
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, anti-proliferative evaluation, docking, and MD simulation studies of new thieno[2,3-d]pyrimidines targeting VEGFR-2†
Souad A. El-Metwallya,
Hazem Elkady *b,
Mohamed Hagrasc,
Dalal Z. Huseind,
Ibrahim M. Ibrahime,
Mohammed S. Taghourb,
Hesham A. El-Mahdyf,
Ahmed Ismailf,
Bshra A. Alsfoukg,
Eslam B. Elkaeedh,
Ahmed M. Metwaly*ij and
Ibrahim H. Eissa*b
*b,
Mohamed Hagrasc,
Dalal Z. Huseind,
Ibrahim M. Ibrahime,
Mohammed S. Taghourb,
Hesham A. El-Mahdyf,
Ahmed Ismailf,
Bshra A. Alsfoukg,
Eslam B. Elkaeedh,
Ahmed M. Metwaly*ij and
Ibrahim H. Eissa*b
aDepartment of Basic Science, Higher Technological Institute, 10th of Ramadan City, Egypt
bPharmaceutical Medicinal Chemistry & Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo, 11884, Egypt. E-mail: Ibrahimeissa@azhar.edu.eg; Hazemelkady@azhar.edu.eg
cDepartment of Pharmaceutical Organic Chemistry, College of Pharmacy (Boys), Al-Azhar University, Cairo 11884, Egypt
dChemistry Department, Faculty of Science, New Valley University, El-Kharja 72511, Egypt
eBiophysics Department, Faculty of Science, Cairo University, Cairo 12613, Egypt
fBiochemistry and Molecular Biology Department, Faculty of Pharmacy (Boys), Al-Azhar University, Nasr City, Cairo 11231, Egypt
gDepartment of Pharmaceutical Sciences, College of Pharmacy, Princess Nourah bint Abdulrahman University, P.O. Box 84428, Riyadh 11671, Saudi Arabia
hDepartment of Pharmaceutical Sciences, College of Pharmacy, AlMaarefa University, Riyadh 13713, Saudi Arabia
iPharmacognosy and Medicinal Plants Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo 11884, Egypt. E-mail: ametwaly@azhar.edu.eg
jBiopharmaceutical Products Research Department, Genetic Engineering and Biotechnology Research Institute, City of Scientific Research and Technological Applications (SRTA-City), Alexandria, Egypt
First published on 4th August 2023
Abstract
In this work, new thieno[2,3-d]pyrimidine-derived compounds possessing potential anticancer activities were designed and synthesized to target VEGFR-2. The thieno[2,3-d]pyrimidine derivatives were tested in vitro for their abilities to inhibit VEGFR-2 and to prevent cancer cell growth in two types of cancer cells, MCF-7 and HepG2. Compound 18 exhibited the strongest anti-VEGFR-2 potential with an IC50 value of 0.084 μM. Additionally, it displayed excellent proliferative effects against MCF-7 and HepG2 cancer cell lines, with IC50 values of 10.17 μM and 24.47 μM, respectively. Further studies revealed that compound 18 induced cell cycle arrest in G2/M phase and promoted apoptosis in MCF-7 cancer cells. Apoptosis was stimulated by compound 18 by increasing BAX (3.6-fold) and decreasing Bcl-2 (3.1-fold). Additionally, compound 18 significantly raised the levels of caspase-8 (2.6-fold) and caspase-9 (5.4-fold). Computational techniques were also used to investigate the VEGFR-2-18 complex at a molecular level. Molecular docking and molecular dynamics simulations were performed to assess the structural and energetic features of the complex. The protein-ligand interaction profiler analysis identified the 3D interactions and binding conformation of the VEGFR-2-18 complex. Essential dynamics (ED) study utilizing principal component analysis (PCA) described the protein dynamics of the VEGFR-2-18 complex at various spatial scales. Bi-dimensional projection analysis confirmed the proper binding of the VEGFR-2-18 complex. In addition, the DFT studies provided insights into the structural and electronic properties of compound 18. Finally, computational ADMET and toxicity studies were conducted to evaluate the potential of the thieno[2,3-d]pyrimidine derivatives for drug development. The results of the study suggested that compound 18 could be a promising anticancer agent that may provide effective treatment options for cancer patients. Furthermore, the computational techniques used in this research provided valuable insights into the molecular interactions of the VEGFR-2-18 complex, which may guide future drug design efforts. Overall, this study highlights the potential of thieno[2,3-d]pyrimidine derivatives as a new class of anticancer agents and provides a foundation for further research in this area.
1. Introduction
The discovery of effective treatments for cancer is a multifaceted challenge that demands a comprehensive understanding of the underlying biological mechanisms responsible for the disease's onset and progression.1 As such, cancer therapy continues to be an active field of research, with investigators diligently striving to develop novel therapies that can optimize patient outcomes and ultimately diminish the incidence of cancer.2Receptor tyrosine kinases are a class of protein kinases that govern inter- and intracellular signaling via signal transduction pathways.3 These proteins play a significant role in the regulation of cellular processes such as growth, proliferation, differentiation, survival, and metabolism.4 Among these, vascular endothelial growth factor receptor-2 (VEGFR-2) is a crucial mediator of endothelial cell migration and proliferation.5 In the context of cancer cells, VEGFR-2 has been identified as a principal driver of cancer cell proliferation, migration, and angiogenesis.6 Additionally, it has been identified through scientific inquiry that a significant upregulation of VEGFR-2 expression is present in a wide range of cancer types.7 Therefore, VEGFR-2 represents a major therapeutic target for the inhibition of cancer growth and metastasis, given its pivotal function in angiogenesis regulation.8
Our laboratory has discovered a plethora of prospective anticancer agents that exhibit inhibitory activity towards VEGFR-2. These agents stem from diverse classes and derivatives, encompassing nicotinamide,9–11 thiazolidine,12,13 naphthalene,14 pyridine,15 quinoline,16 indole,17 and isatin.18
1.1. Rationale
Thieno[2,3-d]pyrimidin-4(3H)-one is a privileged scaffold that exhibited a high interest due to its wide variety of biological effects.19 Thieno[2,3-d]pyrimidin-4(3H)-one shows a broad anticancer activity directing different vital biological targets.20 Moreover, thieno[2,3-d]pyrimidin-4(3H)-one has been utilized as a core scaffold for the design and synthesis of many tyrosine kinase inhibitors.21 Elkaeed et al. reported compound I as promising anticancer agent targeting EGFR.22 El-Metwally et al. designed and synthesized compound II which exhibited promising VEGFR-2 inhibitory activity.23 Dai et al. introduced compound III as good a series of thienopyrimidine-based receptor tyrosine kinase inhibitors that potently inhibited VEGF and PDGF receptor tyrosine kinases.24 El-Sayed et al. discovered compounds IV and V as potential inhibitors of topoisomerase II (ref. 25) (Fig. 1). | ||
| Fig. 1 (A) Reported thieno[2,3-d]pyrimidin-4(3H)-one derivatives as anticancer agents. (B) Reported 1,3,4-oxadiazol derivatives as promising anticancer agents. | ||
Additionally, a considerable attention has been paid to the 1,3,4-oxadiazol moiety. It was emerged in many bioactive agents.26 A wide number of 1,3,4-oxadiazole derivatives showed potent anticancer effects.27 Narsaiah et al. synthesized a new series of 1,3,4-oxadiazol derivatives VI which showed promising cytotoxicity against HeLa, DU145, HepG2, and MBA-MB-231, cancer cell lines.28 Slawinski et al. designed and synthesized series VII as potential antitumor agents against colon cancer, breast cancer, and cervical cancer.29 Valente et al. discovered compound VIII as promising anticancer agent against leukemia cells30 (Fig. 1).
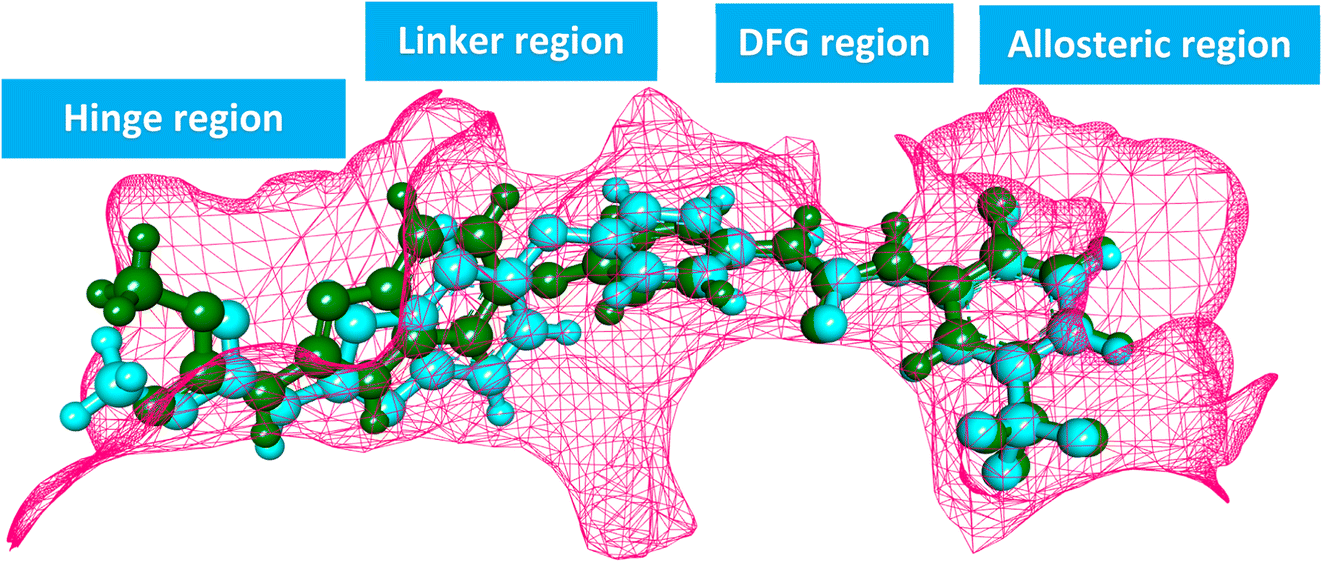
The ATP-binding site is occupied by VEGFR-2 inhibitors which should have four pharmacophoric features (Fig. 2A).31–33 These features are a hetero aromatic system, a spacer moiety, pharmacophore moiety (HBD/HBA), and hydrophobic tail to occupy the hinge region,32 the linker region,34 the DFG motif,35 the allosteric pocket,36 respectively.
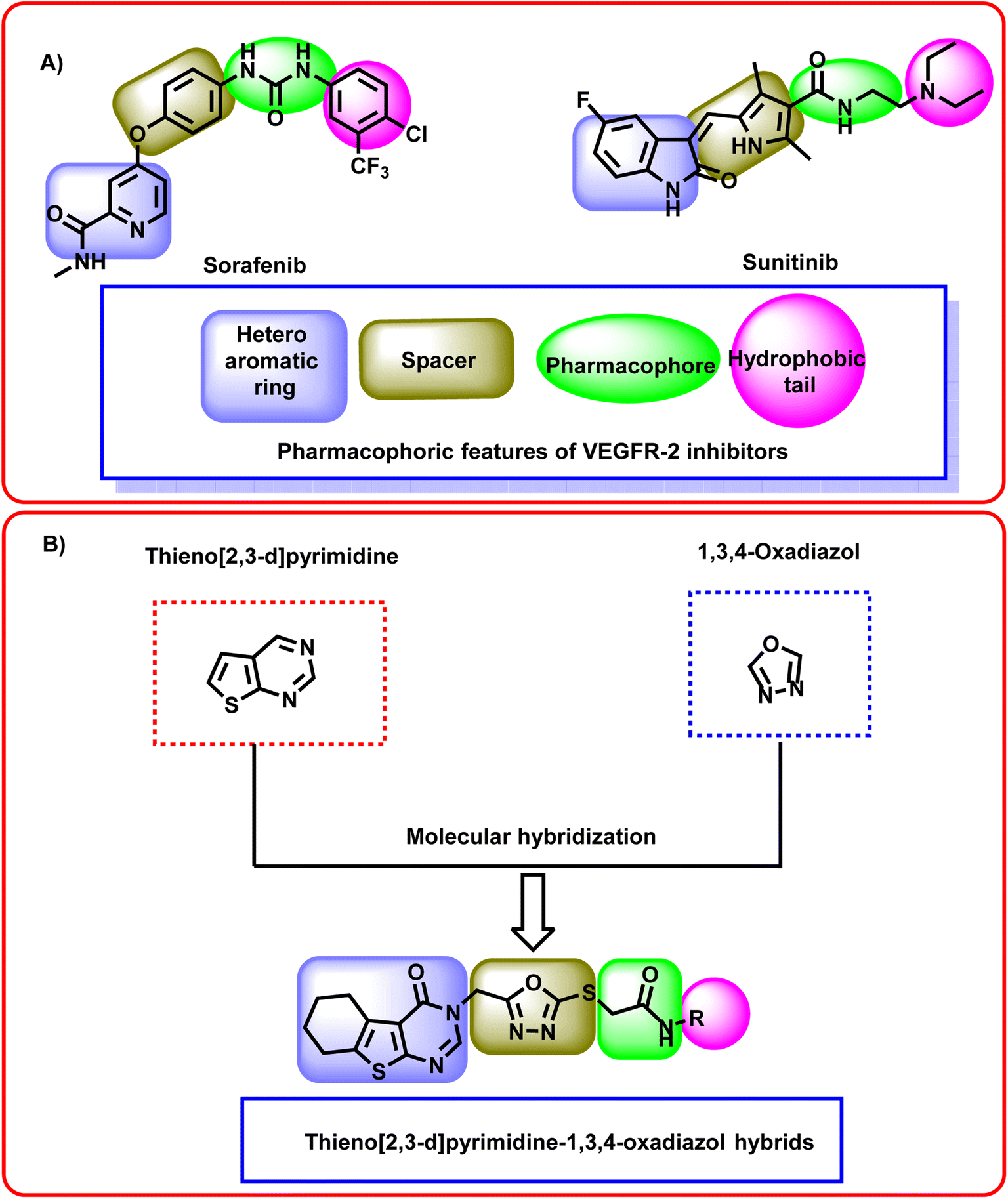 | ||
| Fig. 2 (A) Some reported VEGFR-2 inhibitors showing the essential pharmacophoric features. (B) The hybridization strategy of molecular design. | ||
In this study, a molecular hybridization strategy was applied between thieno[2,3-d]pyrimidin-4(3H)-one and 1,3,4-oxadiazol moieties to reach a new series of VEGFR-2 inhibitors. In which the thieno[2,3-d]pyrimidin-4(3H)-one was used as a hetero aromatic system to occupy the hinge region. 1,3,4-Oxadiazol moiety was used as a spacer moiety to occupy the linker region. The amide moiety was used as a pharmacophore group to occupy the DFG motif region. Finally, different hydrophobic moieties were utilized as a hydrophobic tail to occupy the allosteric binding pocket (Fig. 2B).
2. Results and discussion
2.1. Chemistry
The synthetic itineraries for preparation of the target candidates 18–22 were fully illustrated in Schemes 1–3. According to the reported processes, the starting materials 1 (ref. 37) and 2 (ref. 38) were produced in excellent yields. Then, compound 2 was heated with a potassium hydroxide in absolute ethanol to produce the potassium salt 3. The ester derivative 4 was furnished by refluxing the potassium salt 3 and ethyl chloroacetate in the DMF/KI mixture.39 This ester derivative then reacted with hydrazine hydrate to create the acid hydrazide 5.40 The 5-mercapto-1,3,4-oxadiazole derivative 6 (ref. 40) was obtained via refluxing of acid hydrazide 5 with the clear yellow solution of CS2/KOH in ethanol followed by acidification with HCl. The key starting material 7 was prepared in the present work by refluxing compound 6 with alcoholic KOH while continuous stirring (Scheme 1).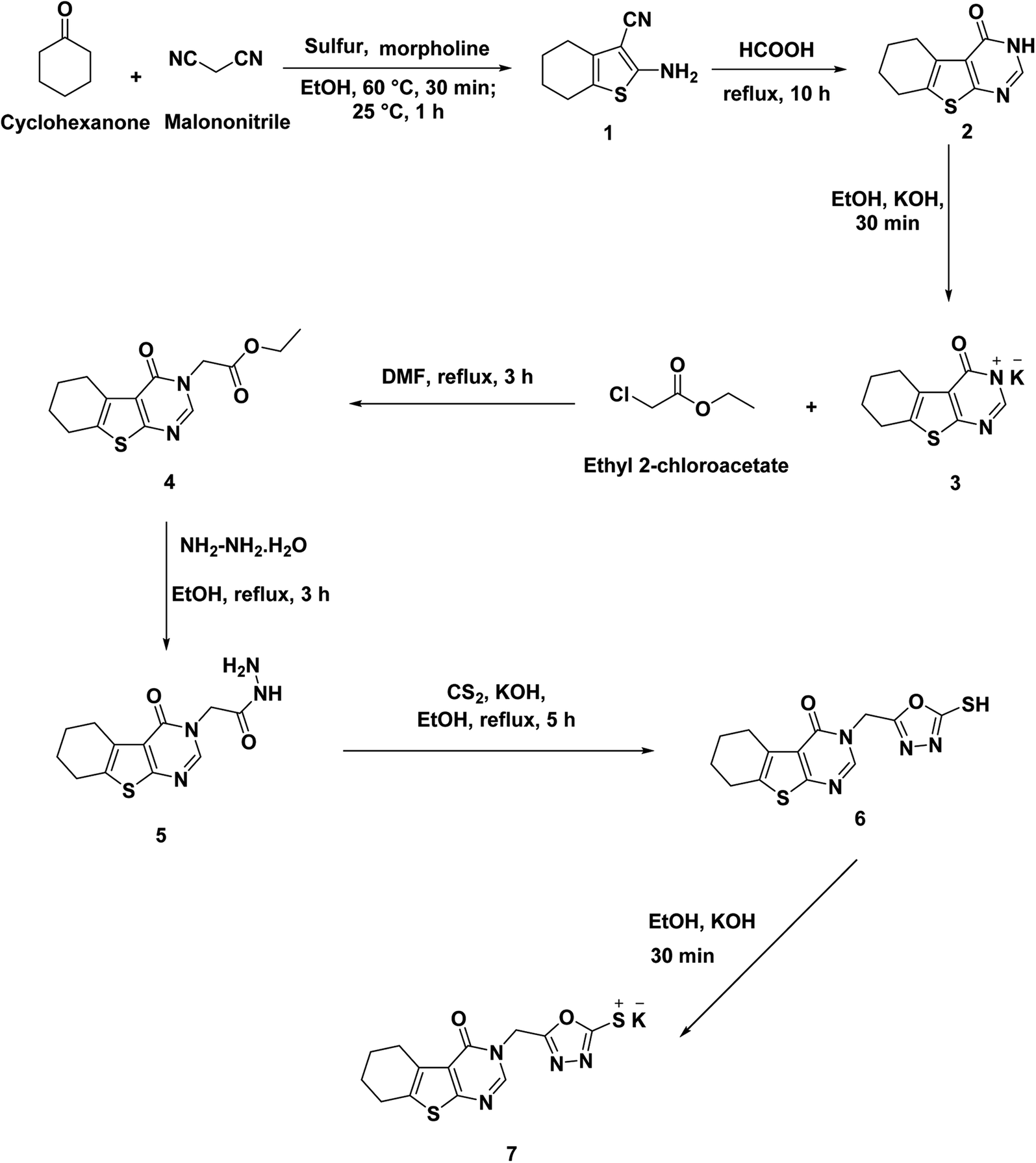 | ||
| Scheme 1 Synthesis of the key starting material 7. | ||
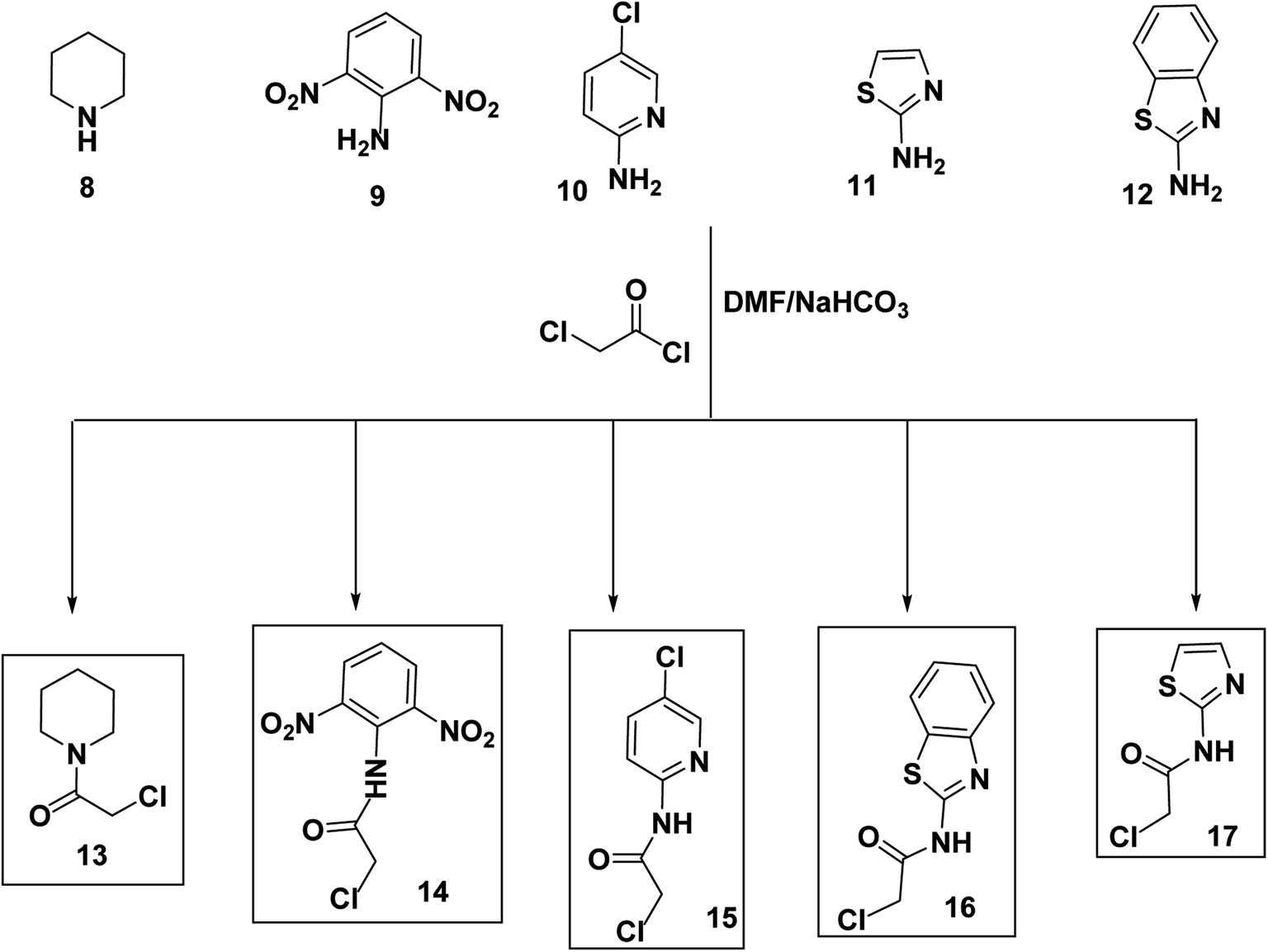 | ||
| Scheme 2 Synthesis of the key intermediates 13–17. | ||
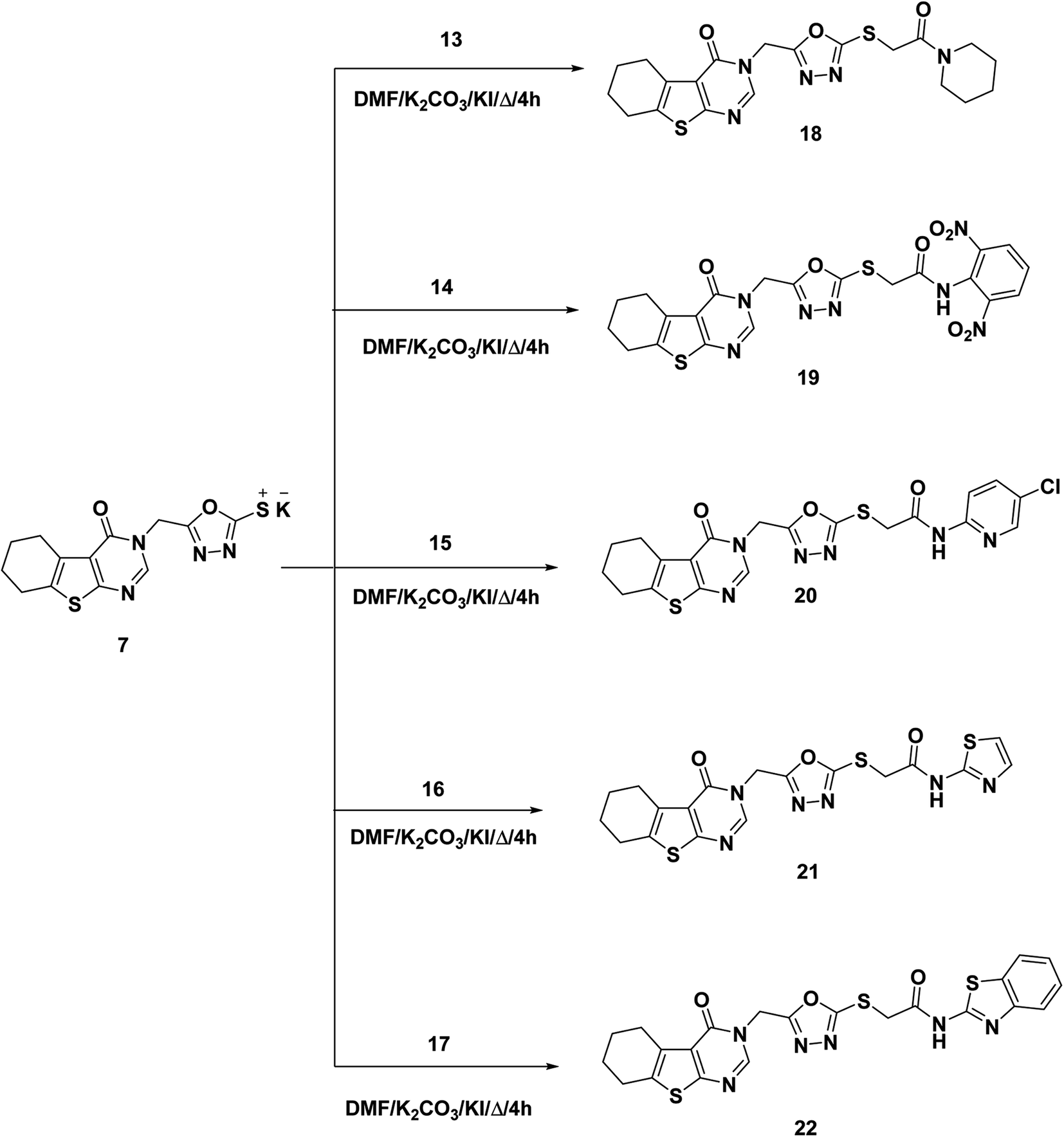 | ||
| Scheme 3 Synthesis of the target compounds 18–22. | ||
Alternatively, chloroacetyl chloride was dropped onto different solutions of DMF each containing the proper amine namely piperidine (8), 2,6-dinitroaniline (9), 5-chloropyridin-2-amine (10), thiazol-2-amine (11), and benzo[d]thiazol-2-amine (12) to produce the corresponding acetamide derivatives namely, 2-chloro-1-(piperidin-1-yl)ethan-1-one (13), 2-chloro-N-(2,6-dinitrophenyl)acetamide (14), 2-chloro-N-(5-chloropyridin-2-yl)acetamide (15), N-(benzo[d]thiazol-2-yl)-2-chloroacetamide (16), and 2-chloro-N-(thiazol-2-yl)acetamide (17) (Scheme 2).
As a final point, compound 7 was heated with the appropriate acetamide derivatives 13–17 in DMF mixtures comprising catalytic KI to produce the final target derivatives 18–22, respectively (Scheme 3).
The IR spectra of the final candidates exhibited the appearance of absorption bands at the ranges of 3291–3400 cm−1 and 1661–1674 cm−1 due to the NH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, respectively. In addition, 1H NMR analyses showed singlet signal appeared at around δ 8.44 ppm corresponding to the pyrimidine proton besides characteristic signals for additional aromatic and aliphatic protons. Taken compound 18 as representative example, it showed many characteristic signals at aliphatic region corresponding to the piperidine moiety. Moreover, 13C NMR spectra of such compound confirmed the previous results as the aliphatic protons of appeared at the aliphatic region.
O groups, respectively. In addition, 1H NMR analyses showed singlet signal appeared at around δ 8.44 ppm corresponding to the pyrimidine proton besides characteristic signals for additional aromatic and aliphatic protons. Taken compound 18 as representative example, it showed many characteristic signals at aliphatic region corresponding to the piperidine moiety. Moreover, 13C NMR spectra of such compound confirmed the previous results as the aliphatic protons of appeared at the aliphatic region.
2.2. Biological investigations
| Comp. | IC50a (μM) | VEGFR-2 IC50a (μM) | |
|---|---|---|---|
| MCF-7 | HepG2 | ||
| a The results are the mean of three experiments. | |||
| 18 | 10.17 ± 1.01 | 24.27 ± 2.08 | 0.18 ± 0.004 |
| 19 | 16.50 ± 1.16 | 31.78 ± 1.15 | 0.29 ± 0.007 |
| 20 | 5.38 ± 0.45 | 4.84 ± 0.38 | 0.83 ± 0.017 |
| 21 | 6.31 ± 0.16 | 11.83 ± 1.05 | 0.42 ± 0.008 |
| 22 | 5.73 ± 0.18 | 5.51 ± 0.25 | 0.26 ± 0.006 |
| Sorafenib | 9.98 ± 0.6 | 13.26 ± 0.3 | 0.12 ± 0.002 |
In details, compounds 20 showed the highest anti-proliferative activities with IC50 values of 5.38 and 4.84 μM against MCF-7 and HepG2 cells, respectively. In addition, compounds 21 (IC50 = 6.31 and 11.83 μM) and 22 (IC50 = 5.73 and 5.51 μM) showed strong anti-proliferative activities against MCF-7 and HepG2 cells, respectively. For compounds 18 (IC50 = 10.17 and 24.27 μM) and 19 (IC50 = 16.50 and 31.78 μM) showed moderate anti-proliferative activities against MCF-7 and HepG2 cells, respectively.
2.2.3.1. Cell cycle analysis. In this test, MCF-7 cells were treated with compound 18 (at a concentration of 10.17 μM) for 72 h. Later, the cells were stained with propidium iodide, and their distribution throughout the various cell cycle phases was determined by using a flow cytometric analysis technique. The results in Table 2 and ESI Data† demonstrated a decrease in the proportion of MCF-7 cells treated with compound 18 at the G0–G1 and S phases. In the G0–G1 phase, it decreased from 58.02% to 56.46%, and at the S phase, it decreased from 30.1% to 28.14%. The proportion of MCF-7 cells, on the other hand, increased in the compound 18-treated cells during the G2/M phase (15.4% vs. 11.88% control cells). These findings supported that compound 18 could halt cell growth at the G2/M phase.
| Sample | Cell cycle distribution (%) | ||
|---|---|---|---|
| %G0–G1 | %S | % G2/M | |
| MCF-7 | 58.02 | 30.1 | 11.88 |
| Compound 18-treated MCF-7 | 56.46 | 28.14 | 15.4 |
2.2.3.2. Apoptosis assay. This method involved staining MCF-7 cells with annexin V and propidium iodide (PI) and then incubating them with compound 18 (at a concentration of 10.17 μM) for 72 h. When compared to untreated control cells, compound 18 caused more MCF-7 cells to undergo apoptosis (Table 3 and ESI Data†). Apoptosis was specifically induced by compound 18 by 23.35% (early apoptosis: 15.96% and late apoptosis: 7.39%), as opposed to 0.72% in control cells (early apoptosis: 0.53% and late apoptosis: 0.19%).
| Sample | Total | Early apoptosis | Late apoptosis | Necrosis |
|---|---|---|---|---|
| MCF-7 | 2.57 | 0.53 | 0.19 | 1.85 |
| Compound 18 treated MCF-7 | 25.76 | 15.96 | 7.39 | 2.41 |
In this experiment, the effects of compound 18 at a concentration of 10.17 μM (IC50 value) on the BAX, Bcl-2, caspase-8, and caspase-9 in MCF-7 cells were investigated. According to the findings in Table 4, compound 18 elevated BAX levels, a protein that promotes apoptosis, by 3.6-fold and significantly decreased Bcl-2 levels by 3.1-fold when compared to control cells. As well, compound 18 revealed 2.6-fold higher levels in caspase-8 compared to the control. Additionally, it achieved a significant increase in caspase-9 levels (5.4-fold) in comparison to the control group.
| Sample | Protein expression | |||
|---|---|---|---|---|
| BAX | Bcl-2 | Caspase-8 | Caspase-9 | |
| Cont. MCF-7 | 1 | 1 | 1 | 1 |
| Compound-18 treated MCF-7 | 3.669 | 0.315 | 2.607 | 5.422 |
| Sample | TNF-α | IL6 |
|---|---|---|
| Cont. MCF-7 | 1 | 1 |
| Compound-18 treated MCF-7 | 0.335 | 0.393 |
2.3. Computational studies
 | ||
| Fig. 3 Superimposition of the native and docked co-crystallized ligand in the active site of VEGFR-2 during the validation of the molecular docking procedure. | ||
The binding energies of the synthesized thieno[2,3-d]pyrimidine derivatives were presented in Table 6. Interestingly, the docking results were harmonious with the biological findings where compound 18 (the most promising VEGFR-2 inhibitor) achieved binding score of −22.71 kcal mol−1 against the VEGFR-2 enzyme. This value were higher than other derivatives and sorafenib (−21.77 kcal mol−1).
| Comp. | ΔG [kcal mol−1] |
|---|---|
| 18 | −22.71 |
| 19 | −22.52 |
| 20 | −21.00 |
| 21 | −19.99 |
| 22 | −20.60 |
| Sorafenib | −21.77 |
As shown in Fig. 4, sorafenib's docking pose involved three crucial H-bonding interactions with Cys917, Glu883, and Asp1044. It also engaged in a number of hydrophobic interactions with the hydrophobic pocket created by Leu887, Leu1017, and Ile890.
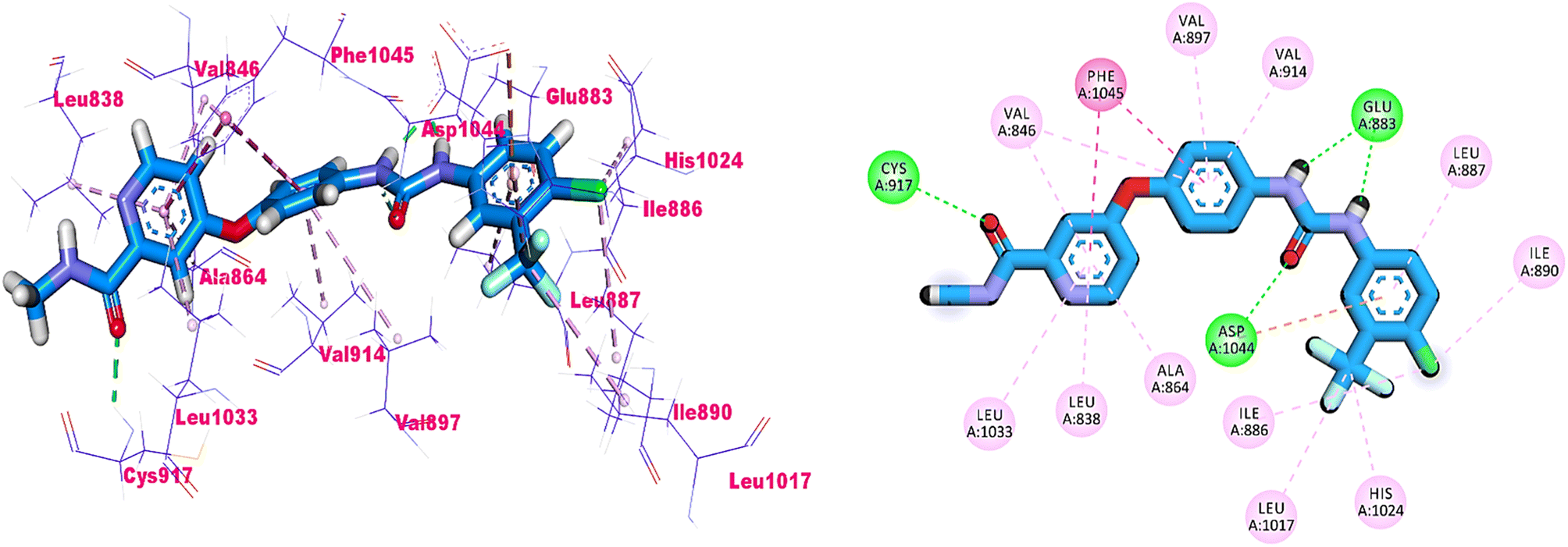 | ||
| Fig. 4 3D and 2D illustrations of sorafeib in the active site of VEGFR-2. | ||
Compounds 18–22 were able to recognize the VEGFR-2 kinase binding site and interact with important amino acids. The thieno[2,3-d]pyrimidine moieties were oriented towards the hinge region and formed several hydrophobic interactions in addition to one H-bond with the Cys917 residue. The 1,3,4-oxadiazole moieties were then incorporated into the linker region and formed hydrophobic interactions with the Val914, Val846, Ala864, Phe1045, and Cys1044. Also, the same compounds interacted via two H-bonds with the NH moiety of Asp1044 and the carboxylate side chain of Glu883 of the conserved DFG motif region. Finally, the terminal heads of compounds 18–22 represented by piperidine (18), 2,6-dinitrophenyl (19), 5-chloropyridine (20), thiazole (21), and benzo[d]thiazole (22) interacted Leu887, Ile886, and Ile890 residues and tightly fit the hydrophobic region (Fig. 5). These findings may gave an explanation for the designed candidates' potent VEGFR-2 inhibitory activities.
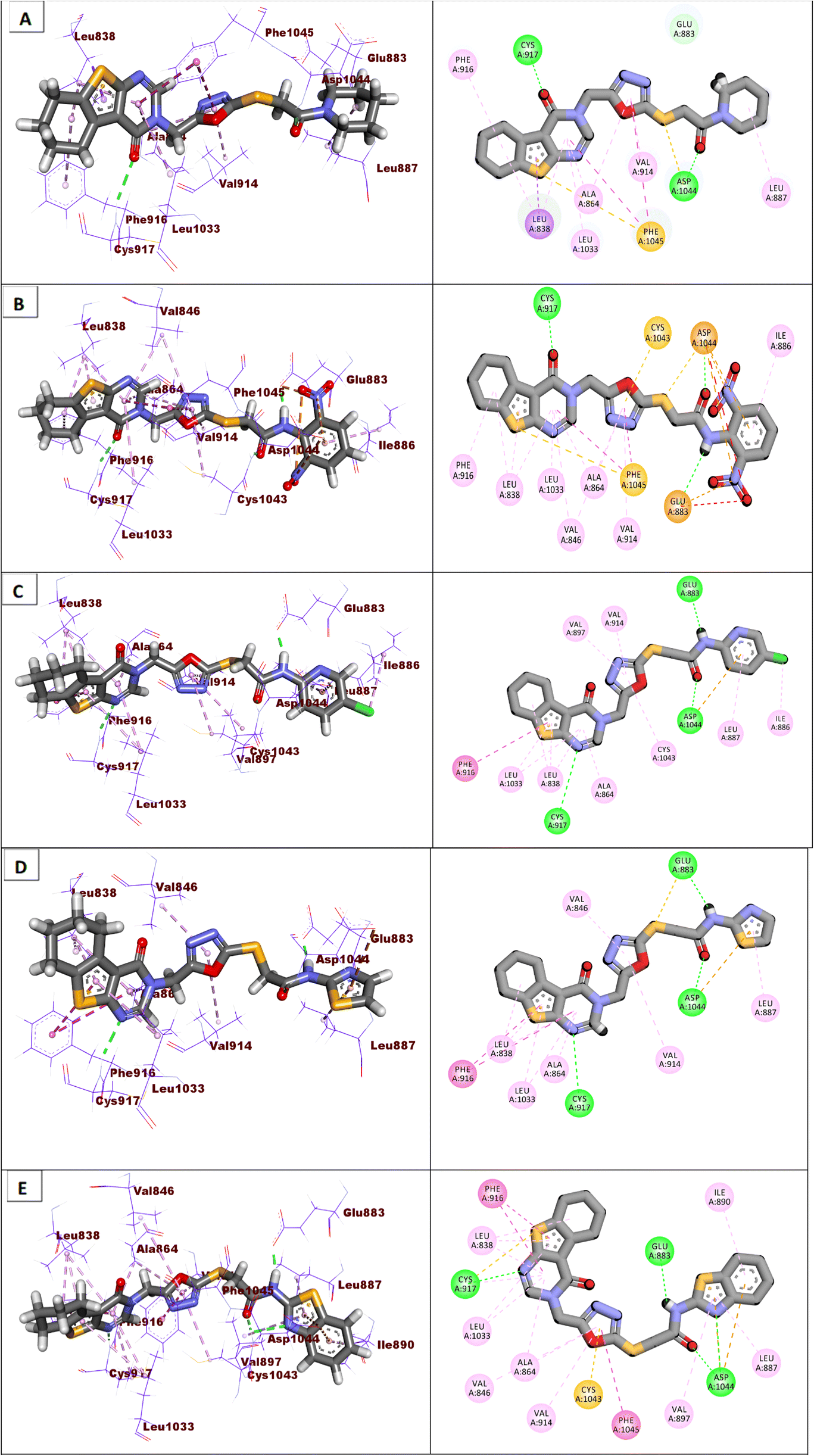 | ||
| Fig. 5 Binding modes of the synthesized compounds against the active site of VEGFR-2. (A) Compound 18, (B) compound 19, (C) compound 20, (D) compound 21, (E) compound 22. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 656 Å2 and 17378 Å2, respectively. Fig. 6E revealed that the overall number of H-bonds is dynamic, with both systems exhibiting fluctuations around 70 bonds. Based on the previous results, the protein's structure is likely to be stable. The fluctuation of amino acids is quite stable (show fluctuation values of less than 2 Å) in the RMSF plot (Fig. 6F), except for the N-terminus (10.5 Å for the holo protein), the Arg840:Phe843 loop of the holo protein (2.9 Å), the Tyr994:Asp996 loop of the holo protein (2.3 Å), the Tyr994:Phe997 loop of the apo protein (2.5 Å), the Gly1046:Leu1065 loop of both systems (6.8 Å), the Pro1106:Ile1112 loop of the apo protein (2.6 Å), and the C terminal (10.3 Å for the apo protein and 6.3 Å for the holo protein). Stable binding occurs between 18 and the VEGFR-2 protein, with an average distance of 8.3 Å between the two centers of masses (Fig. 6G).
656 Å2 and 17378 Å2, respectively. Fig. 6E revealed that the overall number of H-bonds is dynamic, with both systems exhibiting fluctuations around 70 bonds. Based on the previous results, the protein's structure is likely to be stable. The fluctuation of amino acids is quite stable (show fluctuation values of less than 2 Å) in the RMSF plot (Fig. 6F), except for the N-terminus (10.5 Å for the holo protein), the Arg840:Phe843 loop of the holo protein (2.9 Å), the Tyr994:Asp996 loop of the holo protein (2.3 Å), the Tyr994:Phe997 loop of the apo protein (2.5 Å), the Gly1046:Leu1065 loop of both systems (6.8 Å), the Pro1106:Ile1112 loop of the apo protein (2.6 Å), and the C terminal (10.3 Å for the apo protein and 6.3 Å for the holo protein). Stable binding occurs between 18 and the VEGFR-2 protein, with an average distance of 8.3 Å between the two centers of masses (Fig. 6G).
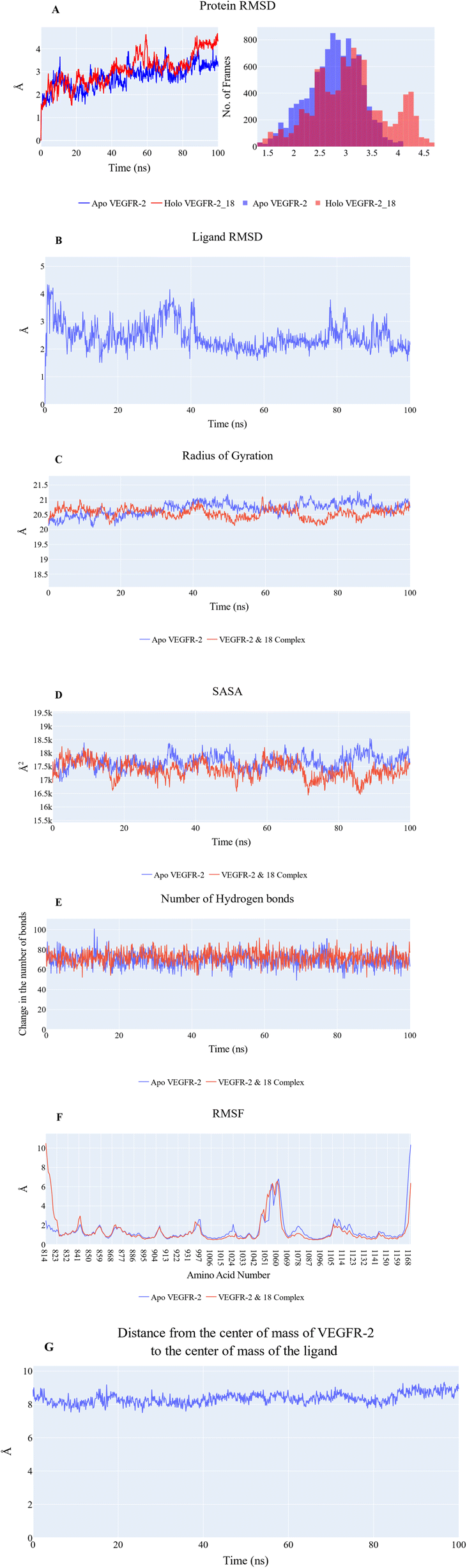 | ||
| Fig. 6 (A) RMSD values from the trajectory for the VEGFR-2 protein in apo (blue line) and holo forms (red line), (B) the ligand RMSD values, (C) radius of gyration for the VEGFR-2 protein in apo (blue line) and holo forms (red line), (D) SASA for the VEGFR-2 protein in apo (blue line) and holo forms (red line), (E) change in the number of hydrogen bonds for the VEGFR-2 protein in apo (blue line) and holo forms (red line), (F) RMSF for the VEGFR-2 protein in apo (blue line) and holo forms (red line), (G) distance from the center of mass of compound 18 and VEGFR-2 protein. | ||
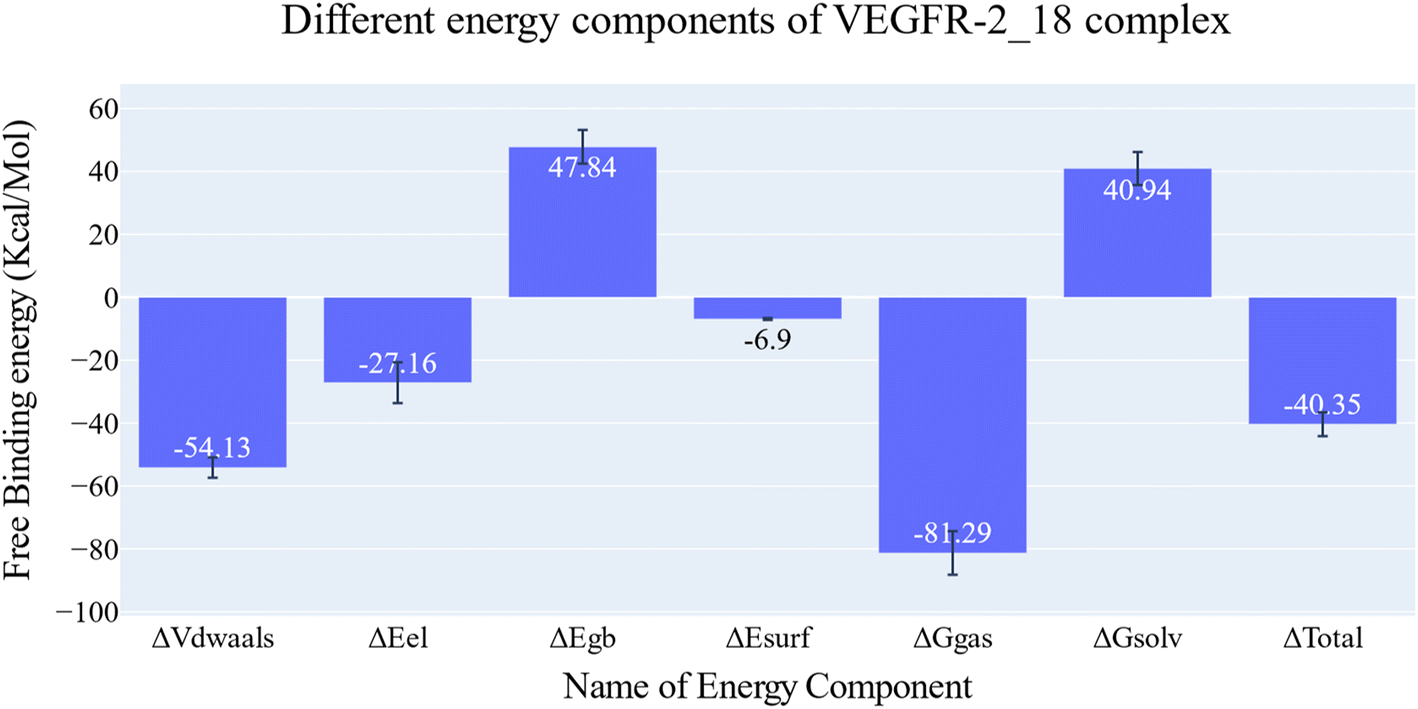 | ||
| Fig. 7 Different energetic components of MM-GBSA and their values. Bars represent the standard deviations. | ||
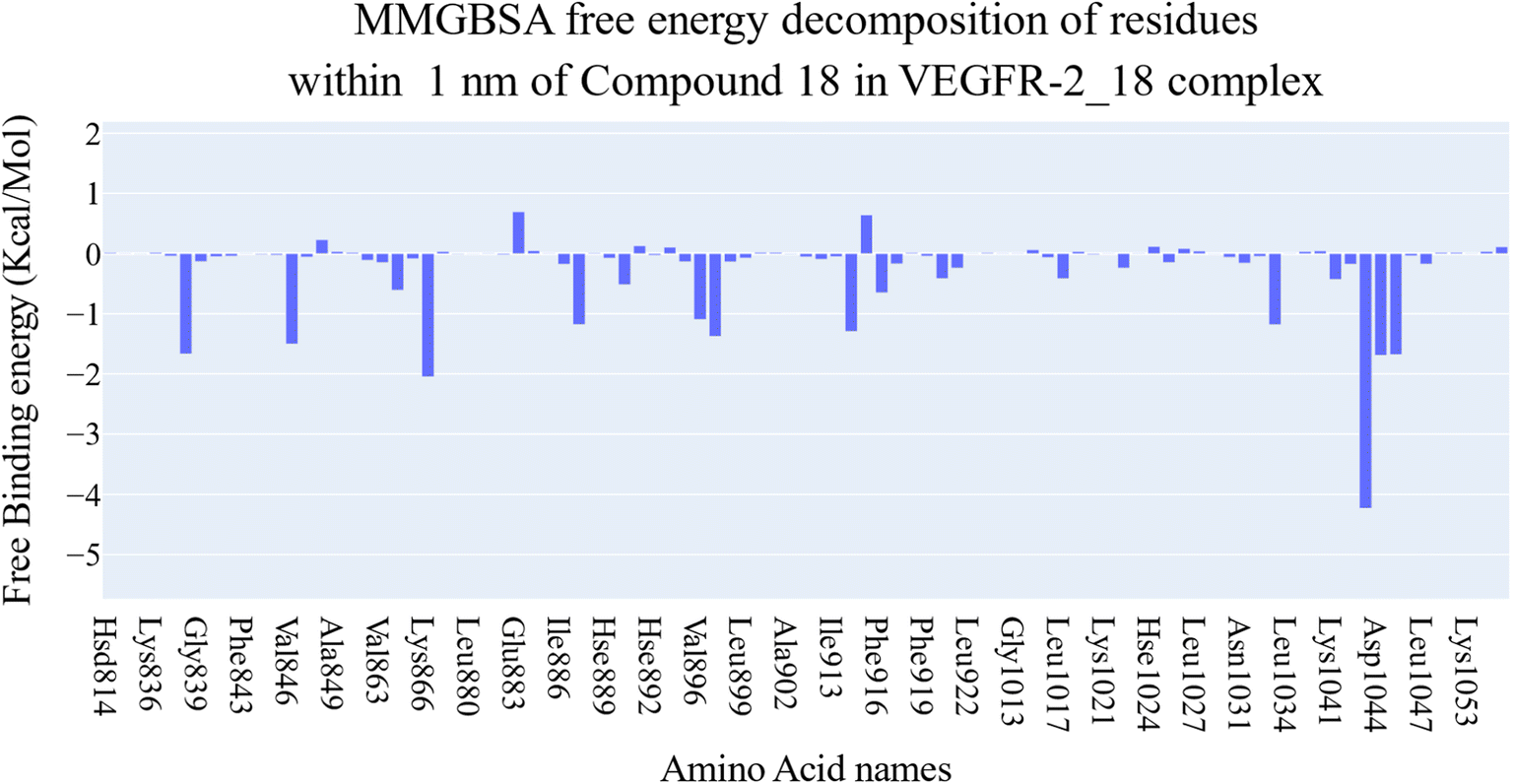 | ||
| Fig. 8 Binding free energy decomposition of the VEGFR-2_18 complex. | ||
Fig. 9A–C indicated that ten amino acids (Leu838, Val846, Leu887, Leu897, Val914, Leu1017, Leu1033, Cys1043, Asp1044, and Phe1045) exhibit extraordinarily persistent hydrophobic interactions (with at least 93.7% incidence). Asp1044 also has a very high H-bond formation efficiency (94.5%). After obtaining representative frames from the trajectory clustering using TTClust, we utilized PLIP to identify contacts and retrieve 3D binding conformations as.pse files (Fig. 10).
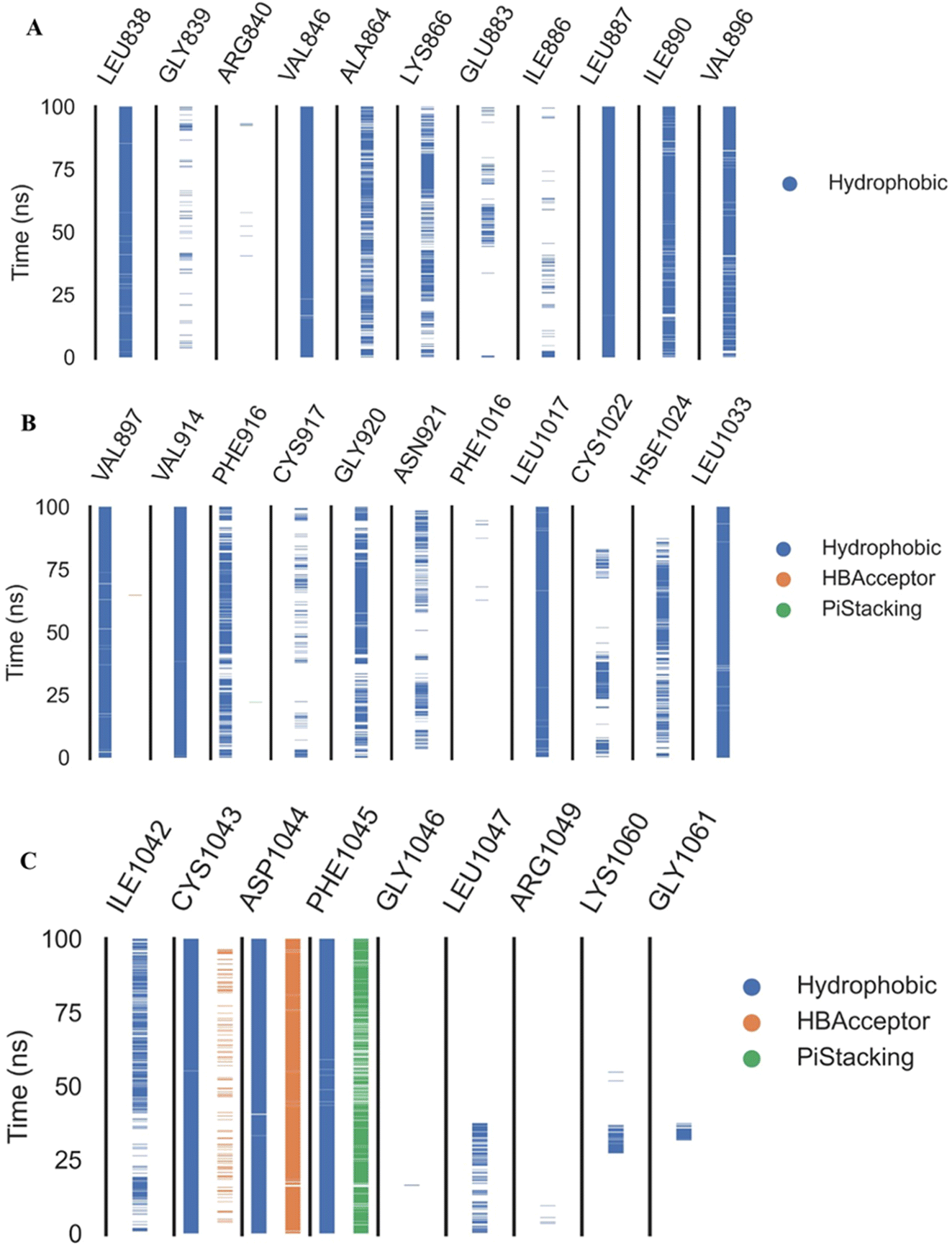 | ||
| Fig. 9 The amino acids, the types of interactions with compound 18, and their occurrence during the whole simulation time using the ProLIF python library. (A) Amino acid sequence from LEU838 to VAL896, (B) amino acid sequence from VAL896 to LEU1033, and (C) amino acid sequence from ILE1042 to GLY1061. | ||
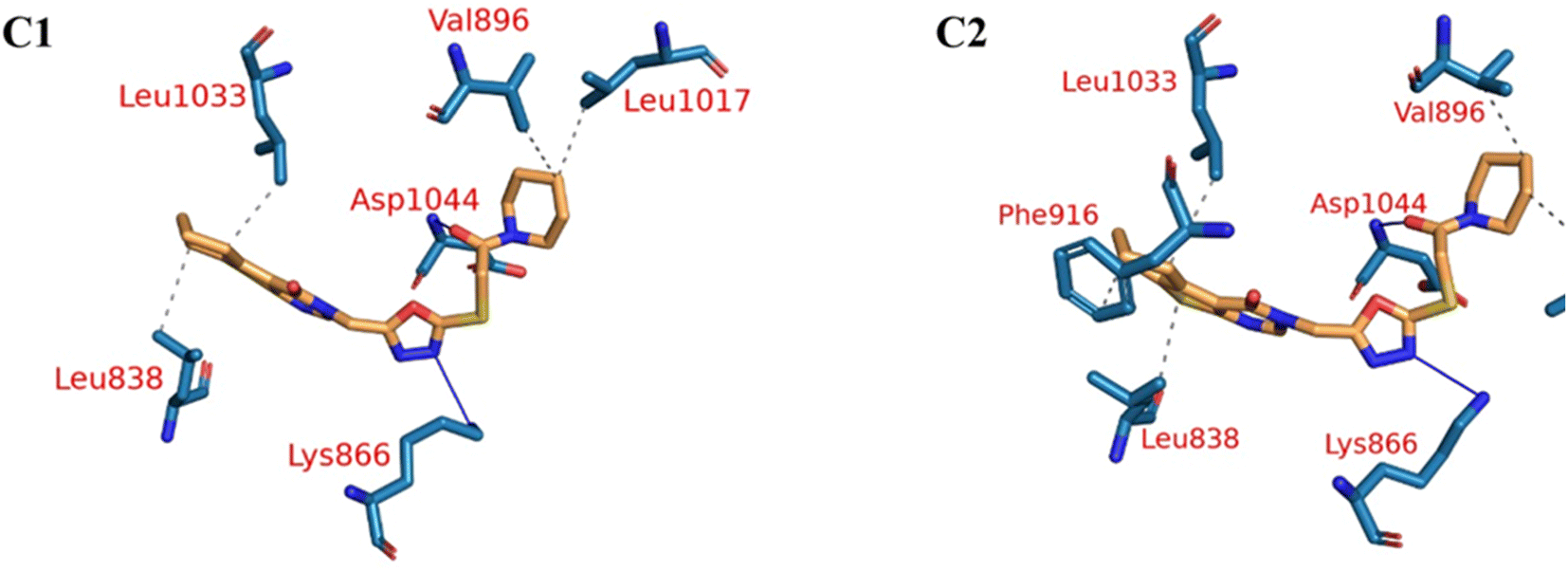 | ||
| Fig. 10 The two clusters representative obtained from TTClust and their 3D interactions with the molecule 18. Grey dashed lines: hydrophobic interactions, blue solid lines: H-bonds, orange sticks: molecule 18, blue sticks: amino acids of VEGFR-2 protein. | ||
PCA was employed to unveil coordinated motion of high amplitude in the trajectory. Using the scree plot (the eigenvalues vs. the eigenvector index) and doing cumulative sums of eigenvalues for each new eigenvector, allowed us to calculate the number of eigenvectors that would represent the essential subspace (Fig. 11). In this case, as can be seen, the first three eigenvectors accounted for almost 87% of the overall variance, with the first eigenvector alone accounting for 75%. Furthermore, the distribution of eigenvectors values of the first PCs is often non-Gaussian. The first two eigenvectors have separated peaks, whereas the third and fourth PCs have peaks that are not totally separated and the remainder starts to show a Gaussian distribution (Fig. 12). The VEGFR-2 cosine content was determined in both the apo and holo simulations to establish the randomness of the motion captured by the first 10 eigenvectors. From what can be seen in Fig. 13, all of the first ten PCs, except for the second one (0.5), in the holo simulation have cosine content values below 0.2, indicating that they are not a random motion. The results of the apo simulation, except for the third eigenvector (cosine content of 0.22), follow the same pattern. Thus, the first three eigenvectors were selected to reflect the essential subspace. The root mean square inner product (RMSIP) shows that there is only a 27.8% overlap between the two subspaces (the first three eigenvectors), indicating that the two trajectories were sampled differently. We also determined the overlap between the apo and holo trajectory C matrices and it was 37% similar.
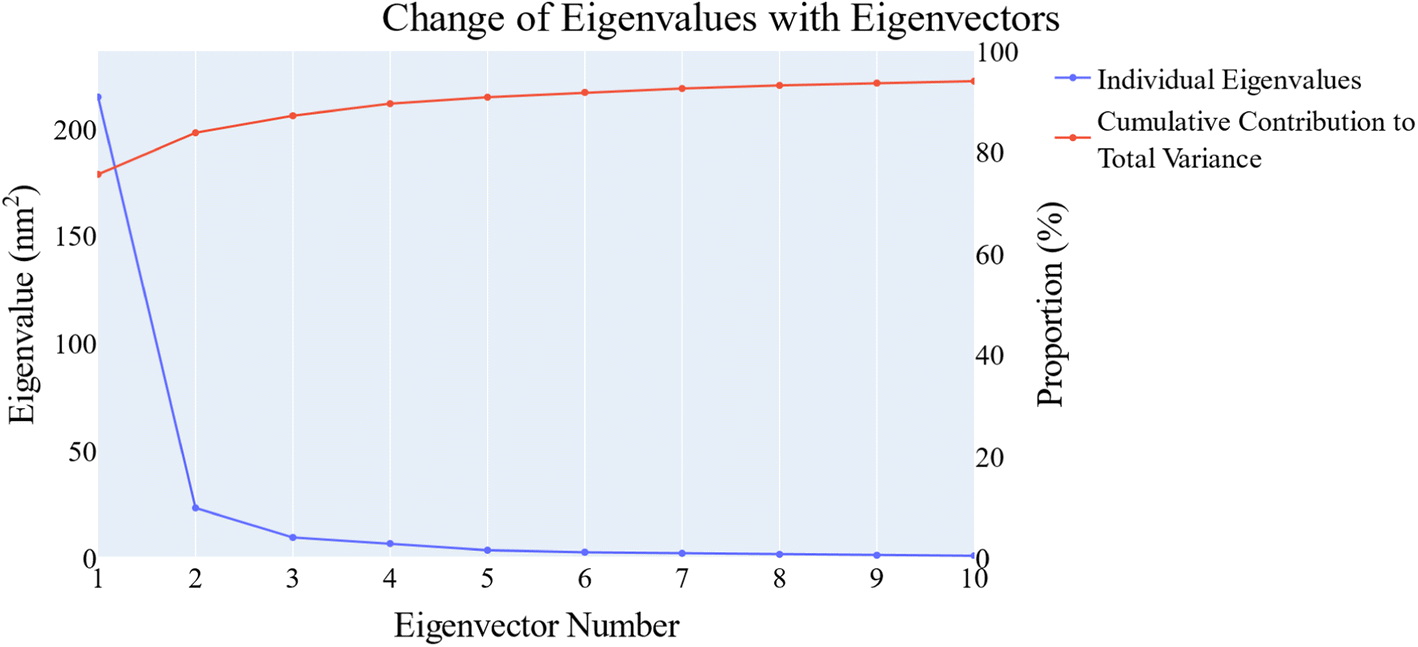 | ||
| Fig. 11 The change in the eigenvalues with increasing the eigenvectors (blue line). In addition, the cumulative variance retained in the eigenvectors is shown (red line). | ||
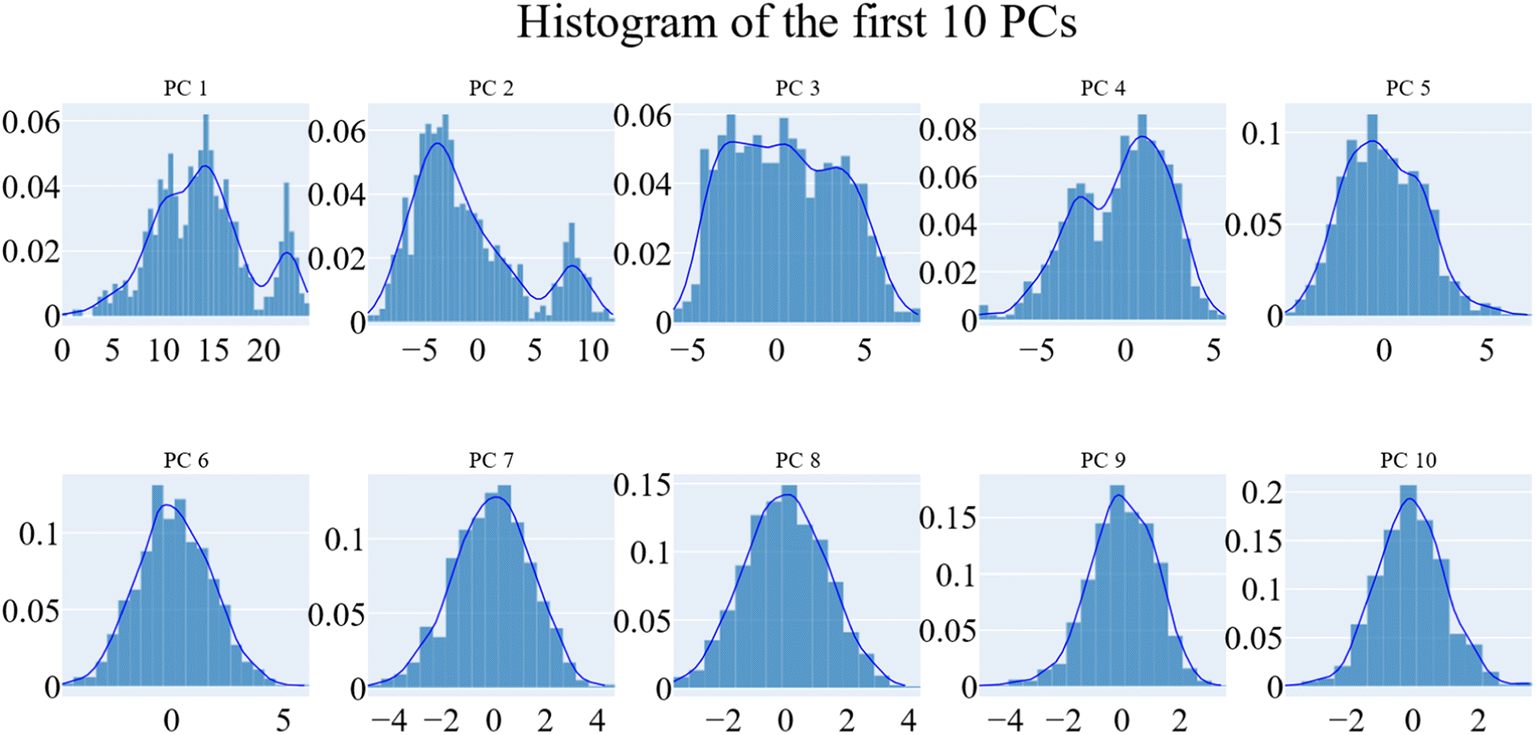 | ||
| Fig. 12 The distribution of the first ten eigenvectors. | ||
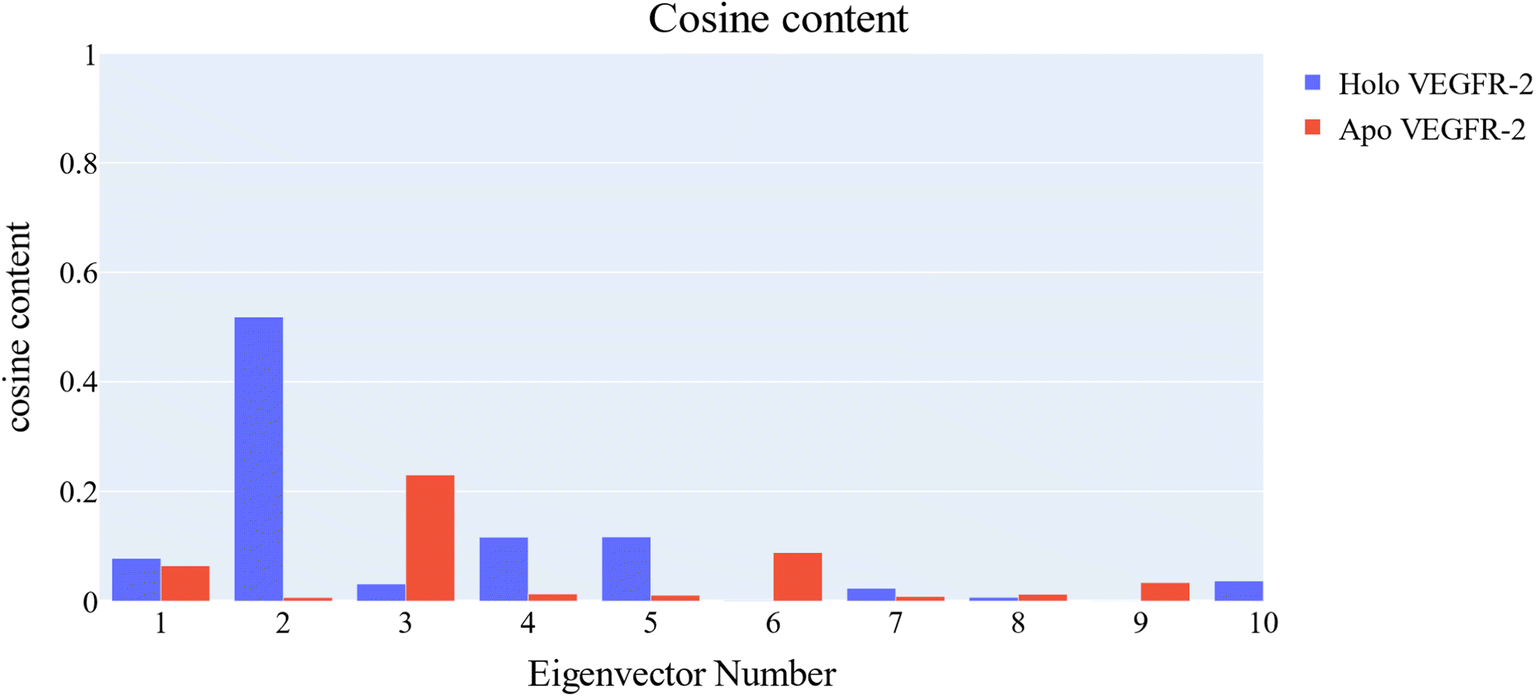 | ||
| Fig. 13 Values of the cosine content of the first ten eigenvectors for the two trajectories. | ||
As a way to evaluate how similarly sampled the two trajectories are, we projected each trajectory onto the first three eigenvectors of the combined C matrix (Fig. 14–16). In these diagrams, the average structure of each trajectory was shown as a bigger marker, and the projections were made using the first three main components in combinations of two. Fig. 14 (projection on the first two eigenvectors) revealed a sparse overlap between the two trajectories and a wide gap between the two average structures. Fig. 15 demonstrated that the average structures of the two trajectories are quite similar and that there is a lot of overlap between the two trajectories when projected on the first and third eigenvectors. Lastly, a projection on the second and third eigenvectors (Fig. 16) revealed a moderate overlap between the two trajectories and a separation between the average structures similar to that of Fig. 14. Porcupine figures were also made to illustrate the motion captured in the first three eigenvectors in the two simulations (Fig. 17). These three eigenvectors all capture motion from the Gly1046:Leu1065 loop opening, but in different directions.
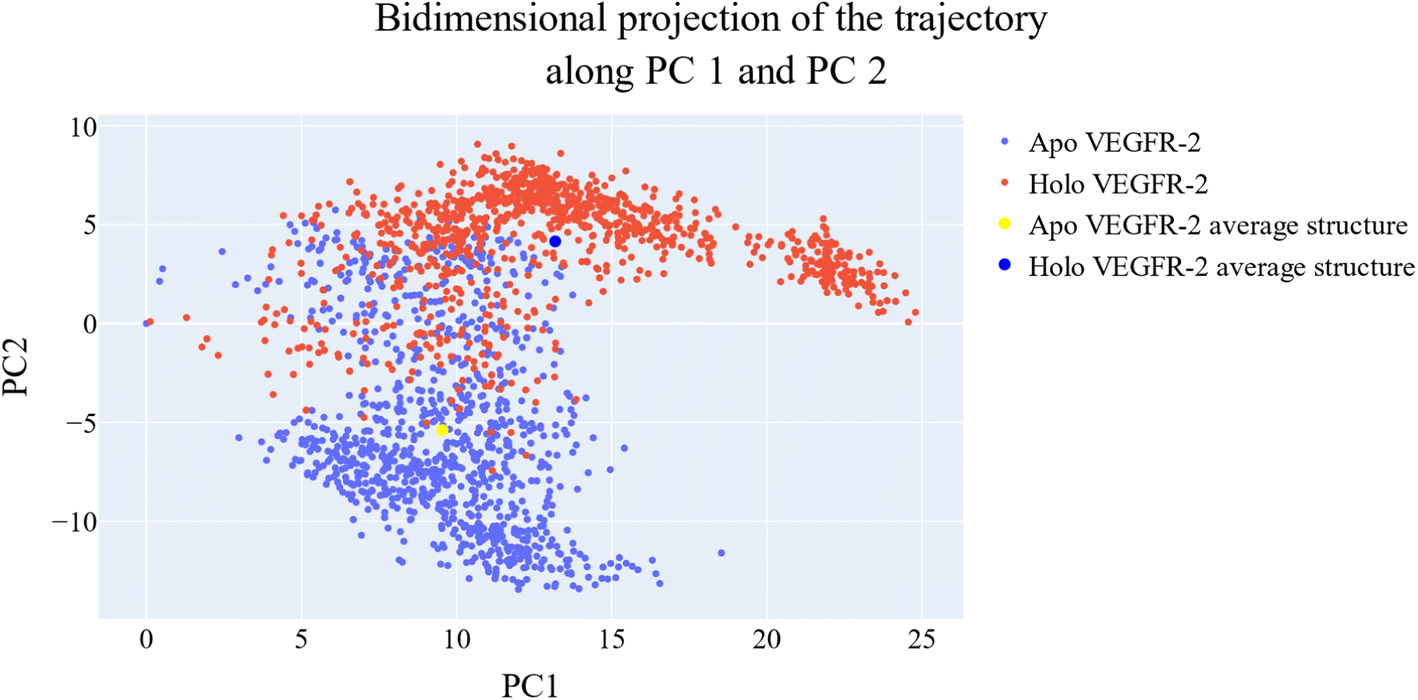 | ||
| Fig. 14 The projection of each trajectory on the first two eigenvectors. Small blue dots are the frames of the VEGFR-2 in the apo simulation, red blue dots are the frames of the VEGFR-2 in the holo simulation, the large yellow dot is the average structure of VEGFR-2 from the apo simulation, and the large blue dot is the average structure of VEGFR-2 from the holo simulation. | ||
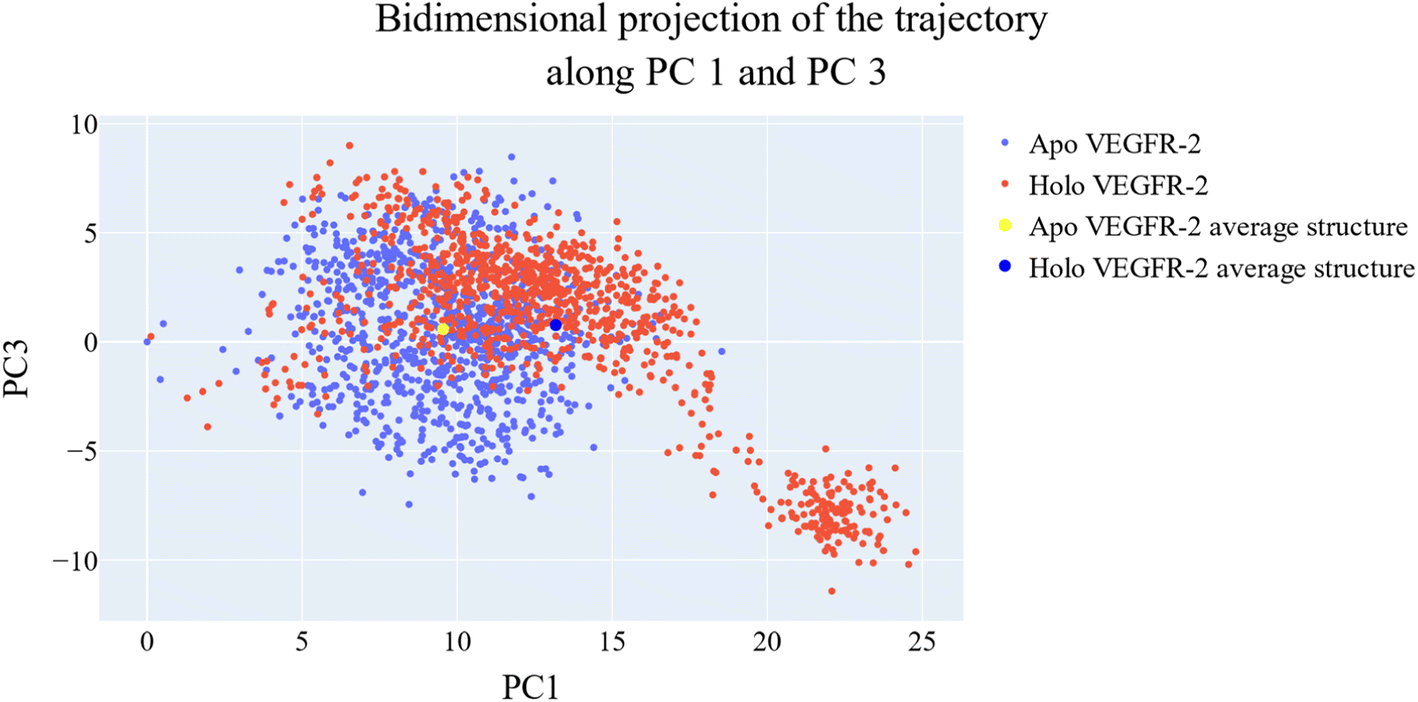 | ||
| Fig. 15 Shows the projection of each trajectory on the first and third eigenvectors. Small blue dots are the frames of the VEGFR-2 in the apo simulation, red blue dots are the frames of the VEGFR-2 in the holo simulation, the large yellow dot is the average structure of VEGFR-2 from the apo simulation, the large blue dot is the average structure of VEGFR-2 from the holo simulation. | ||
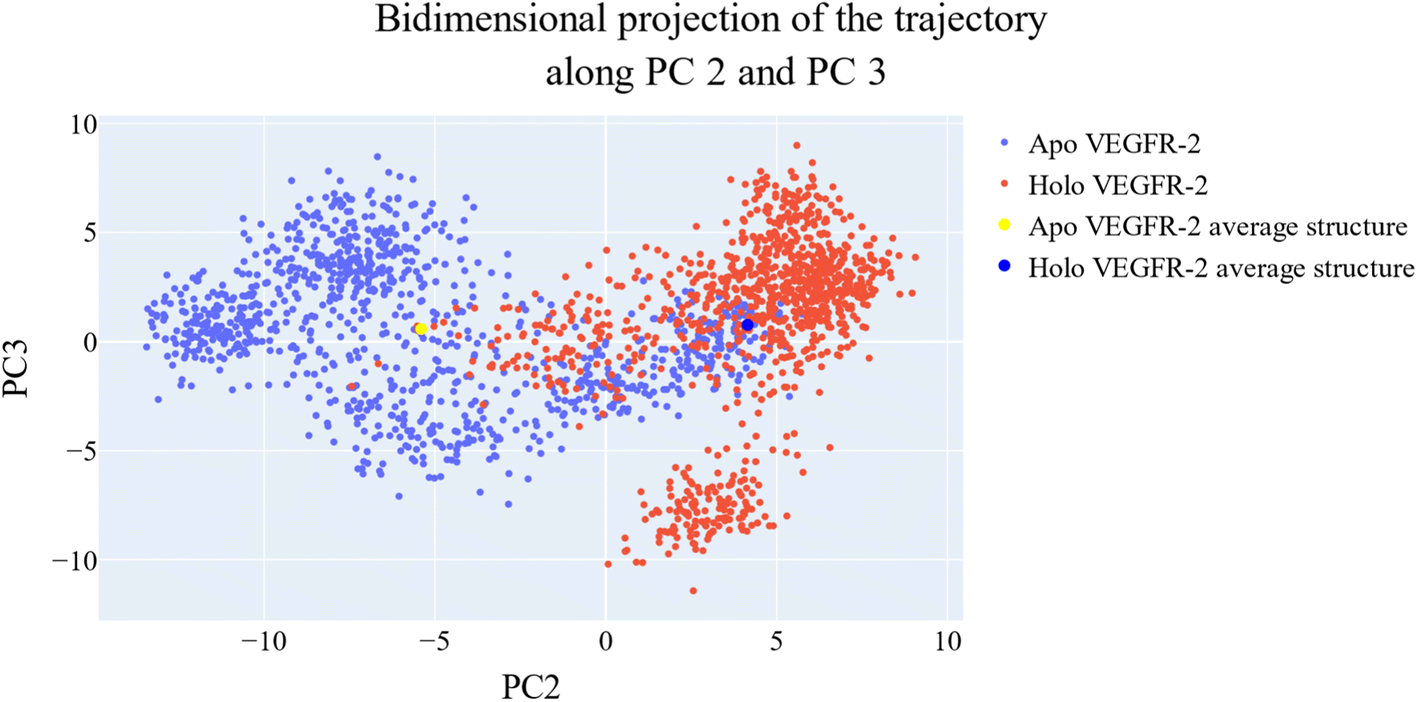 | ||
| Fig. 16 The projection of each trajectory on the second and third eigenvectors. Small blue dots are the frames of the VEGFR-2 in the apo simulation, red blue dots are the frames of the VEGFR-2 in the holo simulation, the large yellow dot is the average structure of VEGFR-2 from the apo simulation, and the large blue dot is the average structure of VEGFR-2 from the holo simulation. | ||
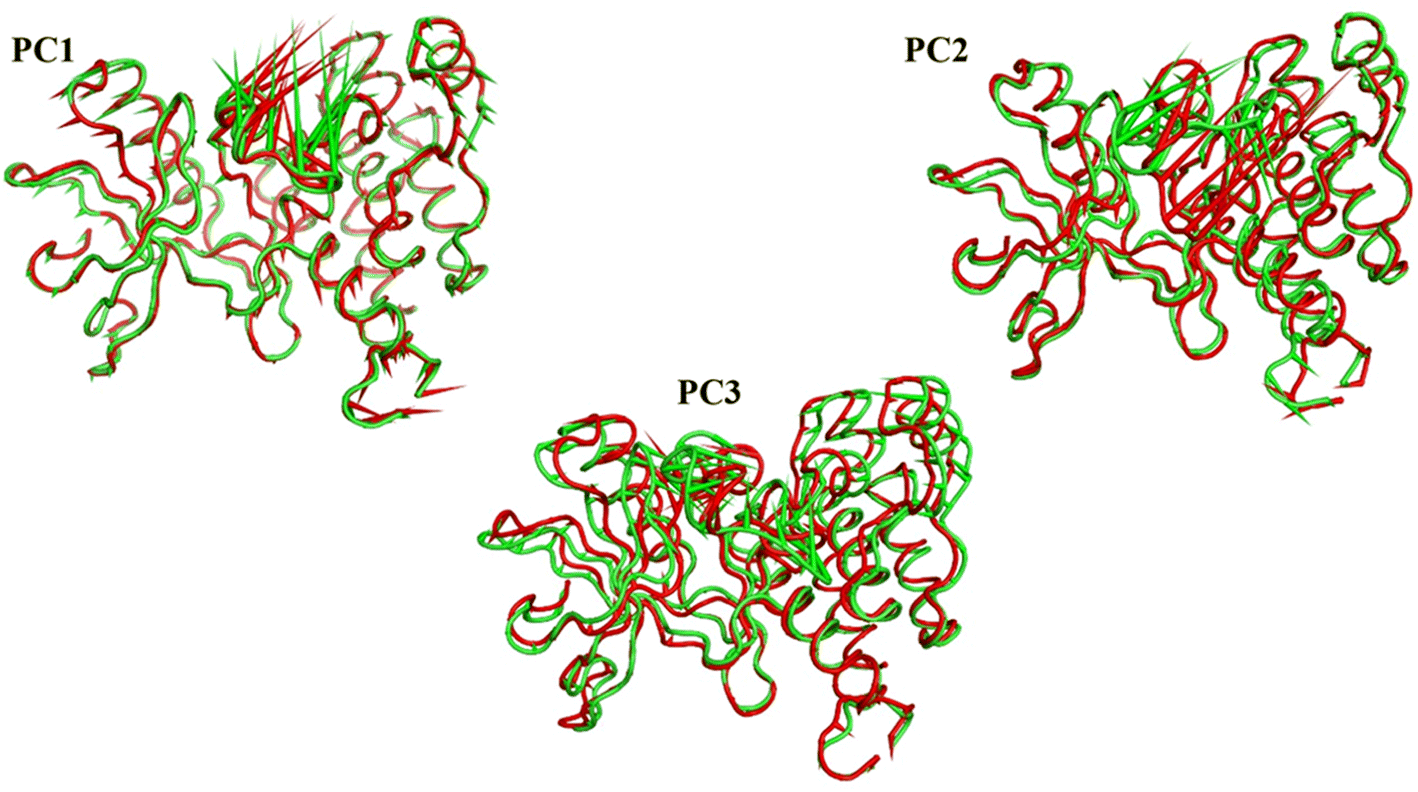 | ||
| Fig. 17 The porcupine figures of each of the first three eigenvectors for both systems. Green cartoon: apo protein trajectory, red cartoon: holo protein trajectory. | ||
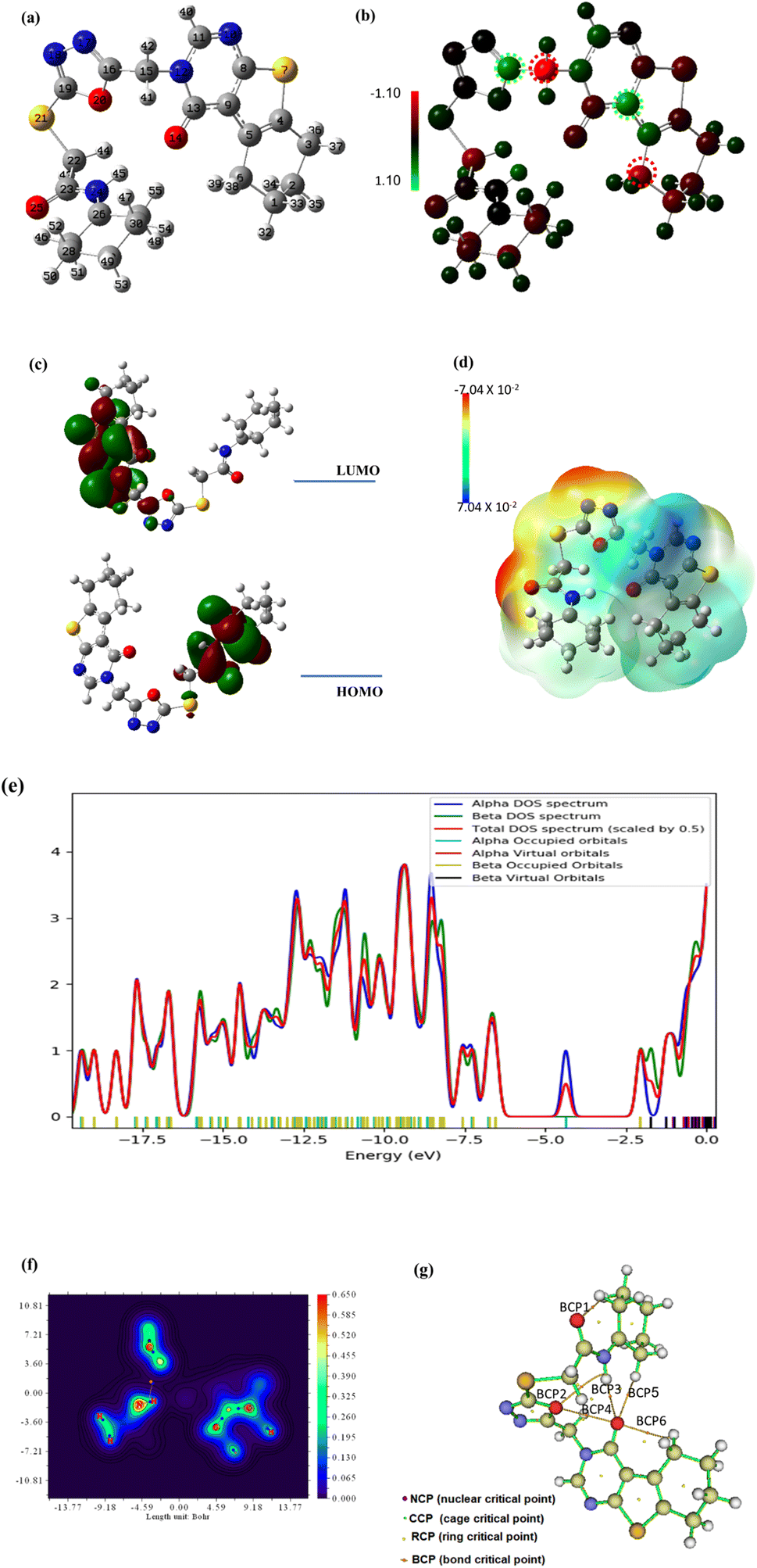 | ||
| Fig. 18 The optimized geometry (a), the Mullikan atomic charge distribution (b), the frontier molecular orbitals (c), the electrostatic potential (d), the total density of states (e), and the QTAIM maps (f and g) at B3LYB/6-311+G(d,p) for compound 18. | ||
The energy of both HOMO and LUMO is estimated in order to broaden the theoretical calculations, and their density distributions are examined and shown in Fig. 18c. The predicted energies of HOMO and LUMO are found to be −4.605 eV and −1.834 eV, respectively, whereas the energy gap between HOMO and LUMO, or Egap, is found to be 2.77 eV which is relatively small. A reasonable Egap value like this shows that the 18 may be bioactive [https://www.sciencedirect.com/science/article/abs/pii/S1476927123001196?via%3Dihub]. The frontier molecular energy gap, Egap, controls the chemical reactivity and stability of the molecule. Ionization ability and electron affinity are both intimately correlated with the HOMO and LUMO energies, respectively. The global reactivity parameters, electron affinity, and ionization potential were theoretically computed using Koopmans' theorem (as detailed in the ESI Data file†) and are shown in Table 7. The electron affinity can be used in combination with ionization energy to give electronic chemical potential, μ, softness (σ) and hardness (η). The value of global softness and hardness suggests that compound 8 is soft and could show potent biological reactivity.47 The findings demonstrated the kinetic stability and potential bioactivity of compound 18.48
| IP | EA | μ (eV) | χ (eV) | η (eV) | σ (eV) | ω (eV) | Dm (Debye) | TE (eV) | ΔNmax | ΔE (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| 4.605 | 1.834 | −3.219 | 3.219 | 1.386 | 0.722 | 7.181 | 7.772 | −57439.9 | 2.323 | −7.181 |
In order to shed light on the theoretical computation of compound 18 structure, the electrostatic potential, or ESP, is examined. Fig. 18d showed the ESP map of the titled compound. The ESP colored map determines compound 18 chemical reactivity sites and how they will respond to the target. According to the results, huge negative ESP zones are centered around oxygen and nitrogen atoms (red zones) on a scale from 7.04 × 10−2 to −7.04 × 10−2, whereas big positive ESP zones are found over hydrogen atoms in the blue zones. The residue of the molecule appears to be almost neutral and is prepared for attacks from hydrophobic centers. Although the positive active sites are hydrogen bond donors, the negative ESP sites are hydrogen bond acceptors. The investigation therefore shows that the bioactive compound may interact with the target in a successful manner.49
Gauss Sum analysis of the overall density of states was shown in Fig. 18e. The compound 18 spectra demonstrated the alpha and beta density of states (DOS) as well as the total density of states (TDOS). The filled orbitals under HOMO have the largest electronic density.
Quantum Theory of Atoms in Molecules (QTAIM) offers a formal method for breaking down molecule attributes into atomic components based on the topological electron density; (ρ) using the assistance of AIMALL and Multiwfn programs. This procedure clarifies the theoretical calculations and interactions within the compound 18 molecule. The overall energy density, H(r), and the Laplacian values, (∇2ρ), of the newly established bond critical points (BCPs), were used as local variables to evaluate the covalent or ionic nature of chemical interatomic interactions within compound 18. Large (ρ), low (negative) ∇2ρ and positive H(r) values at the BCPs were expected for bonds with a high increment of covalent character. Fig. 18f and g, respectively, showed the QTAIM molecular map and filled contour map, while ESI Data† provide the full QTAIM BCPs and routes. The QTAIM parameters in ESI Data† validated the prepared molecule's stability because the most significant BCPs (six BCPs) were produced with (ρ) less than 0.1 and positive (∇2ρ) values, which indicated noncovalent or closed bonding. Moreover, the calculated H(r) confirms that the created BCPs are electrostatic, which contributes to the stability of compound 18.
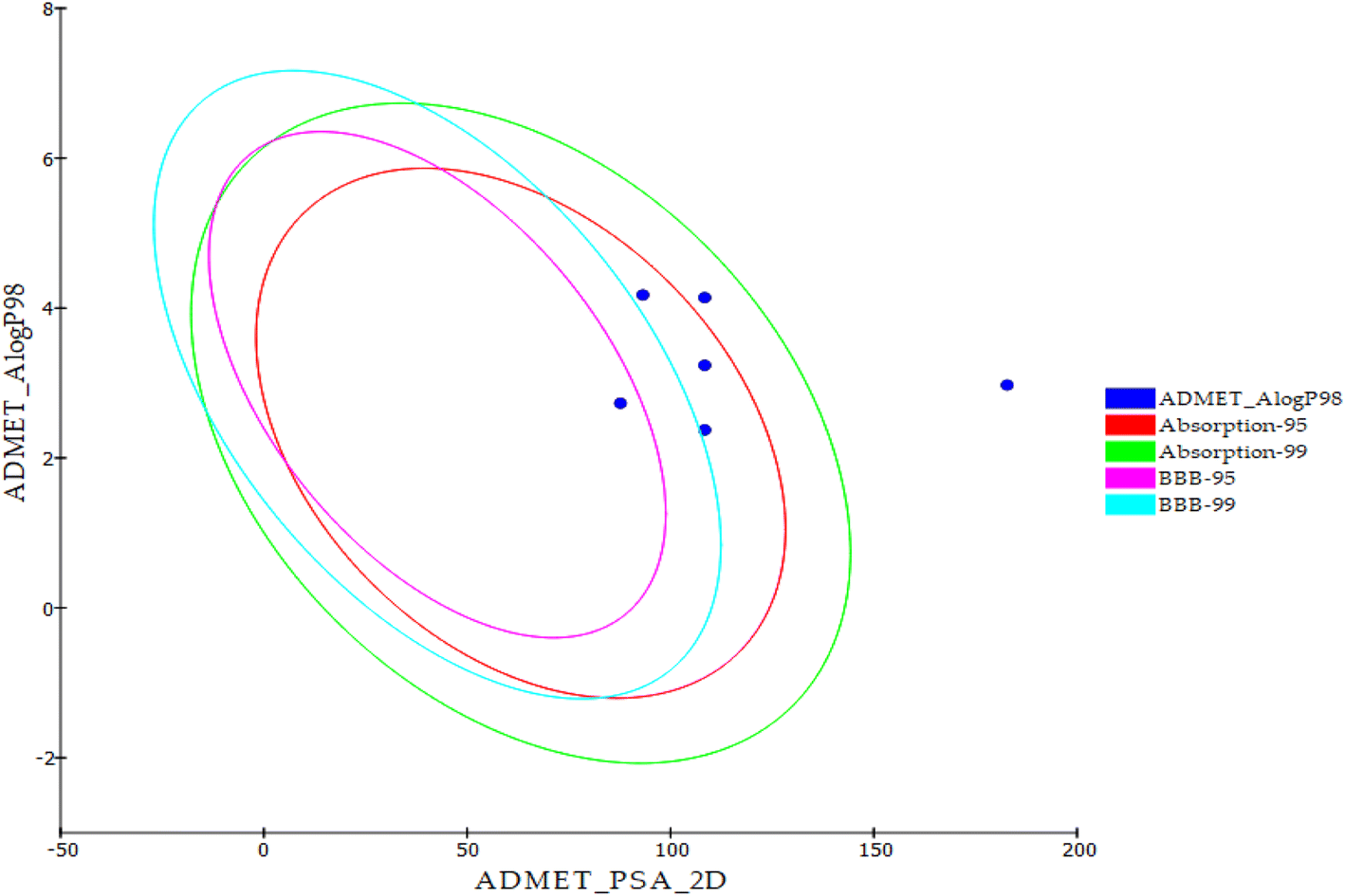 | ||
| Fig. 19 In silico ADME output. | ||
| Code | Comp. | BBB level | Solubility level | Absorption level | CYP2D6 prediction | PPB prediction |
|---|---|---|---|---|---|---|
| 18 | 18 | Low | Low | Good | No inhibition | <90% |
| 19 | 19 | Very low | Very poor | |||
| 20 | 20 | Good | >90% | |||
| 21 | 21 | <90% | ||||
| 22 | 22 | Moderate | >90% | |||
| Sorafenib | Very low | Good |
| Code | Ames prediction | DTP | Carcinogenic potency TD50 (rat)a | Rat maximum tolerated dose (feed)b | Rat oral LD50b | Rat chronic LOAELb | Ocular irritancy |
|---|---|---|---|---|---|---|---|
| a Unit: mg per kg body weight per day.b Unit: g per kg body weight. | |||||||
| 18 | Non-mutagen | Non-toxic | 12.453 | 0.016 | 2.950 | 0.070 | Mild |
| 19 | Mutagen | 30.514 | 0.019 | 3.244 | 0.094 | ||
| 20 | Non-mutagen | 5.036 | 0.044 | 3.652 | 0.043 | ||
| 21 | Mutagen | 8.295 | 0.024 | 5.760 | 0.088 | ||
| 22 | Non-mutagen | 6.388 | 0.026 | 6.240 | 0.114 | ||
| Sorafenib | Toxic | 14.244 | 0.089 | 0.823 | 0.005 | ||
3. Conclusion
In conclusion, this study demonstrates the potential of thieno[2,3-d]pyrimidine derivatives as potent and selective anticancer agents that target VEGFR-2. Compound 18, in particular, was found to have strong anti-VEGFR-2 activity and to effectively inhibit cancer cell growth in vitro in both MCF-7 and HepG2 cancer cell lines. Further investigation showed that compound 18 induced cell cycle arrest at G2/M phase and apoptosis in MCF-7 cancer cells, and was able to modulate the expression of key apoptotic genes such as BAX and Bcl-2, as well as caspase-8 and caspase-9. In addition, computational techniques such as molecular docking, MD simulations, and PLIP provided valuable insights into the structural and energetic features of the VEGFR-2-18 complex, further supporting the potential of this compound as a promising anticancer agent. The use of DFT calculations also provided insights into the geometry and electronic properties of compound 18. Finally, computational ADMET studies suggest that these thieno[2,3-d]pyrimidine derivatives have good drug development potential. Overall, the study provides a foundation for further research, highlighting the potential of thieno[2,3-d]pyrimidine derivatives as a new class of anticancer agents, and offering guidance for future drug discovery efforts.4. Experimental
4.1. Chemistry
4.1.2.1. 3-((5-((2-Oxo-2-(piperidin-1-yl)ethyl)thio)-1,3,4-oxadiazol-2-yl)methyl)-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one 18.
White crystal (yield, 75%); m. p. = 204–206 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.46 (s, 1H, Ar–H pyrimidine), 5.44 (s, 2H, N–CH2), 4.46 (s, 2H, S–CH2), 3.41 (d, 4H, 2CH2 piperidine), 2.86 (m, 2H, CH2), 2.77 (m, 2H, CH2), 1.77 (m, 4H, 2CH2), 1.57 (m, 4H, 2CH2 piperidine), 1.43 (m, 2H, CH2 piperidine); 13C NMR (101 MHz, DMSO-d6) δ 164.57, 164.41, 163.68, 162.07, 156.92, 147.71, 134.23, 131.36, 122.13, 46.85, 43.07, 37.43, 26.18, 25.71, 25.59, 25.00, 24.18 (2C), 22.80, 22.15; for C20H23N5O3S2 (445.56).
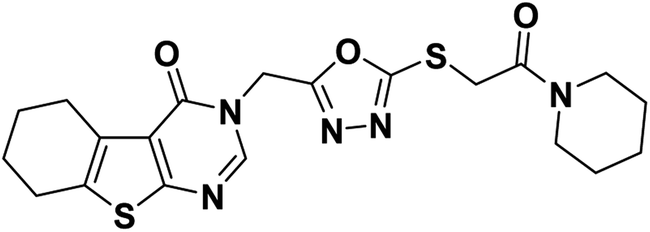
4.1.2.2. N-(2,6-Dinitrophenyl)-2-((5-((4-oxo-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-3(4H)-yl)methyl)-1,3,4-oxadiazol-2-yl)thio)acetamide 19.
Yellow crystal (yield, 70%); m. p. = 238–240 °C; IR (KBr) ν cm−1: 3291 (NH), 2931 (CH aliphatic), 1661 (C
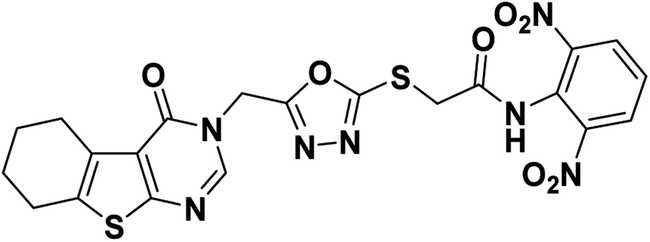 O); 1H NMR (400 MHz, DMSO-d6) δ 8.89 (m, 1H, Ar–H), 8.27 (s, 1H, Ar–H pyrimidine) 8.05 (m, 3H, Ar–H), 4.97 (s, 2H, N–CH2), 4.29 (s, 2H, S–CH2), 2.88 (m, 2H, CH2), 2.77 (m, 2H, CH2), 1.78 (s, 4H, 2CH2); for C21H17N7O7S2 (543.53).
O); 1H NMR (400 MHz, DMSO-d6) δ 8.89 (m, 1H, Ar–H), 8.27 (s, 1H, Ar–H pyrimidine) 8.05 (m, 3H, Ar–H), 4.97 (s, 2H, N–CH2), 4.29 (s, 2H, S–CH2), 2.88 (m, 2H, CH2), 2.77 (m, 2H, CH2), 1.78 (s, 4H, 2CH2); for C21H17N7O7S2 (543.53).
4.1.2.3. N-(5-Chloropyridin-2-yl)-2-((5-((4-oxo-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-3(4H)-yl)methyl)-1,3,4-oxadiazol-2-yl)thio)acetamide 20.
Off-white crystal (yield, 77%); m. p. = 225–227 °C; IR (KBr) ν cm−1: 3291 (NH), 2929 (CH aliphatic), 1666 (C
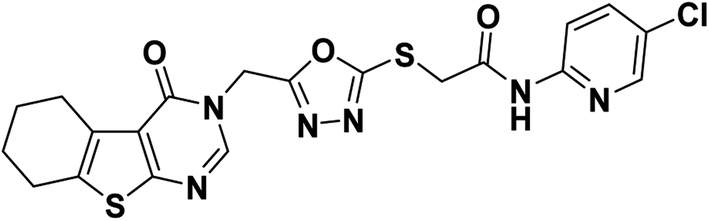 O); 1H NMR (400 MHz, DMSO-d6) δ 11.06 (s, 1H, NH), 8.44 (s, 1H, Ar–H pyrimidine), 8.38 (d, J = 2.7 Hz, 1H, Ar–H), 8.02 (d, J = 8.9 Hz, 1H, Ar–H), 7.89 (dd, J = 8.9, 2.7 Hz, 1H, Ar–H), 5.43 (s, 2H, N–CH2), 4.34 (s, 2H, S–CH2), 2.84 (m, 2H, CH2), 2.76 (m, 2H, CH2), 1.77 (m, 4H, 2CH2); 13C NMR (101 MHz, DMSO-d6) δ 166.21, 164.15, 163.97, 162.05, 156.90, 150.63, 147.64, 146.91, 138.50, 134.17, 131.33, 125.93, 122.08, 114.97, 40.67, 36.97, 25.67, 24.98, 22.78, 22.13; for C20H17ClN6O3S2 (488.97).
O); 1H NMR (400 MHz, DMSO-d6) δ 11.06 (s, 1H, NH), 8.44 (s, 1H, Ar–H pyrimidine), 8.38 (d, J = 2.7 Hz, 1H, Ar–H), 8.02 (d, J = 8.9 Hz, 1H, Ar–H), 7.89 (dd, J = 8.9, 2.7 Hz, 1H, Ar–H), 5.43 (s, 2H, N–CH2), 4.34 (s, 2H, S–CH2), 2.84 (m, 2H, CH2), 2.76 (m, 2H, CH2), 1.77 (m, 4H, 2CH2); 13C NMR (101 MHz, DMSO-d6) δ 166.21, 164.15, 163.97, 162.05, 156.90, 150.63, 147.64, 146.91, 138.50, 134.17, 131.33, 125.93, 122.08, 114.97, 40.67, 36.97, 25.67, 24.98, 22.78, 22.13; for C20H17ClN6O3S2 (488.97).
4.1.2.4. 2-((5-((4-Oxo-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-3(4H)-yl)methyl)-1,3,4-oxadiazol-2-yl)thio)-N-(thiazol-2-yl)acetamide 21.
Orange crystal (yield, 80%); m. p. = 230–232 °C; IR (KBr) ν cm−1: 3307 (NH), 2931 (CH aliphatic), 1673 (C
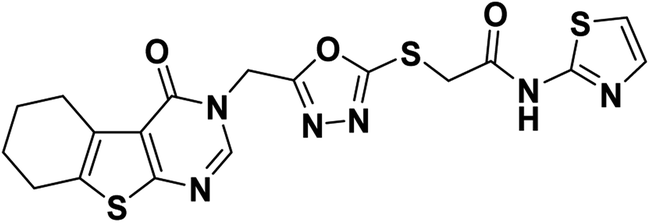 O); 1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 1H, Ar–H pyrimidine), 7.46 (d, J = 3.6 Hz, 1H, Ar–H), 7.18 (d, J = 3.6 Hz, 1H, Ar–H), 5.44 (s, 2H, N–CH2), 4.34 (s, 2H, S–CH2), 2.84 (m, 2H, CH2), 2.74 (m, 2H, CH2), 1.76 (m, 4H, 2CH2); 13C NMR (101 MHz, DMSO-d6) δ 164.23, 164.00, 162.07, 156.92, 147.66, 138.08, 134.17, 131.36, 122.11, 113.98, 113.86, 40.67, 25.69, 25.00, 22.80, 22.15; for C18H16N6O3S3 (460.55).
O); 1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 1H, Ar–H pyrimidine), 7.46 (d, J = 3.6 Hz, 1H, Ar–H), 7.18 (d, J = 3.6 Hz, 1H, Ar–H), 5.44 (s, 2H, N–CH2), 4.34 (s, 2H, S–CH2), 2.84 (m, 2H, CH2), 2.74 (m, 2H, CH2), 1.76 (m, 4H, 2CH2); 13C NMR (101 MHz, DMSO-d6) δ 164.23, 164.00, 162.07, 156.92, 147.66, 138.08, 134.17, 131.36, 122.11, 113.98, 113.86, 40.67, 25.69, 25.00, 22.80, 22.15; for C18H16N6O3S3 (460.55).
4.1.2.5. N-(Benzo[d]thiazol-2-yl)-2-((5-((4-oxo-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-3(4H)-yl)methyl)-1,3,4-oxadiazol-2-yl)thio)acetamide 22.
Yellowish white crystal (yield, 78%); m. p. = 224–226 °C; IR (KBr) ν cm−1: 3400 (NH), 2928 (CH aliphatic), 1674 (C
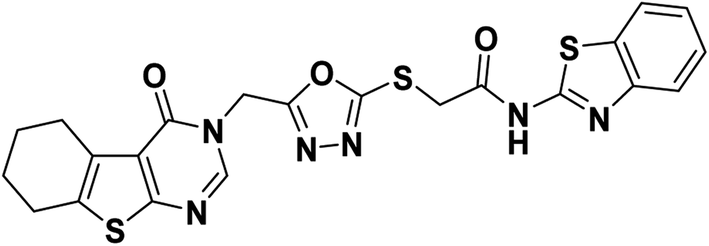 O); 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H, NH), 8.44 (s, 1H, Ar–H pyrimidine), 7.97 (d, J = 7.9 Hz, 1H, Ar–H), 7.76 (d, J = 8.0 Hz, 1H, Ar–H), 7.45 (t, J = 7.6 Hz, 1H, Ar–H), 7.32 (t, J = 7.6 Hz, 1H, Ar–H), 5.44 (s, 2H, N–CH2), 4.42 (s, 2H, S–CH2), 2.84 (d, 2H, CH2), 2.72 (m, 2H, CH2), 1.76 (m, 4H, 2CH2); 13C NMR (101 MHz, DMSO-d6) δ 166.71, 164.08, 163.97, 162.05, 158.26, 156.90, 148.89, 147.61, 134.16, 131.95, 131.31, 126.65, 124.16, 122.20, 122.07, 121.11, 40.66, 36.18, 25.67, 24.93, 22.76, 22.11; for C22H18N6O3S3 (510.61).
O); 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H, NH), 8.44 (s, 1H, Ar–H pyrimidine), 7.97 (d, J = 7.9 Hz, 1H, Ar–H), 7.76 (d, J = 8.0 Hz, 1H, Ar–H), 7.45 (t, J = 7.6 Hz, 1H, Ar–H), 7.32 (t, J = 7.6 Hz, 1H, Ar–H), 5.44 (s, 2H, N–CH2), 4.42 (s, 2H, S–CH2), 2.84 (d, 2H, CH2), 2.72 (m, 2H, CH2), 1.76 (m, 4H, 2CH2); 13C NMR (101 MHz, DMSO-d6) δ 166.71, 164.08, 163.97, 162.05, 158.26, 156.90, 148.89, 147.61, 134.16, 131.95, 131.31, 126.65, 124.16, 122.20, 122.07, 121.11, 40.66, 36.18, 25.67, 24.93, 22.76, 22.11; for C22H18N6O3S3 (510.61).
4.2. Biological examinations
4.3. In silico studies
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R142), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors extend their appreciation to the Research Center at AlMaarefa University for funding this work.References
- F. Biemar and M. Foti, Cancer Biol. Med., 2013, 10, 183 Search PubMed.
- R. A. Ward, S. Fawell, N. Floc'h, V. Flemington, D. McKerrecher and P. D. Smith, Chem. Rev., 2020, 121, 3297–3351 CrossRef PubMed.
- A. Belal, N. M. Abdel Gawad, A. B. Mehany, M. A. Abourehab, H. Elkady, A. A. Al-Karmalawy and A. S. Ismael, J. Enzyme Inhib. Med. Chem., 2022, 37, 1884–1902 CrossRef CAS PubMed.
- R. Trenker and N. Jura, Curr. Opin. Cell Biol., 2020, 63, 174–185 CrossRef CAS PubMed.
- X. Wang, A. M. Bove, G. Simone and B. Ma, Front. Cell Dev. Biol., 2020, 8, 599281 CrossRef PubMed.
- S. J. Modi and V. M. Kulkarni, Med. Drug Discovery, 2019, 2, 100009 CrossRef.
- S. R. Silva, K. A. Bowen, P. G. Rychahou, L. N. Jackson, H. L. Weiss, E. Y. Lee, C. M. Townsend Jr and B. M. Evers, Int. J. Cancer, 2011, 128, 1045–1056 CrossRef CAS PubMed.
- N. Nishida, H. Yano, T. Nishida, T. Kamura and M. Kojiro, Vasc. Health Risk Manage., 2006, 2, 213–219 CrossRef CAS PubMed.
- R. G. Yousef, A. Elwan, I. M. Gobaara, A. B. Mehany, W. M. Eldehna, S. A. El-Metwally, B. A. Alsfouk, E. B. Elkaeed, A. M. Metwaly and I. H. Eissa, J. Enzyme Inhib. Med. Chem., 2022, 37, 2206–2222 CrossRef CAS PubMed.
- R. G. Yousef, A. Ibrahim, M. M. Khalifa, W. M. Eldehna, I. M. Gobaara, A. B. Mehany, E. B. Elkaeed, A. A. Alsfouk, A. M. Metwaly and I. H. Eissa, J. Enzyme Inhib. Med. Chem., 2022, 37, 1389–1403 CrossRef CAS PubMed.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, I. M. Gobaara, B. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, Molecules, 2022, 27, 4606 CrossRef CAS PubMed.
- M. S. Taghour, H. Elkady, W. M. Eldehna, N. M. El-Deeb, A. M. Kenawy, E. B. Elkaeed, A. A. Alsfouk, M. S. Alesawy, A. M. Metwaly and I. H. Eissa, J. Enzyme Inhib. Med. Chem., 2022, 37, 1903–1917 CrossRef CAS PubMed.
- M. S. Taghour, H. Elkady, W. M. Eldehna, N. El-Deeb, A. M. Kenawy, E. B. Elkaeed, B. A. Alsfouk, M. S. Alesawy, D. Z. Husein and A. M. Metwaly, PLoS One, 2022, 17, e0272362 CrossRef CAS PubMed.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, I. M. Gobaara, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, Processes, 2022, 10, 1391 CrossRef CAS.
- R. G. Yousef, H. Elkady, E. B. Elkaeed, I. M. Gobaara, H. A. Al-Ghulikah, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, Molecules, 2022, 27, 7719 CrossRef CAS PubMed.
- M. S. Taghour, H. Elkady, W. M. Eldehna, N. El-Deeb, A. M. Kenawy, A. E. Abd El-Wahab, E. B. Elkaeed, B. A. Alsfouk, A. M. Metwaly and I. H. Eissa, J. Biomol. Struct. Dyn., 2022, 1–16 Search PubMed.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, I. M. Gobaara, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, Processes, 2022, 10, 1391 CrossRef CAS.
- E. B. Elkaeed, M. S. Taghour, H. A. Mahdy, W. M. Eldehna, N. M. El-Deeb, A. M. Kenawy, B. A. Alsfouk, M. A. Dahab, A. M. Metwaly and I. H. Eissa, J. Enzyme Inhib. Med. Chem., 2022, 37, 2191–2205 CrossRef CAS PubMed.
- A. T. Mavrova, D. Vuchev, K. Anichina and N. Vassilev, Eur. J. Med. Chem., 2010, 45, 5856–5861 CrossRef CAS PubMed.
- A. T. Mavrova, S. Dimov, D. Yancheva, M. Rangelov, D. Wesselinova and J. A. Tsenov, Eur. J. Med. Chem., 2016, 123, 69–79 CrossRef CAS PubMed.
- A. G. Golub, V. G. Bdzhola, N. V. Briukhovetska, A. O. Balanda, O. P. Kukharenko, I. M. Kotey, O. V. Ostrynska and S. M. Yarmoluk, Eur. J. Med. Chem., 2011, 46, 870–876 CrossRef CAS PubMed.
- S. A. Elmetwally, K. F. Saied, I. H. Eissa and E. B. Elkaeed, Bioorg. Chem., 2019, 88, 102944 CrossRef PubMed.
- S. A. El-Metwally, M. M. Abou-El-Regal, I. H. Eissa, A. B. Mehany, H. A. Mahdy, H. Elkady, A. Elwan and E. B. Elkaeed, Bioorg. Chem., 2021, 112, 104947 CrossRef CAS PubMed.
- Y. Dai, Y. Guo, R. R. Frey, Z. Ji, M. L. Curtin, A. A. Ahmed, D. H. Albert, L. Arnold, S. S. Arries and T. Barlozzari, J. Med. Chem., 2005, 48, 6066–6083 CrossRef CAS PubMed.
- S. A. El-Metwally, A. K. Khalil and W. M. El-Sayed, Bioorg. Chem., 2020, 94, 103492 CrossRef CAS PubMed.
- H.-Z. Zhang, Z.-L. Zhao and C.-H. Zhou, Eur. J. Med. Chem., 2018, 144, 444–492 CrossRef CAS PubMed.
- M. A. A. Fathi, A. A. Abd El-Hafeez, D. Abdelhamid, S. H. Abbas, M. M. Montano and M. Abdel-Aziz, Bioorg. Chem., 2019, 84, 150–163 CrossRef CAS PubMed.
- R. N. Kumar, Y. Poornachandra, P. Nagender, G. S. Kumar, D. K. Swaroop, C. G. Kumar and B. Narsaiah, Bioorg. Med. Chem. Lett., 2016, 26, 4829–4831 CrossRef PubMed.
- J. Slawinski, K. Szafranski, A. Pogorzelska, B. Zolnowska, A. Kawiak, K. Macur, M. Belka and T. Baczek, Eur. J. Med. Chem., 2017, 132, 236–248 CrossRef CAS PubMed.
- S. Valente, D. Trisciuoglio, T. De Luca, A. Nebbioso, D. Labella, A. Lenoci, C. Bigogno, G. Dondio, M. Miceli and G. Brosch, J. Med. Chem., 2014, 57, 6259–6265 CrossRef CAS PubMed.
- Q.-Q. Xie, H.-Z. Xie, J.-X. Ren, L.-L. Li and S.-Y. Yang, J. Mol. Graphics Modell., 2009, 27, 751–758 CrossRef CAS PubMed.
- K. Lee, K.-W. Jeong, Y. Lee, J. Y. Song, M. S. Kim, G. S. Lee and Y. Kim, Eur. J. Med. Chem., 2010, 45, 5420–5427 CrossRef CAS PubMed.
- R. N. Eskander and K. S. Tewari, Gynecol. Oncol., 2014, 132, 496–505 CrossRef CAS PubMed.
- V. A. Machado, D. Peixoto, R. Costa, H. J. Froufe, R. C. Calhelha, R. M. Abreu, I. C. Ferreira, R. Soares and M.-J. R. Queiroz, Bioorg. Med. Chem., 2015, 23, 6497–6509 CrossRef CAS PubMed.
- Z. Wang, N. Wang, S. Han, D. Wang, S. Mo, L. Yu, H. Huang, K. Tsui, J. Shen and J. Chen, PLoS One, 2013, 8, e68566 CrossRef CAS PubMed.
- J. Dietrich, C. Hulme and L. H. Hurley, Bioorg. Med. Chem., 2010, 18, 5738–5748 CrossRef CAS PubMed.
- K. Gewald, E. Schinke and H. Böttcher, Chem. Ber., 1966, 99, 94–100 CrossRef CAS.
- P. Di Fruscia, E. Zacharioudakis, C. Liu, S. Moniot, S. Laohasinnarong, M. Khongkow, I. F. Harrison, K. Koltsida, C. R. Reynolds and K. Schmidtkunz, ChemMedChem, 2015, 10, 69–82 CrossRef CAS PubMed.
- M. Aruna Kumari, S. Triloknadh, N. Harikrishna, M. Vijjulatha and C. Venkata Rao, J. Heterocycl. Chem., 2017, 54, 3672–3681 CrossRef CAS.
- M. Ghorab and S. Abdel Hamide, Phosphorus Sulfur Silicon Relat. Elem., 1995, 106, 9–20 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- K. M. McKinnon, Curr. Protoc. Immunol., 2018, 120, 5.1.1–5.1.11 Search PubMed.
- A. Adan, G. Alizada, Y. Kiraz, Y. Baran and A. Nalbant, Crit. Rev. Biotechnol., 2017, 37, 163–176 CrossRef CAS PubMed.
- D. Wlodkowic, J. Skommer and Z. Darzynkiewicz, Apoptosis: Methods and Protocols, 2nd edn, 2009, pp. 19–32 Search PubMed.
- N. A. Alsaif, M. A. Dahab, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, H. A. Mahdy and H. Elkady, Bioorg. Chem., 2021, 110, 104807 CrossRef CAS PubMed.
- N. A. Alsaif, M. S. Taghour, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, A. Elwan and H. Elkady, J. Enzyme Inhib. Med. Chem., 2021, 36, 1093–1114 CrossRef CAS PubMed.
- S. A. El-Metwally, A. A. Abuelkhir, H. Elkady, M. S. Taghour, I. M. Ibrahim, D. Z. Husein, A. A. Alsfouk, A. Sultan, A. Ismail and S. Y. Elkhawaga, Comput. Biol. Chem., 2023, 107928 CrossRef CAS PubMed.
- D. Z. Husein, R. Hassanien and M. Khamis, RSC Adv., 2021, 11, 27027–27041 RSC.
- T. Wang and D. Z. Husein, Environ. Sci. Pollut. Res., 2023, 30, 8928–8955 CrossRef CAS PubMed.
- M. J. Waring, J. Arrowsmith, A. R. Leach, P. D. Leeson, S. Mandrell, R. M. Owen, G. Pairaudeau, W. D. Pennie, S. D. Pickett and J. Wang, Nat. Rev. Drug Discovery, 2015, 14, 475–486 CrossRef CAS PubMed.
- L. L. Ferreira and A. D. Andricopulo, Drug discovery today, 2019, 24, 1157–1165 CrossRef CAS PubMed.
- S. Kar and J. Leszczynski, Expert Opin. Drug Discovery, 2020, 15, 1473–1487 CrossRef CAS PubMed.
- G. Moroy, V. Y. Martiny, P. Vayer, B. O. Villoutreix and M. A. Miteva, Drug discovery today, 2012, 17, 44–55 CrossRef CAS PubMed.
- A. R. Kotb, D. A. Bakhotmah, A. E. Abdallah, H. Elkady, M. S. Taghour, I. H. Eissa and M. A. El-Zahabi, RSC Adv., 2022, 12, 33525–33539 RSC.
- A. R. Kotb, A. E. Abdallah, H. Elkady, I. H. Eissa, M. S. Taghour, D. A. Bakhotmah, T. M. Abdelghany and M. A. El-Zahabi, RSC Adv., 2023, 13, 10488–10502 RSC.
- L. Ma, C. Xie, Y. Ma, J. Liu, M. Xiang, X. Ye, H. Zheng, Z. Chen, Q. Xu and T. Chen, J. Med. Chem., 2011, 54, 2060–2068 CrossRef CAS PubMed.
- L. Ma, S. Li, H. Zheng, J. Chen, L. Lin, X. Ye, Z. Chen, Q. Xu, T. Chen and J. Yang, Eur. J. Med. Chem., 2011, 46, 2003–2010 CrossRef CAS PubMed.
- A. Van de Loosdrecht, R. Beelen, g. Ossenkoppele, M. Broekhoven and M. Langenhuijsen, J. Immunol. Methods, 1994, 174, 311–320 CrossRef CAS PubMed.
- M. C. Alley, D. A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589–601 CAS.
- W. M. Eldehna, M. A. El Hassab, Z. M. Elsayed, T. Al-Warhi, H. Elkady, M. F. Abo-Ashour, M. A. Abourehab, I. H. Eissa and H. A. Abdel-Aziz, Sci. Rep., 2022, 12, 12821 CrossRef CAS PubMed.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, A. B. Mehany, B. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, J. Biomol. Struct. Dyn., 2022, 1–16 CrossRef PubMed.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, H. M. Alkahtani, M. M. Alanazi, M. A. Alharbi, I. H. Eissa and M. A. Dahab, RSC Adv., 2021, 11, 30315–30328 RSC.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- S. Jo, X. Cheng, S. M. Islam, L. Huang, H. Rui, A. Zhu, H. S. Lee, Y. Qi, W. Han and K. Vanommeslaeghe, Adv. Protein Chem. Struct. Biol., 2014, 96, 235–265 CrossRef CAS PubMed.
- J. Lee, X. Cheng, S. Jo, A. D. MacKerell, J. B. Klauda and W. Im, Biophys. J., 2016, 110, 641a CrossRef.
- T. Tuccinardi, Expert Opin. Drug Discovery, 2021, 16, 1233–1237 CrossRef CAS PubMed.
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, J. Chem. Theory Comput., 2021, 17, 6281–6291 CrossRef PubMed.
- C. Bouysset and S. Fiorucci, J. Cheminf., 2021, 13, 1–9 Search PubMed.
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, Nucleic Acids Res., 2015, 43, W443–W447 CrossRef CAS PubMed.
- T. Tubiana, J.-C. Carvaillo, Y. Boulard and S. Bressanelli, J. Chem. Inf. Model., 2018, 58, 2178–2182 CrossRef CAS PubMed.
- A. Amadei, A. B. Linssen and H. J. Berendsen, Proteins: Struct., Funct., Bioinf., 1993, 17, 412–425 CrossRef CAS PubMed.
- E. Papaleo, P. Mereghetti, P. Fantucci, R. Grandori and L. De Gioia, J. Mol. Graphics Modell., 2009, 27, 889–899 CrossRef CAS PubMed.
- G. G. Maisuradze and D. M. Leitner, Proteins: Struct., Funct., Bioinf., 2007, 67, 569–578 CrossRef CAS PubMed.
- B. Hess, Phys. Rev. E, 2000, 62, 8438 CrossRef CAS PubMed.
- E. B. Elkaeed, R. G. Yousef, M. M. Khalifa, A. Ibrahim, A. B. Mehany, I. M. Gobaara, B. A. Alsfouk, W. M. Eldehna, A. M. Metwaly and I. H. Eissa, Molecules, 2022, 27, 6203 CrossRef CAS PubMed.
- H. Elkady, A. Elwan, H. A. El-Mahdy, A. S. Doghish, A. Ismail, M. S. Taghour, E. B. Elkaeed, I. H. Eissa, M. A. Dahab and H. A. Mahdy, J. Enzyme Inhib. Med. Chem., 2022, 37, 403–416 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03128d |
| This journal is © The Royal Society of Chemistry 2023 |