DOI:
10.1039/D3RA03340F
(Paper)
RSC Adv., 2023,
13, 21690-21702
First-principles study on the structure and electronic properties of M2CSx (M = Sc, Ti, Y, Zr and Hf, x = 1, 2)†
Received
19th May 2023
, Accepted 30th June 2023
First published on 19th July 2023
Abstract
Two-dimensional (2D) transition metal carbides/nitrides, known as MXenes, have attracted extensive attention due to their rich elemental composition and diverse surface chemistry. In this study, the crystal structure, electronic, mechanical, and electronic transport properties of M2CSx (M = Sc, Ti, Y, Zr, and Hf, x = 1, 2) were investigated by density functional theory (DFT). Our results showed that the studied M2CSx except Y2CS2 are thermodynamically, dynamically, thermally, and mechanically stable. The p–d hybridization between the M-d state and the C/S-p state of M2CS is stronger than that of the corresponding M2CS2. However, the antibonding state would appear near the Fermi level and thus reduce the thermal stability of the material due to the introduction of sulfur vacancies in the Y-free MXenes studied. In contrast, sulfur vacancies would significantly enhance the bonding states of Y–C and Y–S bonds and improve the stability of Y2CSx. This provides an explanation for the experimentally observed formation of non-stoichiometric Ti2CS1.2. The room-temperature electron mobilities of semiconductor Sc2CS (Y2CS) along the x and y directions were determined to be 232.59 (818.51) and 628.22 (552.55) cm2 V−1 s−1, and the room-temperature hole mobilities are only 88.32 (1.64) and 61.75 (17.80) cm2 V−1 s−1. This work is expected to provide theoretical insights for the preparation and application of S-terminated MXenes.
1. Introduction
MXenes are a large family of two-dimensional (2D) transition metal carbides, nitrides, and carbonitrides with diverse structures, compositions, and surface chemistries, and have been widely used in many fields such as energy storage,1,2 electronic devices,3,4 sensors,5,6 catalysts,7,8 the environment,9,10 and biomedicine.11 MXenes are prepared by a top-down synthesis approach, that is, selective etching of A-layer elements from MAX phase precursors. The general formula of MXenes is Mn+1XnTx, where M represents the transition metal element (such as Sc, Ti, V), X is the C or N element, n can vary from 1 to 4, and Tx (x is a variable) indicates the surface group. Since the first MXene (Ti3C2Tx) was obtained by hydrofluoric acid etching in 2011,12 more than 30 stoichiometric MXenes have been prepared and more than 100 compositions of MXenes have been predicted.13 In addition, the surface termination of MXenes is dependent on the synthesis approach and MXene composition.14–16 For example, the surface terminations of MXenes produced by wet chemical etching techniques are generally F, O, and OH.15,17 The halogen-terminated MXenes can be synthesized by etching in Lewis acid CuCl2/CuBr2/CuI2 molten salts.18,19 These terminations can be substituted or eliminated by other functional groups such as S, Se, Te, NH groups through subsequent surface reactions, providing a new route to synthesize new MXenes that cannot be obtained by conventional Lewis acid etching.16,20 The surface terminations of MXenes have a significant impact on the physical, chemical properties and structural stability of MXenes due to the overlapping of non-bonding valence electrons of transition metals through their low-energy electronic states.21–23 Li et al. reported the I-terminated halogenated Ti3C2I2 MXene cathode can activate and stabilize reversible I0/I+ redox couple in the I2–Zn battery to achieve a high voltage plateau.24 Superconductivity was first observed in Nb2CTx terminated with S, Se, or NH groups, while no superconducting transition was found for bare and O-terminated Nb2C.16 Some M2CT2 (T = O, OH or S)-based multifunctional sulfur cathodes were found to effectively suppress the shuttling effect and exhibit excellent cycle performance and high specific capacity in lithium–sulfur batteries.25–27 Zhu et al. showed the O, OH and F groups on Zr2CTx can be seemingly exchanged with S groups through the proposed reaction in CS2 solution.28 A number of S-functionalized MXenes have been reported as electrode materials for potential metal ion batteries due to their lower diffusion barrier and higher storage capacity.29–37 Luo et al. investigated the nitrogen reduction reaction (NRR) of Fe adsorbed on Ti3C2O2 or N/F/P/S/Cl-doped Ti3C2O2−x MXene and found that F and S doping can reduce the energy barrier of the rate-determining step.38 Bae et al. reported a new structural phase of Sc2CO2 containing C3 trimers converted from the hexagonal carbon of the typical trigonal MXene phase, with two very active anion electrons located in the voids between C3 trimers.39 Additionally, Wang et al. demonstrated that the stable Sc2CO2 monolayer is an efficient NH3 gas sensor with high selectivity, high sensitivity and low recovery time through analytical methods based on optical and/or electronic response.40 In comparison, without directly considering the material stability, Hu et al. reported that Sc2CS2 could effectively capture the NH3 and nicotine, while the Y2CS2 exhibits the extremely high sensitivity and selectivity towards NO.41 Importantly, only non-stoichiometric chalcogen-terminated MXenes were observed in the experiment, e.g. x = 1.2 and 1.1 for Ti2CSx, and Ti3C2Sx, respectively,16 despite the dynamical stability of stoichiometric M2CS2 (M = Sc, Ti, V, Cr, Fe, Y, Zr, Nb, Mo, Hf, Ta and W) and Ti3C2S2 have been theoretically confirmed by the calculated phonon dispersions and ab initio molecular dynamics (AIMD) simulations.27,29,42–49 To the best of our knowledge, the non-stoichiometric structures, underlying stability mechanisms and associated physical properties of S-terminated MXenes remain unclear.
In this study, the structural stability, electronic structure, mechanical and electronic transport properties of M2CSx (M = Sc, Ti, Y, Zr and Hf, x = 1, 2) were investigated using density functional theory (DFT). Our results showed that the calculated cohesive energies and bond lengths of the M–C and M–S bonds of M2CS are smaller than those of the counterpart M2CS2 due to the more pronounced electron transfer and stronger p–d overlaying between M-d state and C/S-p state of M2CS. In addition, due to the introduction of sulfur vacancy in the studied MXenes except Y2CSx, the antibonding state would appear near the Fermi level, reducing the thermal stability of the material. Furthermore, both Sc2CS and Y2CS were found to be a thermodynamically stable and dynamically stable semiconductors with appropriate carrier mobility. This work is expected to provide clues for the design and preparation of novel MXenes.
2. Computational details
All the first-principles calculations were carried out by using Vienna Ab initio Simulation Package (VASP). Projector augmented wave (PAW) method50 was employed to describe the electron–core interactions and the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) was used to treat the exchange–correlation potential of valence electrons.51 And the DFT-D3 method with Becke–Jonson damping were included in all calculations to account for the van der Waals (vdW) interaction.52 The cutoff energy of plane waves was set to 500 eV. The Monkhorst–Pack meshes with 12 × 12 × 1 and 9 × 9 × 1 k-points were used to sample the Brillouin zone for the calculations of M2CS2 and M2CS cells, respectively. A large vacuum thickness of 20 Å was used to avoid the spurious interaction between adjacent monolayers. The structures studied were fully optimized using conjugate gradient algorithm, and the energy and force convergences of 10−8 eV and 10−6 eV Å−1 were used for the calculations. In addition, density functional perturbation theory implemented in PHONOPY software was used to calculate phonon spectra to check the dynamical stability of the structures investigated.53,54 And the 4 × 4 × 1 supercell of M2CS2, 2 × 2 × 1 supercell of M2CS, plane wave with a cut-off energy of 650 eV and 4 × 4 × 1 k-point meshes were used for force constant calculations. AIMD simulations were performed for 4 ps in NVT ensemble with a time step of 1.0 fs to assess thermal stability of M2CSx. The Nosé–Hoover thermostat was applied to control the temperature. In order to predict the electronic transport properties, band gap center EBGC was calculated by using GGA-PBE functional and calibrated against vacuum level.55 The band edge of MXene could be accurately evaluated as follows.56–58Here Eg is the calculated band gap of MXene. Carrier mobility of 2D MXenes was studied based on the deformation potential theory according to eqn (3) because the carrier mobility of a 2D intrinsic inorganic semiconductor is mainly dominated by the phonon scattering.59,60| |
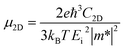 | (3) |
Here, ℏ is the reduced Planck constant, T is the temperature, kB is the Boltzmann's constant, e is the elementary charge. m* = ℏ2/(∂2E/∂k2) is the effective mass of charge, C2D is the elastic modulus under the strain Δa/a0 along the transport direction, determined by C2D = [∂2E/∂[Δa/a0]2]/S0, where E and S0 is the total energy and the area of optimized supercell. And Ei represents the deformation potential constant of valence-band maximum for holes and conduction-band minimum for electrons, calculated by Ei = ∂Eedge/∂(Δa/a0), where Eedge is the energy of ith energy band under a small compressive or tensile strain Δa/a0 along the transport direction. All the structures were visualized by Materials Studio or VESTA3 software.61
3. Results and discussion
3.1. Crystal structure and stability of M2CSx
Four possible configurations for sulfur terminations of a M2CS2 crystal were considered in Fig. S1.† In order to assess the relative thermodynamic stabilities of the configurations studied, cohesive energy Ecoh was calculated by| | |
Ecoh = [Etot(M2CSx) − 2E(M) − E(C) − xE(S)]/(3 + x)
| (4) |
where Etot(M2CSx) is the total energy of M2CSx, E(M), E(C), and E(S) are the energies of isolated M, C, and S atoms, respectively. From the calculated total energies of M2CS2 configurations in Table S1,† it is found that the sulfur layers on both sides of the most stable configuration of M2CS2 (M = Ti, Zr and Hf) (as shown in Fig. 1 and S1B†) are directly projected to the bottom metal layer, while in the most stable configuration of Sc2CS2 and Y2CS2, the sulfur atoms on one side are projected directly to the bottom metal atoms, while the sulfur atoms on the opposite side are projected directly to the carbon atoms in the middle layer (as shown in Fig. 1 and S1C†). Apparently, stable Ti2CS2, Zr2CS2, Hf2CS2 are symmetric type-A structures, while energetically preferred asymmetric type-B structures do exist in Sc2CS2 and Y2CS2. Interestingly, the stable configuration of the studied M2CS2 resembles the counterpart M2CO2.40,62 However, Zhang et al.42 reported that Sc2CS2 and Y2CS2 are actually stabilized in distortion variants of the asymmetric type-B phase depicted in Fig. 1 type-B. Therefore, the relative stability of the type-B phase and the distorted type-B phase of Sc2CS2 and Y2CS2 was compared, and the results are displayed in Table S2 and Fig. S2.† Noteworthily, the dipole-corrected total energies of type-B Sc2CS2 and Y2CS2 calculated in this study are larger than those reported by Zhang et al.42 probably due to the higher convergence criteria used in this study. And the distorted structures of type-B Sc2CS2 and Y2CS2 were optimized back to the type-B phase with the structural symmetry switched off, resulting in their almost identical total energies. This is probably due to the fact that the imaginary phonon modes around the high symmetry point K that lead to the distortion disappear in our calculated phonon spectra (Fig. S6†). This similar distorted structure was also found in transition metal dichalcogenides, but it was determined as a transient state between the 2H phase and the Td phase.63 Therefore, further exploration of this possible distorted phase of type-B Sc2CS2 and Y2CS2 is beyond the scope of this study.
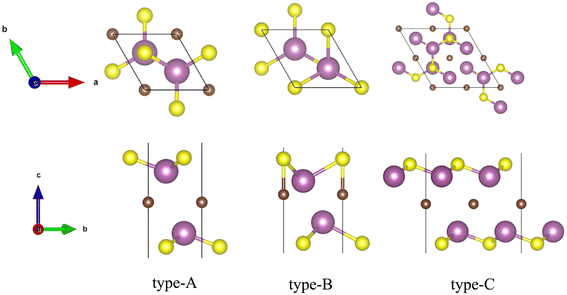 |
| | Fig. 1 The optimized most stable configurations of M2CSx (M = Sc, Ti, Y, Zr and Hf, x = 1, 2). Type-A: the top and side views of the most stable configurations of M2CS2 (M = Ti, Zr and Hf), type-B: the top and side views of the most stable configurations of Sc2CS2 and Y2CS2, type-C: the top and side views of the most stable configurations of M2CS (M = Sc, Ti, Y, Zr and Hf). | |
On the basis of the most stable structure of M2CS2, all possible configurations of M2CS were constructed by 2 × 2 × 1 cell expansion and removal of half of the S terminations, which were done by disorder code.64 There are six possible configures for Ti2CS, Zr2CS, Hf2CS as depicted in Fig. S3,† and nine possible configures for Sc2CS, Y2CS as shown in Fig. S4.† Interestingly, it can be seen from Tables S3 and S4† that the S groups on both sides of all studied M2CS are preferentially arranged in stripes, as shown in Fig. S3 DEF and S4 DEF.† That is, the strip-like S groups on both sides of M2CS2 are removed in turn, so that the number of S atoms removed on both sides is identical. More importantly, among all the arrangements of stripe-like S terminations, the stripe-like S terminations on both sides of M2CS are preferentially parallel and the distance between the two stripe-like S groups is the shortest in the xy plane as displayed in Fig. 1 type-C or S3D and S4F.† This can maintain the crystalline structure of M2CS2 to the greatest extent. Furthermore, it is evident from Table S3† that it is most energetically unfavorable to completely remove the S atoms on one side of the symmetric type-A M2CS2 to expose the MXene, but this is not necessarily the case for the asymmetric type-B M2CS2. As with the symmetric type-A phase, it is most energetically unfavorable to remove all the S atoms projected to the bottom M atoms of the asymmetric type-B M2CS2 (Fig. S4A†). But it can be seen from Table S4† that removing all S atoms projected to C atoms (Fig. S4I†) is easier than removing 1 S atom projected to the bottom M atom and 3 S atoms projected to the C atoms (Fig. S4G and H†). Importantly, considering the small energy differences among the three configurations A, B, and C of the stoichiometric M2CS2 in Table S1,† the most stable non-stoichiometric M2CS configurations derived from these three configurations were also investigated and presented in Fig. S5,† and their corresponding calculated energies are listed in Table S5.† It is evident from Table S5† that the total energy of the most stable configuration of M2CS (M = Sc, Ti, Y, Zr, and Hf) with stripe-like S terminations derived from symmetric type-A M2CS2 (Fig. 1 and S1B†) is the smallest, indicating that regardless of the stable configuration of M2CS2, the stripe-like S terminations of M2CS tend to be located on top of the underlying metal layer as shown in Fig. 1 type-C.
The calculated lattice constants, cohesive energies, space groups and bond lengths of M–S and M–C bonds of the most stable configurations of M2CS2 and M2CS studied are shown in Table 1. For all M2CSx (x = 1 or 2) studied, the cohesive energy of M2CSx is negative, compared to the respective Ecoh values of −6.38 and −5.06 eV per atom for Sc2CO2 (ref. 65) and Ti2CO2,66 which indicates that they are thermodynamically stable to the isolated constituent atoms. The space group of the M2CS2 studied is P3m1, consistent with the experimental observations of traditional stoichiometric MXenes,16 while the space group of M2CS (M = Sc, Ti, Y, Zr, and Hf) is P21/m. The lattice parameter a = 3.16 Å of Ti2CS2 and the lattice constant a = 6.17 Å of Ti2CS are close to the experimentally observed a value of 3.21 Å for Ti2CS.16 And the lattice constants of M2CS2 (M = Sc, Ti, Zr and Hf) are slightly larger than those of the corresponding M2CO2 (i.e. 3.41, 3.04, 3.31 and 3.27 Å for Sc2CO2, Ti2CO2, Zr2CO2 and Hf2CO2)67 due to the weaker M–S bond relative to the M–O bond. Clearly, it can be seen that the lattice constant of Y2CSx is the largest among the studied M2CSx with the same formula due to the weak interatomic bonding of Y–C and Y–S, which will be explained in detail in Section 3.2. The calculated Ti–C bond length of 2.09 Å and Ti–S bond length of 2.36 Å for the Ti2CS are in good agreement with the experimentally observed Ti–C and Ti–S bond lengths of 2.14 Å and 2.41 Å.16 Importantly, the M–C and M–S bond lengths of the M2CS2 studied are longer than those of the counterpart M2CS. And the calculated Ecoh of the M2CS2 studied are greater than those of the counterpart M2CS. In addition, it can be found from Table 2 that the net negative charge of S and C atoms of the M2CS2 studied are smaller than those of the corresponding M2CS, which indicates that more electrons are transferred from the M atom to the C and S atoms of the M2CS than the corresponding M2CS2. The significant charge transfer can also be visually observed from the charge density difference in Fig. S6.† This confirms that the M–C and M–S bonds of M2CS are stronger than the corresponding M2CS2.
Table 1 The calculated Lattice constant (Å), space group, cohesive energy Ecoh (eV per atom) and bond lengths (Å) of M–S and M–C bonds of M2CSx (M = Sc, Ti, Y, Zr and Hf, x = 1, 2)
| Material |
Lattice constant |
Bond length |
Space group |
Ecoh |
Material |
Lattice constant |
Bond length |
Space group |
Ecoh |
| a |
M–S |
M–C |
a |
M–S |
M–C |
| Sc2CS2 |
3.76 |
2.484/2.459 |
2.293/2.650 |
P3m1 |
−6.03 |
Sc2CS |
6.82 |
2.474 |
2.253 |
P21/m |
−6.30 |
| Ti2CS2 |
3.16 |
2.395 |
2.189 |
P3m1 |
−6.70 |
Ti2CS |
6.17 |
2.358 |
2.093 |
P21/m |
−7.00 |
| Y2CS2 |
4.17 |
2.628/2.605 |
2.485/2.877 |
P3m1 |
−6.52 |
Y2CS |
7.37 |
2.656 |
2.429 |
P21/m |
−6.62 |
| Zr2CS2 |
3.44 |
2.531 |
2.394 |
P3m1 |
−8.13 |
Zr2CS |
6.64 |
2.508 |
2.278 |
P21/m |
−8.15 |
| Hf2CS2 |
3.40 |
2.508 |
2.366 |
P3m1 |
−7.92 |
Hf2CS |
6.54 |
2.477 |
2.260 |
P21/m |
−8.14 |
Table 2 Loewdin atomic charge of M2CSx (M = Sc, Ti, Y, Zr and Hf, x = 1, 2)
| Material |
C |
S |
M |
Material |
C |
S |
M |
| Sc2CS2 |
−1.53 |
−0.31/−0.85 |
1.32/1.46 |
Sc2CS |
−1.95 |
−0.94 |
1.45/1.48 |
| Ti2CS2 |
−1.73 |
−0.56 |
1.42 |
Ti2CS |
−1.90 |
−0.78 |
1.30/1.38 |
| Y2CS2 |
−0.85 |
−0.43/−0.10 |
0.75/0.63 |
Y2CS |
−0.91 |
−0.46 |
0.67/0.70 |
| Zr2CS2 |
−1.59 |
−0.54 |
1.36 |
Zr2CS |
−1.63 |
−0.69 |
1.28/1.30 |
| Hf2CS2 |
−1.08 |
−0.31 |
0.85 |
Hf2CS |
−1.02 |
−0.42 |
0.70/0.73 |
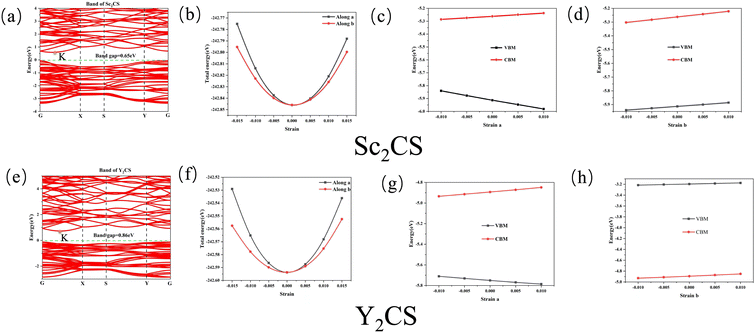
In order to further check the dynamical stability of MXenes, the phonon dispersion curves of M2CSx were calculated. It is worth mentioning that small imaginary frequencies near the Γ point are often observed in the calculated phonon spectra of 2D systems, which could be caused by numerical noise owing to the vacuum region.68–70 As shown in Fig. 2 and S6,† almost no imaginary frequency is observed in the phonon dispersions of M2CS (M = Sc, Ti, Zr, and Y), and some small negative frequencies near the Γ point appear in the phonon dispersion curves of M2CS2 (M = Sc, Ti, Zr, and Hf) and Hf2CS. This is an indication of dynamic stabilities of M2CSx studied except Y2CS2, consistent with the fact that Ti2CS was found in experiments.16 As a comparison, the exception Y2CS2 shows significant imaginary frequencies in the phonon dispersion, consistent with previously reported results by Zhang et al.42 and Li et al.,68 which is a sign that the formation of the Y2CS2 MXene probably needs more inspection from experiments. Then, AIMD simulations were performed to further evaluate the dynamical and thermal stabilities of the M2CSx studied as displayed in Fig. 3. There is no broken M–C and M–S bonds or obvious structural reconstruction within a simulation of 4 ps at 300 K for the M2CS (M = Sc, Y, and Zr) and even at 900 K for the M2CS2 (M = Sc, Ti, Zr, and Hf) and Hf2CS, manifesting the good thermal stability of these S-terminated MXenes. While for Ti2CS, at 300 K, a structural reconstruction of part of the S-terminations moving from the top of bottom metal to the top of the carbon layer is observed, indicating that the strip-like S-terminations of Ti2CS are temperature-sensitive. In contrast, Y2CS2 exhibits a pronounced destruction within 4 ps simulations at 300 K, which can be regarded as evidence of poor thermal stability, consistent with the observation of significant imaginary frequency. From the above analysis, it can be seen that the studied M2CSx except Y2CS2 has good dynamical and thermal stability, and it is particularly noted that the strip-shaped S functional groups of Ti2CS are temperature sensitive and easily disturbed. Chen et al.71 demonstrated that the stabilization of a certain surface configuration of MXene was the statistical average of the adsorption and desorption of surface functional groups. Thus, the total energy of a certain surface configuration is a function of the chemical potential of the functional group. When the chemical potential of the S functional group is high, close to its upper limit (i.e., the chemical potential of elemental S8), the stoichiometric M2CS2 is the thermodynamically most favorable configuration under such condition. As a comparison, when the chemical potential of the S functional group is small, the non-stoichiometric M2CSx may be the thermodynamically preferred configuration, and even M2CSx would decompose into bare M2C and elemental sulfur or sulfide. In the experimental preparation,16,20 sulfur-functionalized MXene was obtained by functional group substitution of halogen-functionalized MXene by Li2S or intercalation of bare MXene by FeS or CuS. Under such preparation conditions, the chemical potential of the S functional group is generally lower, which is more conducive to the formation of non-stoichiometric S-functionalized MXenes, which may be the reason why no stoichiometric S-functionalized MXenes were observed experimentally. Zhu et al.28 theoretically clarified the feasibility of interconversion of Zr2CO2 and excess CS2 to Zr2CS2 under the release of COS. Therefore, under suitable experimental conditions with high chemical potential of the functional group S, stoichiometric S-functionalized MXenes can be prepared.
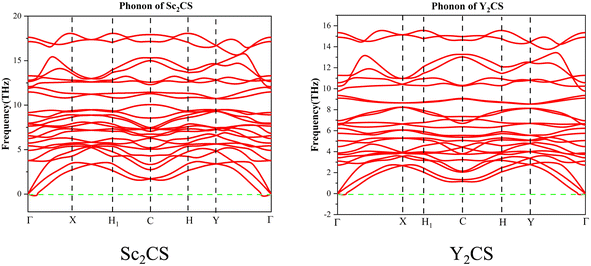 |
| | Fig. 2 The calculated phonon dispersions of the Sc2CS and Y2CS. | |
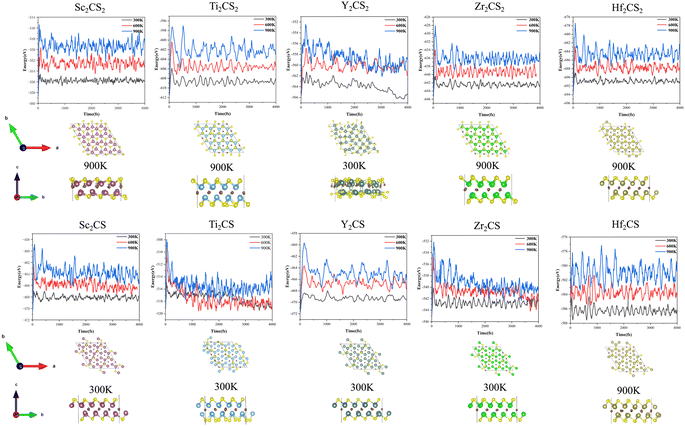 |
| | Fig. 3 Free energy fluctuations with respect to time at 300 K, 600 K and 900 K in AIMD simulations for M2CSx (M = Sc, Ti, Y, Zr, and Hf, x = 1, 2), and the obtained structures of M2CSx after AIMD simulations at specific temperature. The specified temperature in the figure is the highest temperature at which the structure can be kept stable among the three temperatures studied. Noteworthily, the reconstruction of the surface functional groups of Ti2CS and the pronounced destruction of Y2CS2 were observed at the lowest temperature studied, 300 K. | |
3.2. Electronic structure and bonding mechanism of M2CSx
In order to explore the electronic structures of the S-functionalized M2CSx (M = Sc, Ti, Y, Zr, and Hf, x = 1, 2) studied, the calculated electronic band structures of M2CSx as well as the corresponding M2CO2 for comparison are presented in Fig. 4. It can be readily found that the studied M2CO2 are all indirect bandgap semiconductors, in good accordance with the previous reported results.40,67 Interestingly, four sulfur-terminated MXenes Sc2CS2, Y2CS2, Sc2CS, and Y2CS were identified as indirect bandgap semiconductors with bandgaps of 1.56, 1.85, 0.68, and 0.92 eV, respectively, which are larger than the bandgaps of 1.56 and 1.37 eV for the counterparts Sc2CO2 and Y2CO2. In contrast, the energy bands of the studied M2CSx (M = Ti, Zr, and Hf, x = 1, 2) crossing Fermi level exhibit the metallic-like features due to the presence of nonbonding M-d states at the Fermi level in Fig. 5. Since the traditional PBE functional generally underestimates the band gap of the material, the band gap of the sulfur-terminated MXene was corrected by the HSE06 functional.72 After HSE06 correction, the band gaps of Sc2CS2, Y2CS2, Sc2CS and Y2CS were increased to 2.54, 2.75, 1.43 and 1.69 eV, respectively. Their suitable bandgap endows them with great potential as electronic devices. Cautious inspection from the calculated band gaps of M2CSx (M = Sc and Y, x = 1, 2) reveals that sulfur vacancy defects increase the unbonded electrons near the Fermi level of metal atoms on the MXene surface leading to the attenuation of the band gap.
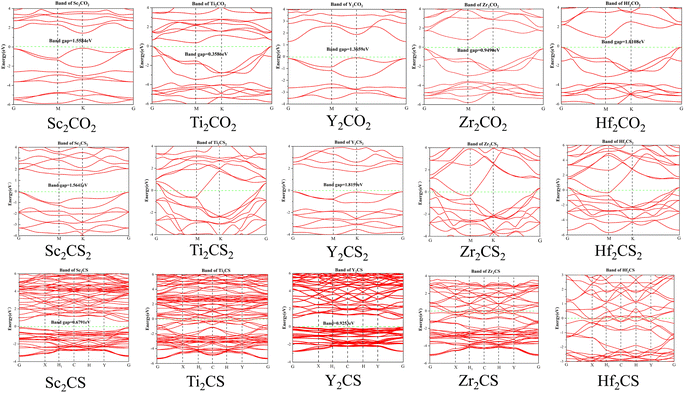 |
| | Fig. 4 The band structures of M2CSx (M = Sc, Ti, Y, Zr, and Hf, x = 1, 2) together with the corresponding M2CO2. The bandgaps of semiconductor materials are shown in the figure. | |
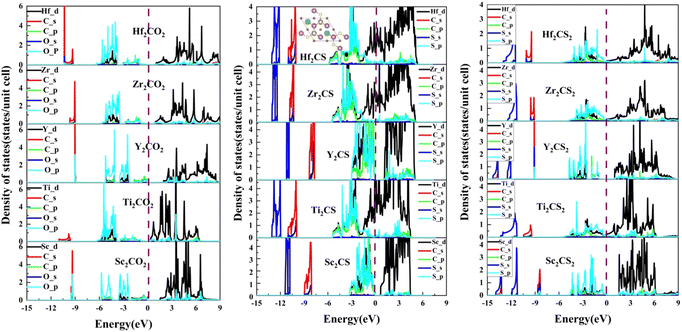 |
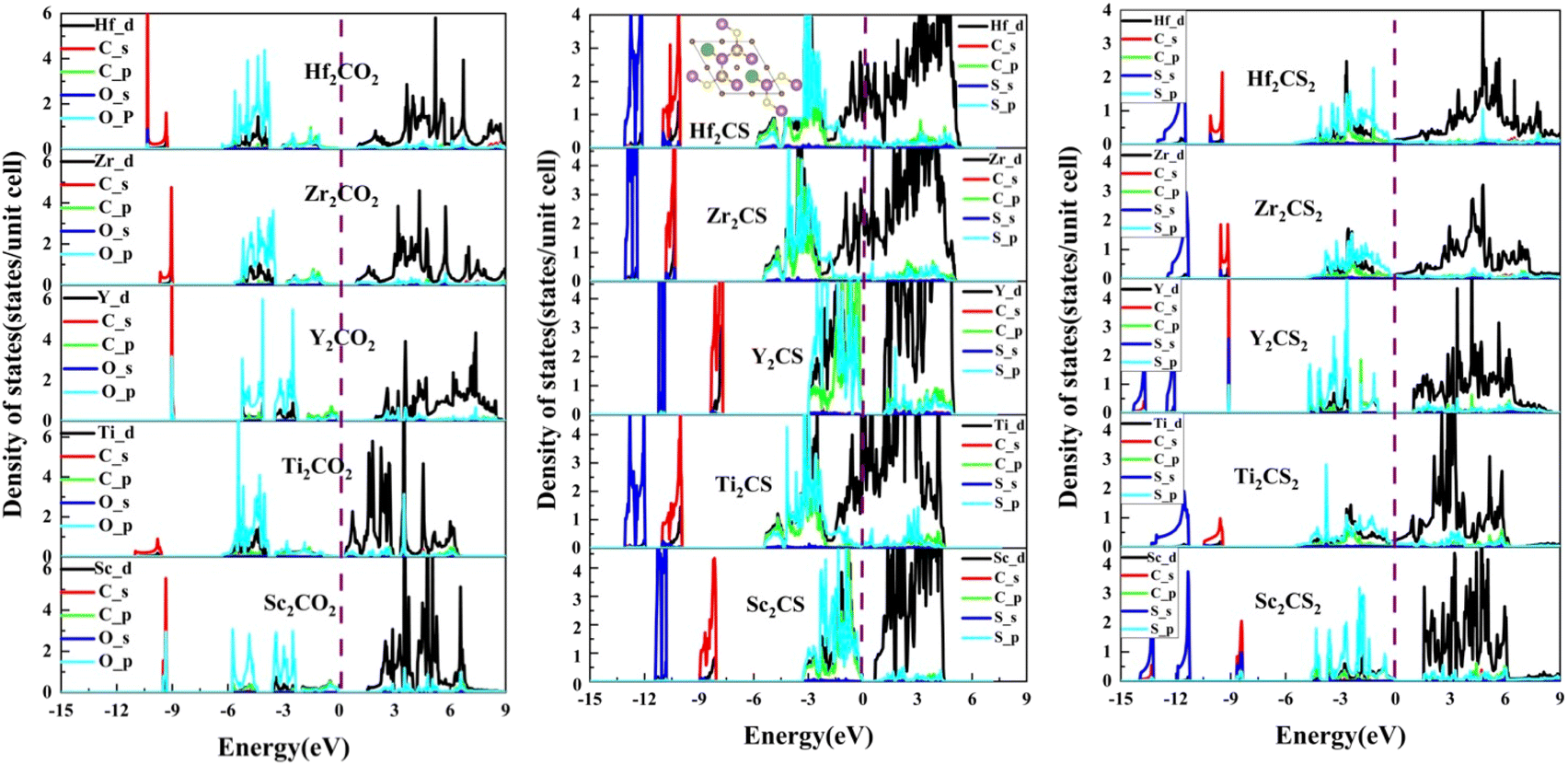
| | Fig. 5 The projected density of states (PDOS) of the M2CSx (M = Sc, Ti, Y, Zr, and Hf, x = 1, 2) as well as the corresponding M2CO2. The green spheres in the insert indicate S vacancies. The black curves represent M-d orbitals, the red and green curves represent C-s and -p orbitals, respectively, and the blue and cyan curves represent S-s and -p orbitals, respectively. | |
In order to study the underlying bonding mechanism of the studied M2CSx, the projected density of states (PDOS) of M2CSx together with the corresponding M2CO2 for comparison were investigated as depicted in Fig. 5. For all MXenes studied, the electronic states above the Fermi level are dominated by nonbonding t2g states and antibonding  states of the M-d orbital. Noteworthily, the S-p state of M2CSx overlaps with the M-d state in the energy range from −4.5 eV to 0 eV, while the O-p state of M2CO2 hybridizes with the M-d state around −6 to −2 eV, which is an implication of the stronger M–O bond than M–S bond. Resembling the corresponding M2CO2, due to the splitting of the 5-fold degenerate M-d orbitals of M2CS2 in the octahedral crystal field of MX6, 5 hybridization peaks of the S-p state are observed, which are more conspicuous in Y2CS2 and Sc2CS2, but this phenomenon disappears in M2CS due to the presence of S vacancies. In particular, the p–d hybridization peaks between S and Y of Y2CS2 are very localized, reflecting the weak nature of the Y–S bond, which are significantly improved in Y2CS. More importantly, the hybridization peaks between the M-d orbital and the C-p or S-p orbital of M2CS are remarkably higher than those of the corresponding M2CS2, which can be regarded as evidence that the M–C and M–S bonds in M2CS are stronger than those in M2CS2.
states of the M-d orbital. Noteworthily, the S-p state of M2CSx overlaps with the M-d state in the energy range from −4.5 eV to 0 eV, while the O-p state of M2CO2 hybridizes with the M-d state around −6 to −2 eV, which is an implication of the stronger M–O bond than M–S bond. Resembling the corresponding M2CO2, due to the splitting of the 5-fold degenerate M-d orbitals of M2CS2 in the octahedral crystal field of MX6, 5 hybridization peaks of the S-p state are observed, which are more conspicuous in Y2CS2 and Sc2CS2, but this phenomenon disappears in M2CS due to the presence of S vacancies. In particular, the p–d hybridization peaks between S and Y of Y2CS2 are very localized, reflecting the weak nature of the Y–S bond, which are significantly improved in Y2CS. More importantly, the hybridization peaks between the M-d orbital and the C-p or S-p orbital of M2CS are remarkably higher than those of the corresponding M2CS2, which can be regarded as evidence that the M–C and M–S bonds in M2CS are stronger than those in M2CS2.
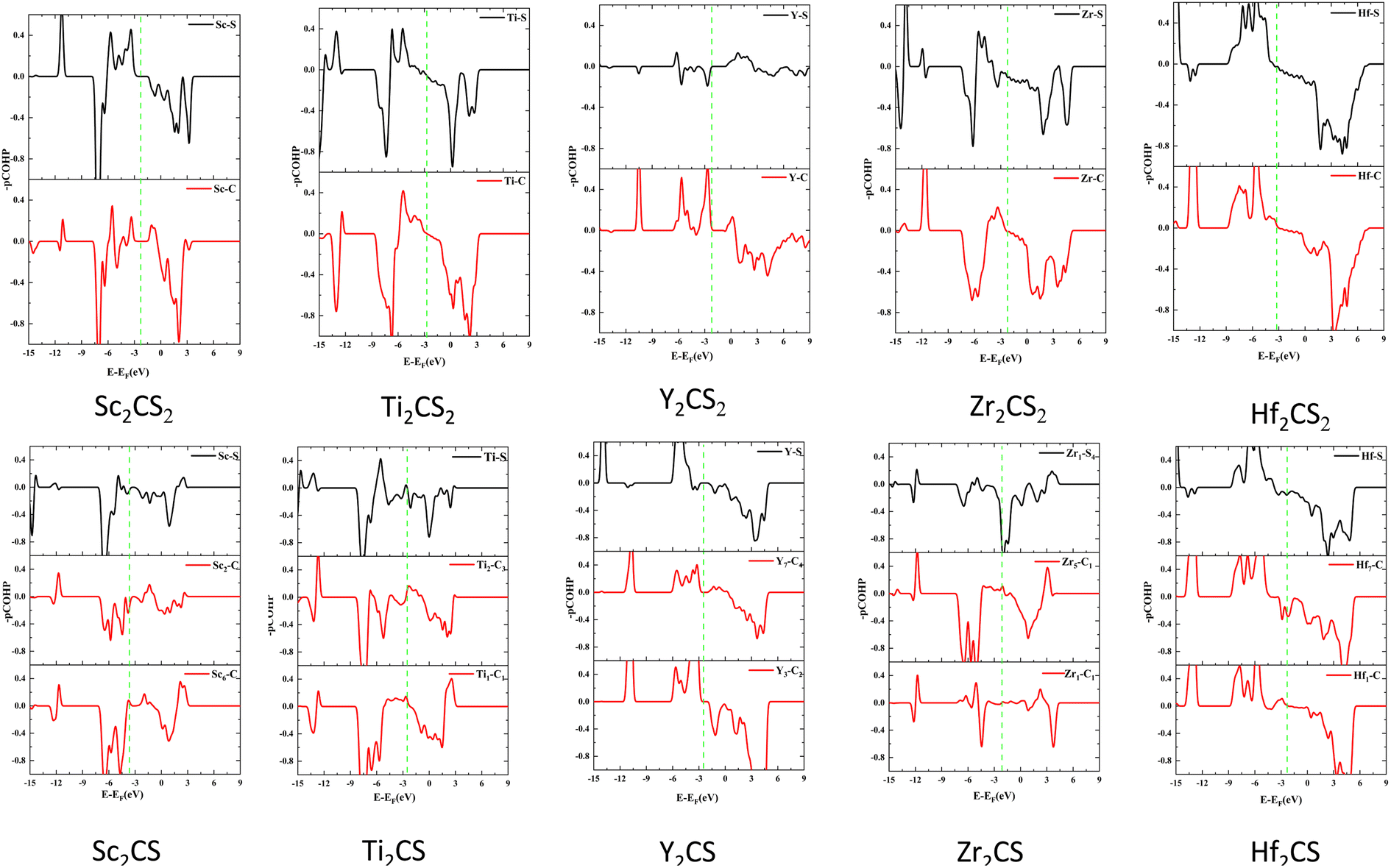
To explore bonding/antibonding states of bonds of the studied M2CSx (M = Sc, Ti, Y, Zr, and Hf, x = 1, 2), the projected crystal orbital Hamilton population (pCOHP) was calculated using the LOBSTER program,73 as shown in Fig. 6. A positive value of −pCOHP indicates a bonding state, and a negative value indicates an antibonding state. The hybridization between M and C and between M and S of the investigated M2CS is stronger than that of the corresponding M2CS2 (seen in Fig. 5) leading to greater splitting of bonding and antibonding states in Fig. 6, i.e. farther away from the Fermi energy level, consistent with the reported results of Ti2CO2.74 However, the introduction of sulfur vacancies will increase the non-bonding eg states of the metal d orbitals, thereby making the antibonding states appear near the Fermi level and significantly reducing the thermal stability of the material, consistent with the AIMD simulation results. Especially, both the bonding and antibonding states of the Y–S bond of Y2CS2 are very small, again manifesting a weak Y–S bond, leading to the largest lattice constant of Y2CS2 among all studied MXenes. On the contrary, the bonding states of the Y–C and Y–S bonds of Y2CS are very obviously enhanced, significantly improving the stability of Y2CSx. Consequently, the d–p hybridization between the metal and C/S atoms of M2CS is significantly higher than that of the corresponding M2CS2, resulting in stronger M–C and M–S bonds in M2CS than in M2CS2. However, due to the introduction of sulfur vacancies in other studied MXenes except Y2CSx, the antibonding state would appear near the Fermi level, reducing the thermal stability of the material. On the contrary, sulfur vacancies would significantly enhance the bonding states of Y–C and Y–S bonds and improve the stability of Y2CSx.
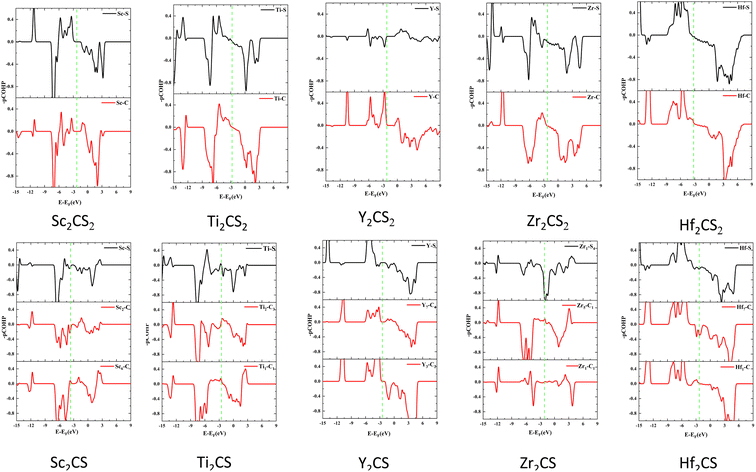 |
| | Fig. 6 The calculated COHP curves of M2CSx (M = Sc, Ti, Y, Zr, and Hf, x = 1, 2). The vertical green dotted line represents the Fermi level. | |
3.3. Mechanical properties
The fabrication of 2D crystal-based electronics generally requires high in-plane stiffness to avoid curling and obtain free-standing membranes. Therefore, the mechanical properties of M2CSx (M = Sc, Ti, Y, Zr, and Hf, x = 1, 2) were investigated by examining their elastic constants. Elastic constants Cij of 2D crystal can be obtained by the first-principles energy-strain method, which is based on the energy variant by applying a small strain ε onto the equilibrium structure. Subsequently, the in-plane Young's modulus Y2D, in-plane shear modulus G2D (in N m−1), and Poisson's ratios v can be obtained by eqn (5)–(7)70| | |
Y2D = (C112 − C122/C11)
| (5) |
The 2D crystal structure is determined to be mechanically stable when all elastic constants satisfy the well-known Born–Huang criteria:75 For a trigonal M2CS2 structure: C11 > 0, C11 > |C12|, C66 > 0, and for a monoclinic M2CS structure: C11 > 0, C11C12 > C12C12, Cij > 0. The calculated elastic constants of M2CSx (M = Sc, Ti, Y, Zr, and Hf, x = 1, 2) in Fig. 7 and Table 3 meet the mechanical stability criteria, indicating they are mechanically stable under ambient conditions. It is evident that all the in-plane stiffnesses of Sc2CSx and Y2CSx are much smaller than those of the M2CSx (M = Ti, Zr, and Hf) probably due to the smaller cohesive energies of Sc2CSx and Y2CSx relative to M2CSx (M = Ti, Zr, and Hf). And the maximum Y2D and G2D values of Sc2CS and Y2CS are larger than those of Sc2CS2 and Y2CS2, respectively, due to the stronger M–C and M–S bonds of M2CS compared with M2CS2, while M2CSx (M = Ti, Zr, and Hf) show the opposite trend, which may be caused by the antibonding states of M–C bond occurring at the Fermi level of M2CS (Fig. 6). The in-plane Young's modulus and in-plane shear modulus of 104.39 and 36.61 N m−1 for Sc2CS2 is the smallest among all the studied M2CS2, which are smaller than those of Sc2CO2.67 Noteworthily, the calculated in-plane Young's moduli of the semiconductors Sc2CS and Y2CS are 125.06–140.35 and 82.77–125.34 N m−1, respectively, which are comparable to those of MoSe2 (125 N m−1) and MoS2 (168 N m−1),76 indicating that Sc2CS and Y2CS monolayer have good mechanical properties. And it is further found that their Poisson's ratios v are all within 0.3. Since the low Poisson's ratio is favorable for the application of MXenes in flexible device materials,66 Sc2CS and Y2CS MXenes have the potential to be used as flexible electronic devices.
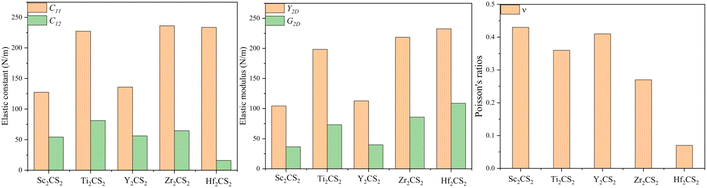 |
| | Fig. 7 The elastic constants C11, C12, in-plane Young's moduli Y2D, in-plane shear moduli G2D (in N m−1), and Poisson's ratios v of the M2CS2 MXenes studied. | |
Table 3 The elastic constants C11, C12, in-plane Young's moduli Y2D, in-plane shear moduli G2D (in N m−1), and Poisson's ratios v of the M2CS MXenes studied
| |
C11 |
C12 |
Y2D (min) |
Y2D (max) |
G2D (min) |
G2D (max) |
v (min) |
v (max) |
| Sc2CS |
138.95 |
42.61 |
125.06 |
140.35 |
50.16 |
50.99 |
0.27 |
0.31 |
| Ti2CS |
194.49 |
55.80 |
175.98 |
196.30 |
70.03 |
75.02 |
0.23 |
0.30 |
| Y2CS |
120.15 |
38.14 |
82.77 |
125.34 |
36.05 |
43.62 |
0.25 |
0.49 |
| Zr2CS |
196.50 |
56.84 |
176.01 |
194.32 |
69.85 |
74.81 |
0.24 |
0.31 |
| Hf2CS |
217.28 |
58.13 |
197.22 |
210.13 |
79.22 |
82.62 |
0.24 |
0.28 |
3.4. Carrier mobility of nonstoichiometric Sc2CS and Y2CS MXenes
Carrier mobility is an important physical quantity to measure the performance of electronic devices. The orthorhombic Sc2CS and Y2CS supercell was adopted to calculate the carrier mobility according to deformation potential theory. As shown in Fig. 8a and e, the valence band maximum (VBM) of Sc2CS and Y2CS at Γ point is observed while the conduction band minimum (CBM) appears at the K point along Γ–X direction in the electronic band structure of nonstoichiometric Sc2CS and Y2CS. As an important band structure-derived property, effective masses of electrons at K point and holes at Γ point of Sc2CS and Y2CS were calculated and shown in Table 4. For both Sc2CS and Y2CS, it can be readily found that the effective hole mass is much larger than the effective electron mass, suggesting that electrons can have higher mobility than holes under an external electric field due to the much smaller effective mass. Interestingly, the effective electron mass and effective hole mass of Sc2CS along x direction are larger than those along y direction, respectively, and the effective hole mass of Y2CS material also shows the same trend, showing effective mass anisotropy. In detail, the effective mass of electrons of Sc2CS along x direction is 0.70me, which is 9.4% larger than that along y direction, while the effective mass of holes along x direction (3.30me) is 61.8% larger than that along y direction. In comparison, for Y2CS, the effective electron masses along the x and y directions are almost equal, but the effective hole mass along the x direction is 1.21 times larger than along the x direction. Besides the effective mass, the deformation potential constant El and elastic modulus C2D of Sc2CS and Y2CS MXene were also calculated. The C2D value of electrons and holes of Sc2CS and Y2CS along x direction is 111.98 and 91.02 J m−2, which is 32.8% and 57.9% larger than those along the y direction, respectively. Additionally, the El values (7.03 eV) of holes at Γ point of Sc2CS along x direction is 1.73 times that along y direction, while the El of electrons at K point along the x direction is only 0.60 times that along the y direction. And the deformation potential constants of holes at Γ point of Y2CS along x and y directions are 4.30 and 3.76 eV, which are 0.35 and 1.75 eV larger than those of electrons at K point along x and y directions, respectively.
 |
| | Fig. 8 (a) and (e) The calculated band structure of Sc2CS and Y2CS monolayer in the orthogonal supercell, (b) and (f) energy–strain relationship along x (Γ–X) and y (Γ–Y) directions of Sc2CS and Y2CS supercell, and (c), (d), (g) and (h) band edges as a function of strain along x and y directions of Sc2CS and Y2CS supercell, respectively. | |
Table 4 The calculated carrier mobilities of nonstoichiometric Sc2CS and Y2CS
| |
Carrier type |
Direction |
C2D (J m−2) |
E1 (eV) |
m* (me) |
μ (cm2 V−1 s−1) |
τ (fs) |
| Sc2CS |
Electron |
x |
111.98 |
7.03 |
0.70 |
232.59 |
92.58 |
| y |
84.33 |
4.06 |
0.64 |
628.22 |
228.62 |
| Hole |
x |
111.98 |
2.42 |
3.30 |
88.32 |
165.72 |
| y |
84.33 |
4.06 |
2.04 |
61.75 |
71.72 |
| Y2CS |
Electron |
x |
91.02 |
4.30 |
0.55 |
818.51 |
255.98 |
| y |
57.66 |
3.95 |
0.58 |
552.55 |
182.23 |
| Hole |
x |
91.02 |
3.76 |
14.04 |
1.64 |
13.11 |
| y |
57.66 |
2.01 |
6.35 |
17.80 |
64.28 |
Therefore, according to eqn (3), the electron mobilities and hole mobilities of Sc2CS and Y2CS are predicted and summarized in Table 4. The room-temperature electron mobilities of Sc2CS along the x and y directions were determined to be 232.59 and 628.22 cm2 V−1 s−1, respectively, compared with the room-temperature hole mobilities of only 88.32 and 61.75 cm2 V−1 s−1 along the x and y directions. That is, the electron mobilities along the x and y directions are about 2.63 and 10.17 times that of the holes, respectively, due to the large difference in effective mass, which exhibits pronounced anisotropy. Resembling the charge transport of Sc2CS, the electron mobilities of Y2CS along the x and y directions (818.51 and 552.55 cm2 V−1 s−1, respectively) are 498 and 30 times higher than the hole mobilities along the x and y directions. Therefore, it can be said that the charge transport of Sc2CS is predominated by electron, which is different from our recently reported Sc2CO2 with similar electron and hole mobilities.40 In addition, the predicted carrier mobility of non-stoichiometric Sc2CS is much higher than that of MoS2 monolayer,77 but much smaller than that of Sc2CO2 (ref. 40) due to defect scattering caused by sulfur vacancies. Therefore, the structurally stable Sc2CS and Y2CS with suitable carrier mobility and mechanical properties may serve as an excellent nanoelectronic material.
4. Conclusion
In this study, the structural stability, electronic structure, elastic and charge transport properties of M2CSx (M = Sc, Ti, Y, Zr, and Hf, x = 1, 2) were studied by using first-principles density functional theory. The studied M2CSx except Y2CS2 were determined to be thermodynamically, dynamically, thermally, and mechanically stable, suggesting that these S-functionalized MXenes can be successfully prepared. In contrast to all studied M2CO2 being indirect bandgap semiconductors, only four S-terminated MXenes (Sc2CS2, Y2CS2, Sc2CS, and Y2CS) were identified as indirect bandgap semiconductors. The p–d hybridization between the M-d state and the S/C-p state of M2CS is stronger than that of M2CS2, indicating that the bonding strength of the M–C/S bond in M2CS is stronger. However, due to the introduction of sulfur vacancies in the Y-free MXene, an antibonding state would occur near the Fermi level, thereby reducing the thermal stability of the material. On the contrary, the sulfur vacancies would significantly enhance the bonding state of Y–C and Y–S bonds and improve the stability of Y2CSx. These provides an explanation for the experimentally observed formation of non-stoichiometric M2CSx. And the room-temperature electron mobilities of Sc2CS (Y2CS) along the x and y directions were determined to be 232.59 (818.51) and 628.22 (552.55) cm2 V−1 s−1, compared with the room-temperature hole mobilities of only 88.32 (1.64) and 61.75 (17.80) cm2 V−1 s−1, which are much higher than that of MoS2 monolayer, but much smaller than that of Sc2CO2 due to defect scattering caused by sulfur vacancies. Therefore, Sc2CS and Y2CS with suitable carrier mobility and mechanical properties are promising as an excellent nanoelectronic materials. This work is expected to provide theoretical insights for the preparation and application of S-terminated MXenes and the discovery of novel MXene materials.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors acknowledge the support of the Key R & D Projects of Zhejiang Province (No. 2022C01236, 2019C01060), the National Natural Science Foundations of China (Grant No. 52250005, 21875271, U20B2021, 21707147, 51372046, 51479037, 91226202, and 91426304), the Entrepreneurship Program of Foshan National Hi-tech Industrial Development Zone, the Major Project of the Ministry of Science and Technology of China (Grant No. 2015ZX06004-001), Ningbo Natural Science Foundations (Grant No. 2014A610006, 2016A610273, and 2019A610106).
References
- D. Xiong, X. Li, Z. Bai and S. Lu, Recent Advances in Layered Ti3C2Tx MXene for Electrochemical Energy Storage, Small, 2018, 14(17), e1703419 CrossRef PubMed.
- X. Xu, Y. Zhang, H. Sun, J. Zhou, F. Yang, H. Li, H. Chen, Y. Chen, Z. Liu, Z. Qiu, D. Wang, L. Ma, J. Wang, Q. Zeng and Z. Peng, Progress and Perspective: MXene and MXene-Based Nanomaterials for High-Performance Energy Storage Devices, Adv. Electron. Mater., 2021, 7(7), 2000967 CrossRef CAS.
- H. Kim and H. N. Alshareef, MXetronics: MXene-Enabled Electronic and Photonic Devices, ACS Mater. Lett., 2019, 2(1), 55–70 CrossRef.
- S. Lee, E. H. Kim, S. Yu, H. Kim, C. Park, S. W. Lee, H. Han, W. Jin, K. Lee, C. E. Lee, J. Jang, C. M. Koo and C. Park, Polymer-Laminated Ti3C2Tx MXene Electrodes for Transparent and Flexible Field-Driven Electronics, ACS Nano, 2021, 15(5), 8940–8952 CrossRef CAS PubMed.
- E. Lee, A. VahidMohammadi, Y. S. Yoon, M. Beidaghi and D. J. Kim, Two-Dimensional Vanadium Carbide MXene for Gas Sensors with Ultrahigh Sensitivity Toward Nonpolar Gases, ACS Sens., 2019, 4, 1603–1611 CrossRef CAS PubMed.
- S. Mehdi Aghaei, A. Aasi and B. Panchapakesan, Experimental and Theoretical Advances in MXene-Based Gas Sensors, ACS Omega, 2021, 6(4), 2450–2461 CrossRef CAS PubMed.
- Y. Li, Y. Chen, Z. Guo, C. Tang, B. Sa, N. Miao, J. Zhou and Z. Sun, Breaking the linear scaling relations in MXene catalysts for efficient CO2 reduction, Chem. Eng. J., 2022, 429, 132171 CrossRef CAS.
- J. Yin, B. Ge, T. Jiao, Z. Qin, M. Yu, L. Zhang, Q. Zhang and Q. Peng, Self-Assembled Sandwich-like MXene-Derived Composites as Highly Efficient and Sustainable Catalysts for Wastewater Treatment, Langmuir, 2021, 37(3), 1267–1278 CrossRef CAS PubMed.
- X. Zhan, C. Si, J. Zhou and Z. Sun, MXene and MXene-based composites: synthesis, properties and environment-related applications, Nanoscale Horiz., 2020, 5(2), 235–258 RSC.
- S. Chertopalov and V. N. Mochalin, Environment-Sensitive Photoresponse of Spontaneously Partially Oxidized Ti(3)C(2) MXene Thin Films, ACS Nano, 2018, 12(6), 6109–6116 CrossRef CAS PubMed.
- K. Huang, Z. Li, J. Lin, G. Han and P. Huang, Two-dimensional transition metal carbides and nitrides (MXenes) for biomedical applications, Chem. Soc. Rev., 2018, 47(14), 5109–5124 RSC.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Two-dimensional nanocrystals produced by exfoliation of Ti3AlC2, Adv. Mater., 2011, 23(37), 4248–4253 CrossRef CAS.
- B. Xu and Y. Gogotsi, MXenes-The fastest growing materials family in the two-dimensional world, Chin. Chem. Lett., 2020, 31(04), 919–921 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, 2D metal carbides and nitrides (MXenes) for energy storage, Nat. Rev. Mater., 2017, 2(2), 16098 CrossRef CAS.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, The world of two-dimensional carbides and nitrides (MXenes), Science, 2021, 372(6547), 1165 CrossRef.
- V. Kamysbayev, A. S. Filatov, H. Hu, X. Rui, F. Lagunas, D. Wang, R. F. Klie and D. V. Talapin, Covalent surface modifications and superconductivity of two-dimensional metal carbide MXenes, Science, 2020, 369(6506), 979–983 CrossRef CAS PubMed.
- Y. Wei, P. Zhang, R. A. Soomro, Q. Zhu and B. Xu, Advances in the Synthesis of 2D MXenes, Adv. Mater., 2021, 33(39), e2103148 CrossRef PubMed.
- M. Li, J. Lu, K. Luo, Y. Li, K. Chang, K. Chen, J. Zhou, J. Rosen, L. Hultman, P. Eklund, P. O. A. Persson, S. Du, Z. Chai, Z. Huang and Q. Huang, Element Replacement Approach by Reaction with Lewis Acidic Molten Salts to Synthesize Nanolaminated MAX Phases and MXenes, J. Am. Chem. Soc., 2019, 141(11), 4730–4737 CrossRef CAS PubMed.
- Y. Li, H. Shao, Z. Lin, J. Lu, L. Liu, B. Duployer, P. O. A. Persson, P. Eklund, L. Hultman, M. Li, K. Chen, X. H. Zha, S. Du, P. Rozier, Z. Chai, E. Raymundo-Pinero, P. L. Taberna, P. Simon and Q. Huang, A general Lewis acidic etching route for preparing MXenes with enhanced electrochemical performance in non-aqueous electrolyte, Nat. Mater., 2020, 19(8), 894–899 CrossRef CAS PubMed.
- H. Ding, Y. Li, M. Li, K. Chen, K. Liang, G. Chen, J. Lu, J. Palisaitis, P. O. Persson, P. Eklund, L. Hultman, S. Du, Z. Chai, Y. Gogotsi and Q. Huang, Chemical-scissor-mediated structural editing of layered transition metal carbides, Science, 2023, 379(6637), 1130–1135 CrossRef CAS PubMed.
- T. Hu, Z. Li, M. Hu, J. Wang, Q. Hu, Q. Li and X. Wang, Chemical Origin of Termination-Functionalized MXenes: Ti3C2T2 as a Case Study, J. Phys. Chem. C, 2017, 121(35), 19254–19261 CrossRef CAS.
- X. Li, Z. Huang, C. E. Shuck, G. Liang, Y. Gogotsi and C. Zhi, MXene chemistry, electrochemistry and energy storage applications, Nat. Rev. Chem., 2022, 6(6), 389–404 CrossRef.
- J. L. Hart, K. Hantanasirisakul, A. C. Lang, B. Anasori, D. Pinto, Y. Pivak, J. T. van Omme, S. J. May, Y. Gogotsi and M. L. Taheri, Control of MXenes' electronic properties through termination and intercalation, Nat. Commun., 2019, 10(1), 522 CrossRef CAS PubMed.
- X. Li, M. Li, Z. Huang, G. Liang, Z. Chen, Q. Yang, Q. Huang and C. Zhi, Activating the I0/I+ redox couple in an aqueous I2–Zn battery to achieve a high voltage plateau, Energy Environ. Sci., 2021, 14(1), 407–413 RSC.
- X. Liang, A. Garsuch and L. F. Nazar, Sulfur cathodes based on conductive MXene nanosheets for high-performance lithium-sulfur batteries, Angew. Chem., Int. Ed., 2015, 54(13), 3907–3911 CrossRef CAS PubMed.
- C. Wei, T. Fang, X. Tang, P. Wang and X. Liu, Non-Negligible Role of Multifunctional MXene Hosts for Li–S Batteries: Anchoring and Electrocatalysis, J. Phys. Chem. C, 2022, 126(40), 17066–17075 CrossRef CAS.
- Y. Wang, J. Shen, L. C. Xu, Z. Yang, R. Li, R. Liu and X. Li, Sulfur-functionalized vanadium carbide MXene (V2CS2) as a promising anchoring material for lithium-sulfur batteries, Phys. Chem. Chem. Phys., 2019, 21(34), 18559–18568 RSC.
- J. Zhu, A. Chroneos, J. Eppinger and U. Schwingenschlögl, S-functionalized MXenes as electrode materials for Li-ion batteries, Appl. Mater. Today, 2016, 5, 19–24 CrossRef.
- Q. Meng, J. Ma, Y. Zhang, Z. Li, C. Zhi, A. Hu and J. Fan, The S-functionalized Ti3C2 MXene as a high capacity electrode material for Na-ion batteries: a DFT study, Nanoscale, 2018, 10(7), 3385–3392 RSC.
- V. Shukla, N. K. Jena, S. R. Naqvi, W. Luo and R. Ahuja, Modelling high-performing batteries with Mxenes: The case of S-functionalized two-dimensional nitride MXene electrode, Nano Energy, 2019, 58, 877–885 CrossRef CAS.
- L. Zhang, W. Zhang, X. Ma, X. Zhang and J. Wen, Computational screening of functionalized MXenes to catalyze the solid and non-solid conversion reactions in cathodes of lithium-sulfur batteries, Phys. Chem. Chem. Phys., 2022, 24(15), 8913–8922 RSC.
- Z. Li, Y. Wu and J. Hou, Exploring the anchoring effect of surface functionalized 2D electrodes Ca2N and Y2C in lithium sulfur battery: First principle study, Appl. Surf. Sci., 2022, 591, 153185 CrossRef CAS.
- S. Wang, Y. Wang, Q. Zhou, X. Li, Y. Li, Y. Liu, Y. Sun, T. Wang, L. C. Xu and Y. Wang, Modelling high performance potassium-ion battery anode materials with two-dimensional vanadium carbide MXene: the role of surface O- and S-terminations, Phys. Chem. Chem. Phys., 2021, 23(6), 3898–3904 RSC.
- K. A. Papadopoulou, A. Chroneos and S.-R. G. Christopoulos, Mg-ion diffusion on the surface of Ti3C2S2 MXene, J. Phys. Chem. Solids, 2022, 166, 110713 CrossRef CAS.
- S.-P. Huang, J.-F. Gu, Y.-R. Ren, K.-N. Ding, Y. Li, Y.-F. Zhang, S.-P. Huang, W. Lin and W.-K. Chen, Investigation of Ordered TiMC and TiMCT2 (M = Cr and Mo; T = O and S) MXenes as High-Performance Anode Materials for Lithium-Ion Batteries, J. Phys. Chem. C, 2022, 126(11), 5283–5291 CrossRef CAS.
- G. Chaney, D. Cakir, F. M. Peeters and C. Ataca, Stability of adsorption of Mg and Na on sulfur-functionalized MXenes, Phys. Chem. Chem. Phys., 2021, 23(44), 25424–25433 RSC.
- Z. Chen, S. Huang, X. Yuan, X. Gan and N. Zhou, A comparative study of M2CS2 and M2CO2 MXenes as anode materials for lithium ion batteries, Appl. Surf. Sci., 2021, 544, 148861 CrossRef CAS.
- H. Luo, X. Wang, C. Wan, L. Xie, M. Song and P. Qian, A Theoretical Study of Fe Adsorbed on Pure and Nonmetal (N, F, P, S, Cl)-Doped Ti3C2O2 for Electrocatalytic Nitrogen Reduction, Nanomaterials, 2022, 12(7), 1081 CrossRef CAS PubMed.
- S. Bae, W. Espinosa-García and Y. G. Kang, et al., MXene Phase with C3 Structure Unit: A Family of 2D Electrodes, Adv. Funct. Mater., 2021, 31(24), 2100009 CrossRef CAS.
- Z. Wang, N. Qiu, E. Wu, Q. Huang, P. An, H. He and S. Du, First-principles study of electronic and optical properties of NH3-adsorbed Sc2CO2 monolayer and its application in gas sensors, J. Mater. Res. Technol., 2023, 24, 173–184 CrossRef CAS.
- C. Hu, X. Yu, Y. Li, J. Cheng and B. Xiao, M2CS2 (M = Sc, Y) with brand-new MXene phase: The promising candidate as the N/O-containing gases sensor and/or capturer, Appl. Surf. Sci., 2023, 607, 155104 CrossRef CAS.
- L. Zhang, C. Tang, C. Zhang and A. Du, First-principles screening of novel ferroelectric MXene phases with a large piezoelectric response and unusual auxeticity, Nanoscale, 2020, 12(41), 21291–21298 RSC.
- E. M. D. Siriwardane, I. Demiroglu, C. Sevik, F. M. Peeters and D. Çakır, Assessment of Sulfur-Functionalized MXenes for Li-Ion Battery Applications, J. Phys. Chem. C, 2020, 124(39), 21293–21304 CrossRef CAS.
- Y. Jing, J. Liu, Z. Zhou, J. Zhang and Y. Li, Metallic Nb2S2C Monolayer: A Promising Two-Dimensional Anode Material for Metal-Ion Batteries, J. Phys. Chem. C, 2019, 123(44), 26803–26811 CrossRef CAS.
- X. Liu, X. Shao, F. Li and M. Zhao, Anchoring effects of S-terminated Ti2C MXene for lithium-sulfur batteries: A first-principles study, Appl. Surf. Sci., 2018, 455, 522–526 CrossRef CAS.
- V. Mehta, H. S. Saini, S. Srivastava, M. K. Kashyap and K. Tankeshwar, S-Functionalized Mo2C Monolayer as a Novel Electrode Material in Li-Ion Batteries, J. Phys. Chem. C, 2019, 123(41), 25052–25060 CrossRef CAS.
- Y. Li, L. Bai, N. Ma and L. Niu, O- or/and S-functionalized Cr2C as electrode material for metal-ion (Li, Na, K, and Mg) batteries: A first principles study, Comput. Theor. Chem., 2022, 1217, 113892 CrossRef CAS.
- J. Yang, A. Wang, S. Zhang, H. Wu and L. Chen, Stability and electronic properties of sulfur terminated two-dimensional early transition metal carbides and nitrides (MXene), Comput. Mater. Sci., 2018, 153, 303–308 CrossRef CAS.
- S. Lee, S. C. Jung and Y. K. Han, Fe2CS2 MXene: a promising electrode for Al-ion batteries, Nanoscale, 2020, 12(9), 5324–5331 RSC.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- E. Caldeweyher, C. Bannwarth and S. Grimme, Extension of the D3 dispersion coefficient model, J. Chem. Phys., 2017, 147(3), 034112 CrossRef PubMed.
- A. Togo and I. Tanaka, First principles phonon calculations in materials science, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- X. Gonze and C. Lee, Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55(16), 10355–10368 CrossRef CAS.
- J. P. Perdew and M. Levy, Physical Content of the Exact Kohn-Sham Orbital Energies: Band Gaps and Derivative Discontinuities, Phys. Rev. Lett., 1983, 51(20), 1884–1887 CrossRef CAS.
- J. Liu, X.-B. Li, D. Wang, H. Liu, P. Peng and L.-M. Liu, Single-layer Group-IVB nitride halides as promising photocatalysts, J. Mater. Chem. A, 2014, 2(19), 6755 RSC.
- M. C. Toroker, D. K. Kanan, N. Alidoust, L. Y. Isseroff, P. Liao and E. A. Carter, First principles scheme to evaluate band edge positions in potential transition metal oxide photocatalysts and photoelectrodes, Phys. Chem. Chem. Phys., 2011, 13(37), 16644–16654 RSC.
- A. K. Singh, K. Mathew, H. L. Zhuang and R. G. Hennig, Computational Screening of 2D Materials for Photocatalysis, J. Phys. Chem. Lett., 2015, 6(6), 1087–1098 CrossRef CAS PubMed.
- J. Dai and X. C. Zeng, Titanium trisulfide monolayer: theoretical prediction of a new direct-gap semiconductor with high and anisotropic carrier mobility, Angew. Chem., Int. Ed. Engl., 2015, 54(26), 7572–7576 CrossRef CAS PubMed.
- J. Bardeen and W. Shockley, Deformation Potentials and Mobilities in Non-Polar Crystals, Phys. Rev., 1950, 80(1), 72–80 CrossRef CAS.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44(6), 1272–1276 CrossRef CAS.
- B. Xiao, Y.-c. Li, X.-f. Yu and J.-b. Cheng, MXenes: Reusable materials for NH3 sensor or capturer by controlling the charge injection, Sens. Actuators, B, 2016, 235, 103–109 CrossRef CAS.
- F. Zhang, H. Zhang, S. Krylyuk, C. A. Milligan, Y. Zhu, D. Y. Zemlyanov, L. A. Bendersky, B. P. Burton, A. V. Davydov and J. Appenzeller, Electric-field induced structural transition in vertical MoTe2- and Mo1−xWxTe2-based resistive memories, Nat. Mater., 2019, 18(1), 55–61 CrossRef CAS PubMed.
- J.-C. Lian, H.-Y. Wu, W.-Q. Huang, W. Hu and G.-F. Huang, Algorithm for generating irreducible site-occupancy configurations, Phys. Rev. B: Condens. Matter Mater. Phys., 2020, 102, 134209 CrossRef CAS.
- K. Xiong, P. Wang, G. Yang, Z. Liu, H. Zhang, S. Jin and X. Xu, Functional Group Effects on the Photoelectronic Properties of MXene (Sc2CT2, T = O, F, OH) and Their Possible Photocatalytic Activities, Sci. Rep., 2017, 7(1), 15095 CrossRef.
- Y. Jiang, Y. Zhang, Q. Huang, L. Hao and S. Du, The compositional dependence of structural stability and resulting properties for Mn+1CnT2 (M = Sc, Ti, V; T = O, OH, F, Cl, Br and I; n = 1, 2): first-principle investigations, J. Mater. Res. Technol., 2020, 9(6), 14979–14989 CrossRef CAS.
- S. Ma, D. Yuan, Z. Jiao, T. Wang and X. Dai, Monolayer Sc2CO2: A Promising Candidate as a SO2 Gas Sensor or Capturer, J. Phys. Chem. C, 2017, 121(43), 24077–24084 CrossRef CAS.
- M. Li, O. Omisakin and J. Young, Effect of chemical substitution and external strain on phase stability and ferroelectricity in two dimensional M2CT2 MXenes, Nanoscale, 2022, 14(18), 6970–6980 RSC.
- J. Young and T. L. Reinecke, Controlling the H to T' structural phase transition via chalcogen substitution in MoTe(2) monolayers, Phys. Chem. Chem. Phys., 2017, 19(47), 31874–31882 RSC.
- V. Wang, G. Tang, Y. C. Liu, R. T. Wang, H. Mizuseki, Y. Kawazoe, J. Nara and W. T. Geng, High-Throughput Computational Screening of Two-Dimensional Semiconductors, J. Phys. Chem. Lett., 2022, 13(50), 11581–11594 CrossRef CAS.
- S. Chen, Z. Fu, H. Zhang, D. Legut, T. C. Germann, Q. Zhang, S. Du, J. S. Francisco and R. Zhang, Surface Electrochemical Stability and Strain-Tunable Lithium Storage of Highly Flexible 2D Transition Metal Carbides, Adv. Funct. Mater., 2018, 28(44), 1804867 CrossRef.
- M. Marsman, J. Paier, A. Stroppa and G. Kresse, Hybrid functionals applied to extended systems, J. Phys.: Condens. Matter, 2008, 20(6), 064201 CrossRef CAS.
- V. L. Deringer, A. L. Tchougreeff and R. Dronskowski, Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets, J. Phys. Chem. A, 2011, 115(21), 5461–5466 CrossRef CAS PubMed.
- Z. Fu, D. Legut, T. C. Germann, C. Si, S. Du, J. S. Francisco and R. Zhang, Phonon-mediated stabilization and softening of 2D transition metal carbides: case studies of Ti2CO2 and Mo2CO2, Phys. Chem. Chem. Phys., 2018, 20(21), 14608–14618 RSC.
- F. Mouhat and F.-X. Coudert, Necessary and sufficient elastic stability conditions in various crystal systems, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
- Z.-Y. Wang, Y.-L. Zhou, X.-Q. Wang, F. Wang, Q. Sun, Z.-X. Guo and Y. Jia, Effects of in-plane stiffness and charge transfer on thermal expansion of monolayer transition metal dichalcogenide, Chin. Phys. B, 2015, 24(2), 026501 CrossRef.
- Y. Cai, G. Zhang and Y.-W. Zhang, Polarity-Reversed Robust Carrier Mobility in Monolayer MoS2 Nanoribbons, J. Am. Chem. Soc., 2014, 136(17), 6269–6275 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Nianxiang Qiu*b,
Gang Fangb and
Shiyu Du
ab,
Nianxiang Qiu*b,
Gang Fangb and
Shiyu Du




 states of the M-d orbital. Noteworthily, the S-p state of M2CSx overlaps with the M-d state in the energy range from −4.5 eV to 0 eV, while the O-p state of M2CO2 hybridizes with the M-d state around −6 to −2 eV, which is an implication of the stronger M–O bond than M–S bond. Resembling the corresponding M2CO2, due to the splitting of the 5-fold degenerate M-d orbitals of M2CS2 in the octahedral crystal field of MX6, 5 hybridization peaks of the S-p state are observed, which are more conspicuous in Y2CS2 and Sc2CS2, but this phenomenon disappears in M2CS due to the presence of S vacancies. In particular, the p–d hybridization peaks between S and Y of Y2CS2 are very localized, reflecting the weak nature of the Y–S bond, which are significantly improved in Y2CS. More importantly, the hybridization peaks between the M-d orbital and the C-p or S-p orbital of M2CS are remarkably higher than those of the corresponding M2CS2, which can be regarded as evidence that the M–C and M–S bonds in M2CS are stronger than those in M2CS2.
states of the M-d orbital. Noteworthily, the S-p state of M2CSx overlaps with the M-d state in the energy range from −4.5 eV to 0 eV, while the O-p state of M2CO2 hybridizes with the M-d state around −6 to −2 eV, which is an implication of the stronger M–O bond than M–S bond. Resembling the corresponding M2CO2, due to the splitting of the 5-fold degenerate M-d orbitals of M2CS2 in the octahedral crystal field of MX6, 5 hybridization peaks of the S-p state are observed, which are more conspicuous in Y2CS2 and Sc2CS2, but this phenomenon disappears in M2CS due to the presence of S vacancies. In particular, the p–d hybridization peaks between S and Y of Y2CS2 are very localized, reflecting the weak nature of the Y–S bond, which are significantly improved in Y2CS. More importantly, the hybridization peaks between the M-d orbital and the C-p or S-p orbital of M2CS are remarkably higher than those of the corresponding M2CS2, which can be regarded as evidence that the M–C and M–S bonds in M2CS are stronger than those in M2CS2.