
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA BODIPY-picolinium-cation conjugate as a blue-light-responsive caged group†
Naoya Ieda *a,
Akira Nakamuraa,
Natsumi Tomitaa,
Kei Ohkubob,
Ryo Izumia,
Yuji Hottac,
Mitsuyasu Kawaguchia,
Kazunori Kimurac and
Hidehiko Nakagawa*a
*a,
Akira Nakamuraa,
Natsumi Tomitaa,
Kei Ohkubob,
Ryo Izumia,
Yuji Hottac,
Mitsuyasu Kawaguchia,
Kazunori Kimurac and
Hidehiko Nakagawa*a
aGraduate School of Pharmaceutical Sciences, Nagoya City University, 3-1, Tanabe-dori, Mizuho-ku, Nagoya, Aichi 467-8603, Japan. E-mail: ieda@pharm.hokudai.ac.jp; deco@phar.nagoya-cu.ac.jp
bInstitute for Open and Transdisciplinary Research Initiatives & Institute for Advanced Co-Creation Studies, Osaka University, 1-6 Yamada-oka, Suita, Osaka, 565-0871, Japan
cGraduate School of Medical Sciences, Nagoya City University, 1, Kawasumi, Mizuho-cho, Mizuho-ku, Nagoya, Aichi 467-8601, Japan
First published on 4th September 2023
Abstract
Caged compounds protected with photolabile protecting groups (PPGs) are useful for controlling various biological events with high spatiotemporal resolution. Most of the commonly used PPGs are controlled by ultraviolet light irradiation, but it is desirable to have PPGs controlled by visible light irradiation in order to minimize tissue damage. Here, we describe a boron-dipyrromethene (BODIPY)-picolinium conjugate (BPc group) that functions as a blue-light-controllable PPG. ESR experiments indicate that the photolysis mechanism is based on intramolecular photoinduced electron transfer. We illustrate the applicability of the BPc group to biologically active compounds by employing it firstly to photocontrol release of histamine, and secondly to photocontrol release of a soluble guanylyl cyclase (sGC) activator, GSK2181236A, which induces photovasodilation. The BPc group is expected to be a useful PPG for controlling various biological events with blue light irradiation.
Introduction
The term “caged” compound was first coined by Hoffman's group to describe a bioactive molecule protected with a photolabile protecting group (PPG). Since then, many PPGs have been developed, not only as tools for controlling biological events with high spatiotemporal resolution,1 but also as candidate therapeutic agents.2–5 However, the most commonly used PPGs, such as the 2-nitrobenzyl-type and coumarin-4-methyl-type PPGs, require harmful ultraviolet to purple light irradiation (<450 nm) for the uncaging reaction.1,6 It would be preferable to use visible light (>500 nm) in order to avoid photodamage to cells or tissues.7 Although some visible-light-driven PPGs have been developed, they require molecular oxygen or have strong photosensitizing activity, restricting their biological applicability.8–14 Very recently, Bojtàr's group reported xanthenium-type PPGs that can be controlled with visible light (>540 nm) and demonstrated an application for caging an anticancer reagent, SN38.15We have developed photoinduced-electron-transfer-driven nitric oxide releasers (PeT-driven NO releasers) which efficiently release NO in response to visible light irradiation up to a wavelength of 660 nm, making them applicable for controlling vasodilation even in vivo.16–19 Here, to expand the utility of this PeT-driven strategy to other bioactive compounds, we focused on the N-alkyl-4-picolinium group. Falvey's group previously demonstrated that the cationic N-alkyl-picolinium group could undergo electron transfer from photoactivated dyes, followed by H2C–O bond scission (Fig. 1a).20–23 However, it has not been established whether this strategy can be applied using visible-light-harvesting dyes, or for biological applications. In this study, we focused on boron-dipyrromethene (BODIPY) as a dye that can harvest 500 nm blue light and we synthesized a BODIPY-picolinium conjugate (BPc group) that can be used as a PPG. To illustrate its utility, we employed the BPc group to cage a soluble guanylyl cyclase activator, GSK2181236A, and demonstrated photocontrol of its vasodilation activity through blue light irradiation.
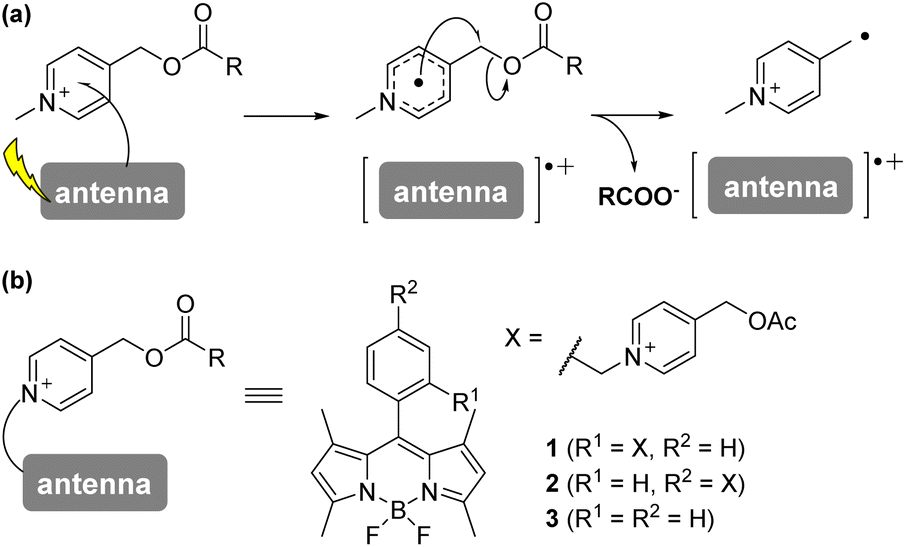 | ||
| Fig. 1 A plausible mechanism of PeT-driven picolinium cation photolysis (a); structure of BODIPY-picolinium conjugate substituted in the ortho-position (1) or the para-position (2), and a reference compound (3). | ||
Results and discussion
Anderson's group reported that N-alkyl-picolinium ester tethered to bis(ethynyl)fluorene dyes (BEF-Pyr) can function as a PPG, driven by PeT from bis(ethynyl)fluorene to the picolinium group.23 However, BEF-Pyr required two-photon excitation (TPE) for visible light uncaging, and UV light irradiation (<400 nm) appeared necessary for one-photon excitation. Although TPE is a powerful method for near-infrared excitation and provides high spatial resolution, it requires pulsed lasers that are expensive and not always readily available. Therefore, we aimed to extend the utility of the picolinium cation by conjugating the cation to a BODIPY dye that absorbs light at around 500 nm, in order to obtain a blue-light-responsive PPG (Fig. 1b). To investigate the importance of the proximity effect between BODIPY and the picolinium cation, we designed two derivatives: one substituted in the ortho-position (1) and the other in the para-position (2).18 Before synthesizing these compounds, we measured the redox potentials of the picolinium cation moiety (S2, synthesized from pyridine-4-methyl acetate (S1)) and the antenna moiety (3) to estimate the Gibbs free energy for PeT (ΔGPeT), which can be calculated using eqn S(1).†24 Cyclic voltammetry measurements showed that the reduction potential of S2 was −0.902 V (vs. SCE) and the oxidation potential of 3 was +1.11 V (vs. SCE) (Fig. S1†). Using eqn S(1),† the Gibbs free energy for PeT (ΔGPeT) of the designed PPG was calculated to be −0.388 eV, indicating that PeT should occur. Compounds 1 and 2 were synthesized as described in the ESI.† In brief, phthalide (S3) was activated with Meerwein's reagent to undergo a Friedel–Crafts-type reaction with 2,4-dimethylpyrrole.25 Trifluoroborate diethyl etherate (BF3·OEt2) and triethylamine (NEt3) were used to obtain a BODIPY derivative (S4). The hydroxyl group was converted to an bromo group via mesylated compound, then an SN2 reaction with S1 was conducted to obtain the ortho-substituted derivative (1). For the synthesis of the para-substituted derivative (2), 4-(chloromethyl)benzoyl chloride (S6) was treated with 2,4-dimethylpyrrole followed by BF3·OEt2 and NEt3 to obtain another BODIPY derivative (S7), and then an SN2 reaction with S1 was conducted. The antenna compound 3 was synthesized according to a previous report.26 The structures of the final products were confirmed by 1H NMR, 13C NMR, and HRMS, and purity was confirmed by HPLC.We first investigated the spectroscopic properties of the compounds. As shown in Fig. 2a, each compound exhibited strong absorption at around 500 nm, while 1 and 2 exhibited lower fluorescence intensity than the antenna compound 3 (Fig. 2b).27 This result indicates that 1 and 2 are relaxed through a different pathway from 3 after photoexcitation, probably through the PeT process. Moreover, 1 exhibited lower fluorescence than 2, suggesting that the short distance between the picolinium cation and the antenna would lead to more efficient PeT in 1 than in 2.28
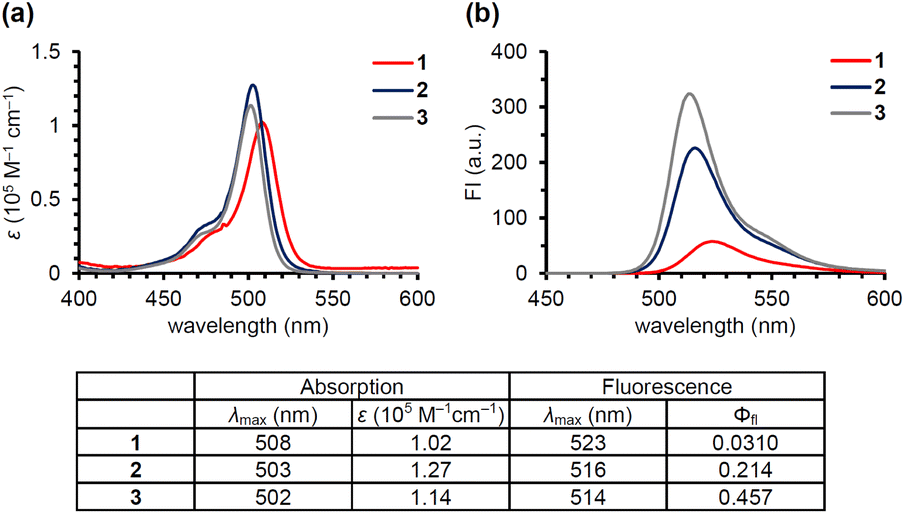 | ||
| Fig. 2 Absorption (a) and fluorescence (b) spectra of 1 (red), 2 (blue) and 3 (gray) in DMSO (10 μM). The spectral data are listed in the table. Ex. 500 nm. The quantum yields were measured using a solution of 3 in MeOH (Φfl: 0.46). | ||
To compare the photodecomposition rates of 1 and 2, a solution of each compound (10 μM) in 100 mM HEPES buffer (pH 7.3, 0.1% DMSO) was photoirradiated with blue light (470–500 nm, 85 mW cm−2) and analyzed by HPLC as shown in Fig. 3. The rate of 1 was faster than 2 in response to blue light irradiation. It is note that no decomposition occurred without photoirradiation as control experiments. To investigate whether the desired ligand release occurred, we also designed and synthesized 4 and 5 (Fig. S2 in ESI†) with protected p-nitroaniline (pNA), which has strong absorbance around 380 nm and is easily detectable by HPLC. As shown in Fig. 4, quicker photodecomposition was again observed with the ortho-substituted derivative 4. Not only quicker photodecomposition but also quicker ligand release was observed for 4, as compared with 5. These results indicated the importance of the proximity effect between the antenna moiety and the picolinium cation moiety for photodecomposition. Furthermore, photodecomposition of the antenna moiety was also observed in LCMS analysis. The m/z value corresponding to S10 was observed, indicating that the radical cation intermediate after photodecomposition undergoes nucleophilic attack by H2O to form an alcohol derivative S10 (Fig. S3 in ESI†).
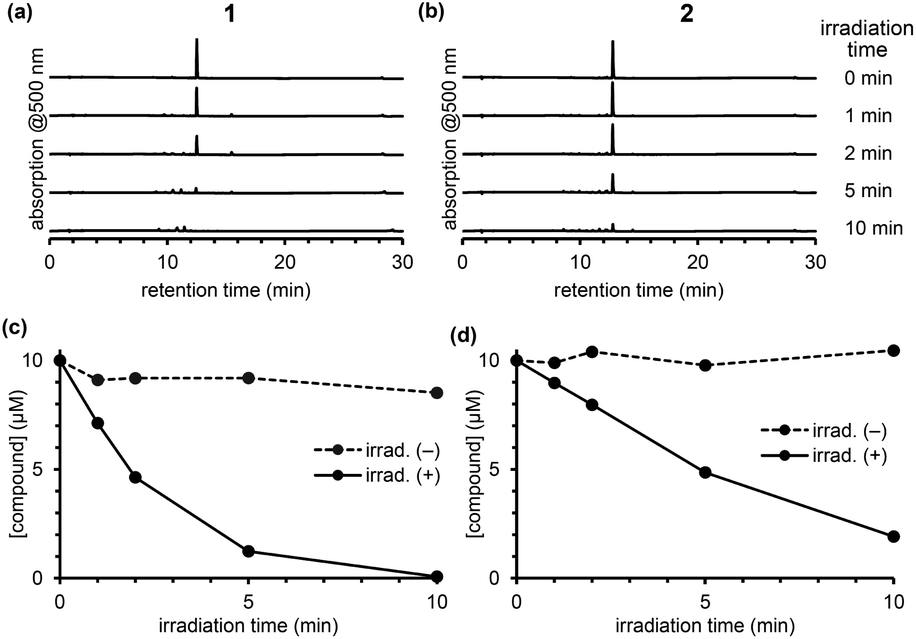 | ||
| Fig. 3 Chromatograms of an irradiated solution of 1 (a) and 2 (b) after photoirradiation. Time courses of concentration of 1 (c) or 2 (d) upon photoirradiation. | ||
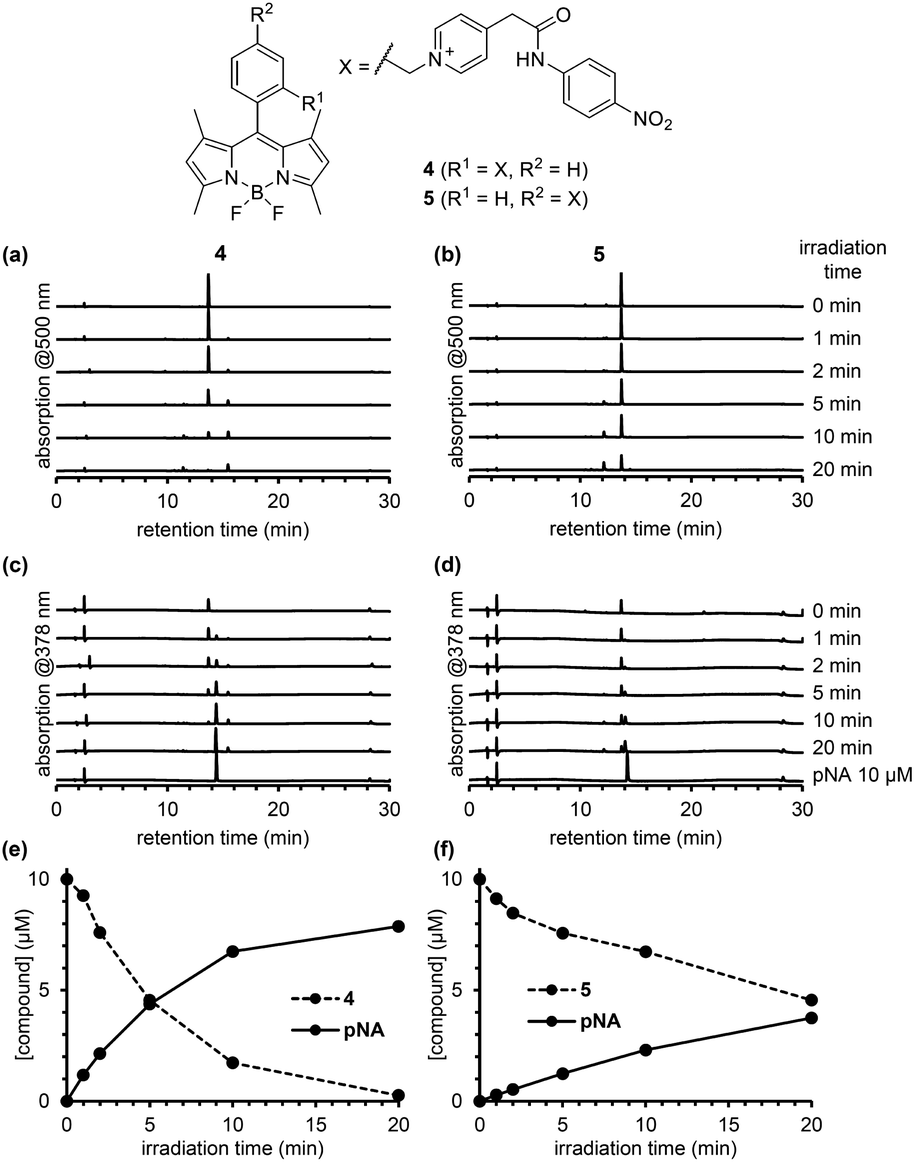 | ||
| Fig. 4 Monitoring the photodecomposition of 4 and 5 and the photorelease of p-nitroaniline (pNA). Chromatograms of a solution of 4 (a) or 5 (b) with detection at 500 nm, and of a solution of 4 (c) or 5 (d) with detection at 378 nm during photoirradiation. Time course of concentration of 4 (e) or 5 (f) during photoirradiation. | ||
To examine the reaction mechanism, we attempted to detect the putative radical intermediates by means of electron spin resonance (ESR) spectroscopy.29 As shown in Fig. 5a and b, we were able to detect the ESR signal due to the radical intermediate 1 or 2 generated by the LED light irradiation (λ = 405 nm) to DMSO glass at −130 °C. The signal observed at g = 2.003 is assigned to the typical organic radicals due to the BODIPY radical cation and the picolinyl radical as the charge-separated state generated by intramolecular PeT from the BODIPY moiety to the picolinium moiety. The signals due to both radicals may be overlapped in this experimental conditions. We also conducted DFT calculations of 1 and 2 to evaluate the possibility of H2C–O bond scission in the one-electron-reduced form (Fig. 5c). As shown in Fig. S4,† the bond length in the one-electron-reduced form was extended compared to that in the non-reduced form for 1 and 2. These experimental results and calculations indicate that one-electron transfer reduction of picolinium moiety by PeT triggers H2C–O bond scission and release of the ligand. It should be noted that while the H2C–O bond length in 2 was longer than that in 1, the efficiency of photodecomposition and ligand release was greater in 1 than 2. This result suggests that the uncaging efficiency is not dependent on the H2C–O bond length but rather on the PeT efficiency.
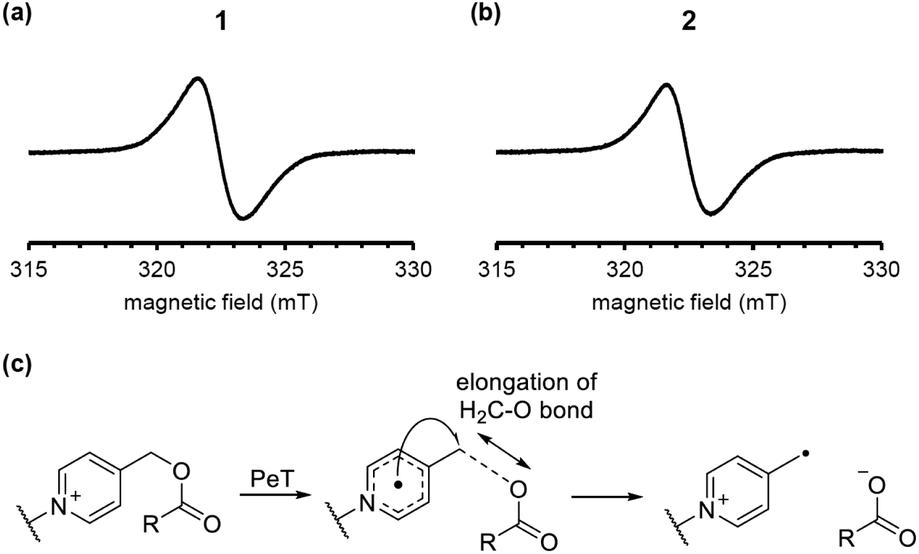 | ||
| Fig. 5 ESR spectra of the charge-separated states generated by the LED-light irradiation of DMSO glass containing 10 μM 1 (a) or 2 (b). A plausible H2C–O bond elongation before the bond scission (c). | ||
In order to confirm the ability of the ortho-substituted BODIPY and picolinium cation conjugate (BPc group) to protect a bioactive compound, we employed it to protect histamine, a messenger that activates histamine receptors (BPc-HA (6); see ESI for synthesis of 6, Fig. S5†). The release of histamine in response to light was detected and quantified using a fluorescence method that employs ortho-phthalaldehyde and 2-mercaptoethanol to form a fluorogenic isoindole derivative with primary amines.30 After photoirradiation of a solution of 6, the derivatizing reagents were added and the solution was analyzed by fluorescence HPLC. As shown in Fig. 6, histamine was released in a light-dependent manner, with a maximum release of 7.0 μM. In Fig. 4, the photodecomposition peak could be observed whereas it was difficult to see it in Fig. 3 and 6. It could be because the strong electron-withdrawing 4-nitroaniline structure improved electron accepting ability which is key to induce mesolytic cleavage for 4 and 5. For other compounds, unexpected degradation pathways, i.e. decomposition of BODIPY structure by singlet oxygen, could be considered preferential. We also calculated the quantum yield of histamine release (ΦHA) from 6, which is the amount of histamine released divided by the number of photons absorbed by 6. The amount of histamine release was quantified using Johnson's method,30 and the absorbed photons were determined using a potassium ferrioxalate actinometer.31 The calculated ΦHA was 2.55 × 10−4 (the detailed data are shown in Table S1†), which is comparable to that of another BODIPY-based caged histamine reported by Urano's group (3.0 × 10−4).32
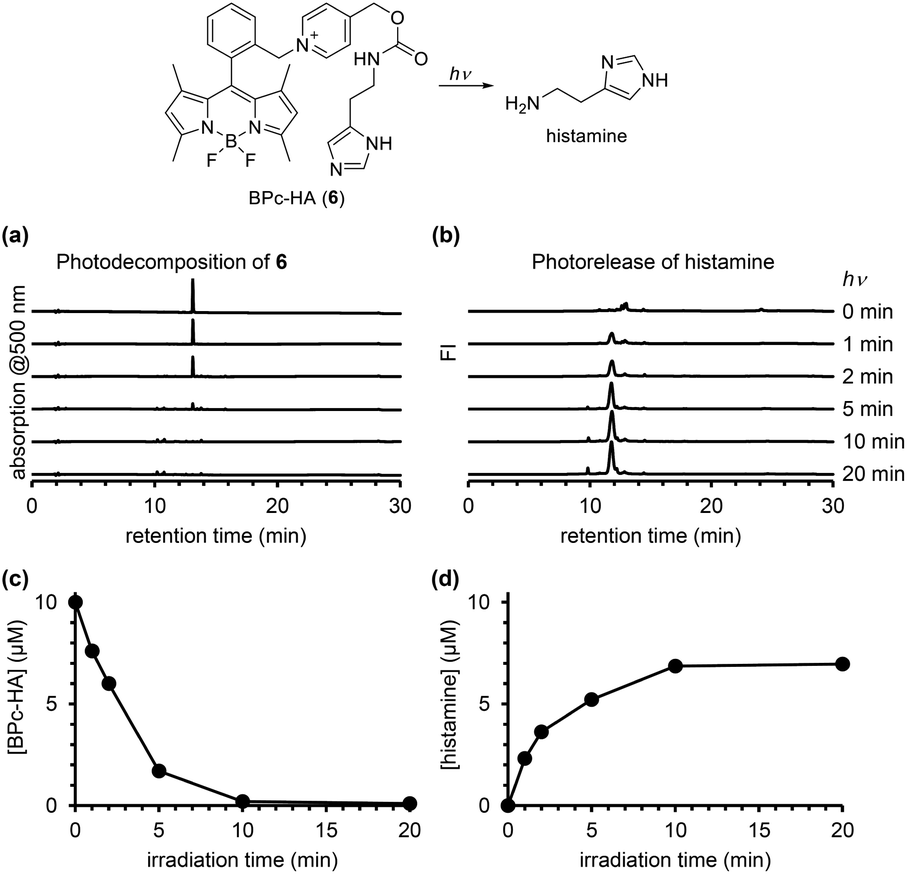 | ||
| Fig. 6 Monitoring the photodecomposition of BPc-HA (6) and photorelease of histamine. Chromatograms of a solution of 6 monitored in terms of absorption at 500 nm (a) and in terms of fluorescence (ex/em: 350/450 nm) after mixing with 2-mercaptoethanol and o-phthalaldehyde (b) during photoirradiation for the indicated time. Time course of concentration of 6 (c) or histamine (d). | ||
To confirm the blue-light responsiveness of the BPc group for controlling biological events, we selected a soluble guanylyl cyclase (sGC) activator, GSK2181236A.33 sGC activators bind to sGC whose heme is oxidized or missing to stimulate the production of cGMP, which in turn promotes smooth muscle relaxation, making them promising candidates for antihypertensive therapy. Although we previously developed PeT-driven NO releasers that induce potent vasodilation through the sGC-cGMP pathway in a light-dependent manner, the vasodilation they produce is transient and limited to the duration of irradiation, due to the short half-life of the NO-sGC complex.34 Thus, to achieve continuous vasodilation for the treatment of chronic hypertension, caged sGC activators would be more suitable than caged NOs. We therefore designed and synthesized BPc-GSK2181236A (7, see ESI for synthesis of 7, Fig. S6†), in which the carboxyl group of GSK2181236A is protected by the BPc group. We evaluated the photovasodilatory effect of 7 on ex vivo rat aorta using the blue-light irradiation. All animal experiments were performed in accordance with the Guiding Principles for the Care and Use of Laboratory Animals of the Science and International Affairs Bureau of the Japanese Ministry of Education, Culture, Sports, Science, and Technology, and were approved by the Animal Experimentation Ethics Committee of Nagoya City University (No. H25-P-09). We placed a strip of rat aorta in a Magnus tube filled with Krebs buffer, and tensioned it by exposing it to noradrenaline (NA). We also administered a nitric oxide synthase inhibitor, L-NAME, to eliminate the effect of endogenous NO. After the tension had reached a plateau, we conducted blue-light irradiation with a 505 nm LED (120 mW cm−2). As shown in Fig. 7a, the smooth muscle tension was decreased after 3 minutes of irradiation, whereas no potent vasodilation was observed without 7 or without irradiation. The slight vasodilation observed in the absence of 7 (Fig. 7b) or light irradiation (Fig. 7c) was probably induced by DMSO, since it has been reported that DMSO induces vasodilation in rat aorta by inhibiting Rho-kinase.35 While the vasodilation produced by caged NOs was transient and only occurred during irradiation, 7 induced prolonged vasodilation even after the irradiation was stopped. These results suggest that 7 could be a candidate photovasodilator for treating chronic hypertensive diseases.
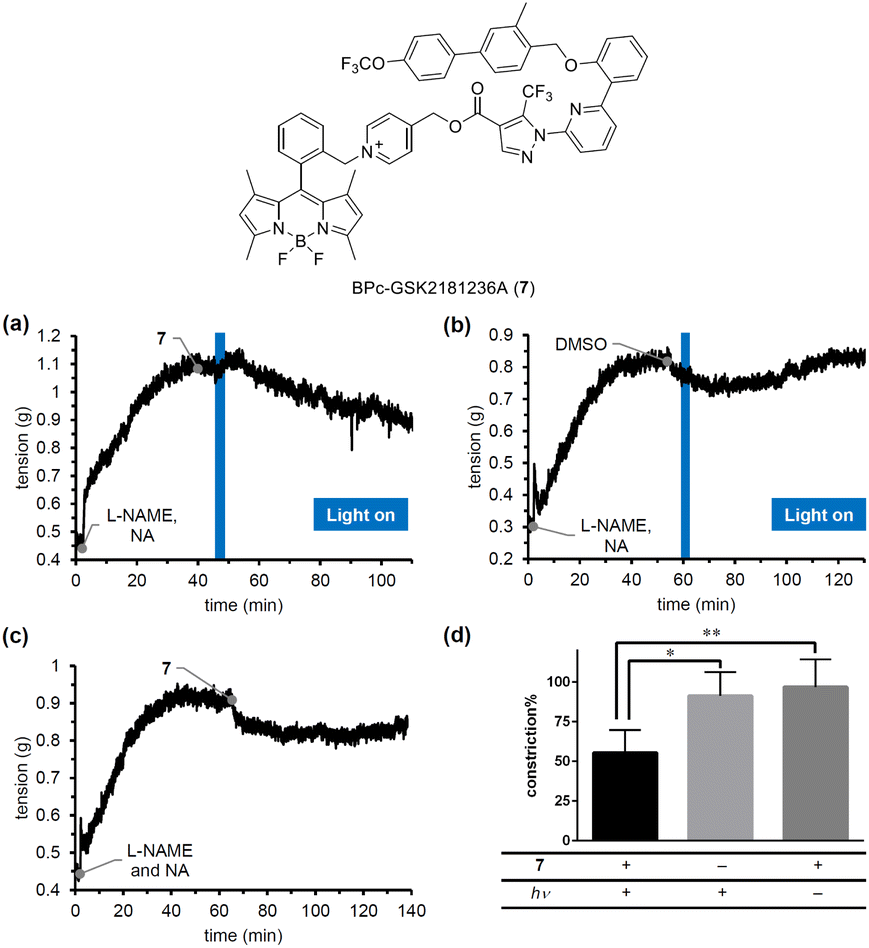 | ||
| Fig. 7 Changes in the tension of rat aorta ex vivo induced by blue-light-mediated drug release from 7 in the presence of a nitric oxide synthetase inhibitor, NG-nitro-L-arginine methyl ester (L-NAME, 10 μM). Rat aorta in a glass tube was treated with noradrenaline (NA, 10 μM). The tube was irradiated with a 505 nm LED (120 mW cm−2) for 3 min in the presence (a) or absence (b) of 7 (50 μM). Another aorta was incubated in the presence of 7 without irradiation (c). The constriction% in each experiment (d) was calculated from the tension value 60 min after photoirradiation. Data are expressed as mean ± SE (shown as error bars, n = 4). Statistical significance was examined by application of Bonferroni's multiple comparison. *p < 0.05, and **p < 0.01. | ||
Conclusion
Based on previous reports of photolysis of picolinium cation conjugates with dyes, we designed a novel visible-light-controllable PPG, the BPc group. The proximity between the antenna and picolinium cation was found to be important for efficient photolysis reaction, which likely occurs through an intramolecular photoinduced electron transfer reaction, as indicated by the detection of radical intermediates as the charge-separated state by ESR analyses. The BPc group has the ability to protect and release bioactive compounds such as histamine and an sGC activator, GSK2181236A. Since previous studies have shown that the photolysis of picolinium cation conjugates with various dyes can occur, we anticipate that a range of functionalized PPGs can be developed, including those absorbing at longer wavelengths and that can be activated in specific cells or diseased organs.Author contributions
N. I. and H. N. conceptualised the project, and wrote, reviewed and edit the manuscript. N. I., A. K., and R. I. synthesized and analysed the compounds. N. T. and Y. H. performed the experiments using rat aorta. K. O. performed ESR experiments and DFT calculations. M. K. and K. K. directed and supported the experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the assistance of the Research Equipment Sharing Center at the Nagoya City University. This work was supported in part by JSPS KAKENHI grant numbers JP20K05752 (N. I.), JP21H05259 (H. N.), JP23H02612 (H. N.), the JST ACT-X Grant Number JPMJAX2011 (N. I.), Takeda Science Foundation (N. I.), and TERUMO Life Science Foundation (N. I.).Notes and references
- J. H. Kaplan, B. Forbush III and J. F. Hoffman, Biochemistry, 1978, 17, 1929–1935 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- L. N. Lameijer, D. Ernst, S. L. Hopkins, M. S. Meijer, S. H. C. Askes, S. E. Le Dévédec and S. Bonnet, Angew. Chem., Int. Ed., 2017, 129, 11707–11711 CrossRef.
- V. H. S. van Rixel, V. Ramu, A. B. Auyeung, N. Beztsinna, D. Y. Leger, L. N. Lameijer, S. T. Hilt, S. E. Le Dévédec, T. Yildiz, T. Betancourt, M. B. Gildner, T. W. Hudnall, V. Sol, B. Liagre, A. Kornienko and S. Bonnet, J. Am. Chem. Soc., 2019, 141, 18444–18454 CrossRef CAS PubMed.
- I. M. Welleman, M. W. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymański, Chem. Sci., 2020, 11, 11672–11691 RSC.
- V. H. Priv-Doz, J. B. Doz, S. F. Priv-Doz, T. E. Dipl-Chem, S. Helm, D. Reuter and U. B. Kaupp, Angew. Chem., Int. Ed., 2001, 40, 1045–1048 CrossRef.
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed.
- P. P. Goswami, A. Syed, C. L. Beck, T. R. Albright, K. M. Mahoney, R. Unash, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2015, 137, 3783–3786 CrossRef CAS PubMed.
- T. Slanina, P. Shrestha, E. Palao, D. Kand, J. A. Peterson, A. S. Dutton, N. Rubinstein, R. Weinstain, A. H. Winter and P. Klán, J. Am. Chem. Soc., 2017, 139, 15168–15175 CrossRef CAS PubMed.
- J. A. Peterson, C. Wijesooriya, E. J. Gehrmann, K. M. Mahoney, P. P. Goswami, T. R. Albright, A. Syed, A. S. Dutton, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 7343–7346 CrossRef CAS PubMed.
- P. Shrestha, K. C. Dissanayake, E. J. Gehrmann, C. S. Wijesooriya, A. Mukhopadhyay, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2020, 142, 15505–15512 CrossRef CAS PubMed.
- R. R. Nani, A. P. Gorka, T. Nagaya, H. Kobayashi and M. J. Schnermann, Angew. Chem., Int. Ed., 2015, 54, 13635–13638 CrossRef CAS PubMed.
- H. Janeková, M. Russo, U. Ziegler and P. Štacko, Angew. Chem., Int. Ed., 2022, 61, e202204391 CrossRef PubMed.
- A. R. Sekhar, Y. Chitose, J. Janoš, S. I. Dangoor, A. Ramundo, R. Satchi-Fainaro, P. Slavíček, P. Klán and R. Weinstain, Nat. Commun., 2022, 13, 3614 CrossRef CAS PubMed.
- A. Egyed, K. Németh, T. Á. Molnár, M. Kállay, P. Kele and M. Bojtár, J. Am. Chem. Soc., 2023, 145, 4026–4034 CrossRef CAS PubMed.
- N. Ieda, Y. Hotta, N. Miyata, K. Kimura and H. Nakagawa, J. Am. Chem. Soc., 2014, 136, 7085–7091 CrossRef CAS PubMed.
- H. Okuno, N. Ieda, Y. Hotta, M. Kawaguchi, K. Kimura and H. Nakagawa, Org. Biomol. Chem., 2017, 15, 2791–2796 RSC.
- N. Ieda, Y. Oka, T. Yoshihara, S. Tobita, T. Sasamori, M. Kawaguchi and H. Nakagawa, Sci. Rep., 2019, 9, 1430 CrossRef PubMed.
- N. Ieda, Y. Hotta, A. Yamauchi, A. Nishikawa, T. Sasamori, D. Saitoh, M. Kawaguchi, K. Kimura and H. Nakagawa, ACS Chem. Biol., 2020, 15, 2958–2965 CrossRef CAS PubMed.
- C. Sundararajan and D. E. Falvey, J. Org. Chem., 2004, 69, 5547–5554 CrossRef CAS PubMed.
- C. Sundararajan and D. E. Falvey, J. Am. Chem. Soc., 2005, 127, 8000–8001 CrossRef CAS PubMed.
- C. Sundararajan and D. E. Falvey, Org. Lett., 2005, 7, 2631–2634 CrossRef CAS PubMed.
- K. A. Korzycka, P. M. Bennett, E. J. Cueto-Diaz, G. Wicks, M. Drobizhev, M. Blanchard-Desce, A. Rebane and H. L. Anderson, Chem. Sci., 2015, 6, 2419–2426 RSC.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- M. del Río, F. Lobo, J. C. López, A. Oliden, J. Bañuelos, I. López-Arbeloa, I. Garcia-Moreno and A. M. Gómez, J. Org. Chem., 2017, 82, 1240–1247 CrossRef PubMed.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, J. Org. Chem., 2011, 76, 7056–7064 CrossRef CAS PubMed.
- Y. Gabe, T. Ueno, Y. Urano, H. Kojima and T. Nagano, Anal. Bioanal. Chem., 2006, 386, 621–626 CrossRef CAS PubMed.
- T. Matsumoto, Y. Urano, T. Shoda, H. Kojima and T. Nagano, Org. Lett., 2007, 9, 3375–3377 CrossRef CAS PubMed.
- N. Ieda, Y. Yoshikawa, N. Tomita, K. Ohkubo, Y. Hotta, M. Kawaguchi, K. Kimura and H. Nakagawa, Chem. Commun., 2022, 58, 8420–8423 RSC.
- S. S. Simons, Jr. and D. F. Johnson, J. Am. Chem. Soc., 1976, 98, 7098–7099 CrossRef.
- C. G. Hatchard and C. A. Parker, Proc. Roy. Soc. Lond. Math. Phys. Sci., 1956, 235, 518–536 CAS.
- N. Umeda, H. Takahashi, M. Kamiya, T. Ueno, T. Komatsu, T. Terai, K. Hanaoka, T. Nagano and Y. Urano, ACS Chem. Biol., 2014, 9, 2242–2246 CrossRef CAS PubMed.
- M. H. Costell, N. Ancellin, R. E. Bernard, S. Zhao, J. J. Upson, L. A. Morgan, K. Maniscalco, A. R. Olzinski, V. L. T. Ballard, K. Herry, P. Grondin, N. Dodic, O. Mirguet, A. Bouillot, F. Gellibert, R. W. Coatney, J. J. Lepore, B. M. Jucker, L. J. Jolivette, R. N. Willette, C. G. Schnackenberg and D. J. Behm, Front. Pharmacol, 2012, 3, 128 CAS.
- A. L. Tsai, V. Berka, I. Sharina and E. Martin, J. Biol. Chem., 2011, 286, 43182–43192 CrossRef CAS PubMed.
- T. Kaneda, N. Sasaki, N. Urakawa and K. Shimizu, Pharmacology, 2016, 97, 171–176 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03826b |
| This journal is © The Royal Society of Chemistry 2023 |