unknown and is being further investigated. Importantly, however, all efficacies from double-element feed solutions were ≥95%, indicating that the metal loading capacity was not fully exploited.
The back-extraction using 0.5 M HNO3 was not very successful (Fig. 5, right). Best recovery was achieved for La/Lu: 75.66 ± 2.03% of La and 68.44 ± 2.09% of Lu. In comparison, Huang et al. achieved better recovery rates, close to 100% of La, Ce, Pr and Nd.48 They used oxalic acid solutions as back-extraction agent, leading to precipitation of REE oxalates and a recovery of the used IL. This selective precipitation with oxalic acid was also used by Liu et al. and appears to be a promising stripping method for REE after extraction with ILs.49 Another recovery strategy used by Zhou et al. enabled “salting out” extracted REE with a 0.5 M sodium chloride solution.24 They used [P66614]2[IOPAA] for extraction and reached a precipitation efficiency of 98.9% in the case of Pr, for example. The REE hereby precipitated as Prx[IOPAA]y and the IL changed into [P66614]Cl. Based on our high extraction efficacies from the aqueous phase, we suggest adding comparable amounts of sodium chloride or oxalic acid for back-extraction experiments in the future. Interestingly, a certain separation between La and Nd occurred during back-extraction: 55.52 ± 3.34% of La and only 37.81 ± 7.45% of Nd were recovered from the IL. Further investigations here could be interesting for REE separation technology.
pH change during extraction
The pH in the feed solution was monitored during extraction experiments. Interestingly, a change over time from initially 2.5 to >4 was hereby recorded in all experiments, whereas the value in the reference feed solutions remained constant at 2.50 ± 0.03, excluding a delayed adaptation after setting. Table S5† presents detailed data. For all experiments, independent of temperature, the correlation between extraction efficacy and pH was positive (Fig. 6).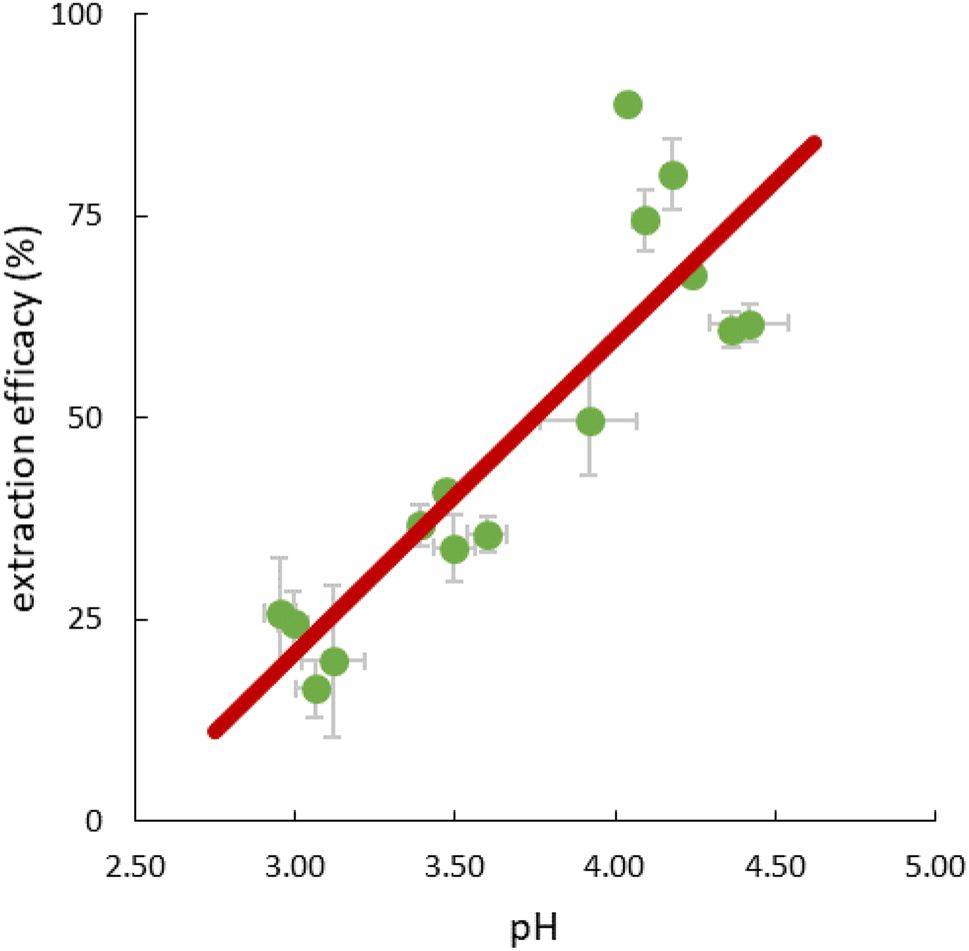 | ||
| Fig. 6 Correlation between pH values (mean ± SD) in the feed solutions and mean extraction efficacies (±SD) during single-element extraction of Ce, Nd and Lu after 1, 2 and 4 hours; initial pH = 2.5, n = 3; R2 = 0.7655. | ||
Assuming anion exchange as the extraction mechanism via formation of penta- and hexanitrato complexes,43 the conjugated base of 3-hydroxy-2-naphthoic acid, 3-hydroxy-2-naphthoate (HNA) appears to suitably modulate the pH value in the aqueous feed solution. The carboxylic acid 3-hydroxy-2-naphthoic acid has a ground-state pKa of 2.6.50 The feed solution is not buffered and therefore susceptible to changes. Literature on pH change during extraction using TSILs is scarce. Tran et al. used 3-butyl-1-methyl-1H-imidazole-3-ium 4-(dioctylamino)-4-oxobutanoate, [N88SA]−[C4min]+, and 1-butylpyridin-1-ium 4-(dioctylamino)-4-oxobutanoate, [N88SA]−[C4Py]+ to extract Ni(II) and Co(II) and observed a pH increase from 4 to 6.8–8 during the extraction.51 They assumed H+ ion uptake from the extraction media into the TSILs. This uptake does not contradict the suggested ion exchange mechanism but is a potential additional factor. Beyond the reported uptake of H+ ions into ILs, the uptake of other ions from the aqueous extraction matrix has also been described, e.g. an extraction of nitrate ions from the matrix via anion exchange, independent from collateral metal extraction.52 For the present study, however, the additional uptake of H+ ions into the IL seems less important because we can explain the extraction mechanism with the release of HNA ions into the aquatic phase as pH modulator. Based on the observed pH change and following literature on the formation of pentanitrato complexes of REE in nitrate media,43 we propose the following extraction mechanism (eqn (5)) for La, Ce, Nd, Ho and Lu in our work:
 | (5) |
Leaching
Investigations on leaching behaviour are fundamental with regard to the (re)usability of ILs as extractants as well as considering the toxicity and possible detrimental effects of ILs on biota and the environment.12,53,54Significant leaching values of the IL [P66614][HNA] during extraction experiments are shown in Fig. 7. Detailed leaching data are available in S6–S8.†
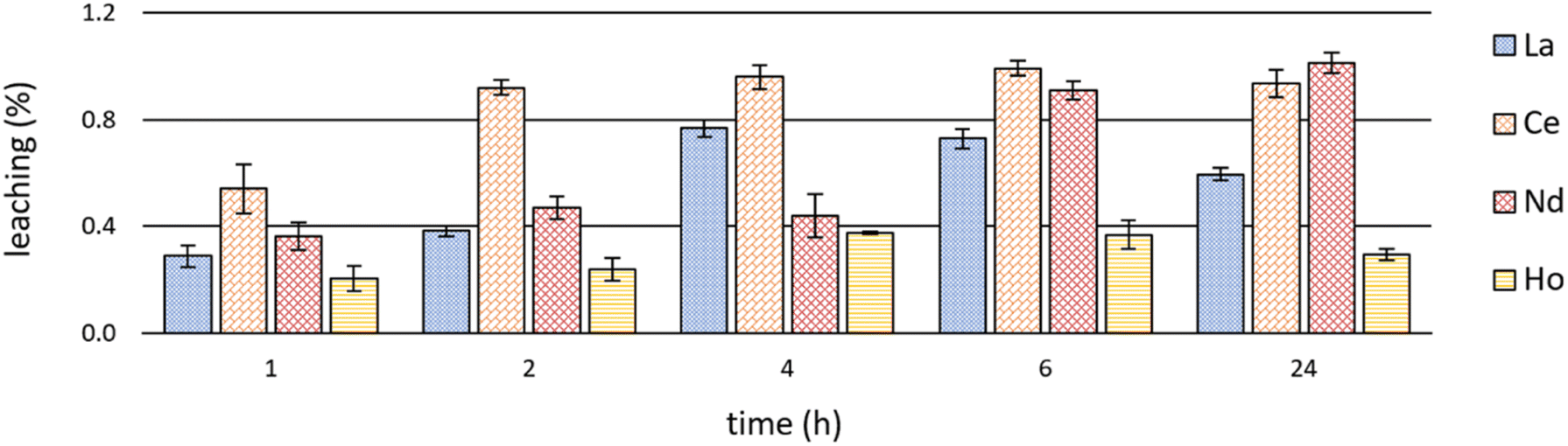 | ||
| Fig. 7 Mean leaching (+SD) of the IL [P66614][HNA] after time-dependent single-element extractions of La, Ce, Nd & Ho at room temperature (23 ± 1 °C) and pH 2.5 (n = 3). | ||
Leaching showed a certain coherence with extraction efficacies (Fig. 3 and Table S1†). After Ce and Nd extraction, with the highest efficacies (>98%), the leaching values were also highest. Ce extraction led to a leaching of 0.92 ± 0.03% after already 2 hours, peaking at 0.99 ± 0.03% after 6 hours. IL leaching during Nd extraction was relatively constant at between 0.36 ± 0.05 and 0.47 ± 0.04% after 1, 2 and 4 hours, yet increased to a maximum of 1.01 ± 0.04% afterwards – the highest value being at room temperature. During La extraction, leaching progressed from 0.29 ± 0.04% after 1 hour to 0.73 ± 0.04% after 6 hours. The values then decreased to 0.60 ± 0.02% after 24 hours. The same trend was observed for Ho extraction, yet with a maximum leaching of 0.38 ± 0.01%. This correlates with the extraction efficacies of about 90% for La and about 60% for Ho. In general, our findings on leaching agree well with those of Pirkwieser et al. for the same IL [P66614][HNA].11 Those authors describe a leaching of 0.07% to 0.23% after 1 hour at pH 8 in different aqueous matrices. Those values are slightly below ours, potentially reflecting the lower, acidic pH values we used for REE extraction and the different extraction mechanism. Pirkwieser et al. further reported a leaching of 0.36% to 0.42% after 2 hours of metal extraction for “pure water” at pH 3.5, an acidic pH value comparable to that in our experiments.12 These values are in good accordance. In general, there are two explanations for the minor differences between Pirkwieser et al.11,12 and our results: (1) the variable solubility of ILs depending on the composition of the aqueous phases,55 (2) a decreased pH value, which influences the solubility of IL and especially the respective anions. Our findings, e.g. pure water versus nitrate-rich water, confirm varying solubilities and leaching values, regardless of potentially ongoing extractions.
The IL leaching during back-extraction experiments ranged from 0.08 ± 0.01% to 0.13 ± 0.01% (the higher value was after the most successful back-extraction of La and Lu). These values are lower than those during REE extraction and agree even better with the leaching values in various matrices reported by Pirkwieser et al.11 This can be explained by our suggested extraction mechanism, which would release two molar equivalents hydroxynaphthoate into the aqueous phase during extraction. During back-extraction, only nitrato complexes of the respective REE are transferred into the aqueous phase, and the leaching can therefore solely be attributed to the solubility of the IL in the aqueous phase.
IL leaching is a fundamental (eco)toxicological parameter regarding aquatic biota in our proposed extraction setup, if the intention is to contribute to greener solutions for REE recycling. IL toxicity generally increases with the hydrophobicity, whereas an increased hydrophobic character of an in itself comparably less toxic compound can contribute to a decreased toxicity, e.g. by using non-aromatic cations instead of aromatic cations.56 The used TSIL [P66614][HNA] represents the 3-hydroxy-2-naphthoate form of the commercially available IL trihexyltetradecyl-phosphonium chloride. The functional anion 3-hydroxy-2-naphthoate is less toxic than similar compounds, especially compared with [P66614][Cl], but must nevertheless be considered an acute toxicant for algae.12,53 This is clearly a limiting factor for the usability of [P66614][HNA].
Conclusions
The task-specific ionic liquid trihexyltetradecylphosphonium 3-hydroxy-2-naphthoate was used successfully to extract Ce, Nd and Lu from both single-element and double-element aqueous feed solutions (>95%) in experiments over 24 hours. High nitrate concentration in the feed solution had a strong positive impact on extraction efficacy, pointing to anion exchange as an extraction mechanism. This was further supported by the positive correlation between pH change during extraction and the degree of leaching. Temperature also significantly impacted the extraction, significantly increasing efficacy and velocity when raised by only 10° to 30 °C. Back-extraction using 0.5 M HNO3 was only partly successful, especially in the case of La and Lu (up to 75%). Further research should therefore focus on alternative recovery methods. Despite the relatively low leaching of the ionic liquid, the (eco)toxicological potential of the compound cannot be belittled. Based on the promising extraction efficacies using our experimental setup, we urge research on reducing this potential (eco)toxicity, possibly by further reducing IL leaching.Author contributions
Conceptualization, A. G. and F. J.; validation, B. K. K. and W. K.; sample preparation and analysis, A. G., J. W. and M. W.; investigation, A. G., F. J. and M. W.; data curation, A. G.; writing—original draft preparation, A. G.; writing—review and editing, A. G., F. J., B. K. K. and W. K.; supervision, F. J. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank Michael Stachowitsch for proofreading the manuscript. Open access funding provided by University of Vienna.References
- K. R. Seddon, J. Appl. Chem. Biotechnol., 1997, 68, 351–356 CrossRef CAS.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- K. N. Marsh, A. Deev, A. C. T. Wu, E. Tran and A. Klamt, Korean J. Chem. Eng., 2002, 19, 357–362 CrossRef CAS.
- D. Kogelnig, A. Stojanovic, F. Jirsa, W. Körner, R. Krachler and B. K. Keppler, Sep. Purif. Technol., 2010, 72, 56–60 CrossRef CAS.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS.
- T. Welton, Biophys. Rev., 2018, 10, 691–706 CrossRef CAS PubMed.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- A. Stojanovic and B. K. Keppler, Sep. Sci. Technol., 2012, 47, 189–203 CrossRef CAS.
- K. Wang, H. Adidharma, M. Radosz, P. Wan, X. Xu, C. K. Russell, H. Tian, M. Fan and J. Yu, Green Chem., 2017, 19, 4469–4493 RSC.
- M. Aghaie, N. Rezaei and S. Zendehboudi, Renewable Sustainable Energy Rev., 2018, 96, 502–525 CrossRef CAS.
- P. Pirkwieser, J. A. Lopez-Lopez, W. Kandioller, B. K. Keppler, C. Moreno and F. Jirsa, Front. Chem., 2018, 6, 172 CrossRef PubMed.
- P. Pirkwieser, J. A. López-López, M. Schagerl, W. Kandioller, B. K. Keppler, C. Moreno and F. Jirsa, Appl. Sci., 2020, 10, 3157 CrossRef CAS.
- N. G. Connelly, T. Damhus, R. M. Hartshorn and A. T. Hutton, Nomenclature of Inorganic Chemistry: IUPAC recommendations 2005, RSC Publishing, Cambridge, 2005 Search PubMed.
- V. Balaram, Geosci. Front., 2019, 10, 1285–1303 CrossRef CAS.
- N. Haque, A. Hughes, S. Lim and C. Vernon, Resources, 2014, 3, 614–635 CrossRef.
- S. M. Jowitt, T. T. Werner, Z. Weng and G. M. Mudd, Curr. Opin. Green Sustainable Chem., 2018, 13, 1–7 CrossRef.
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Cleaner Prod., 2013, 51, 1–22 CrossRef CAS.
- S. A. Wood, Chem. Geol., 1990, 82, 159–186 CrossRef CAS.
- H. Fang, B. E. Cole, Y. Qiao, J. A. Bogart, T. Cheisson, B. C. Manor, P. J. Carroll and E. J. Schelter, Angew. Chem., Int. Ed., 2017, 56, 13450–13454 CrossRef CAS PubMed.
- H. Okamura and N. Hirayama, Anal. Sci., 2021, 37, 119–130 CrossRef CAS PubMed.
- P. Davris, E. Balomenos, D. Panias and I. Paspaliaris, Hydrometallurgy, 2016, 164, 125–135 CrossRef CAS.
- J. E. Quinn, K. H. Soldenhoff and G. W. Stevens, Hydrometallurgy, 2017, 169, 306–313 CrossRef CAS.
- J. E. Quinn, K. H. Soldenhoff and G. W. Stevens, Hydrometallurgy, 2017, 169, 621–628 CrossRef CAS.
- H. Zhou, Y. Wang, X. Guo, Y. Dong, X. Su and X. Sun, J. Mol. Liq., 2018, 254, 414–420 CrossRef CAS.
- N. N. Hidayah and S. Z. Abidin, C. R. Chim., 2019, 22, 728–744 CrossRef CAS.
- L. Maria, A. Cruz, J. M. Carretas, B. Monteiro, C. Galinha, S. S. Gomes, M. F. Araújo, I. Paiva, J. Marçalo and J. P. Leal, Sep. Purif. Technol., 2020, 237, 116354 CrossRef CAS.
- S. Platzer, M. Kar, R. Leyma, S. Chib, A. Roller, F. Jirsa, R. Krachler, D. R. MacFarlane, W. Kandioller and B. K. Keppler, J. Hazard. Mater., 2017, 324, 241–249 CrossRef CAS PubMed.
- D. G. Brookins, in Geochemistry and Mineralogy of Rare Earth Elements, ed. B. R. Lipin and G. A. McKay, De Gruyter, Berlin, 1989, vol. 8, pp. 201–226 Search PubMed.
- K. N. Han, Min. Metall. Explor., 2019, 36, 215–225 Search PubMed.
- N. N. Hidayah and S. Z. Abidin, Miner. Eng., 2018, 121, 146–157 CrossRef CAS.
- T. Liu and J. Chen, Sep. Purif. Technol., 2021, 276, 119263 CrossRef CAS.
- F. Xie, T. A. Zhang, D. Dreisinger and F. Doyle, Miner. Eng., 2014, 56, 10–28 CrossRef CAS.
- W. Li, X. Wang, S. Meng, D. Li and Y. Xiong, Sep. Purif. Technol., 2007, 54, 164–169 CrossRef CAS.
- U. Ray and S. Modak, Indian J. Chem., 1981, 20A, 935–936 CAS.
- C. Basualto, F. Valenzuela, L. Molina, J. Munoz, E. Fuentes and J. Sapag, J. Chil. Chem. Soc., 2013, 58, 1785–1789 CrossRef CAS.
- H. Yuan, W. Hong, Y. Zhou, B. Pu, A. Gong, T. Xu, Q. Yang, F. Li, L. Qiu and W. Zhang, J. Rare Earths, 2018, 36, 642–647 CrossRef CAS.
- K. Shimojo, I. Fujiwara, K. Fujisawa, H. Okamura, T. Sugita, T. Oshima, Y. Baba and H. Naganawa, Solvent Extr. Res. Dev., Jpn., 2016, 23, 151–159 CrossRef CAS.
- R. D. Abreu and C. A. Morais, Miner. Eng., 2014, 61, 82–87 CrossRef CAS.
- J. Su, X. Guo, Y. Gao, S. Wu, R. Xu and X. Sun, J. Rare Earths, 2021, 39, 1273–1281 CrossRef CAS.
- C. H. C. Janssen, N. A. Macías-Ruvalcaba, M. Aguilar-Martínez and M. N. Kobrak, Int. Rev. Phys. Chem., 2015, 34, 591–622 Search PubMed.
- K. Larsson and K. Binnemans, J. Sustain. Metall., 2016, 3, 73–78 Search PubMed.
- T. Van der Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemans, Green Chem., 2013, 15, 919–927 RSC.
- B. Onghena, E. Papagni, E. R. Souza, D. Banerjee, K. Binnemans and T. Vander Hoogerstraete, RSC Adv., 2018, 8, 32044–32054 RSC.
- T. J. Demars, M. K. Bera, S. Seifert, M. R. Antonio and R. J. Ellis, Angew. Chem., Int. Ed., 2015, 54, 7534–7538 CrossRef CAS PubMed.
- M. R. Antonio, R. J. Ellis, S. L. Estes and M. K. Bera, Phys. Chem. Chem. Phys., 2017, 19, 21304–21316 RSC.
- C. A. Buchanan, E. Ko, S. Cira, M. Balasubramanian, B. R. Goldsmith and N. Singh, Inorg. Chem., 2020, 59, 12552–12563 CrossRef CAS PubMed.
- B. B. Mishra and N. Devi, J. Mol. Liq., 2018, 271, 389–396 CrossRef CAS.
- Y. Huang, D. Wang, Z. Duan, J. Liu, Y. Cao and W. Peng, Minerals, 2022, 12, 1592 CrossRef CAS.
- Z. Liu, H. Zhou, W. Li, X. Luo, J. Wang and F. Liu, Sep. Purif. Technol., 2022, 282, 119795 CrossRef CAS.
- P. J. Kovi and S. G. Schulman, Anal. Chem., 1973, 45, 989–991 CrossRef CAS.
- T. T. Tran, N. Azra, M. Iqbal and M. S. Lee, Sep. Purif. Technol., 2020, 238, 116496 CrossRef CAS.
- C. H. C. Janssen, J. Mol. Liq., 2020, 304, 112738 CrossRef CAS.
- S. Platzer, R. Leyma, S. Wolske, W. Kandioller, E. Heid, C. Schroder, M. Schagerl, R. Krachler, F. Jirsa and B. K. Keppler, J. Hazard. Mater., 2017, 340, 113–119 CrossRef CAS PubMed.
- J. Flieger and M. Flieger, Int. J. Mol. Sci., 2020, 21, 6267 CrossRef CAS PubMed.
- D. Dupont, D. Depuydt and K. Binnemans, J. Phys. Chem. B, 2015, 119, 6747–6757 CrossRef CAS PubMed.
- S. P. Ventura, A. M. Gonçalves, T. Sintra, J. L. Pereira, F. Gonçalves and J. A. Coutinho, Ecotoxicology, 2013, 22, 1–12 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information on extraction efficacies, leaching and pH change. See DOI: https://doi.org/10.1039/d3ra03967f |
| This journal is © The Royal Society of Chemistry 2023 |