
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Mizoroki–Heck reaction between in situ generated alkenes and aryl halides: cross-coupling route to substituted olefins
Anushka Korannea,
Shrishty Turakhiaa,
Vikesh Kumar Jhab,
Sangeeta Guptaa,
Rangnath Ravic,
Abhijeet Mishrac,
Anil K. Aggarwalc,
Chandan Kumar Jhad,
Neelu Dheer e and
Abadh Kishor Jha*a
e and
Abadh Kishor Jha*a
aGovt. Shivnath Science College, Rajnandgaon, 491441, Chhattisgarh, India. E-mail: awadhbrave@gmail.com
bGovt. R. R. M. PG College, Surajpur, Chhattisgarh, India
cShivaji College, University of Delhi, Delhi, India
dGovt. Engineering College, Madhubani, Bihar, India
eAcharya Narendra Dev College, University of Delhi, Delhi, India
First published on 25th July 2023
Abstract
This review covers palladium-catalyzed typical Mizoroki–Heck cross-coupling reactions of aryl halides with in situ generated alkenes, by following a typical Heck coupling mechanism to form substituted olefins unlike direct cross-coupling of alkenes with aryl halides in Heck olefination. These reactions solve the issue of alkenes undergoing polymerization at high temperatures and increase reaction efficiency by reducing the reaction time and purification steps.
1. Introduction
Cross-coupling reactions catalyzed by palladium are considered to be the most competent scientific method for the formation of C(sp2)–C(sp2) bonds.1 Most of these reactions proceed through a combination of a nucleophile which is generally an organometallic reagent with an electrophile to provide the corresponding coupling product.2 However, one major limitation of this general approach is the necessity of a stoichiometric amount of the organometallic reagents, which are often prepared via expensive and multistep procedures. The Heck reaction is considered as the earliest example of a C–C bond forming reaction that does not require a stoichiometric amount of the organometallic coupling partner. It is one of the most interesting reaction catalyzed by palladium, involving the chemical reaction of an unsaturated halide (or triflate) with an alkene in the presence of a base to form a substituted alkene3,4 (Scheme 1). The usual mechanism involves mainly four steps which are, oxidative addition, migratory insertion, β-hydride elimination and reductive elimination as shown in Fig. 1. The reaction initiates with oxidative addition of aryl halide to palladium catalyst. Upon introduction of alkene into this, a palladium pie complex gets formed which is then followed by migratory insertion of this alkene into the palladium-aryl bond. Finally β-hydride elimination produces a new substituted olefin. The concluding step of the reaction involves reductive elimination to generate the active palladium catalyst. | ||
| Scheme 1 Palladium catalyzed Mizoroki–Heck coupling reaction. | ||
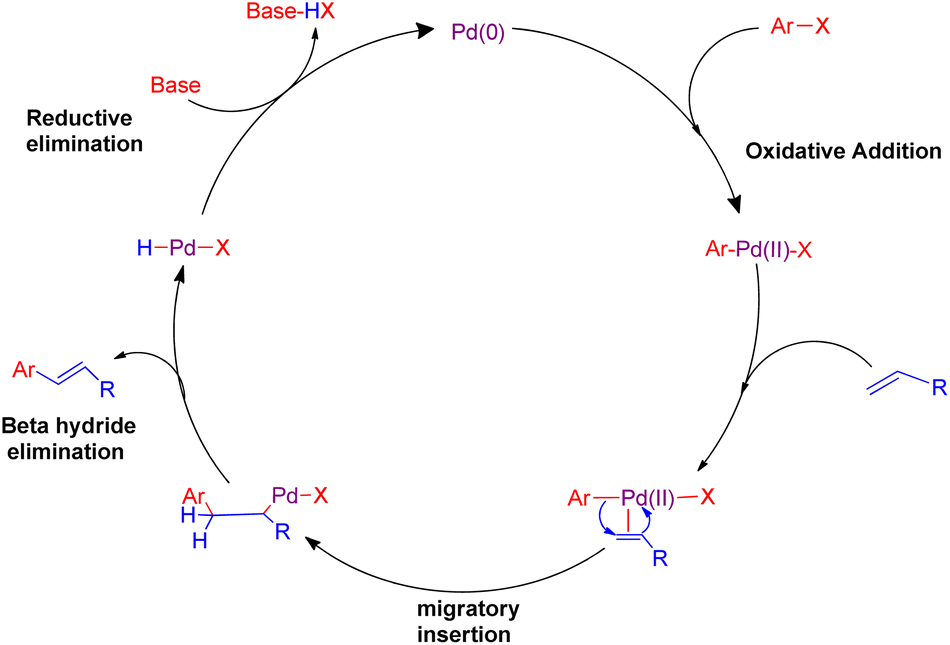 | ||
| Fig. 1 Mechanism of palladium catalyzed Mizoroki–Heck coupling reaction. | ||
This Heck reaction involves various useful transformations which result in the formation of complex organic molecules including bioactive stilbenoids, anticancer agent resveratrol, DMU-212, etc.4 The reaction has been utilized to create a wide range of natural and synthetic compounds, and in most occasions, the transformation occurs in presence of a suitable aryl halide and styrene or similar olefin, including a small amount of palladium catalyst and a stoichiometric amount of a base.
However, the alkenes necessary for the reaction are either difficult to manufacture or hard to purify due to their tendency to polymerize during distillation or storage. Moreover even on employing commercially available olefins, or synthesizing them via various traditional methods available, there remains a possibility of them undergoing polymerization under high reaction temperature conditions and hence are required in higher amount.5
To solve some of these issues, many scientists have reported Heck reaction between in situ generated alkenes and aryl halides. The 1st one of these methods include in situ generation of required alkenes for Heck coupling from unconventional substrates such as 1 or 2-bromo/hydroxy/acetoxy alkyl arenes (Scheme 2) or 1,2-dihaloethane/2-haloethyl acetate/1,2,3,4-tetrazine derivatives/2-chlorosulfonyl chloride/1-iodo-3,3,3-trifluoropropane (Scheme 3). While the 2nd method utilizes Wittig alkenes for sequential Wittig–Heck process (Scheme 4). In literature there are many reports for the in situ synthesis of alkenes by the usage of a carbene precursor such as diazo compounds. However the mechanism when utilizing carbene precursor differs from the traditional Mizoroki–Heck coupling mechanism as it involve a supposed metal-carbene migration process6 and is therefore out of the scope of this review. The 1st method as stated above involves in situ generation of alkenes via various processes such as dehydrohalogenation of 1 or 2-halo ethyl benzene,5,7 dehydration of 1 or 2-hydroxy alkyl arene,7,8 or deacetoxylation of 1 or 2-acetoxy ethyl benzene (Scheme 2).7–9 It also includes the dehydrohalogenation and deacetoxylation of 1,2-dihalo ethane7 and 2-haloethyl acetate7 respectively, followed by their coupling with aryl halide prior to loosing another functional group. In addition, hydrolysis followed by dehydrohalogenation of 2-chloroethanesulfonyl chloride10 or dehydrohalogenation and elimination pathway in 1-iodo-3,3,3-trifluoropropane11 and substituted 1,2,3,4-tetrazine14 respectively were also some of the methods developed to form the desired substituted olefin (Scheme 3).
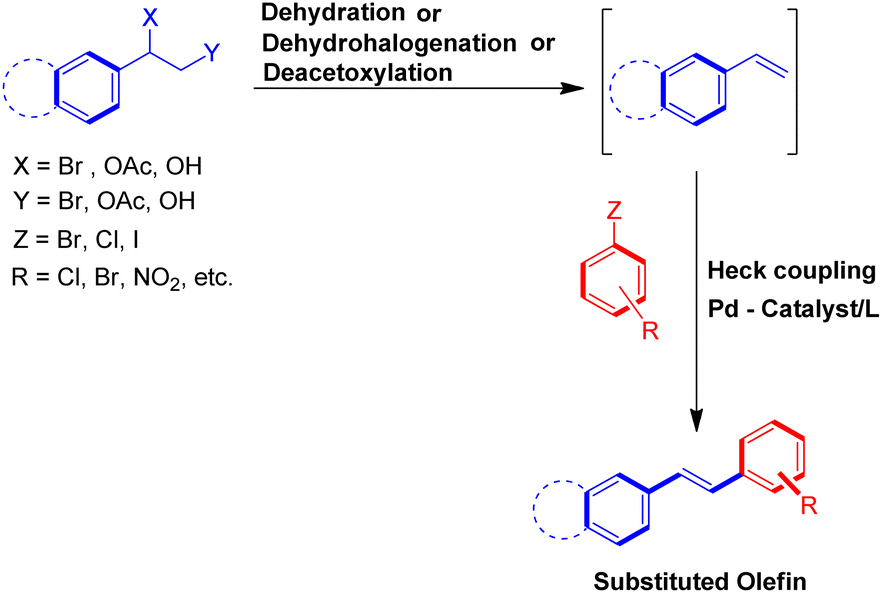 | ||
| Scheme 2 Heck reaction between in situ generated alkenes from 1 or 2-bromo/hydroxy/acetoxy alkyl arenes and aryl halides. | ||
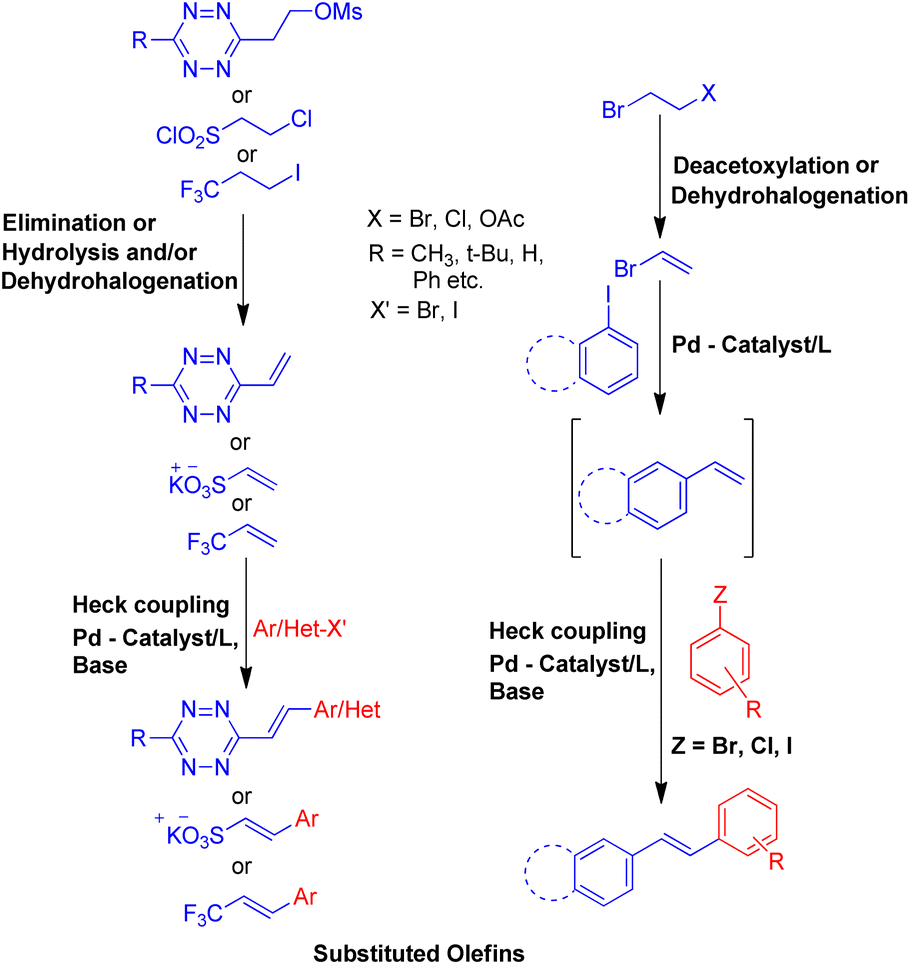 | ||
| Scheme 3 Heck reaction between aryl halide and in situ generated alkenes from 2-chloroethanesulfonyl chloride, 1-iodo-3,3,3-trifluoropropane, 1,2,3,4-tetrazine derivatives, 1,2-dihaloethane and 2-halo ethyl acetate with aryl halides. | ||
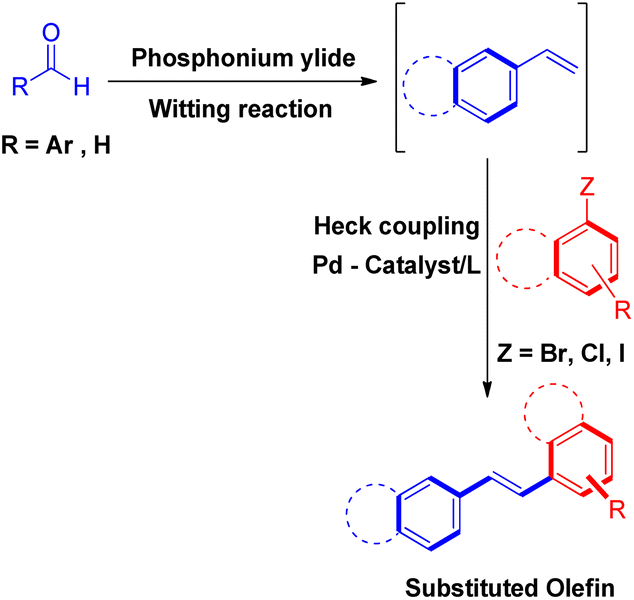 | ||
| Scheme 4 Heck reaction between in situ generated Wittig alkenes and aryl halides. | ||
The second method of tandem Wittig–Heck reaction5,15–27 involves generation of required olefins by reacting aldehyde or ketones with acceptable phosphonium salt and then coupling of these olefin with aryl halide under Heck conditions to result in the formation of desired conjugated species (Scheme 4). Both of these methods of in situ generating the suitable olefins and then proceeding further with Heck coupling have been summarized in this review.
2. Mizoroki–Heck reaction between in situ generated alkenes from 1 or 2-bromo/hydroxy/acetoxy alkyl arenes or 1,2-dihaloethane/2-haloethyl acetate/1,2,3,4-tetrazine derivatives/2-chlorosulfonyl chloride/1-iodo-3,3,3-trifluoropropane and aryl halides
The expansion of Heck reaction using more unusual substrates was 1st performed by Edith J. Parsons and co-workers in 1995. Their primary focus was the in situ generation of alkenes capable of undergoing coupling.7 The usage of “alkene synthons” depends on the unique properties of superheated and supercritical water (SW) which expanded the scope of the Heck reaction to include alkane-based unconventional substrates in addition to the traditional alkene based substrates. They examined a series of alkene synthons in the Heck coupling reaction under supercritical water conditions (Scheme 5). These alkene synthons included, 2-hydroxy ethyl benzene (2a′), 2-bromo ethyl benzene (2a′′), and 2-acetoxy ethyl benzene (2a′′′) (Scheme 5). Apart from them, 1,2-dihaloethane and 2-haloethyl acetate (2aa) were also examined (Scheme 7).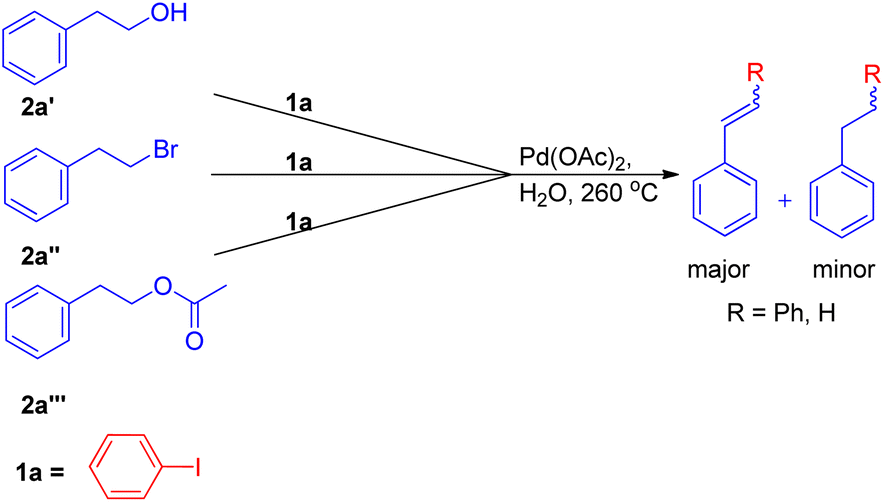 | ||
| Scheme 5 Potential alkene synthons examined in the Heck coupling reaction in superheated water. | ||
They substituted styrenes in the coupling reaction with these synthons and made them to react with iodobenzene, using PdCl2 or Pd(OAc)2 as the catalyst precursor and NaOAc as the base. Each of them were able to form the desired trans-stilbene (3a) and some amount of 1,1-diphenylethylene (3a′) (Scheme 6), however the reported yield of the product was slightly lower from what was observed in the reaction of styrene itself. The production of styrene along with its hydrogenated analogue ethylbenzene (Scheme 6; 3a′′ and 3a′′′ respectively) indicated that synthons were defunctionalized under the applied reaction conditions to form alkenes which then underwent coupling with aryl halide. The presence of significant amount of 2-hydroxy ethyl benzene (2a′) was observed in the reaction mixture when 2-acetoxy ethyl benzene (2a′′) and 2-bromo ethyl benzene (2a′′′) were coupled separately with iodobenzene, which suggested that only 2-hydroxy ethyl benzene was responsible for the formation of styrene and the end products. However the effect of presence of various electron rich and electron poor groups on these alkene synthons (2a′, 2a′′, and 2a′′′) is yet to be explored.
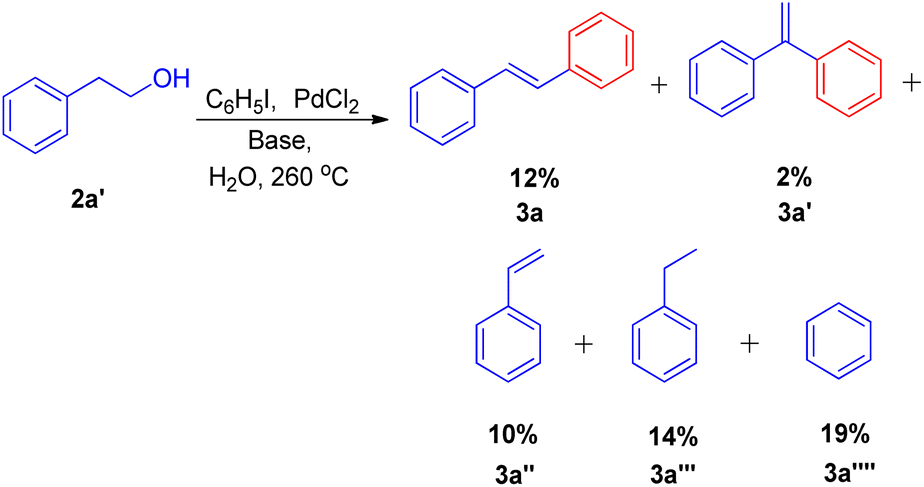 | ||
| Scheme 6 Palladium catalyzed coupling of styrene synthon with iodobenzene. | ||
Vinyl synthons (2aa) were also examined in superheated water coupling system, and the reaction was found to proceed via dehydrobromination first and then coupling with iodobenzene occurred with the loss of other halogen entity to result in styrene which eventually reacted with iodobenzene to form trans-stillbene. However the desired stilbene, was obtained in very low yield (Scheme 7).
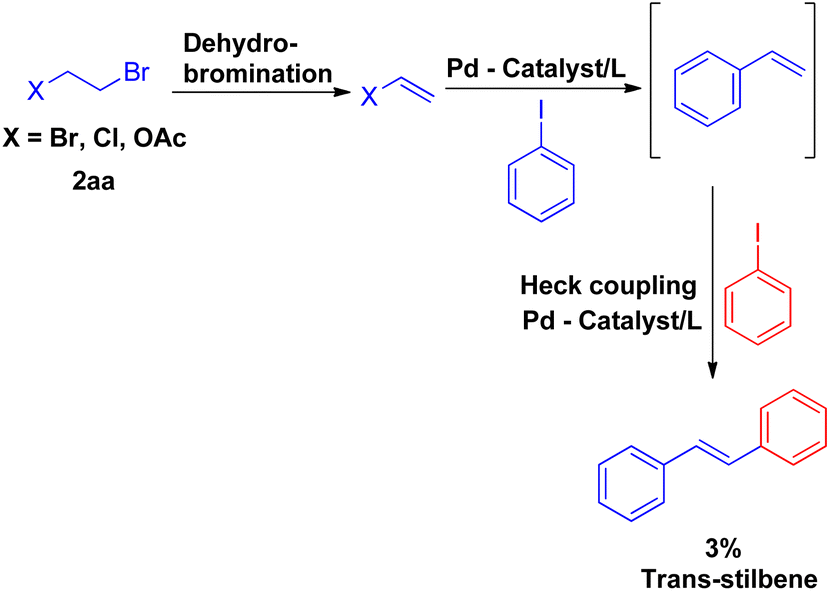 | ||
| Scheme 7 Palladium catalyzed coupling of iodobenzene with alkene generated from 1,2-dihaloethane/2-haloethyl acetate (2aa) and iodobenzene. | ||
In 2010 Akeel S. Saiyed and Ashutosh V. Bedekar5 reported a one pot synthesis which involves olefination followed by the Mizoroki–Heck reaction. The main feature of this method is the in situ synthesis of required olefin (2b) from alkyl halides. Olefins were synthesized by base mediated dehydrohalogenation of 1 or 2-bromoalkyl arenes followed by coupling with aryl halide to form stilbene under Mizoroki–Heck condition (Scheme 8).
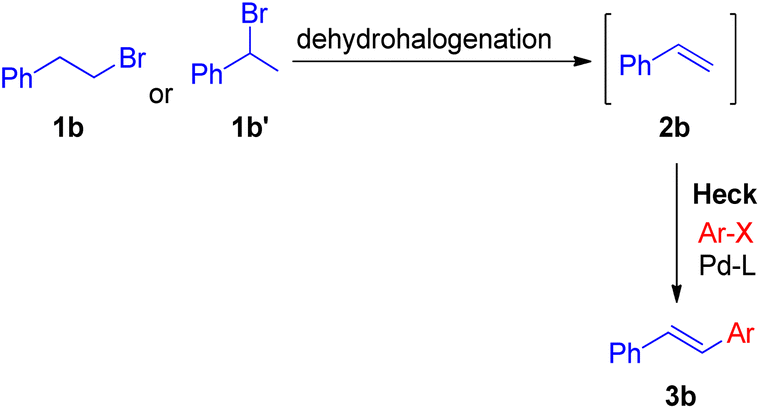 | ||
| Scheme 8 Proposed routes of in situ synthesis of styrene for one-pot Heck reaction. | ||
The optimized conditions (Scheme 9) for this protocol involves reaction between 1.2 equiv. of aryl halide (2b′) and 1 equiv. of alkyl halide (1b–1b′′′) in the presence of Pd(OAc)2 (0.5 mol%) as catalyst, 3 equiv. K2CO3 as base, 0.55 mol% of oxazolinyl ligand in DMA at 140 °C for 40 h to result in formation of substituted olefin in good to excellent yield (54–88%).
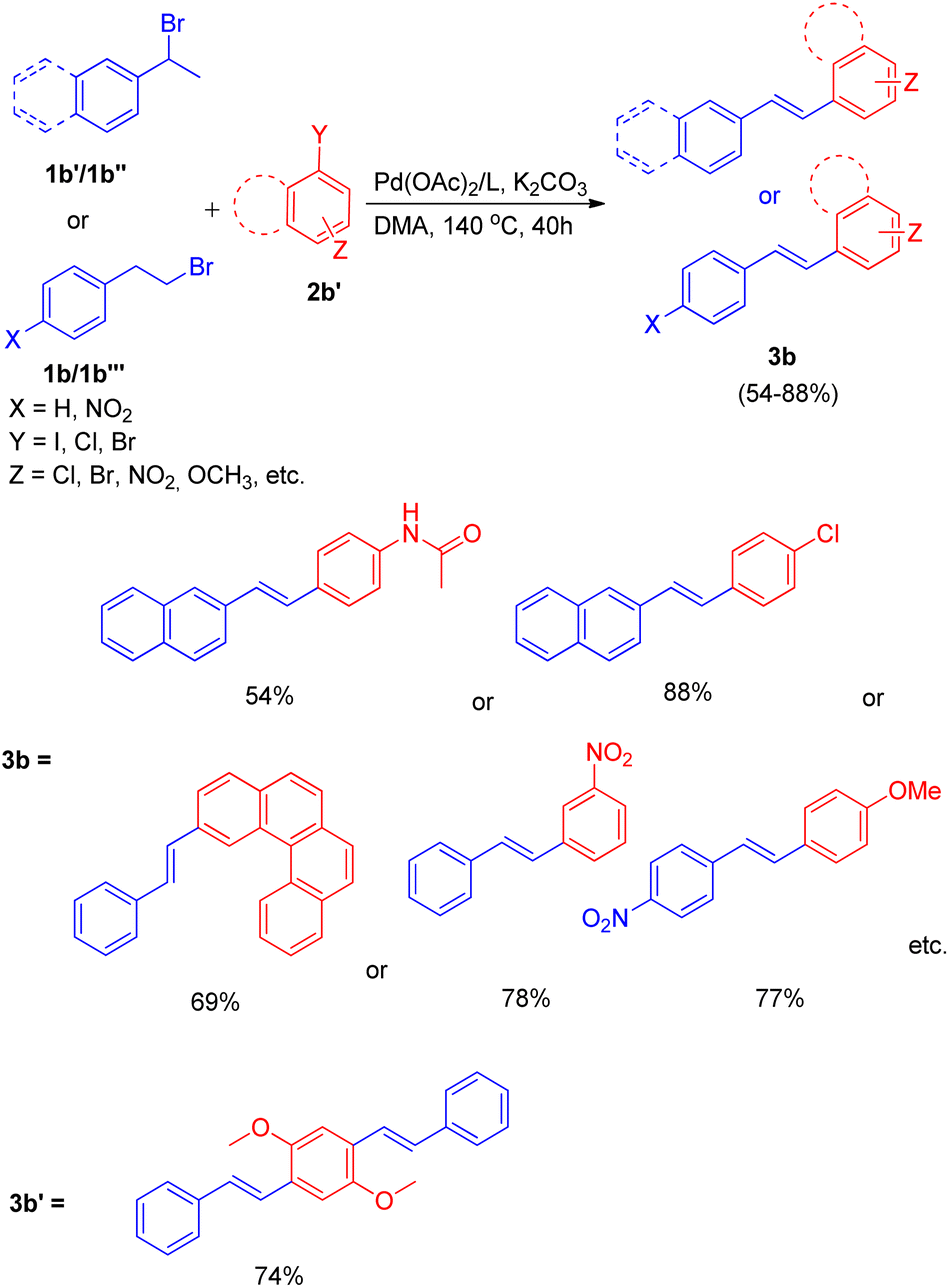 | ||
| Scheme 9 Dehydrohalogenative one-pot Heck reaction. | ||
When under the same reaction conditions, 1,4-diiodo-2,5-dimethoxybenzene was used as an aryl halide to undergo coupling with 2-bromoalkyl arene (1b), double dehydrohalogenative Heck reaction was observed which yielded (3b′) the 74% of corresponding stilbene. To show the generality of the dehydrohalogenative Heck approach, a series of 1 or 2-bromoalkyl arenes (Fig. 2) were treated with suitable aryl halide. The reaction mixture containing suitable aryl halide with bromoalkyl arene was made to react under optimized reaction conditions, which resulted in formation of good yield (54–88%) of stilbene derivatives.
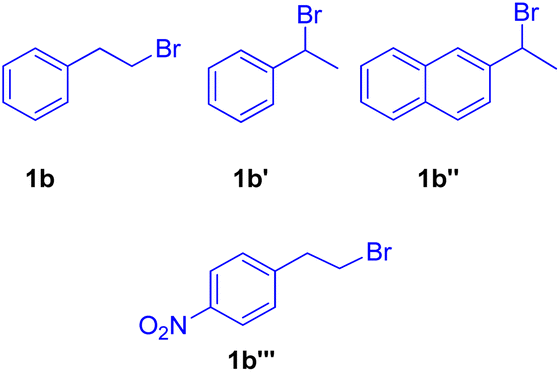 | ||
| Fig. 2 1 or 2-bromoalkyl arenes used for in situ generation of olefins by dehydrohalogenation followed by Mizoroki–Heck reaction. | ||
After the work by Akeel S. Saiyyed and Ashutosh V. Bedekar5 on the dehydrohalogenative Heck reaction, another domino hydrolysis/dehydrohalogenation/Heck reaction was reported by G. K. Surya Prakash et al. in 2011.10 The method employs chloroethanesulfonyl chloride as an alkene synthon which undergoes hydrolysis followed by dehydrohalogenation to form the required olefin for Heck reaction. This alkene then couples with haloarenes to result in the corresponding substituted styrene sulfonate salts. Apart from requiring short reaction time and water as a medium, one of the many advantages offered by this methodology is the utilization of phosphine and additive-free catalytic system. The investigation to attain optimized conditions began by reacting 2-chloroethanesulfonyl chloride (1c) with iodobenzene (2c) in water in the presence of Pd(OAc)2 (2 mol%) as catalyst and potassium carbonate as a base at 180 °C for 10 min in the microwave (Scheme 10). Among the analyzed bases such as NaHCO3, K2CO3, Na2CO3, and triethylamine, K2CO3 (2–3 equiv.) resulted in the highest conversion (85%) of the coupling product (3c). Owing to the absence of ligand in the reaction mixture, the possibility of deactivation of Pd(0) catalyst to form palladium black was avoided by the addition of fresh 1 mol% of the catalyst to the previous reaction mixture after heating it for 10 min in microwave (as mentioned above) and heating it again in the microwave at 180 °C for 10 min.
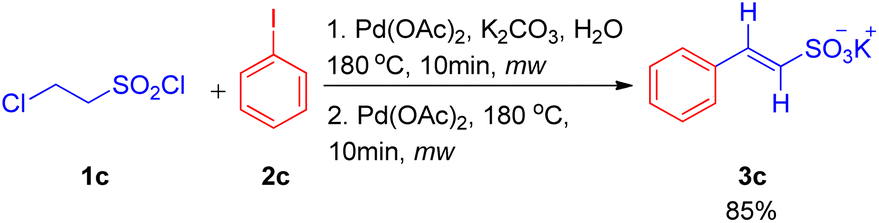 | ||
| Scheme 10 Domino hydrolysis/dehydrohalogenation/Heck coupling pathway for the synthesis of potassium styrene sulfonates. | ||
With the optimized conditions in hand i.e. usage of 2 mol% Pd(OAc)2 as catalyst with 3 equiv. of K2CO3 as base in water at 180 °C for 10 min under microwave, a variety of haloarenes (2c′; 1 equiv.) were analyzed in a reaction with 2-chloroethanesulfonyl chloride (1 equiv.). Apart from iodobenzene which gave 85% of the isolated styrene sulfonate salt, various substituted iodobenzene displayed a good tolerance towards presence of electron withdrawing and electron-donating groups (Scheme 11) giving moderate to excellent yields (25–89%). On the other hand poisoning of Pd catalyst in case of 2-aminoiodobenzene, 2-iodobenzoic acid and ortho substituted heterocycles (2-iodopyridine, 2-iodopyrazine, and 2-iodothiophene) lead to zero to inferior yield of the coupled product (3c′). Other heterocycles such as 5-iodoindole and 3-iodopyridine etc. were able to drive the reaction to completion with moderate yield of the olefinated product (47–66%).
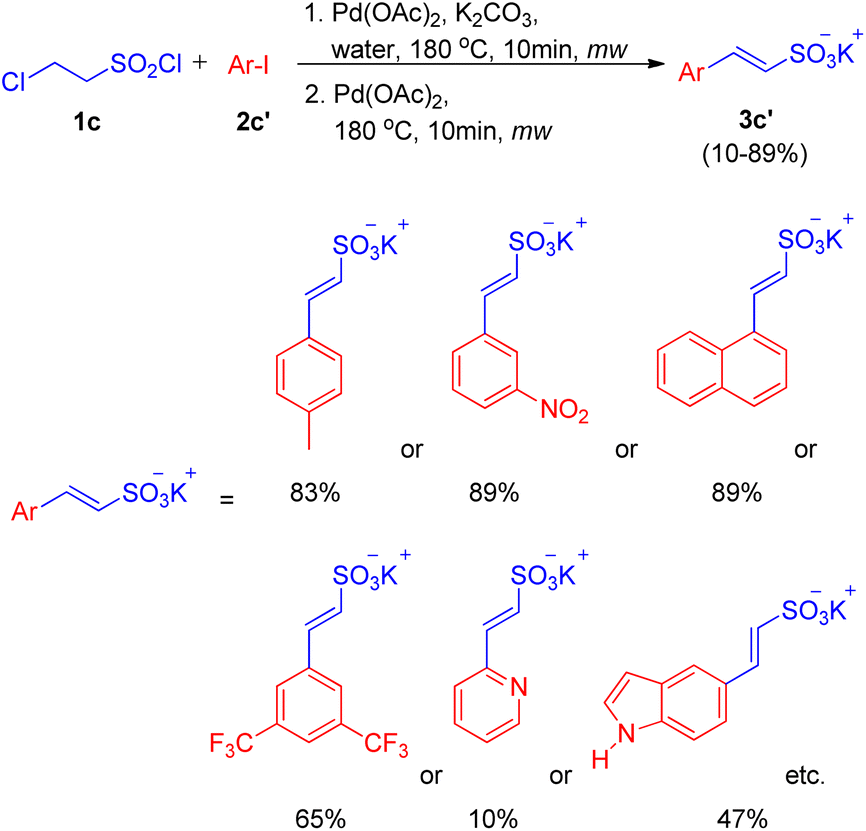 | ||
| Scheme 11 Substrate scope for domino (hydrolysis/dehydrohalogenation/Heck) coupling reaction. | ||
The mechanism being followed in this reaction is proposed to initiate with the hydrolysis of sulfonyl chloride to chlorosulfonate under basic conditions followed by it’s dehydrohalogenation at higher temperature to yield the required vinyl sulfonate in situ which eventually undergoes coupling with haloarene to form the desired Heck-coupled product.
In an attempt to synthesize disulfonate salts (3c′′), the above mentioned protocol was also extended to a number of disubstituted iodobenzenes (Scheme 12; 2c′′), which resulted in generation of disulfonate salts in good yields (29–75%).
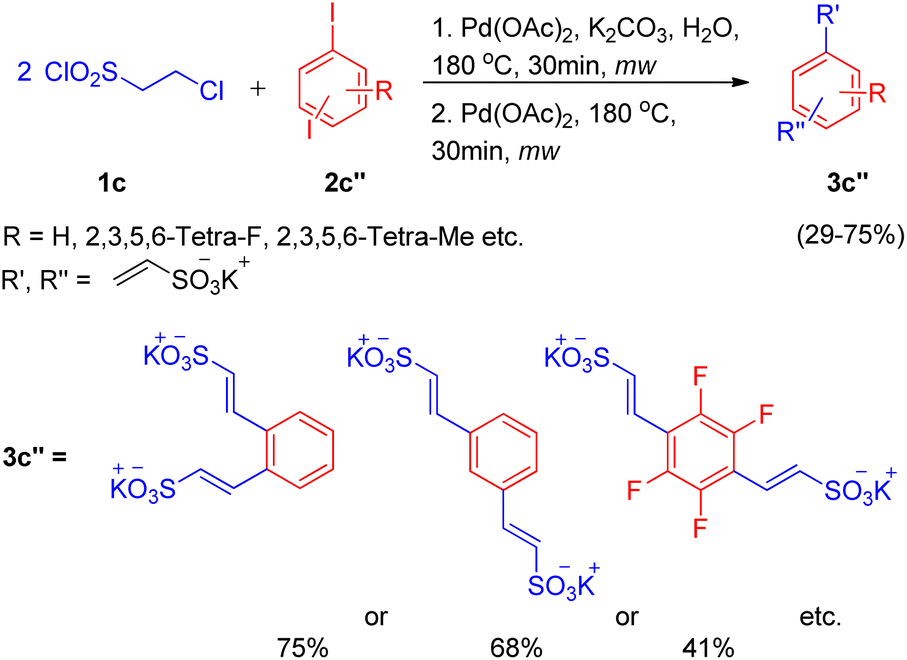 | ||
| Scheme 12 Synthesis of disulfonate salts. | ||
Using a similar domino approach for Heck reaction, G. K. Surya Prakash and co-workers in 2012,11 synthesised β-trifluoromethylstyrene derivatives (3d) via Heck reaction, in moderate to good yields (52–78%) by using 1-iodo-3,3,3-trifluoropropane (1d) (as an alkene synthon) and iodoarenes (2d) under basic conditions. In order to reach the optimized conditions, numerous screening experiments were performed which revealed that, among various palladium catalysts analyzed, such as Pd(OAc)2, Pd2(dba)3 and Pd(PPh3)2Cl2, and bases such as Li2CO3, Na2CO3, K2CO3, and Cs2CO3, using Pd(OAc)2 (2 mol%) as the catalyst and K2CO3 (3 equiv.) as base results in maximum yield of the desired product. Thus the reaction between 3-iodotoluene (1 equiv.) and 1-iodo-3,3,3-trifluoropropane (1 equiv.) under optimized conditions i.e. using Pd(OAc)2 (2 mol%) as catalyst, 3 equiv. K2CO3 as base in DMF as solvent at 200 °C for 1 h in microwave resulted in formation of desired product with 83% yield (Scheme 13).
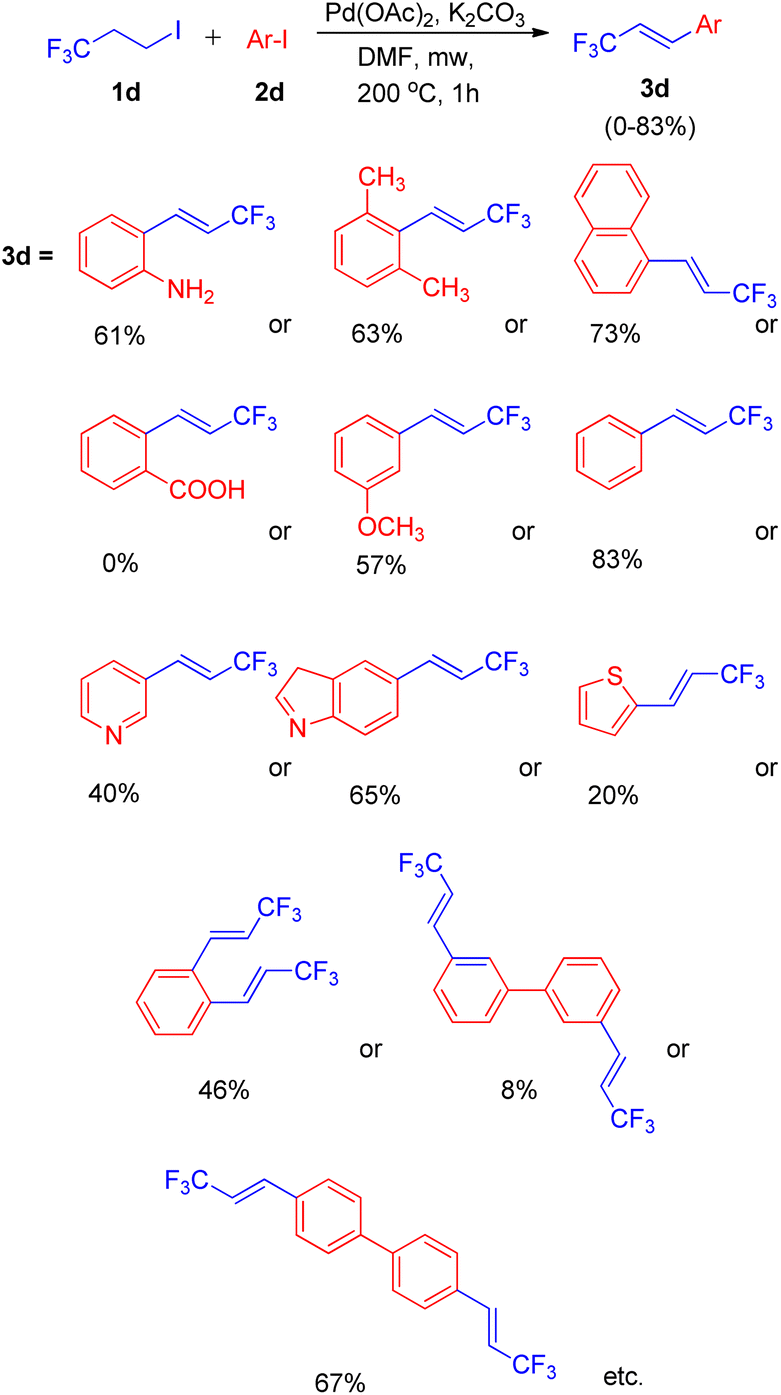 | ||
| Scheme 13 Synthesis of β-trifluoromethylstyrenes, heterocyclic and bis(β-trifluoromethyl)styrenes. | ||
Under the above stated optimized conditions, except chlorobenzene, both bromobenzene and iodobenzene were able to form β-trifluoromethylstyrene in 10% and 83% yield respectively. The higher yield of desired product obtained in case of iodobenzene lead to the analysis of various substituted iodobenzenes under optimized reaction conditions. The presence of electron rich substituents in comparison to electron deficient ones on iodobenzene, resulted in a better yield of the desired product and steric effect was found to be ineffective during this protocol (Scheme 13). 2-Aminoiodobenzene, which poisoned the palladium catalyst in the aforementioned protocol (Scheme 11),10 was surprisingly able to form the corresponding trifluoromethylstyrene in 52% yield. In addition to substituted iodobenzenes, various heterocyclic iodoarenes as well as diiodoarenes also gave the desired product in low to moderate yield (20–65% and 8–69% respectively).
An exclusively-Z minor double Heck product (3d′) was also observed during screening of reaction conditions (Scheme 14), however further studies are needed the attain it's optimized conditions.
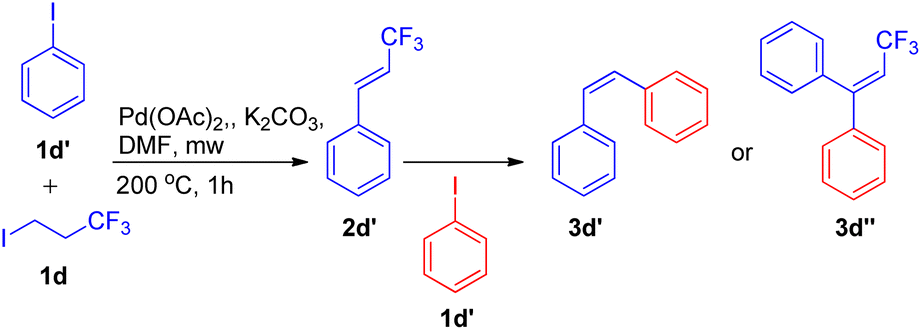 | ||
| Scheme 14 Elimination Heck-domino reaction pathway. | ||
Thus this method using an elimination/Heck domino reaction sequence offers a simple strategy towards synthesis of β-trifluoromethylstyrene derivatives by avoiding employment of additives and gaseous 3,3,3-trifluoropropene reagent in a phosphine free catalytic system.
Since alcohols are readily available and have received great attention as precursors in various tandem oxidative/dehydrative cross-coupling strategies,8 Arun Kumar Sinha and coworkers,9 in 2012 reported an approach for the waste-free dehydrative Heck olefination. This approach involved coupling of in situ formed styrenes with an aryl halide in ionic liquid. The drawback of secondary alcohols getting converted into carbonyls under Heck-type conditions11 and the possibility of cross-contamination while using different medium during dehydration (acidic) of secondary aryl alcohols to convert them into styrene and then using them in Heck (basic) reaction has been overcome by this approach of utilizing ionic liquid where only water is removed as by-product.
Initially the reaction of, 4-iodoanisole (2e, 1 equiv.) with 1-(naphthalen-2-yl)ethanol (1e, 1.5 equiv.) in [hmim]Br using Pd(OAc)2 (4 mol%) as catalyst, PPh3 (5 mol%) as ligand, and Et3N (1.5 equiv.) as a base under microwave irradiation (120 W, 150 °C) for 40 minutes was performed (Scheme 15). Analysis of the crude reaction mixture by HPTLC (over HPLC/GC) showed that 3e was obtained in very low quantity (8%). To improve the yield of 3e, a number of organic and inorganic bases such as Bu3N, piperidine, DBU, Cs2CO3, K2CO3, NaOAc, NH4OAc, and HCOONa were rapidly screened through HPTLC, where HCOONa provided 3e in moderate yield (69%). The collaboration of two bases, HCOONa (1.5 equiv.) and piperidine (1 equiv.), in addition to improving the reaction performance, also proved significant in cutting the reaction time in almost half (from 40 minutes to 15 minutes). The usage of an additive LiCl with catalyst [PdCl2(PPh3)2] also increased the yield of 3e to 78%. The crucial role of ionic liquids was established when no reaction was observed in DMF alone.
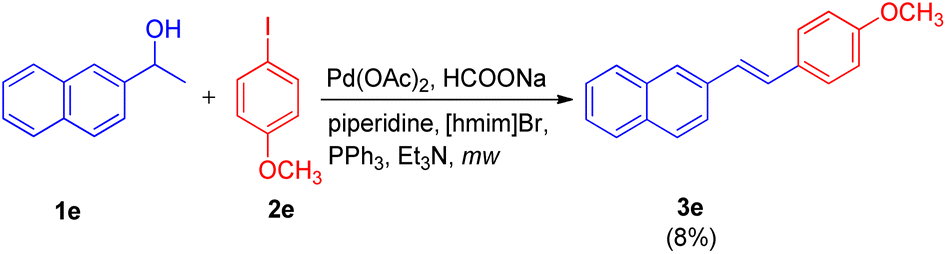 | ||
| Scheme 15 Palladium-catalyzed tendem dehydrative Heck coupling between 4-iodoanisole and 1-(naphthalen-2-yl)ethanol in [hmim]Br ionic liquid. | ||
Under the optimized reaction condition the reaction was tried with different secondary alcohols and reaction gave the desired stilbenoids (3e′) in moderate to good yield (47–89%). The products so obtained showed exclusively E selectivity (Scheme 16). A naphthalene analogue of a PTPIB inhibitor was also synthesized (52% yield) using this protocol (Scheme 16).
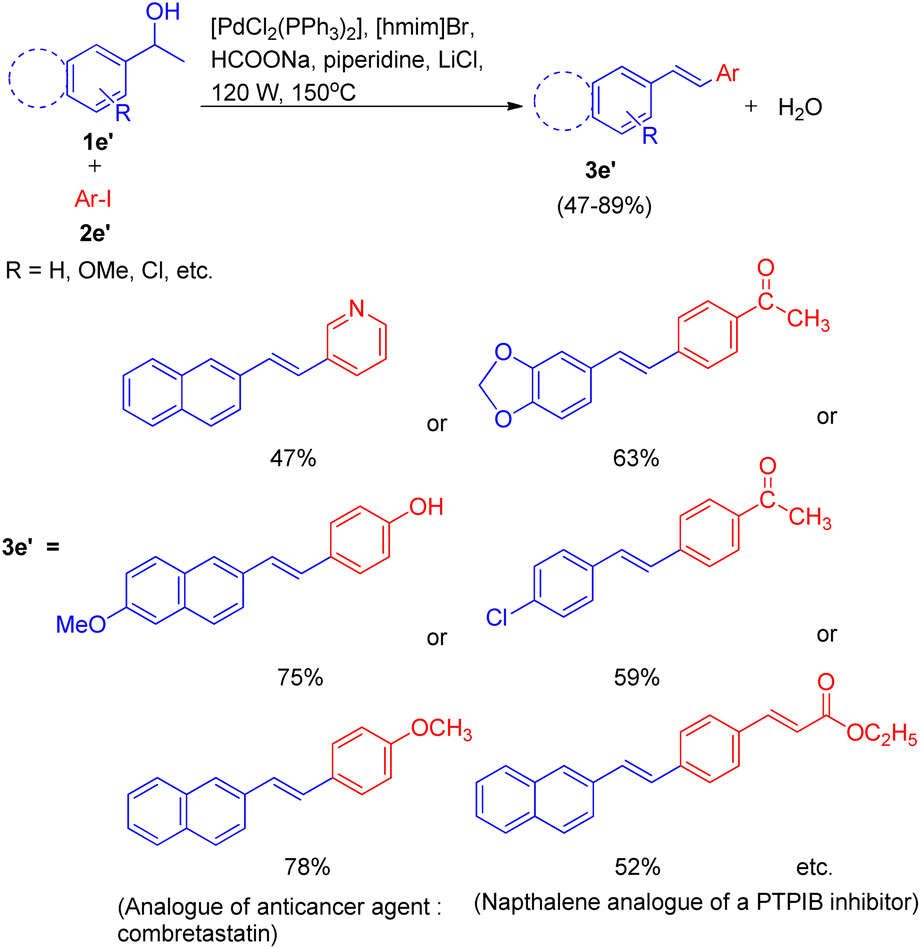 | ||
| Scheme 16 Conversion of secondary aryl alcohols into stilbenoids. | ||
Motivated by these results, they applied the same strategy for the one-pot deacetoxylative Heck coupling of the acetylated derivative of alcohols (1e′′, Scheme 17) and the corresponding stilbenes (3e′′) were obtained in good yields (70–81%).
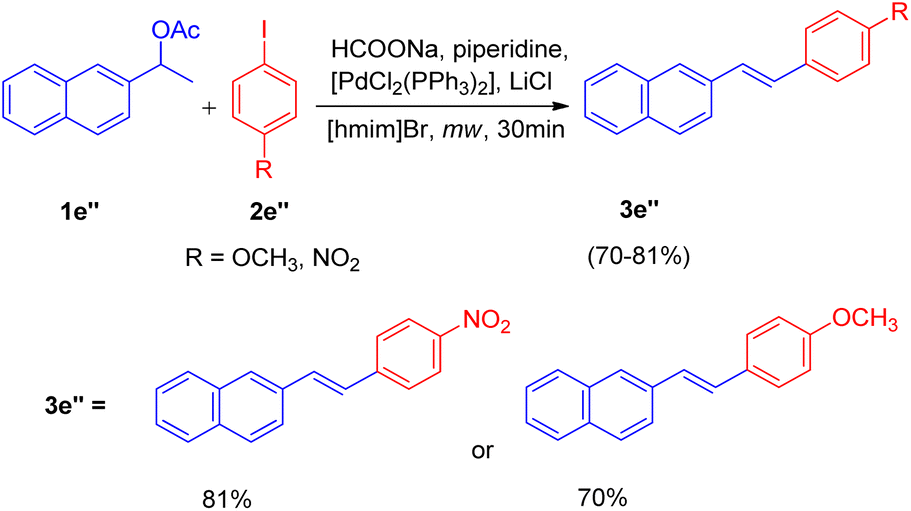 | ||
| Scheme 17 Deacetoxylative Heck coupling. | ||
A diverse range of stilbene possessing electron withdrawing as well as electron donating groups on either of the 2 substrates (secondary aryl alcohol and aryl halide) can be synthesized using the above stated protocol. This approach provided good yields of the desired stillbene when using polyaromatic alcohols, however, due to the polymerization tendency of the intermediate styrene,8 the electron deficient alcohols could only provide required substituted olefins in moderate yield. The proposed mechanism for this protocol has been shown in Fig. 3. The mechanism is proposed to initiate by the microwave and ionic liquid assisted dehydration reaction of 1e′ which leads to formation of styrene (1ee), this styrene then undergoes Pd-catalyzed Heck coupling with aryl halide (2e′) to give the substituted stilbene (3e′). One of the major application of this protocol is it's ability to synthesize numerous analogues of anticancer agent combretastatin12 (3e′, 78%) without the requirement of any protection/deprotection step (Scheme 16).
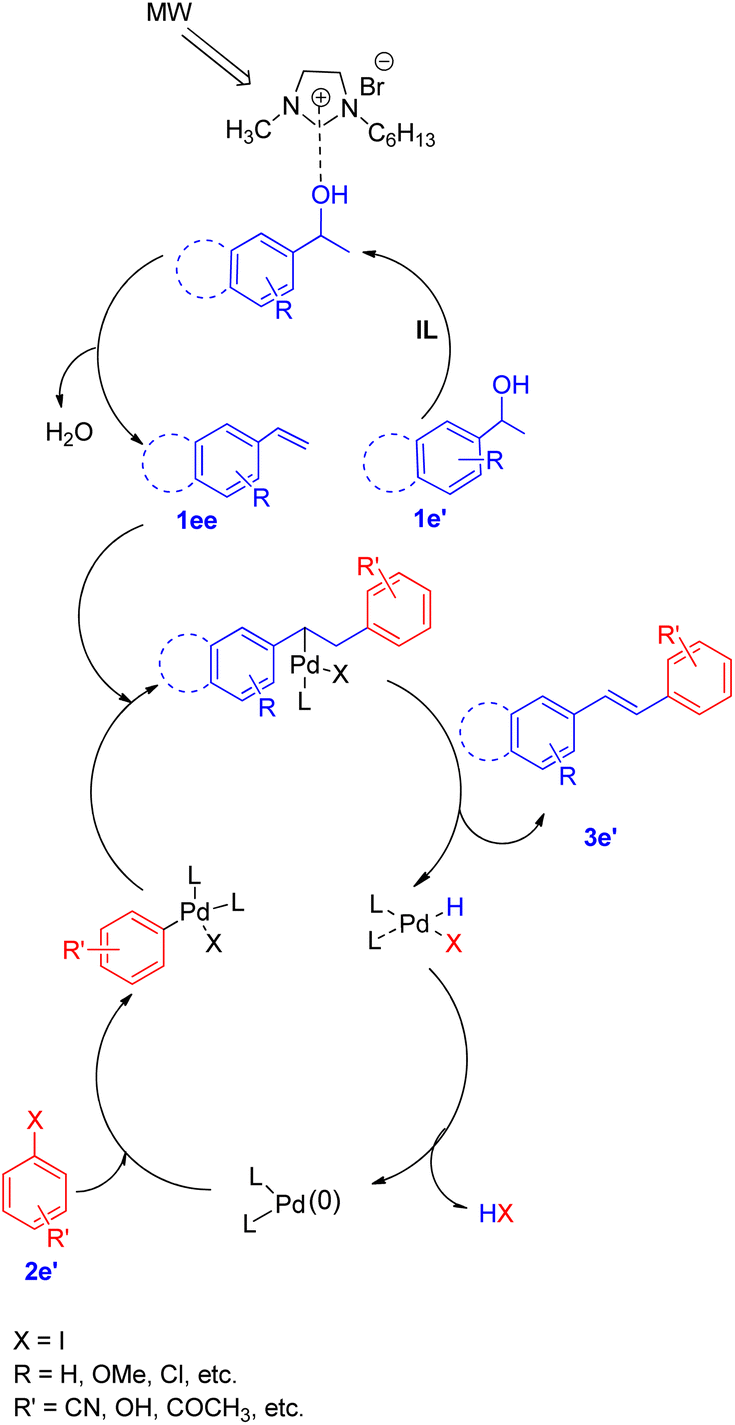 | ||
| Fig. 3 Proposed mechanism for tandem dehydrative Heck coupling in an ionic liquid under microwave irradiation. | ||
After the development of dehydrative Heck reaction in an ionic liquid (acting as solvent) by A. K. Sinha and co-workers,9 a similar method was reported by Jianliang Xiao et al. in the same year (2012)13 with the difference of involving usage of heteropolyacids (HPAs) instead of ionic liquid. The path to achieving optimized conditions began with thermal activation of 1-(4-methoxyphenyl)-ethanol (1f; 1.5 equiv.) in the presence of H3PW12O40 (HPA) in a number of solvents such as DMF, DMA, DMSO, MeCN, hexane, ethanol, anisole, and diglyme etc. among which only DMSO was able to drive the dehydration to completion with 55% isolated yield of the desired alkene (1f′). The development of dehydration conditions was followed by screening of reaction parameters for further coupling with aryl halide (2f; 1 equiv.). The neutralization of reaction media caused by the presence of both HPA and base at the same time was overcome by forming a two-step procedure involving dehydration of aryl alcohol (1.5 equiv.) in the presence of HPA in DMSO at 100 °C for 1 h with the subsequent coupling of the alkene formed with aryl halide (1 equiv.) on addition of a base Et3N (1.5 equiv.), catalyst Pd(dba)2 (2 mol%), and P(t-Bu)3·HBF4 (0.06 equiv.) in DMF at 100 °C for 4 h, resulting in 82% yield of the desired substituted styrene. Various aryl bromides were examined in reaction with benzyl alcohol (1f) under the optimized reaction condition (Scheme 18) and it was found that the aryl bromides containing electron-withdrawing, electron-donating groups as well as heterocyclic substrates were able to form the desired product in good-yields (66–85%).
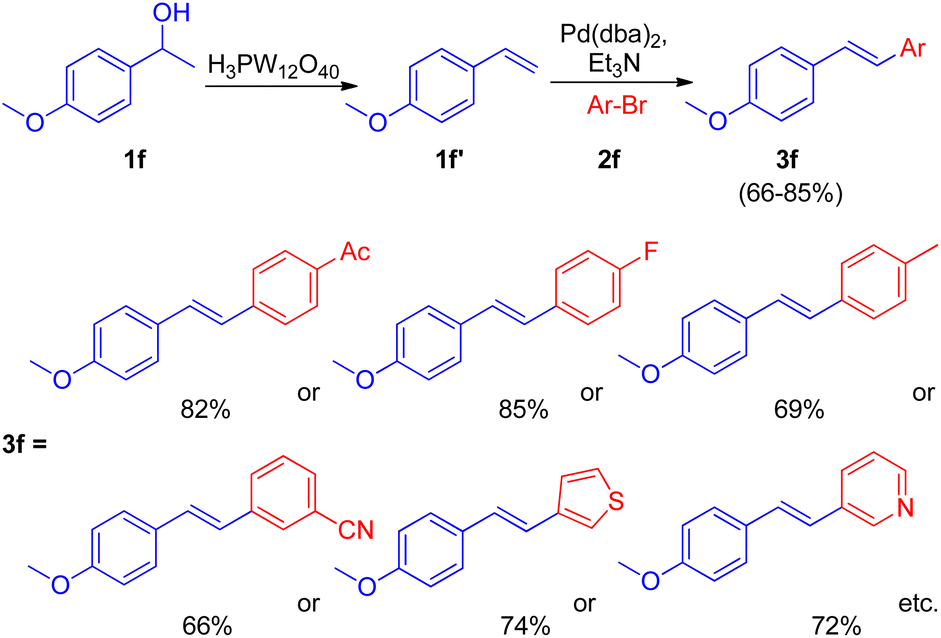 | ||
| Scheme 18 Dehydrative-Heck arylation of benzylic alcohol (1f) with aryl bromides (2f). | ||
Along with them different secondary aryl alcohols (1f′′) were also analyzed (Scheme 19) by coupling them with 4-bromoacetophenone (2f′), which showed that only electron-rich aryl alcohols were undergoing dehydration under the optimized reaction conditions however a switch from DMSO to diglyme and to 1,2-dichloroethane while using electron-neutral and electron deficient aryl alcohols respectively was necessary to afford good to excellent yield (51–91%) of the corresponding stilbenes (3f′).
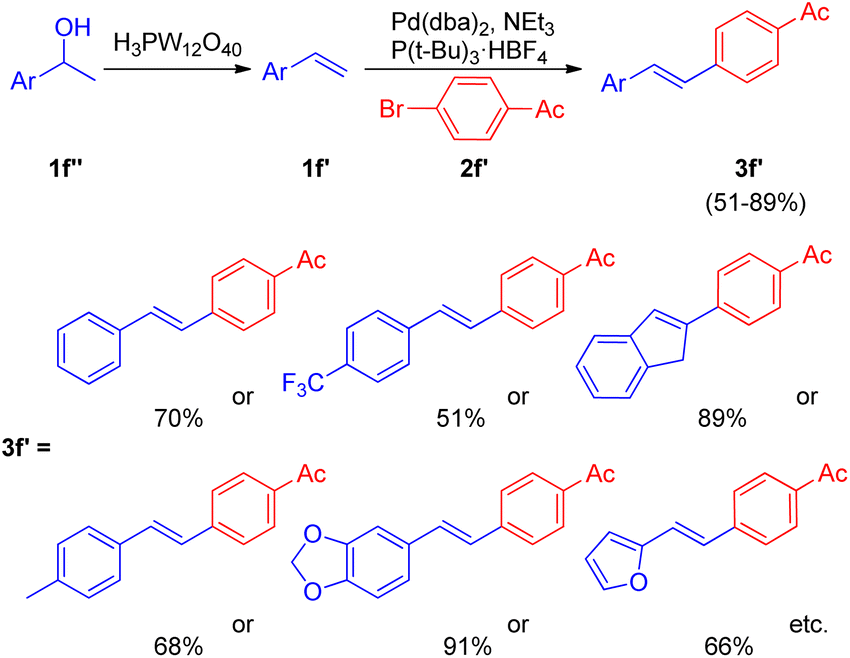 | ||
| Scheme 19 Dehydrative-Heck arylation of alcohols 1f′′ with 4-bromoacetophenone (2f′). | ||
Thus the limited substrate scope of secondary aryl alcohol while using ionic liquid as a solvent was overcome to a certain extent through this protocol.
A practical method for the synthesis of 1,2,4,5-tetrazine derivatives, which plays a significant role in live cell and in vivo imaging, was developed by K. Devaraj and co-workers14 in 2014 by using an elimination-Heck cascade reaction. The screening reaction involved utilization of precursor tetrazine 1g (synthesized by mesylation of 3-methyl-6-hydroxyethyl-1,2,4,5-tetrazine) in place of 3-methyl-6-vinyl tetrazine and reacting it with aryl halide (2g) for an in situ elimination-Heck reaction. Screening reactions revealed that among catalysts (Pd2(dba)3 and Pd(PPh3)4), and among bases such as NEt3 and Cy2NMe, using Pd2(dba)3 (3 mol%), and Cy2NMe (3 equiv.) for reaction between precursor 1,2,4,5-tetrazine (1g; 1 equiv.) and halobenzene (2g; 1–1.5 equiv.) along with ligand A (0.12–0.4 equiv.) at 50 °C for 30 min under microwave resulted in maximum yield of the desired product (3g; R = CH3; 99%) (Scheme 20).
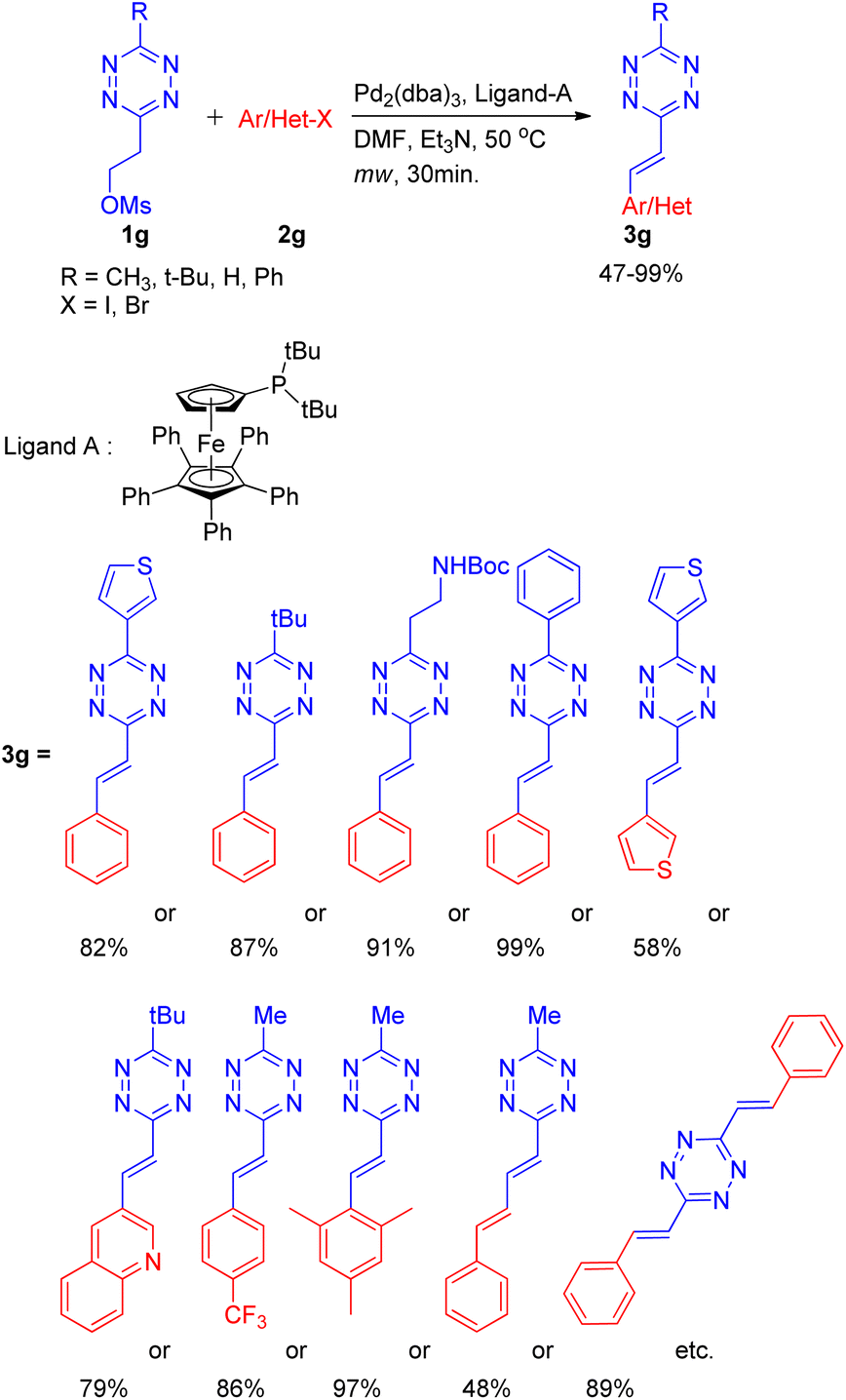 | ||
| Scheme 20 In situ synthesis of alkenyl tetrazines for elimination-Heck cascade reaction. | ||
Under the obtained optimized conditions, the tetrazines with tert-butyl, phenyl, heterocycles, and protected amine group etc. at position-3 were able to drive the reaction to completion (Scheme 20) giving 3-substituted alkenyl tetrazines in good to excellent yield (82–99%). Similarly, various aryl bromides containing electron withdrawing, electron donating, sterically bulky groups, as well as heterocyclic substituents (Scheme 20) also gave desired product in good to excellent yield (58–97%). Conjugated mono-phenylbutadiene, biphenylbutadiene and bistyryl substituted s-tetrazines etc. were also synthesized in moderate yield (48–89%) using the same method.
The overall protocol thus generated facilitates the study of highly conjugated 1,2,4,5-tetrazines with the aim of exploiting them in material science, photovoltaics, chemical biology and specifically in live-cell imaging.
In addition to the all the above methods of in situ generation of alkenes providing the required Heck coupling product in good to excellent yields, the tandem Wittig–Heck sequence because of the greater availability of aromatic aldehyde and ketones as compared to styrene15 and simplicity and efficiency of Wittig reaction, has also been widely researched in the last two decade. Thus the required olefin can either be generated through various procedures given above or by utilizing sequential Wittig–Heck reactions as covered below.
3. Mizoroki–Heck reaction between in situ generated Wittig alkenes and aryl halides
The utilization of sequential Wittig–Heck reaction for formation of substituted olefines began in 2001 when Lutz F. Tietze and co-workers16 reported a 2 two-fold Heck reaction via one Wittig reaction as one of the method for the synthesis of biologically important linear π-conjugated oligomeric pyrrole derivatives (Scheme 21). The efficient synthesis of various oligomeric pyrrole derivatives connected by divinyl units initiates with two-fold Heck reaction of 2,5-dibromopyrrole (1h; 1 equiv.) derivative with p-vinylbenzaldehyde (1h′; 2 equiv.). The pyrrole derivative thus formed (1h′′) undergoes 2-fold Wittig reaction in presence of nBuLi (1.1 mol equiv.), and triphenylphosphonium iodide (1.2 mol equiv.) in THF to yield a substituted alkene (2h) which eventually reacts with the iodopyrrole (2h′; 1 equiv.) again in a 2-fold Heck reaction in the presence of 5 mol% Pd(OAc)2, potassium acetate (4.0 equiv.), and tetrapropylammonium bromide (1.0 equiv.) in DMF at 75 °C for 2 h under nitrogen or argon atmosphere. They were able to synthesize the desired red pentacyclic oligomer (3h) with 28% yield (Scheme 21). Thus this protocol provided a synthetic pathway to linear π-conjugated oligomeric pyrrole derivatives with up to 5 arenes, however the method can also be exploited for the formation of higher oligomers.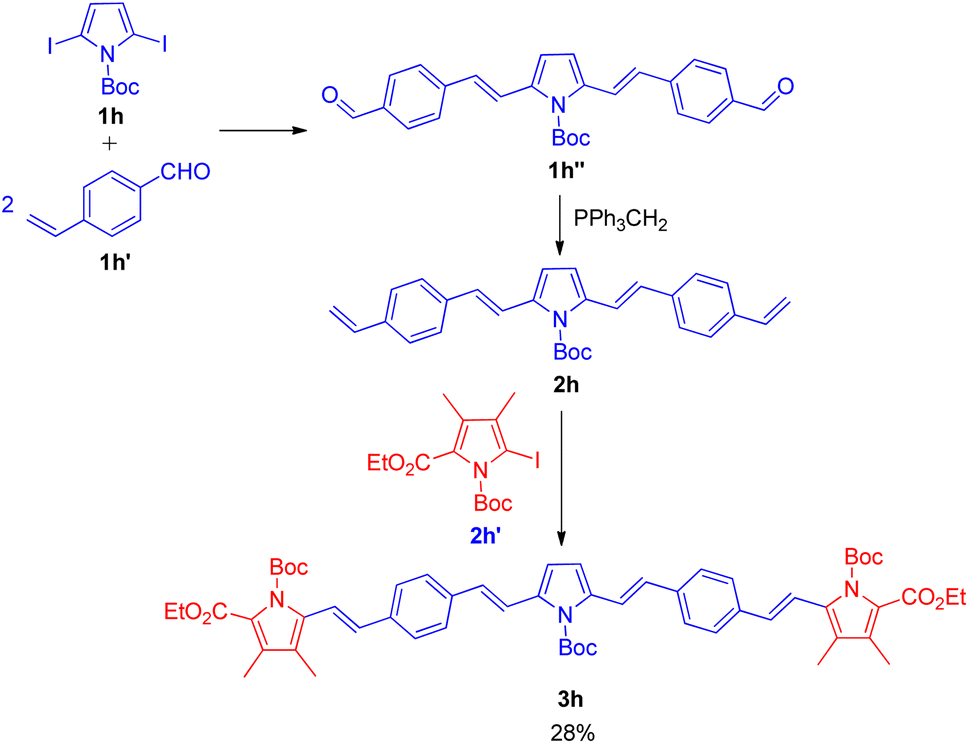 | ||
| Scheme 21 Synthesis of pentacyclic oligomer by a two fold Heck reaction followed by a Wittig reaction and a two fold Heck reaction. | ||
Owing to the presence of medium sized heterocyclic rings fused to aryl rings in many natural products,17 K. C. Majumdar and co-workers17 in 2008, developed an efficient system with high regioselectivity and high yield for the synthesis of oxepin ring (3i), having significance in the preparation of naphthoxepin derivatives (antipsychotic drugs17), by employing sequential Wittig and intramolecular Heck reaction. For the intramolecular Heck reaction to take place, the required substrate (2i) was generated via Wittig reaction of substrate 1ii generated through reaction between hydroxy-aldehydes(1i′) (synthesized via Reimer–Tiemann reaction of 1i), with either 2-bromobenzyl bromide(1i′′) or 2-bromo-5-methoxy benzyl bromide(1i′′′). This reaction proceeded in dry acetone with anhydrous potassium carbonate as base and a small amount of sodium iodide.
The product (1ii) through Wittig reaction generates 2i, which then results in formation of the Z-isomer of eight-membered naphthoxocine compound (3i) with excellent yield (upto 86%) under Heck reaction condition (Scheme 22). Various screening experiments were performed to get optimized reaction condition for Heck reaction revealed that among Pd(PPh3)2Cl2, Pd(OAc)2, PdCl2 and Pd(PPh3)4, Pd(OAc)2 gave the desired product in excellent yield. The presence of TBAB also proved to be necessary for the reaction to go to completion since in the absence of it, no coupling product was obtained.
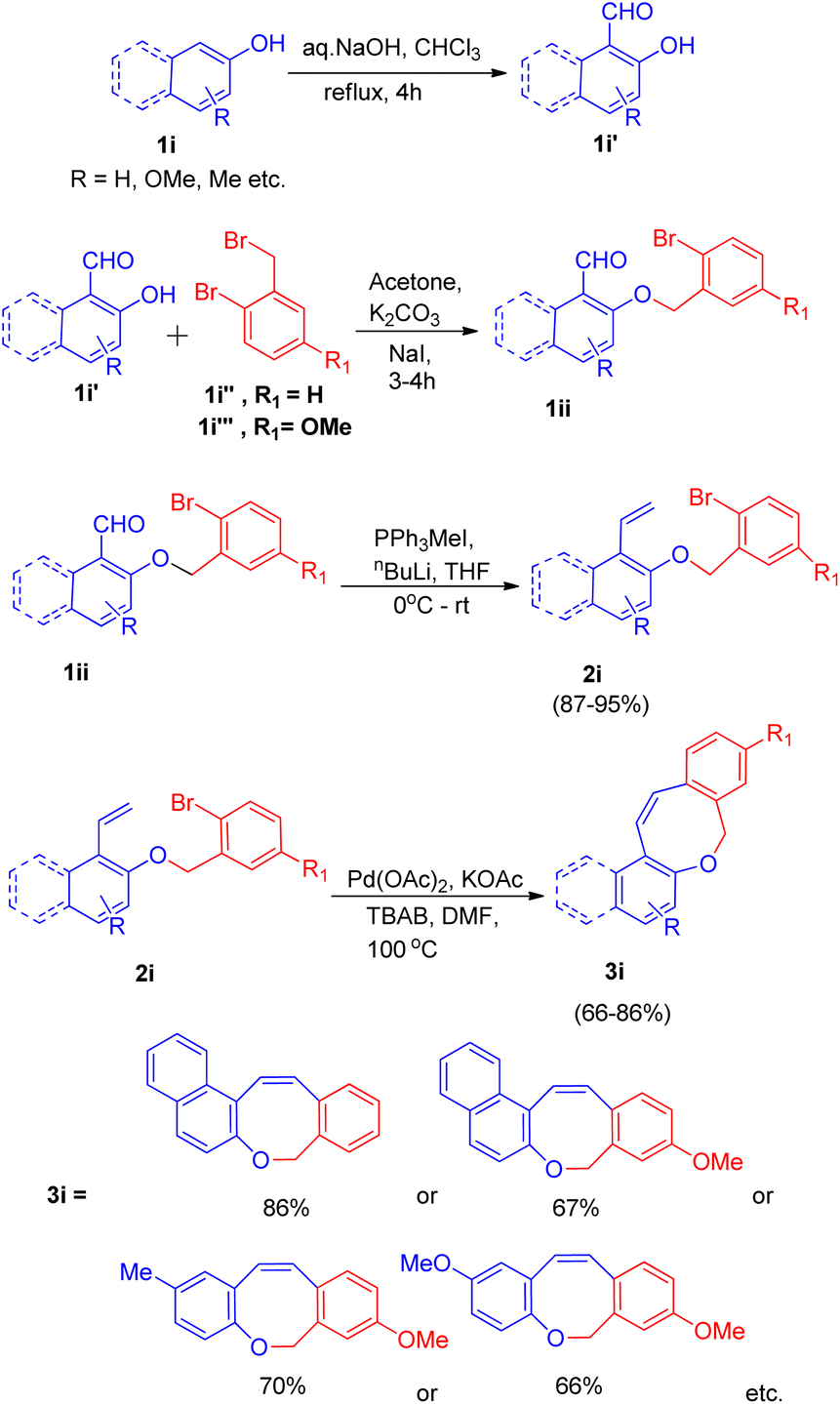 | ||
| Scheme 22 Palladium-catalyzed intramolecular Heck cyclization of 2i (synthesized via Wittig reaction of 1ii) to form 3i. | ||
Among various bases which were analyzed such as K2CO3, Et3N, Ag2CO3, KOAc, Cs2CO3, and solvents such as DMF, CH3CN, Et3N, dioxane, KOAc as base and DMF as solvent was found to be the most efficient ones. Thus for the intramolecular Heck reaction in 2i, on providing a nitrogen atmosphere containing Pd(OAc)2 acting as a catalyst, KOAc as a base, and TBAB as an additive in dry DMF as solvent for 2 h at 100 °C (Scheme 22) the desired eight-membered cyclized oxocine derivatives were obtained in moderate to good yield (66–86%). Similarly Z-isomers of other 8-membered oxocine derivatives were also prepared via 8-endo trig cyclization. The efficiency of these sequential Wittig–Heck reactions were increased by reducing the reaction time and work-up steps.12 Fused oxocine derivatives (3i) can be formed in high yield through this simple cyclization protocol.
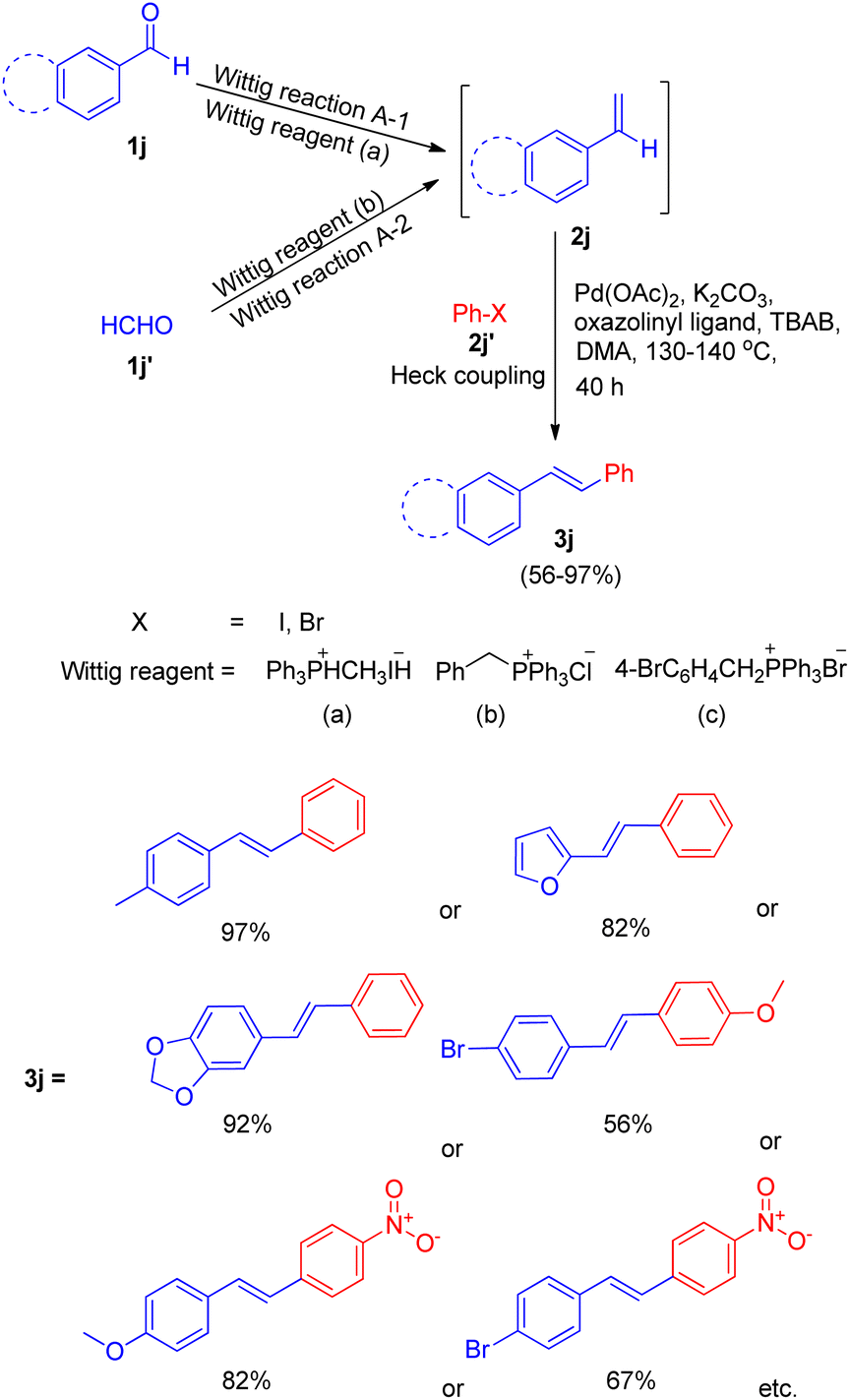
One among the tactic for this sequential-Wittig process was also given by Akeel S. Saiyed and Ashutosh V. Bedekar in 2010 via one pot process.5 The main feature of this method is the in situ synthesis of required olefin. Olefins (2j) were synthesized by Wittig reaction from aldehyde (1j, 1j′) and acceptable phosphonium salt (Scheme 23). The two ways (A-1, A-2) shown in Scheme 23 for in situ synthesis of styrene ends with formation of stilbene on providing Mizoroki–Heck conditions. In this approach, aromatic aldehyde (1j) with phosphonium salt undergo Wittig reaction even with weak base like potassium carbonate, resulting in formation of an olefin (2j) to further couple with an aryl halide (2j′) under Heck reaction conditions (Scheme 23).
 | ||
| Scheme 23 Proposed routes of in situ synthesis of styrene through Wittig reaction for subsequent one-pot Heck reaction. | ||
The optimized conditions for this protocol include employing 1 equiv. of aldehyde with 1 equiv. of phosphonium salt for wittig reaction. The desired alkene thus formed then undergoes Heck coupling with 1 equiv. of an aryl halide in the presence of catalytic quantity of Pd(OAc)2 (0.5 mol%), excess of K2CO3 (3.5 equiv.), 0.01 equiv. of oxazolinyl ligand (1 or 2) (Fig. 4), and TBAB (0.2 equiv.) (as phase transfer catalyst) which was heated in DMA at 130–140 °C for 40 h to result in formation of desired trans-stilbene (3j) in good yields (56–94%). The presence of electron donating or electron withdrawing groups on either of the substrate does not seem to affect the reaction yield to a significant extent making this approach for in situ olefination an attractive option. However when the reaction was carried out using paraformaldehyde and 4-BrC6H4CH2PPh3Br as phosphonium salt, with either 4-iodoanisole or 1-bromo-4-nitrobenzene, the yield of the substituted olefin decreased to 56 and 67% respectively.
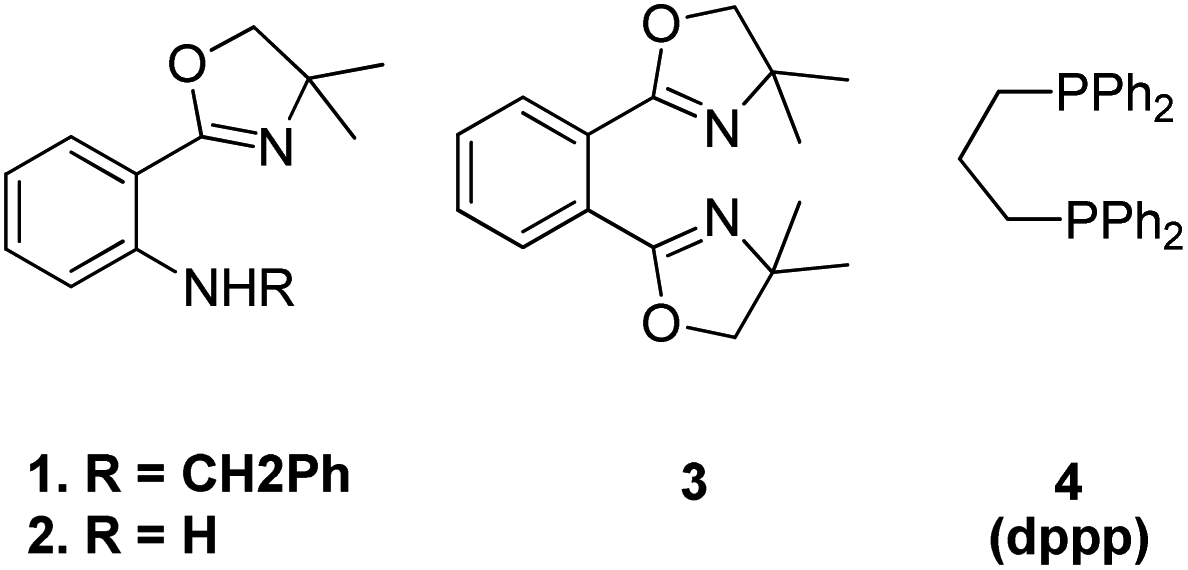 | ||
| Fig. 4 Ligands investigated for the one-pot approach for Wittig–Heck reaction. | ||
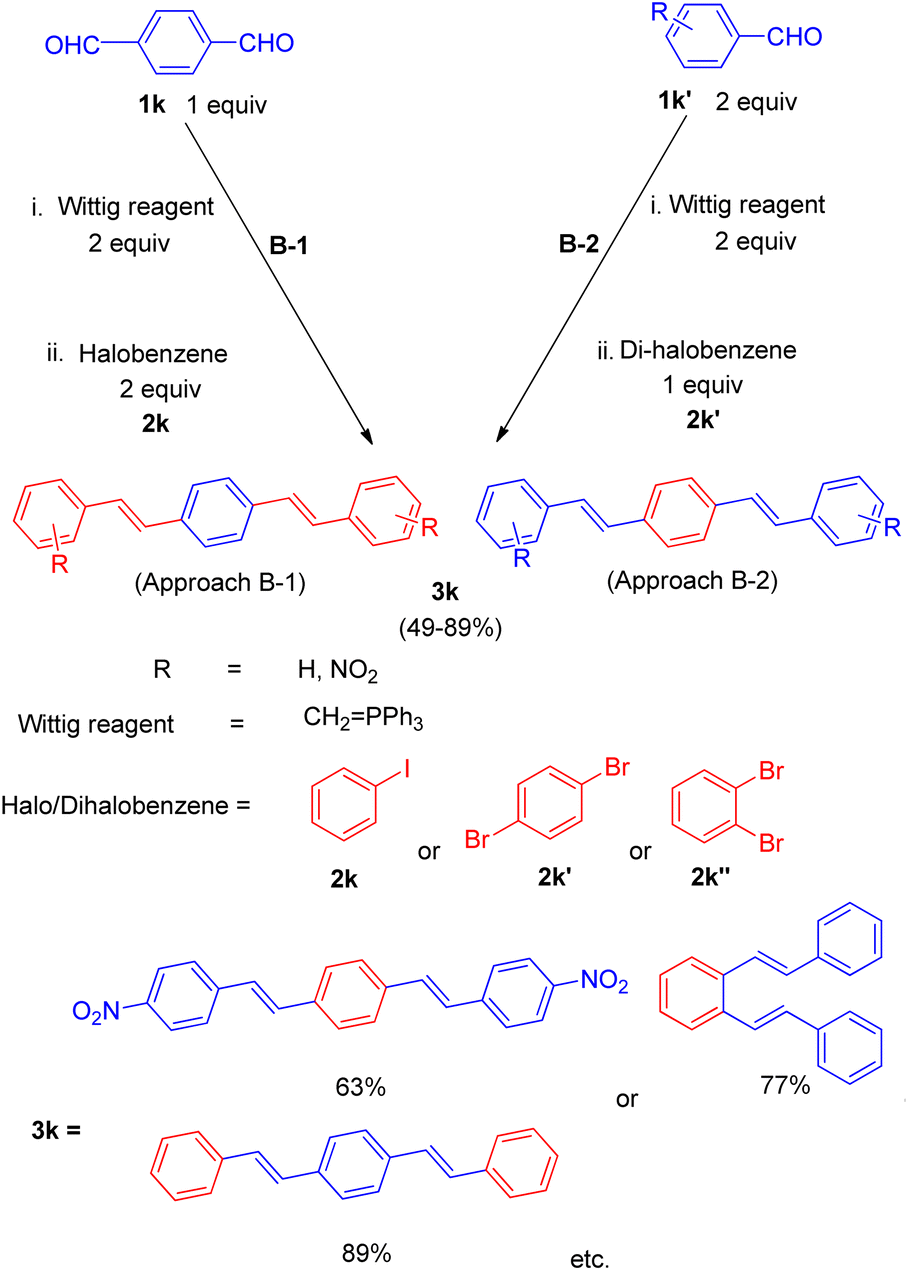
They also gave a one pot five component approach involving simultaneous formation of two double bonds between three aromatic rings through a combination of Wittig and Mizoroki–Heck reaction in a single step process (Scheme 24).
 | ||
| Scheme 24 One-pot five-component approach for the synthesis of distyryl benzene derivatives through Wittig–Heck reaction. | ||
This one-pot five component process was done in two ways under similar reaction conditions: the 1st method involves in situ preparation of 1,4-divinyl benzene by the reaction of 1 equiv. of terephthalaldehyde (1k) and 2 equiv. of Wittig reagent and subsequent introduction of this into the palladium catalyzed Heck reaction for coupling with 2 equiv. of halobenzene (2k) (approach B-1, Scheme 24) while the 2nd method of making 1,4-divinyl benzene involves making two fold excess of styrene by using 2 equiv. of benzaldehyde (1k′) with 2 equiv. of Wittig reagent and subsequent introduction of this into the palladium-catalyzed Heck reaction for coupling with 1,4-dibromo benzene (2k′) (approach B-2, Scheme 24). For 2nd approach, 1,2-dibromobenzene (2k′′) can also be exploited to yield the desired distyryl benzene with 77% yield. The overall yield of the reactions conducted in a single pot was very good, either with ligand 1 or with ligand 2 and with 1,3-bis(diphenylphosphino)propane(dppp) ligand 4 (Fig. 4). Other distyryl benzenes using different aryl halides, and aromatic aldehydes, were also prepared by exploiting the same approach in good to excellent yield (49–89%).
The one-pot methods described here have the advantages of requiring fewer work-up and purification steps, providing appropriate chemical yield, and minimizing waste caused by polymerization of intermediates.
Joan Bosch et al. in 2012,18 developed a synthetic path for the synthesis of olopatadine, an antihistaminic drug and it's E-isomer having sequential highly stereoselective Wittig–Heck reaction as a key step in the process. The protocol for drug development initiates from Williamson reaction to assemble the benzyl aryl ether moiety (3l), which then undergoes Wittig olefination followed by successive intramolecular Heck coupling. The required aldehyde for Wittig reaction was 1st formed by Williamson reaction between 1l and 2l by adding a solution of 2l (1 equiv.) in acetonitrile to a mixture of substituted iodobenzene (1l; 1 equiv.), K2CO3 (1.1 equiv.), and NaI (0.25 equiv.) in acetonitrile, at room temperature for 3 h. The corresponding aldehyde (3l) thus generated was then treated with phosphonium salt to form the olefin (E-isomer; 3l′) which ultimately undergoes intramolecular Heck coupling in presence of Pd(OAc)2 (20 mol%), Bu4NCl (1 equiv.) K2CO3 (2.52 equiv.) as base, in acetonitrile–water (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture followed by alkaline hydrolysis, to yield olopatadine drug (4l; Scheme 25) with complete stereoselectivity. Following a similar reaction pathway and by utilizing aldehyde prepared from 2-formylbenzyl bromide and 4-hydroxy-3-iodophenylacetic ester, trans-olopatadine (E-isomer of 4l) was also synthesized in 70% yield with complete stereoselectivity. On the contrary, the intramolecular Heck reaction using the cis isomer (3l′′) resulted in a mixture of Z and E-isomers of olopatadine drug. The stereoselectivity of the product generated is highly affected by the selection of base taken during ylide formation from phosphonium halide. Using lithium base LHMDS instead of KHMDS during ylide formation from phosphonium iodide shifted the stereoselectivity of the Wittig reaction from obtaining E/Z alkenes in a 1:3 ratio to 9:1 with 73% yield. Thus this methodology involving highly stereoselective sequential Wittig–Heck reaction provided a synthetic route to antihistamine drug (4l) and it's E-isomer.
:1) mixture followed by alkaline hydrolysis, to yield olopatadine drug (4l; Scheme 25) with complete stereoselectivity. Following a similar reaction pathway and by utilizing aldehyde prepared from 2-formylbenzyl bromide and 4-hydroxy-3-iodophenylacetic ester, trans-olopatadine (E-isomer of 4l) was also synthesized in 70% yield with complete stereoselectivity. On the contrary, the intramolecular Heck reaction using the cis isomer (3l′′) resulted in a mixture of Z and E-isomers of olopatadine drug. The stereoselectivity of the product generated is highly affected by the selection of base taken during ylide formation from phosphonium halide. Using lithium base LHMDS instead of KHMDS during ylide formation from phosphonium iodide shifted the stereoselectivity of the Wittig reaction from obtaining E/Z alkenes in a 1:3 ratio to 9:1 with 73% yield. Thus this methodology involving highly stereoselective sequential Wittig–Heck reaction provided a synthetic route to antihistamine drug (4l) and it's E-isomer.
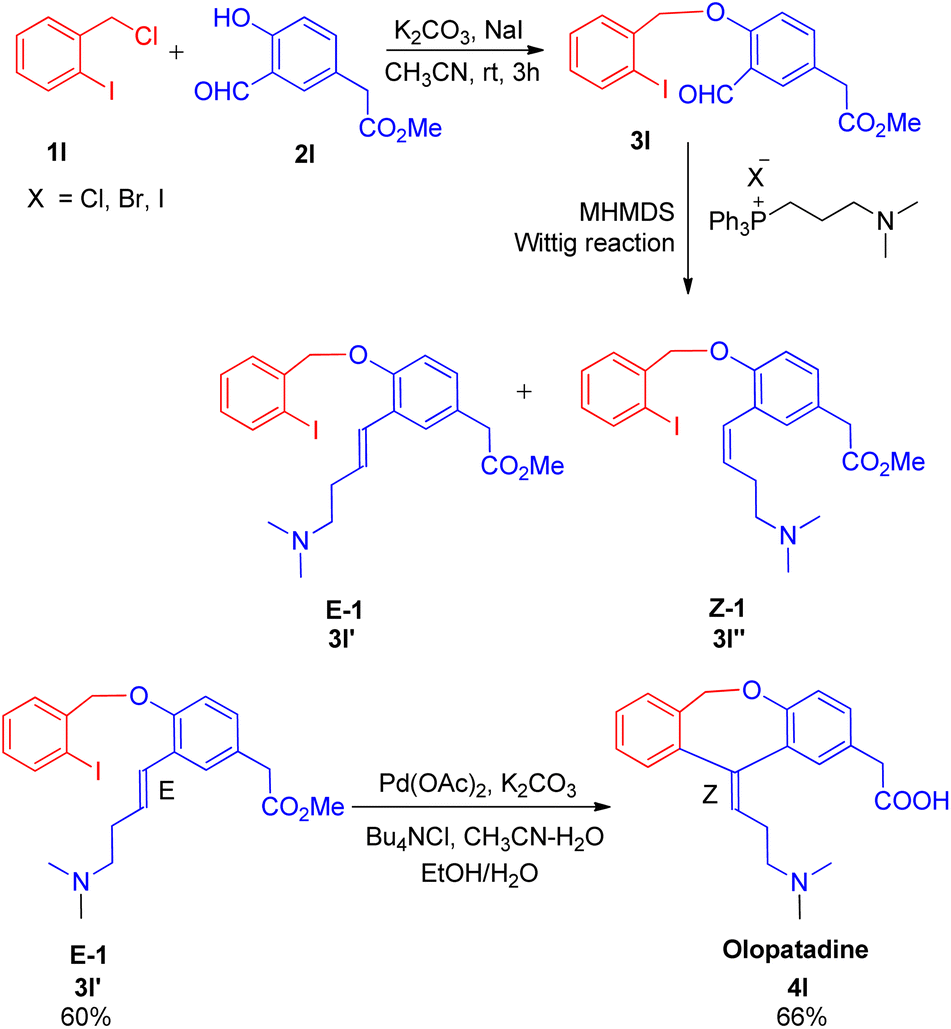 | ||
| Scheme 25 Williamson reaction between 1l and 2l followed by sequential Wittig–Heck reaction of 3l for the synthesis of antihistaminic drug olopatadine (4l). | ||
Ashutosh V. Bedekar and co-workers19 in 2012 also reported one-pot Wittig–Heck reaction for the formation of stilbenes, by utilizing aromatic aldehyde (1m′) synthesized from Kornblum oxidation of benzyl halide (1m) (Scheme 26). The complete reaction sequence involved oxidation of benzyl halide (1m) to aldehyde (1m′), which then undergoes Wittig reaction to produce corresponding olefin (2m) in presence of a phosphonium salt. Further this olefin undergoes palladium-mediated Mizoroki–Heck reaction with an aryl halide (2m′) to produce E-isomers of stilbene (3m) in moderate to good yield (46–76%).
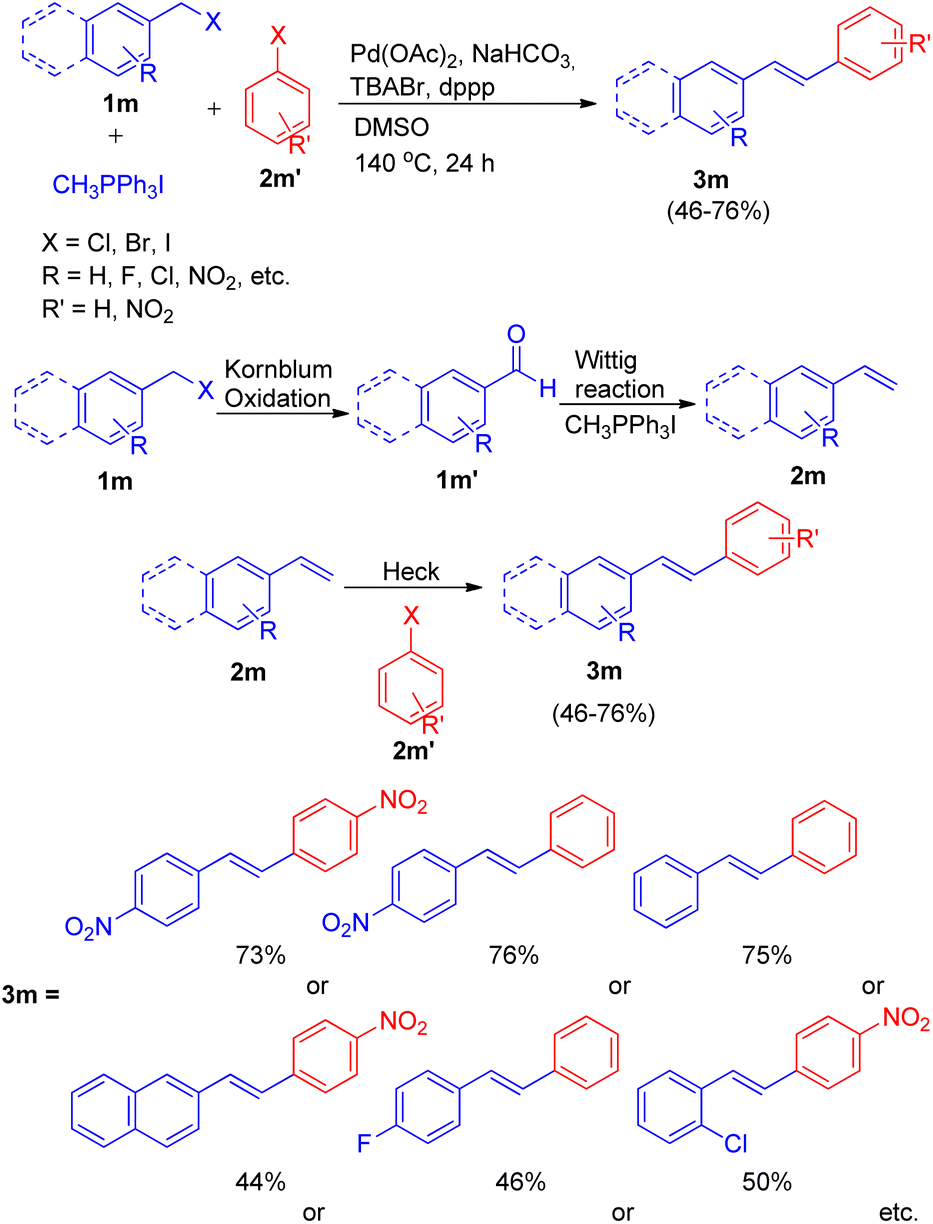 | ||
| Scheme 26 Synthesis of unsymmetrical stilbenes from benzyl halide by one-pot oxidation-Wittig–Heck sequence. | ||
The optimized condition for this protocol involves utilization of 1.5 equiv. of benzyl halide for Kornblum oxidation to result in formation of an aromatic aldehyde. Further reaction of this aromatic aldehyde with 1.5 equiv. of one-carbon Wittig salt along with 1.0 equiv. of aryl halide for sequential Wittig–Heck reaction in the presence of 1 mol% Pd(OAc)2 as catalyst, 6.0 equiv. of NaHCO3 as base, 0.2 equiv. of TBABr, 2% dppp in DMSO as solvent at 140 °C for 24 h yields the required unsymmetrical stilbenes, with primarily E-isomers, in moderate to good yield (46–76%). A variety of benzyl halides and aryl halides bearing EWGs were analyzed under these optimized conditions and it was observed that except for the reaction of 1-(bromomethyl)-4-nitrobenzene with 1-bromo-4-nitrobenzene and with iodobenzene, giving the desired products in 73 and 76% respectively (Scheme 26), the presence of electron withdrawing on either of the two substrate or on both, decreases the yield of the corresponding stilbenes significantly.
In the same year, they were also able to develop a novel one-pot method for the synthesis of highly conjugated alkyloxy stilbenes (3n) from hydroxy benzaldehyde (1n) and one carbon Wittig salt via tandem O-alkylating Wittig–Heck reaction.20 The method involved palladium-catalyzed Mizoroki–Heck reaction (Scheme 27) of alkyloxystyrene (generated in situ via Wittig reaction) with an aryl halide (2n).
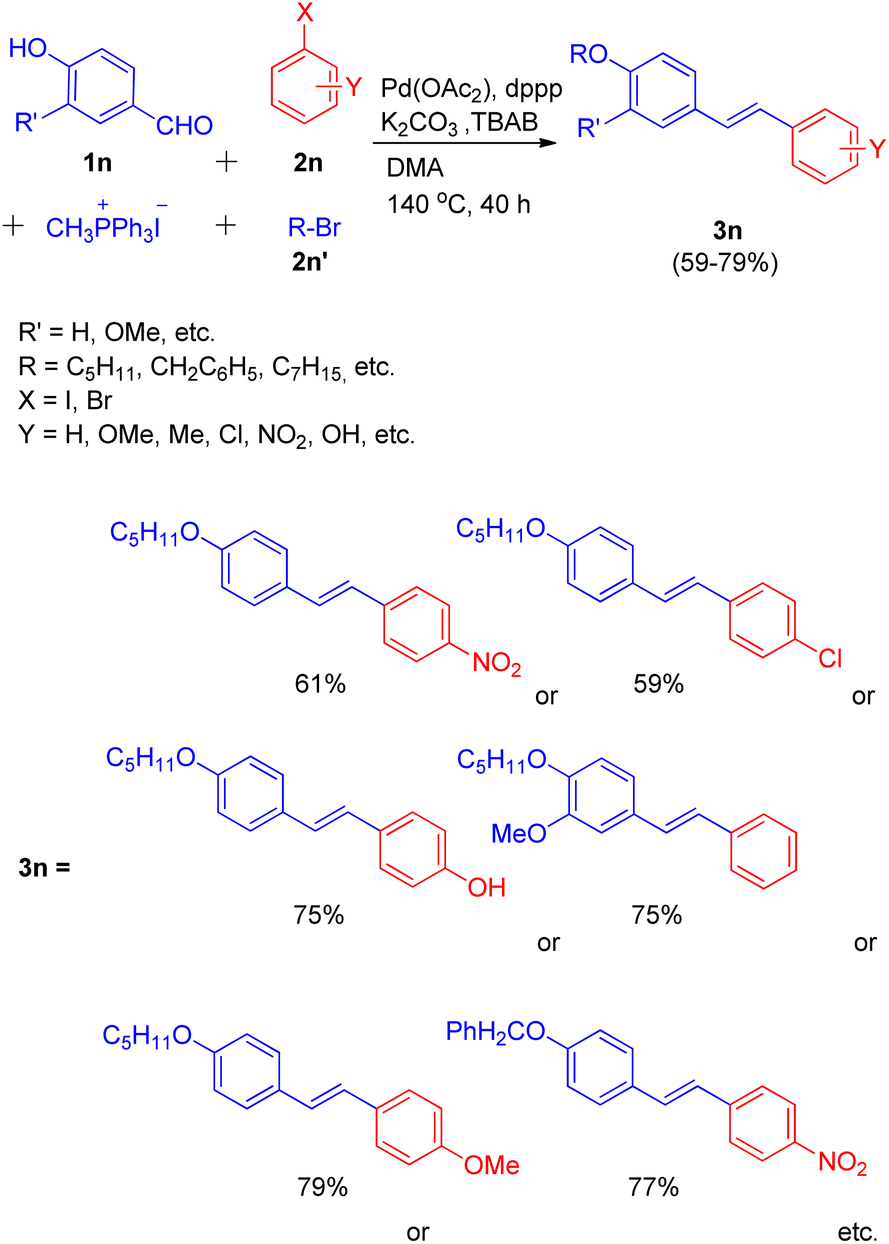 | ||
| Scheme 27 One-pot path for the synthesis of O-alkyloxystilbene through O-alkylating Wittig–Heck reaction. | ||
The given approach involves synthesis of alkyloxystyrene via Wittig reaction of hydroxybenzaldehyde with simultaneous alkylation of the hydroxy group. This is then followed by Heck coupling of the in situ generated alkyloxystyrene with readily available functionalized aryl halide to ultimately result in the formation of corresponding stilbene.
Using different aldehydes and alkyl halides, various screening experiments were performed in order to get an optimized reaction condition which revealed that 1.2 equiv of phosphonium salt for Wittig reaction of 1.2 equiv. of aldehyde, along with 1.2 equiv. alkyl halide for alkylation, and 1.0 equiv. of aryl halide for Heck coupling in the presence of Pd(OAc)2 (0.5%) as catalyst, 6 equiv of K2CO3 as base, 1.0% dppp, 10% TBAB in DMA as solvent at 140 °C for 40 h resulted in moderate to good yield (59–79%) of the mostly E-isomer of corresponding alkyloxystilbene (Scheme 27).
Various aryl halides were analyzed for the above mentioned protocol and it was found that except for the reaction of 1-bromo-4-nitrobenzene and 1-bromo-4-chlorobenzene with 4-hydroxybenzaldehyde and 1-bromopentane, the presence of electron rich and electron poor groups on aryl halide does not affect the overall yield of the reaction.
This method was also employed for the generation of primarily E, E isomers of distyrylbenzenes(3o) having C6–C2–C6–C2–C6 framework (Scheme 28) by reacting 2,5-dimethoxy-1, 4-diiodobenzene (2o) with the same hydroxybenzaldehyde (1n) with one-carbon phosphonium salt and appropriate alkyl halide (1o). This one-pot reaction sequence also gave the required product in good yields (53–71%). Similarly tri- and tetra-stilbene derivatives (4o′ and 5o′ respectively) by exploiting 1,3,5-tribromobenzene (4o) and 1,2,4,5-tetrabromobenzene (5o), were also formed (Scheme 28), but with very low yield (14–17%).
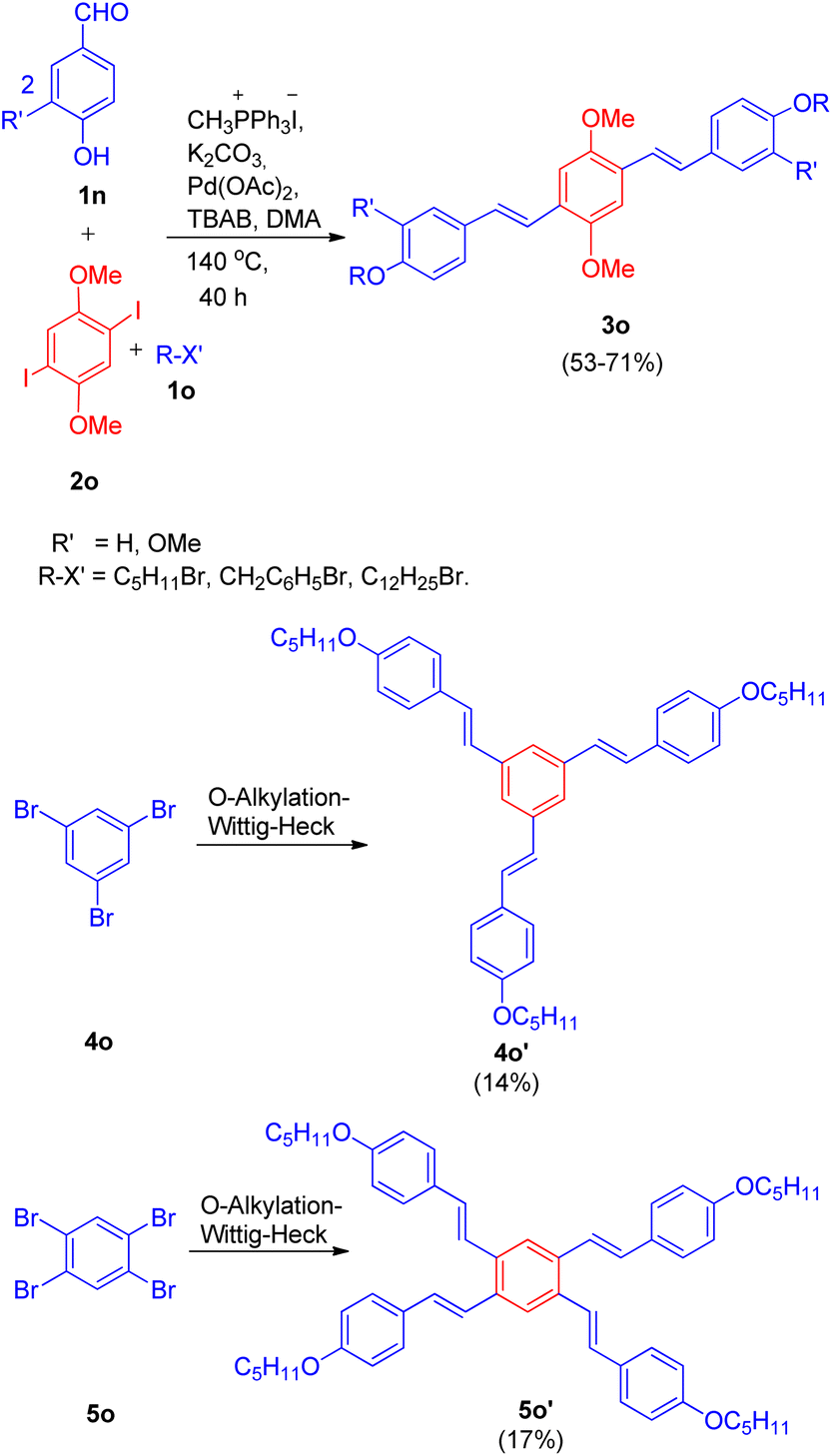 | ||
| Scheme 28 Synthesis of distyrylbenzene, tri- and tetra-stilbene derivatives (3o, 4o′ and 5o′ respectively) through one-pot Wittig–Heck reaction. | ||
Soon after that in 2012, a phosphine free one-pot Wittig–Heck reaction for the synthesis of varied fluorinated styrylbenzene derivatives21 was also developed by them. The reaction between various aromatic aldehyde (1p) with Wittig salt and base provided the required olefin (1p′) (generated in situ) for further carrying out the Mizoroki–Heck reaction with an aryl halide (2p) to form variety of fluorinated stilbene derivatives (Scheme 29; 3p). The optimized conditions for this protocol include employing 1.5 equiv. of aromatic aldehyde, 1.5 equiv of CH3PPh3I for Wittig reaction to yield the desired olefin for further coupling with Ar–X (1.0 equiv.), in the presence of Pd(OAc)2 (0.5 mol%) as catalyst, 1-(a-aminobenzyl)-2-naphthol (6 mequiv.) as ligand (L), K2CO3 (4.0 equiv) as base, with 0.2 equiv. of TBAB, in DMA as a solvent, under N2 atm, at 140 °C for 40 h to synthesize corresponding fluorinated styrylbenzene derivatives (3p) with moderate to good yield (41–89%).
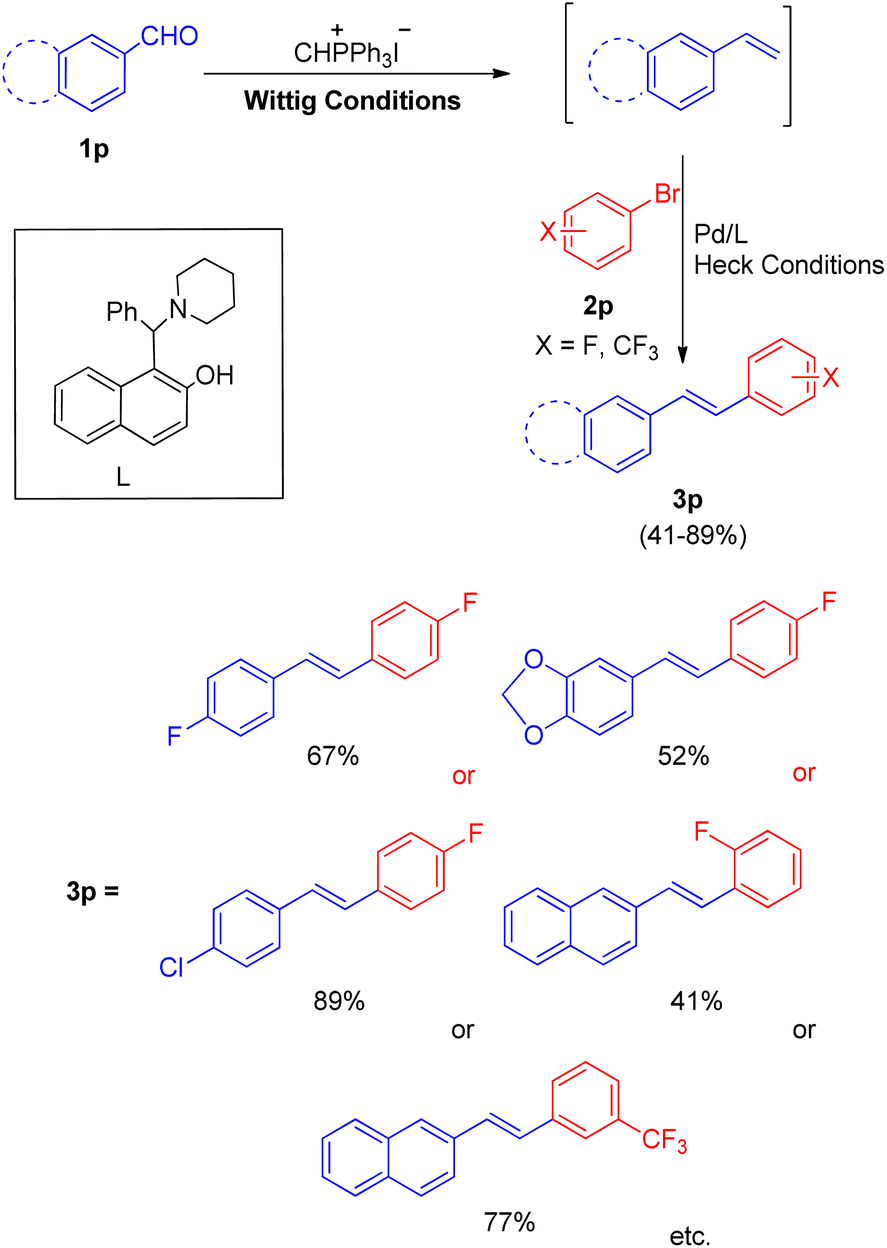 | ||
| Scheme 29 Synthesis of fluorinated stilbenes through Wittig–Heck reaction. | ||
These fluorinated stilbene derivatives were then efficiently used for the synthesis of various fluorinated polyaromatic hydrocarbons. The electronic environment of the groups present on either of the substrate did not seem to affect the reaction yield. Thus this path developed by them provides a wide scope of generating fluorinated polyaromatic hydrocarbons (F-PHCs) having significance in cancer research.17
In 2014, a polyaniline anchored palladium catalyst (PANI-Pd, Fig. 5) was developed and used by Arun L. Patel, Ashutosh V. Bedekar et al.15 in one pot Wittig–Heck reaction for the synthesis of a number of stilbene (3q) derivatives (Scheme 30) with good yields (53–89%). The styrene (2q) generated in situ through Wittig reaction of an aromatic aldehyde (1q) and Ph3PCH3I under basic environment, when subjected to Mizoroki–Heck conditions utilizing PANI-Pd as catalyst was able to give stilbene derivatives (3q) in good to excellent yields (Scheme 30). Since the styrene is generated in situ, it immediately couples with an aryl halide in the presence of PANI-Pd catalyst to result in the formation of corresponding substituted olefin. The optimized condition employed an 1.2 equiv. of aromatic aldehyde (1q) with a wittig salt, methyltriphenylphosphonium iodide (1.4 equiv.) at 60 °C for wittig reaction for in situ synthesis of substituted styrene (2q) which on providing Mizoroki–Heck coupling conditions (presence of PANI-Pd catalyst (0.02 equiv.) with dry potassium carbonate (3 equiv.) in dry DMA at 140 °C for 40 h) couples with aryl iodide to form corresponding olefin (3q) in good yields (53–89%).
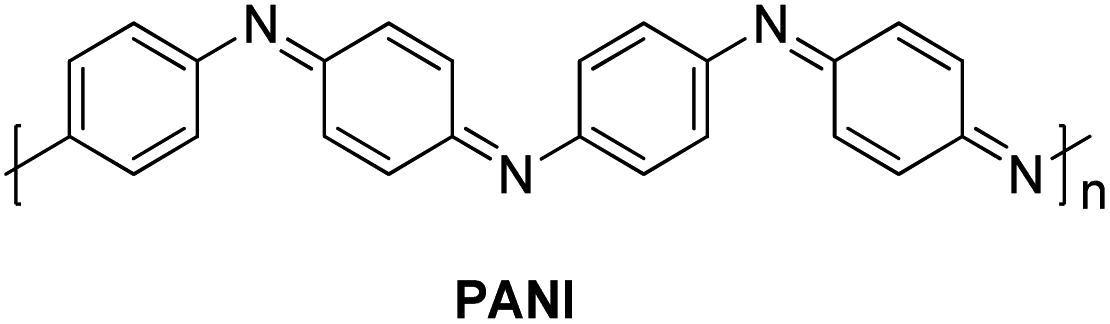 | ||
| Fig. 5 Structure of PANI utilized in one-pot Wittig–Heck reaction. | ||
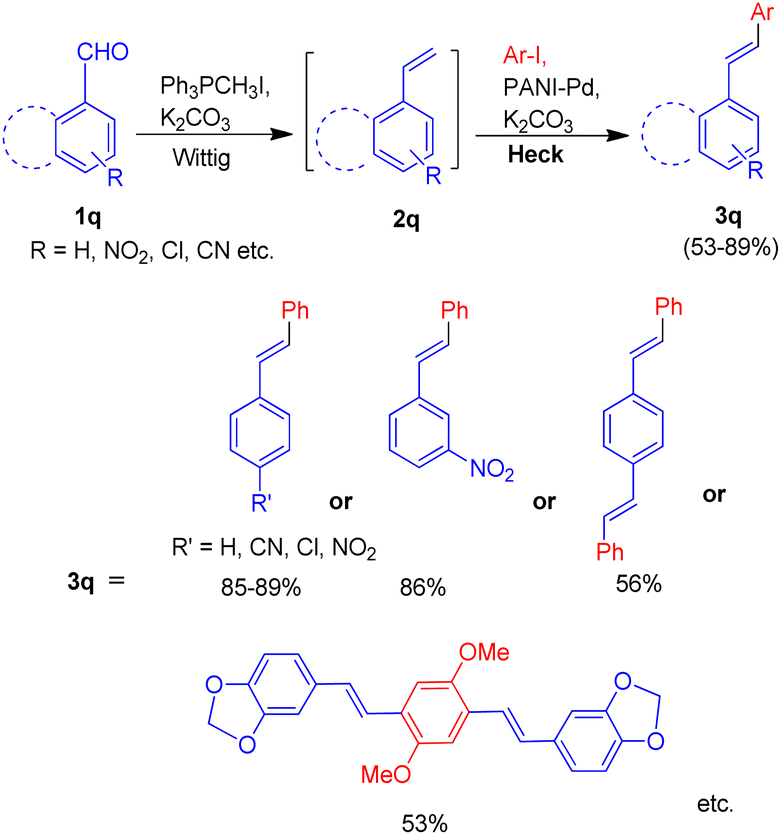 | ||
| Scheme 30 Substrate scope for one-pot Wittig–Heck reaction catalyzed by PANI-Pd. | ||
The catalyst i.e. PANI-Pd, despite only being applicable for aryl bromides and aryl iodides and not for aryl chlorides, displayed high efficiency and provided a wide range of tolerance for functional groups. The presence of electron withdrawing groups on various aryl halides analyzed does not seem to affect the overall yield of the desired product.
Extending their work, in order to synthesize a series of oligo(phenylenevinylenes), carrying different functional groups to study their spectroscopic properties, Ashutosh V. Bedekar and co-worker in 2015 (ref. 22) utilized an almost similar one-pot Wittig–Heck procedure. The one-pot methodology initiates with the in situ generation of substituted styrene via Wittig reaction of 4-substituted benzaldehydes (1r; 1 equiv.) with methyl triphenylphosphonium iodide (1r′; 1 equiv.). The styrene thus formed then subsequently undergoes Mizoroki–Heck reaction with 1,4-diiodo-2,5-dimethoxybenzene (2r; 5 equiv.) by utilizing 5 mol% Pd(OAc)2 as catalyst, K2CO3 as base (3.5 equiv.) with TBAB (2 equiv.) and dppp (0.04 equiv.) in N,N-dimethyl acetamide under nitrogen atmosphere at 140 °C for 24 h, to give the corresponding coupling products 2,5-dimethoxy-1,4-distyryl benzene derivatives (3r) in good yields (28–85%). In similar manner, pyridine containing oligo(phenylenevinylene)s (3r′ and 3r′′) bearing different substituents were also successfully synthesized (64–96%). Thus the one-pot Wittig–Heck methodology efficiently synthesized a number of oligo(phenylenevinylene)'s (OPV) derivatives with various functional groups having different electronic properties in moderate to good yield (28–96%) to study their optical properties (Scheme 31).
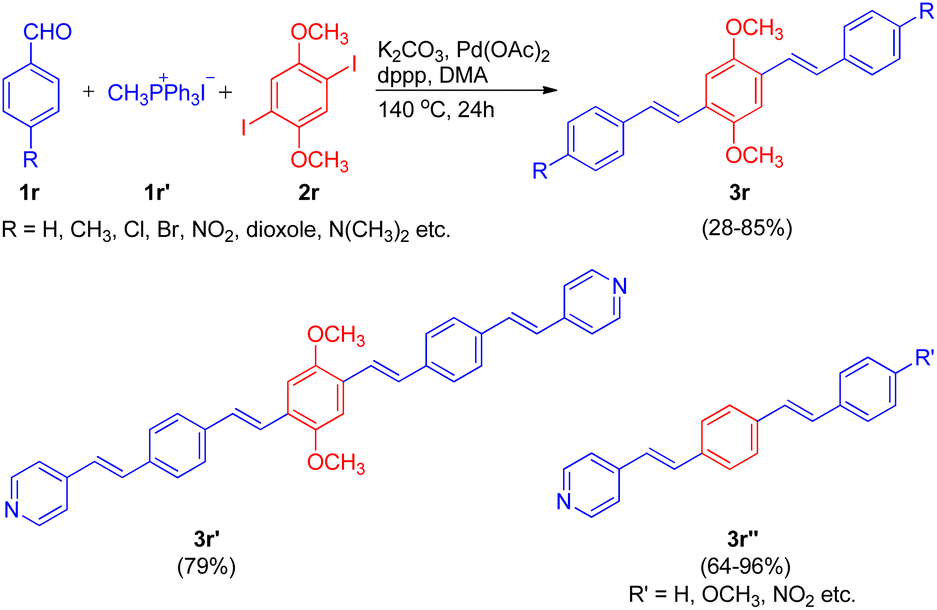 | ||
| Scheme 31 One-pot Wittig–Heck synthetic methodology for the synthesis of OPV's. | ||
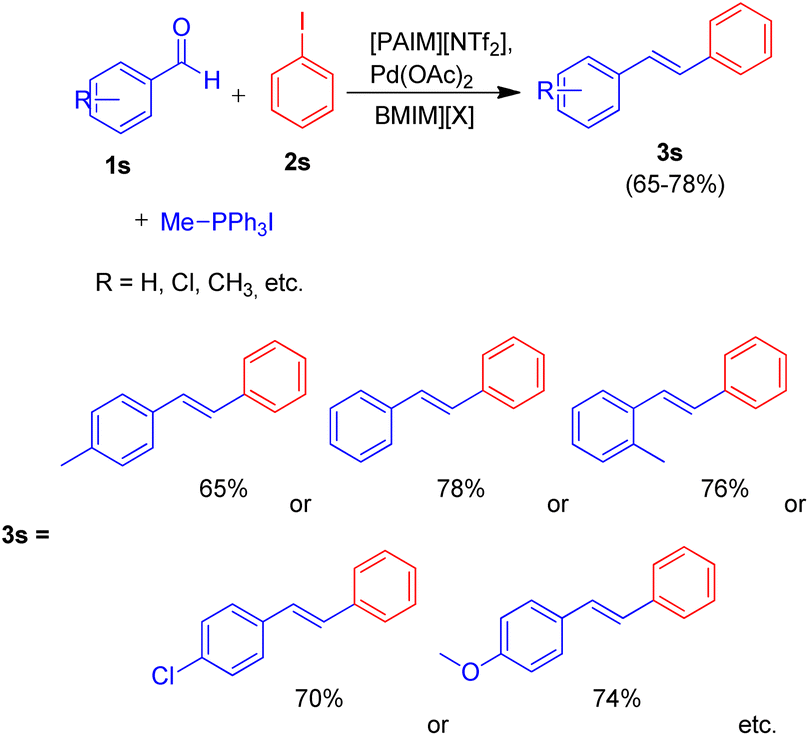
Owing to the various advantages offered by ionic-liquids in palladium-catalyzed cross-coupling reactions, their various classes have been used in Heck reaction. Kenneth K. Laali and co-workers in 2017 (ref. 23) thus utilized the piperidine-appended imidazolium-IL [PAIM][NTf2] (Fig. 6), for the synthesis of substituted diarylethenes via sequential Wittig–Heck reaction. The process first involved Wittig olefination of benzaldehyde (1s) (by utilizing methyltriphenylphosphonium bromide as Wittig reagent), the olefin thus formed then underwent Mizoroki Heck coupling with PhI (2s) in presence of palladium catalyst in order to form substituted trans-diphenyl-ethenes (3s) in good to moderate yield (65–78%) (Scheme 32).
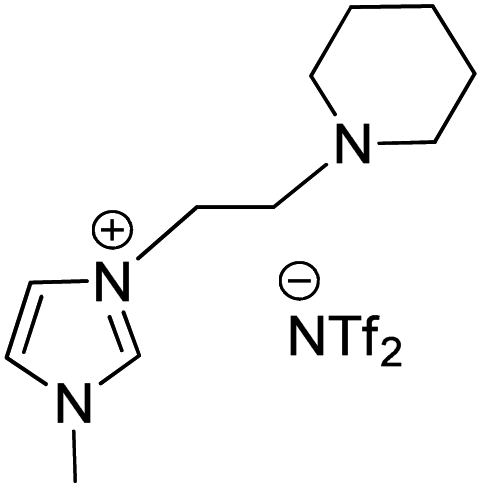 | ||
| Fig. 6 Showing structure of [PAIM][NTf2] ionic liquid. | ||
 | ||
| Scheme 32 Wittig–Heck tandem reactions for the synthesis of substituted trans-diphenyl-ethenes in ionic liquid. | ||
Various aromatic aldehydes were analyzed for the said protocol under optimized condition involving utilization of 1.3 equiv. of aromatic aldehyde, 1.2 equiv. of phosphonium salt as Wittig reagent, 1 equiv. of iodobenzene, in the presence of 3 equiv. of basic ionic liquid (piperidine-appended imidazolium-IL [PAIM][NTf2]) and 10 mol% of Pd(OAc)2 as catalyst in [BMIM][X] (X = PF6 or BF4) as solvent in an oil bath at 65 °C for 8 h resulted in moderate to good yield (65–78%) of the substituted stilbene with primarily E-isomers. The presence of electron withdrawing or electron donating groups on aromatic aldehyde does not affect the product yield significantly.
This method was able to show the efficiency of ionic liquid for the development of simple one pot Wittig–Heck cross-coupling reaction occurring without the addition of any additive.
The Wittig–Heck reaction was also extended for the synthesis of fluorine-containing s-shaped π-conjugated dibenzo[c,l]chrysene derivative by Tetsuji Moriguchi and co-workers in 2017 (ref. 24) in order to study it's photophysical properties as well as determine the structure owing to it's importance in semiconducting material and other electronic devices.
The s-shaped polyaromatic compound was synthesised in 2-step Wittig–Heck reaction. The 1st step involved condensation of a Wittig-salt (1t; 0.5 equiv.) with trifluoromethyl benzaldehyde (2t; 1 equiv.) via Wittig-reaction to generate the desired olefin (2t′) which then in the 2nd step undergoes an intramolecular Heck reaction in presence of Pd(OAc)2 (5 mol%) as catalyst, NaOAc (5 equiv.) as base in DMF at 110 °C for 18 h to result in the formation of desired substituted and highly conjugated cyclic system (3t) however with low yield of 22% (Scheme 33).
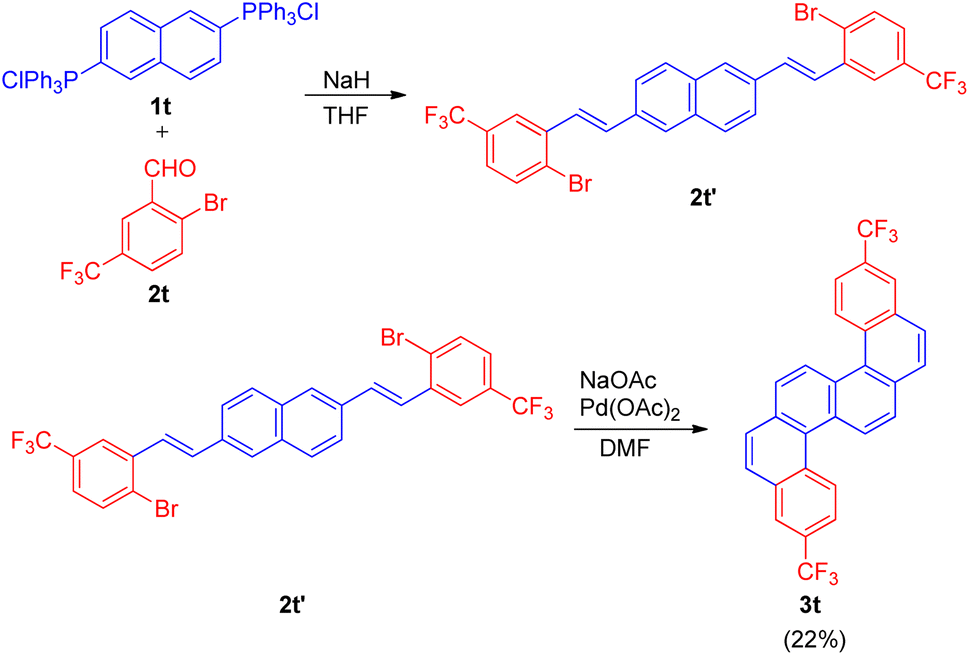 | ||
| Scheme 33 Synthesis of isomeric fluorinated s-shaped polyaromatic dibenzo[c,l]chrysene derivative (3t). | ||
Utilizing the same catalyst developed by Arun L. Patel, Ashutosh V. Bedekar et al.15 in 2014, Arun L. Patel and co-workers in 2019,25 developed an efficient synthetic approach for formation of benzochalcogendiazole based π-conjugated molecules in order to study their photophysical properties. The path leading to the formation of a new class of substituted benzothiazole derivatives exploited the same one-pot Wittig–Heck reaction11 as given by them in 2014. The method involved in situ generation of vinyl arenes via reaction of an aldehyde (2u; 1 equiv.) and Ph3PCH3I (2u′; 2.2 equiv.), which when subjected to Mizoroki–Heck conditions, i.e. on providing a nitrogen atmosphere at 120 °C for 40 h and utilizing potassium carbonate as base (12 equiv.), and PANI-Pd as catalyst (12 mol%) in DMA, couples with dibromobenzothiadiazole (1u; 1 eq.), resulting in formation of bis(vinylarene)-capped benzochalcogendiazoles (3u; Scheme 34). When a series of electron rich and electron poor benzaldehydes were made to react with phosphonium salt to generate the required olefin for undergoing heck coupling with 1u, varied number of conjugated fluorescent heck products (3u) were synthesized in good yields (56–71%).
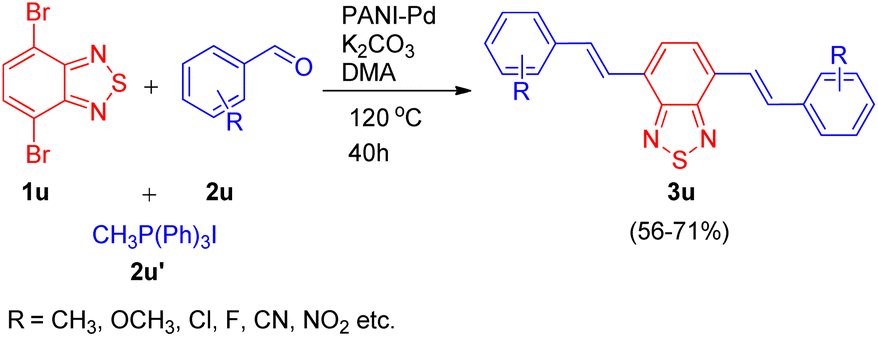 | ||
| Scheme 34 PANI-Pd catalyzed one-pot Wittig–Heck reaction for the synthesis of benzochalcogendiazole-based conjugated molecules. | ||
The one-pot Wittig–Heck reaction given by Ashutosh V. Bedekar was also utilized again in 2021 by Ashutosh V. Bedekar and co-workers26 as one of the key steps for the synthesis of aza[5]helicenes derivatives to study their photophysical properties and effect of methyl substituent in fjord region. Apart from being an important moiety in the area of material science, aza[5]helicenes derivatives also act as an efficient model for regioselectivity, configurational stability, and side reactions, illustrations. The reaction begins with in situ generation of required alkene 3v (4-methoxystyrene) by the Wittig reaction of p-anisaldehyde (2v; 1.5 equiv.), and methyl triphenylphosphonium iodide (2v′; 1.5 equiv.). The alkene thus formed reacts subsequently with halogen substituted 9H-carbazole (1v; 1 equiv.) on providing Heck reaction conditions i.e. using 5 equiv. K2CO3 a base, 0.2 mol% Pd(OAc)2 as catalyst, with 0.02 equiv. dppp and 0.2 equiv. TBAB in N,N-dimethyl acetamide under nitrogen atmosphere at 140 °C for 24 h to result in formation of corresponding carbazole compound 3v (Scheme 35). Further photochemical cyclization of the resulting alkene in presence of I2 and THF lead to the formation of desired cyclized aza[5]helicene (3v′) in 74% yield.
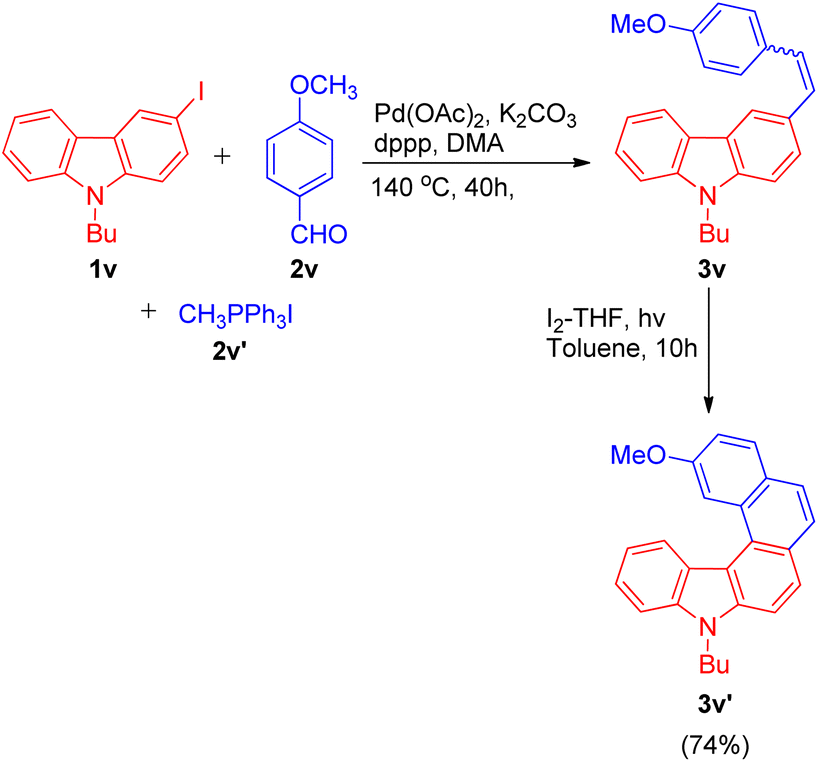 | ||
| Scheme 35 Synthesis of aza[5]helicene. | ||
The aza[5]helicene (3v′) thus generated was investigated further for it's photophysical properties and other structural parameters to gain an insight into the effect of methyl substituent onto their conformation stability in the fjord region.
A recent application of the Wittig–Heck pathway was seen in a successful attempt to synthesize and study the reverse demand voltage fluorophores by Evan W. Miller and co-workers in 2022.27 The development of 3 out of 5 voltage-sensitive fluorophores (VF dyes) exploited Wittig–Heck reaction as a key step. Out of five, three electron withdrawing groups (4-NO2-, 3-NO2-, and 4-CN-) containing substituted stilbenes were synthesized via domino Wittig/Heck/Wittig reaction carried out using Pd(OAc)2 as catalyst, K2CO3 as base in 1,4-dioxane as solvent giving the desired VF dyes (Scheme 36) to study their response to membrane potential. Among the three VF dye developed using Wittig–Heck reaction, 4-NO2-VF (3w) was observed to be voltage sensitive.
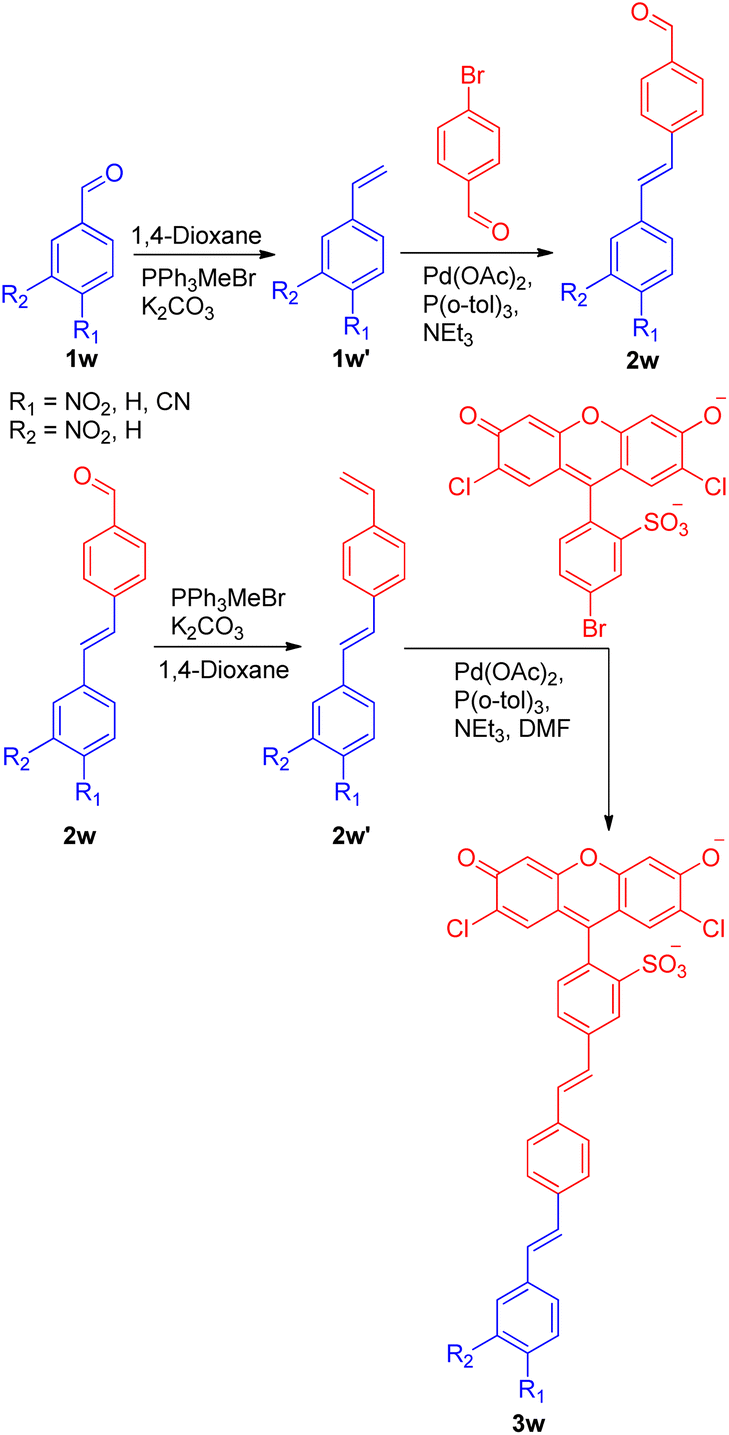 | ||
| Scheme 36 Synthesis routes of 4-NO2-VF, 3-NO2-VF, and 4-CN-VF. | ||
4. Conclusion
Thus summarizing from the above discussion, the difficulties encountered upon direct usage of alkenes during Mizoroki–Heck coupling such as their purification and manufacturing etc., lead to the invention of in situ generation of desired alkenes through various means and then utilizing them in Heck coupling. For the Mizoroki–Heck reaction these alternative ways for generating alkenes apart from employing Wittig reaction, involves hydrolysis followed by dehydrohalogenation, simple dehydrohalogenation, dehydration, elimination or deacetoxylation of various alkenes synthons. However the synthesis of olefins, required for Heck coupling through Wittig reaction has become more appealing in recent years due to substantially greater availability of aromatic aldehyde and ketones as compared to styrene.15These reactions not only did solve the issue of alkenes undergoing polymerization when stored or reacted at high temperature but the one-pot Wittig–Heck sequence also significantly increased the reaction efficiency by reducing the reaction time and purification steps.20 Thus this alternative route to in situ generation of alkenes followed by their coupling with aryl halides to form substituted olefins must be explored further in future owing to their myriad of applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thanks the principal of Govt. Shivnath Science College, Rajnandgaon for providing an opportunity to carry out this study.References
- (a) H. U. Lah, F. Rasool and S. K. Yousuf, RSC Adv., 2015, 5, 78958 RSC; (b) D. P. Ojha and K. R. Prabhu, J. Org. Chem., 2012, 77, 11027 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed. Engl., 2009, 48, 5094 CrossRef CAS PubMed.
- (a) J. Barluenga, P. Moriel, C. Valdes and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587 CrossRef CAS PubMed; (b) J. Radolko, P. Ehlers and P. Langer, Adv. Synth. Catal., 2021, 363, 3616 CrossRef CAS.
- (a) C. Wu and J. Zhou, J. Am. Chem. Soc., 2014, 136, 650 CrossRef CAS PubMed; (b) S. Bräse and A. D. Meijere, Cross-Coupling of Organyl Halides with Alkenes: the Heck Reaction, John Wiley & Sons, Ltd, 2004, vol. 217 Search PubMed; (c) H.-J. Xu, Y.-Q. Zhao and X.-F. Zhou, J. Org. Chem., 2011, 76, 8036 CrossRef CAS; (d) M. L. Kantam, P. Srinivas, J. Yadav, P. R. Likhar and S. Bhargava, J. Org. Chem., 2009, 74, 4882 CrossRef CAS PubMed; (e) L. Yu, Y. Huang, Z. Wei, Y. Ding, C. Su and Q. Xu, J. Org. Chem., 2015, 80, 8677 CrossRef CAS PubMed; (f) D.-H. Lee, A. Taher, S. Hossain and M.-J. Jin, Org. Lett., 2011, 13, 5540 CrossRef CAS; (g) X.-W. Yang, D.-H. Li, A.-X. Song and F.-S. Liu, J. Org. Chem., 2020, 85, 11750 CrossRef CAS PubMed; (h) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307 CrossRef CAS PubMed; (i) C. Wu and J. Zhou, J. Am. Chem. Soc., 2014, 136, 650 CrossRef CAS PubMed; (j) N. Riemer, M. Riemer, M. Krüger, G. J. Clarkson, M. Shipman and B. Schmidt, J. Org. Chem., 2021, 86, 8786 CrossRef CAS.
- (a) R. F. Heck, J. Am. Chem. Soc., 1969, 8, 6707 CrossRef; (b) K. Mori, T. Mizoroki and A. Ozaaki, CSJ, 1973, 46, 1505 CAS; (c) C. C. Perez, J. M. Pena and C. R. D. Correia, New J. Chem., 2014, 38, 3933 RSC; (d) K. Kumar Ponnapalli, A. Kumar Adak, A. Das, J.-S. Song, S.-H. Wu, C.-M. Sun and C.-C. Lin, Asian J. Org. Chem., 2017, 6, 1308 CrossRef CAS; (e) L. Botella and C. Nájera, Tetrahedron Lett., 2004, 60, 5563 CrossRef CAS; (f) G. G. Cross, C. R. Eisnor, R. A. Gossage and H. A. Jenkins, Tetrahedron Lett., 2006, 47, 2245 CrossRef CAS; (g) Z. A. Khan, A. Iqbal and S. A. Shahzad, Mol. Diversity, 2017, 21, 483 CrossRef PubMed.
- S. A. Saiyed and V. A. Bedekar, Tetrahedron Lett., 2010, 51, 6227 CrossRef.
- (a) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed; (b) K. L. Greenman, D. S. Carter and D. L. Van Vranken, Tetrahedron Lett., 2001, 57, 5219 CrossRef CAS; (c) Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef CAS.
- J. Dimminie, S. Metts and E. J. Parsons, Organometallics, 1995, 14, 4023 CrossRef.
- (a) S. Gowrisankar, H. Neumann and M. Beller, Angew. Chem., 2011, 123, 5245 (Angew. Chem., Int. Ed., 2011, 50, 5139) CrossRef; (b) H. Miyamura, T. Yasukawa and S. Kobayashi, Green Chem., 2010, 12, 776 RSC; (c) O. Kose and S. Saito, Org. Biomol. Chem., 2010, 8, 896 RSC; (d) B. Xu, R. J. Madix and C. M. Friend, J. Am. Chem. Soc., 2010, 132, 16571 CrossRef CAS PubMed; (e) K.-I. Shimizu, R. Sato and S. Atsushi, Angew. Chem., 2009, 121, 4042 (Angew. Chem., Int. Ed., 2009, 48, 3982) CrossRef; (f) F.-A. Kang, J. C. Lanter, C. Cai, Z. Sui and W. V. Murray, Chem. Commun., 2010, 46, 1347 RSC.
- (a) R. Kumar, A. Shard, R. Bharti, Y. Thopate and A. K. Sinha, Angew. Chem., Int. Ed., 2012, 51, 2636 CrossRef CAS PubMed; (b) M. L. Crawley, K. M. Phipps, I. Goljer, J. F. Mehlmann, J. T. Lundquist, J. W. Ullrich, C. Yang and P. E. Mahaney, Org. Lett., 2009, 11, 1183 CrossRef CAS PubMed; (c) J. Muzart, Tetrahedron Lett., 2003, 59, 5789 CrossRef CAS.
- G. K. S. Prakash, P. V. Jog, H. S. Krishnan and G. A. Olah, J. Am. Chem. Soc., 2011, 133, 2140 CrossRef CAS.
- G. K. S. Prakash, H. S. Krishnan, P. V. Jog, A. P. Iyer and G. A. Olah, Org. Lett., 2012, 14, 1146 CrossRef CAS PubMed.
- (a) E. A. B. Kantchev, C. J. O. Brien and M. G. Organ, Angew. Chem., 2007, 119, 2824 (Angew. Chem., Int. Ed., 2007, 46, 2768) CrossRef; (b) A. B. S. Maya, C. Prez-Melero, C. Mateo, D. Alonso, J. L. Fernndez, C. Gajate, F. Mollinedo, R. Pelez, E. Caballero and M. Medarde, J. Med. Chem., 2005, 48, 556 CrossRef CAS PubMed.
- P. Colbon, J. H. Barnard, M. Purdie, K. Mulholland, I. Kozhevnikov and J. Xiao, Adv. Synth. Catal., 2012, 354, 1395 CrossRef CAS.
- H. Wu, J. Yang, J. Šečkutė and N. K. Devaraj, Angew. Chem., Int. Ed., 2014, 53, 5805 CrossRef CAS PubMed.
- H. A. Patel, A. L. Patel and A. V. Bedekar, Appl. Organomet. Chem., 2015, 29, 1 CrossRef CAS.
- L. F. Tietze, G. Kettschau, U. Heuschert and G. Nordmann, Chem.–Eur. J., 2001, 7, 368 CrossRef CAS PubMed.
- (a) K. C. Majumdar, B. Chattopadhyay and B. Sinha, Tetrahedron Lett., 2008, 49, 1319 CrossRef CAS; (b) T. S. Kam, Y. M. Tee and G. N. Subramaniam, Prod. Lett., 1998, 12, 307 CrossRef CAS; (c) S. Kotha and K. Mandak, Tetrahedron Lett., 2004, 45, 1391 CrossRef CAS; (d) O. Baudoin, D. Guenard and F. Gueritte, Org. Chem., 2004, 1, 333 CAS; (e) K. Carl and L. Joseph, Chem. Abstr., 1978, 89, 24156 Search PubMed.
- J. Bosch, J. Bachs, A. M. Gómez, R. Griera, M. Écija and M. Amat, J. Org. Chem., 2012, 77, 6340 CrossRef CAS.
- A. S. Saiyed, K. N. Patel, B. V. Kamath and A. V. Bedekar, Tetrahedron Lett., 2012, 53, 4692 CrossRef CAS.
- K. N. Patel, B. V. Kamath and A. V. Bedekar, Tetrahedron Lett., 2013, 54, 80 CrossRef CAS.
- A. V. Bedekar, A. R. Chaudhary, M. Shyam Sundar and M. Rajappa, Tetrahedron Lett., 2013, 54, 392 CrossRef CAS.
- K. N. Patel and A. V. Bedekar, Tetrahedron Lett., 2015, 56, 6617 CrossRef CAS.
- H. M. Savanur, R. G. Kalkhambkar and K. K. Laali, Appl. Catal., 2017, 543, 150 CrossRef CAS.
- T. Moriguchi, D. Tabuchi, D. Yakeya, A. Tsuge, V. Jalli and K. Yoza, J. Mol. Struct., 2018, 1152, 37 CrossRef CAS.
- H. A. Patel, V. J. Bhanvadia, H. M. Mande, S. S. Zade and A. L. Patel, Org. Biomol. Chem., 2019, 17, 9467 RSC.
- B. Rajan and A. V. Bedekar, J. Mol. Struct., 2021, 1234, 130178 CrossRef CAS.
- J. T. McCann, B. R. Benlian, S. K. Yaeger-Weiss, I. J. Knudson, M. He and E. W. Miller, J. Am. Chem. Soc., 2022, 144, 13050 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |