
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization and biological evaluation of pyrazole-based benzene sulfonamides as inhibitors of human carbonic anhydrase II, IX and XII†
Tajamul Hussain ab,
Saif Ullahc,
Salman Alrokayanb,
Salman Alameryd,
Arif Ahmed Mohammeda,
Syeda Abida Ejaze,
Mubashir Azize and
Jamshed Iqbal*cf
ab,
Saif Ullahc,
Salman Alrokayanb,
Salman Alameryd,
Arif Ahmed Mohammeda,
Syeda Abida Ejaze,
Mubashir Azize and
Jamshed Iqbal*cf
aCentre of Excellence in Biotechnology Research, King Saud University, Riyadh, 11451, Saudi Arabia
bResearch Chair for Biomedical Application of Nanomaterials, Biochemistry Department, College of Science, King Saud University, Riyadh, 11451, Saudi Arabia
cCentre for Advanced Drug Research, COMSATS University Islamabad, Abbottabad Campus, Abbottabad 22060, Pakistan. E-mail: drjamshed@cuiatd.edu.pk; Fax: +92-992-383441; Tel: +92-992-383591-96
dBiochemistry Department, College of Science, King Saud University, Riyadh, 11451, Saudi Arabia
eDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, The Islamia University of Bahawalpur, Bahawalpur 63100, Pakistan
fDepartment of Chemistry, COMSATS University Islamabad, Abbottabad Campus, Abbottabad, 22060, Pakistan
First published on 20th June 2023
Abstract
The aberrant level of the carbonic anhydrase isozymes is linked with various disorders which include glaucoma, epilepsy, altitude sickness and obesity. In the present study, a series of the pyrazole-based benzene sulfonamides derivatives (4a–4l) were designed, synthesized and evaluated as the inhibitors of the three isoforms of human carbonic anhydrases (hCAII, hCAIX and hCAXII). A number of the derivatives were found more active inhibitors than acetazolamide used as a standard against the human hCAII, hCAIX and hCAXII. Among the series, the compound 4k inhibited the hCAII to a submicromolar level presenting the IC50 ± SEM concentration of 0.24 ± 0.18 μM, the inhibitor 4j reduced the activity of the hCAIX to the IC50 ± SEM equals 0.15 ± 0.07 μM, whereas, the molecule 4g blocked the catalytic potential of the isozyme hCAXII with as low as IC50 concentration of 0.12 ± 0.07 μM. In addition, compounds 4e and 4k were screened as the preferential inhibitors of the isoform hCAXII as compared to the hCAIX and hCAXII with half of the maximal concentrations of 0.75 ± 0.13 μM, and 0.24 ± 0.18 μM, respectively. Moreover, the compounds 4k, 4j and 4g were docked inside the active pocket of the crystallographic structure of the isoforms hCAXII, hCAIX and hCAXII, respectively. The docked inhibitors showed the binding interactions with the important amino acid residues such as Leu1198, Thr1199, His1094, and Phe1131 in hCAXII isozyme; residues Val121, Thr200, Pro203, and Gln71 in hCAIX; the amino acids Val119, Leu197, Gln89, and Asn64 in the case of hCAXII. In addition, structural geometries, reactivity descriptors, optimization energy and electronic parameters were calculated to predict the activity of the synthesized compounds.
1. Introduction
Carbonic anhydrases (CAs) are zinc-containing enzymes (metalloenzymes) catalyzing the interconversion of CO2 and H2O to bicarbonates (HCO3−) by following a metal hydroxide involving nucleophilic mechanism.1 The active site of CAs contains metal ions (Zn2+) in a tetrahedral geometric shape with the ligands which are three amino acid residues in addition to a H2O/OH−-coordinating the metal ion. Zinc (Zn II) is the metal ion which is present in all the six genetic families of carbonic anhydrases.2 Mononuclear ion (Zn2+) present in the active site of CAs is linked to the 3 histidine residues i.e. His94, His96, His119 and one H2O/OH− ligand to form a tetrahedral linkage. The metallic center having Zn2+ is kinetically labile. Whereas, the metal (Zn2+) free form of carbonic anhydrase i.e. apo CA is stable. Therefore, apo CA is easy to generate relatively by using DPA (pyridine-2,6-dicarboxylic acid), dialysis. The apo CA have 2 (thermodynamically distinctive) Cu2+ (copper) binding sites, one is Cu A and the other is Cu B. Both sites have difference in their affinities for Cu2+. However, binding site Cu B is lower affinity site named as native metal binding site of carbonic anhydrase. While on the contrary, the coordination and location geometry of the Cu A (high affinity site) is unknown.3Eight distinct groups of carbonic anhydrases are known, of which only α-type is dominant in humans which exist in 16 isozymes,4 the α-type CAs have difference in the preference of these enzymes for Zn2+ (inside the active site) responsible for catalysis and their oligomerization state but most of all the three-dimensional fold of the protein. α-class of carbonic anhydrase is present widely in vertebrates, algae, eubacteria and cytoplasm of green plants. β-class is predominantly present in algae, eubacteria and chloroplasts of mono-as well as dicotyledons and the γ-class is mainly present in some eubacteria and archaea.5 Sixteen different isozymes or carbonic anhydrase-related proteins (CARPs) of α class were identified.6 The large differences in these isozymes are their catalytic efficiency and physiological functions depending on their protein sequence, residues in active site and architectures, their quaternary structure, localization and expression and the efficiency of proton-transfer process in the CO2 hydration reaction.7
According to the tissue and cellular localization CAs are responsible for various physiological functions including homeostasis of CO2 and pH regulation, bone resorption, electrolyte secretion in a various tissues and organs, calcification, biosynthetic reactions such as lipogenesis, gluconeogenesis and ureagenesis and tumor progression. The physiological and pathological roles of human carbonic anhydrase (hCA) have been studied extensively and the role of potential CA inhibitors and activators has been investigated. In particular, the implication of CA IX and CA XII in various types of cancers,8 the overexpression of CA II as well as CA XII in glaucoma as well as the significance of CA inhibitors (CAIs) as antiepileptic, diuretics, altitude sickness, painkillers, antiobesity and as an anti-infective drug.9,10
2. Experimental
2.1 General procedure for the synthesis of intermediates (3a–l)
In the first step, chalcones were synthesized by using the different substituted aromatic aldehydes and ketones. In brief, 20 mM of substituted acetophenone (1) and 20 mM substituted benzaldehyde (2) were added in the 20 mL ethanol. The aqueous solution of NaOH (60%) was added in catalytic amount to the reaction mixture and stirred for 24 hours at room temperature. The formation of the intermediate (3) was identified by Thin Layer Chromatography (TLC). Then the reaction mixture was poured to 7–8 mL ice water and neutralized with HCl solution (10%) which resulted in the formation of precipitates. The precipitates of the intermediates were filtered, washed and crystalized. For further purification, recrystallization in 20% ethanol was performed and intermediate was collected for next step.2.2 Procedure for synthesis of benzenesulphonamides (4a–4l)
The chalcones 500 mg (0.9 mM) was dissolved in acetic acid (20 mL) along with 4-hydrazino benzenesulfonamide hydrochloride 900 mg (4 mM). Then, reaction was refluxed for 72 h. Reaction was followed by performing TLC (thin layer chromatography). When the reaction was completed, reaction mixture was poured on ice of 8–10 mL distilled water. The precipitates formed was filtered and dried. For purification column chromatography was performed (LIN and LI, 2012).![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) , cm−1, neat): 3455 (O–H, stretching), 3330 (NH2, stretching), 1660 (C
, cm−1, neat): 3455 (O–H, stretching), 3330 (NH2, stretching), 1660 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, stretching), 1453 (N–N, stretching), 1260 (C–N, stretching); 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 7.62–7.57 (m, 2H), 7.51 (dd, J = 7.7, 1.6 Hz, 1H), 7.29 (ddd, J = 8.5, 7.2, 1.6 Hz, 1H), 7.14 (s, 4H), 7.02 (d, J = 8.9 Hz, 3H), 6.99 (s, 1H), 6.91 (td, J = 7.5, 1.2 Hz, 1H), 5.56 (dd, J = 12.0, 5.3 Hz, 1H), 4.06 (dd, J = 18.0, 12.0 Hz, 1H), 2.23 (s, 3H). 13C NMR (101 MHz, DMSO-d6) 20.74, 44.33, 61.26, 112.56, 116.55, 116.93, 119.71, 125.84, 127.13, 128.48, 129.56, 131.02, 133.93, 137.12, 138.78, 145.40, 151.75, 156.36. HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H21N3O3S: 407.1012., cm−1, neat): 3475 (O–H, stretching), 2970–3475 (N–H, stretching); 1H NMR, (300 MHz) (DMSO-d6) (ppm) δ (s, 1H) 10.21, (m, 4H) 7.95–7.82, (m, 2H) 7.72–7.60, (m, 3H) 7.61–7.53, (s, 2H) 7.48, (m, 3H) 7.35–7.29, (m, 1H) 7.29–7.23, (m, 2H) 7.15–6.87. 13C NMR (101 MHz, DMSO) (ppm) δ 155.71, 151.26, 143.47, 143.08, 141.90, 132.33, 131.24, 130.15, 129.16, 128.03, 127.36, 125.62, 122.94, 119.90, 117.61, 117.04, 108.71, 40.62, 40.41, 40.20, 40.00, 39.79, 39.58, 39.37. HRMS (ESI-TOF) m/z calcd [M + H]+ for C21H18BrN3O3S: 472.0159., cm−1, neat): 3476 (O–H, stretching), 2920–2931 (N–H, stretching); 1H NMR (300 MHz) (DMSO-d6) (ppm) δ (s, 1H) 10.22, (m, 3H) 7.97–7.79, (m, 2H) 7.71–7.62, (m, 2H) 7.61–7.54, (s, 2H) 7.48, (m, 3H) 7.36–7.29, (ddd, J = 9.9, 6.5, 1.7 Hz, 1H) 7.25, (m, 2H) 7.13–6.88. 13C NMR (101 MHz, DMSO) (ppm) δ 155.71, 151.26, 143.47, 143.08, 141.90, 132.33, 131.23, 130.15, 129.16, 128.03, 127.36, 125.62, 122.94, 119.90, 117.05, 108.70, 40.62, 40.41, 40.20, 40.00, 39.79, 39.58, 39.37; HRMS (ESI-TOF) m/z calcd [M + H]+ for C21H18ClN3O3S: 427.0821., cm−1, neat): 3490 (O–H, stretching), 3012–3019 (N–H, stretching); 1H NMR, (300 MHz) (DMSO-d6) (ppm) δ (s, 1H) 9.78, (m, 3H) 8.13–7.77, (m, 2H) 7.69–7.62, (m, 3H) 7.62–7.52, (s, 2H) 7.48, (d, J = 3.0 Hz, 1H) 7.44, (s, 1H) 7.38, (m, 2H) 7.35–7.28, (m, 2H) 6.94–6.83, (s, 3H) 3.76. 13C NMR (101 MHz, DMSO) (ppm) δ 152.76, 149.72, 143.50, 143.10, 141.90, 132.33, 131.21, 129.16, 127.36, 125.68, 122.93, 117.81, 117.79, 112.17, 56.00, 40.62, 40.41, 40.20, 40.00, 39.79, 39.58, 39.37. HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H20BrN3O4S: 501.0431., cm−1, neat): 3504 (O–H, stretching), 3003–3082 (N–H, stretching); 1H NMR (300 MHz) (DMSO-d6) (ppm) δ (s, 1H) 10.35, (m, 2H) 7.70–7.57, (dd, J = 7.8, 1.7 Hz, 1H) 7.52, (dddd, J = 10.7, 9.1, 7.3, 1.7 Hz, 2H) 7.29, (m, 3H) 7.18–7.03, (m, 6H) 7.01–6.81, (dd, J = 12.1, 5.1 Hz, 1H) 5.72, (m, 1H) 4.17–3.97, (s, 3H) 3.90, (q, J = 7.1 Hz, 1H) 1.17. 13C NMR (101 MHz, DMSO) (ppm) δ 156.71, 156.66, 152.70, 145.66, 131.36, 129.53, 128.92, 128.46, 127.84, 121.15, 120.07, 117.08, 116.72, 112.17, 112.12, 56.21, 40.62, 40.41, 40.20, 39.99, 39.79, 39.58, 39.37: HRMS (ESI-TOF) m/z calcd [M + H]+ for C23H23N3O5S: 453.1302., cm−1, neat): 3540 (O–H, stretching), 3395 (NH2, stretching), 1624 (CN, stretching), 1443 (N–N, stretching), 1247 (C–N, stretching), 661 (C–Br); 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 7.72 (d, J = 2.5 Hz, 1H), 7.58 (d, J = 1.8 Hz, 1H), 7.56 (s, 1H), 7.40 (d, J = 2.5 Hz, 0H), 7.38 (d, J = 2.5 Hz, 0H), 7.12 (s, 3H), 7.01 (s, 1H), 7.00 (s, 1H), 6.92 (s, 0H), 6.90 (s, 0H), 5.56 (dd, J = 12.1, 5.2 Hz, 1H), 4.11–3.89 (m, 2H), 2.22 (s, 2H). 13C NMR (101 MHz, DMSO-d6) 21.04, 44.33, 61.26, 111.65, 112.56, 119.08, 119.78, 126.17, 127.64, 130.09, 130.67, 133.93, 134.12, 137.78, 138.40, 145.09, 150.51, 156.36. HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H20BrN3O3S: 485.0412., cm−1, neat): 3554 (O–H, stretching), 3334 (NH2, stretching), 1624 (CN, stretching), 1399 (N–N, stretching), 1190 (C–N, stretching), 1098 (CO, stretching), 789 (C–Br); 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 7.75 (d, J = 2.6 Hz, 1H), 7.64–7.58 (m, 2H), 7.43 (d, J = 2.5 Hz, 0H), 7.41 (d, J = 2.5 Hz, 1H), 7.07–7.02 (m, 4H), 6.93 (d, J = 8.7 Hz, 1H), 6.85 (d, J = 1.2 Hz, 2H), 6.78 (t, J = 1.2 Hz, 0H), 5.59 (dd, J = 12.1, 5.3 Hz, 1H), 5.24 (dd, J = 11.9, 2.9 Hz, 0H), 4.14–4.00 (m, 1H), 4.00 (s, 0H), 3.75 (d, J = 28.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) 44.41, 55.26, 61.8, 110.65, 111.91, 112.26, 112.78, 117.78, 118.17, 119.64, 127.34, 130.09, 130.47, 133.16, 133.75, 143.09, 145.40, 149.92, 155.47, 160.76. HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H20BrN3O4S: 501.0412., cm−1, neat): 3540 (O–H, stretching), 1621 (CN, stretching), 1434 (N–N, stretching), 1260 (C–N, stretching), 765 (C–Br); 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.12 (d, J = 2.5 Hz, 1H), 7.62 (dd, J = 20.7, 8.8 Hz, 4H), 7.39 (td, J = 8.7, 2.5 Hz, 2H), 7.20 (d, J = 8.5 Hz, 2H), 7.08 (s, 2H), 7.03 (s, 2H), 6.90 (s, OH), 5.62 (dd, J = 12.2, 5.1 Hz, 1H), 4.04 (dd, J = 18.2, 12.2 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) 43.87, 60.72, 110.30, 118.81, 113.23, 118.26, 118.96, 120.35, 121.14, 123.03, 126.98, 128.76, 130.12, 131.61, 132.09, 133.37, 149.33; HRMS (ESI-TOF) m/z calcd [M + H]+ for C21H17Br2N3O3S: 548.9412., cm−1, neat): 3479.3 (OH), 2923.9–2991.4 (>NH stretch). 1H NMR (300 MHz) (DMSO-d6) (ppm) δ 11.03 (s, 1H), 7.72–7.60 (m, 3H), 7.50 (d, J = 2.5 Hz, 1H), 7.27 (t, J = 7.9 Hz, 1H), 7.16–7.02 (m, 4H), 6.96–6.78 (m, 3H), 5.68 (dd, J = 12.2, 5.5 Hz, 1H), 4.10 (dd, J = 18.1, 12.3 Hz, 1H), 3.73 (s, 3H), 3.43 (dd, J = 18.1, 5.5 Hz, 1H); 13C NMR (101 MHz, DMSO) (ppm) δ 160.17, 151.23, 150.92, 142.97, 127.77, 127.05, 123.96, 119.42, 112.99, 55.54, 40.62, 40.41, 40.21, 40.00, 39.79, 39.58, 39.37; HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H19Cl2N3O4S: 492.0551., cm−1, neat): 3510, (O–H stretch) 3320, (NH2 stretch) 1599, (CN stretch) 1430, (N–N stretch) 1225, (C–N stretch) 1082, (C–F stretch):1H NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 7.60 (d, J = 8.2 Hz, 3H), 7.15 (s, 4H), 7.08–6.97 (m, 4H), 6.92–6.67 (m, 2H), 5.55 (dd, J = 12.0, 5.3 Hz; 1H), 4.11–4.04 (m, 1H), 4.01 (s, 0H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) 20.74, 44.33, 60.05, 103.36, 112.12, 120.85, 126.95, 128.45, 130.31, 132.12, 133.72, 140.83, 145.90, 150.89, 158.60, 163.45, 165.08; HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H20FN3O3S: 425.1201., cm−1, neat): 3550 (O–H, stretching), 3330 (NH2, stretching), 1630 (CN, stretching), 1449 (N–N, stretching), 1230 (C–N, stretching), 1087 (C–F, stretching), 500–1000 (C–Br); 1H NMR (400 MHz, DMSO-d6) δ 10.72 (s, 1H), 7.62 (s, 1H), 7.57 (d, J = 8.2 Hz, 1H), 7.27 (t, J = 7.8 Hz, 1H), 7.10 (d, J = 8.3 Hz, 1H), 7.05 (s, 2H), 6.94 (d, J = 8.3 Hz, 3H), 6.87–6.80 (m, 2H), 6.80–6.48 (m, 1H), 5.69 (dd, J = 12.1, 5.1 Hz, 1H), 4.05 (dd, J = 18.0, 12.1 Hz, 1H), 3.89 (s, 3H), 3.23 (dd, J = 17.9, 5.1 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) 43.33, 55.82, 56.55, 103.36, 106.90, 114.45, 116.12, 120.85, 126.95, 127.13, 128.48, 129.56, 130.13, 130.31, 130.45, 133.43, 145.12 156.12, 157.72, 162.18; HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H20FN3O4S: 441.1220.N, stretching), 1430, (N–N stretching), 1225 (C–N, stretching), 1082 (C–F, stretching); 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 7.61 (d, J = 7.9 Hz, 3H), 7.55 (d, J = 8.2 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 13.1 Hz, 3H), 6.99 (s, 1H), 6.88–6.69 (m, 2H), 5.61 (dd, J = 12.0, 5.2 Hz, 1H), 4.07 (dd, J = 17.8, 11.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) 44.33, 60.05, 103.36, 106.90, 112.12, 112.12, 120.85, 126.95, 128.48, 130.31, 130.45, 132.12, 133.72, 140.83, 145.90, 150.89, 158.60; HRMS (ESI-TOF) m/z calcd [M + H]+ for C21H17BrFN3O3S: 489.0230.
N, stretching), 1453 (N–N, stretching), 1260 (C–N, stretching); 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 7.62–7.57 (m, 2H), 7.51 (dd, J = 7.7, 1.6 Hz, 1H), 7.29 (ddd, J = 8.5, 7.2, 1.6 Hz, 1H), 7.14 (s, 4H), 7.02 (d, J = 8.9 Hz, 3H), 6.99 (s, 1H), 6.91 (td, J = 7.5, 1.2 Hz, 1H), 5.56 (dd, J = 12.0, 5.3 Hz, 1H), 4.06 (dd, J = 18.0, 12.0 Hz, 1H), 2.23 (s, 3H). 13C NMR (101 MHz, DMSO-d6) 20.74, 44.33, 61.26, 112.56, 116.55, 116.93, 119.71, 125.84, 127.13, 128.48, 129.56, 131.02, 133.93, 137.12, 138.78, 145.40, 151.75, 156.36. HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H21N3O3S: 407.1012., cm−1, neat): 3475 (O–H, stretching), 2970–3475 (N–H, stretching); 1H NMR, (300 MHz) (DMSO-d6) (ppm) δ (s, 1H) 10.21, (m, 4H) 7.95–7.82, (m, 2H) 7.72–7.60, (m, 3H) 7.61–7.53, (s, 2H) 7.48, (m, 3H) 7.35–7.29, (m, 1H) 7.29–7.23, (m, 2H) 7.15–6.87. 13C NMR (101 MHz, DMSO) (ppm) δ 155.71, 151.26, 143.47, 143.08, 141.90, 132.33, 131.24, 130.15, 129.16, 128.03, 127.36, 125.62, 122.94, 119.90, 117.61, 117.04, 108.71, 40.62, 40.41, 40.20, 40.00, 39.79, 39.58, 39.37. HRMS (ESI-TOF) m/z calcd [M + H]+ for C21H18BrN3O3S: 472.0159., cm−1, neat): 3476 (O–H, stretching), 2920–2931 (N–H, stretching); 1H NMR (300 MHz) (DMSO-d6) (ppm) δ (s, 1H) 10.22, (m, 3H) 7.97–7.79, (m, 2H) 7.71–7.62, (m, 2H) 7.61–7.54, (s, 2H) 7.48, (m, 3H) 7.36–7.29, (ddd, J = 9.9, 6.5, 1.7 Hz, 1H) 7.25, (m, 2H) 7.13–6.88. 13C NMR (101 MHz, DMSO) (ppm) δ 155.71, 151.26, 143.47, 143.08, 141.90, 132.33, 131.23, 130.15, 129.16, 128.03, 127.36, 125.62, 122.94, 119.90, 117.05, 108.70, 40.62, 40.41, 40.20, 40.00, 39.79, 39.58, 39.37; HRMS (ESI-TOF) m/z calcd [M + H]+ for C21H18ClN3O3S: 427.0821., cm−1, neat): 3490 (O–H, stretching), 3012–3019 (N–H, stretching); 1H NMR, (300 MHz) (DMSO-d6) (ppm) δ (s, 1H) 9.78, (m, 3H) 8.13–7.77, (m, 2H) 7.69–7.62, (m, 3H) 7.62–7.52, (s, 2H) 7.48, (d, J = 3.0 Hz, 1H) 7.44, (s, 1H) 7.38, (m, 2H) 7.35–7.28, (m, 2H) 6.94–6.83, (s, 3H) 3.76. 13C NMR (101 MHz, DMSO) (ppm) δ 152.76, 149.72, 143.50, 143.10, 141.90, 132.33, 131.21, 129.16, 127.36, 125.68, 122.93, 117.81, 117.79, 112.17, 56.00, 40.62, 40.41, 40.20, 40.00, 39.79, 39.58, 39.37. HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H20BrN3O4S: 501.0431., cm−1, neat): 3504 (O–H, stretching), 3003–3082 (N–H, stretching); 1H NMR (300 MHz) (DMSO-d6) (ppm) δ (s, 1H) 10.35, (m, 2H) 7.70–7.57, (dd, J = 7.8, 1.7 Hz, 1H) 7.52, (dddd, J = 10.7, 9.1, 7.3, 1.7 Hz, 2H) 7.29, (m, 3H) 7.18–7.03, (m, 6H) 7.01–6.81, (dd, J = 12.1, 5.1 Hz, 1H) 5.72, (m, 1H) 4.17–3.97, (s, 3H) 3.90, (q, J = 7.1 Hz, 1H) 1.17. 13C NMR (101 MHz, DMSO) (ppm) δ 156.71, 156.66, 152.70, 145.66, 131.36, 129.53, 128.92, 128.46, 127.84, 121.15, 120.07, 117.08, 116.72, 112.17, 112.12, 56.21, 40.62, 40.41, 40.20, 39.99, 39.79, 39.58, 39.37: HRMS (ESI-TOF) m/z calcd [M + H]+ for C23H23N3O5S: 453.1302., cm−1, neat): 3540 (O–H, stretching), 3395 (NH2, stretching), 1624 (CN, stretching), 1443 (N–N, stretching), 1247 (C–N, stretching), 661 (C–Br); 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 7.72 (d, J = 2.5 Hz, 1H), 7.58 (d, J = 1.8 Hz, 1H), 7.56 (s, 1H), 7.40 (d, J = 2.5 Hz, 0H), 7.38 (d, J = 2.5 Hz, 0H), 7.12 (s, 3H), 7.01 (s, 1H), 7.00 (s, 1H), 6.92 (s, 0H), 6.90 (s, 0H), 5.56 (dd, J = 12.1, 5.2 Hz, 1H), 4.11–3.89 (m, 2H), 2.22 (s, 2H). 13C NMR (101 MHz, DMSO-d6) 21.04, 44.33, 61.26, 111.65, 112.56, 119.08, 119.78, 126.17, 127.64, 130.09, 130.67, 133.93, 134.12, 137.78, 138.40, 145.09, 150.51, 156.36. HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H20BrN3O3S: 485.0412., cm−1, neat): 3554 (O–H, stretching), 3334 (NH2, stretching), 1624 (CN, stretching), 1399 (N–N, stretching), 1190 (C–N, stretching), 1098 (CO, stretching), 789 (C–Br); 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 7.75 (d, J = 2.6 Hz, 1H), 7.64–7.58 (m, 2H), 7.43 (d, J = 2.5 Hz, 0H), 7.41 (d, J = 2.5 Hz, 1H), 7.07–7.02 (m, 4H), 6.93 (d, J = 8.7 Hz, 1H), 6.85 (d, J = 1.2 Hz, 2H), 6.78 (t, J = 1.2 Hz, 0H), 5.59 (dd, J = 12.1, 5.3 Hz, 1H), 5.24 (dd, J = 11.9, 2.9 Hz, 0H), 4.14–4.00 (m, 1H), 4.00 (s, 0H), 3.75 (d, J = 28.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) 44.41, 55.26, 61.8, 110.65, 111.91, 112.26, 112.78, 117.78, 118.17, 119.64, 127.34, 130.09, 130.47, 133.16, 133.75, 143.09, 145.40, 149.92, 155.47, 160.76. HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H20BrN3O4S: 501.0412., cm−1, neat): 3540 (O–H, stretching), 1621 (CN, stretching), 1434 (N–N, stretching), 1260 (C–N, stretching), 765 (C–Br); 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.12 (d, J = 2.5 Hz, 1H), 7.62 (dd, J = 20.7, 8.8 Hz, 4H), 7.39 (td, J = 8.7, 2.5 Hz, 2H), 7.20 (d, J = 8.5 Hz, 2H), 7.08 (s, 2H), 7.03 (s, 2H), 6.90 (s, OH), 5.62 (dd, J = 12.2, 5.1 Hz, 1H), 4.04 (dd, J = 18.2, 12.2 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) 43.87, 60.72, 110.30, 118.81, 113.23, 118.26, 118.96, 120.35, 121.14, 123.03, 126.98, 128.76, 130.12, 131.61, 132.09, 133.37, 149.33; HRMS (ESI-TOF) m/z calcd [M + H]+ for C21H17Br2N3O3S: 548.9412., cm−1, neat): 3479.3 (OH), 2923.9–2991.4 (>NH stretch). 1H NMR (300 MHz) (DMSO-d6) (ppm) δ 11.03 (s, 1H), 7.72–7.60 (m, 3H), 7.50 (d, J = 2.5 Hz, 1H), 7.27 (t, J = 7.9 Hz, 1H), 7.16–7.02 (m, 4H), 6.96–6.78 (m, 3H), 5.68 (dd, J = 12.2, 5.5 Hz, 1H), 4.10 (dd, J = 18.1, 12.3 Hz, 1H), 3.73 (s, 3H), 3.43 (dd, J = 18.1, 5.5 Hz, 1H); 13C NMR (101 MHz, DMSO) (ppm) δ 160.17, 151.23, 150.92, 142.97, 127.77, 127.05, 123.96, 119.42, 112.99, 55.54, 40.62, 40.41, 40.21, 40.00, 39.79, 39.58, 39.37; HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H19Cl2N3O4S: 492.0551., cm−1, neat): 3510, (O–H stretch) 3320, (NH2 stretch) 1599, (CN stretch) 1430, (N–N stretch) 1225, (C–N stretch) 1082, (C–F stretch):1H NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 7.60 (d, J = 8.2 Hz, 3H), 7.15 (s, 4H), 7.08–6.97 (m, 4H), 6.92–6.67 (m, 2H), 5.55 (dd, J = 12.0, 5.3 Hz; 1H), 4.11–4.04 (m, 1H), 4.01 (s, 0H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) 20.74, 44.33, 60.05, 103.36, 112.12, 120.85, 126.95, 128.45, 130.31, 132.12, 133.72, 140.83, 145.90, 150.89, 158.60, 163.45, 165.08; HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H20FN3O3S: 425.1201., cm−1, neat): 3550 (O–H, stretching), 3330 (NH2, stretching), 1630 (CN, stretching), 1449 (N–N, stretching), 1230 (C–N, stretching), 1087 (C–F, stretching), 500–1000 (C–Br); 1H NMR (400 MHz, DMSO-d6) δ 10.72 (s, 1H), 7.62 (s, 1H), 7.57 (d, J = 8.2 Hz, 1H), 7.27 (t, J = 7.8 Hz, 1H), 7.10 (d, J = 8.3 Hz, 1H), 7.05 (s, 2H), 6.94 (d, J = 8.3 Hz, 3H), 6.87–6.80 (m, 2H), 6.80–6.48 (m, 1H), 5.69 (dd, J = 12.1, 5.1 Hz, 1H), 4.05 (dd, J = 18.0, 12.1 Hz, 1H), 3.89 (s, 3H), 3.23 (dd, J = 17.9, 5.1 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) 43.33, 55.82, 56.55, 103.36, 106.90, 114.45, 116.12, 120.85, 126.95, 127.13, 128.48, 129.56, 130.13, 130.31, 130.45, 133.43, 145.12 156.12, 157.72, 162.18; HRMS (ESI-TOF) m/z calcd [M + H]+ for C22H20FN3O4S: 441.1220.N, stretching), 1430, (N–N stretching), 1225 (C–N, stretching), 1082 (C–F, stretching); 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 7.61 (d, J = 7.9 Hz, 3H), 7.55 (d, J = 8.2 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 13.1 Hz, 3H), 6.99 (s, 1H), 6.88–6.69 (m, 2H), 5.61 (dd, J = 12.0, 5.2 Hz, 1H), 4.07 (dd, J = 17.8, 11.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) 44.33, 60.05, 103.36, 106.90, 112.12, 112.12, 120.85, 126.95, 128.48, 130.31, 130.45, 132.12, 133.72, 140.83, 145.90, 150.89, 158.60; HRMS (ESI-TOF) m/z calcd [M + H]+ for C21H17BrFN3O3S: 489.0230.2.3 Transfection of transformed plasmids in HEK 293T cells
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well). 2 mixtures were prepared in this protocol i.e. mixture A and mixture B. Mixture A contained 25 μL of OptiMEM® media and 1 μL of lipofectamine 3000 while mixture B was prepared by adding 25 μL of OptiMEM® media, 1 μL of lipofectamine 3000 and 5 μL of plasmid (100 ng μL−1, plasmids of human carbonic anhydrases hCA II, hCA IX and hCA XII were supplied from Sino Biological (Beijing, China)). Then well A and B was mixed and incubated for 30 min at room temperature. Growth media was removed from cells in each well and 50 μL of that mixture A and B was added. Subsequently, 450 μL of OptiMEM® media was added in each well and incubated in CO2 incubator at 37 °C for 6 h. After 6 hours supernatant was aspirated and fresh prepared culture media (DMEM) was added. Then, cells were incubated for 24 hours in CO2 incubator with 5% CO2.11000 rpm and 4 °C. Supernatant was then transferred to ice chilled clean tubes for performing functional assays and debris was discarded.
000 cells per well). 2 mixtures were prepared in this protocol i.e. mixture A and mixture B. Mixture A contained 25 μL of OptiMEM® media and 1 μL of lipofectamine 3000 while mixture B was prepared by adding 25 μL of OptiMEM® media, 1 μL of lipofectamine 3000 and 5 μL of plasmid (100 ng μL−1, plasmids of human carbonic anhydrases hCA II, hCA IX and hCA XII were supplied from Sino Biological (Beijing, China)). Then well A and B was mixed and incubated for 30 min at room temperature. Growth media was removed from cells in each well and 50 μL of that mixture A and B was added. Subsequently, 450 μL of OptiMEM® media was added in each well and incubated in CO2 incubator at 37 °C for 6 h. After 6 hours supernatant was aspirated and fresh prepared culture media (DMEM) was added. Then, cells were incubated for 24 hours in CO2 incubator with 5% CO2.11000 rpm and 4 °C. Supernatant was then transferred to ice chilled clean tubes for performing functional assays and debris was discarded.2.4 Carbonic anhydrase inhibition assay
The inhibitory activity of synthesized compounds against carbonic anhydrases was determined spectrophotometrically, as previously described.14 The activity assay was performed in tris-base buffer containing 50 mM tris-base and 0.1 mM of ZnCl2. The pH was adjusted to 7.4. The assay was started by the addition of buffer, 1 mM solution of synthesized compound and enzyme solution was added per well. The reaction mixture was then incubated at 25 °C for 10 min and substrate prepared in 4% acetonitrile was added in the reaction mixture. Then, the reaction was incubated at 25 °C for 30 min. Finally, absorbance at 405 nm was measured by using the microplate reader. The 10% DMSO was used as a negative control in the assay while acetazolamide as a positive control for the comparison of inhibition.15,162.5 Density functional theory studies
In this research, Density Functional Theory (DFT) calculations were performed using Gaussian 16 software package with B3LYP functional and 6-31G* basis set for optimization and frequency analysis of the target molecule. The basis set was chosen based on its widespread use and ability to provide a good balance between accuracy and computational cost. The optimization was run until the maximum force on any atom was less than 0.0001 au and the maximum displacement was less than 0.001 Å. The HOMO/LUMO energies, optimization energy, and vibrational frequencies were analyzed to gain insights into the electronic structure and vibrational properties of the target molecule. The obtained results were significant as they helped to understand the system's reactivity, stability, and vibrational modes, which are important for its potential applications. Furthermore, the visualization of the molecular structures and vibrational modes using Gauss View 6 software facilitated the interpretation of the results. Overall, this study demonstrates the importance of DFT calculations with appropriate basis sets and functional for the accurate and reliable prediction of the electronic and vibrational properties of molecular systems.2.6 Molecular docking studies
Docking studies have been performed on the most potent inhibitors 4k, 4j and 4g of the series using PDB IDs 3V7X 1 (hCAII), 5FL4 (hCAIX) and 5MSA (CAXII).17 Molecular docking was performed via Moe and SeeSAR. The ligands were prepared by using the software Molecular Docking Environment (MOE) and energy was minimized by setting the default parameters.18 The protein for each isozyme was prepared, the binding pocket selection, energy minimization and structure preparation and docking was performed by using the automatic software SeeSAR (BioSolveIT GmbH, Germany).19 For the reference ligand, the co-crystallized molecule was selected.20 The most potent inhibitors of the were docked by using the SeeSAR and for each inhibitor 100 poses were generated. The top reliable poses were selected for the HYDE assessment and 3D images were created by using the Discovery Studio Visualizer DS.212.7 Molecular dynamics simulation
In this study, molecular dynamics (MD) simulations were performed on the target protein using Desmond software package (version 2020.2) to investigate its structural dynamics and stability. The crystal structure of the protein was obtained from the Protein Data Bank (PDB) and prepared for the simulation using the Maestro graphical interface. The protein was solvated in an orthorhombic box of TIP3P water molecules and counter ions were added to neutralize the system. The OPLS3 force field was used to model the protein and the solvent. The simulation was performed with an NPT ensemble, using a temperature of 300 K and a pressure of 1 atm, with periodic boundary conditions. The simulation time was 100 ns, with a time step of 2 fs. The trajectory was analyzed using the Desmond Analysis module and PyMOL software. The results of this study provide insights into the conformational changes and stability of the protein and have potential implications for its function and drug design.3. Results and discussion
3.1 Chemistry
The pyrazole-based benzene-sulfonamides (4a–4l) were synthesized as shown in Scheme 1.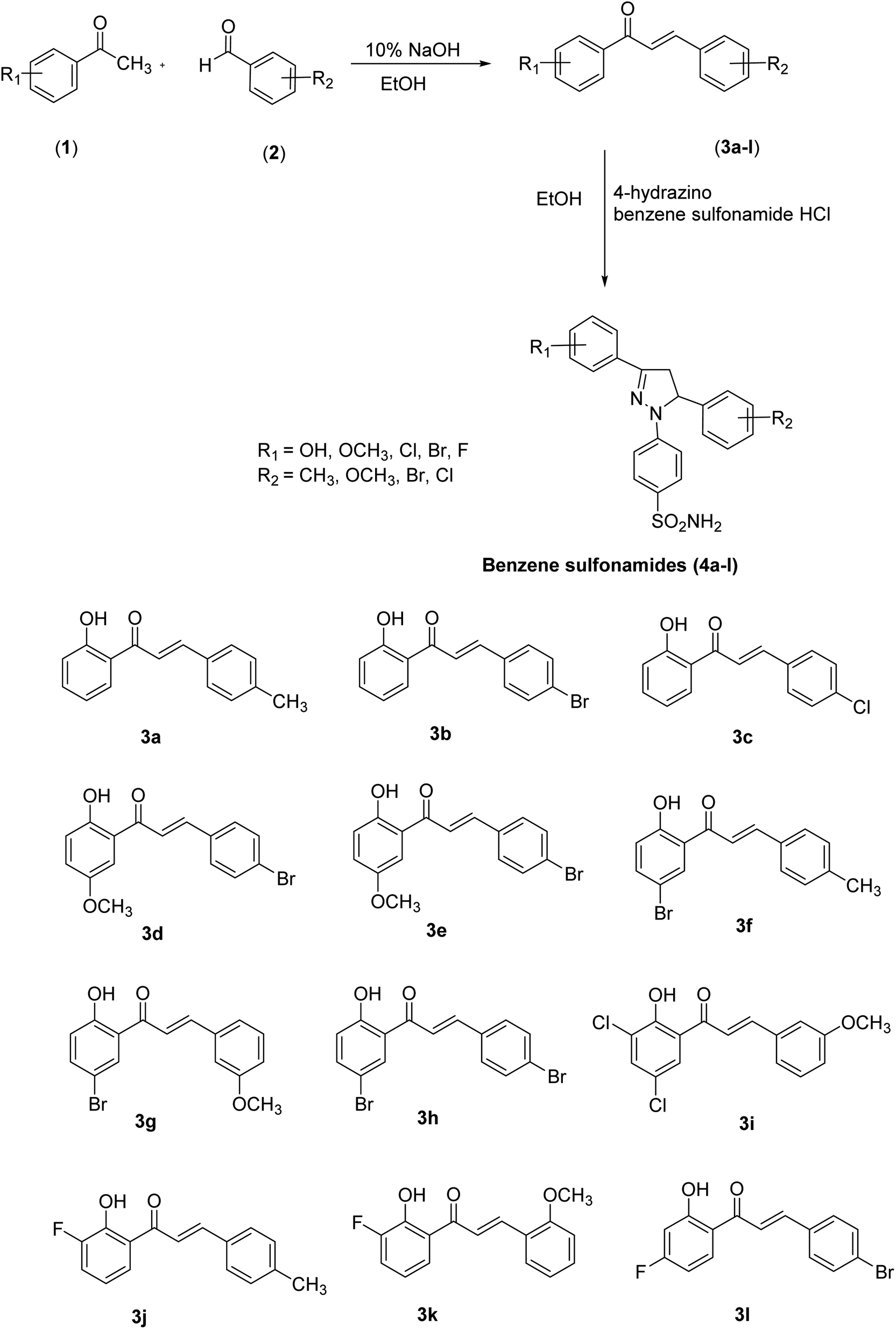 | ||
| Scheme 1 General scheme for the synthesis of intermediates (3a–3l) and pyrazole-based benzene-sulfonamides (4a–4l). | ||
3.2 Carbonic anhydrase inhibition studies
The synthetic compounds and the standard carbonic anhydrase inhibitor (acetazolamide) were tested for the carbonic anhydrase inhibition properties against the recombinant human carbonic anhydrase isozymes (hCAII, hCAIX and hCAXII) by using the reported method. The results are presented in the Table 1.| Codes | Structure | hCAII | hCAIX | hCAXII |
|---|---|---|---|---|
| IC50 ± SEMa (μM) or % inhibition ± SDb | ||||
| a IC50 ± SEM values of the compounds exhibiting over 50% enzyme inhibition.b Percentage inhibition ± SEM. The results are presented as the mean of triplicate assays. SEM = standard error mean; SD = standard deviation. | ||||
| 4a | 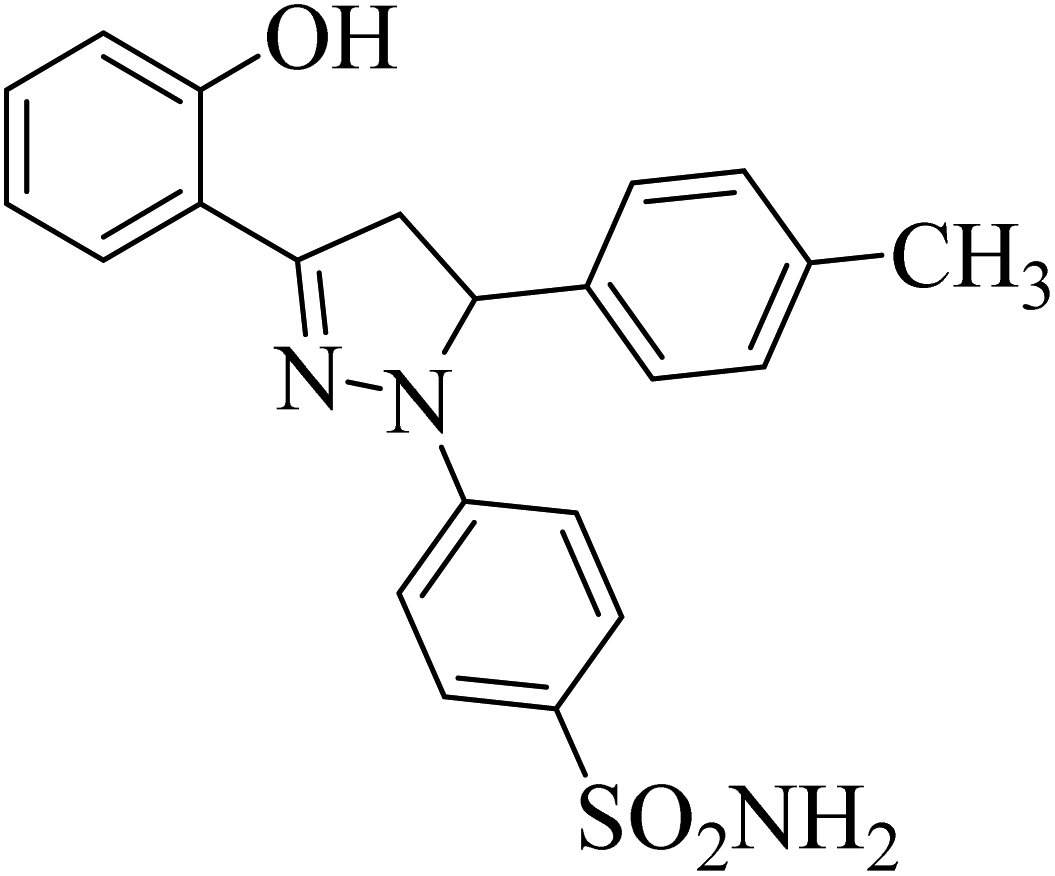 |
39.67 ± 1.4b | 27 ± 3.2b | 25 ± 4.1b |
| 4b | 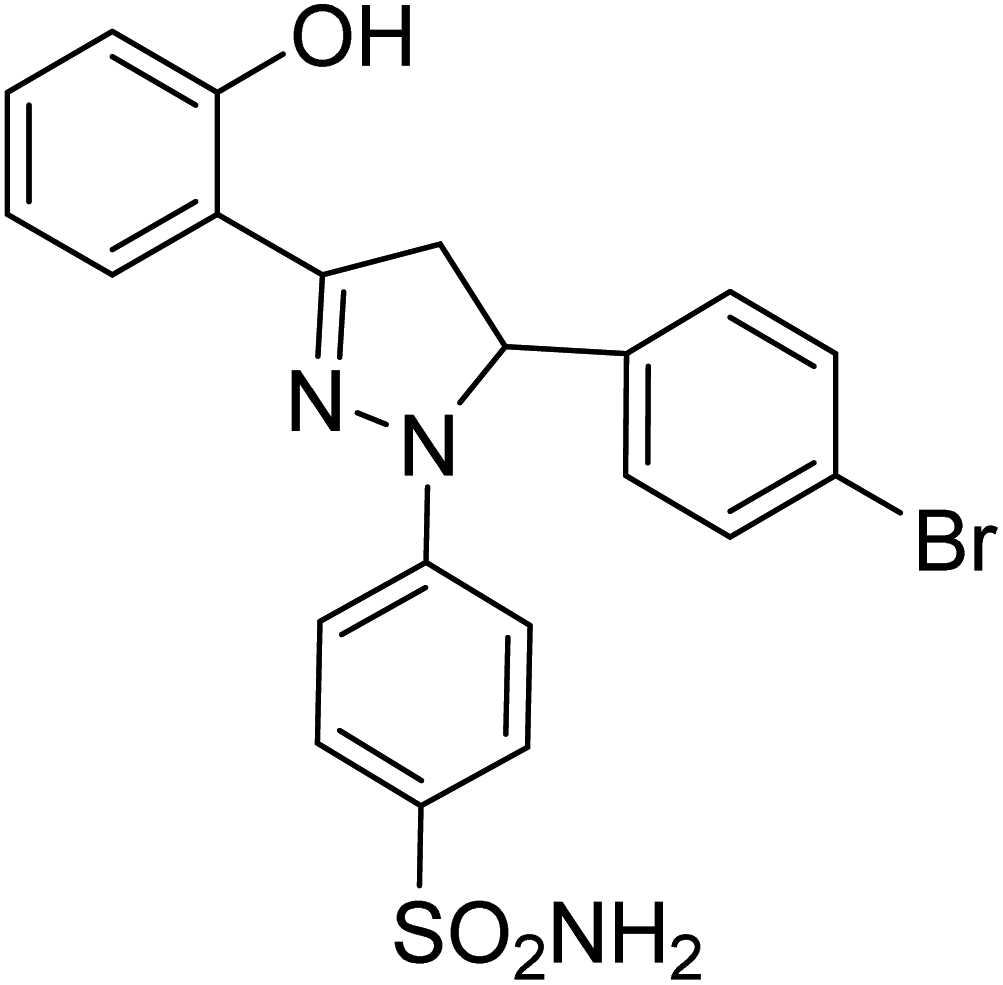 |
1.80 ± 0.22a | 40 ± 2.2b | 8.36 ± 3.88a |
| 4c | 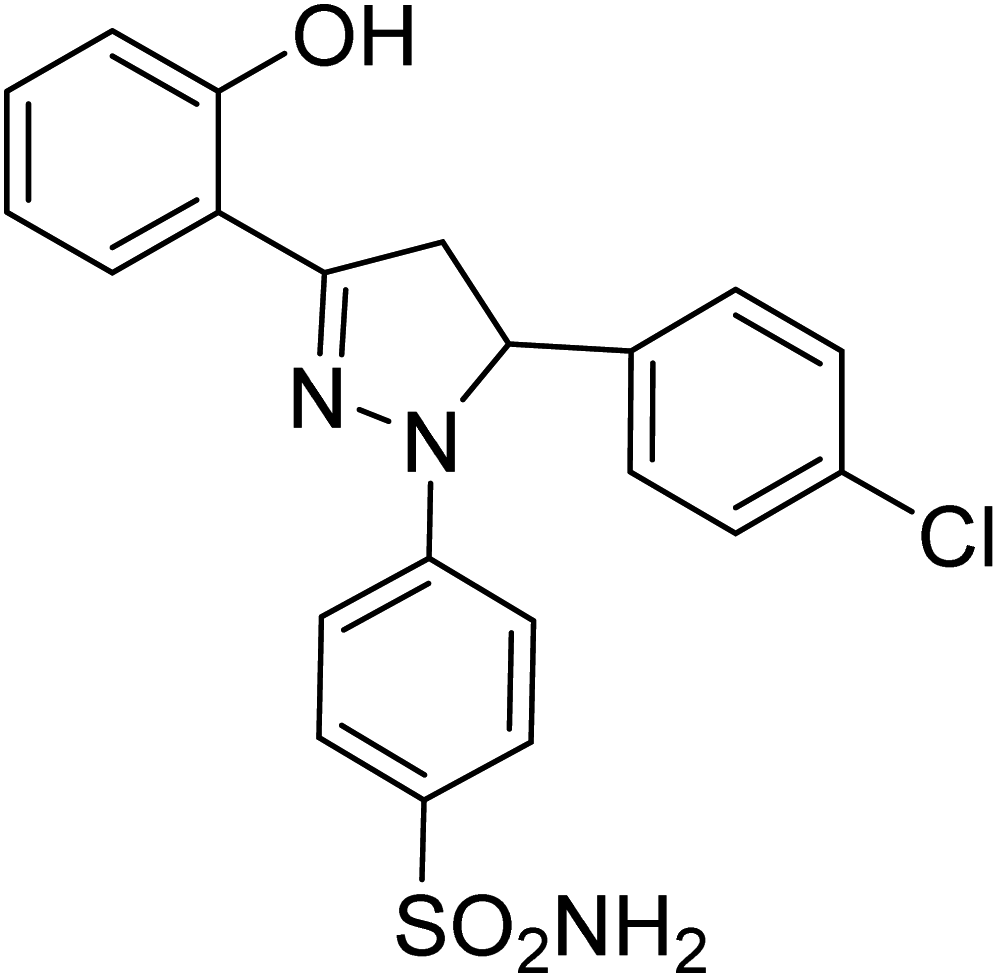 |
1.20 ± 0.40a | 7.08 ± 0.40a | 2.87 ± 0.02a |
| 4d | 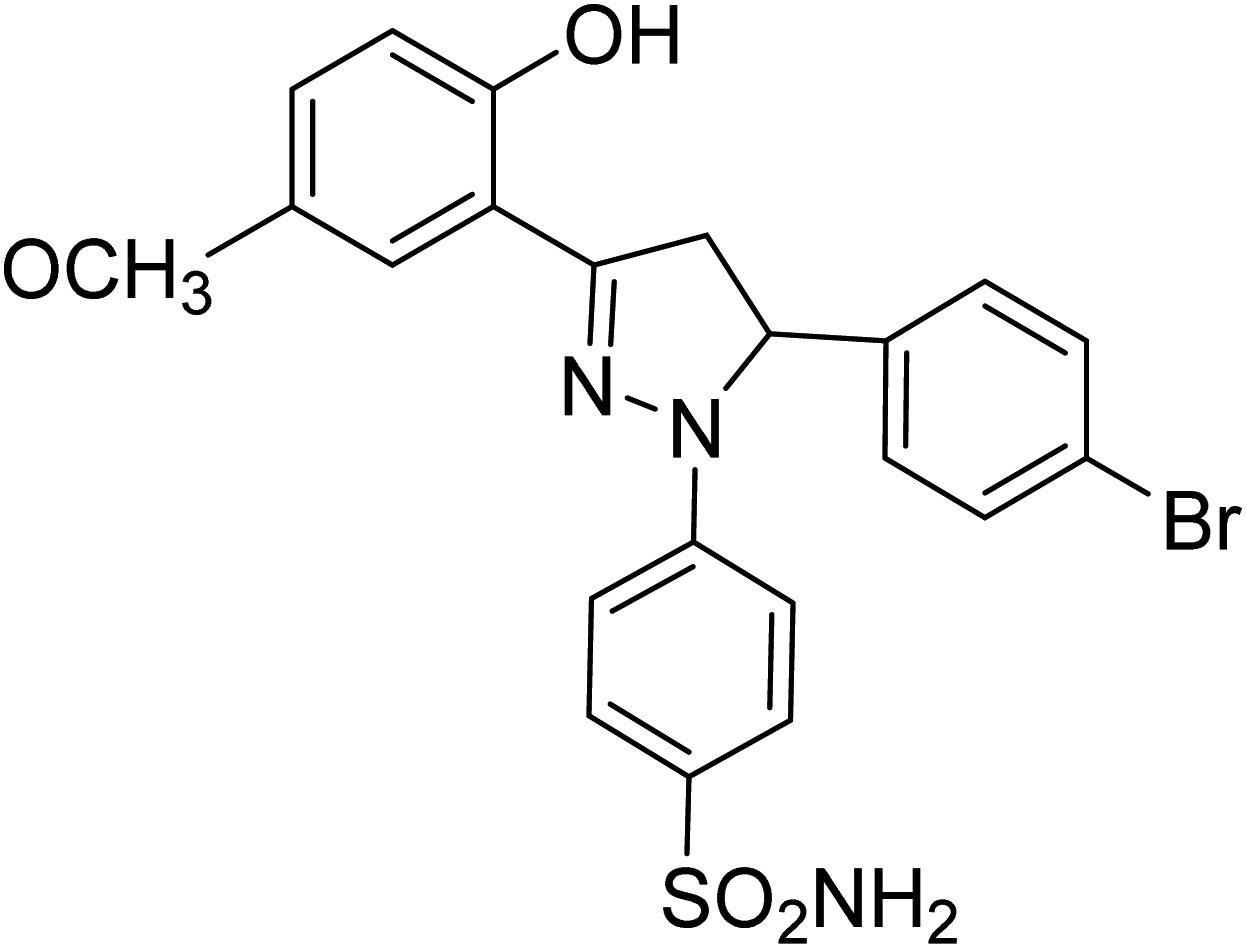 |
1.31 ± 0.11a | 23 ± 4.3b | 4.26 ± 1.5a |
| 4e | 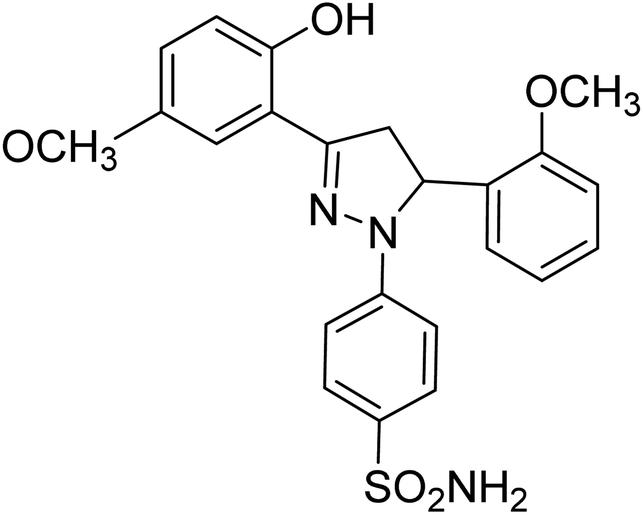 |
0.75 ± 0.13a | 43 ± 1.3b | 34 ± 2.4b |
| 4f | 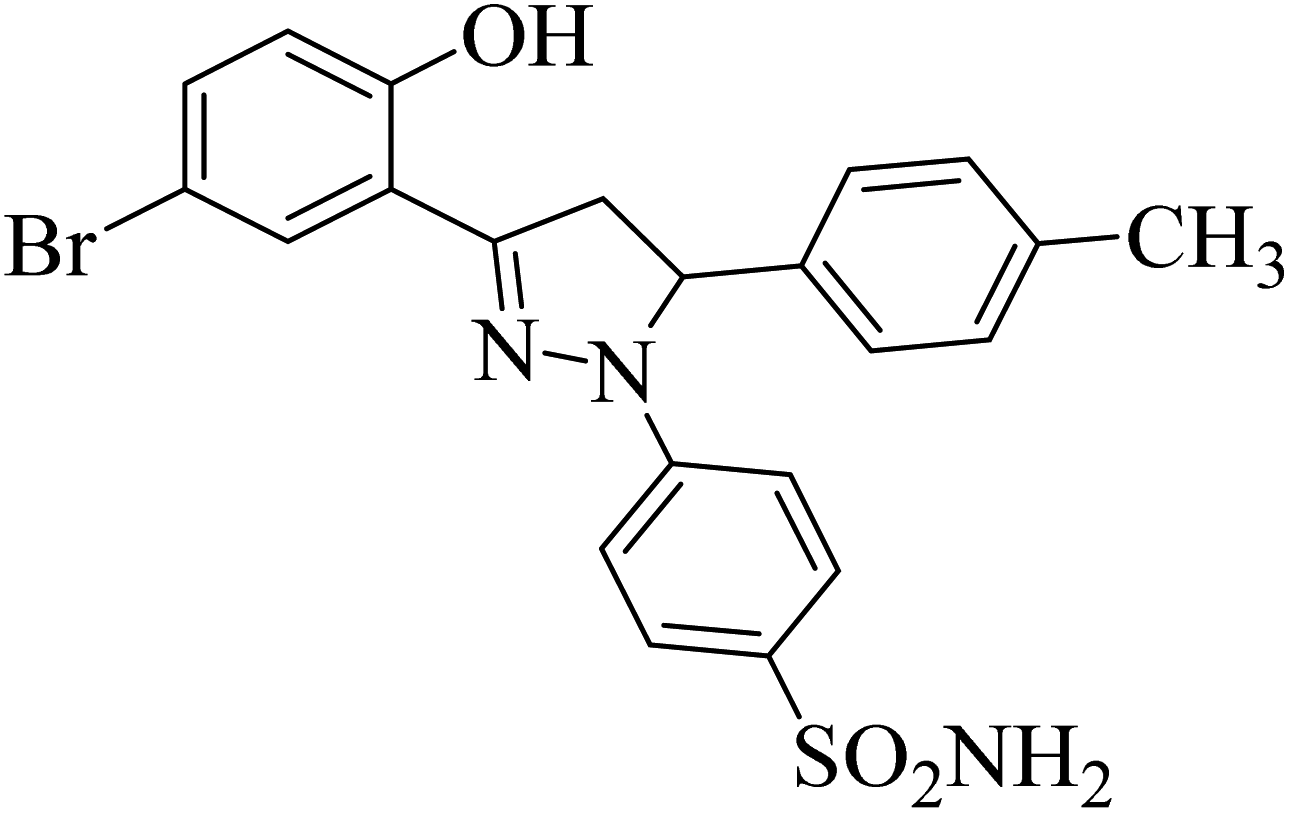 |
31 ± 3.3b | 0.24 ± 0.08a | 6.97 ± 0.14a |
| 4g | 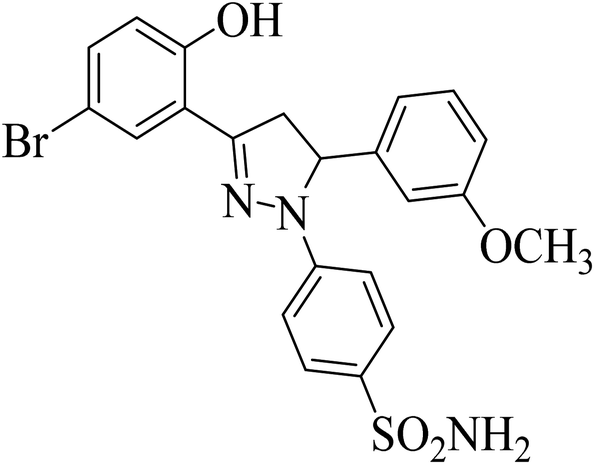 |
8.26 ± 0.99a | 5.28 ± 0.69a | 0.12 ± 0.07a |
| 4h | 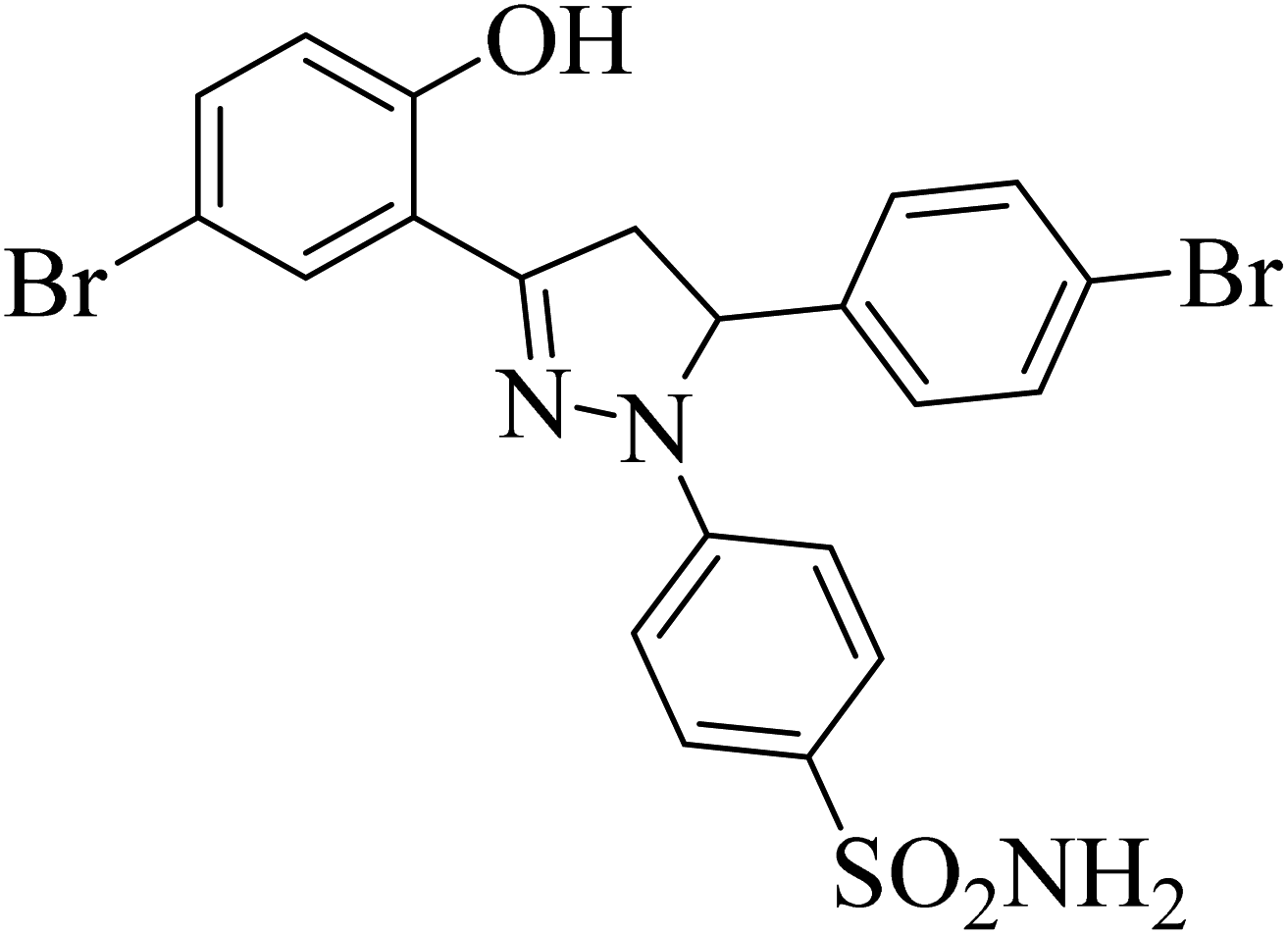 |
17.24 ± 1.77a | 0.26 ± 0.04a | 0.38 ± 0.10a |
| 4i | 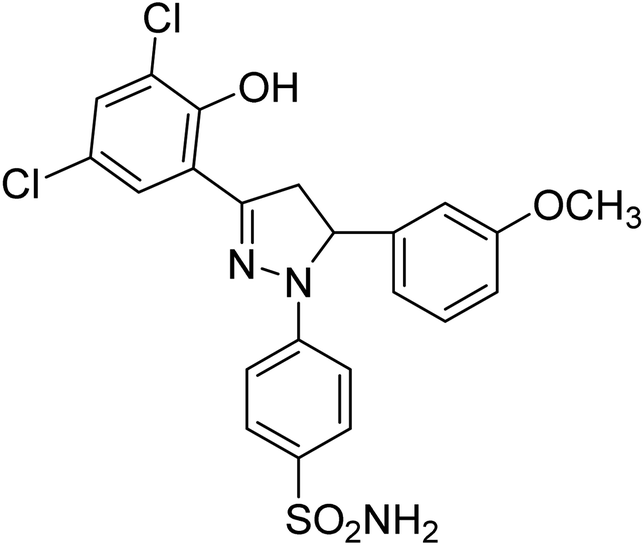 |
8.15 ± 0.19a | 4.51 ± 0.22a | 1.60 ± 0.01a |
| 4j | 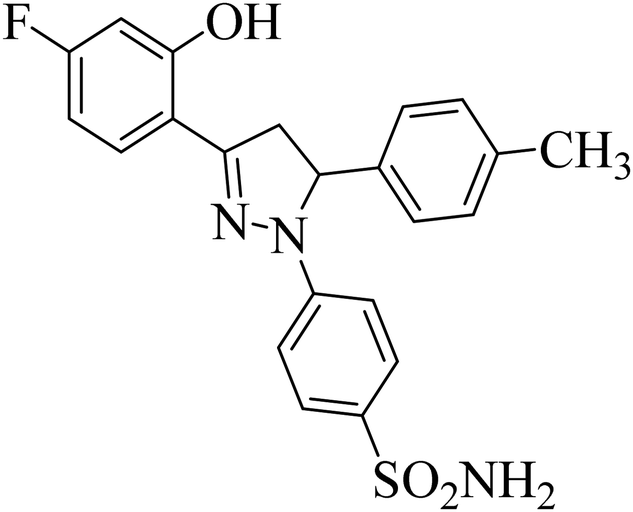 |
0.39 ± 0.05a | 0.15 ± 0.07a | 0.28 ± 0.05a |
| 4k | 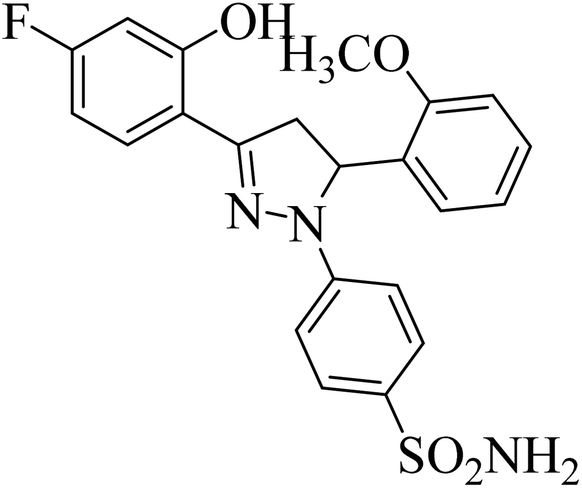 |
0.24 ± 0.18a | 27 ± 2.4b | 1.0 ± 0.09a |
| 4l | 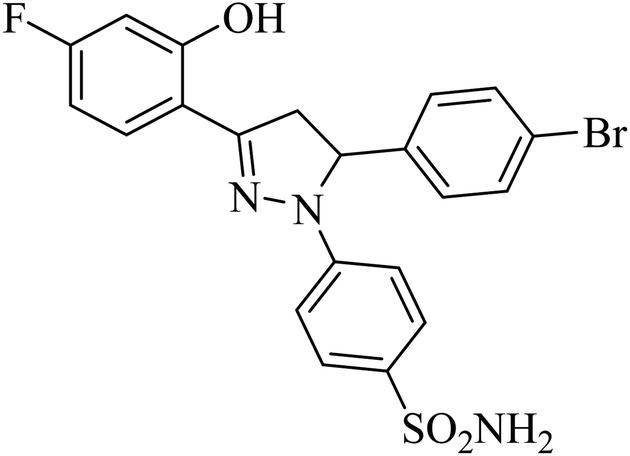 |
26 ± 3.4b | 25 ± 2.5b | 37 ± 1.3b |
| Acetazolamide | 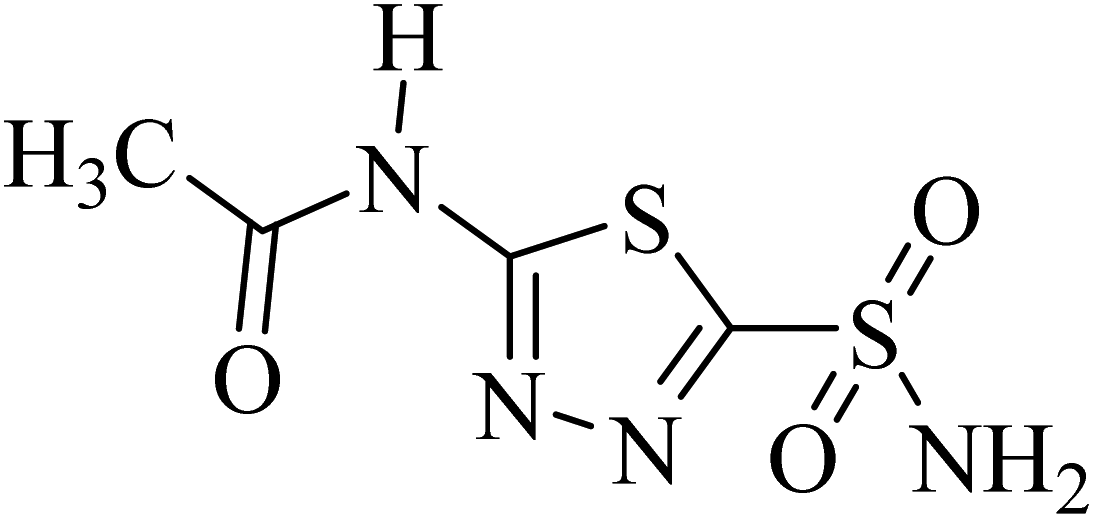 |
1.19 ± 0.04a | 1.08 ± 0.02a | 1.55 ± 0.03a |
3.3 Structure–activity relationship (SAR)
The effect of different substituents towards the inhibition of the activity of the human carbonic anhydrases (hCAII, hCAIX, hCAXII) at the specific positions of the pyrazole based benzene sulfonamides can be explored by studying the structure–activity relationship (SAR). The SAR study was focused on various scaffolds attached to the phenyl rings at positions 3 and 5 of the pyrazole ring.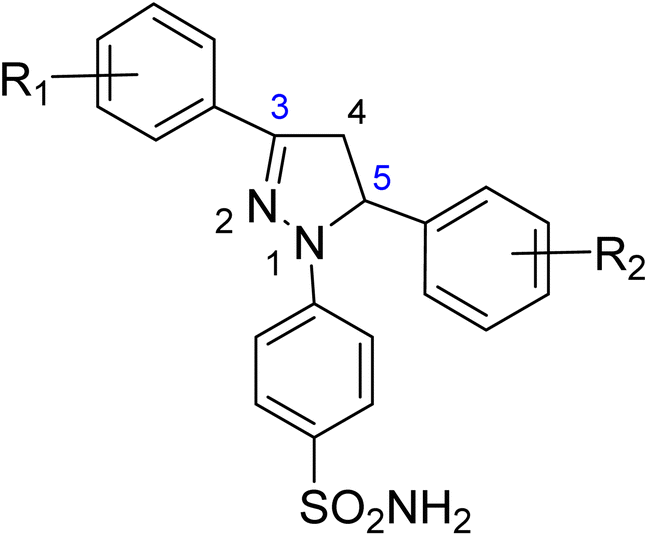
Comparison of the results of the compounds 4a, 4b and 4c with consistent attachment of the 2-hyroxyphenyl at position 3 of the pyrazole ring revealed that substitution at the phenyl ring of position 5 with positive inductive effect group (–CH3) did not reduce the activity however, the presence of negative inductive effect atoms (bromine, chlorine) contributed significantly towards the reduced activity of the hCAII, hCAIX, and hCAXII. The compound 4a with methyl attachment at phenyl ring at position 5 was devoid of any considerable activity (less than 50% inhibition). The compound 4b with bromine attachment at the phenyl ring was found the inhibitor of the human hCAII and hCAXII however, the enhanced enzyme inhibitory potential was obtained when bromine atom of the compound 4b was replaced by chlorine (4c). The compound 4c reduced the activity of the all the three isozymes preferably the hCAII to an IC50 concentration of 1.20 ± 0.40 μM. The results of the compounds 4a–4c suggested that in the presence of 2-hydroxyphenyl ring at the position 3 of the pyrazole ring, 4-chlorophenyl attachment was much favorable for the enzyme inhibition at the position 5. The attachment of the 2-hydroxy-5-methoxyphenyl at the position 3 of the pyrazole ring in compounds 4d and 4e produced interesting results. The compounds 4d with the same bromine substitution as for compound 4b at the phenyl ring attached at the position 5 of the pyrazole ring was found with slightly increased activity against hCAII, and hCAXII as compared to the structure 4b. The increased potency of the compound 4d against hCAII, and hCAXII as compared to the structure 4b may be attributed to the presence of electron withdrawing methoxy group at the para position of hroxyl group with pheny ring at position 3. The inhibitory activity was limited to the selective and very potent against the hCAII (IC50 = 0.75 ± 0.13 μM) for the compound 4e possessing 2-methoxylphenyl attachment at the position 5 of the pyrazole ring. The presence of the 2-hyroxy-4-bromophenyl ring at the position 3 of the pyrazole ring in compounds 4f, 4g and 4h lead the molecules more active against all the tested carbonic anhydrase isoforms. The comparative study of SAR of the compound 4a and 4f showed that the presence of the electron-donating group (methyl) was much favorable for the enzyme inhibition especially hCAII (IC50 = 0.24 ± 0.08 μM) when bromine atom is substituted along with the hydroxyl group with the phenyl ring at position 3 of the pyrazole ring. The activity of the compound was enhanced to all the three isoforms when the electron-donating group methyl was replaced by the electron-withdrawing methoxy group at the phenyl ring attached at the position 5 of the pyrazole ring to yield compound 4g. The compound 4g was screened as the most potent inhibitor of the hCAXII with the IC50 value 0.12 ± 0.07 μM. However, halogenation of the phenyl ring attached at position 5 of the pyrazole scaffold in compound 4h, reduced the activity of the hCAIX and hCAXII to submicromolar level i.e. IC50 = 0.26 ± 0.04 μM and IC50 = 0.38 ± 0.10 μM, respectively.
The SAR study of the compounds 4j, 4k, and 4l demonstrated the more interesting results in the presence of fluorine substitution along with hydroxyl group at the phenyl ring attached at the position 3 of the pyrazole ring. The compound 4j inhibited the activity of the all the three carbonic anhydrase isoforms to low IC50 concentrations of 0.39 ± 0.05 μM, 0.15 ± 0.07 μM and 0.28 ± 0.05 μM against the hCAII, hCAIX and hCAXII, respectively. The activity of the inhibitor 4j might be attributed to the presence of methyl and fluorine groups at the phenyl rings. Similarily, the methoxy group attachment in compound 4k lowered the activity of the hCAII, and hCAXII to a significant level however, it was devoid of the inhibitory effect against the hCAIX i.e., merely 27% inhibition. The activity of the compound 4l was diminished against all the three carbonic anhydrase isoforms. This decline in the activity might be attributed to the presence of both bromine and fluorine atoms at the separate phenyl rings making the aromatic rings more electron deficient on both sides of the molecule 4l.
3.4 Density functional theory studies
The density functional theory studies were conducted to instigate the electronic and stability profile of all synthesized derivatives. The DFT studies were conducted using B3LYP/6-31G level of theory. The structural geometry of each derivative was optimized to steepest energy gradient with no imaginary frequency was notable. In addition, polarizability and dipole moment was also calculated to determine the nature of the compounds. The optimized geometries of all synthesized derivatives are shown in Fig. 1.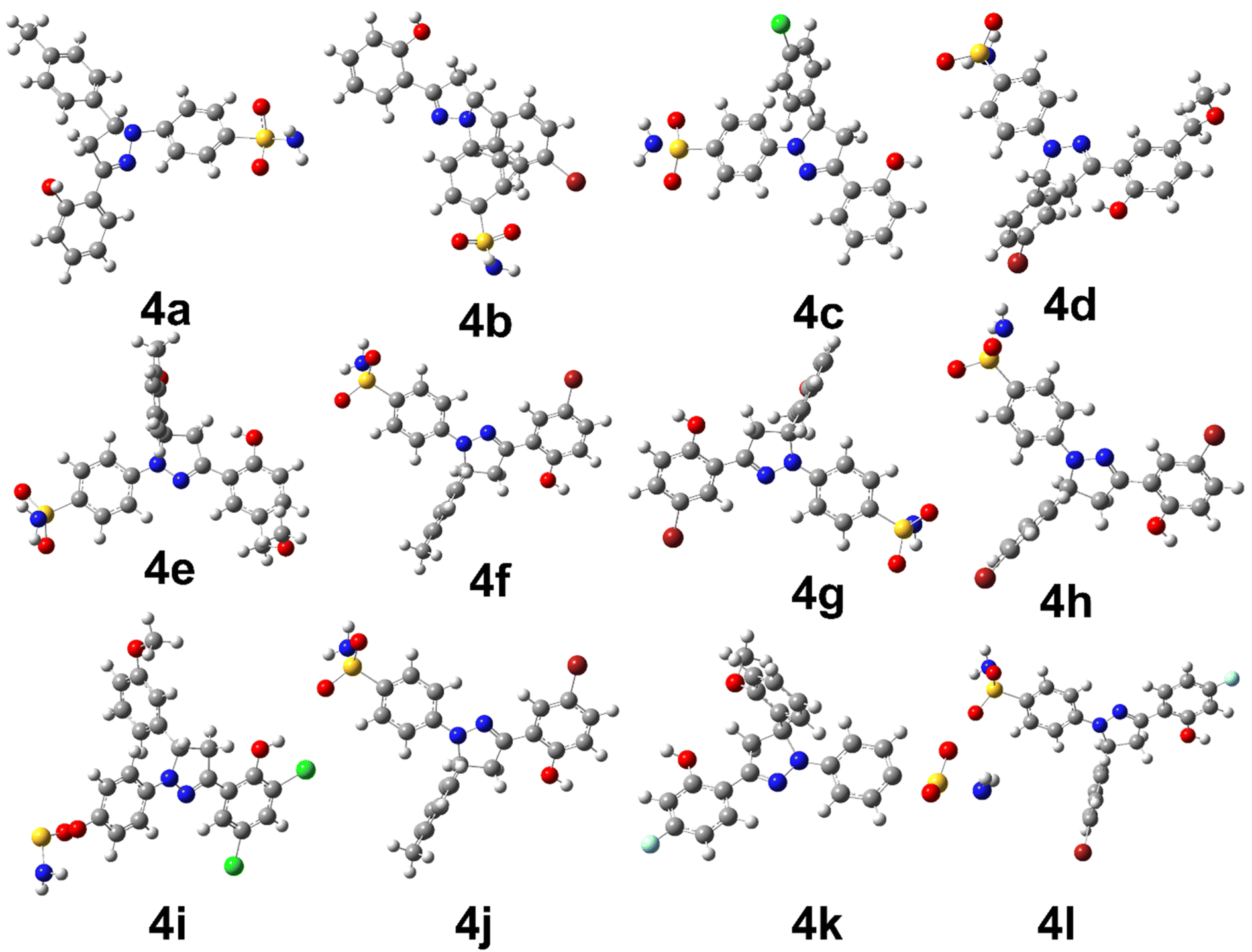 | ||
| Fig. 1 The optimized structural geometries of synthesized derivatives. | ||
The Table 2 is providing the information regarding optimization energy and electronic parameters of all derivatives.
| Codes | Optimization energy | Dipole moment | Polarizability (α) | HOMO (eV) | LUMO (eV) | HOMO–LUMO (ΔeV) |
|---|---|---|---|---|---|---|
| 4a | −1636.769 | 7.144577 | 371.066 | −0.179 | −0.163 | 0.016 |
| 4b | −1853.874 | 5.861 | 391.346 | −0.209 | −0.161 | 0.048 |
| 4c | −2058.793 | 9.767 | 311.056 | −0.201 | −0.132 | 0.069 |
| 4d | −4323.964 | 5.207 | 341.577 | −0.209 | −0.068 | 0.141 |
| 4e | −1867.480 | 8.681 | 341.950 | −0.204 | −0.060 | 0.144 |
| 4f | −4209.497 | 10.736 | 330.187 | −0.200 | −0.065 | 0.135 |
| 4g | −4284.673 | 10.061 | 331.098 | −0.203 | −0.067 | 0.136 |
| 4h | −6741.160 | 11.300 | 332.431 | −0.206 | −0.069 | 0.137 |
| 4i | −2632.920 | 5.299 | 341.932 | −0.193 | −0.111 | 0.082 |
| 4j | −2166.893 | 6.931 | 323.031 | −0.203 | −0.051 | 0.152 |
| 4k | −1803.608 | 9.970 | 296.695 | −0.191 | −0.049 | 0.142 |
| 4l | −4269.401 | 9.569 | 316.231 | −0.199 | −0.056 | 0.143 |
The above data represents the optimization energy, dipole moment, polarizability, HOMO energy, LUMO energy, and HOMO–LUMO energy gap of 12 compounds (4a–4l). The HOMO and LUMO energy levels range from −0.211 eV to −0.049 eV and −0.163 eV to 0.152 eV, respectively. The HOMO–LUMO energy gap (ΔeV) ranges from 0.016 eV to 0.152 eV. Among these compounds, 4k, 4g, and 4j have unique features that make them interesting candidates for further investigation. Compound 4k stands out in this dataset with the lowest optimization energy (−1803.608 Hartree) compared to the rest of the compounds. This compound also exhibits the highest polarizability (296.695 Bohr3), which is a measure of the ease with which the electron cloud can be distorted by an external electric field. The high polarizability of 4k may suggest that it is a relatively polarizable molecule, which could have implications for its interaction with other molecules in a biological system.
Compound 4g has the optimization energy of (−4284.673) and a relatively high polarizability (331.098), indicating that it is also a stable and polarizable compound. It has a slightly larger HOMO–LUMO energy gap (0.136) than 4k, indicating that it may have slightly different electronic properties. However, its dipole moment (10.061) is the second-highest in the set, which may make it more challenging to work with experimentally. Nevertheless, the combination of stability and polarizability makes 4g an interesting candidate for further investigation.
Compound 4j has a relatively low optimization energy (−2166.893) and a small HOMO–LUMO energy gap (0.152), indicating that it is a stable compound with electronic properties similar to 4k. However, its dipole moment (6.931) is lower than that of 4g, which may make it a more attractive experimental candidate. Additionally, it has a relatively low polarizability (323.031), which may make it less susceptible to electronic perturbations. These properties make 4j an interesting candidate for further investigation as well. Briefly, compounds 4k, 4g, and 4j all have unique features that make them interesting candidates for further investigation. Compound 4k is the most stable and easily polarizable, while 4g is also stable and polarizable but has a higher dipole moment. Compound 4j is stable with properties similar to 4k but has a lower dipole moment and polarizability. The FMOs orbitals are illustrated in Fig. 2. It is notable that nearly entire molecules are localized with HOMO and LUMO orbitals which represent efficient electron transfer in intra and inter molecule. It is also predictive of reactive nature of different parts of synthesized derivatives especially diazole and benzene rings.
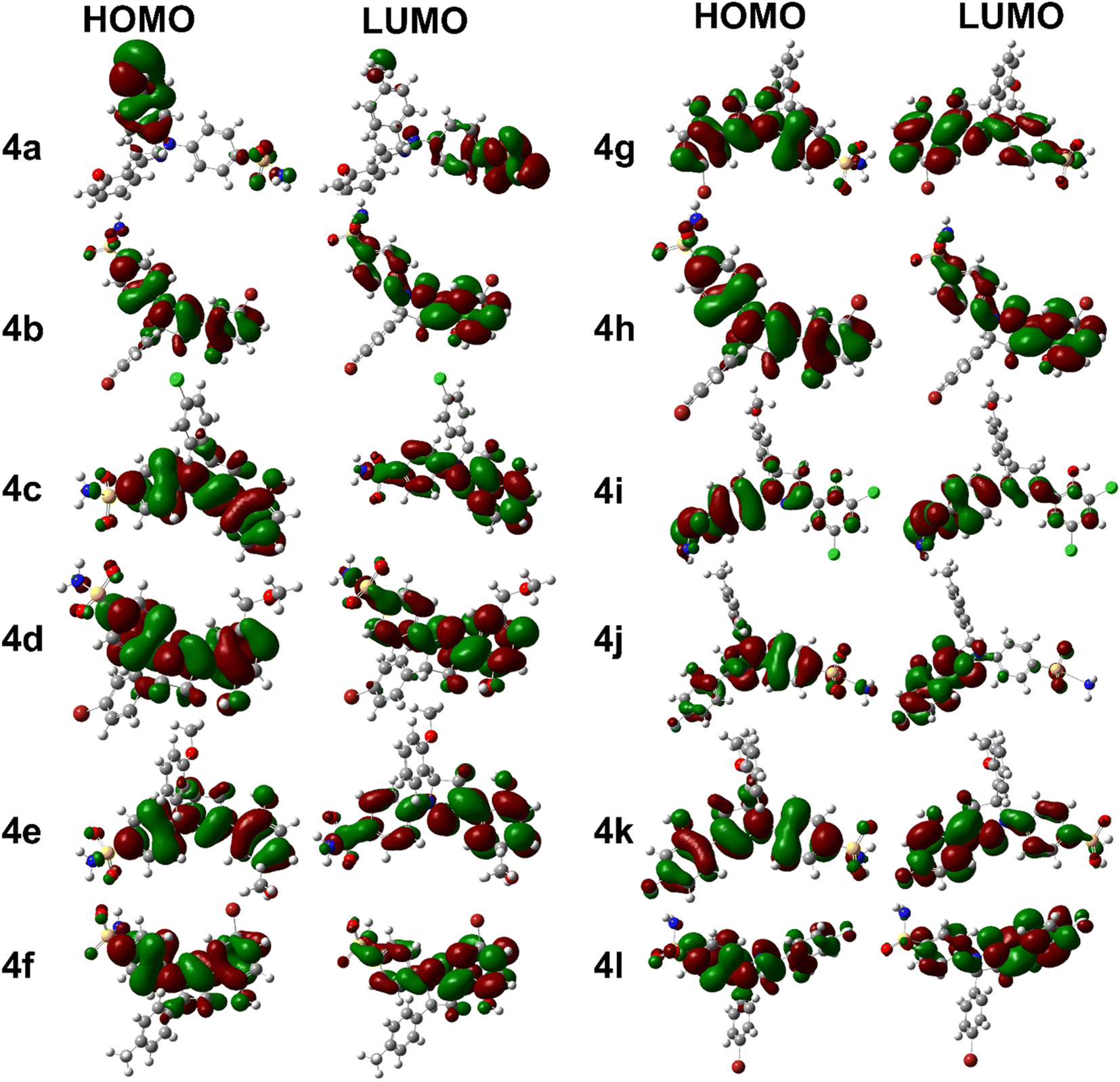 | ||
| Fig. 2 FMOs orbitals of synthesized derivatives. | ||
The chemical potential (μ), electronegativity (χ), hardness (η), softness (S), and electrophilicity index (ω) values for the 12 compounds are also provided in the data. The chemical potential is a measure of the potential energy of a molecule, and it indicates the ability of the molecule to donate or accept electrons. The electronegativity measures the ability of an atom to attract electrons towards itself in a molecule. The hardness represents the resistance of a molecule to undergo a chemical reaction, while the softness is the inverse of hardness, indicating the ease with which a molecule can undergo a reaction. The electrophilicity index measures the ability of a molecule to act as an electrophile, i.e., to accept electrons.
In general, the chemical potential values for the compounds are negative, indicating that they are stable and have lower potential energy. The electronegativity values for all compounds are equal, indicating that they have similar electron-attracting abilities. The hardness values are relatively low, which suggests that the compounds are reactive and can easily undergo chemical reactions. The softness values are relatively high, indicating that the compounds are relatively soft and can easily undergo deformation. The electrophilicity index values for all compounds are relatively high, indicating that the compounds are highly reactive and can easily accept electrons.
Based on the values of η and S, we can conclude that compounds 4a, 4b, and 4i are relatively harder and less polarizable compared to other compounds. On the other hand, compounds 4d, 4e, 4j, and 4k are relatively softer and more polarizable compared to other compounds. Compounds 4g and 4h have intermediate values of η and S. The electrophilicity index (ω) values provide an indication of the stability and reactivity of the molecule towards nucleophiles, with higher values indicating a greater tendency towards nucleophilic attack. In this case, the ω values of all the compounds are relatively low, which indicates that they are less likely to undergo nucleophilic attack.
Overall, based on the properties analyzed, compounds 4d, 4e, 4j, and 4k appear to be relatively more polarizable and softer, which may make them more reactive towards electrophiles. Conversely, compounds 4a, 4b, and 4i appear to be relatively harder and less polarizable, which may make them less reactive towards electrophiles. The Table 3 is providing the detailed data about reactivity descriptors of the compounds.
| Codes | Chemical potential μ (eV) | Electronegativity χ (eV) | Hardness η (eV) | Softness S (eV−1) | Electrophilicity index ω (eV) |
|---|---|---|---|---|---|
| 4a | −0.171 | 0.171 | 0.008 | 62.500 | 1.828 |
| 4b | −0.185 | 0.185 | 0.024 | 20.833 | 0.713 |
| 4c | −0.167 | 0.167 | 0.035 | 14.493 | 0.402 |
| 4d | −0.139 | 0.139 | 0.071 | 7.092 | 0.136 |
| 4e | −0.132 | 0.132 | 0.072 | 6.944 | 0.121 |
| 4f | −0.133 | 0.133 | 0.068 | 7.407 | 0.130 |
| 4g | −0.135 | 0.135 | 0.068 | 7.353 | 0.134 |
| 4h | −0.138 | 0.138 | 0.069 | 7.299 | 0.138 |
| 4i | −0.152 | 0.152 | 0.041 | 12.195 | 0.282 |
| 4j | −0.127 | 0.127 | 0.076 | 6.579 | 0.106 |
| 4k | −0.120 | 0.120 | 0.071 | 7.042 | 0.101 |
| 4l | −0.128 | 0.128 | 0.072 | 6.993 | 0.114 |
3.5 Molecular docking studies
Human carbonic anhydrases hCAII, hCAIX, and hCAXII enzymes play crucial roles in various physiological processes such as pH regulation, CO2 transport, and bicarbonate secretion. These enzymes are also involved in several diseases such as glaucoma, cancer, and osteoporosis. Therefore, targeting these enzymes can be significant in the development of potential therapies for these diseases. Molecular modeling studies of potent inhibitors can provide insights into the relative selectivity and potency of designed analogues, which can aid in the development of more effective drugs with fewer side effects. Molecular modeling studies were conducted on human carbonic anhydrase II (PDB: 3V7X), hCAIX (PDB: 5FL4), and hCAXII (PDB: 5MSA) enzymes to analyze the relative selectivity and potency of our designed analogues. To ensure the accuracy and efficiency of the modeling studies, high-resolution crystal structures of the enzymes were downloaded from the PDB server at https://www.rcsb.org/. The crystal structures of hCAII, hCAIX, and hCAXII with 1.03 Å, 1.8 Å, and 1.2 Å resolution were selected for in silico studies of potent inhibitors. The crystal structure of hCAII contains the co-crystallized ligand N-[2-(3,4-dimethoxyphenyl)ethyl]-4-sulfamoylbenzamide in its active site, while the crystal structures of hCAIX and hCAXII contain a homo tetramer chain. For docking studies, chain A was considered, which contains co-crystallized ligands 5-(1-naphthalen-1-yl-1,2,3-triazol-4-yl) thiophene-2-sulfonamide (9FK) and 2,3,5,6-tetrafluoro-4-(propylthio)benzenesulfonamide 3TV. The co-crystallized ligands were selected as a reference for docking, and the docking protocol was validated after successfully reproducing the co-crystallized pose. Using MOE, and SeeSAR software, the RMSD values for the reference ligand of hCAII, hCAIX, and hCAXII were found to be 0.58 Å and 0.83 Å, respectively. These findings provide valuable insights into the selectivity and potency of the designed analogues, and their potential as inhibitors for these enzymes.In this study, the binding of compound 4k and acetazolamide (AZM) was investigated in the active site of hCAII (Fig. 3). The HYDE assessment score, which represents the binding energy of the compound, was found to be higher for compound 4k (−32.21 kJ mol−1) compared to AZM (−28.59 kJ mol−1), indicating a stronger binding affinity for the former. The zinc ion in the active site of hCAII was found to interact with the deprotonated nitrogen of the sulfonamide group in compound 4k, and Thr199 formed a hydrogen bond with the oxygen of the free sulfonamide group (SO2NH2). Additionally, Trp209 showed -sulfur interaction with the sulfur of the sulfonamide group, and Phe131, Val135, Pro202, and Leu198 exhibited π-alkyl and π-sigma interactions, all of which are comparable to the interactions observed with the standard inhibitor AZM. These findings suggest that compound 4k has the potential to be a potent inhibitor of hCAII, and further in vitro and in vivo studies are warranted to evaluate its efficacy and safety.
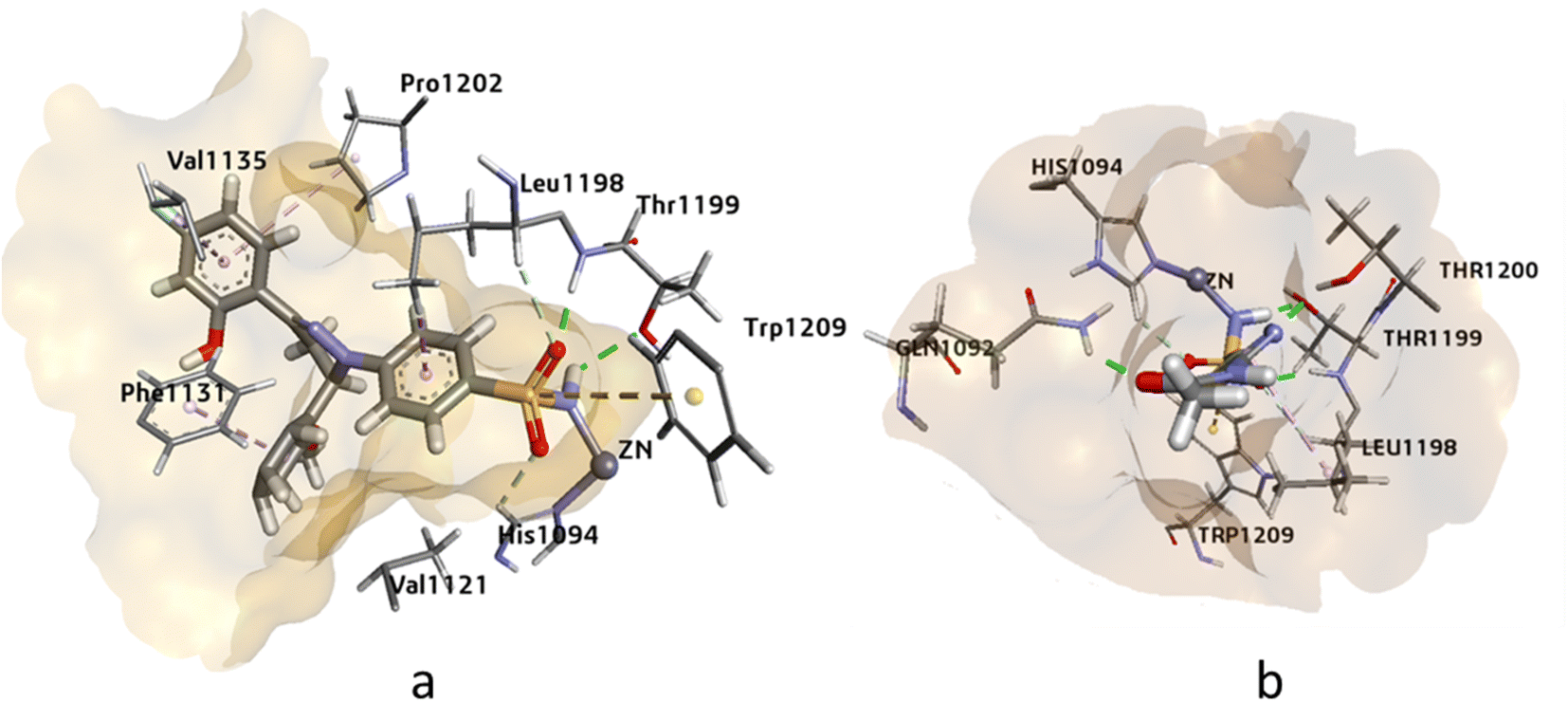 | ||
| Fig. 3 The 3D representation of the binding interactions of compound (a): 4k and (b): AZM with the active site residues of hCAII. | ||
The docking analysis of compound 4j and the standard inhibitor at the active site of hCAIX revealed that the HYDE assessment score of compound 4j was −31.97 kJ mol−1, which was higher than that of the standard inhibitor (−24.72 kJ mol−1). The key interactions observed between the docked ligand and the active amino acid residues included the cofactor Zn++ binding with the deprotonated nitrogen of the sulphonamide, which was consistent with the interaction observed with the standard inhibitor. Additionally, Thr201 and Gln71 showed hydrogen bonding interactions with the oxygen of the sulphonamide and the hydroxyl group attached at the ortho position of the aromatic ring. Other notable interactions were observed with Leu134, Val130, Pro203, Leu91, Val121, and Leu199, which showed π-alkyl and π-sigma interactions with the docked ligand. Overall, these interactions were similar to those observed with the standard inhibitor at the active site of compound 4j (Fig. 4).
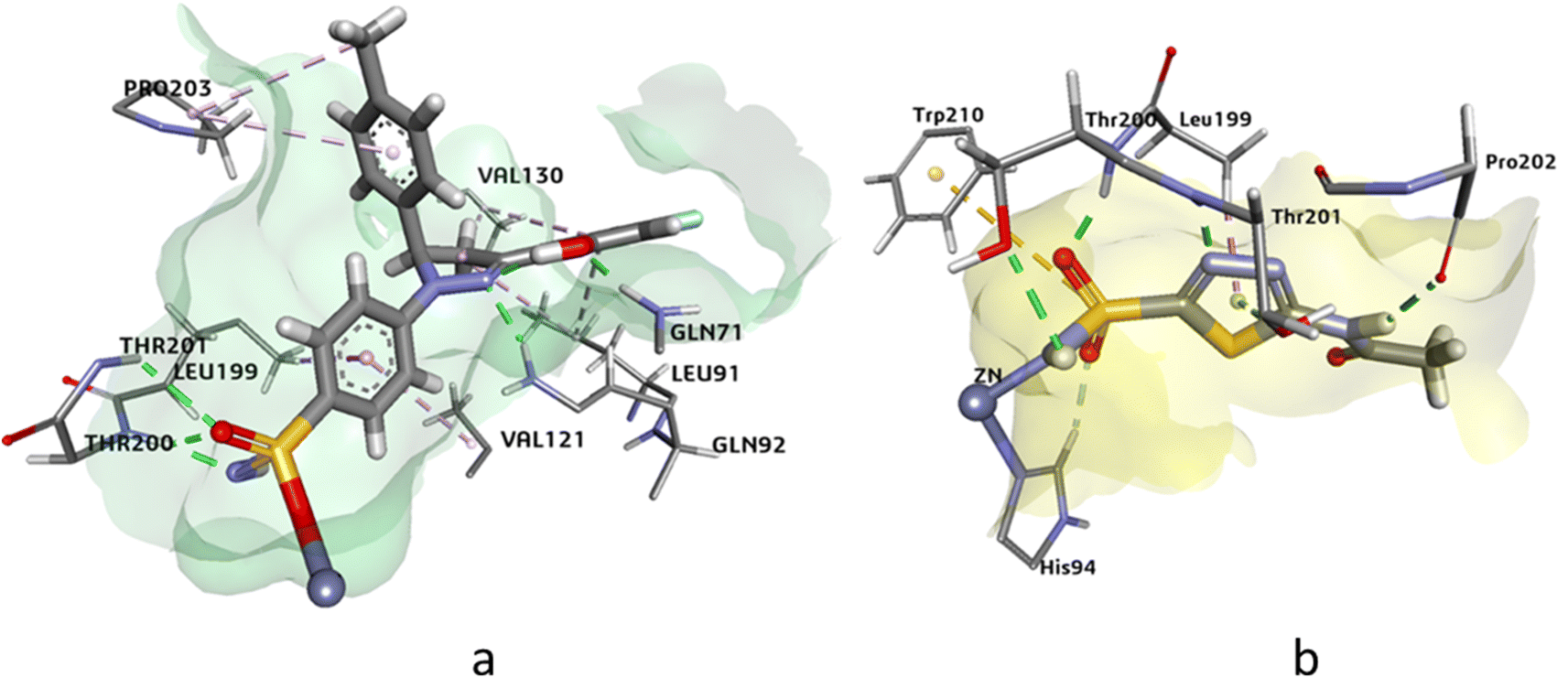 | ||
| Fig. 4 The 3D representation of the binding interactions of compound (a): 4j and (b): AZM with the active site residues of hCAIX. | ||
The docking analysis of compound 4g against hCAXII yielded promising results with a higher HYDE assessment score of −33.05 kJ mol−1 compared to the standard inhibitor AZA with −28.20 kJ mol−1. The compound 4g showed strong binding affinity towards the active site of hCAXII with key interactions observed between the inhibitor and active amino acid residues. Notably, the nitrogen atom of the sulfonamide exhibited interaction with Zn++ which is crucial for enzyme inhibition. Furthermore, the compound 4g formed favorable hydrogen bonding interactions with Gln89, Asn64, and Thr198 with oxygen of the hydroxyl, methoxy, and sulphonamide functional groups respectively. These interactions were found to be critical for stabilizing the inhibitor in the active site of the enzyme. Additionally, the residues Ala129, Val119, and Val197 showed π-alkyl and pi-sigma interactions with the inhibitor which further contributed to the inhibitor's strong binding affinity towards hCAXII as shown in Fig. 5. Overall, these results suggest that compound 4g has great potential as a potent inhibitor of hCAXII and can serve as a starting point for the development of novel therapeutics for diseases associated with this enzyme.
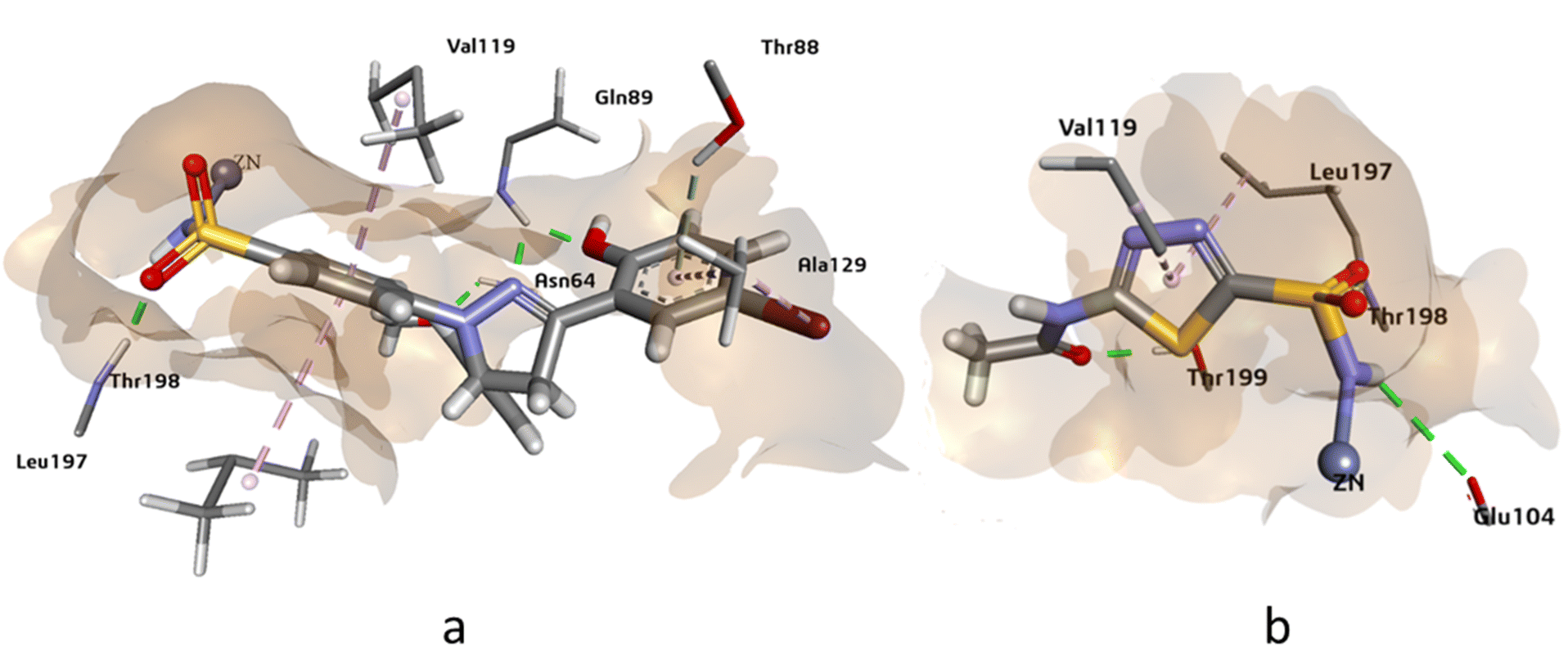 | ||
| Fig. 5 The 3D representation of the binding interactions of compound (a): 4g and (b): AZM with the active site residues of hCAXII. | ||
3.6 Molecular dynamics simulations
Molecular dynamics (MD) simulation is a computational technique that has proven to be invaluable in the field of scientific research. It is a powerful tool for studying the behavior of atoms and molecules over time, and for gaining insight into the complex physical and chemical phenomena that govern the behavior of materials. MD simulations involve numerically solving the equations of motion for a set of interacting atoms or molecules, and calculating their positions, velocities, and forces at each time step. The simulation allows for the exploration of the thermodynamics and kinetics of molecular systems, as well as the structural and dynamic properties of materials.In current study we have investigated the strength of molecular interactions of potent derivatives against respective proteins. It notable that compound 4k has exhibited strong in vitro and in silico inhibitory potential against hCAII. Whereas, compound 4j and 4g was strongly inhibiting the hCAIX and hCAXII respectively. The findings of in silico and experimental studies were further assisted by MD simulations.
The stability of molecular interactions of hCAII-4k was revealed by 100 ns simulations. It was notable that apo protein (hCAII) demonstrated the significant stability pattern with average RMSD of 2 Å. In addition, equilibrated trajectory revealed the minimum structural variations inside crystallographic structure of the protein which is predictive of strong stability pattern. The RMSD of Apo protein was evolved from 1 Å to 2 Å with average RMSD of 1.84 Å. In terms of liganded protein, the stability pattern of the hCAII-4k complex, as observed in the RMSD (root-mean-square deviation) analysis, indicates that the complex remains relatively stable throughout the simulation, with some minor fluctuations. During the first 20 ns of the simulation, the RMSD of the complex remains below 2 Å, indicating that the overall structure of the complex is maintained with minimal deviation from the starting structure. Between 20 ns and 80 ns, the RMSD rises to 2.5 Å, suggesting that the complex undergoes some structural rearrangement or fluctuation during this period. However, the RMSD remains relatively stable within this range, indicating that the overall structure of the complex is still well-maintained. After 80 ns, the RMSD slightly fluctuates, but eventually re-equilibrates and stabilizes at around 3.5 Å. The minor fluctuations observed during this period are likely due to the inherent flexibility of the protein–ligand complex, as well as thermal fluctuations and establishment of new contacts.
Overall, the stability pattern of the hCAII-4k complex as observed in the RMSD analysis suggests that the complex remains structurally stable throughout the simulation, with some minor fluctuations and rearrangements occurring over time. The deviations observed are relatively small and do not indicate any major disruptions or instabilities in the complex. The average RMSD of liganded protein was calculated to be 2.5 Å as depicted in Fig. 6.
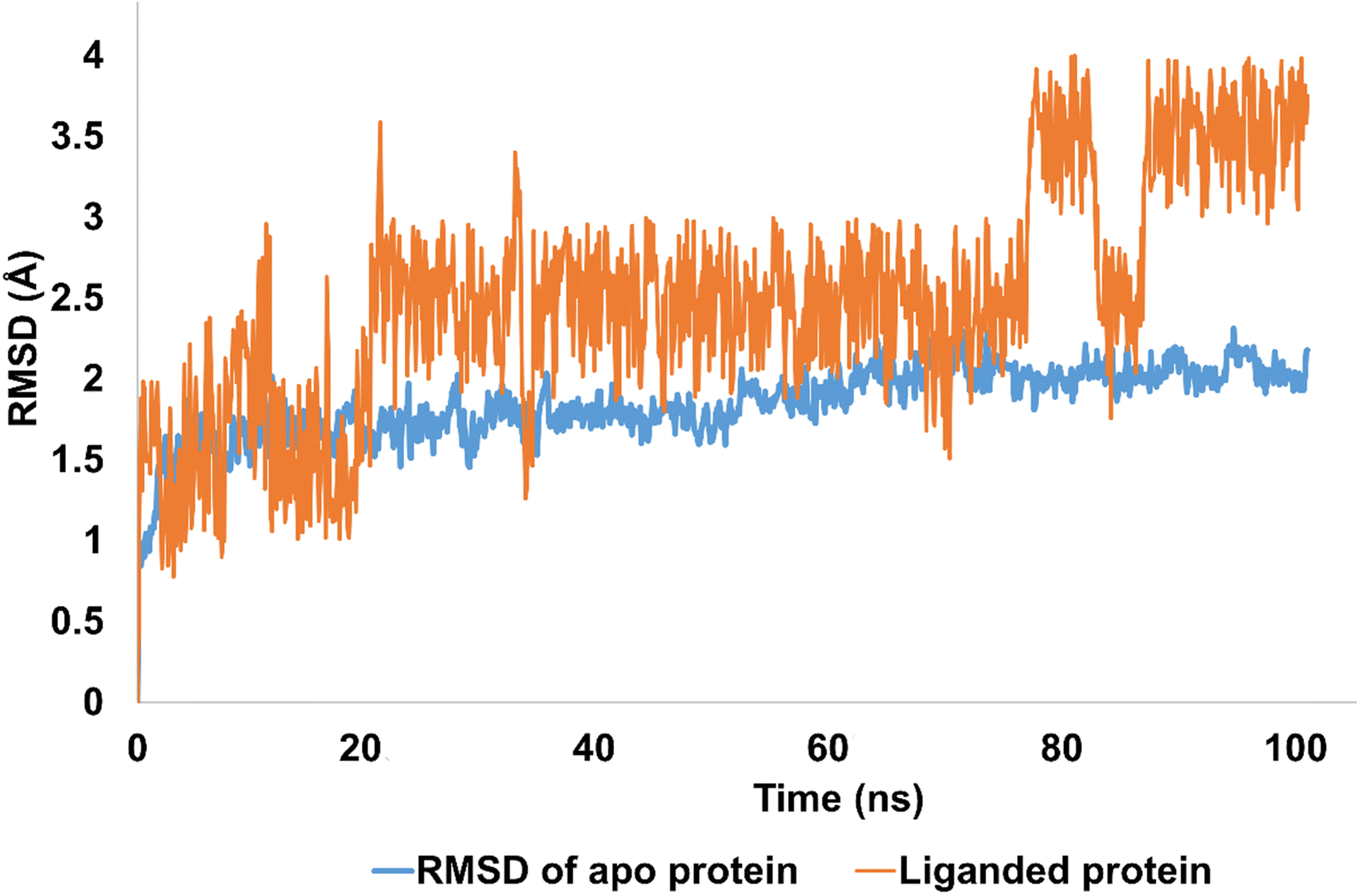 | ||
| Fig. 6 The evolution of RMSD pattern for apo protein (hCAII) and liganded protein (hCAII-4k complex). | ||
RMSF is a measure of the average deviation of each atom's position from its mean position, in a given simulation or set of structures. It provides information about the flexibility or mobility of the residue, where a high RMSF value indicates a more flexible or mobile residue. Analyzing the RMSF data, it was deduced that majority of amino acid residues demonstrated significant stability. However, some residues have higher RMSF values than others, indicating that they are more flexible and perturbed significantly. For example, HIS1003 has an RMSF value of 4.4, which is the highest in the dataset, indicating that this residue is highly flexible. On the other hand, some residues have low RMSF values, indicating that they are less flexible. For example, Arg1089 has an RMSF value of 0.483, which is one of the lowest values in the dataset, indicating that this residue is relatively rigid.
It was notable that important residues of targeted protein were remained in contact with compound 4k. The amino acid residues are in contact with a ligand (indicated by “green lines” in the Fig. 7), while others are indicated without any dashes. Comparing the RMSF values of residues in contact with a ligand to those not in contact, it was notable that there is no clear pattern or trend. Some residues in contact with a ligand have high RMSF values, while others have low RMSF values. Similarly, some residues not in contact with a ligand have high RMSF values, while others have low RMSF values. Overall, the RMSF provided the information about the flexibility of different residues in a protein molecule, which can be useful in understanding the structural and functional properties of the protein. However, without more context about the specific protein and simulation used to generate this data, it is difficult to draw more specific conclusions.
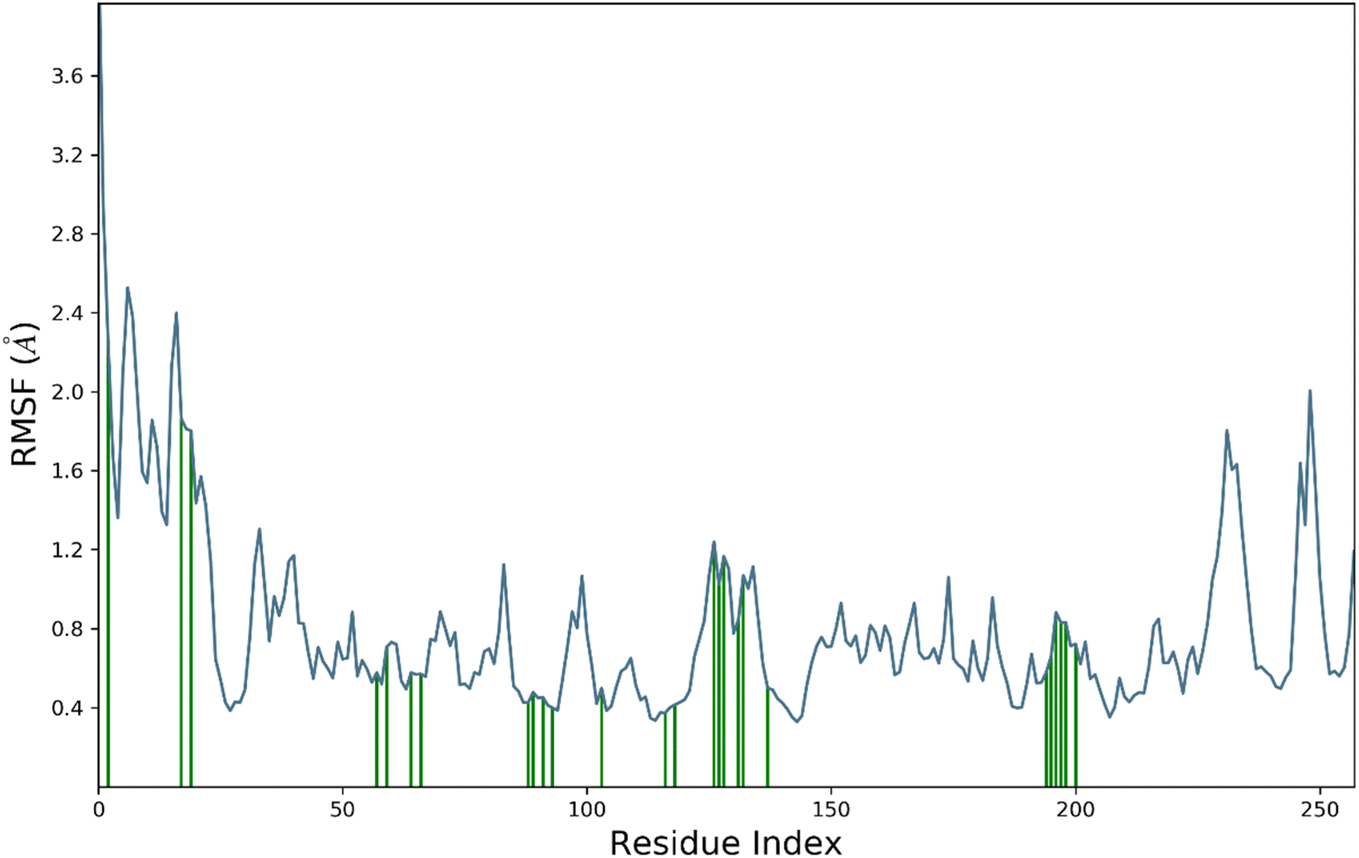 | ||
| Fig. 7 The residue wise fluctuations of hCAII in complex with compound 4k. | ||
The contact profile of 4k against hCAII reveals the residues in contact with the ligand and their frequency of occurrence during the simulation time. The hydrogen bonding occurs with Gln1092, His1094, His1119, Thr1199, and Thr1200 for 80%, 60%, 10%, 10%, and 15% of simulation time respectively. The percentage of simulation time for each interaction suggests that hydrogen bonding with Gln1092 and His1119 occurs more frequently than with the other residues. This information could be useful in designing future drug candidates to target these specific residues.
Furthermore, hydrophobic interactions with Ile1091, Phe1131, Val1135, Leu1198, Pro1202, and Leu1204 were also observed during the simulation time for 5%, 58%, 30%, 20%, 40%, and 5% of simulation time respectively. The results indicate that hydrophobic interactions with Phe1131 and Leu1204 occur most frequently. These findings could be useful in designing small molecules that can exploit these hydrophobic interactions for stronger binding affinity with hCAII.
Additionally, water bridges were observed to stabilize the protein–ligand complex. Water bridges are formed when water molecules bridge between the protein and ligand, and they have been shown to contribute significantly to ligand binding affinity. Therefore, the presence of water bridges in the simulation suggests that these interactions could play a crucial role in the binding affinity of 4k to hCAII. Briefly, the findings suggest that 4k interacts with hCAII through a combination of hydrogen bonding, hydrophobic interactions, and water bridges. These interactions are dynamic and occur at varying frequencies throughout the simulation time, which could be exploited to design more effective drug candidates targeting hCAII. The Fig. 8 is illustrating the contact profile of 4k against hCAII.
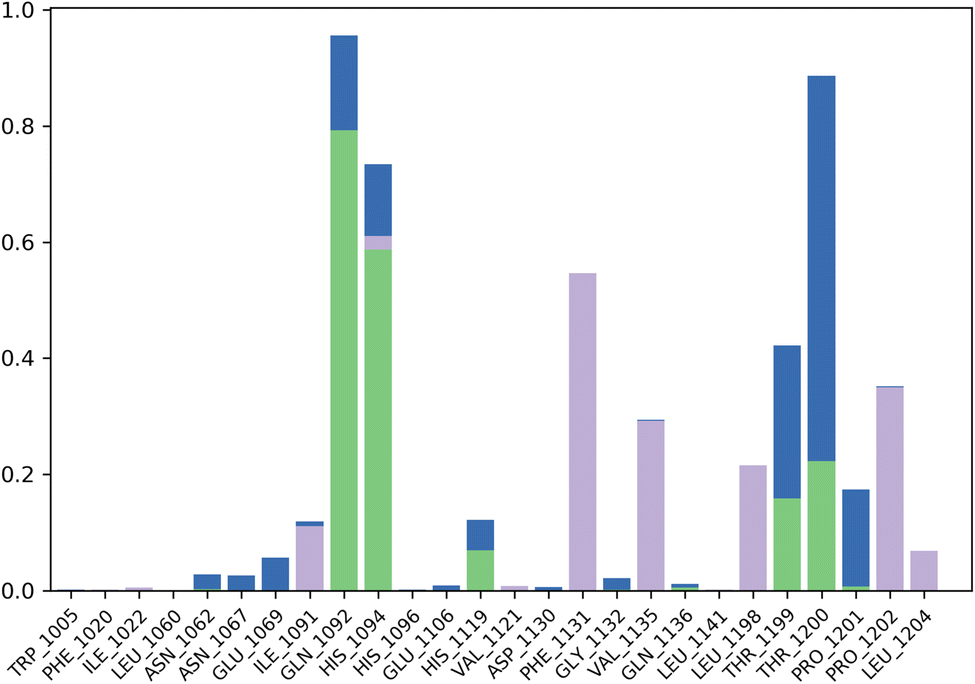 | ||
| Fig. 8 Contact map profile of compound 4k against hCAII. | ||
The compound 4j was the significant inhibitor of hCAIX revealed by in vitro and molecular docking studies. The further assistance of these findings is provided by MD simulations studies. In which apo protein and liganded protein was assessed for their stability over 100 ns of time period.
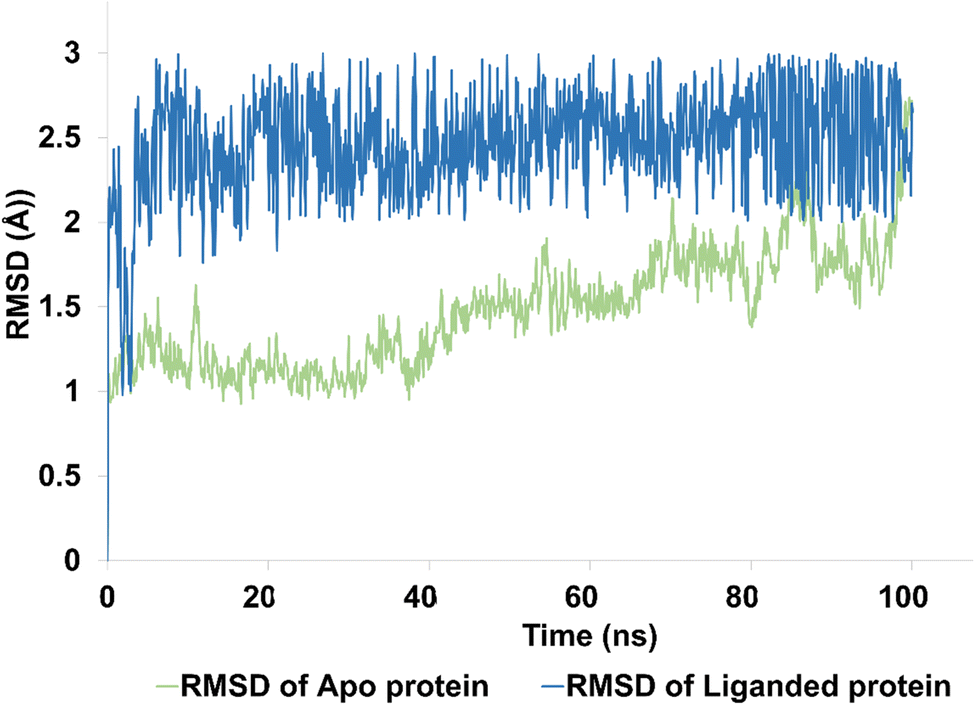 | ||
| Fig. 9 The evolution of RMSD pattern for apo protein (hCAIX) and liganded protein (hCAIX-4j complex). | ||
The apo protein showed an RMSD ranges between 1–2.5 Å with an average RMSD of 1.45 Å, indicating that the protein structure remained relatively stable throughout the simulation (Fig. 9). On the other hand, the liganded protein showed a slightly higher RMSD range of 2–2.8 Å with an average RMSD of 2.45 Å, indicating that the protein structure may have undergone some conformational changes upon binding to the ligand (Fig. 9). Despite these changes, the trajectory of the simulation remained quite stable and equilibrated, indicating that the protein–ligand complex remained in a stable state throughout the simulation. Overall, these results suggest that the hCAIX-4j complex is stable and the ligand is likely to remain bound to the protein in a stable conformation. These findings could be useful in the development of potential therapeutic agents targeting the hCAIX protein.
This RMSF analysis provides important insights into the flexibility and interactions of amino acid residues, which are crucial in understanding protein structures and functions. The RMSF analysis for 4j-hCAIX complex reveals that Trp9, Tyr11, and Arg62 have ligand contacts for significant period of simulation, while the remaining residues do not. The RMSF values of the amino acid residues range from the lowest value of Cys24 to the highest value of Gly13 (4.12 Å). Therefore, Cys24 is the least flexible, while Gly13 is the most flexible amino acid residue. In addition, the average RMSF was calculated to be 0.7 Å which demonstrate significant stability profile of targeted protein. The figure is illustrating the residue wise fluctuation of hCAIX in contact with compound 4j (Fig. 10).
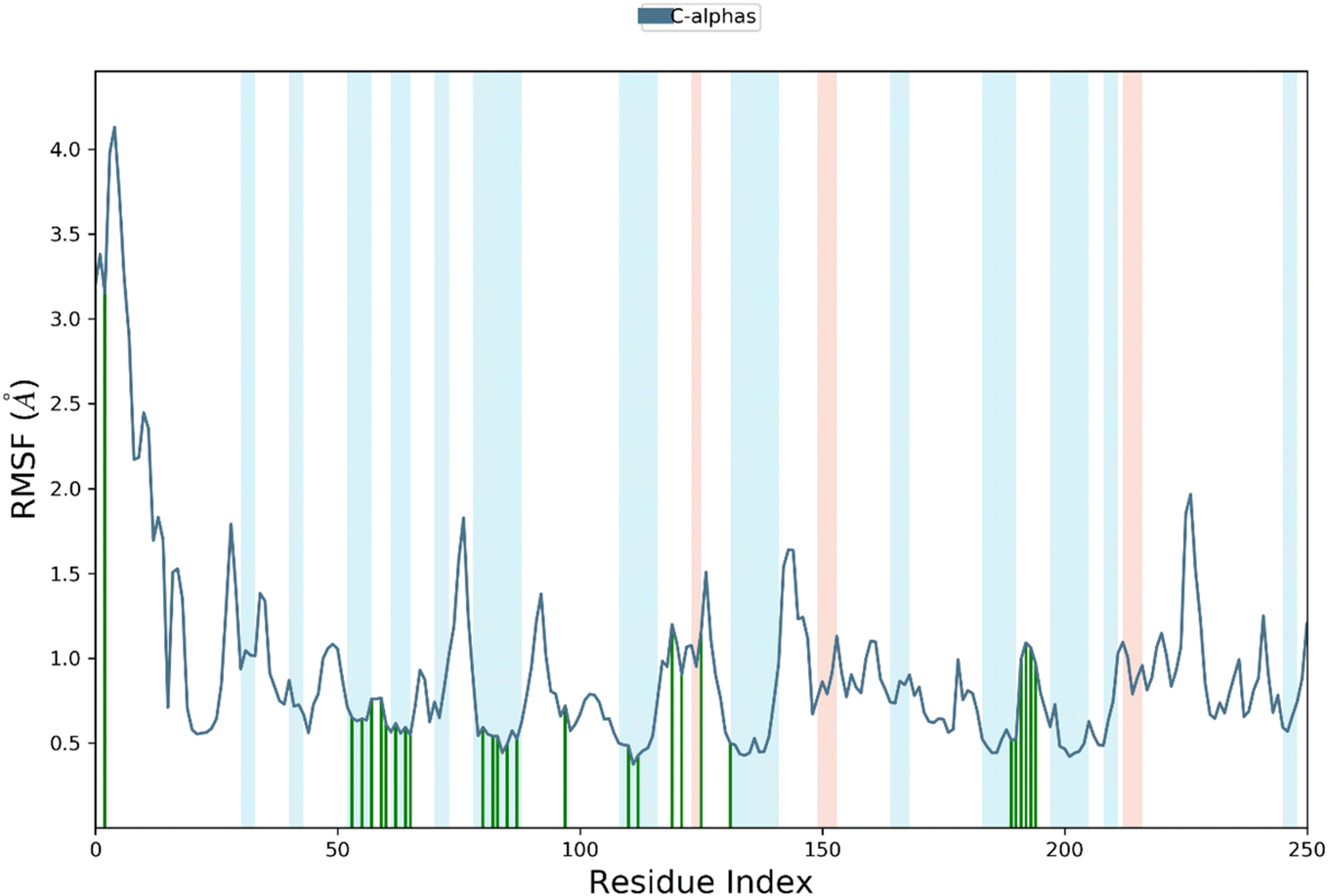 | ||
| Fig. 10 The residue wise fluctuations of hCAIX in complex with compound 4j. | ||
Contact map analysis of compound 4j with hCAIX revealed several key interactions between the ligand and the active site residues. Hydrogen bonds were formed between Gln71, Gln92, His94, His119, Thr200, and Thr201, with interaction fractions ranging from 20% to 100%. These hydrogen bonds indicate a strong binding affinity between the ligand and the active site of hCAIX.
In addition to the hydrogen bonds, hydrophobic interactions were observed between the ligand and His94, Val121, Val130, Leu199, and Pro203. These hydrophobic interactions are important for stabilizing the ligand in the active site of the enzyme, and are likely to contribute to the overall binding energy of the compound.
Taken together, the hydrogen bonds and hydrophobic interactions observed in the contact map analysis suggest that compound 4j has a strong binding affinity for hCAIX, and could potentially serve as an effective inhibitor of the enzyme. Further experimental validation will be necessary to confirm these findings and to determine the efficacy of compound 4j as an inhibitor of hCAIX activity. The Fig. 11 is illustrating the contact map analysis for 4j compound against hCAIX.
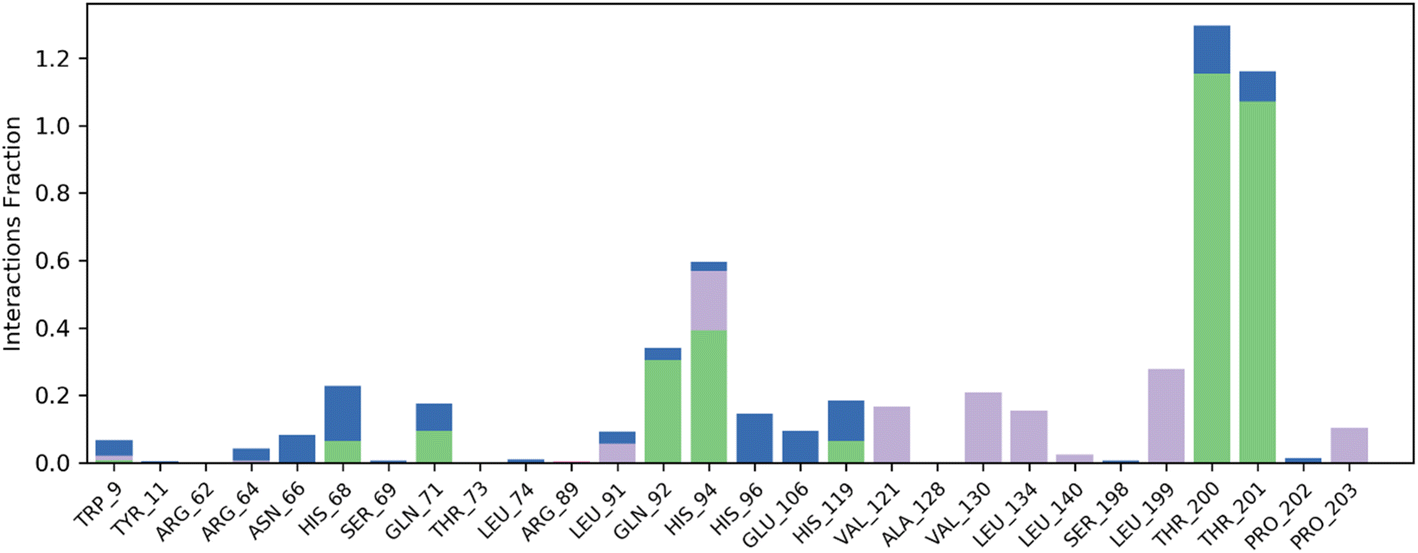 | ||
| Fig. 11 Contact map profile of compound 4j against hCAIX. | ||
Molecular dynamics (MD) simulation analysis of compound 4g was performed against both hCAXII and the apo protein to investigate the stability and conformational changes in the protein–ligand complex. The RMSD analysis revealed that the apo protein remained stable throughout the simulation with an average RMSD of 1 to 1.5 Å. On the other hand, the RMSD of the liganded protein showed some fluctuations initially but eventually stabilized with an average RMSD of 2.5 Å over the course of the simulation as shown in Fig. 12.
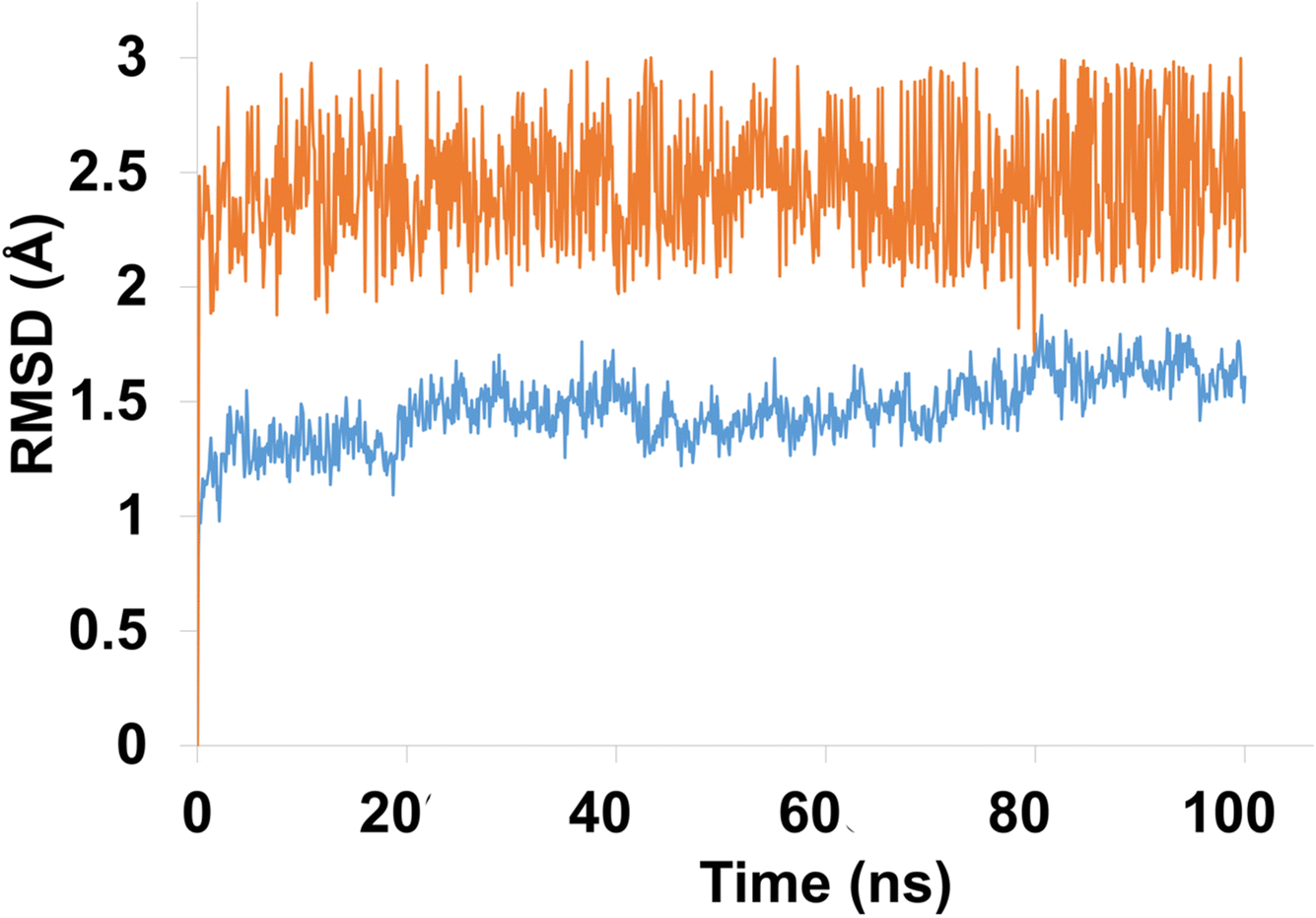 | ||
| Fig. 12 The evolution of RMSD pattern for apo protein (hCAXII) and liganded protein (hCAXII-4g complex). | ||
Further analysis showed that compound 4g was able to maintain stable interactions with the active site of hCAXII throughout the simulation. The hydrogen bond analysis revealed that Thr199, Asn64, and Thr198 were consistently forming hydrogen bonds with the oxygen atoms of the hydroxyl, methoxy, and sulfonamide groups of compound 4g, with an interaction fraction ranging from 20% to 100%. In addition, hydrophobic interactions were observed between Lys69, Leu197, and His66 residues of the protein with the ligand.
Root mean square fluctuation (RMSF) analysis is a method to investigate the flexibility of protein residues over the course of molecular dynamics simulations. Here, we performed RMSF analysis of hCAXII and 4g complex to understand the stability of the complex. The RMSF values of the CAXII protein in the complex with 4g ranged from 0.4 to 2 Å, indicating that the protein is relatively stable and does not undergo significant structural fluctuations. The regions with higher RMSF values are mostly located in the flexible loops and turns, which are expected to be more mobile than the structured regions of the protein. In contrast, the RMSF values of the 4g ligand showed a relatively stable profile throughout the simulation, with RMSF values ranging from 0.4 to 1.5 Å. This indicates that the ligand is well accommodated within the active site of the protein and does not undergo significant structural changes during the simulation. Overall, the RMSF analysis suggests that the hCAXII and 4g complex is a stable system and the ligand is well accommodated within the active site of the protein as depicted in Fig. 13.
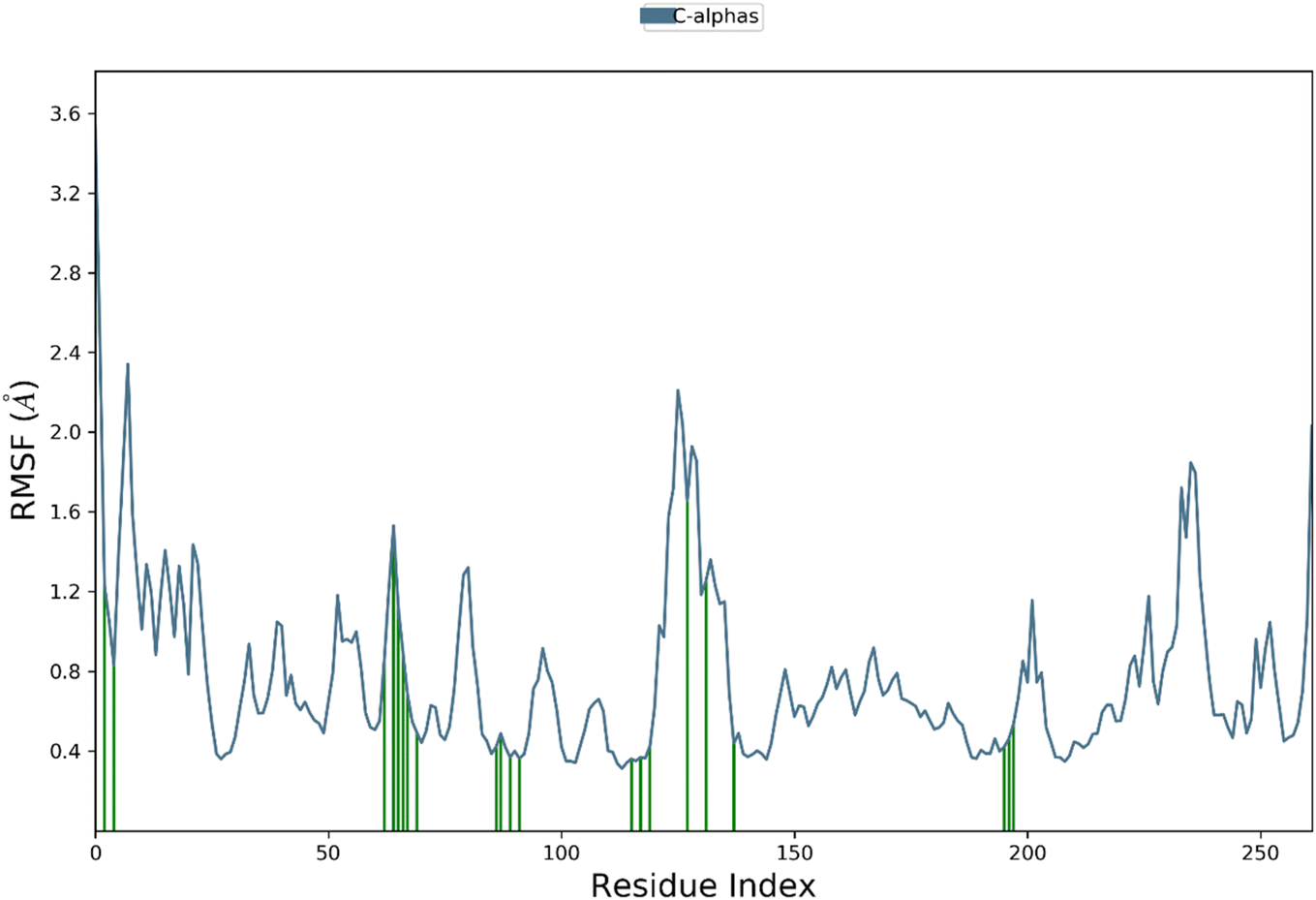 | ||
| Fig. 13 The residue wise fluctuations of CAXII in complex with compound 4g. | ||
4. Conclusion
Based on the findings of the present study of pyrazole-based benzene sulfonamides 4a–4l, it can be concluded that a number of the molecules inhibited the activity of all the three isozymes to submicromolar levels.The compounds 4e selectively blocked the activity of the hCAII to an IC50 values of 0.75 ± 0.13 μM however, the molecules 4j and 4k were also found very potent but non-selective inhibitors of hCAII showing the IC50 values of 0.39 ± 0.05 μM and 0.24 ± 0.18 μM. The compounds 4f, 4h and 4j preferably reduced the activity of hCAIX to an IC50 concentrations of 0.24 ± 0.08 μM, 0.26 ± 0.04 μM, and 0.15 ± 0.07 μM. Similarly, the compounds 4g, 4h, and 4j significantly inhibited the activity of hCAXII presenting the IC50 values of 0.12 ± 0.07 μM, 0.38 ± 0.10 μM, and 0.28 ± 0.05 μM, respectively. Moreover, an extensive computational study with different parameters of each compounds (i.e. optimization energy, electronic parameters, optimized structural geometries) was carried out to predict the binding mode of interactions and the comparative potency of the compounds to interact with the enzyme active sites residues. The molecular docking studies of most active compounds showed the interactions with the important amino acid residues of active pockets of hCAII, hCAIX and hCAXII crystallographic structures. The obtained inhibitors may be further evaluated as therapeutic leads against the hCAII, hCAIX and hCAXII associated ailments.
Conflicts of interest
The authors declared no conflict of interest.Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research (IFKSURC-1-3703).References
- B. N. Saglık, D. Osmaniye, U. A. Cevik, S. Levent, B. K. Çavuşoglu, O. Buyukemir, D. Nezir, A. B. Karaduman, Y. Ozkay, A. S. Koparal, S. Beydemir and Z. A. Kaplancıklı, Eur. J. Med. Chem., 2020, 198, 112392 CrossRef PubMed.
- C. T. Supuran, Expert Rev. Neurother., 2016, 16, 961–968 CrossRef CAS PubMed.
- W. L. Nettles, H. Song, E. R. Farquhar, N. C. Fitzkee and J. P. Emerson, Inorg. Chem., 2015, 54, 5671–5680 CrossRef CAS PubMed.
- A. Bonardi, A. Nocentini, S. Bua, J. Combs, C. L. Lomelino, J. Andring, L. Lucarini, S. Sgambellone, E. Masini, R. McKenna, P. Gratteri, C. T. Supurana and P. Gratteri, J. Med. Chem., 2020, 63, 7422–7444 CrossRef CAS PubMed.
- A. Ahmed, P. A. Channar, A. Saeed, M. Kalesse, M. A. Kazi, F. A. Larik, Q. Abbas, M. Hassan, H. Raza and S. Y. Seo, Bioorg. Chem., 2019, 86, 624–630 CrossRef CAS PubMed.
- R. Meleddu, S. Distinto, F. Cottiglia, R. Angius, P. Caboni, A. Angeli, C. Melis, S. Deplano, S. Alcaro, F. Ortuso, C. T. Supuran and E. Maccioni, ACS Med. Chem. Lett., 2020, 11, 852–856 CrossRef CAS PubMed.
- A. Nocentini, M. Ceruso, S. Bua, C. L. Lomelino, J. T. Andring, R. McKenna, C. Lanzi, S. Sgambellone, R. Pecori, R. Matucci, L. Filippi, P. Gratteri, F. Carta, E. Masini, S. Selleri and C. T. Supuran, J. Med. Chem., 2018, 61, 5380–5394 CrossRef CAS PubMed.
- A. M. Alafeefy, R. Ahmad, M. Abdulla, W. M. Eldehna, A. M. S. Al-Tamimi, H. A. Abdel-Aziz, O. Al-Obaid, F. Carta, A. A. Al-Kahtani and C. T. Supuran, Eur. J. Med. Chem., 2016, 109, 247–253 CrossRef CAS PubMed.
- C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2012, 27, 759–772 CrossRef CAS PubMed.
- P. Kumar, A. Nagarajan and P. D. Uchil, Cold Spring Harbor Protoc., 2019, 95414 Search PubMed.
- V. Mirzaie, T. Eslaminejad, H. Babaei and S. N. Nematollahi-Mahani, Drug Des., Dev. Ther., 2020, 14, 3589 CrossRef CAS PubMed.
- X. Wang, M. Wang, Z. Jia, L. Qiu, L. Wang, A. Zhang and L. Song, Mar. Biotechnol., 2017, 19, 22–35 CrossRef CAS PubMed.
- G. P. Subedi, R. W. Johnson, H. A. Moniz, K. W. Moremen and A. Barb, J. Visualized Exp., 2015, 106, 53568 Search PubMed.
- S. Jalil, S. Ullah, S. M. Zaraei, R. M. Sbenati, A. I. Shahin, J. Pelletier, J. Sevigny, T. H. Al-Tel, J. Iqbal and M. I. El-Gamal, Med. Chem. Res., 2023, 32, 869–883 CrossRef CAS.
- C. Turkes, M. Arslan, Y. Demir, L. Cocaj, A. R. Nixha and S. Beydemir, Bioorg. Chem., 2019, 89, 103004 CrossRef CAS PubMed.
- A. Saeed, M. Al-Rashida, M. Hamayoun, A. Mumtaz and J. Iqbal, J. Enzyme Inhib. Med. Chem., 2014, 29, 901–905 CrossRef CAS PubMed.
- K. Buran, S. Bua, G. Poli, F. E. Önen Bayram, T. Tuccinardi and C. T. Supuran, Int. J. Mol. Sci., 2019, 20, 1208 CrossRef CAS PubMed.
- MOE (Molecular Operating Environment), version 2019.0201, Chemical Computing Group; CCG, 2019 Search PubMed.
- SeeSAR version 12.1, BioSolveIT GmbH, Sankt Augustin, Germany Search PubMed.
- N. Schneider, G. Lange, S. Hindle, R. Klein and M. A. Rarey, J. Comput.-Aided Mol. Des., 2013, 27, 15–29 CrossRef CAS PubMed.
- Dassault Systemes BIOVIA Discovery Studio Modeling Environment, Release Dassault Systems, San Diego, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03276k |
| This journal is © The Royal Society of Chemistry 2023 |