
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoselective Nozaki–Hiyama–Takai–Kishi and Grignard reaction: short synthesis of some carbahexopyranoses†
Allam Vinaykumar*a,
Banothu Surenderb and
Batchu Venkateswara Rao *b
*b
aFluoro-Agrochemicals, CSIR-Indian Institute of Chemical Technology, Hyderabad, India. E-mail: vinaykumar@iict.res.in
bOrganic Synthesis and Process Chemistry, CSIR-Indian Institute of Chemical Technology, Hyderabad, India. E-mail: drb.venky@gmail.com
First published on 28th July 2023
Abstract
A common, divergent, efficient, stereoselective and short approach for the total syntheses of some carbahexopyranoses namely, MK7607, (−)-gabosine A, (−)-conduritol E, (−)-conduritol F, 6a-carba-β-D-fructopyranose and other carbasugars using chemoselective Grignard or Nozaki–Hiyama–Takai–Kishi (NHTK) reactions and RCM. Herein, the Grignard and NHTK reactions are able to differentiate the reactivity difference between lactol or lactolacetate and aldehyde of 2 & 6 under given conditions to give the desired skeleton chemoselectivity.
Introduction
Carbasugars, the carbocyclic analogues of sugars, constitute an important class of natural products. These carbasugars were stable to enzymatic hydrolysis in biological systems and display a diverse range of biological activities.1 Their inhibition activity has enormous potential for the treatment of many diseases.2 Carbohydrates are involved in various cellular signaling pathways, therefore there is a great interest in synthesis and biological exploration of carbasugars and their synthetic analogues. Many carbasugar-type natural products have polyoxygenated methyl or hydroxymethyl cyclohexane as a common structural feature, which are classified under C7-cyclitols. Gabosines belongs to this class and they are secondary metabolites isolated from several Streptomyces strains and also known as ketocarbasugars. They are known to display a wide range of interesting bioactivities, such as antibiotic,3 anticancer,4 inhibition of cholesterol biosynthesis,3b and DNA binding properties.5 Gabosine derivatives are also considered as very potent and emerging antitumor agents due to their glutathione S-transferases (GST) inhibition activity.6 In addition gabosine related derivatives have been used as intermediates for the synthesis of biologically active compounds such as an L-fucosyltransferase inhibitor,7a valienamine and its derivatives,7b and pseudo sugar C-disaccharides.7c Some secondary metabolites with the gabosine structural pattern were isolated from natural sources and shown to have important biological activity. These are MK7607, streptol (also named valienol), uvamalol A and uvacalol A (Fig. 1).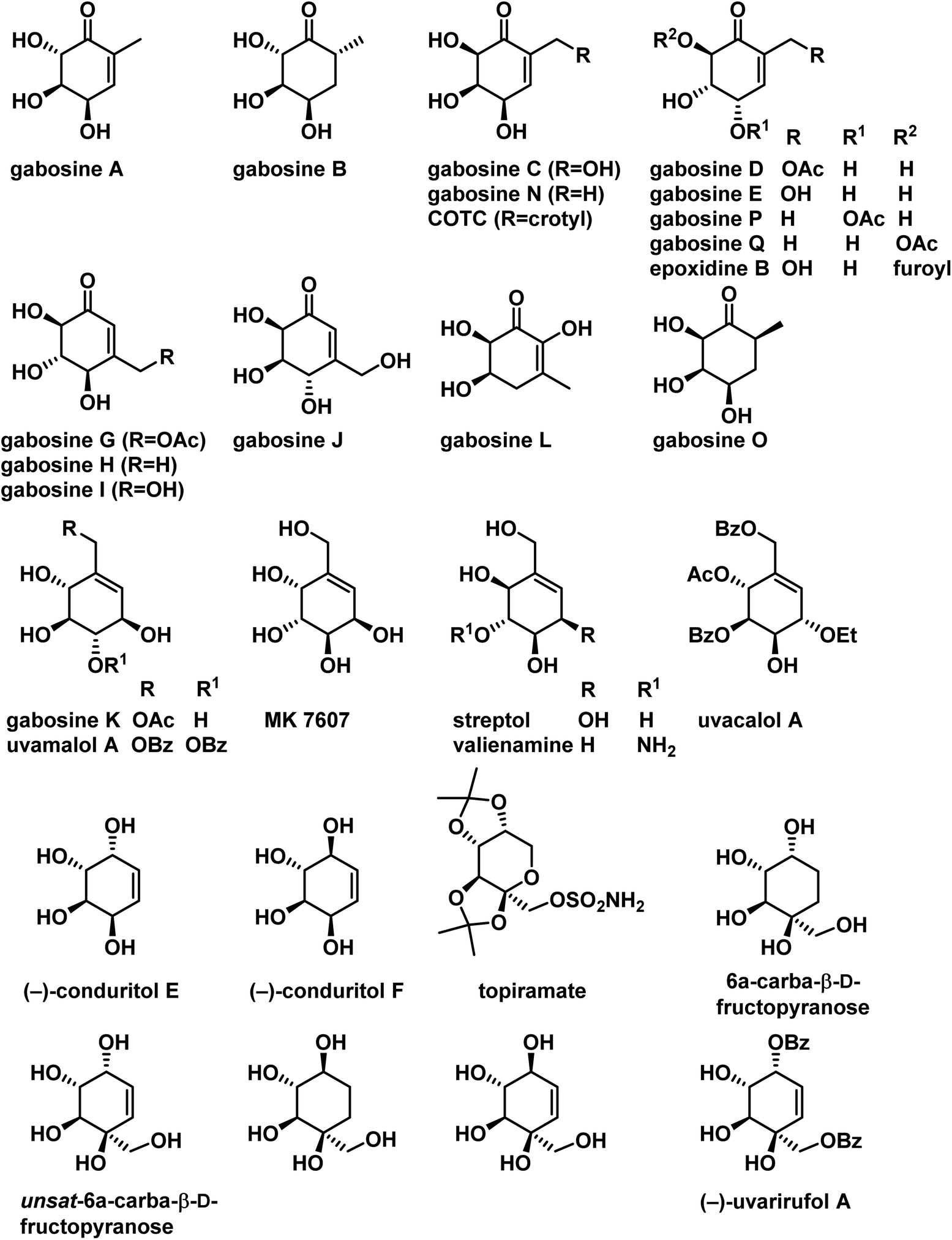 | ||
| Fig. 1 Some of the naturally occurring carbasugars belongs to gabosine family and its related compounds. | ||
Another interesting class of cyclitols are conduritols or 1,2,3,4 cyclohexene tetrols. Several conduritol derivatives possess antifeedant, antibiotic, antileukemic, and growth-regulating activity.8 Conduritol epoxides and aminoconduritols act as inhibitors of glycosidases,9 Also, conduritols have been widely used as intermediates in chemical syntheses of inositols,10 deoxyinositols,11 conduritol epoxides,9a,c aminoconduritols,9a,c,12 cyclophellitol,13 pseudosugars,14 amino sugar analogs,15 etc. Some more interesting compounds under this class are 6a-carba-β-D-fructopyranose which could be used as a non-nutritive sweetener since it has a sweet taste16 which is also active carba-isostere of topiramate, a useful antiepileptic drug.17 (−)-Uvarirufol A isolated from Uvariarufa and similar polyhydroxy cyclohexene derivatives showed a wide range of biological activities such as anti-tumor, anti-malarial, anti-leukemic, and pesticidal.18
The structural diversity and promising biological activities of these molecules have triggered several approaches for their synthesis. Synthesis of gabosines was reviewed by Mac19a and co-workers. The isolation, structural determination, biogenetic studies, biological evaluation, and synthesis of gabosines and related molecules have been reviewed by Bayon and Figueredo.19b Later, recently some novel interesting approaches for these molecules have been reported.20 In continuation of our interest in this area, the structural diversity and promising biological activities of MK7607, (−)-gabosine A, (−)-conduritol E, (−)-conduritol F, 6a-carba-β-D-fructopyranose and other carbasugars motivated us to develop a common strategy for their synthesis. These molecules have a common stereochemical arrangement, therefore a common strategy was envisaged for their synthesis.
(−)-Gabosine A was isolated from the Streptomyces strain,3a,b shows DNA binding activity and it is an important synthetic target.20b,c,21 (+)-MK7607 is a natural isomer, an α-galactose mimic, isolated from the culture of Curvularia eragrostidis D2452 and was found to have effective herbicidal and antimicrobial activities. The first total synthesis of MK7607 was reported in racemic form by Mehta et al.22a in 2000 from norbornane system. Later, many synthetic chemists focused their attention for the synthesis of (+)-MK7607,22a (−)-MK7607 (ref. 20b and 20c) and its epimers.20b,22 The synthesis of (−)-MK7607 was reported by Shing et al.22c from (−)-shikimic acid and Sureshan et al.20b from D-pinitol. The synthesis of conduritol from cyclohexadiene-cis-1,2-diols have been accomplished in both racemic and enantioselective manner. Hudlicky group reported the enantiocontrolled synthesis of conduritols (+)-E and (−)-F from bromobenzene in 1991.23a Next year, Carless23b applied a similar sequence of chemical transformations to reach conduritol E and reported optical rotation for (−)-conduritol E and pointed out the erroneous assignment in the Hudlicky synthesis. Also, some enantiopure conduritols have been prepared by employing chiral starting materials such as carbohydrates23c and diethyl L-tartrate.23d
Enantiomerically pure carba-β-D-fructopyranose has been synthesized previously from a chemically resolved Diels–Alder adduct of furan and acrylic acid,24a also from (−)-quinic acid,24b L-arabinose24c and an enzymatically resolved homochiral building block derived from cyclohexene.24d Additionally, the enantiomer of carba-β-D-fructopyranose ie 6a-carbab-L-fructopyranose has been targeted by different groups.24e,f So far there is no report on unsaturated analog of carba-β-D-fructopyranose 30 and other carbasugars 31 & 35. Only few reports are present on enantiomer of 30 i.e. unsaturated analog of carba-β-L-fructopyranose from microbial oxidation of benzoic acid24c and chiral starting material (−)-shikimic acid.24f Herein, we would like to present the chemoselective nucleophilic addition methodology for the synthesis of above molecules. Our approach is a short and efficient which involves chemoselective Grignard and NHTK reactions from a single common intermediate. Our basic strategy is depicted in Scheme 1.
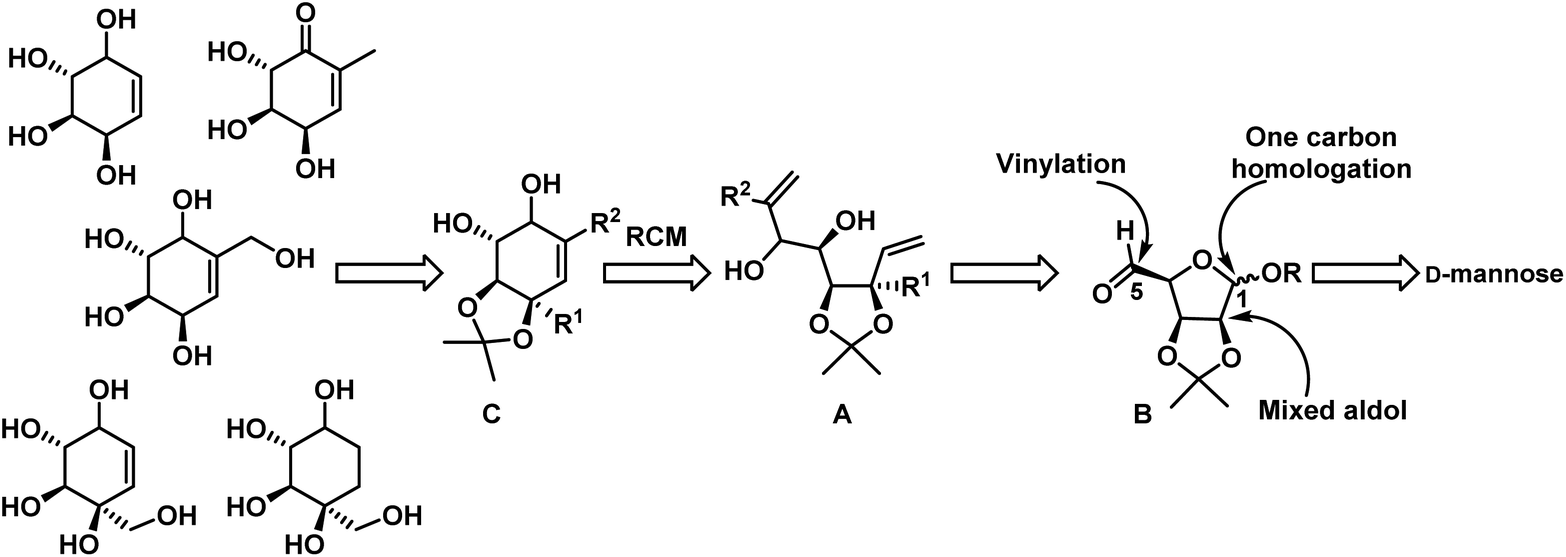 | ||
| Scheme 1 Retrosynthetic analysis. | ||
To synthesize RCM precursor A, from D-mannose in a classical way generally we need to carry protection and deprotection steps at C-1 and C5 positions. To minimize these steps in the synthesis and to avoid harsh conditions in deprotection steps, we envisaged a chemoselective NHTK reaction and vinyl Grignard addition on C-5 aldehyde of B in presence of hemiacetal followed by one carbon homologation at C-1 will give the required diene precursor for RCM. This can be elongated to the cyclohexene core C, a key intermediate for the synthesis of target molecules.
As per our strategy, we started our synthesis from D-mannose which is a cheap and commercially available. D-Mannose was converted to diol 1 using reported method25a in 65% overall yield. When diol 1 was subjected to oxidative degradation with NaIO4 in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (4:1) for 0.5 h afforded compound 2 (Scheme 2). The next step is the chemoselective nucleophilic addition at C-5 aldehyde in presence of anomeric acetate. Later the anomeric acetate of the resultant adduct can be easily hydrolysed to lactol and Wittig reaction of the resultant lactol should give the diene for RCM reaction.
:H2O (4:1) for 0.5 h afforded compound 2 (Scheme 2). The next step is the chemoselective nucleophilic addition at C-5 aldehyde in presence of anomeric acetate. Later the anomeric acetate of the resultant adduct can be easily hydrolysed to lactol and Wittig reaction of the resultant lactol should give the diene for RCM reaction.
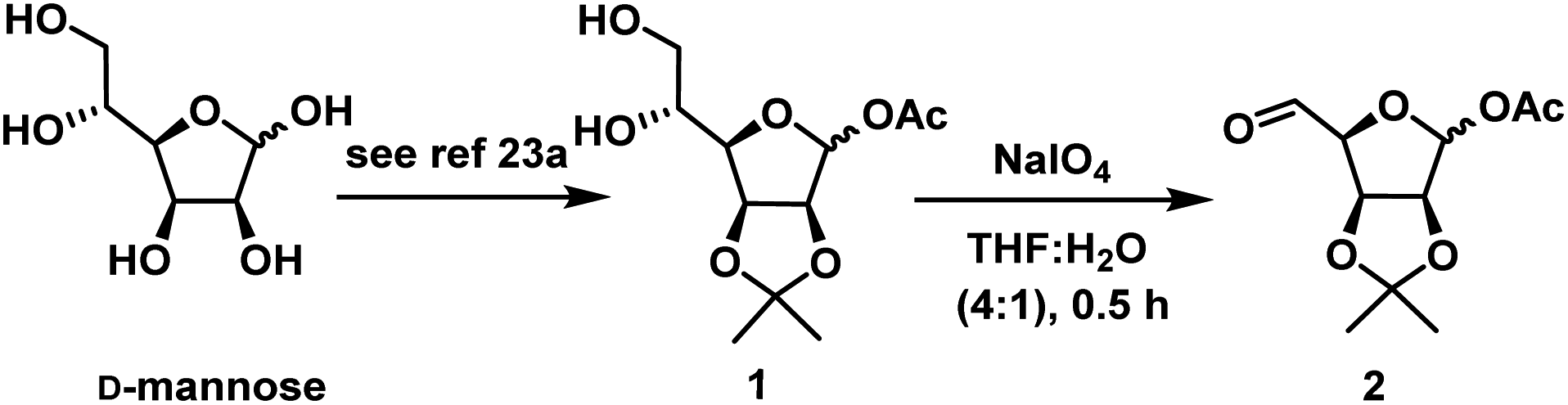 | ||
| Scheme 2 Construction of acetate aldehyde intermediate 2. | ||
Chemoselective vinyl Grignard addition on intermediate 2
As mentioned above, the compound 2 has two functional groups which are reactive towards Grignard reagents one is free aldehyde and the other is anomeric acetate. It was felt that the selectivity can be achieved by reducing the reagent reactivity and minimizing its concentration in the reaction medium. After studying some conditions, it was found that the slow addition of Grignard reagent (1 equiv.) to 2 at −78 °C for 1 h gave the desired products in good yields.Vinylmagnesium bromide addition gave a mixture of ene compounds 3a & 3b as an inseparable mixture in the ratio of 1.5:1 (anti:syn) in 51% yield. When compound 2 was subjected to isopropenylmagnesium bromide for the synthesis of (−)-gabosine A under the above-mentioned conditions gave the mixture of ene compounds 4a & 4b as an inseparable mixture in the ratio of 2.5:1 (anti:syn) in 68% yield (Scheme 3).
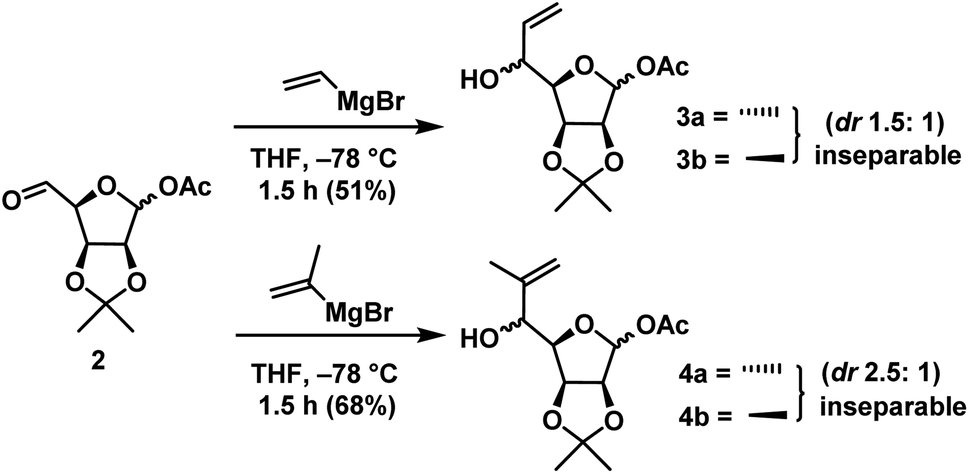 | ||
| Scheme 3 Chemoselective vinyl Grignard addition on intermediate 2. | ||
A possible chelation controlled transition state for the Grignard addition was given in Fig. 2. Where one face is blocked by the acetonide group. Also it was found that the structurally bulky isopropenyl group gave better facial selection when compared to vinyl.
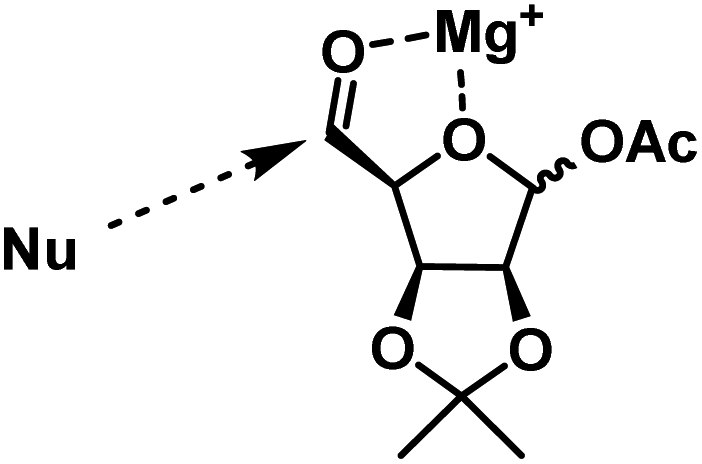 | ||
| Fig. 2 Chelation controlled transition state.The configuration of the newly generated chiral centers in 3a & 3b were confirmed after completing the synthesis of target molecules and comparing the spectral data with the reported data. | ||
For the synthesis of (−)-MK7607, to introduce ‘3C’ unit the compound 7 having protected hydroxymethyl part is not a practical choice for Grignard reaction. Therefore, it was thought to use NHTK reaction for the chemoselective introduction of 3C unit. The vinylchromium reagent generated in NHTK reaction is less reactive when compared to Grignard reagent. Therefore, it was further envisaged that on the substrate like 6 having aldehyde and lactol, the vinyl chromium should able to add chemoselectivity on the aldehyde. This strategy further eliminates the protection and deprotection steps at anomeric positions.For this, D-mannose was converted to diol 5 using the reported literature method25b in two steps in 50% overall yield. Compound 5 on oxidative cleavage with NaIO4 in THF and H2O in 4:1 ratio at 0 °C gave the aldehyde 6 (Scheme 4).
 | ||
| Scheme 4 Construction of hemiacetal aldehyde intermediate 6. | ||
Compound 6 has two aldehyde groups, one is in free form at C5 and the other is in hemiacetal form at C1. When we treat the compound 6 with strong nucleophiles, such as alkyl lithium, magnesium, copper and other organometallic compounds may result in C–C bond formation at both the places. The NHTK reaction26 is a very mild and highly chemoselective method for forming carbon–carbon bonds27 with aldehydes in natural product synthesis. The NHTK reaction shows exceptional chemoselectivity in nucleophilic addition towards aldehydes over other functional groups. But to the best of our knowledge, we haven't find chemo selectivity addition of NHTK nucleophile on aldehydes in presence of lactol as in 6. When, we carried out NHTK reaction on compound 6, the addition of alkenyl chromium nucleophile (generated from 7)28 has taken place selectively on the free aldehyde to afford the alkenes 8 and 9 in the ratio of 4:1 (anti:syn) in 65% yield (from NMR) for two steps (Fig. 3). This is due to pronounced chemoselectivity and poor nucleophilicity of organochromium reagents. In NHTK reaction, the nucleophilic addition has taken place in a nonchelation mode due to the weak chelating property of chromium salt to give anti isomer 8 as the major. Only major isomer 8 was separated by column chromatography, whereas the minor isomer 9 was not separated in pure form, every time it was eluted as a mixture along with major isomer (Scheme 5). In order to see validity again we tried with 2-bromopropene under NHTK condition on compound 6 for the synthesis of (−)-gabosine A which gave 10a and 10b as an inseparable mixture in the ratio of 4:1 (anti:syn) in 69% yield for two steps.
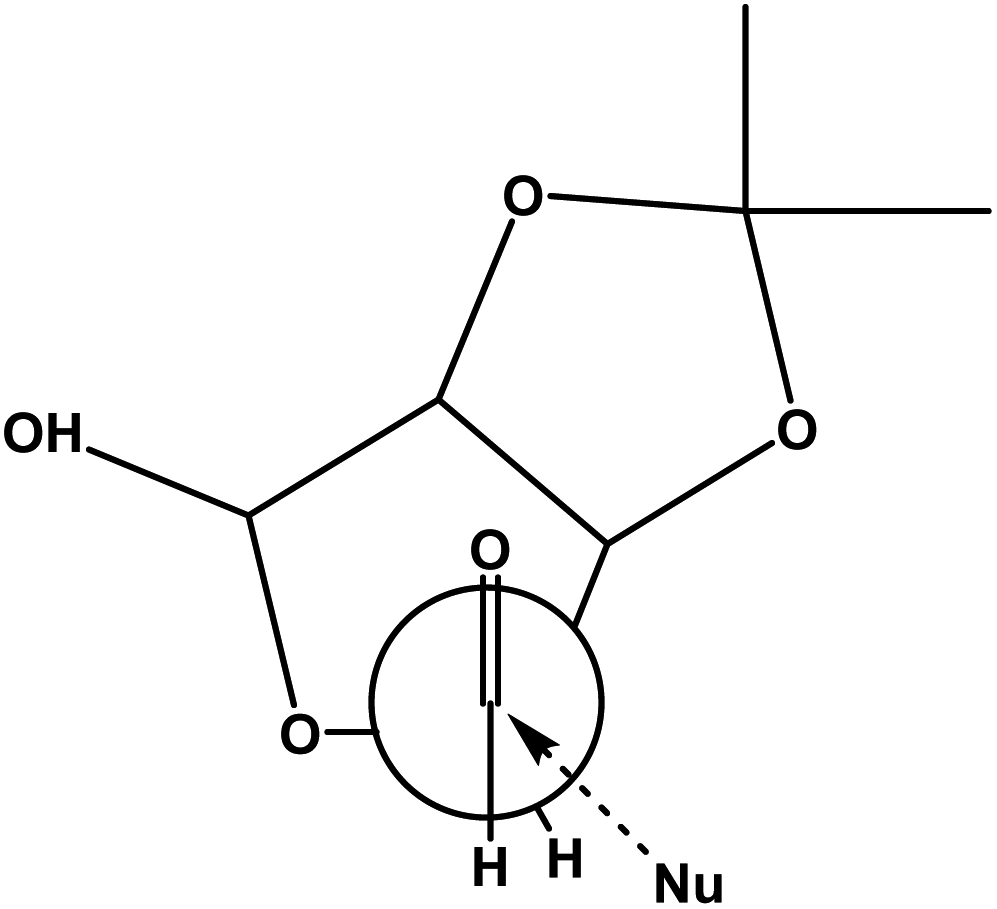 | ||
| Fig. 3 Felkin–Anh model where nucleophile is organochromium. | ||
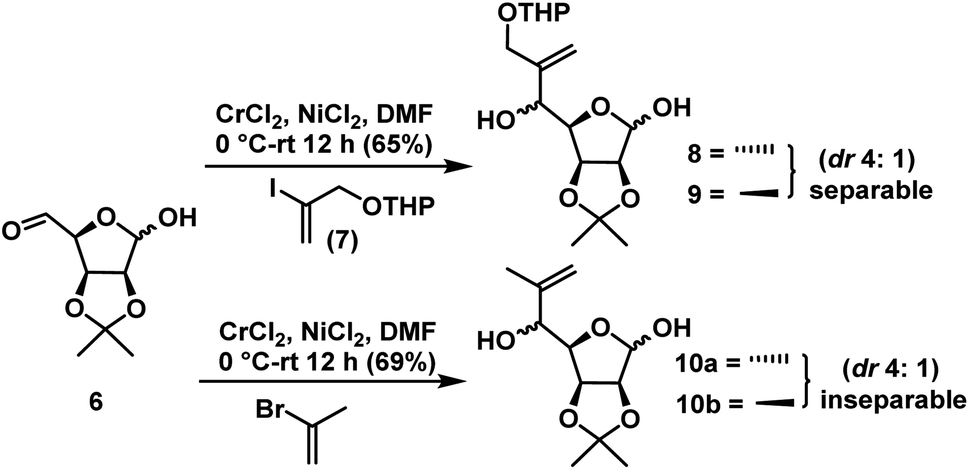 | ||
| Scheme 5 Chemoselective organochromium addition on intermediate 6. | ||
The major anti27a isomer was further confirmed after completing the synthesis of (−)-MK7607 2 by comparing the spectral data with the reported one.
The next step is to utilize addition products for the synthesis of target compounds. To make diene precursor for RCM for the synthesis of (−)-MK7607 one-carbon homologation of lactol 8 using Wittig olefination29 afforded the terminal olefin compound 11 in 76% yield. Ring-closing metathesis reaction was carried out on 11 using Grubbs II generation catalyst28 to give substituted cyclohexene 12 in 85% yield. Finally, deprotection of acetonide in 12 under acidic conditions gave the target molecule (−)-MK7607 in 70% yield (Scheme 6).
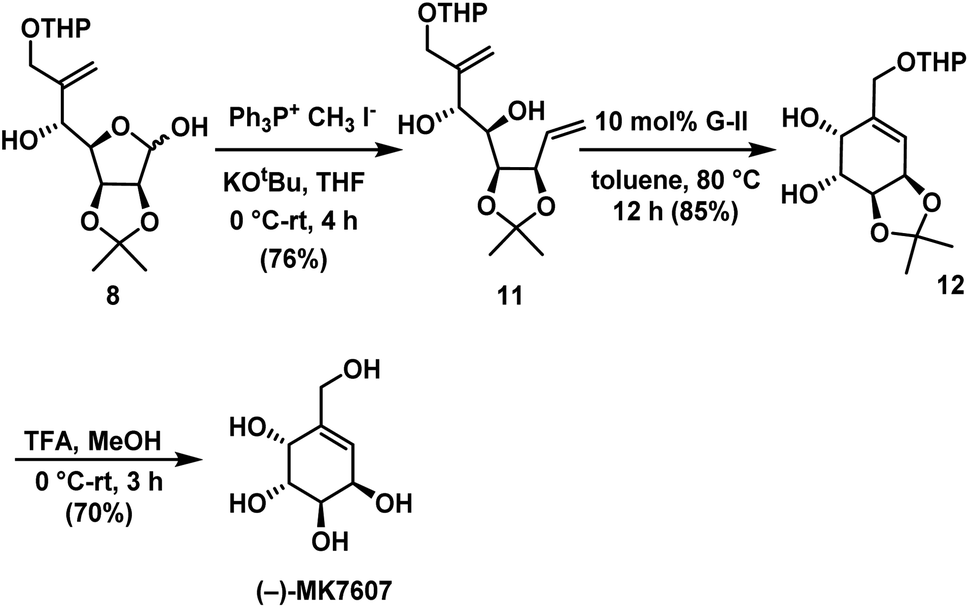 | ||
| Scheme 6 Synthesis of (−)-MK7607. | ||
One carbon Wittig olefination on the mixture of 10a & 10b afforded the diolefins compounds 13a & 13b as an inseparable mixture in the ratio of 4:1 in 82% yield. Ring-closing metathesis was carried out on a mixture of 13a & 13b using Grubbs II generation catalyst30 to give substituted cyclohexenes as an inseparable mixture of 14a & 14b in the ratio of 4:1 in 85% yield. Allylic alcohol was selectively oxidized to ketone in compound 14a & 14b with DMP in DCM for a period of 1 h to give ketone compound 15. Deprotection of acetonide in 15 under acidic conditions gave the target molecule (−)-gabosine A in 80% yield (Scheme 7).
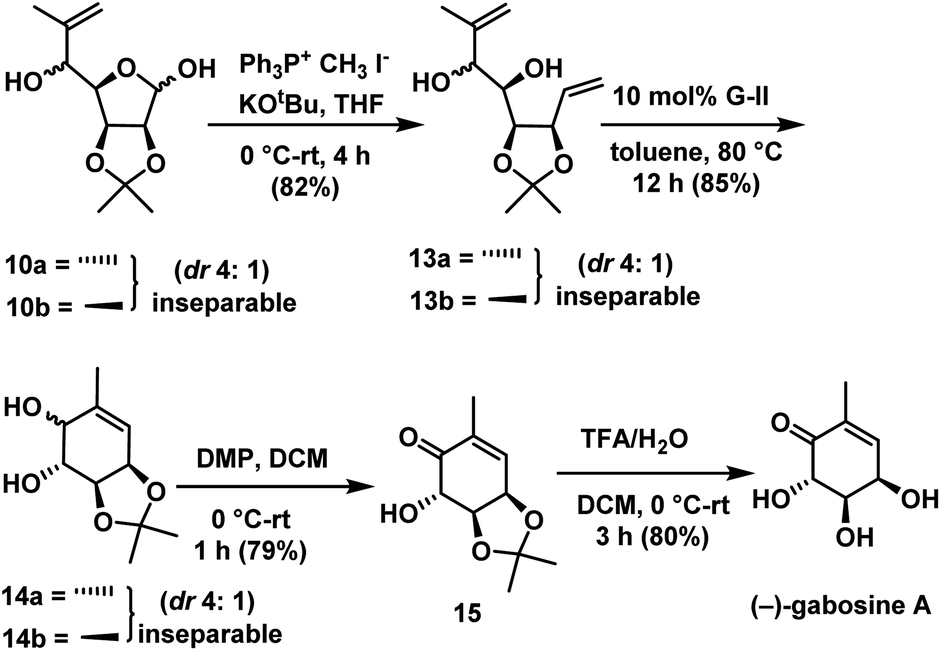 | ||
| Scheme 7 Synthesis of (−)-gabosine A. | ||
Deacylation in compounds 4a & 4b was done with K2CO3 in MeOH to give the inseparable mixture of hydroxyl compounds 10a & 10b in the ratio of 2.5:1 (anti:syn) in 91% yield. Then mixture was carried to the synthesis of (−)-gabosine A as shown in Scheme 7.
Deacylation of compounds 3a & 3b gave lactol 16 (52.5%) & 17 (37.5%) as separable diastereomers in the ratio of 1.5:1 (anti:syn). Wittig olefination on compounds 16 & 17 independently gave dienes 18 (90%) & 19 (88%). Next we focused our attention on its ring-closing olefin metathesis. Compounds 18 and 19 were subjected to RCM using Grubbs 2nd generation catalyst30 independently to give cyclohexenes 20 (72%) and 21 (80%). Finally, removal of protecting groups in 20 & 21 was achieved independently by treating with TFA/H2O in methanol, for a period of 3 h to give the (−)-conduritol E (96%) and (−)-conduritol F (96%) (Scheme 9), whose spectral data were in good agreement with the reported data.
For the synthesis of 6a-carba-β-D-fructopyranose & other three carbasugars we need to introduce hydroxymethyl unit. For this, compounds 16 & 17 were subjected to the mixed aldol reaction31 independently with excess 37% aqueous formaldehyde to give the 1,3 diols 22 (85%) & 23 (80%). To make diene precursor, we tried Wittig olefination on compounds 22 & 23 which failed to give the desired products and every time the starting materials were recovered. The inertness of substrates 22 & 23 towards Wittig olefination even with excess reagent might be due to the presence of a free hydroxyl group adjacent to the lactol (see Table 1). To circumvent this, we protected the primary alcoholic group in 22 & 23 as TBDPS ether to get 24 & 25 by treating with TBDPS-Cl, imidazole, and DMAP (cat.) for 6 h in 86% & 88% yields respectively (Scheme 8). Wittig olefination32 under forced conditions on compounds 24 & 25 independently gave the dienes 26 (86%) & 27 (88%).
| Compound | Wittig salt (equivalents) | Potassium tert-butoxide (equivalents) | Temp (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 22 | 6 | 5 | rt | 12 | nr |
| 23 | 6 | 5 | rt | 12 | nr |
| 24 | 6 | 5 | 100 | 3 | 86 |
| 25 | 6 | 5 | 100 | 4 | 88 |
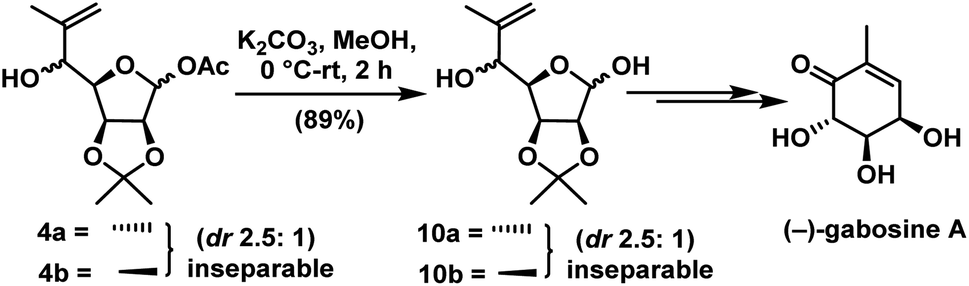 | ||
| Scheme 8 Synthesis of (−)-gabosine A. | ||
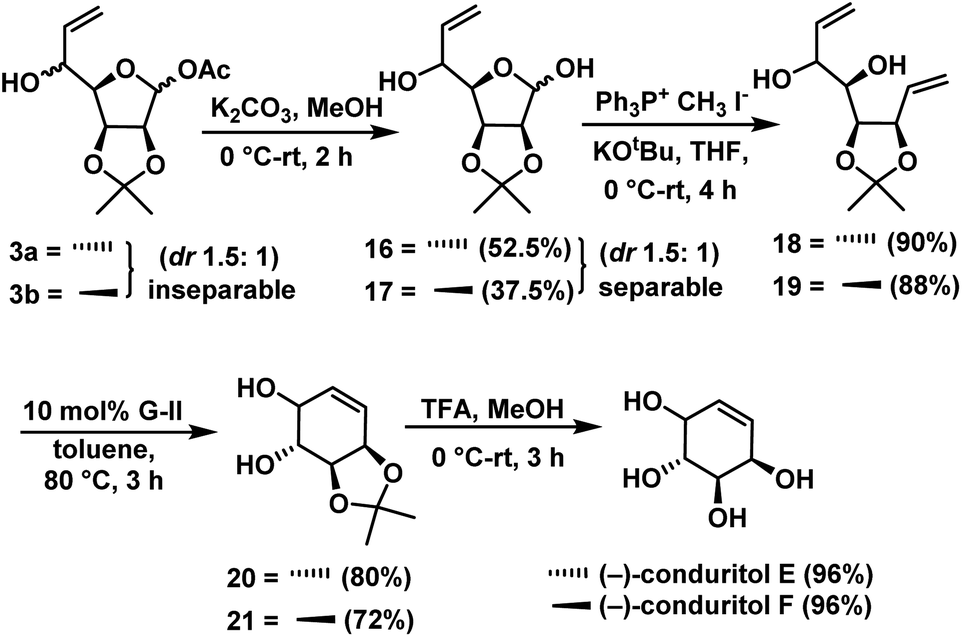 | ||
| Scheme 9 Synthesis of (−)-conduritol E and (−)-conduritol F. | ||
Compounds 26 and 27 were subjected to RCM using Grubbs 2nd generation catalyst independently to give cyclohexene intermediates 28 (80%) and 29 (75%). Finally, removal of protecting groups in cyclohexene derivative was achieved by treating the compounds 28 & 29 independently with TFA/H2O in methanol, for a period of 3 h to give the carbasugar 30 (90%) and carbasugar 31 (92%) (Scheme 10), whose spectral data were in good agreement with the reported data in the case of 30 and assigned structure in the case of 31.
 | ||
| Scheme 10 Synthesis of carbasugars 30 and 31. | ||
Hydrogenation of cyclohexene compounds 28 & 29 independently over palladium on activated carbon gave cyclohexane derivatives 32 (98%) & 33 (95%). Finally, deprotection of the acetonide in 32 & 33 with TFA/H2O in methanol for a period of 3 h afforded compound 6a-carba-β-D-fructopyranose 34 (98%) and carbasugar 35 (95%) (Scheme 11), whose spectral and physical data were in agreement with the reported data in the case of 34 and assigned structure in the case of 35.
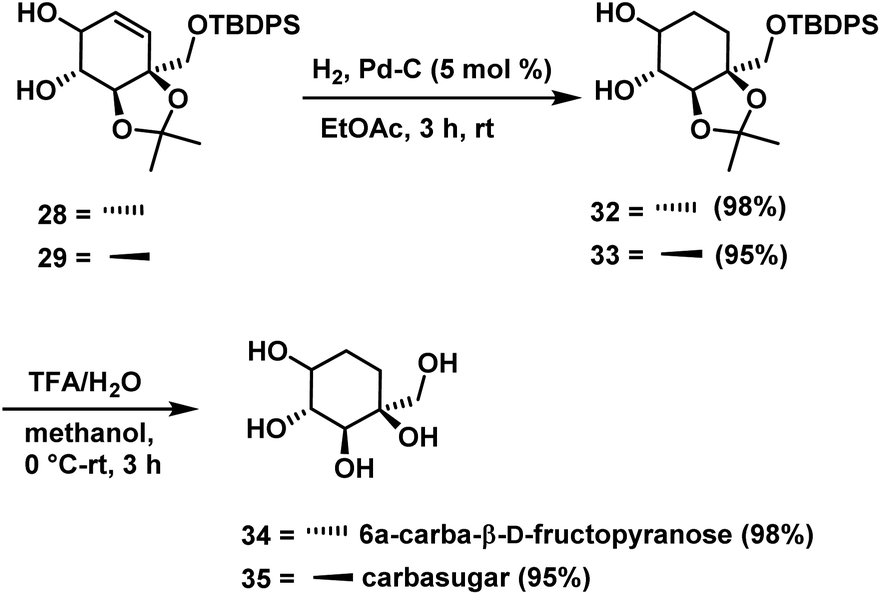 | ||
| Scheme 11 Synthesis of 6a-carba-β-D-fructopyranose 34 and carbasugar 35. | ||
Conclusions
We have successfully developed a common and short strategy for the synthesis of MK7607, (−)-gabosine A, (−)-conduritol E, (−)-conduritol F, 6a-carba-β-D-fructopyranose 34 and carbasugars 30, 31 & 35 using chemoselective nucleophilic addition (Grignard, NHTK) reaction and RCM. To best of our knowledge there is no report so far such type of chemoselectivity, where Grignard or NHTK reaction can differentiate the reactivity between lactol or lactolacetate and aldehyde. This approach is useful to make different carbasugars in a short time.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AVK thanks, the Council of Scientific and Industrial Research (CSIR), New Delhi for a Research Fellowship, BS thank the University Grants Commission for a Research Fellowship, and Dr B. V. Rao, Emeritus Scientist (CSIR), thanks CSIR-New Delhi for financial support under the CSIR-Emeritus Scientist Scheme (No. 21(1109)/20/EMR-II) and the Director of CSIR-IICT for constant support and encouragement. CSIR-IICT manuscript communication number: IICT/Pubs./2023/116.Notes and references
- (a) O. Arjona, A. M. Gómez, J. C. López and J. Plumet, Chem. Rev., 2007, 107(5), 1919–2036 CrossRef CAS PubMed; (b) M. Das and K. Manna, Curr. Bioact. Compd., 2015, 11(4), 239–248 CrossRef CAS; (c) J. Marco-Contelles, Eur. J. Org. Chem., 2001, 2001(9), 1607–1618 CrossRef; (d) M. D. Witte, W. W. Kallemeijn, J. Aten, K.-Y. Li, A. Strijland, W. E. Donker-Koopman, A. M. C. H. van den Nieuwendijk, B. Bleijlevens, G. Kramer and B. I. Florea, et al., Nat. Chem. Biol., 2010, 6(12), 907–913 CrossRef CAS PubMed; (e) W. W. Kallemeijn, K.-Y. Li, M. D. Witte, A. R. A. Marques, J. Aten, S. Scheij, J. Jiang, L. I. Willems, T. M. Voorn-Brouwer and C. P. A. A. van Roomen, et al., Angew. Chemie Int. Ed., 2012, 51(50), 12529–12533 CrossRef CAS PubMed; (f) S. Roscales and J. Plumet, Int. J. Carbohydr. Chem., 2016, 2016, 1–42 CrossRef.
- (a) N. Asano, Glycobiology, 2003, 13(10), 93R–104 CrossRef CAS PubMed; (b) E. Borges de Melo, A. da Silveira Gomes and I. Carvalho, Tetrahedron, 2006, 62(44), 10277–10302 CrossRef CAS.
- (a) T. Tsuchiya, N. Mikami, S. Umezawa, H. Umezawa and H. Naganawa, J. Antibiot., 1974, 27(8), 579–586 CrossRef PubMed; (b) G. Bach, S. Breiding-Mack, S. Grabley, P. Hammann, K. Hütter, R. Thiericke, H. Uhr, J. Wink and A. Zeeck, Liebigs Ann. Chem., 1993, 1993(3), 241–250 CrossRef.
- (a) C. F. M. Huntley, D. S. Hamilton, D. J. Creighton and B. Ganem, Org. Lett., 2000, 2(20), 3143–3144 CrossRef CAS PubMed; (b) D. Kamiya, Y. Uchihata, E. Ichikawa, K. Kato and K. Umezawa, Bioorg. Med. Chem. Lett., 2005, 15(4), 1111–1114 CrossRef CAS PubMed.
- (a) Y.-Q. Tang, C. Maul, R. Höfs, I. Sattler, S. Grabley, X.-Z. Feng, A. Zeeck and R. Thiericke, Eur. J. Org. Chem., 2000, 2000(1), 149–153 CrossRef; (b) A. Maier, C. Maul, M. Zerlin, S. Grabley and R. Thiericke, J. Antibiot., 1999, 52(11), 952–959 CrossRef CAS PubMed.
- (a) T. K. M. Shing, H. T. Wu, H. F. Kwok and C. B. S. Lau, Bioorg. Med. Chem. Lett., 2012, 22(24), 7562–7565 CrossRef CAS PubMed; (b) C.-H. Wang, H. T. Wu, H. M. Cheng, T.-J. Yen, I.-H. Lu, H. C. Chang, S.-C. Jao, T. K. M. Shing and W.-S. Li, J. Med. Chem., 2011, 54(24), 8574–8581 CrossRef CAS PubMed.
- (a) S. Cai, M. R. Stroud, S. Hakomori and T. Toyokuni, J. Org. Chem., 1992, 57(25), 6693–6696 CrossRef CAS; (b) S. Horii, H. Fukase, T. Matsuo, Y. Kameda, N. Asano and K. Matsui, J. Med. Chem., 1986, 29(6), 1038–1046 CrossRef CAS PubMed; (c) C. Barbaud, M. Bols, I. Lundt and M. R. Sierks, Tetrahedron, 1995, 51(33), 9063–9078 CrossRef CAS.
- (a) A. Kornienko and A. Evidente, Chem. Rev., 2008, 108(6), 1982–2014 CrossRef CAS; (b) M. Gultekin, M. Celik and M. Balci, Curr. Org. Chem., 2004, 8(13), 1159–1186 CrossRef.
- (a) M. Balci, Pure Appl. Chem., 1997, 69(1), 97–104 CrossRef CAS; (b) T. Hudlicky, D. A. Entwistle, K. K. Pitzer and A. Thorpe, Jpn. Chem. Rev., 1996, 96(3), 1195–1220 CrossRef CAS PubMed; (c) M. Balci, Y. Sütbeyaz and H. Secen, Tetrahedron, 1990, 46(11), 3715–3742 CrossRef CAS.
- (a) L. E. Brammer and T. Hudlicky, Tetrahedron: Asymmetry, 1998, 9(12), 2011–2014 CrossRef CAS; (b) M. Desjardins, L. E. Brammer and T. Hudlicky, Carbohydr. Res., 1997, 304(1), 39–42 CrossRef CAS; (c) T. Hudlicky, M. Mandel, J. Rouden, R. S. Lee, B. Bachmann, T. Dudding, K. J. Yost and J. S. Merola, J. Chem. Soc. Perkin Trans. 1, 1994, 12, 1553 RSC; (d) M. Mandel and T. Hudlicky, J. Chem. Soc. Perkin Trans. 1, 1993, 7, 741 RSC.
- C. Le Drian, J.-P. Vionnet and P. Vogel, Helv. Chim. Acta, 1990, 73(1), 161–168 CrossRef CAS.
- (a) C. Sanfilippo, A. Patti, M. Piattelli and G. Nicolosi, Tetrahedron: Asymmetry, 1997, 8(10), 1569–1573 CrossRef CAS; (b) T. Hudlicky, H. Luna, H. F. Olivo, C. Andersen, T. Nugent and J. D. Price, J. Chem. Soc. Perkin Trans. 1, 1991, 12, 2907 RSC.
- (a) B. M. Trost and E. J. Hembre, Tetrahedron Lett., 1999, 40(2), 219–222 CrossRef CAS; (b) H. Takahashi, T. Iimori and S. Ikegami, Tetrahedron Lett., 1998, 39(38), 6939–6942 CrossRef CAS.
- (a) D. A. Entwistle and T. Hudlicky, Tetrahedron Lett., 1995, 36(15), 2591–2594 CrossRef CAS; (b) L. Pingli and M. Vandewalle, Tetrahedron, 1994, 50(24), 7061–7074 CrossRef CAS.
- T. Hudlicky, J. Rouden, H. Luna and S. Allen, J. Am. Chem. Soc., 1994, 116(12), 5099–5107 CrossRef CAS PubMed.
- T. Suami, S. Ogawa, M. Takata, K. Yasuda, A. Suga, K. Takei and Y. Uematsu, Chem. Lett., 1985, 14(6), 719–722 CrossRef.
- B. E. Maryanoff, S. O. Nortey, J. F. Gardocki, R. P. Shank and S. P. Dodgson, J. Med. Chem., 1987, 30(5), 880–887 CrossRef CAS PubMed.
- L. Yong-Hong, X. Li-Zhen, Y. Shi-Lin, D. Jie, Z. Yong-Su, Z. Min and S. Nan-Jun, Phytochemistry, 1997, 45(4), 729–732 CrossRef.
- (a) D. H. Mac, S. Chandrasekhar and R. Grée, Eur. J. Org. Chem., 2012, 2012(30), 5881–5895 CrossRef CAS; (b) P. Bayón and M. Figueredo, Chem. Rev., 2013, 113(7), 4680–4707 CrossRef.
- (a) X. Liang, Y. Zhao, X. Si, M. Xu, J. Tan, Z. Zhang, C. Zheng and C. Zheng, Angew. Chemie Int. Ed., 2019, 58(41), 14562–14567 CrossRef CAS; (b) S. Mondal and K. M. Sureshan, J. Org. Chem., 2016, 81(23), 11635–11645 CrossRef CAS PubMed; (c) X. Yang, P. Yuan, F. Shui, Y. Zhou and X. Chen, Org. Biomol. Chem., 2019, 17(16), 4061–4072 RSC; (d) W. Ren, R. Pengelly, M. Farren-Dai, S. Shamsi Kazem Abadi, V. Oehler, O. Akintola, J. Draper, M. Meanwell, S. Chakladar and K. Świderek, et al., Nat. Commun., 2018, 9(1), 3243 CrossRef PubMed; (e) N. Biduś, P. Banachowicz and S. Buda, Tetrahedron, 2020, 76, 131397 CrossRef; (f) Z. Zhang, Y. Zhou and X.-W. Liang, Org. Biomol. Chem., 2020, 26(6), 5558–5566 RSC; (g) P. Banachowicz and S. Buda, RSC Adv., 2019, 9(23), 12928–12935 RSC.
- (a) M. G. Banwell, A. M. Bray and D. J. Wong, New J. Chem., 2001, 25, 1351–1354 RSC; (b) D. H. Mac, R. Samineni, A. Sattar, S. Chandrasekhar, J. S. Yadav and R. Grée, Tetrahedron, 2011, 67, 9305–9310 CrossRef CAS; (c) V. Kumar, P. Das, P. Ghosal and A. K. Shaw, Tetrahedron, 2011, 67, 4539–4546 CrossRef CAS.
- (a) G. Mehta and S. Lakshminath, Tetrahedron Lett., 2000, 41, 3509–3512 CrossRef CAS; (b) C. Lim, D. J. Baek, D. Kim, S. W. Youn and S. Kim, Org. Lett., 2009, 11, 2583–2586 CrossRef CAS PubMed; (c) C. Song, S. Jiang and G. Singh, Synlett, 2001, 2001, 1983–1985 CrossRef; (d) L. Fitjer and U. Quabeck, Synth. Commun., 1985, 15, 855–864 CrossRef CAS; (e) J. Sardinha, A. P. Rauter and M. Sollogoub, Tetrahedron Lett., 2008, 49, 5548–5550 CrossRef CAS.
- (a) T. Hudlicky, J. D. Price and H. F. Olivo, Synlett, 1991, 1991, 645–646 CrossRef; (b) H. A. J. Carless, Tetrahedron Lett., 1992, 33, 6379–6382 CrossRef CAS; (c) L. Ackermann, D. El Tom and A. Fürstner, Tetrahedron, 2000, 56, 2195–2202 CrossRef CAS; (d) W.-W. Lee and S. Chang, Tetrahedron: Asymmetry, 1999, 10, 4473–4475 CrossRef CAS.
- (a) S. Ogawa, Y. Uematsu, S. Yoshida, N. Sasaki and T. Suami, J. Carbohydr. Chem., 1987, 6, 471–478 CrossRef CAS; (b) T. K. M. Shing and Y. Tang, Tetrahedron, 1991, 47, 4571–4578 CrossRef CAS; (c) S. M. Totokotsopoulos, A. E. Koumbis and J. K. Gallos, Tetrahedron, 2008, 64, 3998–4003 CrossRef CAS; (d) S. M. Totokotsopoulos, A. E. Koumbis and J. K. Gallos, Synlett, 2000, 2000, 0971–0974 CrossRef; (e) M. H. Parker, B. E. Maryanoff and A. B. Reitz, Synlett, 2004, 2095–2098 CrossRef CAS; (f) C. Song, S. Jiang and G. Singh, Synlett, 2001, 2001, 1983–1985 CrossRef.
- (a) S. Huang, H. Yu and X. Chen, Angew. Chem., Int. Ed., 2007, 46, 2249–2253 CrossRef CAS; (b) J. Everson and M. J. Kiefel, J. Org. Chem., 2019, 84, 15226–15235 CrossRef CAS PubMed.
- (a) H. Jin, J. Uenishi, W. J. Christ and Y. Kishi, J. Am. Chem. Soc., 1986, 108, 5644–5646 CrossRef CAS; (b) K. Takai, M. Tagashira, T. Kuroda, K. Oshima, K. Utimoto and H. Nozaki, J. Am. Chem. Soc., 1986, 108, 6048–6050 CrossRef CAS PubMed.
- (a) A. Fürstner, Chem. Rev., 1999, 99, 991–1046 CrossRef PubMed; (b) A. Gil, F. Albericio and M. Álvarez, Chem. Rev., 2017, 117, 8420–8446 CrossRef CAS PubMed.
- N. Kamiya, Y. Chikami and Y. Ishii, Synlett, 1990, 1990(11), 675–676 CrossRef.
- D. Naveen Kumar, B. V. Rao and G. S. Ramanjaneyulu, Tetrahedron: Asymmetry, 2005, 16(9), 1611–1614 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1(6), 953–956 CrossRef CAS PubMed.
- P. Ho, Branched-Chain Sugars, Can. J. Chem., 1979, 57(4), 381–383 CrossRef CAS.
- L. Fitjer and U. Quabeck, Synth. Commun., 1985, 15(10), 855–864 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03704e |
| This journal is © The Royal Society of Chemistry 2023 |