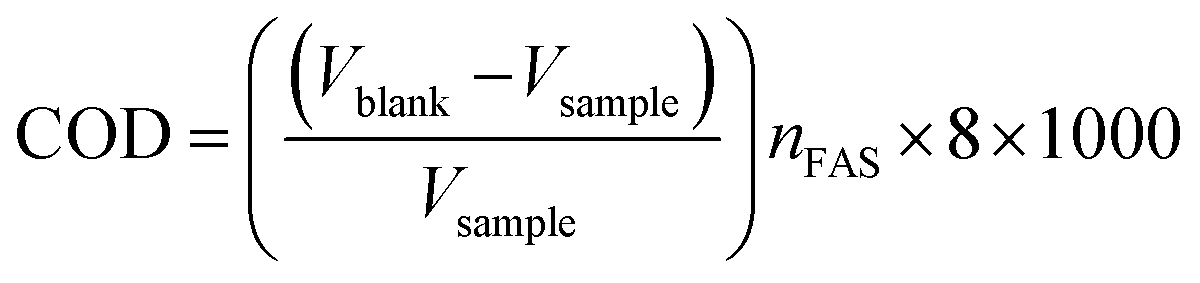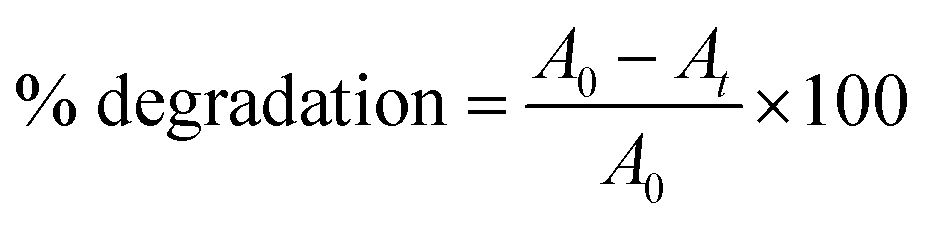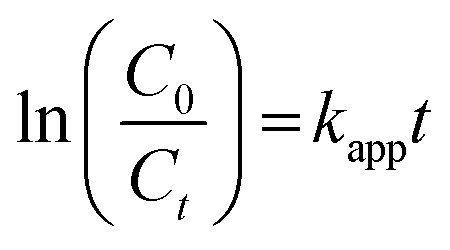DOI:
10.1039/D3RA03315E
(Paper)
RSC Adv., 2023,
13, 23181-23196
Structural and optical properties of silver supported α-Fe2O3 nanocomposite fabricated by Saraca asoca leaf extract for the effective photo-degradation of cationic dye Azure B
Received
19th May 2023
, Accepted 13th July 2023
First published on 1st August 2023
Abstract
In recent decades, several nanocomposites developed by chemical synthetic routes, have been demonstrated as efficient photocatalysts for the photodegradation of hazardous organic dyes. The present investigation reports the sonochemical-assisted fabrication of silver-supported α-Fe2O3 nanocomposites (SA@Ag@IONCs) using the Saraca asoca leaf extract. The magnetic nanocomposites can be easily removed from the reaction mixture. The morphology of these materials was characterized by field emission scanning electron microscopy (FESEM), high-resolution transmission electron microscopy (HRTEM), XPS, BET surface area analyzer, UV-visible spectroscopy, photoluminescence, X-ray diffraction (XRD), and VSM techniques. The XRD and electron microscopy analyses revealed the small size and well-crystalline SA@Ag@IONC particles with spherical and buckyball structures. The large surface area of SA@Ag@IONCs was confirmed by BET analysis. The absorption edge in UV-visible spectra appeared to migrate towards high wavelengths for the SA@Ag@IONC composite, causing a change in the bandgap energy. In the case of the sonication assisted composite, the bandgap energy was 2.1 eV, making it easier for the electron to transfer from the valence band to conduction band. The decoration of ultrasmall silver onto the surfaces of the α-Fe2O3 nanocomposite, which considerably increases the capacity to absorb sunlight, enhances the efficiency of charge carrier separation, and inhibits the electron–hole recombination rate as confirmed by the reduced PL intensity, is responsible for the excellent photocatalytic degradation performance. Outcomes shown SA@Ag@IONCs have a high photodegradation rate as well as high-rate constant value at an optimized condition that is at pH 9 and 0.5 g L−1 dose of nanocomposite, photodegradation rate of Azure B is ∼94%. Trap experiment results indicated that O2˙− and h+ are the active species responsible for the photodegradation of AzB.
1. Introduction
The increasing global significance of semiconducting nanomaterials worldwide has drawn great interest due to their wide applicability in multiple fields of research and the developing new technologies. The numerous potential applications of novel semiconductor nanocatalysts used in a variety of fields such as environment purification, solar energy conversion, biomedical, sensors, and water splitting have made them an important area of research.1–4 Due to their capacity to decontaminate environmental pollutants and split water using visible light, semiconducting heterogeneous nanomaterials have gained much attention as a result of rising environmental pollution and global energy shortage.3,5–7
Wastewater treatment via photocatalysis is a low-temperature, inexpensive, and environmentally friendly technique that uses a variety of wide-bandgap semiconductor materials. In this procedure, nano photocatalysts are exposed to different sources of radiation, such as visible light, ultraviolet radiation as well as solar energy comparable to the band gap energy of semiconductor materials, to generate electron–hole (e−–h+) pairs.3,8–10 They are transferred to the material surface, where they produce radicals such as hydroxyl (OH˙−), oxygen atoms, and superoxide anion radicals (O2˙−). These radicals attack the dye compound that is adsorbed onto the surface and decomposes into different anions, cations, and gases.11,12 Therefore, a significant factor in determining the photocatalytic activity of semiconductors is the lifetime of photogenerated e–h pairs. The average size of nanomaterials, dopants, and noble metal co-catalyst surfaces are some of the variables shown in previous studies, which play a significant role in the lifetime of photogenerated electron–hole pairs. It has been proved that for these systems, the photoluminescence (PL) analysis methods to study the change in photo induced electron hole pairs are associated with the photocatalytic activity of semiconductor materials, as summarized in brief by other researchers.13–15
Several methods, such as replacing the bulk material with nanosized semiconducting materials and doping metal ions into metal oxide photocatalysis materials, have been used to enhance the photodegradation process.14,16 These techniques can significantly boost the detachable rate of photogenerated electron–hole charge species in semiconductor compounds, which will raise photocatalytic efficiency. To study the structural characteristics of the active sites present on the surface of metal oxide nanomaterials, the photoluminescence approach has been employed significantly.17,18 This is due to its extremely sensitive and non-destructive properties. The PL techniques have also helped to understand the surface process of the semiconductor in photocatalysis reactions. The photoluminescence (PL) spectra are a useful tool to examine optical properties, electronic composition, and photochemical features of semiconducting substances. They can be used to understand the substrate oxygen vacancies and irregularities along with the effectiveness of charge capturing, immigration, as well as transportation.14,15,19
Heterojunction photocatalysts have a wide range of applications, including photocatalytic water splitting, environmental purification, pharmaceutical wastes, and photodegradation of hazardous dyes.16,20–22 The drawback of the photocatalytic procedure is the complete separation of nanocatalyst particles from the treated water, which causes secondary contamination and loss of reusability of the catalyst. To overcome this above problem, a catalyst is used that is easily removed from treated water and can be used for further processes. Metal nanocomposites, which possess magnetic properties, have received attraction due to their excellent performance as well as easy removal from the reaction mixture.
Among all the other semiconductor materials, iron oxide (Fe2O3) nanomaterial is considered one of the efficient photocatalysts because of their affordable price, excellent catalytic property, high photosensitivity, good stability, nontoxicity, biocompatibility, unique, electrical, optical, and magnetic properties.23,24 Iron oxide has low band gap energy and it is a semiconducting material. Iron oxide NPs being magnetically separable gives an additional advantage over other supports. It avoids the classical filtration method of separation. When α-Fe2O3 nanoparticle is encored with other metal nanoparticles, the overall magnetic and recyclability properties are enhanced in photocatalytic applications.25,26 When iron oxide nanoparticle is exposed to sunlight, their fast recombination rates of electron–hole pairs lower the photocatalytic activity. To decrease the e–h pair recombination rates, the band gap energy is lowered and extends the optical absorption band without affecting the efficiency of photocatalytic activity, and the nanocatalyst must be modified. Several effective approaches were investigated for this purpose.27–30 The technique used to prevent the short lifetime of photogenerated (e−–h+) pairs and decrease its band gap energy is to incorporate various types of metal into the surface of iron oxide nanoparticles. These techniques can significantly boost the separation rate of photogenerated charge carriers in semiconductor materials, which will raise the efficiency of the photocatalytic process.10,19
Noble metals have excellent photocatalytic efficiency, wide biomedical applications as well as antimicrobial and antibacterial properties because they display enhanced absorption in the visible range as a result of their surface plasmon resonance and serve as a superficial charge trapper to activate the surface sites in the crystallite lattice, favoring to increase the electron–holes pair separation and decreasing the recombination rate.31 Various noble metals (Pt, Rh, Pd, and Au) are too expensive to be employed on a large scale. As a result, Ag supported iron oxide nanoparticles have generated interest for the photocatalytic application, particularly Ag-containing NCs are considered to have enormous photocatalytic applications due to distinctive physicochemical properties as compared to other noble metals.28,32,33
The green chemistry approach is a proven path for the synthesis of nanocomposite materials and it is explored as one of the most innovative processes of synthesis of the nanomaterials. For example, the different parts of plant extract comprising amino acids, alkaloids, polyphenols, flavonoids, and many more bioactive components are accountable for reducing the metal ions and also stabilize the nanomaterials in the form of capping agents in the aqueous medium.34–36 Bio-conjugated metal NPs have shown excellent antimicrobial, anticancer, drug delivery, and catalytic activity.37–39 Therefore, the conjugation of biologically active phytochemicals of medicinal plants with metal nanocomposites is supposed to enhance the photocatalytic properties of metal/metal oxide nanocomposites. Sonochemical treatment has gained popularity recently as a technique to engineer environment-friendly materials. A number of metallic nanoparticles (NPs) have been created using ultrasound as part of research into the synthesis of photocatalytic materials.28,40,41 Sonochemical surface modification of nanocomposites improves the physico-chemical parameters, such as particle size and specific surface area.42
Inspiring from a previous literature review, in the present work, sonication assisted silver supported α-Fe2O3 binary nanocomposite has been prepared via the green synthetic approach using an aqueous extract of leaves of the plant Saraca asoca. The as-formed sonication-assisted SA@Ag@IONCs and without sonication-assisted Ag@Fe2O3 nanocomposite have been used for the photocatalytic degradation of dye Azure B in aqueous media under solar light irradiation. SA@Ag@IONC nanocomposites were characterized by XRD, SEM, HRTEM, PL, and UV-visible spectrophotometer to study the impact of optical property, photoluminescence PL on photodegradation of dye molecules.
2. Experimental detail
2.1. Material and synthesis method
All the chemicals used in this work, like ferrous sulfate heptahydrate (FeSO4·7H2O), silver nitrate (AgNO3), potassium dichromate (K2Cr2O7), mercuric sulphate (HgSO4), ferrous ammonium sulphate, silver sulphate (Ag2SO4), ferroin indicator, and Azure B were analytic grade and used without purification. Ethanol, acetone, and double distilled water were used as solvents. Fresh leaves of Saraca asoca collected from campus were washed two–three times to remove dust, sun-dried, and crushed to make a fine powder.
2.2. Preparation of leaf extract
Powdered leaves of Saraca asoca (15 g) were transferred into 250 mL of double distilled water in a 500 mL round bottom flask and refluxed at 80 °C for 1.5 h. The extract was allowed to cool at 27 °C, and Whatman No. 1 filter paper was used to filter the extract. Filtered leaf extracts were stored at a temperature of 4 °C for further usage.
2.3. Sonochemical fabrication of SA@Ag@IONCs
Silver-supported iron oxide binary nanocomposite has been synthesized by a facile one-pot green synthesis method. 0.2 M FeSO4·7H2O was prepared in 100 mL deionized water and 25 mL plant extract was added. The solution mixture was stirred with a magnetic stirrer for 6 h at room temperature. The greenish color of the solution mixture turned to brownish-green color, indicating the formation of iron oxide. The produced solution was ultrasonicated for 60 min, centrifuged at 5000 rpm, and washed 2–3 times with double distilled water, followed by ethanol to remove impurities. In the second step, 100 mL AgNO3 (0.5 mM) in deionized water was mixed with 100 mL FeSO4·7H2O (0.2 M) solution in deionized water. 25 mL plant extract was transferred to the mixture, and the solution was stirred for six h at room temperature. The dark brownish-green color thus formed was ultrasonicated for 60 min. The as-synthesized SA@Ag@IONC nanocomposite and with the same method without sonication Ag@Fe2O3 nanocomposite was centrifuged at 5000 rpm and washed several times with deionized water and ethanol for the removal of impurities. Samples were dried at 80 °C and stored in a vial for characterization and photocatalytic activity.
2.4. Photo-driven activity of SA@Ag@IONCs
The photo-driven activity of synthesized SA@Ag@IONC nanocomposite was investigated for degradation of Azure B (AzB) under direct sunlight irradiation. In this experiment, a freshly prepared 50 mL aqueous Azure B (AzB) solution having 20 mg L−1 concentration was taken in five separate tubes, and 20, 30, 40, 50, and 60 mg of SA@Ag@IONCs catalyst was added to each tube. In a separate tube, the same volume of AzB solution without catalyst was taken, which is treated as blank or control. All the reaction mixtures were thoroughly mixed at 30 °C and sonicated. Each tube containing the reaction mixture was placed for 30 min in the dark to complete the adsorption–desorption equilibrium process. Then, under the influence of solar light, a photocatalytic reaction was carried out. At various time intervals, the reaction was monitored spectrophotometrically. The effect of pH (3–11) on AzB photodegradation was also observed. The kinetic study of the photodegradation reaction was carried out to determine the rate constant on different parameters, such as catalyst dose and the effect of pH from the graph between ln (C0/Ct) versus time.
2.5. Photocurrent measurements
In this experiment, firstly SA@Ag@IONC nanocomposite was dispersed in ethanol and ultrasonicated. While doing this, a glass substrate was properly cleaned with acetone to eliminate grease. The cleaned glass film was heated over a hot plate for 1 h at 50 °C. With a dropper, dispersed materials were simultaneously deposited onto glass film. The device was coated with silver electrodes at a 2 mm distance and once again left to be dry over the hot plate at 50 °C for 40 min. The photocurrent response measurement was done using these constructed devices.
2.6. COD analysis
The dichromate reflux technique was applied to evaluate the COD of samples. A specific amount of oxidant was injected into the sample solution and the solution was boiled for 2 h until the sample was properly digested. In this dichromate reflux process, the Azure B solutions were digested for 2 h with HgSO4, a specific amount of standard K2Cr2O7, and Ag2SO4 in the presence of concentrated H2SO4 solution and then titrated against a standard solution of ferrous ammonium sulphate (FAS) using ferroin as an indicator. Double distilled water was utilized instead of the analytes in blank condition. The COD of samples was calculated using the formula:
3. Characterization techniques
XRD analysis system (PANalytical, Netherlands). The FE-SEM pictures of SA@Ag@IONCs have been recorded on Zeiss (Merlin Compact equipment). Scanning electron microscope (SEM) graphs and energy dispersive X-ray analysis (EDAX) of the SA@Ag@IONCs were observed on JEOL equipment (JSM 6490 LB). UV-vis absorption spectra of samples were recorded on a dual-beam spectrophotometer (Carry 100) operated between 200-800 nm. PL spectra were recorded on (FLS980 D2D2, Edinburg) instrument, a double monochromatic light using 450 W xenon lamp source, and 360 nm excitation wavelength at ambient temperature. FTIR spectra were recorded by PerkinElmer Spectrum II and Thermo-Scientific (Nicole 6700) palletization machine was used to make potassium bromide pellets. The morphology and size of hematite and SA@Ag@IONC nanocomposite were investigated via HRTEM using JEOL-2100 equipment. The zeta potential of SA@Ag@IONCs has been evaluated via Zetasizer ZS90 (Nano series Malvern Instrument) at absolute room temperature. XPS measurement was performed on Prob II, FEI Inc. (PHI 5000 Versa). BET (Brunauer Emmett Teller), nitrogen adsorption–desorption, and pore size have been determined by Belsorp mini-II instrument. Room temperature magnetic property measurement was acquired by using MPMS-03 SQUID (Quantum Design) magnetometer. The degradation products were characterized using Agilent LC/Q-Tof 1260 at 30 °C using methanol/0.1 N formic acid (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) with a flow rate of 0.2 mL min−1 in the C-18 column.
:10) with a flow rate of 0.2 mL min−1 in the C-18 column.
4. Results and discussion
4.1. XRD study
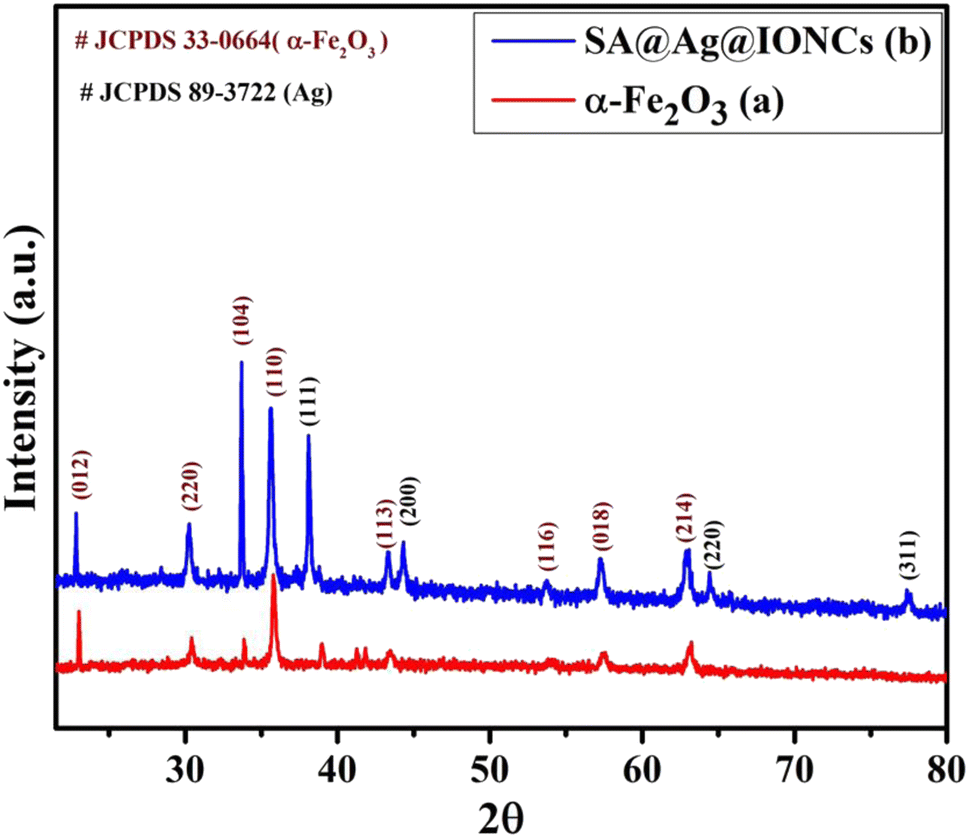
To examine the crystallinity of biogenic synthesized materials, XRD analysis was carried out in the 2θ range of 20–90°. The prominent characteristic peaks in the XRD pattern of the iron oxide sample (Fig. 1a) at different 2θ values indicate the formation of α-Fe2O3. The strength of diffraction spikes with (hkl) values (012), (220), (104), (110), (113), (116), (018), and (214) were examined with the acceptable value (JCPDS card no.: 33-0664) and the structure was determined to be rhombohedral α-Fe2O3.43 Fig. 1b shows the XRD spectrum of the above synthesized sonochemically loaded silver onto the α-Fe2O3 surface via Saraca asoca leaf SA@Ag@IONC nanocomposite and Saraca asoca leaf mediated Ag@Fe2O3. In the XRD pattern apart from diffraction peaks of α-Fe2O3, the additional peaks corresponding to Ag crystals with (hkl) values (111), (311), (220), and (200) were observed, which is attributed due to the Ag phase (JCPDS 89-3722). Sonication-assisted SA@Ag@IONCs have a sharp crystalline peak with a small particle size. Similar results have been reported in previous reports.32,44,45 The average crystallites size (nm) was evaluated using the Debye Scherrer formula,
| D = 0.9 λ/βcosθ |
Herein, D represents the mean crystalline size, λ is the X-ray wavelength (0.1546 nm), β corresponds to the FWHM (full-width half maximum value) in radian, and θ denotes the Bragg's angle. The mean crystallite size of Ag@α-Fe2O3 and SA@Ag@IONCs nanocomposite were found to be 81 and 25.9 nm. This result suggests that the decoration of Ag nanoparticles increased the uniformity and decreased the crystallite size by preventing the agglomeration caused by the phytochemicals present in Saraca asoca extract. The synthesis of highly dispersed Ag/α-Fe2O3 nanocomposite was also confirmed from the BET surface area analysis. The improved surface area as well as the porosity of the as-fabricated SA@Ag@IONCs have caused enhancement in the photocatalytic performance of SA@Ag@IONC nanocomposite.
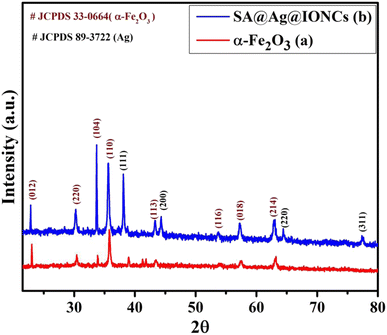 |
| | Fig. 1 XRD pattern of α-Fe2O3 nanoparticles (a) and SA@Ag@IONCs (b). | |
4.2. Morphological analysis
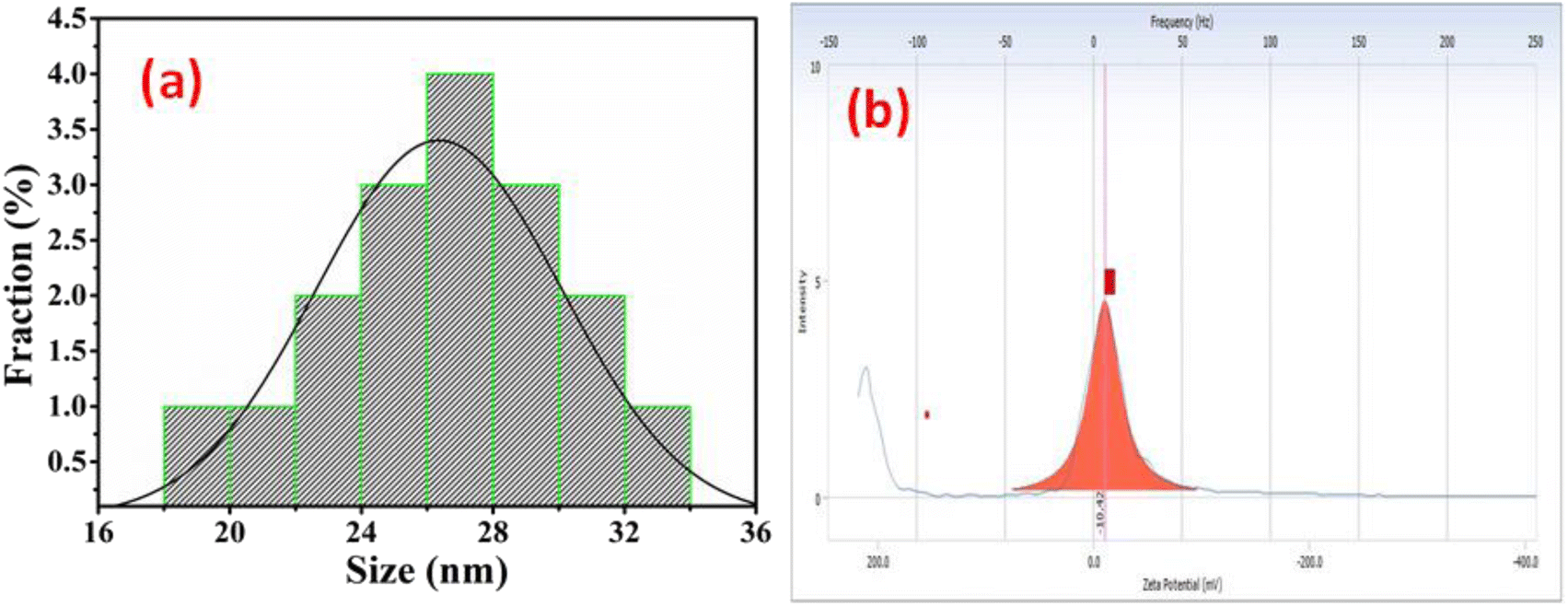
The morphology of Ag@Fe2O3 and SA@Ag@IONCs has been analyzed by SEM and FESEM images. In the SEM images of Ag@Fe2O3, more agglomeration was observed as compared to sonochemically assisted nanocomposite (Fig. 2a and b). We observed some spherical shape particles in SA@Ag@IONCs but due to aggregation, particle shape was not clear in Ag@Fe2O3. In the FESEM images (Fig. 3a–e) more clear view of Ag@Fe2O3 and SA@Ag@IONCs is observed. These images indicate that when silver is loaded sonochemically on the surface of Fe2O3, the particles are evenly distributed as nano-sized spherical shape as compared to Ag@Fe2O3 nanocomposite, biomolecules included in the SA extract wrap these nanoscales, maintaining them apart from one another and preventing clumping. Because of the delayed nucleation development and subsequent creation of nanoparticles with distinct nanoscale, low temperature, and ultrasonication process also helps to generate a non-agglomerative form of SA@Ag@IONCs. But in the case of Ag@Fe2O3 composite, silver ion is not uniformly distributed on the surface of Fe2O3, the agglomerated microparticles form a bunch's structure, and as a result of this, nanocomposite has large particle size and several irregular shapes embedded on the surface of iron oxide nanoparticles. The EDS analysis of SA@Ag@IONCs revealed the presence of silver, iron, oxygen, and traces of carbon in their atomic weight percent (Fig. 2c). Morphology, size, and structure of SA@Ag@IONCs have been further analyzed by TEM and HRTEM studies. In the bright field/dark field TEM images (Fig. 4a–c), a beautiful topology of spherical particles is observed; however, in some images (Fig. 4d and e), variable structures like spherical and buckyball structures are observed. The slight appearance of layering (Fig. 4g and h) is probably due to the adsorbed phytochemicals present in the Saraca asoca plant extract. In the SAED pattern (Fig. 4j), all the observed crystal planes of SA@Ag@IONCs are in good agreement with diffraction planes (113) and (214) for Fe2O3 as well as (111) and (311) for Ag matched with the XRD pattern, which further supports the formation of well crystalline NPs. In the HRTEM image (Fig. 4i) well-resolved lattice fringes with 0.231 and 0.268 nm spacing are observed, which correspond to the (111) plane of Ag NPs and (104) plane of α-Fe2O3.46,47 This is in good agreement with XRD results. The result indicates that Ag particles are nucleated over α-Fe2O3. The diameter/dimension of SA@Ag@IONCs has been deduced to be 15–27 nm. The particle size distribution curve (Fig. 5a) revealed that the particle size distributes between 18 to 34 nm while the highest population appears at 26 nm. This lies on the higher side than those of XRD/TEM results because of the experimental process of particle size distribution. In the particle size distribution experiment, the adsorbed molecules of the media solution make a boundary at the solid-solution interface; thus observed size always lies on the higher side. Morphological assessment of SA@Ag@IONCs revealed that the synthesized particles have spherical or buckyball structures, possessing a porous coating of phytochemical over the exterior surface of SA@Ag@IONCs nanostructures. The highly porous surface of SA@Ag@IONCs nanostructures is supposed to have high adsorption capability, thereby improving photocatalytic performance.
 |
| | Fig. 2 SEM image of Saraca acosa mediated Ag@Fe2O3 at different scales (a) SA@Ag@IONCs, (b) and EDS (c). | |
 |
| | Fig. 3 FESEM image of Saraca acosa mediated Ag@Fe2O3 (a–c) and SA@Ag@IONCs (d–f). | |
 |
| | Fig. 4 TEM and HR-TEM images of SA@Ag@FNCs at different scales (a–i), selected area electron diffraction pattern (j). | |
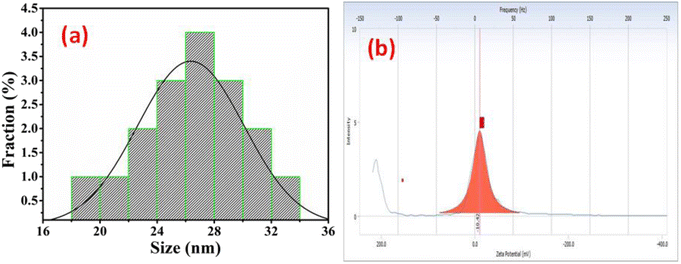 |
| | Fig. 5 Particle size distribution curve of SA@Ag@IONCs (a) zeta potential of SA@Ag@IONCs (b). | |
4.3. Zeta potential
The surface charge of SA@Ag@IONCs was also examined by the zeta potential@DLS dynamic light scattering technique based on Rayleigh scattering from the suspended nanostructures that undergo Brownian motion. The zeta potential of the nanocomposite was found at −10.42 mV, as shown in Fig. 5b. The negative charge of the zeta potential value of nanocomposite reveals that the repulsion among the particle makes the nanocomposite stable. The zeta potential value of SA@Ag@IONCs represents the negative charge around the nanostructure surface, which shows a strong affinity towards cationic dye molecules. Therefore, electrostatic interaction among nano photocatalyst and AzB molecules attract them closer, and therefore the movement of charge carriers takes place, resulting in the degradation of AzB molecules.
4.4. XPS analysis
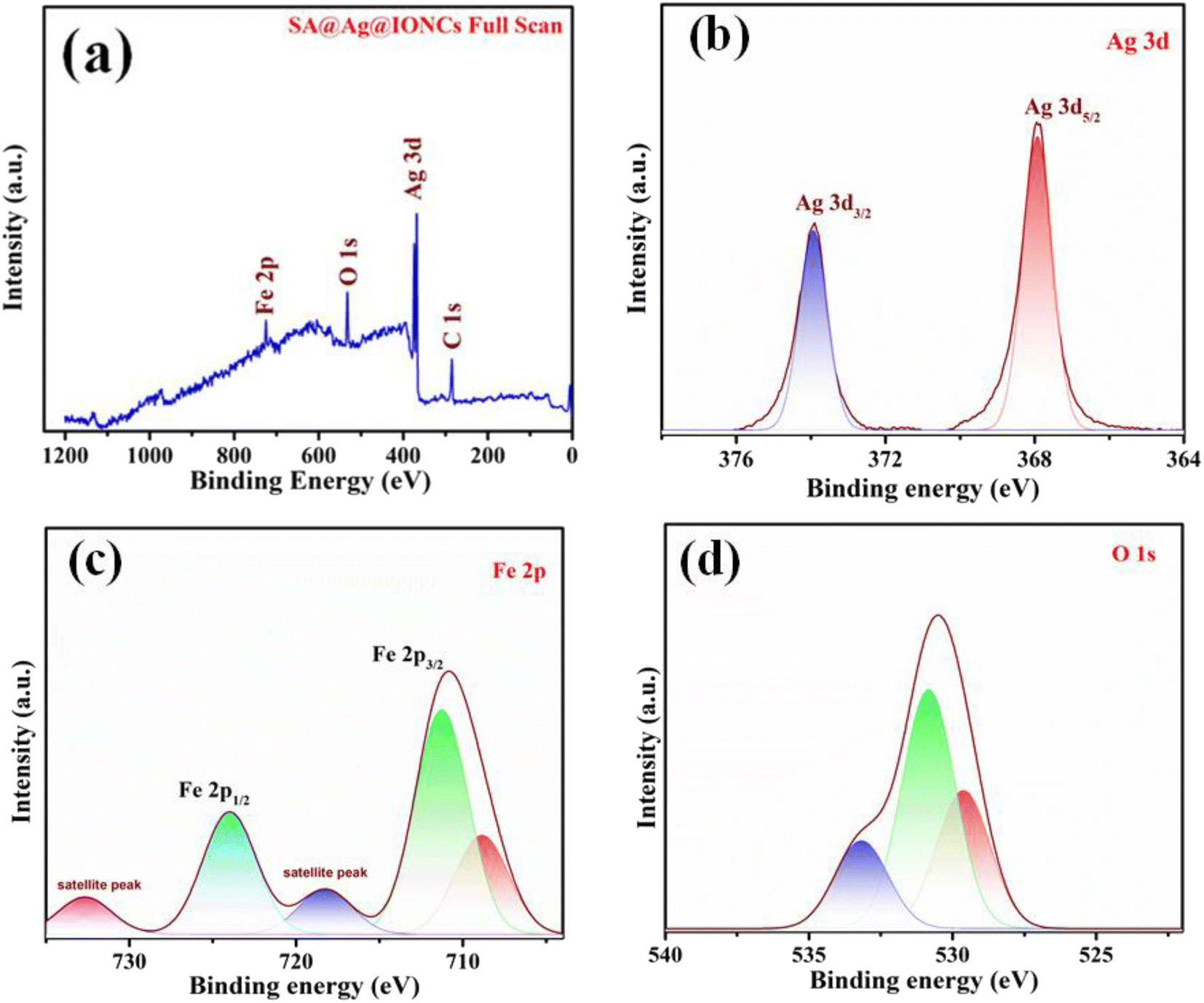
XPS spectral analysis has been done to confirm the chemical composition and oxidation states of the SA@Ag@IONCs composite. The characteristic peaks Ag 3d5/2 and Ag 3d3/2 of Ag in SA@Ag@IONCs are found at 367.9 and 373.8 eV, respectively, as shown in Fig. 6b. The energy difference observed between Ag 3d5/2 and Ag 3d3/2 is 5.9 eV. Thus, the binding energy of Ag 3d5/2 shifts to a low value in comparison to bulk Ag0. This is due to the reduced electron density value of silver, and this reduction in the electron density of silver is observed because of the e− transfer from silver to α-Fe2O3 or due to the formation of few silver oxides,29 Fe 2p spectrum of SA@Ag@IONCs contains three peaks at 709.3, 711.3, and 724.4 eV, which attributes to the peak of iron Fe 2p3/2 and Fe 2p1/2, respectively (Fig. 6c). The predetermined value of energy difference between these two bands is about 13.1 eV, which is characteristics of the Fe3+ state, which indicates the generation of α-Fe2O3. An observable peak in the spectra of Fe at 718.3 eV and 732.4 are the two Fe3+ shake-up satellite peaks in α-Fe2O3.23,48 The spectrum of O 1s deconvolution spikes have been split into three, that is at 529.8 (Fe–O), 530.8 (Fe–OH surface hydroxyl), and 532.9 eV corresponding to oxygen bonded in SA@Ag@IONCs, as shown in Fig. 6d, which is ascribed to Fe–O–Ag bonding.36,49 The spectral results indicate the presence of silver (Ag), iron (Fe), and oxygen (O), as depicted in Fig. 6a. The atomic% composition of silver, iron, and oxygen was found to be 7.25, 38.33, and 54.42%. As we reported the atomic weight percent in EDS also, we found a little bit of difference from EDS result. This is because XPS, in contrast to SEM-EDS, collects information utilizing secondary and backscattered electrons from deep regions, whereas XPS only provides information for the outermost layer (at least 10 nm). This is why electron microscopy consistently provides better information regarding the atomic weight percentage of compounds than XPS. The results indicated silver nanoparticles are successfully layered on the surface of α-Fe2O3 nanoparticles.
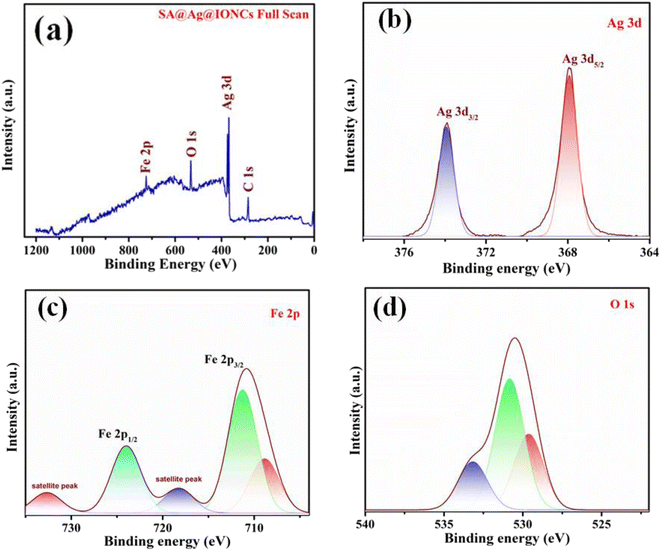 |
| | Fig. 6 Full XPS scan of SA@Ag@IONCs (a), narrow scan spectra of Ag 3d (b), Fe 2p (c), and O 1s (d). | |
4.5. Optical analysis
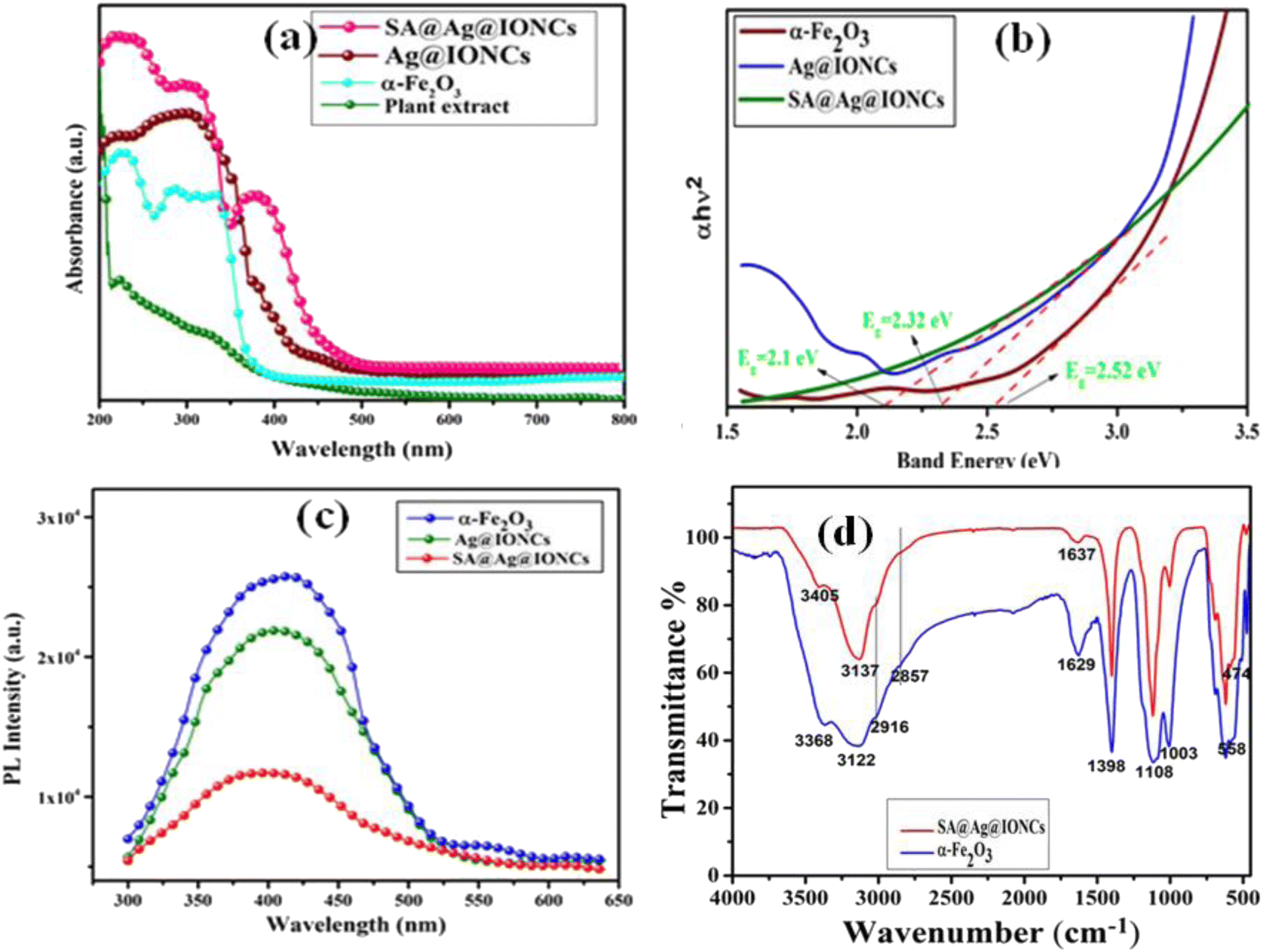
UV-visible absorption spectra of plant extract, α-Fe2O3, Ag@Fe2O3, and SA@Ag@IONCs are depicted in Fig. 7a. The absorption peaks of leaf extract observed at 230 nm and 330 nm are due to benzoyl systems and cinnamoyl rings, which suggested the existence of a polyphenolic group in Saraca asoca leaf extract.42 A strong absorption peak at 320 nm shows the formation of iron oxide nanoparticle.50,51 However, in the Ag@Fe2O3 and SA@Ag@IONCs along with iron oxide, the distinctive additional peak at 410 nm is due to the surface plasmon resonance of Ag NPs.24,52 The SPR pattern depends upon the particle shape, size, and dielectric constant. The observed SPR peak at 410 nm is due to the formation of very small-size Ag NPs in plant extract medium.53 The dimensions, geometry, and dielectric constant of the metal nanoparticles themselves, as well as the dielectric constant of the neighboring medium, all influence both the frequency and width of the surface plasmon absorption.54 The average diameter of the nanomaterials has a significant impact on the SPR band in an aqueous phase, presuming almost the same crystalline structure, medium dielectric constant, and temperature.31
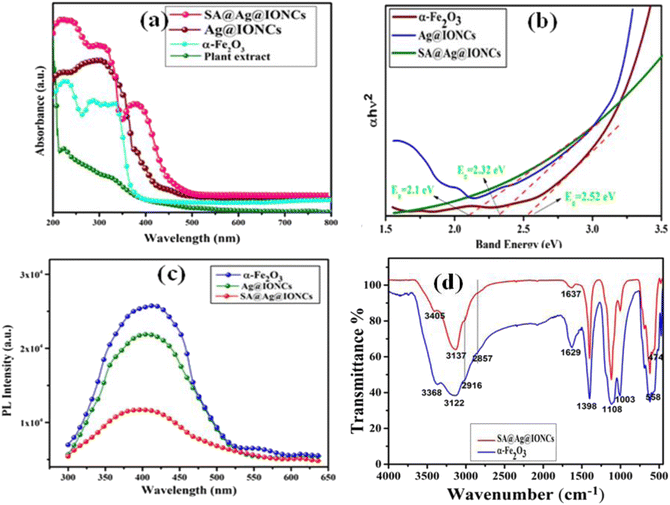 |
| | Fig. 7 UV-visible spectra of plant extract, α-Fe2O3 and SA@Ag@IONCs (a), Tauc plot (b), PL spectra (c), and FTIR spectra (d). | |
The band gap energy of pure Fe2O3 and SA@Ag@IONCs might be determined by using the Tauc plot formula: hv = A (h−Eg)n/2, where a, h, v, A, and Eg are the absorption coefficient, plank constant, light frequency, constant, and band gap energy, respectively. Fig. 7b demonstrates the band gap energy of α-Fe2O3, Ag@Fe2O3, and SA@Ag@IONCs are 2.52 eV, 2.32, and 2.1 eV, respectively. The absorption edge appears to migrate towards a higher wavelength for the SA@Ag@IONCs composite, causing a change in bandgap energy. In the case of sonication-assisted composite, the bandgap energy is 2.1 eV, making it easier for an electron transfer from the valence band to the conduction band. But in the case of Ag@Fe2O3, the distribution of Ag over the surface of iron oxide is not evenly distributed, so the proper transportation of the charge carrier is not possible because of the agglomeration of nanoparticles. Whereas, with the use of sound waves, the clusters are broken up into smaller particles by the sonication technique, which also stabilizes the phytochemical in the extract medium. As a result, agglomeration of a particle can be prevented so that the decoration of Ag is on the surface of the iron oxide nanoparticle and provide better availability for capturing light and generating e−–h+ for the transfer of electron. As a result, the size of the crystallite shrinks and increases its surface area as verified by the BET plot. These two factors significantly improve the photocatalytic degradation of Azure B dye under solar light irradiation.
4.6. Photoluminescence analysis
Photoluminescence was used to examine the rate of separation and recombination of photoexcited electrons and holes. The photocatalytic reaction process is a form of photochemical process, whereas the photoluminescence phenomenon is a form of photophysical process. The stochastic behavior of photo-generated charge carriers in semiconducting substances is strongly related to PL and photocatalysis reaction processes depending on their mechanism. PL analysis can be used to build the relationship between PL intensity and photocatalytic efficiency.13,15 The efficiency of pure Fe2O3, Ag@Fe2O3, and SA@Ag@IONCs for photocatalytic application under solar light irradiation is demonstrated by the change in absorption band edge and band gap energies. Fig. 7c displays the PL spectra of pure Fe2O3, Ag@Fe2O3, and SA@Ag@IONCs. Pure Fe2O3 shows intense emission near 410 nm at an excitation wavelength of 350 nm.55,56 In contrast to pure iron oxide, Ag@Fe2O3, and SA@Ag@IONCs have a lower PL intensity at 408 nm. The blue shift of the PL peak after decorating silver into Fe2O3 as well as layering of phytochemical of Saraca asoca leaf extract, decreasing the overall diameter of SA@Ag@IONCs. It is known that Ag-doped semiconductors do not result in new PL phenomena; however, it decreases the excitonic PL intensity, which is mainly ascribed due to the capturing of noble Ag+ ions. This indicates that the photoelectrons and holes are efficiently separated in Ag-supported Ag@Fe2O3 and SA@Ag@IONCs. This result indicates that after the decoration of silver with iron oxide, the Ag@Fe2O3 and SA@Ag@IONCs show a slower rate of recombination of photo-induced electrons and holes. The slower rate of recombination of electrons and holes pair is the result of a synergic effect when silver is doped with iron oxide.57 In the case of Ag@Fe2O3, the rate of recombination of photogenerated electron and hole pairs is fast in comparison to sonication-assisted SA@Ag@IONCs, which might be due to not properly distributing on the surface because of agglomeration of particles. On the other hand, SA@Ag@IONCs PL intensity is lower in comparison to both iron oxide as well as Ag@Fe2O3, which inhibits the recombination of electron–hole pair, and this ultimately increases the photocatalytic performance. On irradiation of light, the electrons in the VB are excited to the CB of Fe2O3. The photo induced electrons from the CB of Fe2O3 gets transferred to CB of Ag and the photo generated holes from Ag goes to the VB of Fe2O3. This ensures effective charge separation, which further results in lower rate of recombination of electron and holes and this significantly enhances the photocatalytic efficiency of SA@Ag@IONCs.
4.7. FTIR analysis
In the biogenic synthesis, it is known that the phytochemicals of plant extract play a dual role as reducing and capping agents for the formation of NPs.42 In the FTIR spectrum of α-Fe2O3 Fig. 7d, the intense peaks are observed at 3368 and 3122 cm−1, which are to O–H along with N–H stretching vibrations of phenols58 amino groups, and amide links of proteins or enzymes. In the case of SA@Ag@IONCs, these peaks were observed at 3405 and 3137 cm−1, which indicates that Ag is anchored on α-Fe2O3. Small peaks in the FTIR spectra of both samples at 2916 cm−1 and 2857 cm−1 are related to –C–H stretching vibrations. The intense bands at 1629 cm−1 (in α-Fe2O3) and 1637 cm−1 (in SA@Ag@IONCs) are due to the stretching of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of polyphenols and CC bond stretching of the aromatic ring.59,60 The absorption spike at 1398 cm−1 in Fe2O3 and SA@Ag@IONCs indicates vibrational stretching of C–N groups of amines and several heterocyclic molecules (flavones and alkaloids). The strong peaks in the FTIR spectra of both samples at 1108 cm−1 are due to C–O–C vibration while the peak at 1003 cm−1 is due to C–O stretching of esters. In the FTIR spectra of α-Fe2O3 and SA@Ag@IONCs, two characteristic peaks appeared at 474 cm−1 and 558 cm−1 due to Fe–O vibration. The high-frequency peak at 558 cm−1 is associated with the deformation of Fe–O at the tetrahedral and octahedral sites, whereas the lower frequency peak at 474 cm−1 is related to the deformation of Fe–O at the octahedral centers of hematite.61 The FTIR results showed that Saraca asoca leaf extract plays a dual role, i.e., the generation of NPs and their relative stabilization by adsorption or capping on the nanoparticle surface.
O bond of polyphenols and CC bond stretching of the aromatic ring.59,60 The absorption spike at 1398 cm−1 in Fe2O3 and SA@Ag@IONCs indicates vibrational stretching of C–N groups of amines and several heterocyclic molecules (flavones and alkaloids). The strong peaks in the FTIR spectra of both samples at 1108 cm−1 are due to C–O–C vibration while the peak at 1003 cm−1 is due to C–O stretching of esters. In the FTIR spectra of α-Fe2O3 and SA@Ag@IONCs, two characteristic peaks appeared at 474 cm−1 and 558 cm−1 due to Fe–O vibration. The high-frequency peak at 558 cm−1 is associated with the deformation of Fe–O at the tetrahedral and octahedral sites, whereas the lower frequency peak at 474 cm−1 is related to the deformation of Fe–O at the octahedral centers of hematite.61 The FTIR results showed that Saraca asoca leaf extract plays a dual role, i.e., the generation of NPs and their relative stabilization by adsorption or capping on the nanoparticle surface.
4.8. VSM analysis
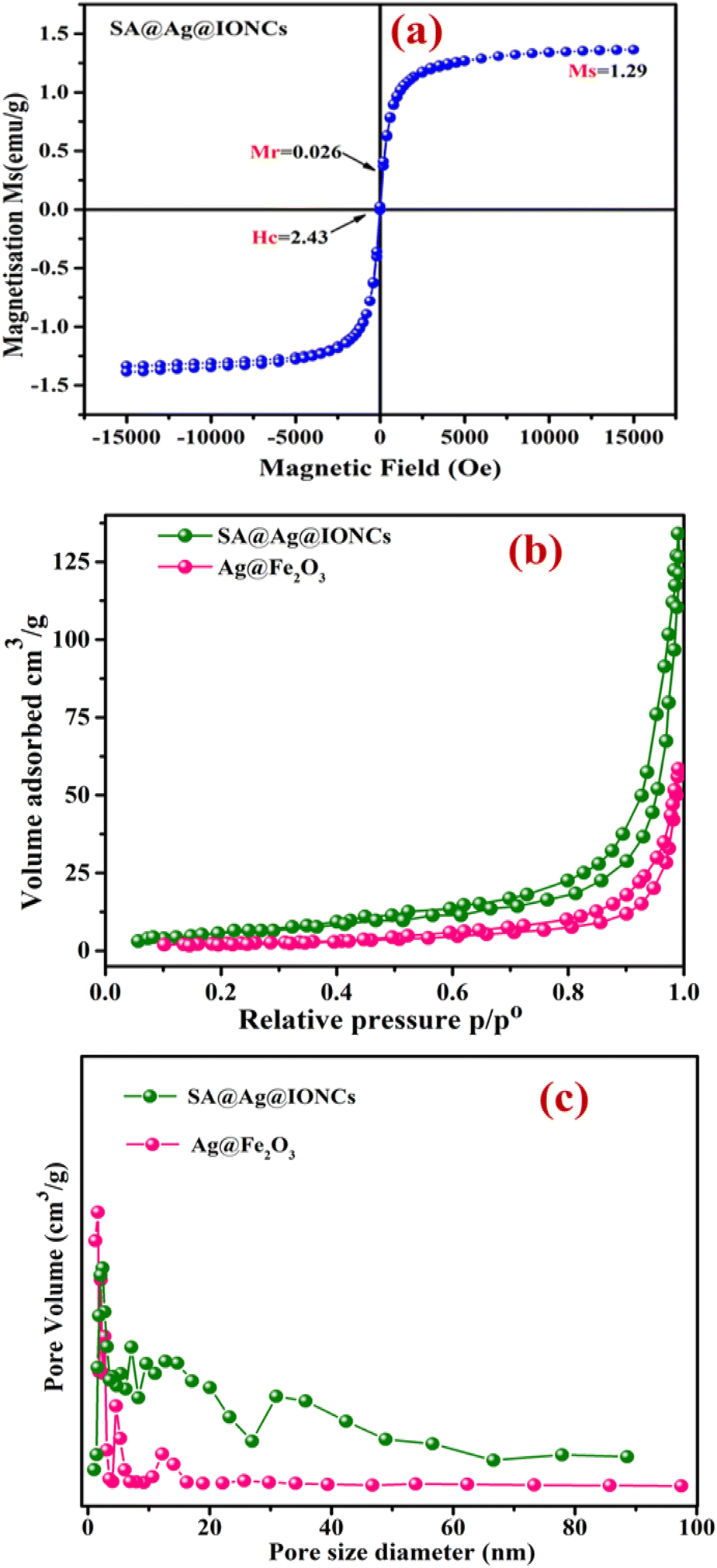
By using a vibrating sample magnetometer at ambient temperature with an incorporated magnetic field sweeping from −15000 to +15000 oersted, the value of saturation magnetization (Ms), remanence magnetization (Mr), and coercivity (Hc) can be obtained. The magnetic behavior of SA@Ag@IONCs as prepared was investigated. The VSM curve of SA@Ag@IONCs is displayed in Fig. 8a. It exhibits superparamagnetic behavior, and the SA@Ag@IONCs saturation magnetization (Ms) is measured at 1.29 emu g−1, remanence magnetization (Mr) is 0.028, and coercivity (Hc) is 2.43 Oe.49,62 Ms, which is commonly measured in emu g−1, is an essential parameter for characterizing iron oxide nanocomposite and it is significantly dependent on the chemical composition of the nanocomposite. Low values of coercive field Hc observed in Oe, as well as remnant magnetization Mr, are crucial markers of developing superparamagnetic characteristics. The overall magnetic moment of the nanocrystals can be thought of as one huge magnetic moment made up of all the individual magnetic moments of the atoms. These nanoparticles exhibit a specific preference for the magnetic axes (or directions of easy magnetization) in which they align.63 The transition from ferromagnetic to superparamagnetic material occurs as particle size continues to fall below a certain limit of size.64 The existence of Ag on the surface of α-Fe2O3 nanoparticles may be the cause of the low saturation magnetization.65 Based on this analysis, SA@Ag@IONCs is a magnetic photocatalyst that can be effortlessly extracted from the aqueous media by using an external magnetic field. This fast recovery is essential for its practical use in the further utilization of photocatalyst.66
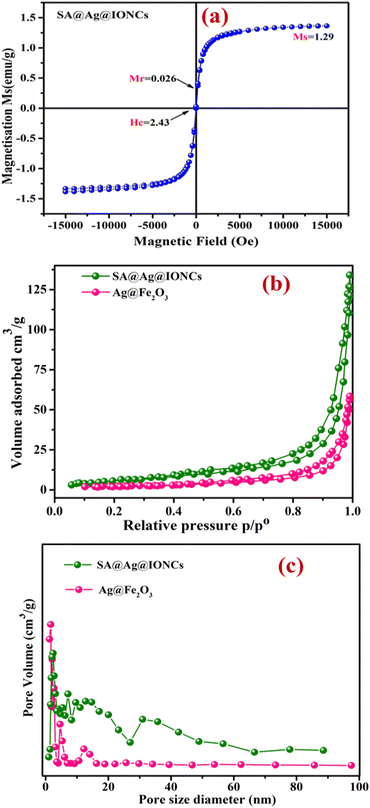 |
| | Fig. 8 Vibrating sample magnetometer curve of SA@Ag@IONCs (a) BET N2 adsorption–desorption isotherm of Ag@Fe2O3, SA@Ag@IONCs (b) BJH pore size distribution curve (c). | |
4.9. BET analysis
The porosity of Ag@Fe2O3 and SA@Ag@IONCs was estimated by the N2 gas adsorption–desorption isotherm process. The Barrett–Joyner–Halenda technique was used to determine the average pore diameter, which is observed in the range of 18 nm and 42 nm for Ag@Fe2O3, SA@Ag@IONCs, respectively, as shown in Fig. 8c. This result indicates that SA@Ag@IONCs has 2 folds with a more porous surface edge as compared to Ag@Fe2O3. The BET-specific surface areas (SBET) of Ag@Fe2O3 and sonochemically synthesized SA@Ag@IONCs is 8.27 m2 g−1 and 27.33 m2 g−1, respectively, as shown in Fig. 8b, as compared to lower surface areas reported in the literature.67,68 The pore volume of sonochemically assisted SA@Ag@IONCs and Ag@Fe2O3 is 0.2482 cm3 g−1 and 0.1059 cm3 g−1, respectively. The number of active sites and surface area of nanoparticles rise with increasing pore volume,69,70 which raises adsorption performance and consequently boosts photocatalytic performance. Compared to Ag@Fe2O3 nanocomposite, the sonochemically assisted SA@Ag@IONCs has a greater surface area and pore volume. Hence, SA@Ag@IONCs has a better choice for photocatalytic uses due to their large pore volume, large surface area, and mesoscopic properties.
4.10. Photo-driven activity of SA@Ag@IONCs against Azure B (cationic blue dye)
The photo-driven activity of synthesized α-Fe2O3, Ag@Fe2O3, and SA@Ag@IONC nanocomposite was investigated for degradation of Azure B (AzB) under direct sunlight irradiation. We have noticed the maximum photocatalytic activity for SA@Ag@IONCs in comparison to pure α-Fe2O3, Ag@Fe2O3 by further examining the efficiency of the photocatalytic activity of SA@Ag@IONCs and the influence of important factors (such as pH and catalyst dose). For a dose-dependent study, a freshly prepared 50 mL aqueous Azure B (AzB) solution having 20 mg L−1 concentration was taken in five separate tubes. 20, 30, 40, 50, and 60 mg of SA@Ag@IONCs catalyst was added to each tube. In a separate tube, the same volume of AzB solution without catalyst was taken, which is treated as blank or control. All the reaction mixtures were thoroughly mixed at 30 °C and sonicated to establish equilibrium; thereafter, each reaction mixture was put under direct sunlight irradiation. Rapid decoloration of blue AzB solutions was observed in the presence of catalyst SA@Ag@IONCs while virtually no change in color was noticed in the absence of a catalyst. The progress of photocatalytic degradation AzB was monitored spectro-photometrically in the wavelength range 200–800 nm using the supernatant part of filtered aliquots of reaction suspension at different time intervals. The absorption spectra of AzB consist of maximum absorbance at 643 nm, which is characteristic of the AzB dye. The percentage photocatalytic degradation of the AzB solution was determined by the relative change in the optical density of the dye solution.
A0 and At represent the optical density of the AzB solution at t = 0 and different intervals. The photocatalytic degradation of different amounts of nanocomposite dose was obtained by plotting Ct/C0 versus time. The best-fitted, pseudo-first-order kinetics of photo-assisted degradation of AzB using SA@Ag@IONCs was investigated using the expression:
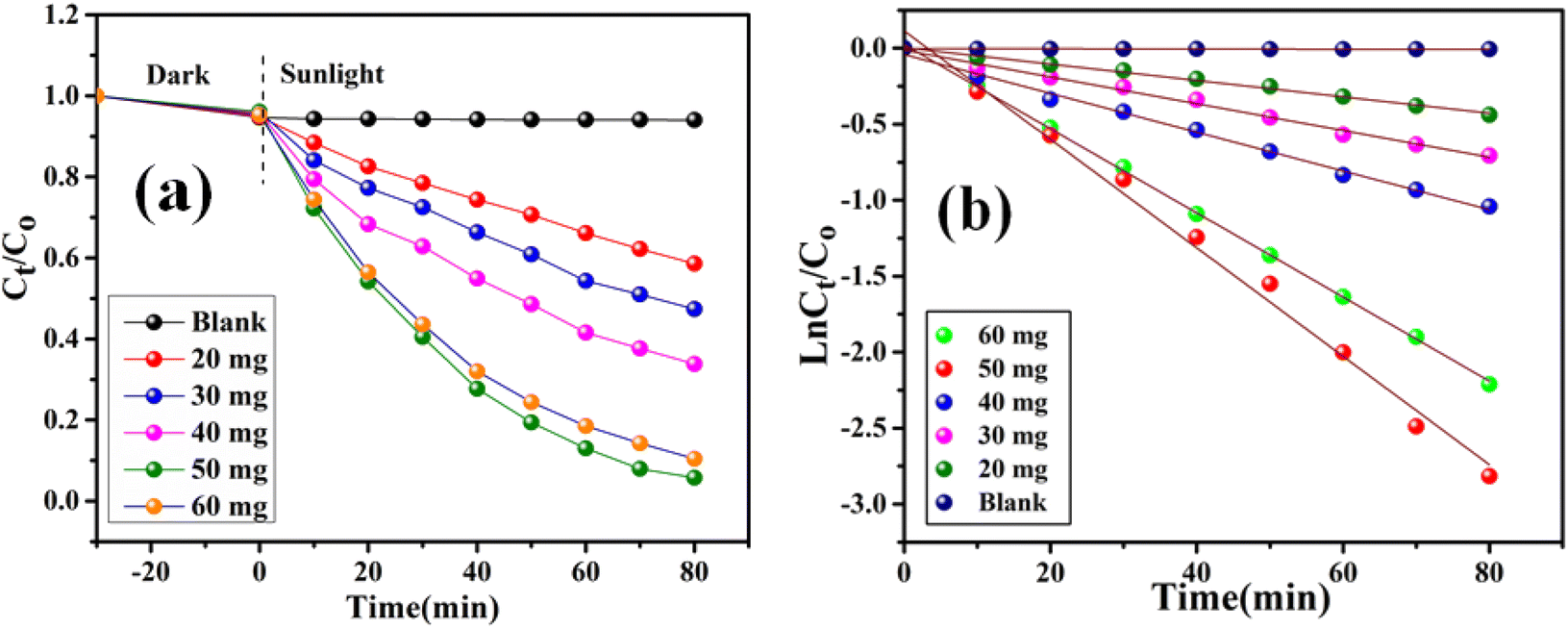
Here, Kapp represents the apparent rate constant, C0 and Ct indicate the initial concentration and after time ‘t’ respectively. The photodegradation efficiency of AzB was analyzed by Ct/C0 versus time and ln(Ct/C0) versus time plots as shown in Fig. 9a and b.
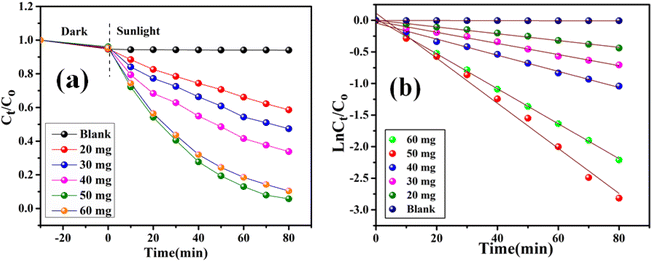 |
| | Fig. 9 Dose dependent photocatalytic degradation performance of SA@Ag@IONCs (a) kinetics plot of SA@Ag@IONCs nanocomposite (b). | |
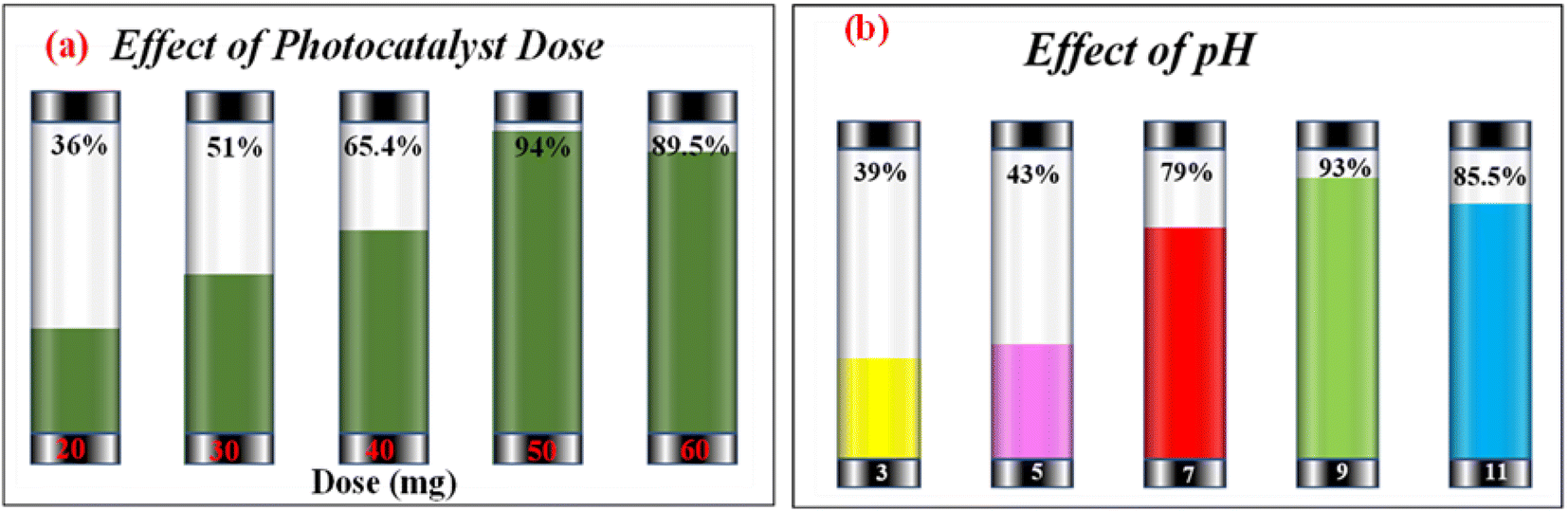
Investigating the impact of nanocatalyst dose on the photo-assisted degradation of AzB dye under solar light exposure using different doses of SA@Ag@IONC nanocomposite is depicted in Fig. 10a. 36, 51, 65.4, 94, and 89.5% degradation of AzB has been found using 20, 30, 40, 50, and 60 mg of SA@Ag@IONC nanocomposite, respectively. The obtained results were best supported by pseudo-first-order kinetics and the rate constant was found to be 0.005, 0.008, 0.012, 0.036, and 0.028 min−1 for 20, 30, 40, 50, and 60 mg of SA@Ag@IONC nanocomposite, respectively. As the amount of photocatalyst is increased, the rate of photocatalytic degradation increases until we obtain optimized condition at 50 mg of catalyst dose; after that, it shows a decreased rate at 60 mg. Initially, the rate of dye degradation increases rapidly throughout the increase of nanocatalyst dose because of the increase in the availability of active sites on the catalyst's surface. Simultaneously, as the turbidity of the suspension increases with a high dose of photocatalyst, sun-light penetration reduces, and therefore the photoactivated volume of the suspension drops.71 As a result, higher doses of the catalyst may be ineffective due to aggregation as well as lower irradiation field due to light scattering.
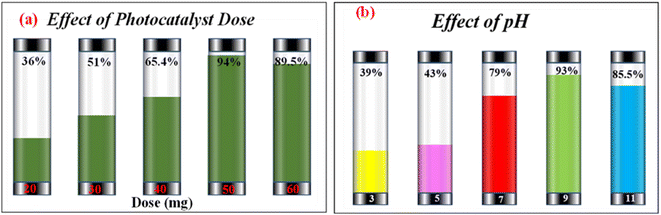 |
| | Fig. 10 Photocatalytic degradation percentage of Azure B at different doses of SA@Ag@IONCs (a) effect of pH on photocatalytic degradation of Azure B (b). | |
The concentration of H+ ions in the reaction mixture is another significant factor that affects the rate of photodegradation. The presence of active species in the reaction mixture, which affects the adsorption–desorption characteristics of AzB on the photocatalyst surface can be influenced by pH value. Taking into account this aspect, we observed the impact of pH on the rate of reaction using an optimized dose (50 mg) of nanocatalyst. The impact of pH on degradation efficiency was found to be rather significant. The order of AzB degradation efficiency was found to be pH 9 > pH 11 > pH 7 > pH 5 > pH 3 and photodegradation percentages are 93.13%, 83.8%, 78.8%, 43%, and 39% as shown in Fig. 10b. The rate of degradation is found to be highest at basic pH, then neutral pH, and acidic pH. Degradation efficiencies are observed to be low in acidic medium. In an acidic medium, the concentration of H+ at the binding site of SA@Ag@IONCs makes it positively charged, causing the molecules of cationic dye (AzB) to repel each other, and therefore, the degradation of AzB is decreased. However, the nanocomposite surface acquires more negative charge at higher pH, which increases the electrostatic interaction between the negatively charged nanocatalyst and cationic dye molecule, which speeds up the reaction. The production of ˙OH radicals as a result of a large number of hydroxyl ions in the alkaline medium is one potential explanation for the accelerated photocatalytic breakdown of AzB at higher pH levels. The potential to break down when exposed to sunlight increased when the pH is changed, from neutral to basic.
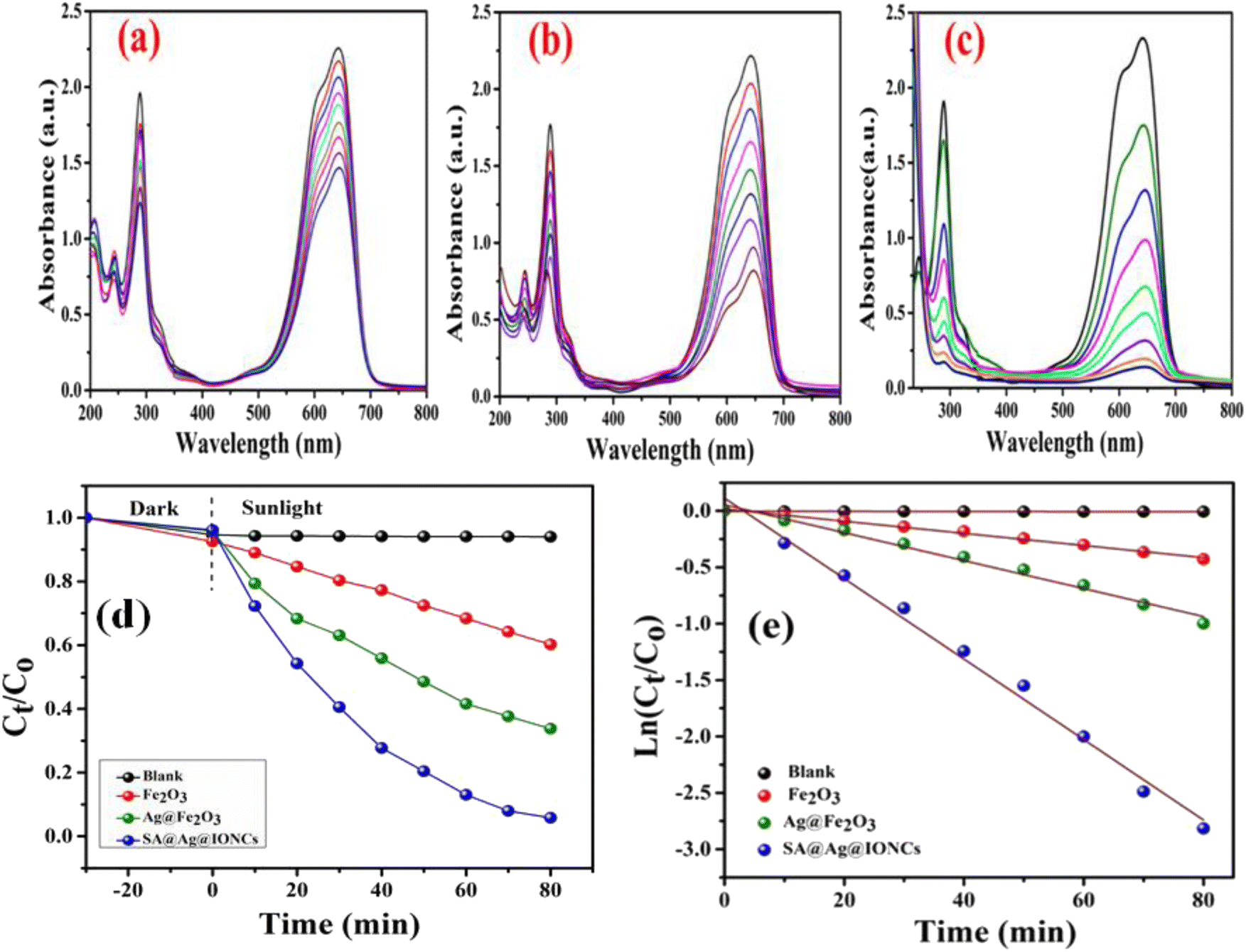
The efficiency of photocatalytic activity at the same optimized condition for comparable degradation for pure α-Fe2O3, Ag@Fe2O3, and SA@Ag@IONCs is shown in Fig. 11a–c. 50 mg of α-Fe2O3 NPs and Ag@Fe2O3, as in the case of SA@Ag@IONC nanocomposite, was added to 50 mL (20 mg L−1) aqueous Azure B (AzB) solution, and at pH 9, degradation of dye AzB has been found to 39% and 63.3% for α-Fe2O3 and Ag@Fe2O3 respectively, while in the same condition, degradation percentage using SA@Ag@IONCs, nanocomposite was 94%. Comparative kinetic study of α-Fe2O3, Ag@Fe2O3, and SA@Ag@IONCs for the photodegradation efficiency of AzB was analyzed by Ct/C0 versus time and ln(Ct/C0) versus time plots as shown in Fig. (11d and e). The obtained results were best fitted by pseudo-first-order kinetics and the rate constant was found to be 0.005, 0.012, and 0.036 min−1 respectively, whereas t1/2 half-life 138.6, 57.75, and 19.25 min. We observed that the rate constant of SA@Ag@IONC nanocomposite is 6 folds and 3 folds higher than the α-Fe2O3 nanoparticle and Ag@Fe2O3 nanocomposite. The sonication technique uses sound waves to break up the large clusters of molecules into smaller particles, stabilizing the phytochemical present in the extract medium. As a result, aggregated particles can be avoided, leading to proper Ag loaded on the surface of α-Fe2O3 and improved availability for absorbing solar light and generated electron–hole pair for e− transfer. It also possesses low bandgap energy, lower PL intensity, and large surface area, significantly enhancing the photocatalytic activity efficiency of SA@Ag@IONC nanocomposite (Table 1).
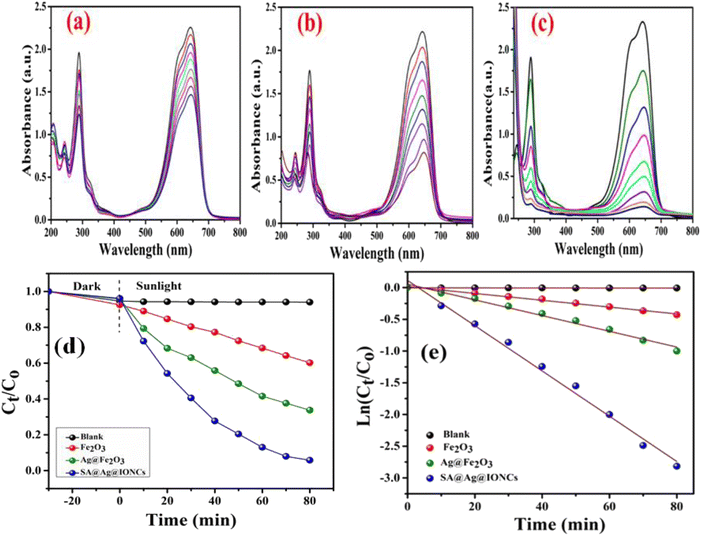 |
| | Fig. 11 Photocatalytic degradation performance of α-Fe2O3, Ag@Fe2O3, and SA@Ag@IONCs UV-vis plot (a–c), kinetics plot of SA@Ag@IONCs nanocomposite (d and e). | |
Table 1 Comparison of photocatalytic performance in the degradation of cationic dye
| Photo-catalyst |
Synthesis methods |
Dyes |
Rate constant (min) |
Irradiation light |
Time (min) |
Degradation (%) |
Ref. |
| Fe3O4@ZnO |
Hydrothermal/calcination |
MB |
2.4 × 10−2 |
300 W Xe lamp |
90 |
90 |
72 |
| Fe2O3/Ag |
Aloe vera gel extract |
MB |
— |
Visible light |
140 |
88 |
66 |
| CoFe2O4/Cr2O3/SiO2 |
Chemical |
MB |
2.5 × 10−2 |
UV light (12 mW Hg) lamp |
120 |
90 |
73 |
| CNFO/g-C3N4 |
Chemical |
MB |
1.7 × 10−2 |
Sunlight |
180 |
97 |
74 |
| WO3/Fe2O3 |
Hydrothermal |
MB |
— |
Visible light 500 W Xe lamp |
240 |
73 |
75 |
| PANI/CdO |
Calcination |
MB |
1.9 × 10−2 |
Sunlight |
240 |
97 |
76 |
| ZIF-8 |
Chemical |
MB |
1.7 × 10−2 |
UV light |
120 |
82 |
77 |
| FNZ/PANI |
Chemical |
MB |
2.2 × 10−2 |
Sunlight |
300 |
99 |
78 |
| Chitosan/PANI/Co3O4 |
Chemical |
MB |
0.08 × 10−2 |
UV light |
180 |
88 |
79 |
| CuO@CQD |
P. Peels |
AzB |
— |
Visible light |
120 |
97 |
80 |
| GO/CuO |
Hydrothermal |
AzB |
1.38 × 10−2 |
200 W tungsten lamp |
120 |
80 |
81 |
| SA@Ag@IONCs |
Saraca asoca |
AzB |
3.6 × 10−2 |
Sunlight |
80 |
94 |
This work |
4.11. Investigation of photodegradation products using LC-MS
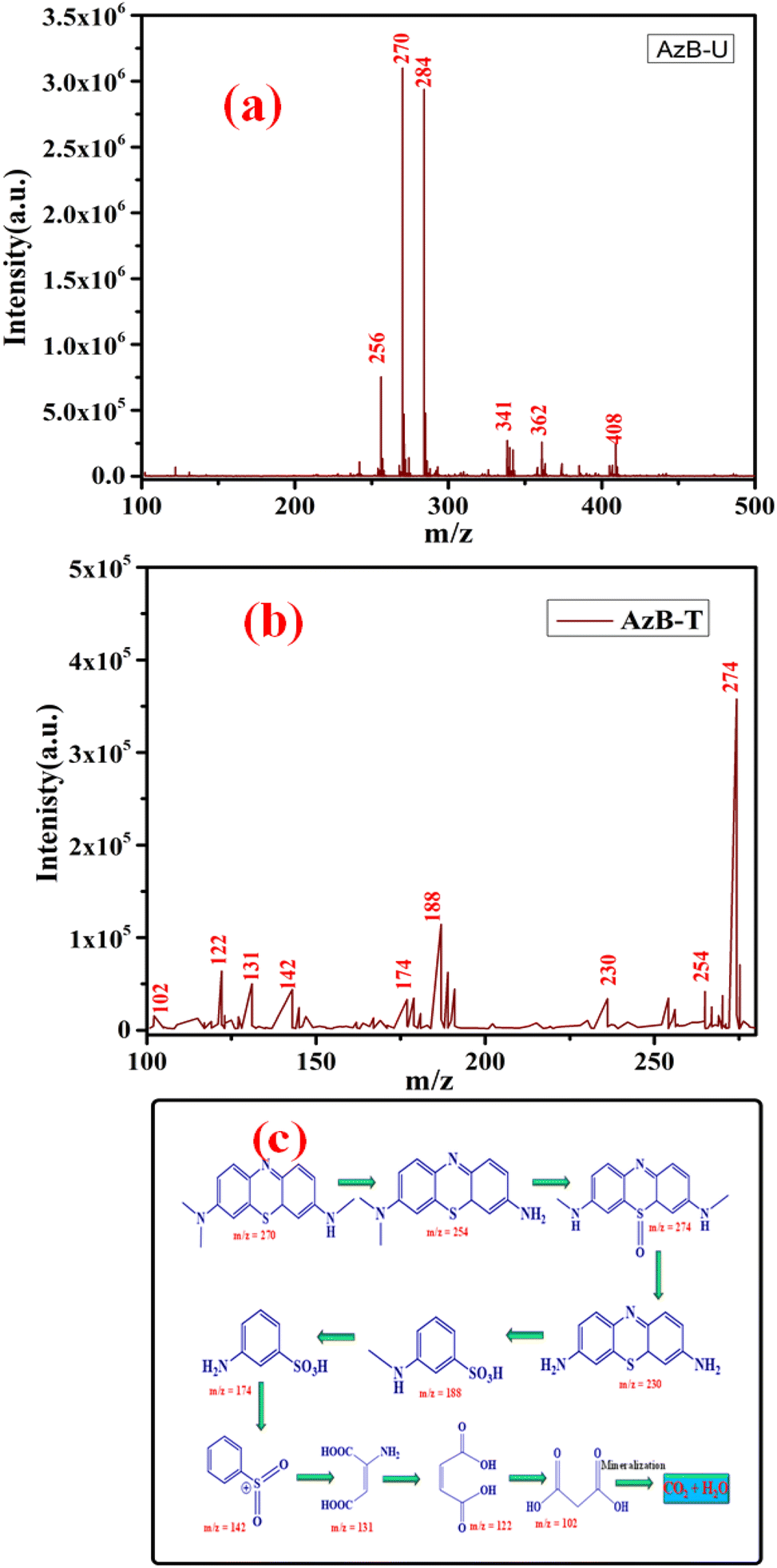
To investigate the photodegradation mechanism of AzB by using SA@Ag@IONCs, LC-MS was performed to investigate the various degradation intermediates. The result revealed that AzB molecules were split into smaller degradation products. LC-MS spectrum of untreated AzB solution revealed its presence as the main component (Fig. 12a), however in the presence of SA@Ag@IONCs catalyst, a distinctive peak corresponding to m/z at 270 was not seen, indicating that the AzB molecules completely broke down. Meanwhile, several peaks were found, which were probably the degradation intermediate products formed when AzB molecules degraded, as shown in Fig. 12b. The AzB molecule was broken down into fragments having m/z values of 254, 274, 230, 188, 174, 142, 131, 122 and 102, which were attributed due to C14H15N3S, C14H15N3SO, C12H11N3S C7H9NO3S, C6H7NO3S, C6H5O2S+, C4H5NO4, C4H4O4, and C3H4O4 species.82–84 The LC-MS findings show that the photodegradation of AzB occurred in the presence of SA@Ag@IONCs rather than adsorption. LC-MS results showing the degraded byproducts or intermediates formed by sunlight-driven degradation of Azure B and its degradation pathway are given in Fig. 12c.
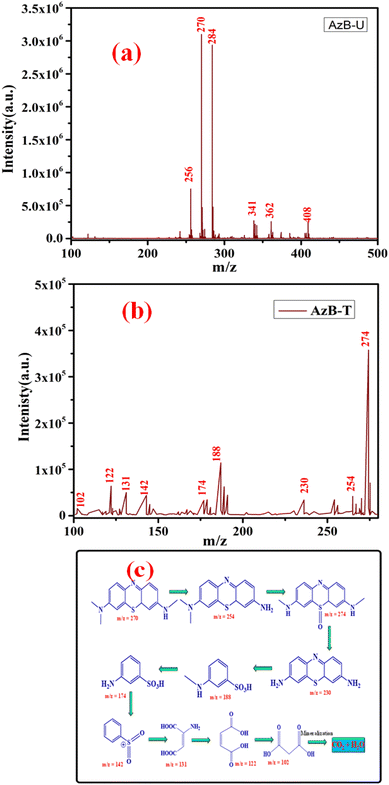 |
| | Fig. 12 LC-MS spectrum of Azure B untreated (a) Azure B treated (b) and intermediates formed after degradation of Azure B (c). | |
4.12. Trap experiment
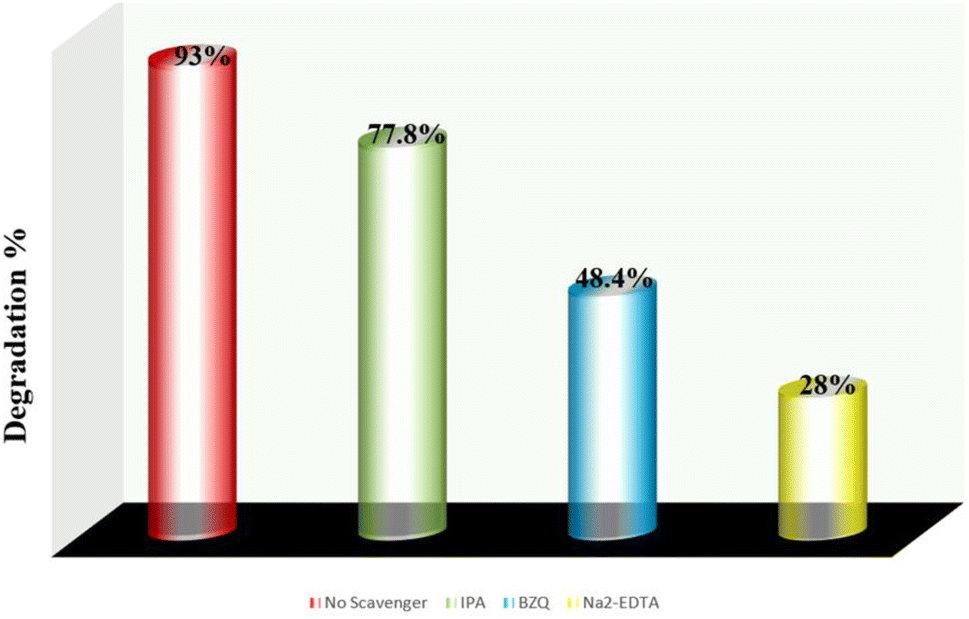
In the photocatalysis process, when electromagnetic radiation from solar light or photon source with energy equivalent to or greater than that of the photocatalysts energy band gap incident, electron (e−) and holes (h+) are generated in the conduction band and valence band, respectively. Dissolved oxygen is reduced by an electron in the conduction band to form superoxide radical (˙O2−), while water molecule is oxidized in the presence of a hole in the valence band to create ˙OH radical. The hazardous material is quickly decomposed by these reactive oxygen species (ROS) into smaller, nontoxic metabolites. By using trap experiments, the underlying photocatalytic process for the deterioration of AzB has been further investigated. As efficient scavengers for ˙OH, h+, and ˙O2−, respectively, isopropyl alcohol (IPA), disodium-EDTA, and para-benzoquinone (BZQ) have been employed to identify the primary active radical species participating in sunlight-driven deterioration process.42,66 The photocatalytic reaction is observed to be suppressed by all scavengers; however, the scavenging capacity of the species is reduced in the order Na2-EDTA > BZQ > IPA. Using the scavengers IPA, BZQ, and Na2-EDTA, the percentage photocatalytic degradation of AzB was determined to be 77.8%, 48.4%, and 28%, respectively (Fig. 13). This result indicates that h+ and ˙O2− are the active radical species involved in the photodegradation of AzB.
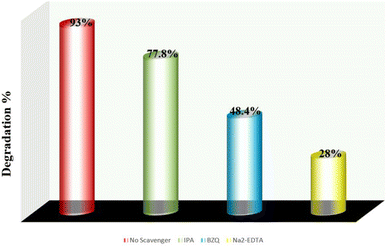 |
| | Fig. 13 Photocatalytic degradation percentage of Azure B using scavengers IPA, Na2EDTA, and BZQ. | |
4.13. Photocatalytic mechanism
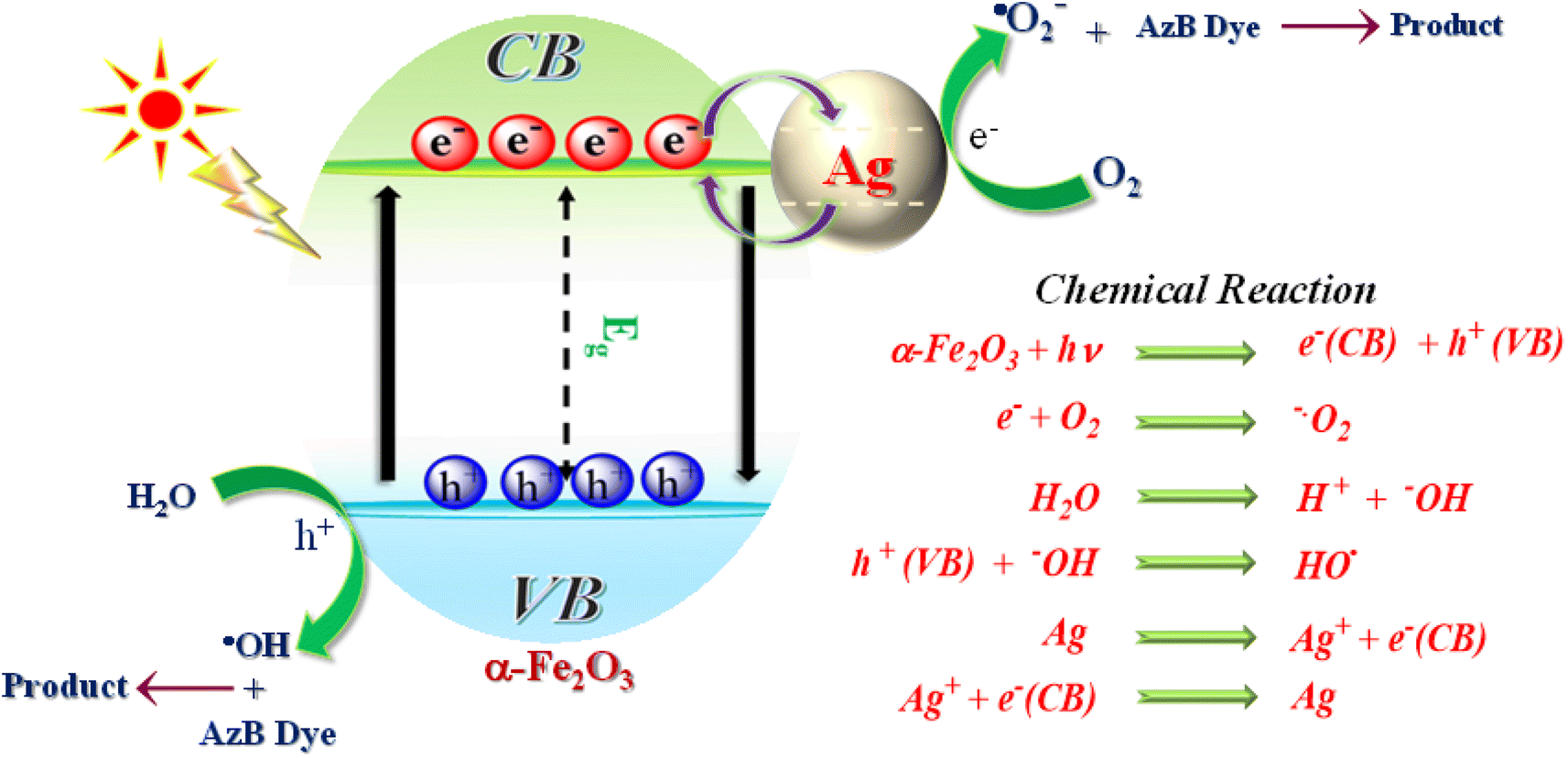
Based on the above findings and published literature, a feasible photocatalytic mechanism has been proposed for the photodegradation of AzB dye using SA@Ag@IONCs photocatalyst in the presence of solar light, as shown in Fig. 14. On exposure to sunlight radiation, α-Fe2O3 nanoparticles act as photosensitizers, producing electron–hole pairs. Noble metal Ag is added onto the surface of α-Fe2O3 to improve their photocatalytic efficiency. When exposed to solar light, α-Fe2O3 emits photoinduced electrons and holes on CB and VB, respectively. The aforementioned factors result in photoinduced free electrons that interact with dissolved O2 molecules to form superoxide anion radical and holes that interact with a water molecule to form hydroxyl radical, both of which contribute to the photodegradation of AzB dye molecules. The generated charge carrier undergoes rapid recombination because of its minimal lifetime, which decreases the efficiency of a photocatalyst. The silver nanoparticles serve as an electron absorber, preventing the recombination of charge carriers and capturing the photoinduced electrons. The photoinduced electrons then move to the surface of the Ag and reduce O2 to ˙O2−, while the photogenerated holes on the α-Fe2O3 oxidize H2O to ˙OH. Finally, during the oxidation–reduction by ˙OH, ˙O2−, and h+, Azure B dye is degraded into CO2, H2O, and NH4+.
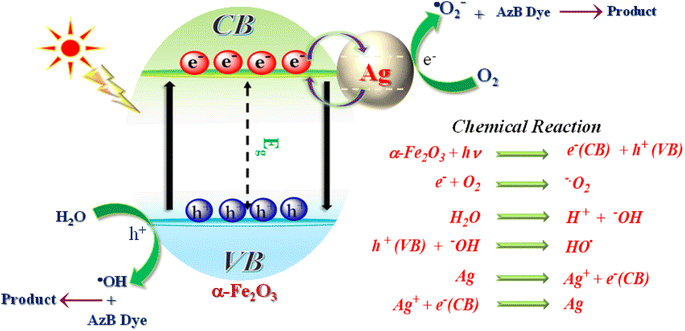 |
| | Fig. 14 Plausible mechanism of photocatalytic degradation process. | |
4.14. Photocurrent analysis
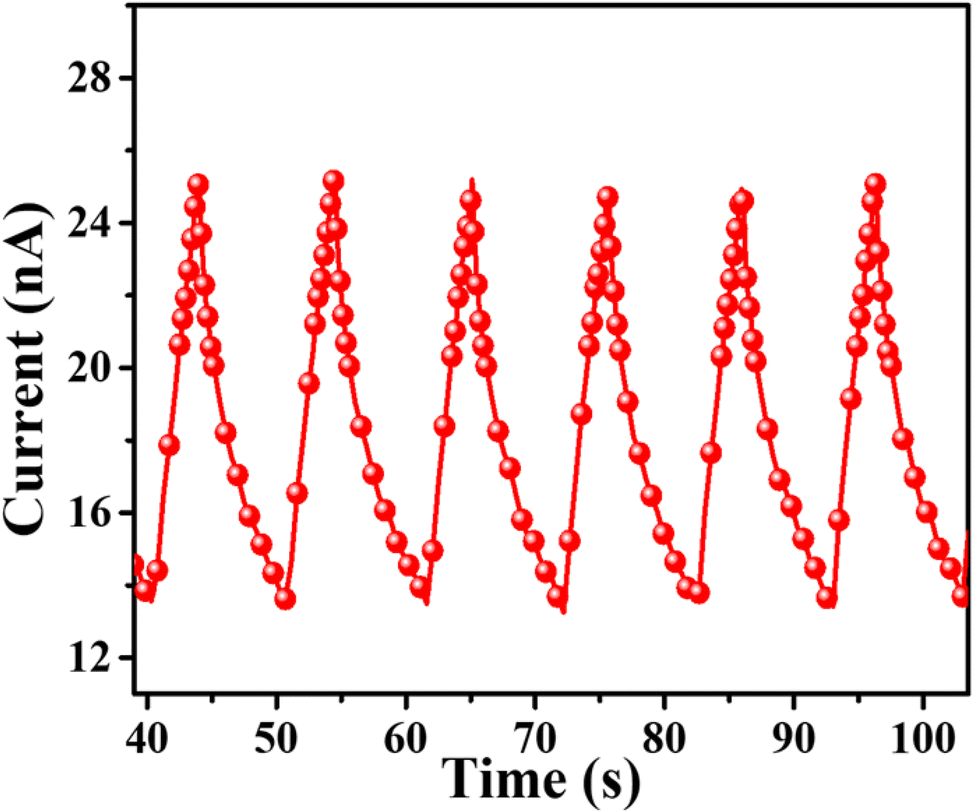
The photocurrent measurement of SA@Ag@IONCs as produced with the numerous on–off phases of visible light are given in (Fig. 15), which demonstrates that photocurrent is significantly improved on exposure to visible light, the photocurrents generated by SA@Ag@IONCs is 2.5 × 10−8 A. Once the irradiation is finished, SA@Ag@IONCs photocurrent responses have reverted to their initial level. When photo-induced electrons ionize in the conducting band of the semiconductor, leaving holes in the valence band, which causes the photocurrent to flow.
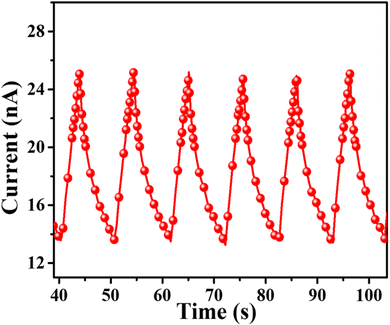 |
| | Fig. 15 Photocurrent of SA@Ag@IONCs. | |
4.15. COD analysis
UV-visible spectrophotometry results could not be used to verify whether AzB was entirely degraded into CO2 and H2O. The COD measurement is an important method for finding out how mineralization works. COD analyses the quantity of oxygen necessary to decompose organic molecules in water contents. The COD experiment was performed for both the solution of AzB dye before and after the photo-degradation reaction. The dichromate reflux techniques were used to examine the COD. The starting dye concentration and catalyst loading for COD testing were both 15 mg L−1. COD values for untreated and treated AzB were found to be 416 and 37.3 mg L−1, respectively. The COD results show that the photodegradation efficiency of nanocomposite for AzB is approximately 91%, which indicates almost complete mineralization of AzB.
4.16. Recyclability of photocatalyst
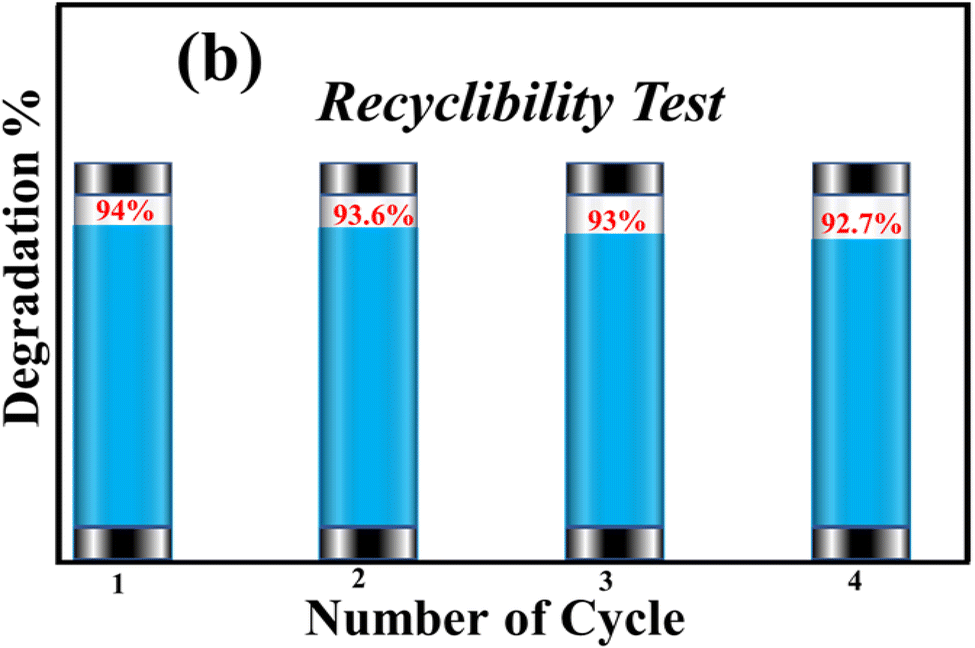
The recyclability of synthesized SA@Ag@IONCs was evaluated up to four times when the AzB dye was completely degraded. The SA@Ag@IONCs were isolated from the dye solution, washed multiple times, oven dried at 85–90 °C, and reused. The stability of SA@Ag@IONCs in terms of percentage degradation was evaluated for four further cycles, and the results are presented in Fig. 16. The results showed that the photocatalytic efficiency decreased slightly in the third cycle but not significantly in cycles 1 or 2. This slight decrease in the degradation of dye is due to the loss of some active site present on the surface of the photocatalyst. Overall, 1.3% degradation is seen after the 3rd cycle, which is quite low. These findings demonstrate that the photocatalyst is extremely stable for the degradation of AzB dye.
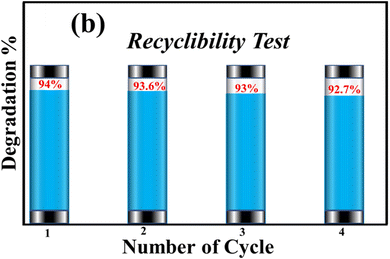 |
| | Fig. 16 Recyclability test of SA@Ag@IONCs for degradation of AzB. | |
5. Conclusion
The present work has a one-pot green, rapid, and cost-effective synthesis method where Saraca asoca leaf extract acts as a reducing, stabilizing, and capping agent. The synthesized SA@Ag@IONCs has spherical shape morphology, with an average particle size of Ag/α-Fe2O3, and SA@Ag@IONC nanocomposite at 81 nm and 25.9 nm calculated using Debye Scherrer equation form XRD pattern. The nanocomposite has a spherical-shaped morphology confirmed by HRTEM images with an average particle size of ∼25 nm. The presence of Fe, Ag, and O in SA@Ag@IONCs was confirmed by EDX and XPS analyses. By sonochemical approach, ultrasmall silver was decorated onto the surfaces of the α-Fe2O3 nanocomposite, which have a large surface area and pore volume as compared to Ag@Fe2O3. The optical band gap energy of SA@Ag@IONCs is 2.1 eV. The decoration of silver on iron oxide, Ag@Fe2O3, and SA@Ag@IONCs shows lower PL intensity as compared to pure Fe2O3 and also a slower rate of recombination of photo-induced electrons and holes. The slower rate of recombination of electrons and holes pair is the result of a synergic effect when silver is loaded with iron oxide. The lower the PL intensity of photocatalyst material, the more quickly the dissociation rate of photogenerated charge carriers, which can significantly increase the efficiency of photocatalytic activity. Photocatalytic degradation of Azure B at different photocatalyst doses revealed the highest degradation at 50 mg of nanocomposite with pH 9. We have observed the maximum rate of photodegradation (94%) for SA@Ag@IONCs, it follows pseudo-first-order kinetics and the rate constant was found to be 0.036 min−1, whereas t1/2 half-life is 19.25 min. The active species for the degradation of AzB using nanocomposite were ˙O2−, and h+, respectively. Due to its magnetism feature, the nanocomposite could be readily removed from the reaction solution and thereafter applied repeatedly. Four regeneration runs of the nanocomposite were completed without any deterioration in photocatalytic activity. Overall, it can be said that using a sonochemical-assisted synthesis of SA@Ag@IONC nanocomposite is a better choice rather than Ag@Fe2O3 breakdown hazardous dye in wastewater treatment.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The author is thankful to the UGC-JRF, New Delhi for providing financial support with fellowship, authors would also like to acknowledge the Sophisticated Instrument Laboratory (SIL), Department of Chemistry (BBAU), USIC facility (BBAU) and IIT Kanpur for providing furnished instrumental laboratory and characterization support.
References
- X. Hu, G. Li and J. C. Yu, Langmuir, 2010, 26, 3031–3039 CrossRef CAS PubMed.
- A. Hernández-Ramírez and I. Medina-Ramírez, Photocatalytic semiconductors, Springer, 2016 Search PubMed.
- M. Tahir, S. Tasleem and B. Tahir, Int. J. Hydrogen Energy, 2020, 45, 15985–16038 CrossRef CAS.
- K. Yan, P. Li, H. Zhu, Y. Zhou, J. Ding, J. Shen, Z. Li, Z. Xu and P. K. Chu, RSC Adv., 2013, 3, 10598–10618 RSC.
- S. Li, S. Shan, S. Chen, H. Li, Z. Li, Y. Liang, J. Fei, L. Xie and J. Li, J. Environ. Chem. Eng., 2021, 9, 105967 CrossRef CAS.
- K. Kaur, R. Badru, P. P. Singh and S. Kaushal, J. Environ. Chem. Eng., 2020, 8, 103666 CrossRef.
- D. Saini, R. Aggarwal, S. R. Anand and S. K. Sonkar, Sol Energy, 2019, 193, 65–73 CrossRef CAS.
- S. Liu, Y. Chen and L. Dong, ECS Trans., 2015, 66, 45 CrossRef CAS.
- T.-J. Whang, M.-T. Hsieh and H.-H. Chen, Appl. Surf. Sci., 2012, 258, 2796–2801 CrossRef CAS.
- G. G. Bessegato, T. T. Guaraldo, J. F. de Brito, M. F. Brugnera and M. V. B. Zanoni, Electrocatalysis, 2015, 6, 415–441 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. Pérez-González, S. Tomás, J. Santoyo-Salazar, S. Gallardo-Hernández, M. Tellez-Cruz and O. Solorza-Feria, J. Alloys Compd., 2019, 779, 908–917 CrossRef.
- J. Liqiang, Q. Yichun, W. Baiqi, L. Shudan, J. Baojiang, Y. Libin, F. Wei, F. Honggang and S. Jiazhong, Sol. Energy Mater. Sol. Cells, 2006, 90, 1773–1787 CrossRef.
- M. Anpo, I. Tanahashi and Y. Kubokawa, J. Phys. Chem, 1980, 84, 3440–3443 CrossRef CAS.
- A. Lassoued, M. S. Lassoued, B. Dkhil, S. Ammar and A. Gadri, Phys. E: Low-Dimens. Syst. Nanostructures, 2018, 101, 212–219 CrossRef CAS.
- N. A. Barakat, M. A. Kanjwal, I. S. Chronakis and H. Y. Kim, J. Mol. Catal. A: Chem., 2013, 366, 333–340 CrossRef CAS.
- X. Li and F. Li, Environ. Sci. Technol., 2001, 35, 2381–2387 CrossRef CAS PubMed.
- J. C. Yu, J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Mater., 2002, 14, 3808–3816 CrossRef CAS.
- R. Justin, K. Tao, S. Román, D. Chen, Y. Xu, X. Geng, I. M. Ross, R. T. Grant, A. Pearson and G. Zhou, Carbon, 2016, 97, 54–70 CrossRef CAS.
- A. Mirzaei, K. Janghorban, B. Hashemi, M. Bonyani, S. G. Leonardi and G. Neri, Ceram. Int., 2016, 42, 18974–18982 CrossRef CAS.
- D. Mohanta and M. Ahmaruzzaman, J. Hazard. Mater., 2020, 397, 122685 CrossRef CAS PubMed.
- M. Nasrollahzadeh, M. Maham and S. M. Sajadi, J. Colloid Interface Sci., 2015, 455, 245–253 CrossRef CAS PubMed.
- M. A. Bhosale, D. Ummineni, T. Sasaki, D. Nishio-Hamane and B. M. Bhanage, J. Mol. Catal. A: Chem., 2015, 404, 8–17 CrossRef.
- E. A. Kwizera, E. Chaffin, Y. Wang and X. Huang, RSC Adv., 2017, 7, 17137–17153 RSC.
- J. Kaushik, Twinkle, S. R. Anand, S. K. Choudhary and S. K. Sonkar, ACS ES&T Water, 2022, 3, 227–235 Search PubMed.
- J. Lian, X. Duan, J. Ma, P. Peng, T. Kim and W. Zheng, ACS Nano, 2009, 3, 3749–3761 CrossRef CAS.
- S. Rajamohan, V. Kumaravel, A. Abdel-Wahab, S. Ayyadurai and R. Muthuramalingam, Can J Chem Eng, 2018, 96, 1713–1722 CrossRef CAS.
- Y. Hayashi, H. Takizawa, M. Inoue, K. Niihara and K. Suganuma, IEEE Trans. Electron. Packag. Manuf., 2005, 28, 338–343 CAS.
- G. B. Hoflund, Z. F. Hazos and G. N. Salaita, Phys. Rev. B, 2000, 62, 11126 CrossRef CAS.
- Twinkle, J. Kaushik, T. Singla, N. K. Lamba, M. Jain, N. Sharma, S. K. Choudhary and S. K. Sonkar, Ind. Eng. Chem. Res., 2023, 62, 9091–9103 CrossRef CAS.
- B. J. Wiley, S. H. Im, Z.-Y. Li, J. McLellan, A. Siekkinen and Y. Xia, J. Phy. Chem. B, 2006, 110, 15666–15675 CrossRef CAS PubMed.
- T. Wu and M. R. Zachariah, RSC Adv., 2019, 9, 1831–1840 RSC.
- V. Vidhu and D. Philip, Micron, 2014, 56, 54–62 CrossRef CAS PubMed.
- S. Iravani, Green Chem., 2011, 13, 2638–2650 RSC.
- M. Herlekar, S. Barve and R. Kumar, J. Nanopart Res., 2014, 2014 Search PubMed.
- Á. de Jesús Ruíz-Baltazar, Inorg. Chem. Commun., 2020, 120, 108148 CrossRef.
- S. Ghosh, P. More, R. Nitnavare, S. Jagtap, R. Chippalkatti, A. Derle, R. Kitture, A. Asok, S. Kale and S. Singh, J. Nanomed. Nanotechnol., 2015, 6, 1–9 CAS.
- N. Patra, A. C. Taviti, A. Sahoo, A. Pal, T. K. Beuria, A. Behera and S. Patra, RSC Adv., 2017, 7, 35111–35118 RSC.
- M. Sardar, A. Mishra and R. Ahmad, Biosens. Nanotechnol., 2014, 239–266 CAS.
- Y. Maeda, Y. Hayashi, J. Fukushima and H. Takizawa, Ultrason. Sonochem., 2021, 73, 105476 CrossRef CAS PubMed.
- H. Friedman, S. Reich, R. Popovitz-Biro, P. Von Huth, I. Halevy, Y. Koltypin, A. Gedanken and Z. Porat, Ultrason. Sonochem., 2013, 20, 432–444 CrossRef CAS PubMed.
- K. B. Singh, N. Gautam, D. D. Updhyay and G. Pandey, J. Mol. Struct., 2022, 1269, 133756 CrossRef CAS.
- L. Liu, H.-Z. Kou, W. Mo, H. Liu and Y. Wang, J. Phys. Chem. B, 2006, 110, 15218–15223 CrossRef CAS.
- J.-R. Chiou, B.-H. Lai, K.-C. Hsu and D.-H. Chen, J. Hazard. Mater., 2013, 248, 394–400 CrossRef PubMed.
- S. A. Alzahrani, M. A. Malik, S. A. Al-Thabaiti and Z. Khan, Appl. Nanosci., 2018, 8, 255–271 CrossRef CAS.
- P. Singh, Y. J. Kim, C. Wang, R. Mathiyalagan and D. C. Yang, Artif Cells Nanomed Biotechnol, 2016, 44, 1150–1157 CAS.
- G. Murugadoss, G. Manibalan, S. Hazra, M. Rajesh Kumar, J. R. Rajabathar and H. M. Yadav, J. Mater. Sci.: Mater. Electron., 2022, 1–11 Search PubMed.
- S. Hao, H. Wang, R. Yang, D. Liu, X. Liu, Q. Zhang and X. Chen, Appl. Phys. A, 2021, 127, 1–11 CrossRef.
- B. Paul, D. D. Purkayastha, S. S. Dhar, S. Das and S. Haldar, J. Alloys Compd., 2016, 681, 316–323 CrossRef CAS.
- V. Niraimathee, V. Subha, R. E. Ravindran and S. Renganathan, Int. J. Environ. Sustain. Dev., 2016, 15, 227–240 CrossRef.
- S. Lakshminarayanan, M. F. Shereen, K. Niraimathi, P. Brindha and A. Arumugam, Sci. Rep., 2021, 11, 1–13 CrossRef PubMed.
- K. Tahir, S. Nazir, B. Li, A. U. Khan, Z. U. H. Khan, P. Y. Gong, S. U. Khan and A. Ahmad, Mater. Lett., 2015, 156, 198–201 CrossRef CAS.
- A. Aykaç, M. C. Martos-Maldonado, J. M. Casas-Solvas, I. Quesada-Soriano, F. García-Maroto, L. García-Fuentes and A. Vargas-Berenguel, Langmuir, 2014, 30, 234–242 CrossRef PubMed.
- A. Rai, A. Singh, A. Ahmad and M. Sastry, Langmuir, 2006, 22, 736–741 CrossRef CAS PubMed.
- H. Fei, X. Ai, M. Gao, Y. Yang, T. Zhang and J. Shen, J. Lumin., 1995, 66, 345–348 CrossRef.
- Q. Wang, Y. Chen, J. Xu, Y. Situ and H. Huang, Res. Chem. Intermed., 2018, 44, 2365–2378 CrossRef CAS.
- Y. Shi, H. Li, L. Wang, W. Shen and H. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 4800–4806 CrossRef CAS.
- S. B. Aziz, M. Brza, M. Hamsan, M. Kadir, S. Muzakir and R. T. Abdulwahid, J. Mater. Res. Technol., 2020, 9, 3734–3745 CrossRef CAS.
- S. Senthilkumar and T. Sivakumar, Int. J. Pharm. Sci., 2014, 6, 461–465 Search PubMed.
- R. Dubey, A. Gunasekaran, T. Papadopoulos, S. J. Childe, K. Shibin and S. F. Wamba, J. Clean. Prod., 2017, 142, 1119–1130 CrossRef.
- A. Mirzaei, K. Janghorban, B. Hashemi, S. R. Hosseini, M. Bonyani, S. G. Leonardi, A. Bonavita and G. Neri, Process. Appl. Ceram., 2016, 10, 209–217 CrossRef CAS.
- N. S. Zaidi, J. Sohaili, K. Muda and M. Sillanpää, Sep. Purif. Rev., 2014, 43, 206–240 CrossRef CAS.
- Y. Zhang, Z. Yang, B.-P. Zhu, W. Yu, S. Chen, X.-F. Yang, F. Jin and J. Ou-Yang, Ceram. Int., 2014, 40, 3439–3443 CrossRef CAS.
- M. A. Busquets, A. Espargaró, R. Sabaté and J. Estelrich, Nanomaterials, 2015, 5, 2231–2248 CrossRef CAS PubMed.
- M. Khatami, H. Q. Alijani, M. Haghighat, M. Bamrovat, S. Azhdari, M. Ahmadian, M. Nobre, M. Heidari, M. Sarani and S. Khatami, Iran. J. Biotechnol., 2019, 10, 33–39 Search PubMed.
- A. Saranya, A. Thamer, K. Ramar, A. Priyadharsan, V. Raj, K. Murugan, A. Murad and P. Maheshwaran, J. Photochem. Photobiol., B, 2020, 207, 111885 CrossRef CAS PubMed.
- S. Tang, S. Vongehr and X. Meng, J. Phys. Chem. C, 2010, 114, 977–982 CrossRef CAS.
- N. Singh, S. Riyajuddin, K. Ghosh, S. K. Mehta and A. Dan, ACS Appl. Nano Mater., 2019, 2, 7379–7392 CrossRef CAS.
- D. Li, H. Song, X. Meng, T. Shen, J. Sun, W. Han and X. Wang, Nanomaterials, 2020, 10, 546 CrossRef CAS.
- A. P. Naik, H. Mittal, V. S. Wadi, L. Sane, A. Raj, S. M. Alhassan, A. Al Alili, S. V. Bhosale and P. P. Morajkar, J. Environ. Manage., 2020, 258, 110029 CrossRef CAS.
- N. Daneshvar, D. Salari and A. Khataee, J. Photochem. Photobiol. A, 2004, 162, 317–322 CrossRef CAS.
- S. Huang, J. Zhao, C. Wu, X. Wang, S. Fei, Q. Zhang, Q. Wang, Z. Chen, K. Uvdal and Z. Hu, Chem. Eng. Sci., 2019, 209, 115185 CrossRef CAS.
- K. K. Senapati, C. Borgohain, K. C. Sarma and P. Phukan, J. Mol. Catal. A: Chem., 2011, 346, 111–116 CrossRef CAS.
- I. F. Waheed, M. A. Hamad, K. A. Jasim and A. J. Gesquiere, Diamond Relat. Mater., 2023, 133, 109716 CrossRef CAS.
- R. Lei, H. Ni, R. Chen, B. Zhang, W. Zhan and Y. Li, J. Mater. Sci.: Mater. Electron., 2017, 28, 10481–10487 CrossRef CAS.
- H. Gulce, V. Eskizeybek, B. Haspulat, F. Sarı, A. Gülce and A. Avcı, Ind. Eng. Chem. Res., 2013, 52, 10924–10934 CrossRef CAS.
- H.-P. Jing, C.-C. Wang, Y.-W. Zhang, P. Wang and R. Li, RSC Adv., 2014, 4, 54454–54462 RSC.
- S. Kant, S. Kalia and A. Kumar, J. Alloys Compd., 2013, 578, 249–256 CrossRef CAS.
- S. Shahabuddin, N. M. Sarih, F. H. Ismail, M. M. Shahid and N. M. Huang, RSC Adv., 2015, 5, 83857–83867 RSC.
- Y. Vyas, P. Chundawat, P. B. Punjabi and C. Ameta, ChemistrySelect, 2021, 6, 8566–8580 CrossRef CAS.
- R. Banu, N. Salvi, S. Gupta, C. Ameta, R. Ameta and P. B. Punjabi, Arab J Sci Eng, 2022, 47, 365–378 CrossRef CAS.
- S. Nekouei and F. Nekouei, J. Photochem. Photobiol., A, 2018, 364, 262–273 CrossRef CAS.
- D. Matsunami, K. Yamanaka, T. Mizoguchi and K. Kojima, J. Photochem. Photobiol., A, 2019, 369, 106–114 CrossRef CAS.
- P. Jia, H. Tan, K. Liu and W. Gao, Mater. Res. Bull., 2018, 102, 45–50 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*