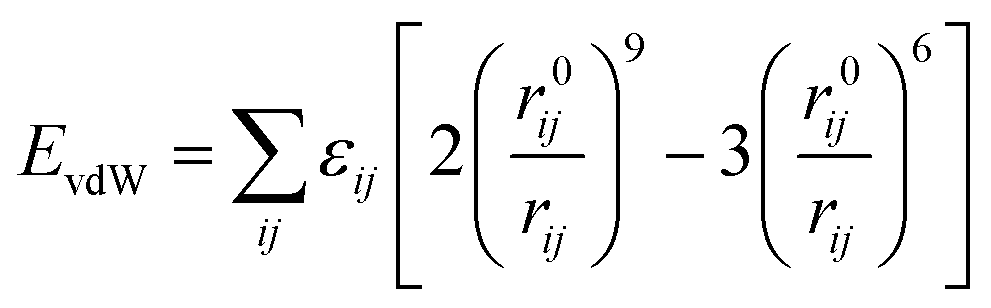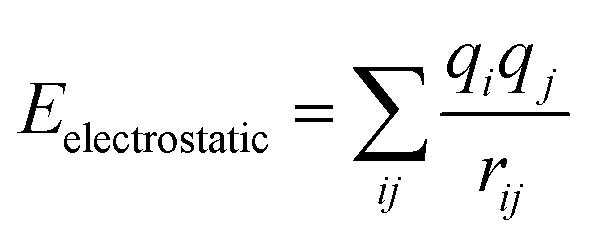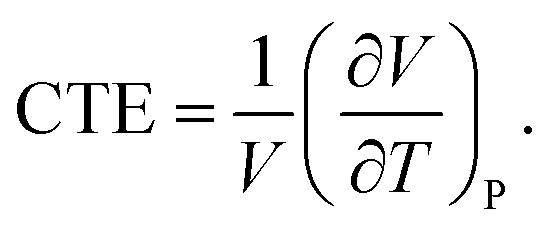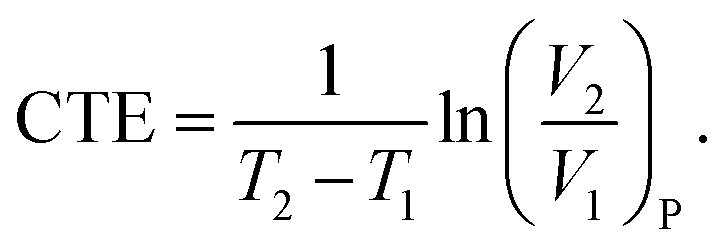DOI:
10.1039/D3RA03000H
(Paper)
RSC Adv., 2023,
13, 21080-21087
Exploring thermodynamic and structural properties of carbon nanotube/thermoplastic polyurethane nanocomposites from atomistic molecular dynamics simulations†
Received
6th May 2023
, Accepted 7th July 2023
First published on 12th July 2023
Abstract
Carbon nanotubes (CNTs) and thermoplastic polyurethane (TPU) nanocomposites have emerged as promising materials for various applications in the field of nanotechnology. An understanding of the thermodynamic and structural properties is of fundamental significance in designing and fabricating CNT/TPU nanocomposites with desired properties. To this end, this work has employed atomistic molecular dynamics (MD) simulations to study the thermal properties and interfacial characteristics of TPU composites filled with pristine or functionalized single-walled carbon nanotubes (SWNTs). Simulations reveal that the introduction of SWNTs suppresses TPU chain dynamics and favors the hydrogen bond formation induced by the wrapping of TPU chains around SWNTs, leading to an increase of glass transition temperature (Tg) and a reduction of volumetric coefficient of thermal expansion (CTE) in the rubbery state. Compared to pristine and hydrogenated SWNTs, SWNTs featuring polar groups, such as carboxyl (–COOH), oxhydryl (–OH) and amine (–NH2) groups, show improved affinity for TPU molecules, suppressing polymer mobility. Analysis of SWNT/TPU binding energy and solubility parameter suggests that electrostatic interactions are responsible for such a functionalized SWNT/TPU interface enhancement. Furthermore, the amine groups exhibit the highest potential for forming hydrogen bonds with the urethane carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of TPU chains, resulting in lowest polymer mobility and highest Tg. In general, this research work could provide some guidance for material design of polymer nanocomposites and future simulations relevant to TPUs.
O) of TPU chains, resulting in lowest polymer mobility and highest Tg. In general, this research work could provide some guidance for material design of polymer nanocomposites and future simulations relevant to TPUs.
1 Introduction
Carbon nanotubes (CNTs) and thermoplastic polyurethane (TPU) nanocomposites have recently emerged as promising materials in the field of nanotechnology. CNTs are known for their unique physical and chemical properties, such as high mechanical strength, electrical conductivity, low thermal conductivity, and high surface area to volume ratio.1,2 Consisting of alternating hard and soft segments, TPU is a lightweight and flexible thermoplastic elastomer that combines the advantages of both thermoplastic and elastomeric properties. TPU has excellent physico-chemical properties, such as excellent resistance to wear, tear and abrasion, good chemical stability, and facility of processing.3 The development of CNT/TPU nanocomposites has been motivated by the need for materials with a wide range of properties, from improved thermal stability and flame retardant to enhanced electrical and mechanical properties,4–6 and for their potential use in sensors, biomedical, smart textiles, and many others.7–9
Despite progress, there are still obstacles that impede the advancement of CNT/TPU composite. A major complication lies in managing the CNT-TPU interface and controlling the dispersion of CNTs within TPU matrix. For example, Novikov et al.10 employed the coagulation (antisolvent) precipitation technique to fabricate SWCNT/TPU composites, and with a homogeneous distribution of SWCNTs, the composites showed excellent performance in mechanical, piezoresistive, and EMI shielding properties. A common solution to this issue is the surface modification of CNTs to introduce functional groups.11 Using solution mixing and melt mixing, Sui et al.12 prepared TPU composites with carboxyl and unmodified MWCNTs, and their findings highlighted the significance of CNT surface modification. Zhuang et al.13 prepared TPUs filled with pre-oxidized CNTs, and their results also confirms the important role of CNT surface modification on the dispersion of CNT.
The interfacial characteristics of CNT/TPU composites are critical for their structure and properties; however, direct experimental exploration at a molecular level is challenging.14–16 In this context, molecular dynamics (MD) simulation technique serves a powerful tool for studying the behavior of materials on an atomic level, as well as the overall properties of the material. By applying MD simulations, it is possible to predict the behavior of polymer nanocomposites under various conditions. In the past decades, MD simulations have been utilized to examine the interfacial properties between CNTs and a range of polymers, such as epoxy,17,18 nylon 6,19 chitosan,20 and polymethyl methacrylate,21 with the majority of studies focusing on the interfacial binding energy. In recent past, some MD simulation research work of PU-based or TPU-based nanocomposites have also been carried out. For example, Aliyev et al.22 investigated the interactions between PU and graphene (Gr) and graphene oxide (GO), and their results showed that highly functionalized GO exhibited better adhesion with PU due to the higher polarization and more hydrogen bonds. By establishing three models of functionalized defective Gr/TPU nanocomposites, Talapatra et al.23 examined the effects of Gr on the elastic moduli and strength properties, and they found that the interfacial normal strength was much higher than interfacial shear strength. By adopting MD simulations, the compatibility between PU and asphalt has been studied by Huang et al.24 and Lu et al.25 through analyzing the cohesive energy density, hydrogen bonding, and radial distribution function.
Although quite a few simulations have been conducted to study the structure and properties of polymer nanocomposites related to CNTs or TPUs, only one simulation research work done by Madkour et al.26 to the best of our knowledge, has focused on the CNT/TPU nanocomposite systems. They employed MD simulations to calculate the structure and interfacial interactions between pristine MWCNT and TPU to interpret their experimental observations. To this end, the aim of this study is to explore the effects of pristine or functionalized single-wall carbon nanotube (SWNT) on the thermodynamic and structural properties of thermoplastic polyurethane (TPU) through atomistic MD simulations, as the behavior of CNT/TPU composites has yet to be fully understood. Specifically, the glass transition and thermal expansion of SWNT/TPU composites, the TPU diffusion behavior, the interfacial binding strength, the solubility parameter, and the hydrogen bonds affected by SWNTs are carefully analyzed. Some of our simulation results have been shown to be in line with experimental values.
2 Methodology
2.1 Simulation model
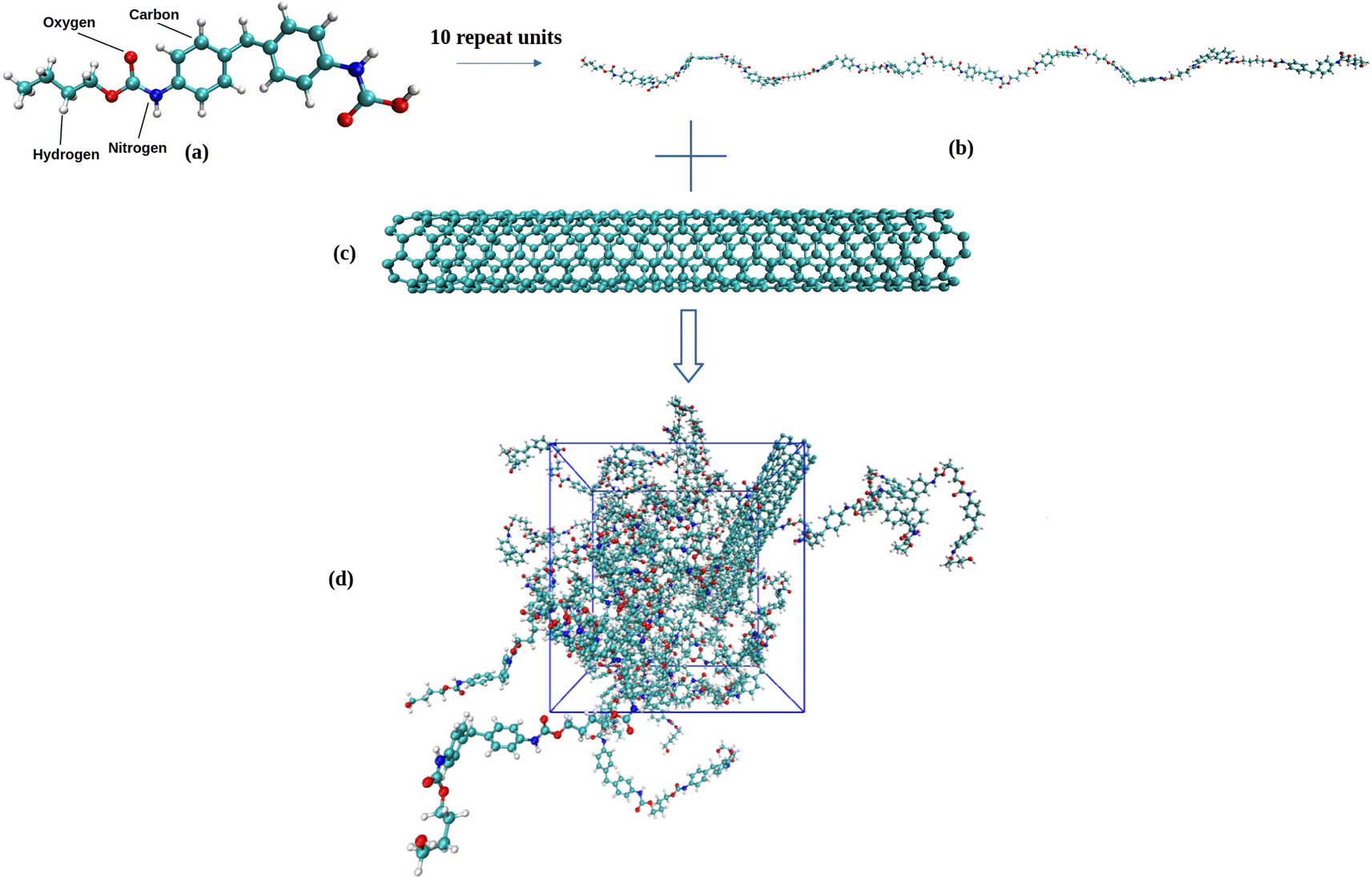
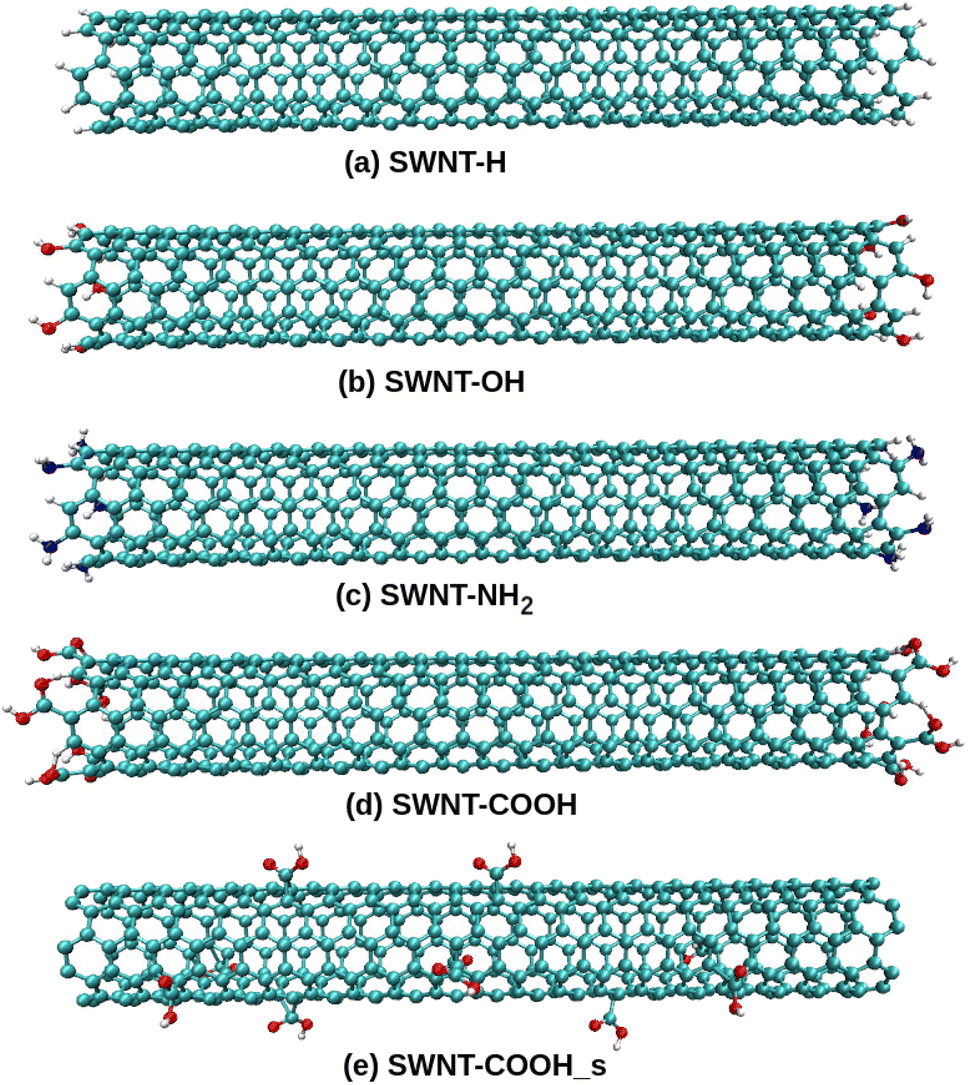
All-atom models of TPU nanocomposites filled with either pristine SWNT or functionalized SWNT are established in the present work. The widely used TPU, poly[(1,4-butanediol)-alt-(4,4′-diphenylmethane diisocyanate)] (MDI/BDO), is chosen for the polymer matrix. The BDO and MDI molecules are used to construct a TPU repeat unit, as seen in Fig. 1a, and each TPU chain is composed of 10 repeat units. The BDO part of the TPU acts as the soft segment and the MDI part as the hard segment. Similar TPU models have been used by Lempesis and Rutledge et al.27,28 and Talapatra et al.23 and their simulation results have confirmed the validity of such TPU model. An armchair (5,5) single-walled carbon nanotube (SWNT) with a diameter of 6.8 Å and a length of 49.2 Å is used as the nanofiller. Additionally, various models of functionalized SWNTs, including (end-functionalized) SWNT-H, SWNT-OH, SWNT-NH2, SWNT-COOH, and (side functionalized) SWNT-COOH_s, are constructed herein due to the necessity of introducing functional groups in various experiments involving SWNTs, as displayed in Fig. 2. Each of these functionalized SWNTs has 10 functional groups bonded to the carbon atoms.
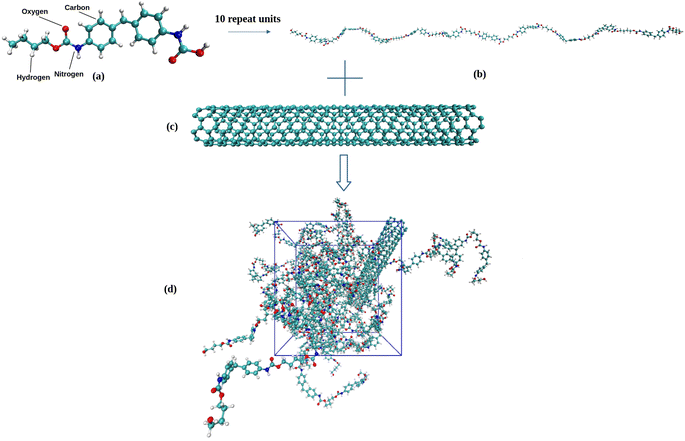 |
| | Fig. 1 (a) Chemical structure of a TPU repeat unit, (b) a TPU chain consisting 10 repeat units (460 atoms), (c) armchair (5,5) SWNT model, and (d) snapshot of SWNT/TPU simulation system. | |
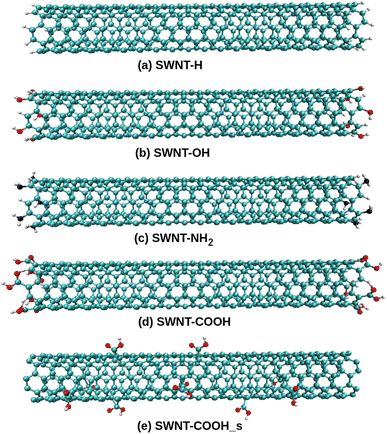 |
| | Fig. 2 The SWNTs functionalized with the (a) –H, (b) –OH, (c) –NH2, and –COOH groups (d) to the ends and (e) to the body surface. | |
In this study, the classic COMPASS (condensed-phase optimized molecular potentials for atomistic simulation studies) force field29 is employed to describe all the interactions between atoms in the atomistic model. COMPASS force field, using ab initio and empirical parametrization techniques, has been parameterized and validated for a broad range of molecules, such as polymers, in condensed phases, and is thus believed to enable accurate prediction of structural and thermodynamic properties of SWNT/TPU nanocomposites. The valence energy Evalence is accounted for by the bond stretching, valence angle bending, dihedral angle torsion, and improper interactions,
| | |
Evalence = Ebond + Eangle + Etorsion + Eimproper
| (1) |
and the non-bond interaction potential is accounted for by the van der Waals and electrostatic interactions,
| | |
Enon-bond = EvdW + Eelectrostatic
| (2) |
| |
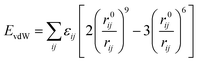 | (3) |
| |
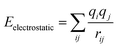 | (4) |
where both the van de Waals and electrostatic interactions are cut off at the distance of 12.5 Å. Ewald summation method is employed to compute the electrostatic energy. The bond, angle, dihedral, improper, and pair styles for the COMPASS force field are supported by the CLASS2 package in the large-scale atomic/molecular massively parallel simulator (LAMMPS),
30 where more detailed descriptions about the force field can be found. The COMPASS CLASS2 package has already proven to be effective and accurate in handling polymer materials such as graphene/PMMA composites.
31 In our simulations, each SWNT/TPU composite system contains ten TPU chains and one SWNT molecule (more than 5000 atoms in total), as shown in
Fig. 1d.
2.2 Molecular dynamics simulation
All the MD simulations are executed through LAMMPS molecular dynamics packages. To begin, all the TPU chains and the SWNT (pristine or functionalized) are placed in a large simulation box. Energy minimization is performed using the steepest descent method. We then equilibrate our simulation systems for 1 ns at 400 K under an NPT ensemble (0.1 MPa), followed by cooling to 300 K, 320 K, 340 K, 360 K, 380 K, and 400 K, and another 1 ns of NPT equilibration (0.1 MPa). Periodic boundary conditions are applied in all three directions to remove the edge effects. The Nosé–Hoover thermostat and barostat are employed to control the temperature and the pressure respectively. The Newtonian equations of motions are integrated using the velocity-Verlet algorithm, with a time step of 1 fs. For each simulated composite system, we have carefully monitored that the thermodynamic equilibrium state has been reached, and for statistical significance, five NPT runs (carried out separately) are averaged over. As depicted in Fig. S1 of ESI,† the total potential energy and the system density all quickly reach equilibrium within 50 ps of the 1 ns simulation time, and then remain relatively steady around their respective equilibrium values, indicating that the system is adequately converged. Simulation data are collected every 5 ps for 100 ps after sufficient equilibrium of the simulation system, yielding pertinent structures and dynamical features of interest.
3 Results and discussion
3.1 Glass transition
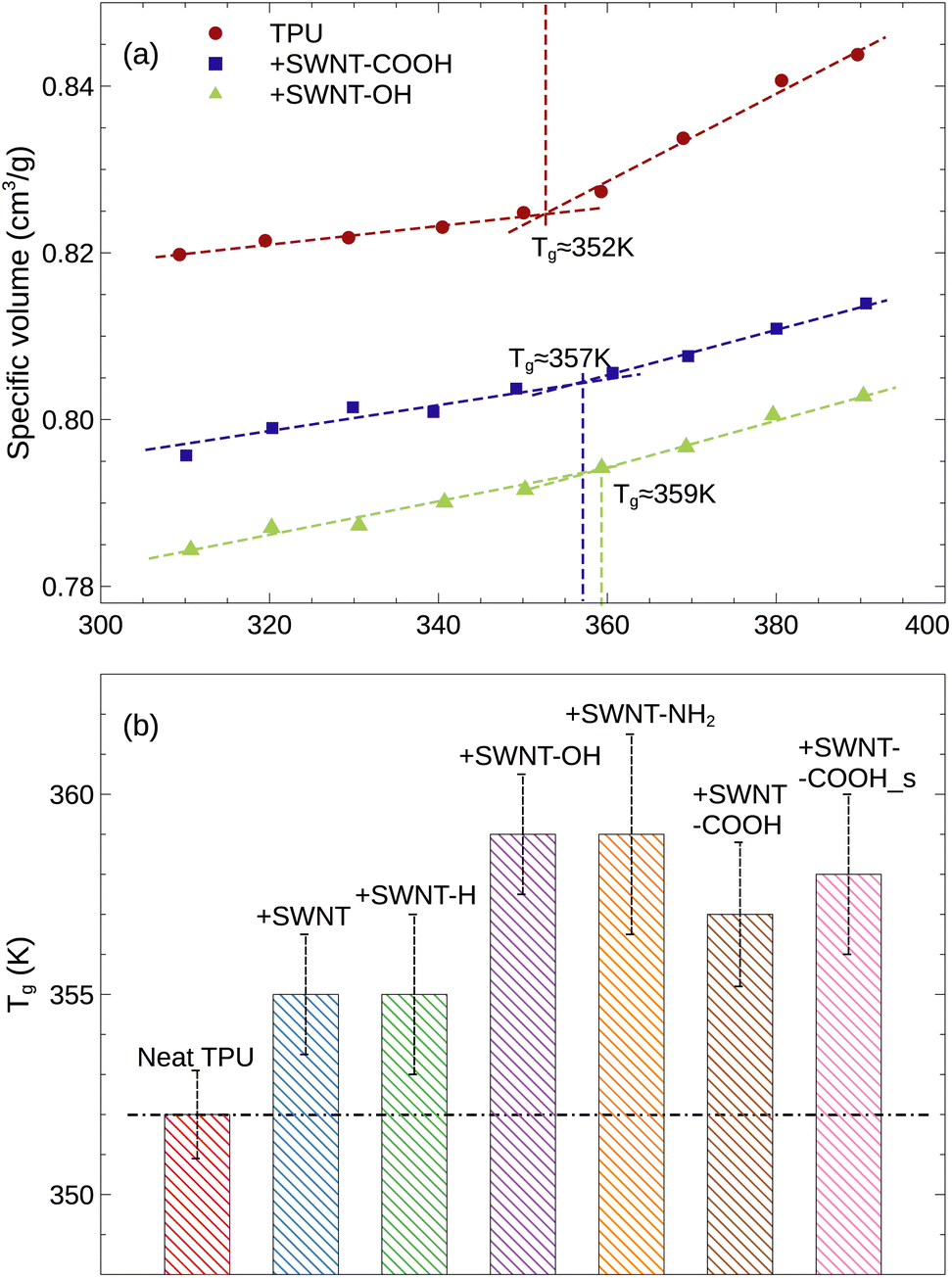
The glass transition temperature (Tg) distinguishes the transition of an amorphous material from a hard and brittle glassy state into a soft and rubbery state; through the glass transition range, the material will exhibit a distinct change in many properties, such as the thermal expansion, the heat capacity, the viscosity, and shear modulus. In the current simulation, the Tg can be identified by the intersection of two straight lines on the plot of specific volume32,33 versus temperature, as illustrated in Fig. S2 of ESI.† We note that the large uncertainty in specific volume is due to the finite size effect and the thermal fluctuations. Below the glass transition, the specific volume (the reciprocal of material density) increases slowly with increasing temperature, indicating the glassy state, whereas above the glass transition, it increases rapidly, indicating the rubbery state. For the neat TPU system, the Tg is found to be approximately 351–352 K, which is consistent with the experimental range of 342–382 K. In addition to the Tg, the TPU density and solubility parameter extracted from our MD simulations are also in close agreement with experimental values, as seen in Table 1, validating that our simulation models and methods are adequate to reproduce the structure and properties of TPU. By the way, although the density and solubility parameter from our simulation are slightly lower than the experimental values, which is likely a result of our shorter chain model compared to realistic TPU chains, our qualitative findings remain unchanged.
Table 1 Comparison of TPU properties extracted from our simulation with experimental values
| |
Glass transition temperature Tg (K) |
Density at 300 K (g cm−3) |
Solubility parameter δ (MPa0.5) |
| Data was collected from https://polymerdatabase.com/. |
| Current simulation |
351–352 |
1.18–1.19 |
22.7–23.0 |
| Experimental valuea |
342–382 |
1.20–1.27 |
23.2–24.1 |
Fig. 3 illustrates how the glass transitions of TPUs are impacted by the incorporation of SWNTs. For visual clarity, only three representative curves are shown. The uncertainty due to the five independent MD runs and the curve fitting is reflected in the error bars on the Tg. It is observed that the Tg is increased with the addition of SWNTs, e.g., from 352 K for neat TPU to 355 K for pristine SWNT/TPU system. This can be attributed to the large SWNT molecules impeding the mobility of TPU chains, which is corroborated by a reduction in diffusivity of TPU molecules and an increase in hydrogen bonds formation (as discussed in detail below). For polymer composites with a low loading of CNTs, the Tg variation induced by SWNT inclusions is typically ∼10 K, which agrees well with experimental results.34–36 It is noteworthy that the polar groups (i.e., –OH, –NH2, and –COOH) functionalized SWNT-filled TPU composites exhibit relatively higher Tgs, indicating stronger interfacial interactions between SWNTs and TPUs, in agreement with experimental observations.37,38 In particular, SWNT-OH and SWNT-NH2 exhibit the highest enhancement (∼8 K increment) in Tg due to the hydrogen bonds, which is qualitatively consistent with existing experiments.39
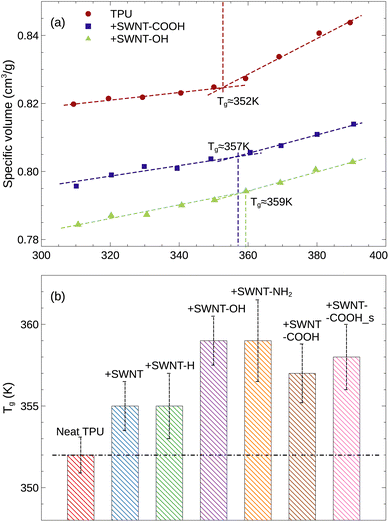 |
| | Fig. 3 (a) Specific volume as a function of temperature for various simulation systems. For visual clarity, only three representative curves are shown in this plot. (b) Glass transition temperature (Tg) for various TPU materials. | |
3.2 Polymer mobility
To demonstrate the effects of functionalized SWNTs on the Tg, the molecular mobility of TPU chains is further examined. The mean squared displacement (MSD(t)) of polymer atoms is employed to characterize the diffusion behavior of molecules through| | |
MSD(t) = 〈|xt − x0|2〉
| (5) |
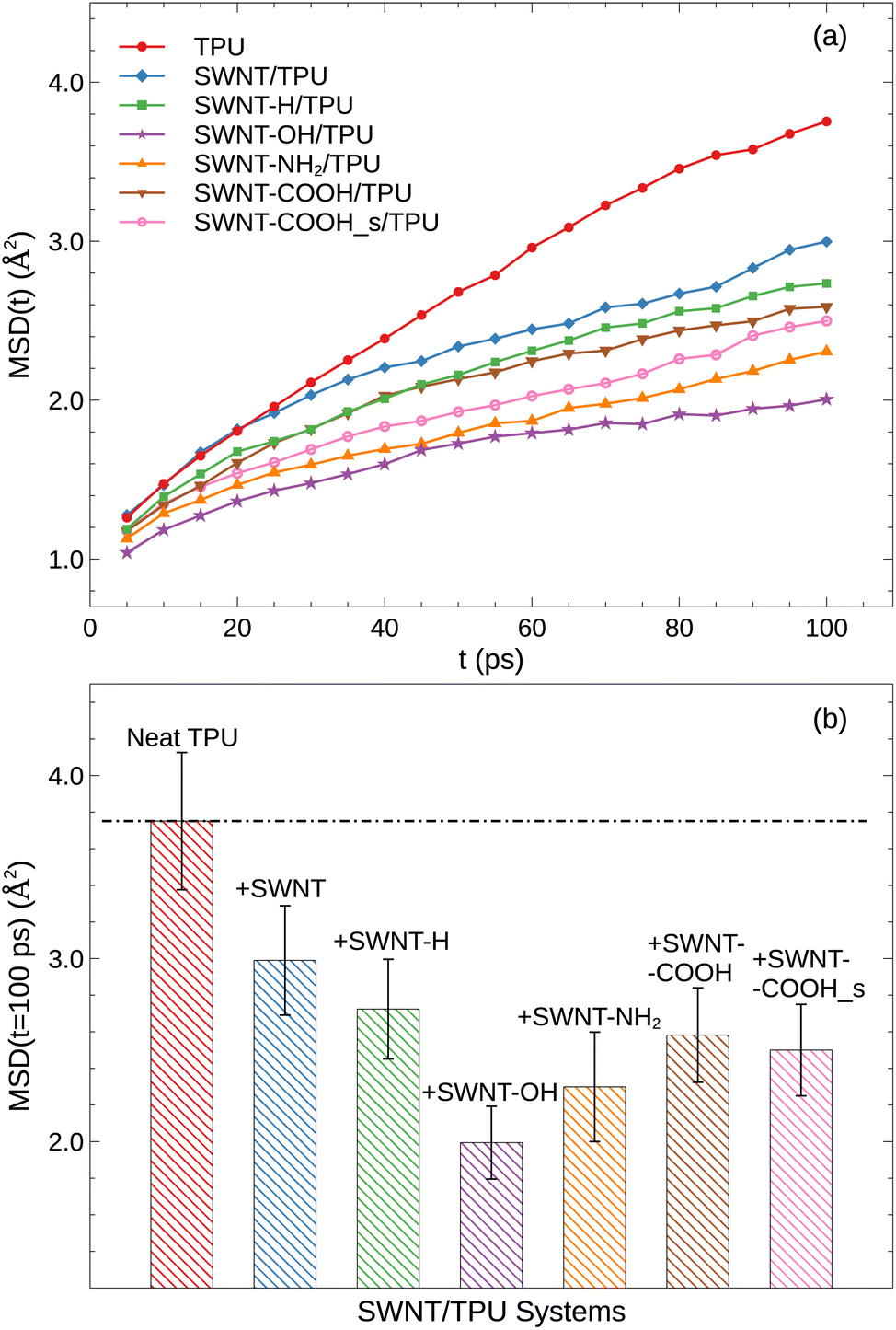
where vectors x0 and xt denote the atom positions at the reference time of 0 and t, respectively, and 〈…〉 represents the ensemble average. Fig. 4a displays the MSD(t) of TPU chains as a function of time at the temperature of 400 K well above Tg. Each MSD(t) curve increases with time. As one would anticipate, the incorporation of SWNT into TPU matrix results in a substantial decrease in the polymer mobility. Notably, SWNTs functionalized with polar groups exhibit the most pronounced effect, which verifies our explanation for the observed differences in the Tg (Fig. 3).
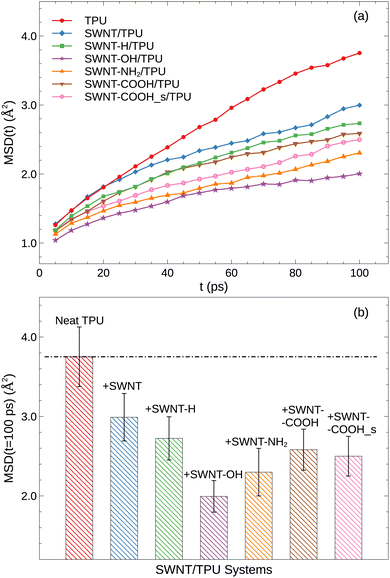 |
| | Fig. 4 (a) The mean squared displacement (MSD(t)) of TPU atoms as a function of time at 400 K; (b) comparison of MSD(t) = 100 ps for various SWNT/TPU composites. | |
Specifically, we present the MSD at 100 ps in Fig. 4b to quantitatively assess polymer mobility in this sub-diffusive regime. As can be seen, the presence of SWNTs in the TPU blend reduces the mobility of TPU chains at the examined temperatures compared to the neat TPU system, aligning with the observed Tg results. Of note, the polar groups modified-SWNTs systems exhibit even lower MSD(t) due to the enhanced hydrogen bonding formation (Fig. 5) and stronger interfacial interactions (Fig. 6) that hinder the dynamics and flexibility of the chains near SWNT surface.40,41
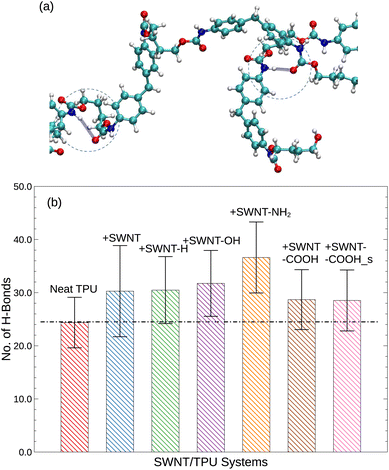 |
| | Fig. 5 (a) Illustration of the formation of hydrogen bonds. The blue, white, red and cyan spheres represent the nitrogen, hydrogen, oxygen, and carbon atoms, respectively. (b) Average number of hydrogen bonds in various SWNT/TPU systems. | |
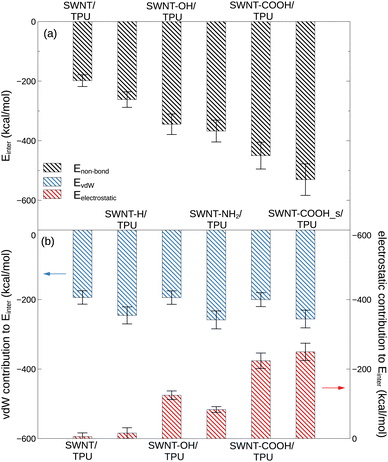 |
| | Fig. 6 (a) The interfacial binding energy between TPU and various SWNTs at 400 K. (b) Van de Waals and electrostatic contributions to the SWNT/TPU interfacial binding energy. | |
3.3 Hydrogen bonding
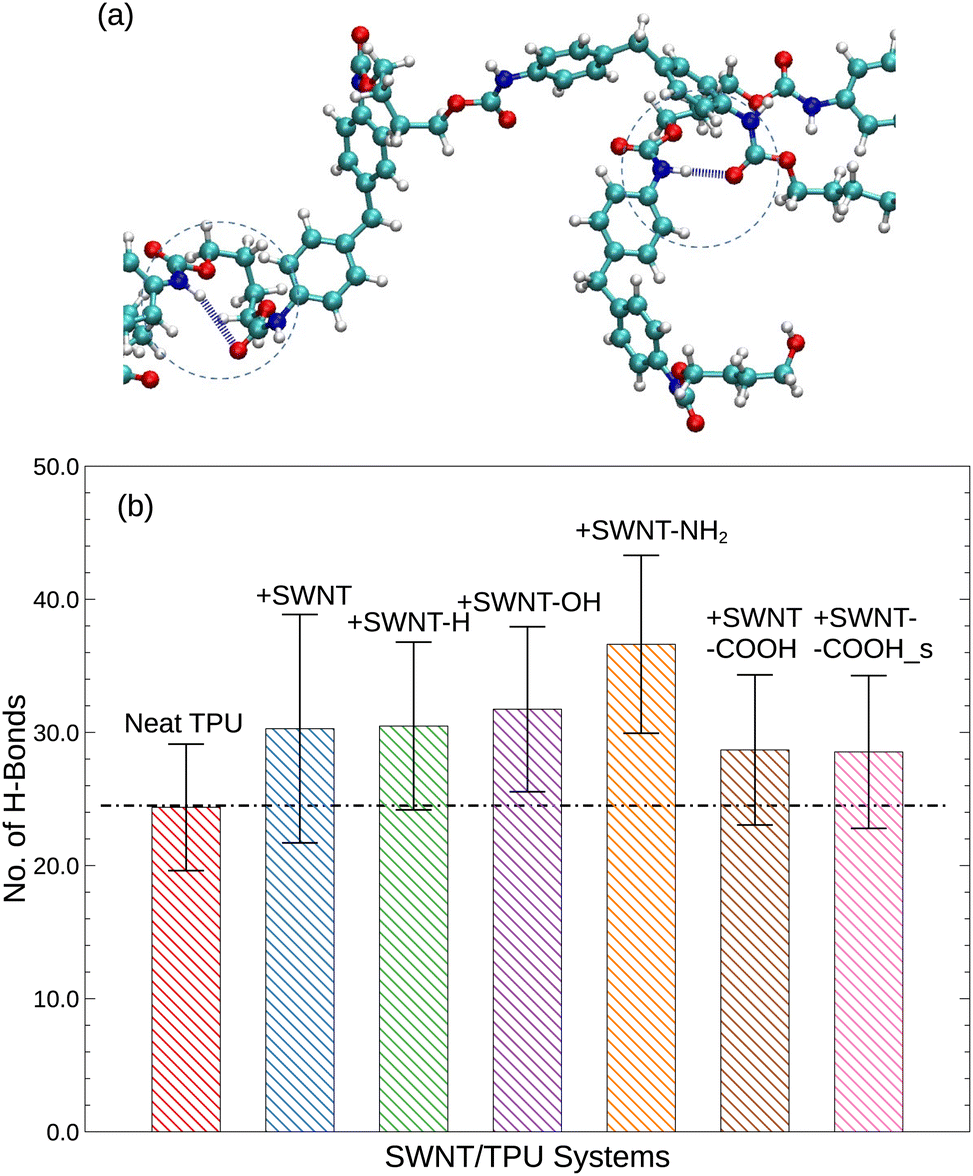
In order to gain deeper insights into the observed discrepancies in Tg and MSD(t) for different TPU materials, we further investigate the effects of functionalized SWNTs on the hydrogen bond formation. Although the hydrogen bonding is not specifically treated in our simulation models, we still would like to make a brief analysis into the influences of SWNTs on the hydrogen bonds of TPUs. Using VMD (visual molecular dynamics42) HBonds plugin, the average number of hydrogen bonds is estimated, and the results are shown in Fig. 5. Here, the criteria by Ippolito et al.43 and McDonald and Thornton44 for identifying hydrogen bonds in protein-ligand systems is adopted. In particular for the current simulation, a hydrogen bond is considered to be formed between a polar atom (N, O) with a hydrogen bonded to it (the donor, D) and another polar atom (N, O) (the acceptor, A) provided that the D–A distance is less than the cut-off distance of 3.5 Å and the D–H–A angle is less than the cut-off angle of 40°, as illustrated by Fig. 5a. Although the cut-off distance and angle are a little arbitrary at a reasonable level and thus may lead to some quantitative variations, the qualitative aspects of our results will not be changed.
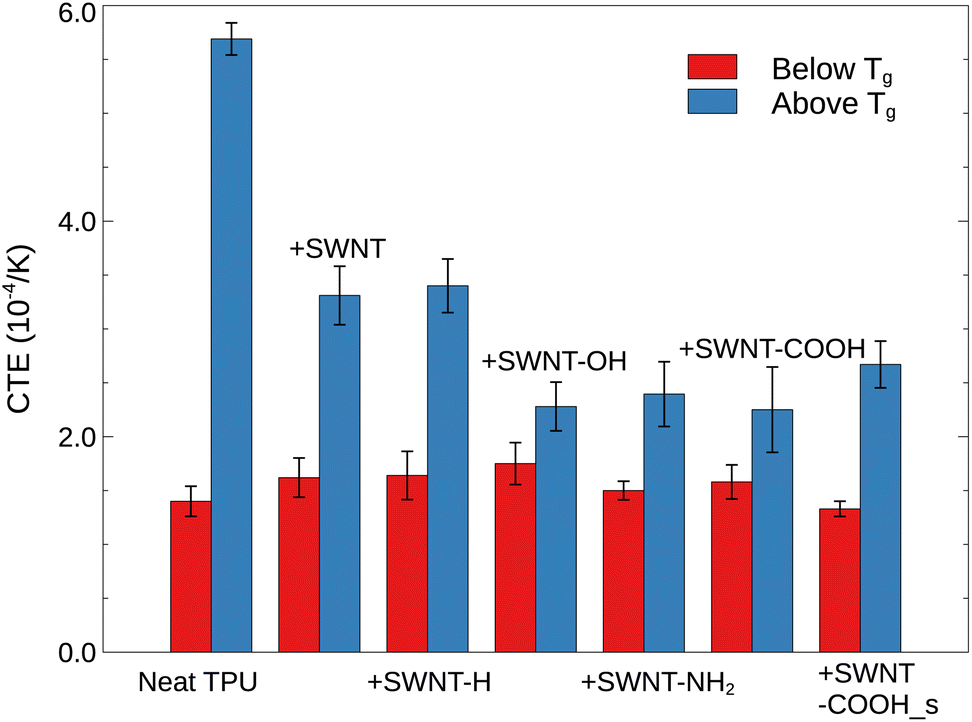
According to Fig. 5, the introduction of SWNTs, either pristine or functionalized, leads to an increase in hydrogen bonds than neat TPU. This effect can be attributed to the favorable van de Waals interactions (π–π and CH–π interactions) between the graphitic walls of SWNTs and the TPU backbones, which stimulate the wrapping of TPU chains around SWNTs and thus favor hydrogen bonding formation.45 Additionally, polar groups tethered to SWNTs, such as carboxyl (–COOH), oxhydryl (–OH) and amine (–NH2) groups, can further form hydrogen bonds with the urethane carbonyl (–CO) of TPU chains, with amine groups exhibiting the greatest potential and carboxyl groups the least. The increased hydrogen bonds are expected to lead to the observed enhancement of glass transition temperature Tg (Fig. 3) and a reduction of thermal expansion (Fig. 7) of polymer systems, in consistent with experimental findings.46
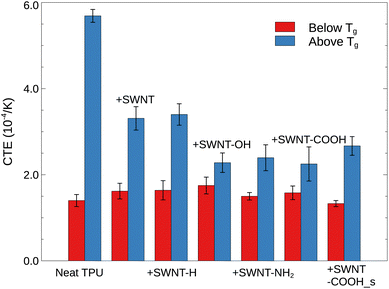 |
| | Fig. 7 Coefficient of thermal expansion (CTE) below and above Tg for a range of TPU materials. | |
3.4 Interfacial binding energy
Furthermore, the interfacial interactions between the functionalized SWNTs and the TPU matrix are evaluated to elucidate the effects of functionalized SWNTs on the structural and thermodynamic properties of TPU. The interfacial binding energy (Einter) is employed to characterize the interfacial bonding property in this work. The Einter can be estimated from the change in potential energy of the polymer material with and without SWNTs,18,19| | |
Einter = Etotal − (ESWNT + ETPU)
| (6) |
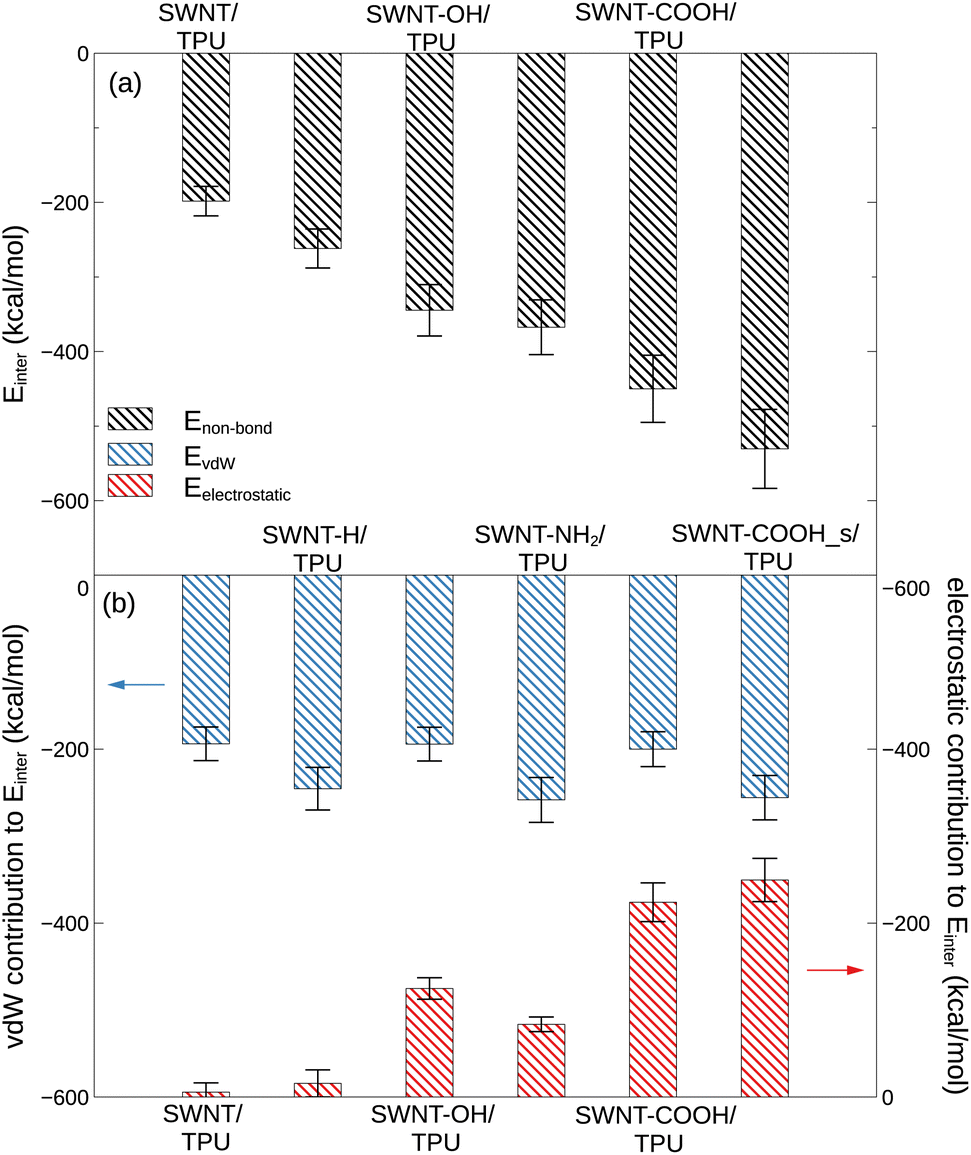
where Etotal is the total non-bond potential energy of the composite, and ESWNT and ETPU denote the potential energies of the SWNTs and the neat TPU system separately. The van de Waals and electrostatic contributions to the interfacial interaction are estimated likewise. Fig. 6 displays the total interfacial binding energy between various SWNTs and TPU matrix at 400 K, as well as the van de Waals and electrostatic contributions to this binding energy.
From Fig. 6, it can be observed that the polar groups-modified SWNTs (e.g. SWNT-COOH) have a much stronger interfacial interaction with TPU molecules than the pristine ones, as evidenced by the much lower Einter. In particular, the interfacial binding strength of SWNT-COOH is the strongest among the composite systems examined, followed by SWNT-NH2, SWNT-OH, SWNT-H, and pristine SWNT in order, indicative of a significant role for functional groups in the interfacial properties of SWNT/TPU nanocomposites. This is further corroborated by electrostatic interactions (6b), which highlight the contribution from polar atoms such as oxygen19 and nitrogen. The side-functionalized COOH groups (-SWNT-COOH_s) are found to confer more improvement in SWNT-TPU interfacial binding than end-functionalized ones (-SWNT-COOH), likely due to a more homogeneous distribution on the SWNT surface without steric hindrance, allowing for better contacts and interaction between COOH groups and TPU chains. In practice, however, such sidewall functionalization is not always recommended, as the conversion of sp2 hybridized carbon atoms to sp3 ones and the simultaneous loss of the π-conjugation system on the graphene layer would lead to a decrease in the intrinsic properties of SWNTs, such as electrical conductivity stemming from π-electrons.47–49 By the way, it has been reported50 that the interfacial binding interaction of nanocomposites is not much affected by temperature at low filler concentration.
Non-bond interfacial interactions between SWNT and TPU, such as CH–π, π–π stacking,16 polymer wrapping around SWNT41 and hydrogen bonding, is described as a combination of van der Waals and electrostatic interactions in our simulation model (eqn (2)). As indicated by Fig. 6, the interfacial interactions between TPU and pristine and hydrogenated SWNTs are predominantly driven by van der Waals forces, while the electrostatic interactions play an important role in the binding between SWNT-COOH and TPU chains. Despite similar van der Waals interactions for all modified SWNT/TPU composites in comparison to the pristine SWNT/TPU composite, the improved binding strength observed for the modified SWNTs is primarily attributed to electrostatic interactions stemming from the various functional groups. It is observed that, although the SWNT-OH and SWNT-NH2 exhibit relatively weaker affinity to TPU polymer compared to SWNT-COOH, the oxhydryl (–OH) and amine (–NH2) groups are more likely to form hydrogen bonds with the urethane carbonyl (–CO) of TPU chains, as evidenced by Fig. 5, resulting in, accompanied by lower chain mobility (Fig. 4), a relatively higher Tg in Fig. 3b. In addition, these results concerning the interaction between SWNT and TPU are further validated by the corresponding solubility parameter analysis, as detailed in the ESI.† Generally, the introduction of polar groups with electrostatic interactions and hydrogen bonds into SWNTs can enhance the SWNT/TPU interface strength and consequently improve the stress transfer efficiency and the mechanical properties.51
3.5 Thermal expansion
Additionally, the influences of SWNTs on the thermal expansion of TPUs are investigated. The volumetric coefficient of thermal expansion (CTE), as one of the most thermal properties, is defined as| |
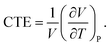 | (7) |
The CTE is supposed to show a sharp change at Tg, and can thus be assumed as a constant below or above Tg regardless of its slight variations with temperature in either glassy or rubbery state.50 For the specific volume vs. temperature plot in this work, the averaged CTE (below or above Tg)52 can be written as follows,
| |
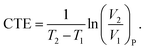 | (8) |
Provided that V1 is close to V2, the CTE can be further simplified as the slope of specific volume vs. temperature in glassy and rubbery region normalized by the volume at a reference temperature (commonly Tg). From Fig. 3a, the slopes of specific volume vs. temperature of all TPU systems below Tg are quite close to each other, with the CTE of ≈1.57 × 10−4 K−1. This is due to the inherent extremely low thermal expansion of SWNTs (CTE ≈ 0 K−1)53 and the restraint of TPU segment movement in glassy state, limiting the contribution of SWNTs to thermal expansion. However, the CTE of TPUs in rubbery region (above Tg) is greatly affected by the presence of SWNTs. Specifically, pristine and hydrogenated SWNTs alike have been observed to reduce the volumetric thermal expansion by approximately half, likely owing to decreased TPU segmental dynamics and increased hydrogen bonding. SWNTs featuring polar groups (–OH, –COOH, and –NH2) are demonstrated (above) to interact more strongly with TPU molecules, suppressing polymer mobility and inducing extra hydrogen bonds, thus resulting in even lower coefficients of thermal expansion (CTEs). Parenthetically, the averaged CTE of all SWNT/TPU composites is raised from ≈1.57 × 10−4 K−1 of glassy state to ≈2.79 × 10−4 K−1 of rubbery state, owing to the differences in segmental mobility across the glass transition.
4 Conclusion
In this study, atomistic MD simulations are performed to investigate the thermodynamic and structural properties of SWNT/TPU nanocomposites. Specifically, the effects of functionalized SWNTs on the glass transition and thermal expansion of SWNT/TPU composites, the interfacial binding energy, and the hydrogen bonds are examined in detail. The glass transition temperature Tg is found to increases with the addition of SWNT, due to the restrained TPU dynamics and the increased hydrogen bonds. In comparison to pristine SWNT, SWNTs modified by polar groups (e.g., –OH, –NH2, and –COOH) have a stronger effect on the decrease of polymer mobility and the increase of Tg, due to the improved SWNT-TPU interfacial interaction strength and extra hydrogen bonds with the urethane carbonyl (–CO) of TPU chains. Analysis of the SWNT/TPU binding energy and solubility parameter δ suggests that the van der Waals interactions contribute a little to such functionalized SWNT/TPU interface, yet the electrostatic aspects are likely to dominate the interfacial interactions and their compatibility. Hydrogen bonds are shown to be increased by SWNT inclusion, likely due to the wrapping of TPU chains around SWNT stimulated by favorable van der Waals interactions. Polar groups tethered to SWNTs, such as carboxy (–COOH), oxhydryl (–OH) and amine (–NH2) groups, can further form hydrogen bonds with the urethane carbonyl (–CO) of TPU chains, with SWNT-NH2 system exhibiting the most hydrogen bonds. The volumetric thermal expansion coefficient (CTE) in rubbery region (above Tg) is drastically reduced by the SWNT inclusion, especially for polar groups-functionalized SWNTs.
In conclusion, the increase of Tg and the reduction of CTE can be attributed to the impeded polymer dynamics, resulting from increased hydrogen bonds induced by SWNT inclusion and strengthened SWNT-TPU interfacial interactions, especially for SWNTs functionalized with polar groups. Lastly, it is important to note that although our simulated results agree well with experimental value for density, glass transition temperature Tg, and solubility parameter δ, the simulation models are still somewhat simplified. For instance, the modeled TPU chains are too short comparing to realistic chains; moreover, parameters such as the size and volume fraction of SWNTs, the molar mass distribution of TPU chains, and more, are not taken into account. In general, this research work may have potential applications in the design and fabrication of polymer nanocomposites, and can also provide some guidance for future simulations related to TPUs.
Author contributions
Jianxiang Shen: conceptualization, formal analysis, funding acquisition, investigation, methodology, project administration, visualization, writing – review & editing; Xue Li: data curation, funding acquisition, investigation, visualization, writing – original draft; Ping Li: formal analysis, validation; Baoqing Shentu: conceptualization, formal analysis, supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by Natural Science Foundation of Zhejiang Province, China (LY22B040002), and National Natural Science Foundation of China (52203091). Computations were performed at Wuzhen Supercomputer Center of China.
References
- I. A. Kinloch, J. Suhr, J. Lou, R. J. Young and P. M. Ajayan, Science, 2018, 362, 547–553 CrossRef CAS PubMed
 .
. - M. N. Norizan, M. H. Moklis, S. Z. N. Demon, N. A. Halim, A. Samsuri, I. S. Mohamad, V. F. Knight and N. Abdullah, RSC Adv., 2020, 10, 43704–43732 RSC .
- J. Datta and P. Kasprzyk, Polym. Eng. Sci., 2018, 58, E14–E35 CrossRef CAS .
- L. Wan, C. Deng, H. Chen, Z.-Y. Zhao, S.-C. Huang, W.-C. Wei, A.-H. Yang, H.-B. Zhao and Y.-Z. Wang, Chem. Eng. J., 2021, 417, 129314 CrossRef CAS .
- D. Martin, A. Osman, Y. Andriani and G. Edwards, in Advances in Polymer Nanocomposites, Woodhead Publishing, 2012, pp. 321–350 Search PubMed .
- T. A. Phung Hai, M. Tessman, N. Neelakantan, A. A. Samoylov, Y. Ito, B. S. Rajput, N. Pourahmady and M. D. Burkart, Biomacromolecules, 2021, 22, 1770–1794 CrossRef CAS PubMed .
- T. Chen, Y. Xie, Z. Wang, J. Lou, D. Liu, R. Xu, Z. Cui, S. Li, M. Panahi-Sarmad and X. Xiao, ACS Appl. Polym. Mater., 2021, 3, 5317–5338 CrossRef CAS .
- W.-J. Sun, L. Xu, L.-C. Jia, C.-G. Zhou, Y. Xiang, R.-H. Yin, D.-X. Yan, J.-H. Tang and Z.-M. Li, Compos. Sci. Technol., 2019, 181, 107695 CrossRef CAS .
- J. Tang, Y. Wu, S. Ma, T. Yan and Z. Pan, Composites, Part B, 2022, 232, 109605 CrossRef CAS .
- I. V. Novikov, D. V. Krasnikov, A. M. Vorobei, Y. I. Zuev, H. A. Butt, F. S. Fedorov, S. A. Gusev, A. A. Safonov, E. V. Shulga and S. D. Konev, et al., ACS Appl. Mater. Interfaces, 2022, 14, 18866–18876 CrossRef CAS PubMed .
- N. Mohd Nurazzi, M. M. Asyraf, A. Khalina, N. Abdullah, F. A. Sabaruddin, S. H. Kamarudin, S. Ahmad, A. M. Mahat, C. L. Lee and H. Aisyah, et al., Polymers, 2021, 13, 1047 CrossRef PubMed .
- G. Sui, D. Liu, Y. Liu, W. Ji, Q. Zhang and Q. Fu, Polymer, 2019, 182, 121838 CrossRef .
- Y. Zhuang, Y. Guo, J. Li, K. Jiang, Y. Yu, H. Zhang and D. Liu, RSC Adv., 2020, 10, 23644–23652 RSC .
- M. Rahmat and P. Hubert, Compos. Sci. Technol., 2011, 72, 72–84 CrossRef CAS .
- J. Chen, L. Yan, W. Song and D. Xu, Composites, Part A, 2018, 114, 149–169 CrossRef CAS .
- J. Chen, B. Liu, X. Gao and D. Xu, RSC Adv., 2018, 8, 28048–28085 RSC .
- W. Jian and D. Lau, Compos. Sci. Technol., 2020, 191, 108076 CrossRef CAS .
- W. Zhang, X. Deng, G. Sui and X. Yang, Carbon, 2019, 145, 629–639 CrossRef CAS .
- J.-S. Yang, C.-L. Yang, M.-S. Wang, B.-D. Chen and X.-G. Ma, RSC Adv., 2012, 2, 2836–2841 RSC .
- D. Aztatzi-Pluma, E. O. Castrejon-Gonzalez, A. Almendarez-Camarillo, J. F. Alvarado and Y. Duran-Morales, J. Phys. Chem. C, 2016, 120, 2371–2378 CrossRef CAS .
- A. Farazin and M. Mohammadimehr, Adv. Nano Res., 2020, 9, 83–90 Search PubMed .
- I. Aliyev, S. Hasanov, E. Rasulov, R. Moradi and A. R. Vakhshouri, Mater. Today: Proc., 2021, 42, A27–A35 CAS .
- A. Talapatra and D. Datta, Mol. Simul., 2021, 47, 602–618 CrossRef CAS .
- T. Huang, Z. Zhang, L. Wang, J. Sun, Z. Wang, H. Liu and L. Chen, Case Stud. Constr. Mater., 2022, 17, e01424 Search PubMed .
- P. Lu, S. Huang, Y. Shen, C. Zhou and L. Shao, Polym. Eng. Sci., 2021, 61, 2323–2338 CrossRef CAS .
- T. M. Madkour, F. M. Hagag, W. Mamdouh and R. A. Azzam, Polymer, 2012, 53, 5788–5797 CrossRef CAS .
- N. Lempesis and G. C. Rutledge, et al., Polymer, 2016, 107, 233–239 CrossRef CAS .
- S. Zhu, N. Lempesis, P. J. in‘t Veld and G. C. Rutledge, Macromolecules, 2018, 51, 1850–1864 CrossRef CAS .
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS .
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS .
- F. Lin, Y. Xiang and H.-S. Shen, Composites, Part B, 2017, 111, 261–269 CrossRef CAS .
- J. B. Buchholz, W. Paul, F. Varnik and K. Binder, J. Chem. Phys., 2002, 117, 7364–7372 CrossRef CAS .
- J. Shen, X. Li, L. Zhang, X. Lin, H. Li, X. Shen, V. Ganesan and J. Liu, Macromolecules, 2018, 51, 2641–2652 CrossRef CAS .
- T. Sterzyński, J. Tomaszewska, K. Piszczek and K. Skórczewska, Compos. Sci. Technol., 2010, 70, 966–969 CrossRef .
- A. Allaoui and N. eddine El Bounia, Express Polym. Lett., 2009, 3, 588–594 CrossRef CAS .
- A. S. Babal, R. Gupta, B. P. Singh and S. R. Dhakate, RSC Adv., 2015, 5, 43462–43472 RSC .
- F.-L. Jin, C. Ma and S. Park, Mater. Sci. Eng., A, 2011, 528, 8517–8522 CrossRef CAS .
- C. Jiang, D. Jiang, J. Zhang, S. Lin, X. Shang and S. Ju, Polym. Compos., 2018, 39, 1129–1138 CrossRef .
- S. Roy, R. S. Petrova and S. Mitra, Nanotechnol. Rev., 2018, 7, 475–485 CrossRef CAS PubMed .
- J. Shen, X. Lin, J. Liu and X. Li, Phys. Chem. Chem. Phys., 2020, 22, 16760–16771 RSC .
- D. A. Vega, A. Milchev, F. Schmid and M. Febbo, Phys. Rev. Lett., 2019, 122, 218003 CrossRef CAS PubMed .
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed .
- J. A. Ippolito, R. S. Alexander and D. W. Christianson, J. Mol. Biol., 1990, 215, 457–471 CrossRef CAS PubMed .
- I. K. McDonald and J. M. Thornton, J. Mol. Biol., 1994, 238, 777–793 CrossRef CAS PubMed .
- A. Llanes-Pallás, K. Yoosaf, H. Traboulsi, J. Mohanraj, T. Seldrum, J. Dumont, A. Minoia, R. Lazzaroni, N. Armaroli and D. Bonifazi, J. Am. Chem. Soc., 2011, 133 39, 15412–15424 CrossRef PubMed .
- T. Yiyao, L. Luo, Q. Yang, L. Zhang, M. Wang, D. Wu, X. Wang and X. Liu, Polymer, 2020, 188, 122100 CrossRef .
- A. Jorio, R. Saito, G. F. Dresselhaus and M. S. Dresselhaus, in The sp2 Nanocarbons: Prototypes for Nanoscience and Nanotechnology, John Wiley Sons, Ltd, 2011, ch. 1, pp. 1–15 Search PubMed .
- P. Ma, N. A. Siddiqui, G. Marom and J. Kim, Composites, Part A, 2010, 41, 1345–1367 CrossRef .
- A. B. S. Diekmann, M. C. V. Omelan and U. Giese, Polymers, 2021, 13, 1355 CrossRef CAS PubMed .
- J. Yang, Z. Zhu, D. Huang and Q. Cao, Chin. Phys. B, 2020, 29, 023104 CrossRef CAS .
- Z. pitálský, D. Tasis, K. Papagelis and C. Galiotis, Prog. Polym. Sci., 2010, 35, 357–401 CrossRef .
- M. Hadipeykani, F. Aghadavoudi and D. Toghraie, Phys. A, 2020, 546, 123995 CrossRef CAS .
- H. Jiang, B. Liu, Y. Huang and K. C. Hwang, J. Eng. Mater. Technol. Trans. ASME, 2004, 126, 265–270 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Xue Li*d,
Ping Lic and
Baoqing Shentu*a
abc,
Xue Li*d,
Ping Lic and
Baoqing Shentu*a