
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnchoring ultrasmall Pd nanoparticles by bipyridine functional covalent organic frameworks for semihydrogenation of acetylene†
Ji-qiu Zhanga,
Yu-Hao Wanga,
Shu-jing Zhanga,
Yang-qian Lina,
Qing-qing Guan b and
Xi-meng Xu*a
b and
Xi-meng Xu*a
aFaculty of Civil Engineering and Mechanics, Kunming University of Science and Technology, Kunming 650500, PR China. E-mail: ximeng-Xu@kust.edu.cn
bSchool of Chemical Engineering and Technology, Xinjiang University, Urumqi, Xinjiang 830046, PR China
First published on 17th August 2023
Abstract
Acetylene hydrogenation is a well-accepted solution to reduce by-products in the ethylene production process, while one of the key technical difficulties lies in developing a catalyst that can provide highly dispersed active sites. In this work, a highly crystalline layered covalent organic framework (COF) material (TbBpy) with excellent thermal stability was synthesized and firstly applied as support for ultrasmall Pd nanoparticles to catalyze acetylene hydrogenation. 100% of C2H2 conversion and 88.2% of C2H4 selectivity can be obtained at 120 °C with the space velocity of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The reaction mechanism was elucidated by applying a series of characterization techniques and theoretical calculation. The results indicate that the coordination between Pd and N atom in the bipyridine functional groups of COFs successfully increased the dispersibility and stability of Pd particles, and the introduction of COFs not only improved the adsorption of acetylene and H2 onto catalyst surface, but enhanced the electron transfer process, which can be responsible for the high selectivity and activity of catalyst. This work, for the first time, reported the excellent performance of Pd@TbBpy as a catalyst for acetylene hydrogenation and will facilitate the development and application of COFs materials in the area of petrochemicals.
000 h−1. The reaction mechanism was elucidated by applying a series of characterization techniques and theoretical calculation. The results indicate that the coordination between Pd and N atom in the bipyridine functional groups of COFs successfully increased the dispersibility and stability of Pd particles, and the introduction of COFs not only improved the adsorption of acetylene and H2 onto catalyst surface, but enhanced the electron transfer process, which can be responsible for the high selectivity and activity of catalyst. This work, for the first time, reported the excellent performance of Pd@TbBpy as a catalyst for acetylene hydrogenation and will facilitate the development and application of COFs materials in the area of petrochemicals.
1. Introduction
The production of ethylene (C2H4), which is at the core of the petrochemical industry, is generally accompanied by the production of a small amount of acetylene (C2H2). This is one of the key reasons for catalyst poisoning and products oligomerization, which may exert negative effects on the production of C2H4. The selective hydrogenation of C2H2 has been considered as an effective method for removing the trace C2H2 due to the simple process, low energy consumption and low C2H4 loss.1–4 The core mechanism of this method is the complete conversion of C2H2 to C2H4 via an addition reaction. However, the side reactions with oligomer production under improper control have always been the main bottleneck of C2H2 hydrogenation. One of the solutions to solve this problem is the development of high selective catalysts that could effectively reduce the occurrence of side reactions.Although Fe, Cu, Zn and other non-noble metal-based catalysts have long been used in hydrogenation reactions,5–7 satisfactory activity is hard to obtain at low temperatures. In contrast, the substitution by Pd, a typical noble metal, overcomes the limit of temperature successfully.8 Pd-based catalysts with unique electronic properties often exhibit outstanding advantages in catalytic activity for selective hydrogenation of alkynes.9,10 Especially the research on the electronic structure between Pd atoms and carriers has always been a hot topic pursued by people. For instance, Yang et al. dispersed single atom Pd onto nanoscale TiO2 through self-assembly, resulting in a TOF of over 8000 h−1 and a selectivity of over 91% for styrene.11 At the same time, the doping of Pd atoms also contributes to the dissociation of H2 on the catalyst surface, improving hydrogenation activity.12,13 However, the high cost and easy agglomeration during reaction still remain as major concerns in the real application. A well-recognized strategy to solve these issues is the impregnation of Pd onto high-surface-area carrier which can not only increase the dispersibility of particles, but also enhance the thermal and chemical stability. For the selective hydrogenation of C2H2, the most studied catalyst carrier for Pd was alumina due to its low cost.14–16 However, the lacking of porous structure brings certain difficulties in improving mass transfer, dispersing active sites, as well as enhancing anti-coking properties.
Recently, a new class of materials i.e. metal–organic frameworks (MOFs) have attracted increasing attention as the catalyst carrier in many catalytic reactions due to their unique crystalline porosity, flexible and tailorable properties.17,18 Metal–organic frameworks (MOFs) with unique regular channels are more likely to show strong adsorption capacity for C2H2 in mixed systems containing unsaturated hydrocarbon.19,20 Recent studies have shown that MOFs usually need to be combined with other mesoporous materials to improve the reusability and stability of catalysts. For instance, Li et al. have used mesoporous silica for stabilization Pd@ZIF-8 to achieve a conversion rate of 98% for phenylacetylene and a selectivity of 96% for styrene.21 It can be seen that the instability of metal coordination bonds in the structure of MOFs still makes their widespread application in industry impossible. In addition, graphene, as carbon materials with porous structures and large specific surface areas, are often used as carriers for fixing metal atoms through local coordination, effectively solving the problem of metal atom dispersion. By way of illustration, Zhou et al. successfully introduced Pd atoms onto nitrogen-doped graphene using freeze-drying assisted method. The local coordination from Pd–N hindered the aggregation of Pd particles, achieving a result of 99% acetylene conversion rate and 93.5% ethylene selectivity.22 Furthermore, Huang et al. dispersed Pd atoms onto Graphene through Pd–C bond anchoring, obtaining 100% conversion and 90% ethylene selectivity.23 However, the underdeveloped pore structure of graphene limits its mass transfer efficiency in heterogeneous reactions. Therefore, it is impressive that covalent organic frameworks (COFs), as an emerging crystalline organic porous material, not only have the advantages of the aforementioned properties,24,25 but also are more unique in terms of thermal and chemical stability. More significantly, as a well-defined and predictable functional material, COFs can be endowed with different catalytic functions by varying the means of post-synthesis. Therefore, it's reasonable to conduct that COFs might be an ideal catalyst carrying material for Pd in the reaction of C2H2 hydrogenation.
In this work, a novel bipyridine-functionalized COFs (TbBpy) was developed via the reaction of 1,3,5-triformylbenzene (Tb) with 2,2′-bipyridine-5,5′-diamine (Bpy), applying a solvothermal synthesis method.26 Pd supported on TbBpy (Pd@TbBpy) was then obtained by a simple impregnation process and applied as catalyst for the selective C2H2 hydrogenation. A series of techniques were carried out to characterize the structure of Pd@TbBpy. The catalytic activity and long-term stability were systematically investigated under different conditions. To further elucidate the reaction mechanism and the function of TbBpy, in situ diffusion reflectance infrared Fourier transform spectra (DRIFTS) and the quantum chemical calculations were used to probe the surface processes. This work for the first time reported the excellent properties of TbBpy as carrier for Pd catalyst, which will facilitate the development and application of Pd in industrial C2H2 hydrogenation process.
2. Experimental section
2.1 Chemical and materials
All the chemicals used in the experiments were at A.R. grade. 1,3,5-Triformylbenzene and 2,2′-bipyridyl-5,5′-diamine were obtained from Jilin Chinese Academy of Sciences-Yanshen Technology Co., Ltd. Ethanol, mesitylene, acetic acid, N,N-dimethylformamide, tetrahydrofuran, and acetone were obtained from Aladdin. Palladium acetate (Pd(OAc)2) and NaBH4 were purchased from Tianjing Fuchen Chemical Reagents Factory. And the water used in the experiment was deionized water.2.2 Catalyst preparation
The preparation process of Pd@TbBpy was shown in Fig. 1.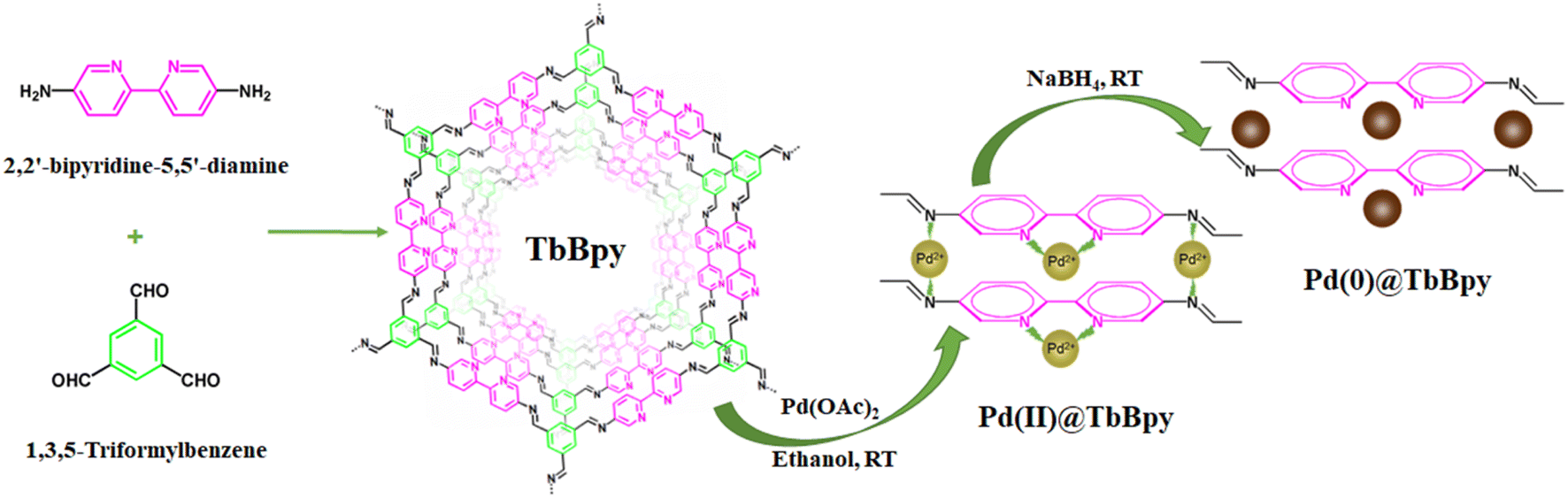 | ||
| Fig. 1 Schematic diagram of preparation process of Pd@TbBpy. | ||
:4) and transferred into a glass ampule, then aqueous acetic acid (AcOH) of 6.0 mol L−1 was added, and the glass ampoule was quickly frozen in a liquid nitrogen bath. The internal pressure of the bottle is evacuated and flame-sealed, and after defrosting at room temperature, it was further heated to 120 °C. After three days, the yellow solid in the ampule was collected and washed with excess N,N-dimethylformamide (DMF), tetrahydrofuran (THF), and acetone, and then dried under vacuum at 80 °C for 24 h to yield the as-prepared TbBpy. The details about TbBpy synthesis are provided in the ESI (Tables S1–S3†).The method using NaBH4 as reducing agent is as follows: TbBpy (100 mg) was dispersed in 10 mL of ethanol, and then 0.76 mL of Pd(OAc)2 (1 mg mL−1) was added slowly under continuous stirring. After 24 h, NaBH4 (2 mol L−1, 2 mL) was added dropwise into the mixture and stirred for 2 h. Then, the residue was collected through centrifugation, washed three times with ethanol (10 mL), and dried at 60 °C under vacuum for 24 h.
The method using H2 as reducing agent is as follows: TbBpy (100 mg) was dispersed in 10 mL of ethanol, and then 0.76 mL of Pd(OAc)2 (1 mg mL−1) was added slowly under continuous stirring. After 24 h, the residue was collected through centrifugation, washed three times with ethanol (10 mL), and dried at 60 °C under vacuum for 24 h. The catalyst was reduced in H2/N2 (20% H2, 50 mL min−1) gas flow at 80 °C for 1 h, and then Pd@TbBpy was obtained.
2.3 Catalyst characterization
The crystalline phase of materials was analyzed by Powder X-ray diffraction (PXRD, X'Pert PRO MPD, PANalytical) with Cu Kα radiation (λ = 1.54 Å). The structure of materials can be characterized by The Fourier transform infrared (FT-IR, Nicolet IS 10, Thermo Scientific) spectroscopy. The specific surface area and pore-size distribution were calculated using the Brunauer–Emmett–Teller (BET) method and non-local density functional theory (NLDFT) modelling based on N2 adsorption data. The nitrogen adsorption and desorption isotherms were tested at 77 K on a Micromeritics TriStar II system. The samples were degassed under vacuum at 150 °C for 6 h before the measurements. The surface morphology of TbBpy and Pd@TbBpy was analyzed by high-resolution transmission electron microscopy (TEM, JEM-2100, JEOL). The sample should be pre-treated before the test. Firstly, it was ground and sonicated, and then the sample was dispersed and casted on a thin carbon-coated TEM grid (copper), and finally, it can be transferred to the filter and the solvent (i.e. ethanol) will evIaporate. The STEM image was recorded with a JEOL JEM-2100 F high-resolution transmission electron microscope equipped with energy dispersive X-ray spectroscopy (EDS) operating at 200 kV. The chemical properties of Pd(II)@TbBpy and Pd@TbBpy were analyzed by X-ray photoelectron spectra (XPS, K-Alpha, Thermo Scientific) equipped with Mono Al Kα. The binding energy calibration based on surface contamination carbon C1s (284.8 eV). The thermal properties of TbBpy and Pd@TbBpy can be analyzed by Thermogravimetric Analyzer (TGA, 5500, TA). The temperature was raised from 26 °C to 800 °C in N2 atmosphere at a heating rate of 10 °C min−1. The quantitative elemental analysis for Pd was performed by inductively coupled plasma spectrometry (ICP-AES) on an Agilent 720ES ICP-OES/MS. Temperature programmed desorption of H2 (H2-TPD) experiments were carried out on xianquan TP-5080. Typically, 100 mg catalyst was pretreated in 10% H2–90% Ar at 120 °C for 2 h and then returned to room temperature. The temperature programmed desorption experiment was carried out in pure Ar atmosphere (flow rate of 30 ml min−1) at a heating rate of 10 °C min−1 to 600 °C.The material transformation mechanism of C2H2 hydrogenation on Pd@TbBpy was explored by in situ diffusion reflectance infrared Fourier transform spectra (DRIFTS, Nicolet iS50, Thermo Scientific) equipped with a high-temperature in situ diffuse reflection cell and a liquid nitrogen cooled MCT detector. The small porcelain crucible is filled with Pd@TbBpy powder and carefully leveled, and the solid powder were first pretreated in 20% H2/N2 at 150 °C for 2 h, and cooled to 120 °C in pure He at a flow rate of 10 mL min−1 for 30 min to ensure a stable condition and the system purged of H2, then the background spectrum was recorded. Thereafter, the Pd@TbBpy powder was exposed to reactant stream (20% H2/N2, 20% C2H2/N2 and pure He) flow for 100 min. During the process, a series of spectral data in the range of 4000–650 cm−1 at different minutes were obtained.
All density functional theory (DFT) calculations have been performed by using the Perdew–Burke–Ernzerhof (PBE)29 formula in generalized gradient approximation (GGA) using VASP.30,31 The ion nucleus was described by selecting the projection enhanced wave (PAW) potential.32 Take valence electrons into account using a plane wave basis set with a kinetic energy cutoff of 450 eV. Partial occupancies of the Kohn–Sham orbitals were allowed using the Gaussian smearing method and a width of 0.05 eV. The electronic energy was considered self-consistent when the energy change was smaller than 10−5 eV. A geometry optimization was considered convergent when the energy change was smaller than 0.05 eV Å−1. The Brillouin zone is sampled with 1 × 1 × 1 Gamma k-points.33
2.4 Acetylene hydrogenation
The gas phase selective hydrogenation of C2H2 was performed in a heated fixed-bed microreactor under continuous flow conditions. After volume controlling by gas pressure reducer and flowmeter, the gases (i.e., 20% C2H2/N2 acetylene, 20% H2/N2 hydrogen and pure He with a H2/C2H2-ratio of 2:1) have been fed into the reaction pipe (Φ8 mm × 200 mm) loaded with Pd@TbBpy. In a typical experiment, 20 mg of the catalyst was diluted with 100 mg crushed quartz sand and filled in the middle of the reactor pipe. Before the reaction, Pd@TbBpy was activated for 2 h at 20% H2/N2 with a flow rate of 50 mL min−1 and 150 °C, and then cooled to room temperature at pure He with a flow rate of 10 mL min−1. The outlet gas stream has been collected for analysis by a gas chromatography (GC7890A, Agilent, USA) equipped with a thermal conductivity detector and a long silica packed column (Φ 0.25 mm × 50 mm, Al2O3/Na2SO4 coating). The conversion of C2H2 and selectivity of C2H4 for the catalytic reactions can be calculated as bellow, i.e.,
 | (1a) |
 | (1b) |
3. Results
3.1 Characterization of Pd@TbBpy catalysts
The long-range order of TbBpy can be characterized by XRD. At first, the effect of solvent species on the crystal structure of TbBpy were explored (Fig. S1†), it's clear that no obvious characteristic peaks of TbBpy can be observed when 1,4-dioxane and n-butanol were applied as solvents. While the replacement by mesitylene and ethanol clearly promoted the production of TbBpy, with the characteristic peak showing at 2θ = 24.8°, which can be assigned to the (001) plane of TbBpy, corresponding to the layered stacking structure in the COFs. It might be resulted from the fact that the solvents would determine the solubility as well as the condensation reaction rate of the two organic monomers.34 Besides, the ratio of solvent components would influence the reaction balance between skeleton formation and crystallization,35 and a much better crystal structure has been observed in Fig. S2† at volume ratio of ethanol/mesitylene of 1:4. Further, the concentration of acetic acid aqueous solution was adjusted to improve the crystallinity of TbBpy. As can be seen in Fig. S3,† when the concentration was 6 mol L−1, the crystal morphology is relatively complete. While as it further increased, the crystallinity of TbBpy decreased instead. This is probably because the high concentration of acetic acid disrupted the rate of reversible polycondensation in the process of linking organic monomers through polycondensation and orderly assembly of organic monomers, which led to a incomplete self-healing of the structure.35
The XRD patterns of TbBpy and Pd@TbBpy synthesized under the optimal condition are shown in Fig. 2a, the typical diffraction peaks of TbBpy at 2θ of 3.53° and 24.3° can be clearly seen, which can be attributed to the crystal planes of (100) and (001), respectively. For Pd@TbBpy, only diffraction peak of Pd corresponding to (111) plane at 2θ of 39.94° can be observed,36 suggesting the successful loading of Pd particles onto the TbBpy. While the disappearance of peaks of (100) and (001) planes might be ascribed to the partial collapse of internal structure of TbBpy after the loading of Pd.37
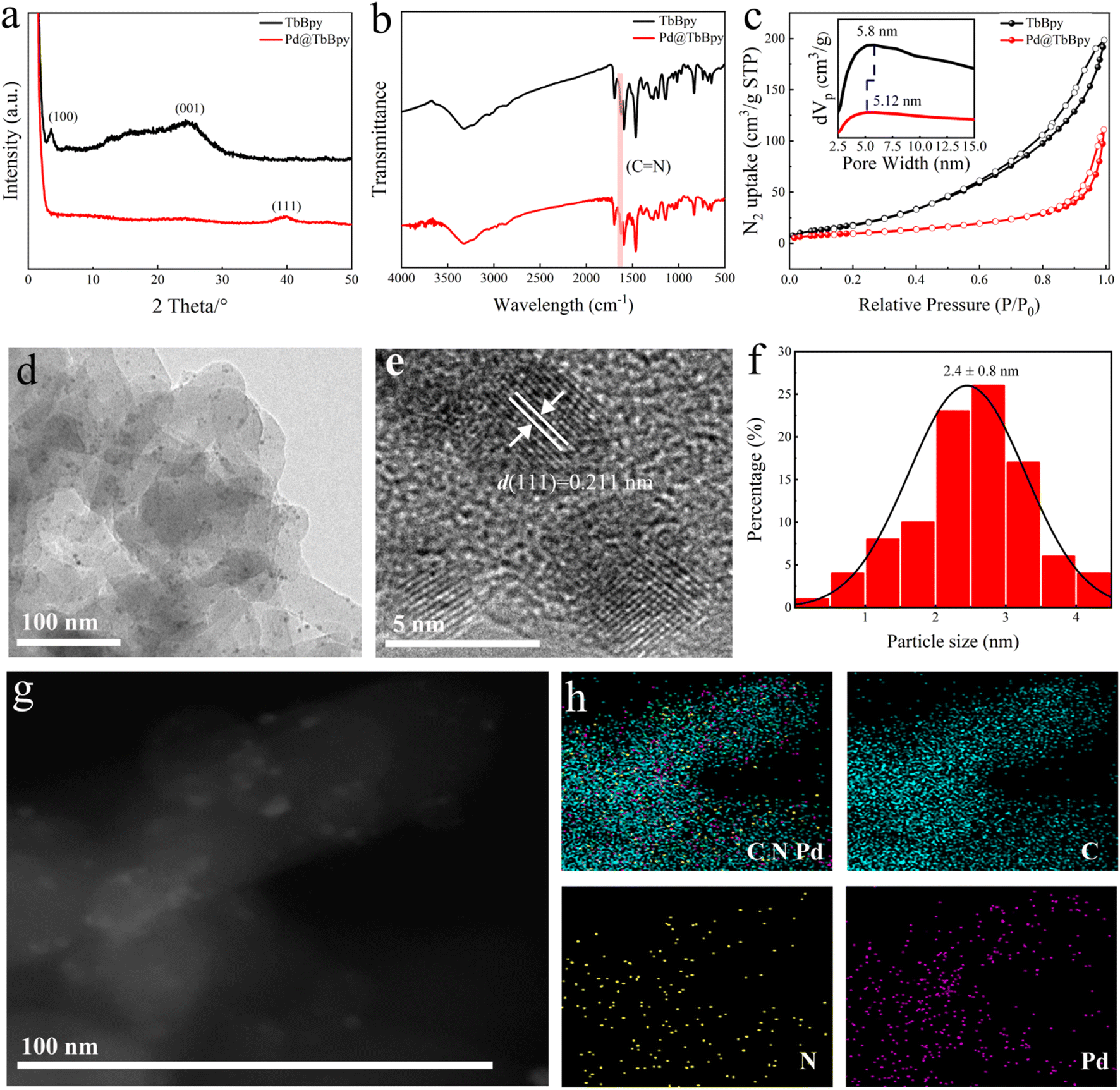 | ||
| Fig. 2 The (a) XRD profile, (b) FT-IR spectra, and (c) N2 adsorption–desorption isotherms of TbBpy and Pd@TbBpy; the (d) TEM image and (e) HR-TEM image of Pd@TbBpy. (f) The Pd particle size frequency distribution histogram; (g) the STEM image and (h) corresponding elemental mapping image of Pd@TbBpy. | ||
To further confirm that TbBpy is composed of 1,3,5-triphenylcarbaldehyde and 5,5-diamino-2,2-bipyridine connecting to each other through Schiff base reaction, the functional group structure of TbBpy was determined by FT-IR (Fig. S4†). The FT-IR spectrum of TbBpy shows a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration peak at 1618 cm−1, which means that the polycondensation reaction has occurred. When Pd nanoparticles were embedded in the pores of TbBpy, as shown in Fig. 2b, a strong vibrational peak (CN stretching vibration) was still observed at a wavelength of 1618 cm−1, and the intensity was weakened, indicating that the immersion of Pd may cause the collapse of layered structure inside TbBpy, which is in accordance with the results of XRD analysis.
N stretching vibration peak at 1618 cm−1, which means that the polycondensation reaction has occurred. When Pd nanoparticles were embedded in the pores of TbBpy, as shown in Fig. 2b, a strong vibrational peak (CN stretching vibration) was still observed at a wavelength of 1618 cm−1, and the intensity was weakened, indicating that the immersion of Pd may cause the collapse of layered structure inside TbBpy, which is in accordance with the results of XRD analysis.
The surface areas (BET) of TbBpy and Pd@TbBpy have been calculated by the BET method, which were 76.10 and 36.32 m2 g−1, respectively (Fig. 2c). The pore size distribution embedded in Fig. 2c shows that the average pore size of TbBpy and Pd@TbBpy are 5.8 nm and 5.12 nm, respectively. The decrease in pore size may resulted from the block by Pd particles, which further demonstrates the successful loading onto TbBpy.26 Comparing with other COFs, such as COF-LZU1 (410 m2 g−1)26 and TpPa-1 (484 m2 g−1),38 TbBpy has a much lower BET, which might be resulted from its unique structure with inhomogeneous mesopores and lower long-range ordered.
The morphological structures of the Pd@TbBpy were characterized by TEM and STEM scanning. The random distribution of Pd particles can be clearly observed in Fig. 2d, and the average diameter of the Pd loaded is about 2.4 nm (Fig. 2f). Moreover, a 0.211 nm of interplanar crystalline space was observed in HRTEM image (Fig. 2e), which matched well with (111) crystal plane of Pd. The EDS elemental mapping of Pd@TbBpy were also analyzed to reveal the distribution of Pd, C and N. The EDS mapping clearly showed that Pd homogeneously distributed on the TbBpy surface, which further suggested the well dispersion of Pd nanoparticles. In addition, inductively coupled plasma spectroscopy (ICP) analysis indicated that the Pd content of Pd@TbBpy is 0.72 wt%, which deviates slightly from the theoretical Pd content (0.75 wt%) we applied.
The thermal stability of TbBpy and Pd@TbBpy was evaluated by TGA. As shown in Fig. S5,† the mass loss of the material was relatively small when the temperature was lower than 400 °C. Then, the heat cracks started to appear in TbBpy and Pd@TbBpy at 400–550 °C, with the mass loss reaching to 20–30%. And the further rising of temperature to 800 °C gradually led to the continuous increase in ash content. These phenomenon show that TbBpy can maintain good thermal stability below 400 °C, and the load of Pd will not affect the thermal stability.
The XPS analysis was conducted to elucidate the interactions between Pd and N groups in the TbBpy. At first, from the total survey scan of XPS (Fig. 3a), the peaks of O, C, N, and Pd can be found. Specifically, in the N 1s spectrum shown in Fig. 3b, the peaks at 399.08 eV and 398.53 eV can be ascribed to pyrrole-N and pyridine-N, respectively, which are basic units of N groups in TbBpy. Besides, in the Pd 3d spectrum (Fig. 3c), the peaks at binding energy of 335.78 eV and 340.88 eV corresponded to Pd 3d5/2 and Pd 3d3/2, respectively. The results indicate that Pd(II) has been successfully reduced to Pd(0), which was then loaded onto TbBpy.39 Notably, the binding energy peak at 338.0 eV can be attributed to the formation of PdO2 caused by Pd@TbBpy contacting with air, which is consistent with the result of PdO/Cel-xPdxO2-δ reported in the literature.40 For comparison, the XPS analysis of un-reduced Pd(II), i.e. Pd(II)@TbBpy was also conducted, with the Pd 3d spectrum showing in Fig. 3d, with a binding energy of 337.58 eV assigning to the Pd 3d5/2 peak and a binding energy of 342.81 eV assigned to the Pd 3d3/2 peak. Compared with the characteristic peak of Pd(OAc)2 (338.4 eV), the binding energy peak of Pd(II)@TbBpy at 337.58 eV is reduced by about 0.62 eV, which indicates that the Pd(II) species accepts the lone pair of electrons from the N atom in the imine group to form a strong coordination bond.26
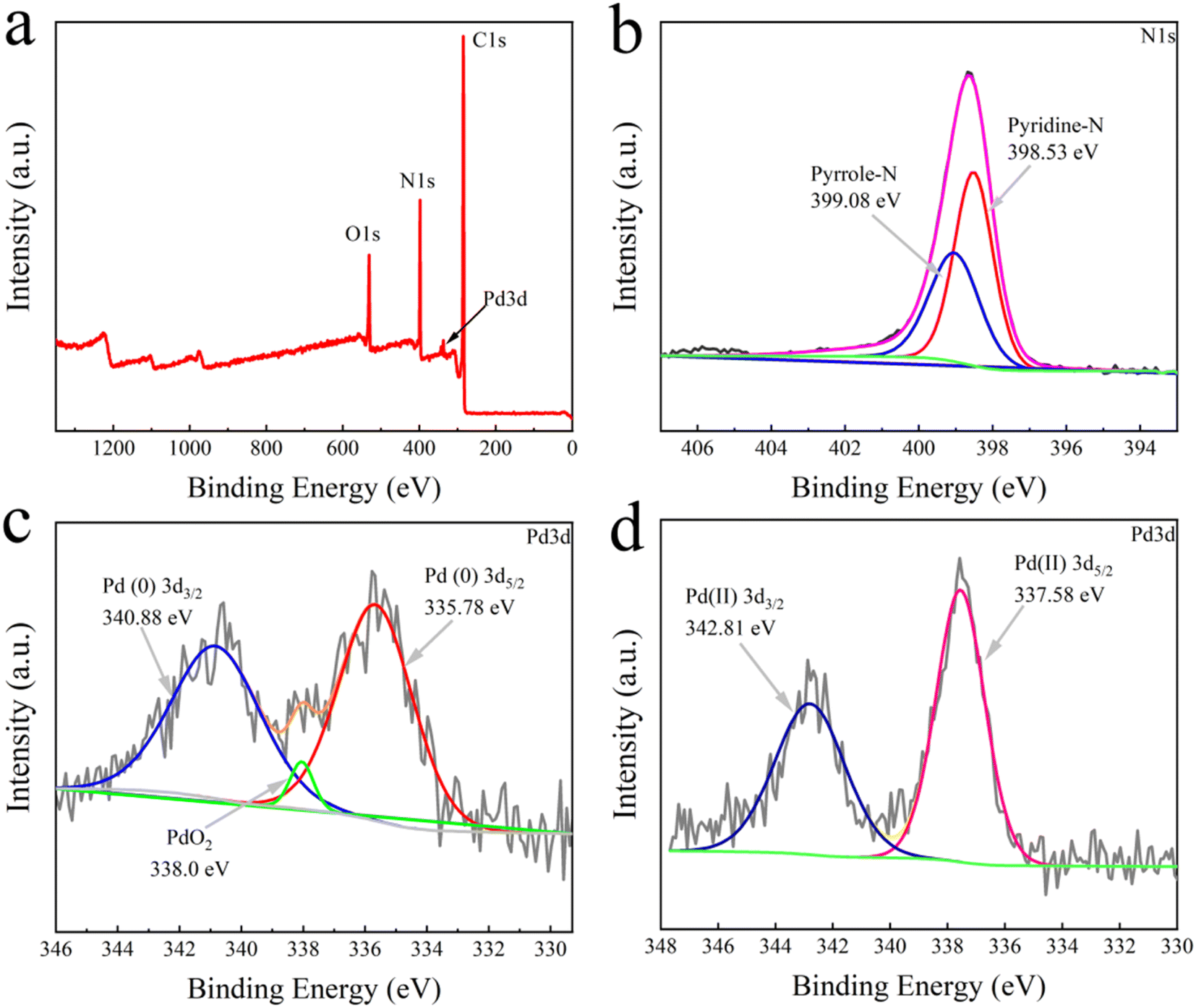 | ||
| Fig. 3 (a) XPS total spectrum, (b) N 1s spectrum, and (c) Pd 3d spectrum of Pd@TbBpy; (d) Pd 3d XPS spectrum of Pd(II)@TbBpy. | ||
3.2 Catalytic performance over Pd@TbBpy catalysts
Normally, an ideal C2H2 hydride Pd-based catalyst should meet the following requirements: (1) the lower Pd loading; (2) the complete conversion of C2H2 at lower temperature and higher C2H4 selectivity; (3) the stable catalytic activity. With these indicators as reference, we systematically investigated the catalytic performance of the catalyst for C2H2 hydrogenation.At first, H2 and NaBH4 were applied as Pd(II) reductants to investigate the effect of reductant phase on the catalytic activity (Fig. S6†). It turns out that both of the C2H2 conversion rates reached 100% with two reductants. While the C2H4 selectivity of NaBH4-reduced catalyst was 52.2%, being higher than that of H2-reduced catalyst, which was only 43.5%. This might be explained by the fact that NaBH4 could penetrate into the pores of the TbBpy via capillary force more easily, and reduced the Pd precursor with higher efficiency.41 Meanwhile, Pd particles can be restricted by the bipyridine functional group and would not overflow again, which may guarantee the uniform distribution of Pd. On the other hand, the reduction by H2 has to be conducted under high temperature (120 °C), which can be inclined to result in aggregation of Pd particles.42,43
Moreover, the effect of Pd loading on the catalytic activity of Pd@TbBpy was explored. As shown in Fig. 4a, when the loading of Pd nanoparticles increased from 0.25 wt% to 1 wt%, the conversion remained at 100%, while the C2H4 selectivity increased first and then decreased, which were 67.8%, 69.3%, and 75.1%, and 52.2%, respectively. This trend implies that a lower Pd loading may not guarantee enough active sites for the reactants, while an exorbitant Pd loading may lead to the agglomeration of Pd nanoparticles. The Pd particle size analysis (Fig. S7†) after the reaction also demonstrates that the Pd particle size is much larger than before the reaction. Therefore, based on the comparative results, a Pd content of 0.75 wt% was selected for subsequent experiments.
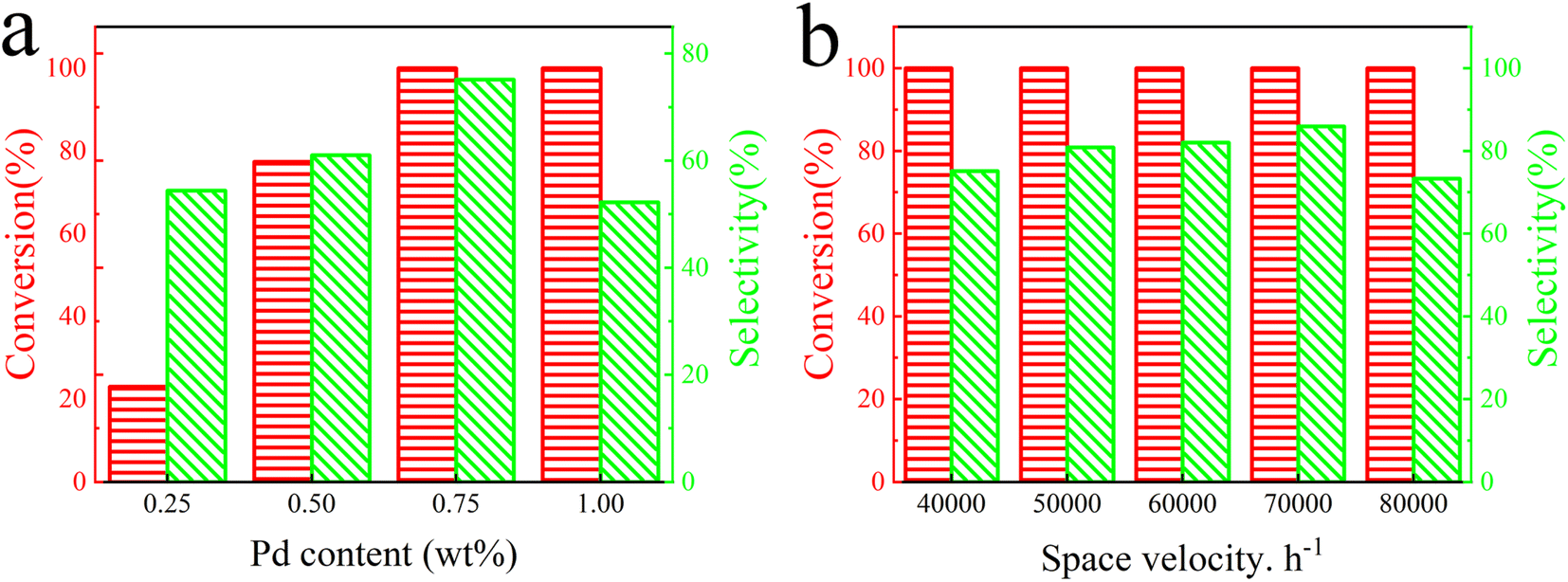 | ||
| Fig. 4 Catalytic activities of Pd@TbBpy (a) with different Pd contents and (b) at different space velocities. | ||
Besides, the effect of space velocity on the catalytic performance of Pd@TbBpy was also investigated (Fig. 4b). Over 120 °C, the C2H2 conversion remained at 100% at all selected space velocities, which were 40000 h−1, 50000 h−1, 60000 h−1, 70000 h−1, and 80000 h−1. As for the C2H4 selectivity, it was 73.4%, 78.5%, 81.02%, 88.2%, and 82.3%, respectively. The highest selectivity was obtained at velocity of 70000 h−1. Apparently, when the space velocity is too low, the products may not be blown away from the catalyst surface in time, resulting in an excessive hydrogenation or formation of other intermediates.44 On the other hand, it would also be hard for the reactants to steadily adsorb on the active sites for reaction at higher space velocities. In addition, as shown in Fig. S8,† the C2H2 conversion of the Pd@TbBpy catalyst achieves complete conversion at 100 °C, which is about 40% higher than that of the Pd precursor. The C2H4 selectivity of Pd@TbBpy catalyst reached 88.2% at 120 °C and the efficiency of C2H4 selectivity was twice that of Pd precursor at the same temperature. Therefore, the difference in reaction efficiency is very remarkable between Pd@TbBpy and Pd precursor. Based on the above results, the Pd loading of 0.75 wt% and the space velocity of 70000 h−1 was selected as the optimal condition for following activity tests. At a low reaction temperature of around 120 °C, The catalytic performance of Pd@TbBpy is comparable to other highly selective Pd based catalysts supported on carbon materials listed in Table S4.† In fact, compared to other single atom Pd based catalysts, our developed catalyst has more advantages in experimental results and Pd distribution. By comparison, it was found that in this work, the Pd content we used was slightly higher than other catalytic groups, and the experimental parameters were not strict and harsh. In addition, it is important that the high ethylene selectivity is due to the layered stacking structure and the N-site anchoring of COFs, which enables the uniform distribution and stability of Pd nanoparticles.
The reactions involved in industrial production usually need to be carried out for a long time, which puts forward strict requirements on the life of the catalyst. Therefore, the long-term stability of the Pd@TbBpy was evaluated under the optimal reaction condition. As shown in Fig. 5, during the 1500 min of continuous reaction, the catalytic activity kept excellent with the C2H2 conversion being 100% and the C2H4 selectivity being ∼90%. Compared with other reported Pd-based catalysts,45–48 Pd@TbBpy shows more remarkable long-term stability. However, a gradual formation of green oil, which was resulted from the deposit of by-products during the continuous hydrogenation, has been observed. It might has exerted some negative effects on the process activity as well as the catalyst stability.44,49
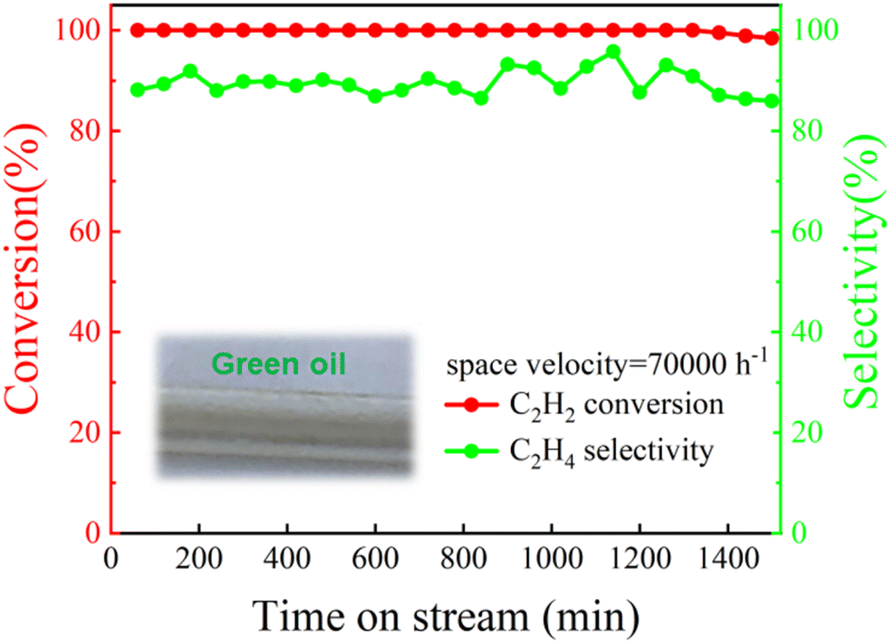 | ||
| Fig. 5 Life test of Pd@TbBpy for the hydrogenation of C2H2 (temperature = 120 °C; space velocity = 70000 h−1; time ≥ 1500 min; 20% C2H2/N2, 20% H2/N2 and He). | ||
3.3 Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) analysis
To further analyze the reaction mechanism, in this section, the dynamic adsorption–desorption processes of C2H2 and C2H2/H2 over Pd@TbBpy were investigated by in situ FT-IR spectra. Notably, before the reactions, Pd@TbBpy has been reduced by the mix of H2 (20%)/N2 (80%) at 120 °C for 60 min, and then purged with pure He to remove all adsorbates on the surface. Firstly, the adsorption behaviors of C2H2 onto Pd@TbBpy were analyzed under 80 °C, 100 °C, 120 °C, 150 °C, and 200 °C. As can be seen in Fig. 6a, the intensity of peaks didn't vary much as the temperature changing, demonstrating that the saturation adsorption of C2H2 could be obtained at low temperature. Specifically, the characteristic peaks of C2H2 at 3308–3264 cm−1, which are attributed to the![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH bending mode (R(δJ = +1), P(δJ = −1)), and the central Q branch peaks (δJ = 0) of CH (∼730 cm−1), as well as the IR-active CH bending vibrations (717 and 756 cm−1) can be clearly observed, indicating a typical π-bond adsorption.33 The C2H2 hydrogenation over zeolite-supported bimetallic catalyst has also been reported in Huang's work, in which the π-bond adsorption was confirmed as the main adsorption mode, and it was the dominant factor affecting the higher hydrogenation activity.50
CH bending mode (R(δJ = +1), P(δJ = −1)), and the central Q branch peaks (δJ = 0) of CH (∼730 cm−1), as well as the IR-active CH bending vibrations (717 and 756 cm−1) can be clearly observed, indicating a typical π-bond adsorption.33 The C2H2 hydrogenation over zeolite-supported bimetallic catalyst has also been reported in Huang's work, in which the π-bond adsorption was confirmed as the main adsorption mode, and it was the dominant factor affecting the higher hydrogenation activity.50
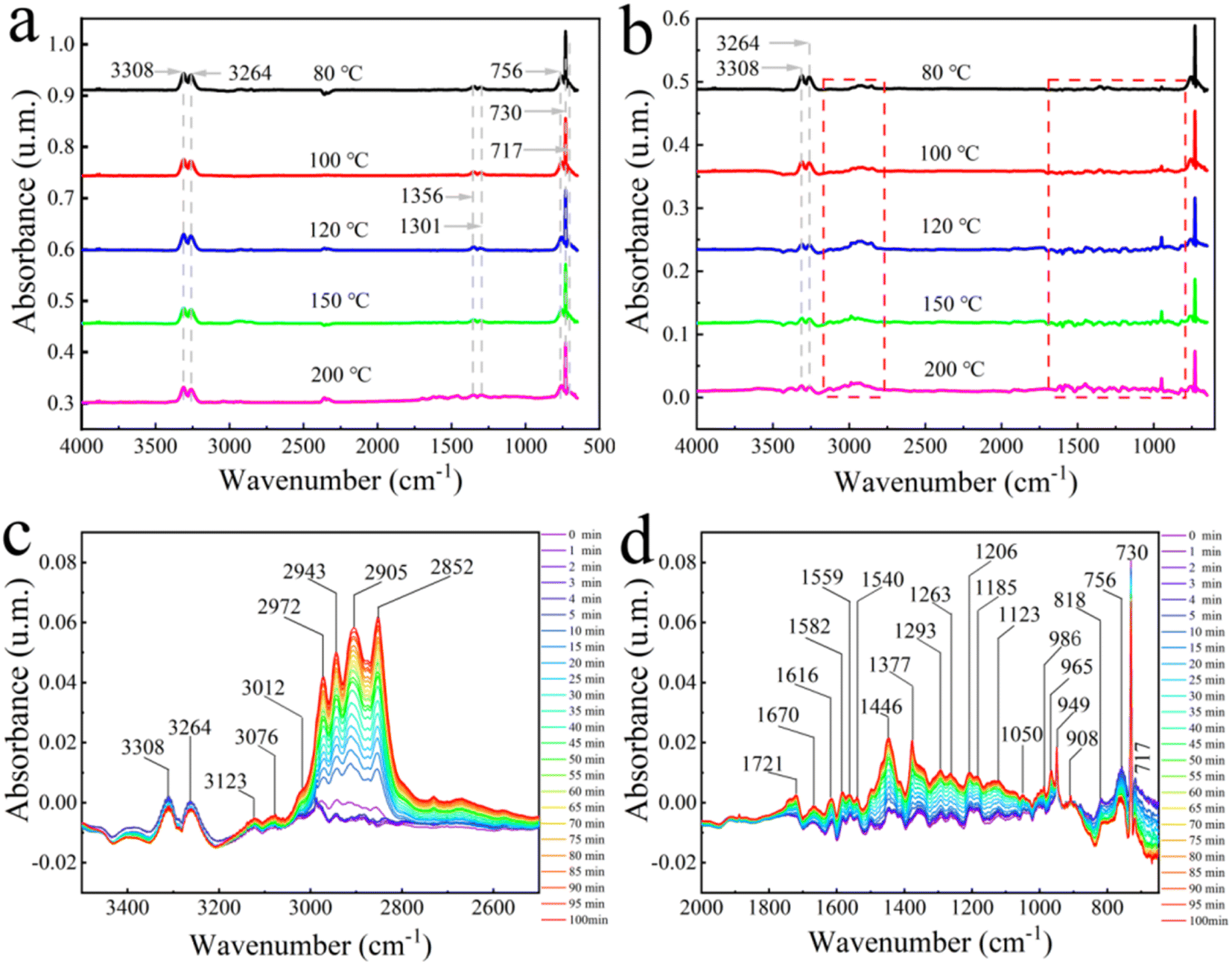 | ||
| Fig. 6 (a) In situ DRIFTS spectra of C2H2 adsorption on Pd@TbBpy. (b) In situ DRIFTS spectra of C2H2 hydrogenation on Pd@TbBpy. The real-time in situ DRIFTS spectra (c) 3500–2500 cm−1 and (d) 2000–650 cm−1 of C2H2 hydrogenation on Pd@TbBpy (temperature = 120 °C; space velocity = 70000 h−1; 20% C2H2/80% N2, 20% H2/80% N2). | ||
Besides, the C2H2 adsorption over Pd@TbBpy under 120 °C for 100 min was also continuously monitored by FT-IR spectroscopy (Fig. S9a†). It's clear that the characteristic peaks of C2H2 at 3308–3264 cm−1 remained almost same during the whole period, which indicated that the C2H2 adsorption reached saturation at the very beginning and the adsorbed C2H2 has stayed at stable form without transforming into oligomeric state. In addition, the products transformation process of C2H2 hydrogenation over Pd@TbBpy was also investigated under 80 °C, 100 °C, 120 °C, 150 °C, and 200 °C, as shown in Fig. 6b. During the adsorption–desorption processes, the peaks of C2H2 gradually diminished as the temperature increased, while some characteristic peaks at 3100–2750 cm−1 and 1700–800 cm−1, which can be attributed to the vibration of related functional groups of C2H4 and saturated alkanes, gradually showed up. The detailed dynamic adsorption process of feed gas has been evaluated by the real-time in situ DRIFTS spectra, in which, different from FT-IR spectra, the structural changes of functional groups during the reaction can be recorded, as shown in Fig. 6c and d.
The absorption peaks from 3123 cm−1 to 3012 cm−1 can be assigned to the stretching vibration of the C–H bond on the olefinic double bond (CC–H). Thereinto, the characteristic peak at 3076 cm−1 was assigned to the stretching vibration of the terminal double bond hydrogen (〉CCH2). Besides, the peaks at 1670 cm−1, 1616 cm−1 and 1582 cm−1 were corresponded to the stretching vibration of alkene CC. Meanwhile, the peaks around 986 cm−1, 965–949 cm−1, and 908 cm−1 were attributed to the torsional vibration of alkene CH2, the out-of-plane bending vibrations of trans-configuration CH (R1-HCCH-R2), and the out-of-plane rocking vibrations of alkene CH2 in alkyl alkenes, respectively. These results confirmed the alkene production during the C2H2 hydrogenation over Pd@TbBpy. However, the typical peaks belonging to alkane can also been observed, including the asymmetric stretching vibration of –CH3 (2972 cm−1) and –CH2 (2943 cm−1), and the symmetric stretching vibrations of –CH3 (2905 cm−1) and –CH2 (2852 cm−1). It suggested that the saturated alkanes were also generated inevitably during the hydrogenation process.50
Finally, after 100 min of reaction, the desorption experiment was carried out with pure He purging over Pd@TbBpy surface. As shown in Fig. S9c,† the characteristic peaks of C2H2 (3308–3264 cm−1, 756–717 cm−1) disappear very quickly, while the peaks belonging to the products (C2H4 and alkanes) still existed, which indicated that C2H2 was more easily desorbed from the Pd@TbBpy surface than the products. However, the presence of C2H4 may lead to the further hydrogenation, resulting in the formation of carbon layers depositing on the catalyst surface.51 The similar result has also been reported in Zhang's work,52 in which, a carbonaceous layer was observed on the surface of Pd/Al2O3 catalyst after the hydrogenation of C2H2.
3.4 DFT analysis
To further elucidate the role of TbBpy in the reaction and the interaction between Pd and TbBpy, DFT calculation was further conducted. As can be seen in Fig. 7, after the loading of Pd onto TbBpy, the adsorption energies (Eads) of C2H2 and H2 increased from −2.12 eV and −0.73 eV to −2.92 eV and −0.88 eV, respectively. The higher Eads indicated that the adsorption of C2H2 and H2 onto Pd were both easier with the presence of TbBpy, which is in accordance with the results of DRIFTS. In addition, there are two peaks displayed in H2-TPD (Fig. S10†) at 135 °C and 482 °C, which are attributed to the desorption of chemically adsorbed hydrogen on the Pd phase and the desorption of overflow hydrogen, respectively.53,54 Therefore, it can be seen that H2 can be well adsorbed on the Pd@TbBpy when the temperature is below 135 °C, which provides strong evidence for obtaining good reaction activity. Clearly, the interaction of Pd and TbBpy benefited the adsorption of reactants, which is the very first step to initiate the whole reaction. Besides, the charge transfer value of C varied from −0.11e to −0.15e from the unsupported to the supported Pd, indicating the electron-grabbing power of C in C2H2 was enhanced with the presence of TbBpy. Meanwhile, the charge transfer value of H changed from 0.027e to 0.41e, implying that the electron donating ability of H2 was also greatly improved. It indicated that the electron transfer process between the reactants was obviously enhanced with the presence of TbBpy. Above results show that the interaction between Pd and COFs material helped to increase the catalytic activity from the atomic level.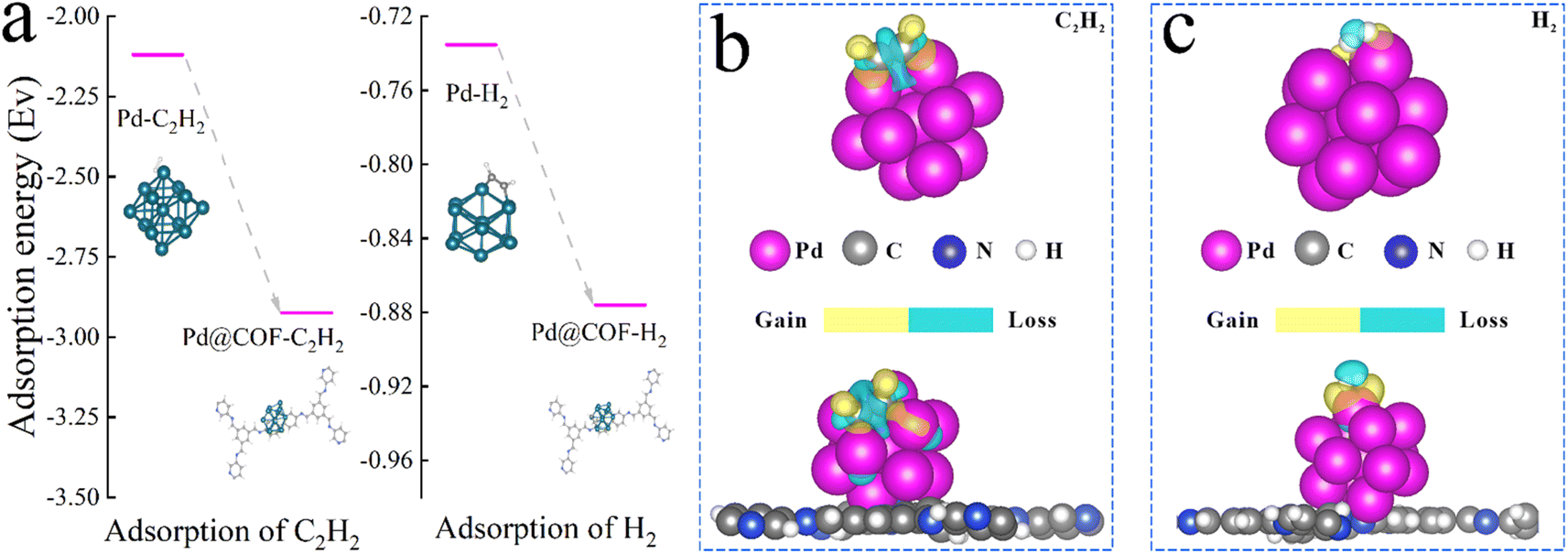 | ||
| Fig. 7 (a) Adsorption energy diagrams of Pd and Pd@TbBpy in C2H2 and H2 absorption process. Charge transfer of the adsorbed (b) C2H2 and (c) H2 around the Pd and Pd@TbBpy. | ||
4. Conclusions
In this study, a novel 2D covalent organic framework (TbBpy) was synthesized via the Schiff base reaction of 1,3,5-triformylbenzene and 2,2′-bipyridyl-5,5′-diamine, accompany with the formation of CN bond. Pd nanoparticles were loaded onto the surface and into the pores of TbBpy with uniform particle sizes (i.e. ∼2.4 nm), which coordinated with N atoms in the bipyridine functional group of TbBpy. The 100% of C2H2 conversion and 88.2% of C2H4 selectivity can be obtained at the temperature of 120 °C and the space velocity of 70000 h−1. Under the this optimal condition, Pd@TbBpy showed remarkable stability as well as life (>1500 min) in the durability experiments. Over 150 °C, the over-hydrogenation of intermediate products was more inclined to happen with the production of green oil, which was composed of hydrocarbon derivatives and might exerted negative effects on catalyst activity. In addition, based on the results of DRIFTS, it was found that C2H2 can quickly achieve equilibrium of adsorption on the surface of Pd@TbBpy without transformation into oligomer. And a typical form of π-bond adsorption has been observed, which can be beneficial for the catalytic reactivity. However, the intermediate products were found out to be more difficult to desorb from the Pd@TbBpy surface than C2H2, thus leading to the generation of saturated alkanes inevitably, which further explain the production of green oil. The DFT analysis revealed that, after the loading of Pd onto TbBpy, the adsorption of C2H2 and H2 onto the catalyst were both improved, and the electron transfer between C and Pd was also enhanced, which might further demonstrate the promotion effects of TbBpy as support toward the catalytic activity of Pd. The above results demonstrate and elucidate the high activity and selectivity of Pd catalyst supported on TbBpy in the C2H2 hydrogenation, promoting the application of COFs materials in this area.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was jointly supported by the National Natural Science Foundation of China (42177464), the Yunnan Fundamental Research Projects (140520210219), and the National Key Research and Development Program of China (Grant No. 2019YFC1805904).References
- L. Sun, L. Jiang and X. Hua, et al., Preparation of Au/xCeO2-Al2O3 catalysts with enhanced catalytic properties for the selective acetylene hydrogenation, J. Alloys Compd., 2019, 811, 152052, DOI:10.1016/j.jallcom.2019.152052.
- L. Yanan, A. J. Mccue and M. Chenglin, et al., Palladium phosphide nanoparticles as highly selective catalysts for the selective hydrogenation of acetylene, J. Catal., 2018, 364, 406–414, DOI:10.1016/j.jcat.2018.06.001.
- B. Huang, T. Wang and Z. Yang, et al., Iron-Based Bimetallic Nanocatalysts for Highly Selective Hydrogenation of Acetylene in N,N-Dimethylformamide at Room Temperature, ACS Sustainable Chem. Eng., 2017, 5(2), 1668–1674, DOI:10.1021/acssuschemeng.6b02413.
- F. Studt and F. Abild-Pedersen, et al., On the Role of Surface Modifications of Palladium Catalysts in the Selective Hydrogenation of Acetylene, Angew. Chem., Int. Ed., 2008, 120(48), 9439–9442, DOI:10.1002/anie.200802844.
- L. He, X. Gong and L. Ye, et al., Synergistic effects of bimetallic Cu-Fe/SiO2 nanocatalysts in selective hydrogenation of diethyl malonate to 1,3-propanediol, J. Nat. Gas Chem., 2016, 025(006), 1038–1044, DOI:10.1016/j.jechem.2016.11.007.
- T. Abdollahi and D. Farmanzadeh, et al., Graphene-supported Cu_(11) nanocluster as a candidate catalyst for the selective hydrogenation of acetylene: a density functional study, J. Alloys Compd., 2017, 735, 117–130, DOI:10.1016/j.jallcom.2017.11.051.
- C. Lu, A. Zeng and Y. Wang, Enhanced Hydrogenation Activity over a Zn-Modified Cu-Based Catalyst in Acetylene Hydrogenation, Ind. Eng. Chem. Res., 2022, 61(51), 18696–18702, DOI:10.1021/acs.iecr.2c03502.
- R. Gao, J. Xu and J. Wang, et al., Pd/Fe2O3 with Electronic Coupling Single-Site Pd-Fe Pair Sites for Low-Temperature Semihydrogenation of Alkynes, J. Am. Chem. Soc., 2022, 144(1), 573–581, DOI:10.1021/jacs.1c11740.
- Q. Guan, C. Yang and S. Wang, et al., Reactive Metal-Biopolymer Interactions for Semihydrogenation of Acetylene, ACS Catal., 2019, 9(12), 11146–11152, DOI:10.1021/acscatal.9b04042.
- Q. Guan, J. Zhang and L. He, et al., Selective hydrogenation of acetylene to ethylene over the surface of sub-2nm Pd nanoparticles in miscanthus sinensis-derived microporous carbon tubes, ACS Sustain. Chem. Eng., 2020, 8(31), 11638–11648, DOI:10.1021/acssuschemeng.0c03043.
- F. Yang, S. Ding and H. Song, et al., Single-atom Pd dispersed on nanoscale anatase TiO2 for the selective hydrogenation of phenylacetylene, Sci. China Mater., 2020, 63(6), 982–992, DOI:10.1007/s40843-020-1271-x.
- J. Chang, M. Hülsey and S. Wang, et al., Electrothermal Water-Gas Shift Reaction at Room Temperature with a Silicomolybdate based Pd Single-Atom Catalyst, Angew. Chem., Int. Ed., 2023, e202218265, DOI:10.1002/ange.202218265.
- K. Lee, U. Anjum and T. Araújo, et al., Atomic Pd-promoted ZnZrOx solid solution catalyst for CO2 hydrogenation to methanol, Appl. Catal., B, 2022, 304, 120994, DOI:10.1016/j.apcatb.2021.120994.
- S. Komhom, O. Mekasuwandumrong and P. Praserthdam, et al., Improvement of Pd/Al2O3 catalyst performance in selective acetylene hydrogenation using mixed phases Al2O3 support, Catal. Commun., 2008, 10(1), 86–91, DOI:10.1016/j.catcom.2008.07.039.
- Y. Q. Cao, Z. J. Sui and Y. Zhu, et al., Selective Hydrogenation of Acetylene over Pd-In/Al2O3 Catalyst: Promotional Effect of Indium and Composition-dependent Performance, ACS Catal., 2017, 7(11), 7835–7846, DOI:10.1021/acscatal.7b01745.
- J. T. Feng, X. Y. Ma and D. G. Evans, et al., Enhancement of Metal Dispersion and Selective Acetylene Hydrogenation Catalytic Properties of a Supported Pd Catalyst, Ind. Eng. Chem. Res., 2011, 50(4), 1947–1954, DOI:10.1021/ie101508z.
- M. Tejeda-Serrano, M. Marta and R. Bethany, et al., Isolated Fe(III)–O Sites Catalyze the Hydrogenation of Acetylene in Ethylene Flows under Front–End Industrial Conditions, J. Am. Chem. Soc., 2018, 140(28), 8827–8832, DOI:10.1021/jacs.8b04669.
- S. C. Xiang, Z. Zhang and C. G. Zhao, et al., Rationally tuned micropores within enantiopure metal-organic frameworks for highly selective separation of acetylene and ethylene, Nat. Commun., 2011, 2, 204, DOI:10.1038/ncomms1206.
- L. N. Ma, Z. H. Wang and L. Zhang, et al., Extraordinary Separation of Acetylene-Containing Mixtures in a Honeycomb Calcium-Based MOF with Multiple Active Sites, ACS Appl. Mater. Interfaces, 2023, 15(2), 2971–2978, DOI:10.1021/acsami.2c19321.
- L. Liu, S. Wu and D. Li, et al., Partial Linker Substitution Strategy to Construct a Quaternary HKUST-like MOF for Efficient Acetylene Storage and Separation, ACS Appl. Mater. Interfaces, 2022, 14(32), 36882–36889, DOI:10.1021/acsami.2c10346.
- Z. Li, M. Hu and J. Liu, et al., Mesoporous silica stabilized MOF nanoreactor for highly selective semi-hydrogenation of phenylacetylene via synergistic effect of Pd and Ru single site, Nano Res., 2022, 15, 1983–1992, DOI:10.1007/s12274-021-3849-2.
- S. Zhou, L. Shang and Y. Zhao, et al., Pd Single-Atom Catalysts on Nitrogen-Doped Graphene for the Highly Selective Photothermal Hydrogenation of Acetylene to Ethylene, Adv. Mater., 2019, 31(18), 1900509, DOI:10.1002/adma.201900509.
- F. Huang, Y. Deng and Y. Chen, et al., Atomically Dispersed Pd on Nanodiamond/Graphene Hybrid for Selective Hydrogenation of Acetylene, J. Am. Chem. Soc., 2018, 140(41), 13142–13146, DOI:10.1021/jacs.8b07476.
- Q. Fang, J. Wang and S. Gu, et al., 3D Porous Crystalline Polyimide Covalent Organic Frameworks for Drug Delivery, J. Am. Chem. Soc., 2015, 137(26), 8352–8355, DOI:10.1021/jacs.5b04147.
- Q. Fang, Z. Zhuang and S. Gu, et al., Designed synthesis of large-pore crystalline polyimide covalent organic frameworks, Nat. Commun., 2014, 5, 4503, DOI:10.1038/ncomms5503.
- S. Y. Ding, J. Gao and Q. Wang, et al., Construction of Covalent Organic Framework for Catalysis: Pd/COF-LZU1 in Suzuki-Miyaura Coupling Reaction, J. Am. Chem. Soc., 2011, 133(49), 19816–19822, DOI:10.1021/ja206846p.
- V. D'Elia and A. W. Kleij, Surface science approach to the heterogeneous cycloaddition of CO2 to epoxides catalyzed by site-isolated metal complexes and single atoms: a review, Green Chem. Eng., 2022, 3, 210–227, DOI:10.1016/j.gce.2022.01.005.
- Y. Guo, Y. Li and X. Du, et al., Pd single-atom catalysts derived from strong metal–support interaction for selective hydrogenation of acetylene, Nano Res., 2022, 15, 10037–10043, DOI:10.1007/s12274-022-4376-5.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set - ScienceDirect, Comput. Mater. Sci., 1996, 6(1), 15–50, DOI:10.1016/0927-0256(96)00008-0.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775, DOI:10.1103/PhysRevB.59.1758.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13(12), 5188–5192, DOI:10.1103/PhysRevB.13.5188.
- S. Ding and W. Wang, Covalent organic frameworks (COFs): from design to applications, Chem. Soc. Rev., 2013, 42, 548–568, 10.1039/c2cs35072f.
- X. Feng, X. S. Ding and D. L. Jiang, Covalent organic frameworks, Chem. Soc. Rev., 2012, 41, 6010–6022, 10.1039/c2cs35157a.
- W. X. Niu, L. Zhang and G. B. Xu, Shape-Controlled Synthesis of Single-Crystalline Palladium Nanocrystals, ACS Nano, 2010, 4(4), 1987–1996, DOI:10.1021/nn100093y.
- K. Wang, L. M. Yang and X. Wang, et al., Covalent Triazine Frameworks via a Low-Temperature Polycondensation Approach, Angew. Chem., 2017, 56, 14149–14153, DOI:10.1002/anie.201708548.
- P. Pachfule, M. K. Panda and S. Kandambeth, et al., Multifunctional and robust covalent organic framework-nanoparticle hybrids, J. Mater. Chem. A, 2014, 2(21), 7944–7952, 10.1039/c4ta00284a.
- P. G. Tsyrul'nikov, T. N. Afonasenko and S. V. Koshcheev, et al., State of palladium in palladium-aluminosilicate catalysts as studied by XPS and the catalytic activity of the catalysts in the deep oxidation of methane, Kinet. Catal., 2007, 48(5), 728–734, DOI:10.1134/S0023158407050187.
- L. Meng, J. J. Lin and Z. Y. Pu, et al., Identification of active sites for CO and CH4 oxidation over PdO/Ce1-xPdxO2-delta catalysts, Appl. Catal., B, 2012, 119, 117–122, DOI:10.1016/j.apcatb.2012.02.036.
- Q. L. Zhu, J. Li and Q. Xu, Immobilizing metal nanoparticles to metal-organic frameworks with size and location control for optimizing catalytic performance, J. Am. Chem. Soc., 2013, 135(28), 10210–10213, DOI:10.1021/ja403330m.
- Y. Yan, H. Li and Z. H. Lu, et al., Effects of reduction temperature and content of Pd loading on the performance Pd/CeO2 catalyst for CO oxidation, Chin. Chem. Lett., 2019, 30(6), 1153–1156, DOI:10.1016/j.cclet.2019.03.030.
- K. Ahmad and S. Upadhyayula, Influence of reduction temperature on the formation of intermetallic Pd2Ga phase and its catalytic activity in CO2 hydrogenation to methanol, Greenhouse Gases: Sci. Technol., 2019, 9(3), 529–538, DOI:10.1002/ghg.1872.
- I. Y. Ahn, H. L. Ji and S. K. Sang, et al., Formation of C4 species in the deactivation of a Pd/SiO2 catalyst during the selective hydrogenation of acetylene, Catal. Today, 2007, 123(1–4), 151–157, DOI:10.1016/j.cattod.2007.02.011.
- L. Y. Zhang, Y. X. Ding and K. H. Wu, et al., Pd@C coreshell nanoparticles on carbon nanotubes as highly stable and selective catalysts for hydrogenation of acetylene to ethylene, Nanoscale, 2017, 9, 14317–14321, 10.1039/C7NR04992G.
- Y. Luo, S. A. Villaseca and M. Friedrich, et al., Addressing electronic effects in the semi-hydrogenation of ethyne by InPd2 and intermetallic Ga-Pd compounds, Catalysis, 2016, 338, 265–272, DOI:10.1016/j.jcat.2016.03.025.
- L. D. Shao, W. Zhang and M. Armbruester, et al., Nanosizing Intermetallic Compounds Onto Carbon Nanotubes: Active and Selective Hydrogenation Catalysts, Angew. Chem., Int. Ed., 2011, 50, 10231–10235, DOI:10.1002/anie.201008013.
- Q. Feng, S. Zhao and Q. Xu, et al., Mesoporous Nitrogen-Doped Carbon-Nanosphere-Supported Isolated Single-Atom Pd Catalyst for Highly Efficient Semihydrogenation of Acetylene, Adv. Mater., 2019, 31(36), 1901024, DOI:10.1002/adma.201901024.
- B. Yang, R. Burch and C. Hardacre, et al., Mechanistic Study of 1,3-Butadiene Formation in Acetylene Hydrogenation over the Pd-Based Catalysts Using Density Functional Calculations, J. Phys. Chem. C, 2014, 118(3), 1560–1567, DOI:10.1021/jp408807c.
- W. Huang, J. R. Mccormick and R. F. Lobo, et al., Selective hydrogenation of acetylene in the presence of ethylene on zeolite-supported bimetallic catalysts, J. Catal., 2007, 246, 40–51, DOI:10.1016/j.jcat.2006.11.013.
- J. Wood, M. J. Alldrick and J. M. Winterbottom, et al., Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) study of ethyne hydrogenation on Pd/Al2O3, Catal. Today, 2007, 128, 52–62, DOI:10.1016/j.cattod.2007.04.016.
- Q. Zhang, W. Dai and W. Mu, et al., Pd-Ag/Al2O3 and Pd/Al2O3 catalysts for selective hydrogenation of ethyne with in-situ DRIFTS, CIESC J., 2011, 62, 71–77, DOI:10.3724/SP.J.1077.2011.00165.
- J. Zhao, L. Ma and X. L. Xu, et al., Synthesis of carbon-supported Pd/SnO2 catalyst for highly selective hydrogenation of 2,4-difluoronitrobenzene, Chin. Chem. Lett., 2014, 25, 1137–1140, DOI:10.1016/j.cclet.2014.01.024.
- X. Li, Z. Jin and Q. Dai, et al., Liquid phase hydrodechlorination of chlorinated aromatics at lower temperature on highly defective Pd sites, Res. Chem. Intermed., 2018, 45(3), 1087–1104, DOI:10.1007/s11164-018-3670-z.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03552b |
| This journal is © The Royal Society of Chemistry 2023 |