
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-resolution imaging and analysis of the intestinal bacterial load of Caenorhabditis elegans during early adulthood†
Farzad Rezaeianaran * and
Martin A. M. Gijs
* and
Martin A. M. Gijs
Laboratory of Microsystems, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: rezaeianaran.farzad@epfl.ch
First published on 8th June 2023
Abstract
We study the presence within the worm Caenorhabditis elegans (C. elegans) of a fluorescent strain of the worm's bacterial food (Escherichia coli (E. coli) OP50) during early adulthood. Use of a microfluidic chip based on a thin glass coverslip substrate allows investigation of the intestinal bacterial load using a Spinning Disk Confocal Microscope (SDCM) equipped with a high-resolution objective (60×). High-resolution z-stack fluorescence images of the gut bacteria in adult worms, which were loaded in the microfluidic chip and subsequently fixed, were analyzed using IMARIS software and 3D reconstructions of the intestinal bacterial load in the worms were obtained. We present an automated bivariate histogram analysis of the volumes and intensities of the bacterial spots for each worm and find that, as the worms age, the bacterial load in their hindguts increases. We show the advantage of single-worm resolution automated analysis for bacterial load studies and anticipate that the methods described in our work can be easily implemented in existing microfluidic solutions to enable thorough studies of bacterial proliferation.
Introduction
The nematode C. elegans is a bacterivorous organism with considerable genetic similarity to humans and is used as a model for studying the relation between microbiota and aging and health/diseases.1–7 In particular, this roundworm, owing to its genetic tractability and evolutionarily conserved innate immunity is an established model organism for the investigation of virulence mechanisms of pathogens.5In order to discover the virulence determinants, different mutants of a pathogen of interest as well as the worms are generated and the interactions between the bacteria and the host (worm) are studied through survival curves and bacterial colonization assays. However, these assays typically cannot capture the interactions between the host and the bacteria on a microscopic scale. For example, the bacterial colonization itself can only measure the number of viable bacteria and it cannot determine whether the bacteria are merely accumulating in the gut or if they are proliferating there. Additionally, this assay cannot reveal the spatio-temporal dynamics of bacterial colonization in the gut as this requires real-time in vivo fluorescence imaging of the gut. Furthermore, the impact of pathogenic bacteria on the worm is not limited to the colonization of the gut. For example, it has been reported that both Salmonella typhimurium (S. typhimurium) and Pseudomonas aeruginosa (P. aeruginosa) reduce the motility and the rate of pharyngeal pumping of the worm over the course of infection.8,9 Intestinal infection with Serratia marcescens (S. marcescens) leads to the lysis and vacuolation of intestinal cells and a reduction of the volume of the epithelium.10 Therefore, a complete description of bacterial pathogenesis also requires imaging, to extract the relevant phenotypes, and the use of/designing of specific assays that target a certain interaction between the host and the pathogen (e.g., using competition and shifting assays to investigate whether a certain pathogen is capable of establishing a persistent infection8,11). While we mainly discussed the requirements for studying bacterial pathogenesis in the worm, it should be noted that the same requirements apply when it comes to investigating the beneficial aspects of commensal and probiotic bacteria or the role of the worm's native microbiome.
Based on the analysis above, it can be seen that a comprehensive characterization of the host–microbiota interactions, if done manually, is extremely laborious. However, this is exactly what makes microfluidics such a powerful approach. Microfluidic devices offer:12 (i) precise control over experimental conditions, (ii) facilitating manipulations of worms permitting high throughput screening studies, (iii) ease-of-doing long-term studies, (iv) reversible immobilization for both low- and high-resolution imaging, (v) laser microsurgery.13–15 As a result, the use of microfluidics for studying C. elegans has been extensively practiced16 and different devices for low-throughput long-term studies at single worm resolution17–21 as well as high-throughput and high-content screening studies have been reported.22–24
However, only a few microfluidic devices have been reported for studying the interactions between bacteria and C. elegans.25–28 Yang et al.,25 fabricated a microfluidic chip featuring 32 chambers arranged radially and 4 concentration gradient generators for evaluation of in vivo antimicrobial activity of several compounds. Each concentration generator supplies 8 unique chambers with 8 different concentrations of a given antibiotic. The device enables on-chip establishment of an infection model (in this case Staphylococcus aureus after 6 hours) and antibiotic treatment of the infection (in this case amoxicillin and some natural compounds for 48 hours) and, compared to conventional methods, requires less analysis time and less reagent consumption. However, the evaluation of antimicrobial activity was mostly limited to survival curves (within a 72 hours period). Fluorescence imaging of the worms were feasible but, since each chamber contained around 10 worms that were not in immobilized, neither high-resolution imaging nor single-worm resolution analysis were possible.
In another study by Lee et al.,26 a microfluidic chip for implementing a C. elegans killing assay was reported. The chip featured large chambers where the worm and the pathogen of interest (in this case P. aeruginosa) could be loaded. Survival curves were obtained with high accuracy and reproducibility. Due to lack of an immobilization mechanism on the chip, only low-resolution imaging could be performed. Imaging in the brightfield channel was used for the automated detection of dead worms and motility analysis, while fluorescence imaging was implemented for measuring the expression of irg-1::GFP (infection response gene 1). Since, the worms were freely moving in the chamber without any compartments, they could not be differentiated from each other, and thus individual worms could not be monitored over the entire course of the experiment.
In the more recent works by Viri et al.,27,28 the dynamics of bacterial transition, load and absorption in the gut of the worms are studied in an automated fashion through fluorescence imaging. The first microfluidic device27 contained 4 channels, each of which comprised 5 consecutive chambers, separated by filter structures. These chambers, which could hold 1–3 worms, included a culturing area where the worms could freely move and feed on fluorescent E. coli and tapered traps that immobilize the worm and stop it from eating (due to the application of strong fluid flow required for immobilization). Low-resolution fluorescent imaging over the entire body of the worms was performed, while they were freely moving and feeding on the fluorescent bacteria. Analyzing the resulting image sequence yielded the spatio-temporal distribution of the bacteria in the gut, albeit at a low resolution. In addition, the duration of a full cycle of bacterial ingestion until intestinal clearance was determined. Similar imaging and analysis were also performed while the worms were immobilized. In this case, the time constant of the transition of recently ingested bacteria could be obtained (as the worms could not eat while they were immobilized).
The second microfluidic device28 was similar in scope to the first one but had a different design. The device still consisted of 4 channels, but each channel had 4 parallel chambers instead. As before, these chambers included a culturing area where the worms were free to move and feed but the design of the trapping structure was modified so that the worm could continue eating while it was immobilized. As a result, the dynamics of bacterial load and digestion could be studied on immobilized worms using fluorescent bacteria at relatively higher imaging resolution. The higher resolution made it possible to distinguish the ingested intact bacteria from the ingested disrupted ones and as a result, and the time constant for food digestion was determined. As the immobilization could be maintained for 30 hours, long-term studies at single-worm resolution were also possible. While the newly improved immobilization and imaging procedure allows the spotting of intact bacteria, the bacterial dynamics load assay described was not capable of answering questions on bacterial colonization of the gut. Granting that the intact bacteria are indeed viable and have the potential to forming colonies, it is by no means guaranteed that this effectively happens in the gut; it is very much possible that the viable bacteria merely accumulate in the gut and do not proliferate.
Therefore, in this work, we intend to bring the analysis to a higher level of resolution, by acquiring for the first time high-resolution z-stack fluorescence images of fluorescent E. coli bacteria in the gut of fixed worms using a SPDM. The resulting images are subsequently imported in IMARIS software to allow visualization bacterial load of the gut in 3D. Such representation facilitates the investigation of eventual bacterial colonization of the gut at high-spatial resolution. We could study whether there are preferential regions in the gut for colonization and how intestinal bacterial load advanced with time.
Materials and Methods
Chip materials and chemicals
4 inch 550 μm Si wafers, 5-inch Chromium/soda–lime glass masks, polydimethylsiloxane (PDMS) Sylgard 184 (Dow®) and trimethylchlorosilane (TMCS) (Sigma-Aldrich) were obtained from the Center of MicroNanoTechnology (CMi) at EPFL (Lausanne, Switzerland). Kayaku Advanced Materials (KAM) SU-8 3050 was purchased from Micro Resist Technology GmbH (Berlin, Germany). 45 mm × 70 mm glass coverslips with a thickness of 170 μm were purchased from Biosystems Switzerland AG (Muttenz, Switzerland). Saint-Gobain Tygon™ ND 100-80 Tubing (inner diameter and thickness of 0.02 inch and outer diameter of 0.06 inch) was bought from Fisher Scientific (Reinach, Switzerland). Nematode growth medium (NGM) plates were ordered from the Solution Preparation Facility at EPFL. S-basal was prepared by first dissolving 5.85 g of NaCl, 6 g of KH2PO4 and 1 g of K2HPO4 in H2O until a final volume of 1 l was reached. The resulting solution was then autoclaved, and, once it cooled down, 1 ml of cholesterol solution (5 mg ml-1 in ethanol) was added aseptically to obtain S-basal. S-medium was obtained by adding aseptically 500 μl of 1 M potassium citrate (pH 6), 500 μl of trace metals solution, 150 μl of 1 M CaCl2 and 150 μl of 1 M MgSO4 to 50 ml of S-basal. The materials involved in the preparation of S-basal and S-medium, lysogeny broth (LB) for bacteria culture and tetracycline were ordered from Sigma-Aldrich Chemie GmbH (Buchs, Switzerland). 4% paraformaldehyde (PFA) solution in phosphate buffered saline (PBS) for the fixation of C. elegans was purchased from Fisher Scientific (Reinach, Switzerland).Worm and bacteria culture and preparation
C. elegans wild-type (WT) Bristol N2 strain were maintained 22 °C on NGM plates (55 mm in diameter) seeded with E. coli OP50. In order to synchronize the worm populations for experiments, gravid adult worms are suspended in S-medium in a falcon tube overnight. During this period, the adults lay eggs which will hatch into L1 larvae. Due to the absence of food, the hatched L1s will enter developmental arrest resulting in synchronized L1 populations. The next day, L1s were aspirated from the falcon tube and thus separated from dead adults (as the adults sediment while the L1s remain floating in S-medium) and were placed inside 1.5 ml Eppendorf tubes to be centrifuged for 4 min at 2000 RPM. Afterwards, the L1s were aspirated from the bottom of the Eppendorf tube and about 100 of them were dispensed on NGM plates seeded with E. coli OP50.E. coli OP50 was grown overnight in LB at 37 °C on a shaker. E. coli OP50 expressing Red Fluorescent Protein (RFP) was provided by the C. elegans Ageing Laboratory at the University College London. E. coli OP50 was transformed with plasmid pRZT3::dsRED to construct RFP-expressing E. coli OP50. The plasmid also contains genes for tetracycline resistance. As a result, LB RFP E. coli OP50 was grown in LB containing 10 μg ml−1 of tetracycline overnight at 37 °C on a shaker.
Fabrication of the microfluidic chip
The layout of the microfluidic device was designed with Clewin 4.0 (WieWeb software, Hengelo, The Netherlands) and then transferred to a chromium/soda-lime glass mask using standard mask fabrication processes. Afterwards, the mask was employed in standard soft photolithography processes to fabricate a 75 μm high SU-8 mold on a Si wafer. Next, the SU-8 mold was treated with TMCS to promote the detachment of PDMS from SU-8. Subsequently, PDMS with base-to-curing agent ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was casted onto the mold and cured at 80 °C for 2 hours. Afterwards, the cured PDMS device was cut and separated from the SU-8 mold and punched to create inlets and outlets. Next, the PDMS device was bonded to a 45 mm × 70 mm coverslip with a thickness of 170 μm using oxygen plasma. To enhance the bonding between the PDMS device and the cover slide, the microfluidic chip was kept on a hotplate at 80 °C for 10 minutes. Lastly the tubing for the inlet and outlet were connected.
:1 was casted onto the mold and cured at 80 °C for 2 hours. Afterwards, the cured PDMS device was cut and separated from the SU-8 mold and punched to create inlets and outlets. Next, the PDMS device was bonded to a 45 mm × 70 mm coverslip with a thickness of 170 μm using oxygen plasma. To enhance the bonding between the PDMS device and the cover slide, the microfluidic chip was kept on a hotplate at 80 °C for 10 minutes. Lastly the tubing for the inlet and outlet were connected.
Image processing and statistical analysis
IMARIS (version 9.9.1) was used to process the raw optical microscopy images. The results of image analysis by IMARIS were extracted to datasheets which were further processed in MATLAB (version 2022a) and Graphpad Prism (version 9.5.0) software. MATLAB was used for basic arithmetic operations on the datasets and in particular, to create histograms in an automated fashion. Furthermore, MATLAB was also used to extract certain quantitative results and prepare them for further processing for GraphPad Prism. GraphPad Prism was mainly used to elaborate on the statistical variation and the statistical significance of the data. The number of worms analyzed for each experiment is shown by the parameter “n”. Statistical significance was determined using a one-tailed Mann–Whitney test.Microscopy platform and the imaging parameters
Two different microscopes were used in our experiments. A Zeiss Axio Imager.M2 with a 2.5X Zeiss objective was used in brightfield mode to prepare the microfluidic chip for the experiment and to load and fix worms on the chip. We used a Visitron CSU-W1, a SDCM for high-resolution fluorescence imaging. It was equipped with a Hamamatsu ImagEMX2 electron-multiplying CCD (EMCCD) camera. The different lasers used for the excitation of the fluorophores for imaging in the RFP and the Green Fluorescent Protein (GFP) channels were operating at wavelengths 561 nm and 488 nm, respectively. We used Chroma Technology (Vermont, USA) ET605/70m and ET525/50m emission filters for the RFP and GFP channels, respectively. Lastly an Olympus U PLAN S APO 60X/1.42 NA objective was employed for high-resolution imaging. The imaging was done with the following parameters: exposure time: 60 ms, laser power for both the RFP and the GFP channels: 50%, gain of the EMCCD camera: 200, pinhole size: 50 μm, z-stack imaging: 50 μm range with step size of 0.2 μm.Experimental
Operation of the microfluidic device
We have designed our microfluidic device (Fig. 1a) such that we can load an adult worm population of interest on the device, contain them within an area in the channel and fix them for the purpose of high-resolution imaging with a SDCM. The device comprises 5 lanes where each lane is 2 cm in length, 1300 μm wide and 75 μm high. This height was chosen since the diameter of a young adult worm is on average 48 μm and can reach up to 80 μm during adulthood. Each lane also features 2 sets of filter structures, the purpose of which is to keep the worms on the right side of the channel while a fluid flow towards the inlet is maintained. Fig. 1b shows an example of the fabricated microfluidic chip. The filters are each 20 μm wide and the spacing between them is 15 μm, except in the middle area where it is 30 μm instead (Fig. 1c). As the worms are compressible, especially if high flow rates are used, the filter sizes should be smaller than the diameter of the worms.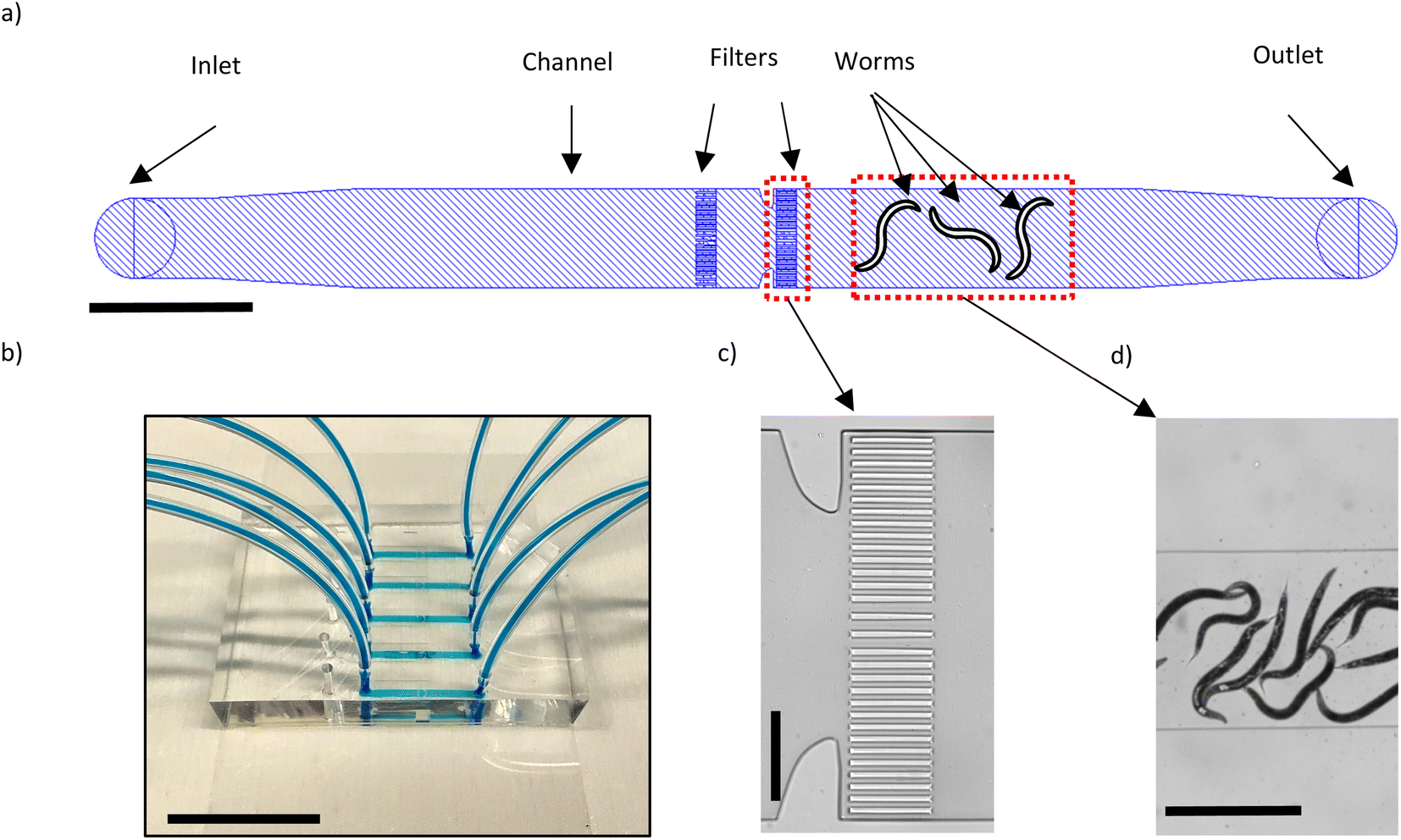 | ||
| Fig. 1 Microfluidic device for high-resolution imaging of E. coli OP50 in the C. elegans gut. (a) Schematic representation of one of the five lanes in each device. The lanes are 1300 μm wide and 75 μm high. The filters serve to contain the worms in the channel. Scale bar = 2 mm. (b) Image of a fabricated microfluidic chip where the channels are filled with a blue dye. Scale bar = 2 cm. (c) Brightfield image of the filter structures. Scale bar = 300 μm. (d) Brightfield image of the worms in the channel. Scale bar = 2 mm. | ||
Before a worm population of interest can be loaded on the chip, the channels should be filled by S-medium. The chip therefore is placed on the stage of the Zeiss Axio Imager.M2 equipped with a 2.5X Zeiss objective to be observed in brightfield mode. To introduce S-medium in the chip, we connect its inlet tube to a neMESYS syringe pump (CETONI GmbH, Korbuβen, Germany) where a 1 ml BD l Luer-Lok syringe containing S-medium is present. When the chip is filled with S-medium for the first time, bubbles of air are bound to form in the channel. To remove the air bubbles, the tube of the outlet is clipped, 10 μl of S-medium is dispensed in the channel which pressurizes the air bubbles and thus facilitates their disappearance via the outward diffusion of air through the PDMS. In our case, the debubbling process takes around 15–20 minutes. Next, the worms suspended in S-medium (inside an Eppendorf tube) can be placed at the outlet of the chip and by aspirating with a flow rate of 500 nl s−1, the worms are loaded in the chip (Fig. 1d). It is preferred to have around 10–15 worms in the channel. In the case of overcrowding/undercrowding, further aspirations/dispensing of S-medium can be performed to adjust the number of worms present in the channel. In order to fix the worms, a 4% PFA solution is placed at the outlet and by aspiration with a flow rate of 80 nl s−1, it is introduced inside the channel. We use a relatively low flow rate to avoid excessive pushing of the worms against the filters. The worms are fixed within 5–10 minutes of being in contact with the 4% PFA solution. At this point, the inlet and outlet tubing are cut and clipped, and the worms are ready for the high-resolution imaging.
Experiment planning
The aim of our experiment is to observe the bacterial presence in the intestine of the worm for young adult worms of different age via high-resolution fluorescence imaging. Fig. 2 shows the steps involved in our experiment. To obtain a synchronized L1 population, an adult population of wild-type C. elegans is suspended overnight in S-medium in a 50 ml falcon tube. During this period, the adults lay eggs that will hatch into L1 larvae. Due to lack of food, these larvae will be in a state of developmental arrest resulting in a synchronized population. The day after, ∼100 hatched L1s were placed on a NGM plate seeded with E. coli OP50 (Fig. 2a and b). The L1s are cultured on these plates until they reach adulthood (46 hours at 22 °C) and afterwards, they are transferred to an NGM plate seeded with RFP labelled E. coli OP50. Before this transfer takes place, the number of E. coli OP50 present in the S-medium is reduced to a minimum by serial dilutions (1:1010 total dilution) (Fig. 2c). This allows us to safely assume that, after the transfer, the adult population is only feeding on RFP-expressing bacteria (Fig. 2d). The transfer is done after the worms become adults because the bacterial colonization of the gut starts in adulthood. We plan to image the adult worms that have fed on RFP labelled bacteria for 1, 2, 3 and 4 days. Since our imaging relies on fixing the worms to enable highest possible resolution, it's not possible to follow an identical worm at different times. In our case, for each experimental condition (1, 2, 3 and 4 days of feeding on RFP-expressing bacteria), we used separate worm populations fed over variable times with RFP-labelled E. coli OP50. Since we are using WT worms in our experiment, naturally the adults will lay eggs over time. This will cause two problems, namely, the fast depletion of food on the plates and a confusion in selecting the original adult worms (and not their progenies) for imaging. Therefore, only the adults should be transferred to a new plate seeded with RFP-labelled E. coli OP50 every 24 hours (Fig. 2e). The transfer process is done in the same way as explained earlier (Fig. 2c), however, the main aim is instead removing the L1 progenies floating in S-medium by serial dilutions. Furthermore, before the adult worms are loaded on the chip for fixation and imaging, progenies and in particular, RFP-labelled E. coli OP50 should be reduced to a minimum. Therefore, considering these two aspects, steps in Fig. 2d and e should be repeated in this manner: 0×, 1×, 2× and 3× for 1 day, 2 days, 3 days and 4 days of feeding, respectively (0 meaning that steps in Fig. 2d and e are performed once). After the worms have spent the required time feeding on RFP-labelled E. coli OP50 and before they are loaded on chip to be imaged, the worms must stay in the Eppendorf tube containing S-medium (Fig. 2e) for an hour. Based on the work by Viri et al.,27 in WT worms the time interval between bacterial ingestion and intestinal clearance is about 70 to 90 s. Therefore, it is reasonable to assume that after one hour of starving the worms, any bacteria present in the gut which are observed by fluorescence microscopy may be at least persistent and eventually are colonizing the intestine. The chip is prepared for worm loading as explained in the materials and methods section, one day before the experiment. As the starvation time approaches 1 hour, the worms are loaded on the chip where they are fixed. Afterwards the tubings are cut and clipped and the chip is ready to be taken to a SDCM for high-resolution imaging. The RFP-labelled E. coli OP50 was previously used in lifespan and ageing studies and there were no mentions of potential effects of the fluorescent tag on the worm.29–31 As a result, we expect in good faith, that the RFP-labelled E. coli OP50, other than being fluorescent, is identical to the unlabelled E. coli OP50. Consequently, no control experiments were conceived.
 | ||
| Fig. 2 The experimental steps involved in high-resolution imaging of fluorescent E. coli in the C. elegans gut. (a) Suspending adult worms in S-medium overnight yields numerous synchronized L1s in developmental arrest the next day. (b) ∼100 synchronized L1s are transferred to a NGM plate seeded with E. coli OP50 and kept there until they become adults (46 hours at 22 C°). (c) The adult worms are taken from the NGM plate and are washed in S-medium in a 1.5 ml Eppendorf tube; since adults naturally sediment at the bottom of the tube, the supernatant is removed (1:10 dilution) and the process is repeated until the number of E. coli OP50 present is reduced to a minimum (a total of 1:1010 dilution). (d) The adults are now transferred to a NGM plate seeded with E. coli OP50 expressing RFP and kept there for 24 h. (e) To remove the progenies of the adult worms, every 24 h, the adults are taken from the plate and are suspended in S-medium and a total of 1:1010 dilution is performed (similar to “step c”). Afterwards, the adult worms are transferred to new NGM plates seeded with E. coli OP50 expressing RFP. Given the number of the days, adult worms are planned to be fed on E. coli OP50 expressing RFP (1 day, 2 days, 3 days and 4 days), steps d and e are repeated as many times as required (0×, 1×, 2× and 3× for 1 day, 2 days, 3 days and 4 days feeding, respectively). Once the last repetition of “step e” is done, the worms are instead kept in the Eppendorf tube containing S-medium for one hour to ascertain that recently ingested bacteria are fully processed. (f) The worms are loaded on the microfluidic chip and are fixed with a 4% PFA solution. Afterwards, the tubings are cut and clipped and the microfluidic chip is transferred to a SDCM for high-resolution imaging of the gut bacteria. | ||
Imaging procedure and analysis
Fig. 3a shows a typical brightfield image of fixed worms that have fed on RFP-labelled E. coli OP50. In order for the worms to be properly imaged in the fluorescence channel, overcrowding of the worms should be avoided, as it can cause two problems. The first problem is the overlapping of the worms on top of each other, which makes it impossible to distinguish the signals originating from each worm. The second problem is fluorophore photobleaching that takes place during fluorescence imaging.32 When the channel is overcrowded, the worms might not be overlapping but still they are in close proximity of each other such that, during the fluorescence imaging of one worm, the worm next to it is also being exposed and thus is being subjected to photobleaching. | ||
| Fig. 3 Imaging of typical fixed worms that have fed on RFP E. coli OP50 (for 1 day in figure a and 4 days in figures b, c and d). (a) Brightfield image of fixed worms, in which, the hindgut, as area of interest, is delineated. High-resolution (b) Brightfield image, (c) RFP channel image where the bacteria in the hindgut can be seen and (d) GFP channel image where the worm autofluorescence can be observed. (a: 10× objective, scale bar = 250 μm; b, c and d: high-resolution imaging using an oil-immersive 60× objective, scale bar = 40 μm). | ||
Our previous observations (not shown here) revealed that the hindgut is most often the location where bacterial presence is seen and, thus, we limit our observations to the hindgut (Fig. 3a). High resolution brightfield images, RFP channel and GFP channel images are shown in Fig. 3b, c and d, respectively. The imaging parameters (as mentioned in the Materials and Methods section) have been optimized such that signal/noise ratio is high enough in the fluorescence channels, photobleaching is limited and signal saturation is avoided. The z-range of the z-stacks (50 μm) is also large enough to cover the intestine entirely. We use the brightfield mode (Fig. 3b) to avoid photobleaching while locating the hindgut and adjusting the center of focus for z-stack imaging; afterwards fluorescence imaging is performed (Fig. 3c and d). The presence of fluorescent bacteria in hindgut can be clearly observed in Fig. 3c while the worm autofluorescence that originates mainly from the intracellular lysosome-derived granules within the intestinal cells33 is seen in Fig. 3d. As the field of view allows only a length of 150 μm of the worm to be imaged at a time, each worm needs to be imaged twice to observe the entire hindgut. In our case, the region closest to the tail was imaged first and it was followed by imaging the second half of the hindgut which is further away from the tail.
We can see in Fig. 4 several slices from z-stack fluorescence imaging of a typical fixed worm that has fed on RFP labelled E. coli OP50 for 4 days. As shown schematically in (Fig. 4a), the bacteria can be present in different focal planes (z numbers) within a cross-section of the intestinal lumen of the worm. In conventional widefield microscopy the light is received from many focal planes some of which are out of focus and thus leading to poor resolution (blurry image). In contrast, in SDCM, the pinholes block the signal from out-of-focus planes and as a result the bacteria in each focal plane are resolved.
 | ||
| Fig. 4 Z-stack fluorescence imaging of typical fixed worms that have fed on RFP E. coli OP50 for 4 days. The z-stacks are taken using a 60× oil-immersion objective in both RFP and GFP channels. They include 250 slices with a step size of 200 nm and total focal range of 50 μm. (a) The three example slices with their associated planes of focus, positioned relative to a schematic worm cross-section. Typical images acquired at these slices are shown both in the RFP channel (b–d) and the GPF channel (e–g), where the z numbers of images e, f and g are the same as those of images b, c and d, respectively. Focusing reveals RFP E. coli OP50 in the different regions of the hindgut, as highlighted by the white dashed lines in (b–d). The signal in the GFP channel originates from autofluorescence in the adult worms. In the presence of strong autofluorescence, the latter signal can also be observed in the RFP channel, as delineated by the yellow dashed lines in (b and e) and (d and g). In these cases, the RFP signal present in the yellow dashed line-limited area in (b and d) should not be attributed to the existence of bacteria and therefore should be omitted from any visualization/analysis pertaining to bacterial load. (Scale bar = 40 μm). | ||
Fig. 4b–d are slices with certain z numbers in the RFP channel, where each focal plane reveals the presence of bacteria in different positions (not only in plane but also along the z-axis) within the hindgut. Furthermore, while individual bacterial spots can be observed in Fig. 4a and c, in Fig. 4b, the bacteria appear to form clusters having high density. Fig. 4e–g show the same slices (the z numbers of e, f and g are the same as b, c and d, respectively) imaged in the GFP channel. It can be seen that, in the presence of strong worm autofluorescence, the signal appears both in the GFP (Fig. 4e and g) channel and the RFP channel (Fig. 4b and d). As a result, in these cases the latter RFP signals should be omitted from bacterial load analysis.
We use IMARIS for visualization and processing of our raw z-stack fluorescence images. IMARIS assembles all the slices belonging to the same channel (e.g. the RFP channel) of a given raw image and thereby creates a 3D image. In our case, since our raw images have two channels, namely, the RFP and the GFP channel, IMARIS creates two 3D images from each raw images-stacks. We can also display the two 3D images at the same time as shown in Fig. 5a. In this image, a worm that has fed on RFP E. coli OP50 for 3 days is displayed. The green signal originates from the autofluorescence of the worm, while the orange signal is due to RFP E. coli. A magnified view of the purple rectangle is shown in Fig. 5b. We can now observe more clearly the orange spots. We also see 3 yellow spots, as indicated by the white arrows, that are due to strong autofluorescence. The strong autofluorescence leads to the presence of signals both in the GFP and the RFP channels. The overlap of the stronger signal in the GFP channel with the weaker signal in the RFP channel results in the yellow spots. These signals should be omitted from our bacterial analysis.
 | ||
| Fig. 5 Visualization and processing of z-stack fluorescence images in IMARIS. (a) 3D presentation of a z-stack image by assembling all the slices from each channel together (RFP and GFP) and displaying both channels at the same time. In this case, a worm that has fed on RFP E. coli OP50 for 3 days is being shown. Scale bar = 15 μm (b) E. coli OP50 are seen as orange spots, while the yellow spots originate from the worm's autofluorescence. Since the worm's autofluorescence is strong in these spots, the signal is present both in the GFP channel and the RFP channel and thus the spots appear yellow (as shown by the arrows) due to the overlap of the green color and the red color. A proper bacterial spot detection procedure should identify the orange spots and ignore the yellow ones. Scale bar = 10 μm. (c) The spot detection relies on an estimated of the smallest spot size and an IMARIS parameter called “Quality”. In our case, we used 1 μm in x and y directions and 2 μm in z direction. The parameter “Quality” is related to the intensity at the center of a spot while including corrections for the background intensity around that spot. This parameter acts as threshold and thus only spots with higher intensities than this threshold value will be accepted. The optimized “Quality” parameter value was 150 and it was such that the detected spots matched the actual spots we could observe, and no false positives were present. In addition, a threshold limit (3500–4000) was also set on the intensity of the GFP channel to reject the spots that originate from the worm autofluorescence. The white ellipsoids shown are the spots detected using the above-mentioned parameters. It should be noted that at this stage, all the spots are assumed to have the same size that is set by the smallest spots size we used for the spot detection algorithm. It's only due to the perspective projection that the farther spots are seen smaller. Scale bar = 10 μm. (d) The centers of the detected spots are used as seeds and by using a “local contrast” method the spot size is determined. In this approach, the intensity distribution around the center of the detected spot is first adjusted by subtracting from it the intensity of the background in the vicinity of that spot. After this correction, only the region that has intensity higher than a threshold value is considered to be a part of the spot. The optimized threshold value was 80.4 and at this value the finalized spots matched visually to the actual spots. The white ellipsoids seen are the finalized detected spots. Since an actual bacterial region may not be always ellipsoid, the size determination algorithm uses the volume of this region to construct an ellipsoid that has the same volume as this region. Therefore, there is not always a perfect overlap between the actual bacterial spot and the finalized detected spot (as shown by the arrows in d). Scale bar = 10 μm. | ||
By using the built-in spot detection algorithm of IMARIS, we can detect bacterial spots and obtain their sizes and intensities in a consistent manner. The algorithm begins with setting the RFP channel as the channel of interest for spot detection. Afterwards, we need to provide an initial estimate for the smallest spot size. Based on our observation of bacterial spots across different slices of the raw z-stack fluorescence images, we found the smallest bacteria spot to be 1 μm in the “X” and the “Y” directions. We assumed the bacterial spots to be spherical but due to the point spread function (PSF) of the microscope, the spherical spots become elongated in the “Z” direction and turn into ellipsoids. We thus used 2 μm in the “z” direction to account for the effect of PSF. IMARIS then uses the spot size estimation together with an internal parameter called “Quality” to detect spots. The “Quality” parameter is related to the intensity at the center of a spot in the RFP channel while including correction for the background intensity around that spot. This parameter sets a threshold on the detected spots, meaning spots with lower values are discarded and the spots with higher value are accepted. Based on one typical image, we optimized the value of this parameter such that the vast majority of the spots that we observe (in that one image) are detected by IMARIS and no false positives are present. We found the optimized value for the “Quality” to be 150 and we used this value for the detection of the spots in all the images to make sure that the detection of spots is consistent (i.e. the results obtained from different images can be meaningfully compared).
The spot detection algorithm also allows the additional filtering of the detected spots by other parameters such as the central intensity of the spots in the GFP channel. This is extremely useful, since, by setting an upper limit (3500–4000, depending on the image) for the central intensity we can discard those spots (in the RFP channel) that originate from the strong autofluorescence of the worm. Fig. 5c shows the detected spots after applying the said constraints i.e. estimation of smallest spot size, “Quality” factor, and central intensity in the GFP channel. As it can be seen, all the bacterial spots are detected while the yellow spots, as shown by the white arrows, are discarded from further analysis.
It should be noted that, at this step, only for the purpose of representation, the algorithm assigns to all the detected spots the same ellipsoid that we defined when we provided the dimensions of the smallest spot size. More importantly, it should be emphasized that up to this step, IMARIS treats these detected spots merely as seed points, meaning that it is only the centers of the spots that are meaningful, while their sizes still need to be determined in a consistent manner. The reason that those ellipsoids appear to have different sizes in Fig. 5c is due to the perspective projection.
Next, we used the “local contrast” method in IMARIS to find the spot size. In this method, the intensity distribution in the region around the center of a detected spot is first adjusted by subtracting from it the intensity of the background in the vicinity of that spot. After the adjustment, only the region around the center of that spot that has an intensity higher than a certain threshold value is considered to be belonging to the spot. If this threshold value is too low, the finalized detected spot will be larger than the actual bacterial spot while if the threshold value is too high, the finalized detected spot will be smaller than the actual bacterial spot. Based on analyzing typical images, we found the optimized value threshold value to be 80.4 in order that the finalized detected spot matched the actual bacterial spot. Fig. 5d shows the finalized detected spots where the sizes of the detected spots now match the size of the actual bacterial spots. The fine-tuning of this threshold value is subjective, as it is somewhat arbitrary to exactly determine where the border of the actual bacterial spot is and in turn how large the finalized detect spot should be. In addition, sometimes the actual bacterial spot is not perfectly ellipsoidal, which makes it impossible to have a perfect overlap between the actual bacterial spot and the finalized detected spot (Fig. 5d). As a result, in these cases, it is harder to find the exact value for the threshold parameter. In fact, in all cases, the diameters of the finalized spots are calculated such that the volumes of the finalized detected spots are equal to the volumes of the regions around the center of the spots that have higher intensity than the chosen threshold value. Despite these slight difficulties, we used the same threshold value (80.4) for all the images to ensure that the spot sizes are determined in a consistent manner.
Results
Fig. 6 shows the extracted data after image analysis by IMARIS, and the post-processing performed to visualize the volume distribution and the intensity distribution of the bacterial spots within the hindgut of the worm. In this example, the data from a single worm that has fed on RFP labelled E. coli OP50 for 4 days is shown. This particular worm was chosen because its hindgut contained only a few bacteria and thus is suitable for illustration purposes. The first two columns of the table (“Vol” and “Int”) in Fig. 6a are the raw outputs obtained from image analysis in IMARIS. “Vol” is the volume of each spot while” Int” is its average intensity in the RFP channel. To account for the change in background intensity across different experiments (2780–2965 arbitrary units (au)), we modified the average intensity by subtracting from it the background intensity to obtain “C. Int”. The background intensity was obtained by averaging the intensities over an area where the worm is not present. In this case the background intensity was 2900 au. The total bacterial load in the worm is proportional to the sum of the product of “C. Int” and “Vol” (Vol · C. Int) across all the bacterial spots. | ||
| Fig. 6 Analysis of the detected bacterial spots in a single worm that has fed on RFP labelled E. coli OP50 for 4 days. (a) The table contains the volume (Vol) and the average intensity of the detected bacterial spots (Int), as extracted from IMARIS. C. Int is the average spot intensity corrected for the background. The product of the volume and the corrected average intensity of each spot (Vol · C. Int) is proportional to the number of bacteria within that spot. The summation of (Vol · C. Int) over all the bacteria spots in one worm is proportional to the total bacterial load in that worm. (b) Histogram of the volume of the detected bacterial spots. (c) Histogram of the average intensity of the detected bacterial spots. (d) The bivariate histogram showing the distribution of the volume and the average intensity of the bacterial spots. | ||
The volume and the average intensity distribution across all the bacterial spots can be presented with histograms (Fig. 6b–d). While the histograms in Fig. 6b and c are simpler and easier to grasp, in principle, they exclude the possibility of revealing any trends or correlation between the volumes of the bacterial spots and their average intensities. Therefore, we prefer to use a bivariate histogram (Fig. 6d) to visualize the data in a more precise manner. This method of post-processing can be applied to each worm within an experiment to obtain single-worm resolution statistics of the bacterial presence within the hindgut. Additionally, the volumes and the average intensities of all the bacterial spots from all the worms belonging to one experiment, e.g., all the worms that have fed on RFP labelled E. coli OP50 for 4 days, can be pooled together. These data can be then visualized with a bivariate histogram normalized by the number of worms in that particular experiment (Fig. 7d). This bivariate histogram thus shows an average distribution of the volumes and the average intensities of the bacterial spots of worms in one experiment. Additionally, normalizing the sum of Vol · C. Int across all the pooled bacterial spots by the total number of worms in the experiment yields the average bacterial load per worm (Fig. 7e).
 | ||
| Fig. 7 Bivariate histograms displaying the average distribution of the volumes and the average intensities of the bacterial spots in the hindgut of worms that have fed on RFP-labelled E. coli OP50 for (a) one day (n = 10), (b) two days (n = 16), (c) three days (n = 11) and (d) four days (n = 12). (e) The total bacterial load of each worm after each day of feeding on fluorescent bacteria. The horizontal bar shows the average total bacterial load for each day while the error bars represent the range of bacterial load in each day. *p ≤ 0.05. | ||
It can be seen that, as the worms age, the bacterial load in their hindguts increases (Fig. 7). This is in agreement with the general dynamics of intestinal bacterial load of the worm with age.34 It is known that the function of the grinder in the pharynx and the innate immunity of the worm decline with age and that older worms have less control over the number of bacteria present in their intestine, thus this trend is to be expected.34 Furthermore, over time, the number of bacterial spots, their sizes and their average intensity also increase. Therefore, not only there are more bacterial spots within the hindgut of the worm but also that each bacterial spot contains more bacteria. This suggests that there are preferential places in the hindgut where the bacteria accumulate over time, or it may also be possible that the ingested bacteria colonize the gut and proliferate there. It should be emphasized that these two phenomena are not mutually exclusive. Additionally, we can observe a significant increase in the number of small bacterial spots (volume ranging from 0 to 2 μm3) in day 3 and day 4. Perhaps due to the weakening of the innate immune system35 and the loss of function in the grinder in the pharynx36 in older worms, it becomes easier for the recently ingested bacteria to accumulate in the gut. As before, another possible explanation would be that, in older worms, due to the weakening of the immune system, the bacteria can more easily proliferate and spread in the gut.
The bacterial load in each individual worm in each day can be seen in Fig. 7e. A bacterial load of 1 was attributed to the worms for which no bacteria could be detected, so that the bacterial load can be displayed using a logarithmic axis. It can be seen that the average load in the hindgut of the worms is increasing with the age of worm similar to what was observed with the bivariate histograms. Even though the average bacterial load can be useful for identifying general trends, it does not provide an accurate representation of all the worms involved in a given experiment and in fact can be quite misleading. Additionally, by using average values to represent the entire population, the subpopulations that exhibit a significantly different phenotype (than the average of the entire population) will remain undiscovered. For example, while the average bacterial load for day 2 is quite high and comparable to that of day 3 and day 4, the large majority of the worms at day 2 have no or very little bacterial in their hindguts. Similar representation problems can also exist for the worms in day 1 and day 3. Even in the case of worms of day 4, which have the least worm-to-worm variations, the worm with highest bacterial population has 200 times more bacteria in hindgut than the one with the lowest bacterial population.
Previous studies done via conventional techniques on agar plates are affected by the same issue.8,11,34,37 In these studies, 10 worms were taken from a given experiment, washed, mechanically disrupted and number of colony forming bacteria were determined by culturing them on suitable agar plates. Therefore, this method inherently pools together the bacteria of all the worms and as a result can only provide an average bacterial load to represent the state of bacterial colonization in the gut of the worms. Moreover, the few worms with bacterial load that are greatly different from the majority of the worms (e.g. in day 2 the worms having bacterial load measuring around 106–107 compared with the majority that have few or no bacteria) cannot be simply discarded a priori. The experiments therefore need to be repeated and, in case of persistence of the “outlying” results, further systematic investigations should take place to discover causal relations/correlations.
Lastly, considering the bacterial loads in individual worms, there seems to be a saturating limit for the number of bacteria present in the hindgut. If the bacteria are simply accumulating in the gut, it is possible that the accumulation becomes balanced with defecation. In the case of proliferation, perhaps the bacterial population reaches an equilibrium with the immune system of the worm and/or with the hindgut milieu. Regardless of the exact explanation, this saturating limit has been previously reported by Portal and Blaser.11 Based on the discussions above, we also plotted the bivariate histograms (Fig. S1†) of selected worms that have fed on RFP-labelled E. coli OP50 for 4 days (highlighted by red circle in Fig. 7e). It can be seen in Fig. S1a† that the volumes, the intensities and the number of the bacterial spots in the gut are low, which implies a relatively small bacterial load. However, considering the worms in Fig. S1b and c,† the increase in bacterial load is accompanied by the formation of larger bacterial spots with higher intensities and the appearance of numerous small bacterial spots. This shows that, while with time the number of bacteria within the gut increases, an adequate explanation of the variation observed among the worms with the same feeding conditions requires further investigation.
Discussion
C. elegans, as a genetically tractable organism with evolutionary conserved pathways of innate immunity, is an interesting model for understanding bacterial pathogenesis. In the conventional methods of studying bacterial pathogenesis, mutant worms and bacteria are generated. Afterwards the interaction between these two is observed by culturing the mutant worms on the mutant bacteria and obtaining the survival curves and the average numbers of colony-forming bacteria. However, survival curves cannot provide a complete description of the said interactions. As mentioned earlier, the average number of colony-forming bacteria does not necessarily represent the state of all the worms and one may miss subpopulations that have few or no bacterial load. Additionally, this measurement cannot determine whether the viable bacteria are merely accumulating in the intestine or that they are actively proliferating within the gut and as a result shifting assays using fluorescent and non-fluorescent bacteria can be utilized to tackle this issue. For example in the study by Aballay et al.,8 WT adults were fed on a mixed lawn containing GFP-tagged S. typhimurium SL1344 and E. coli DH5α with a ratio of 1:1000 for 5 hours. Afterwards, the worms are washed and transferred to E. coli OP50 plates and every day, 10 of these worms are randomly selected and the number of S. typhimurium SL1344 bacteria was determined. It was observed that the number of S. typhimurium SL1344 bacteria increased a few bacteria in day 0 (day the transfer took place) to around 104 in day 4. This clearly shows that S. typhimurium SL1344 can persist and proliferate in the intestine. Additionally, since S. typhimurium SL1344 is fluorescent, in principle, it is possible to follow its proliferation dynamics.
The effect of the bacteria on the host is not only limited to the colonization of the gut. For example, P. aeruginosa and S. typhimurium reduce the motility and the rate of pharyngeal pumping of the worm overtime. Both of these phenotypes can be tracked in vivo using microfluidic solutions.21,28 Moreover, infection with bacteria can also damage the intestinal cells. For example, it has been reported that S. marcescens is a pathogen of the worm that over time causes vacuolation of the intestinal cells and reduces the volume of the intestinal epithelium.10 Overall, these examples show that interactions between the worm and the bacteria can have various aspects that need to be monitored.
In this work, for the first time to the best of our knowledge, we have carried out high-resolution z-stack imaging of fluorescent E. coli OP50 bacteria using a SDCM. Utilizing IMARIS, we constructed a 3D representation of the bacteria within the hindgut of each worm and managed to identify their sizes and their (fluorescent) intensities in an automated way. We showed that the number of bacterial spots, their sizes and their intensities increase as the worm ages, in agreement with previous studies.4,34 Interestingly, our single-worm bacterial analysis showed that the conventional sampling method for evaluating the bacterial presence within the gut of the worms may be less adequate and can be misleading in some cases. While the main innovation in our work is the establishment of a high-resolution imaging and analysis pipeline, our work also demonstrated that on-chip high-resolution imaging of intestinal bacteria is not hindered by the optical distortions that the PDMS layer and the liquid media can potentially introduce. Consequently, the methods we developed in this work can be readily implemented in existing microfluidic chips that are aimed at studying host–microbiota interactions.
To better highlight the novelty of our approach and the new possibilities offered by our imaging and analysis pipeline, we briefly compare our work with the previously reported microfluidic devices applied to host–microbiota interactions. The first reported microfluidic only considered the life span of the bacteria-infected worms that were being treated on-chip with antimicrobial compounds.25 The imaging possibilities were only briefly explored and were limited to fluorescence imaging at low magnifications. The HandKAchip microfluidic device for on chip hands-free killing assay improved on the previous work.26 Brightfield and fluorescence images, albeit with low-resolution and acquired at a low-magnification, were used to monitor the life span of the worms as well as their motility and infection response (irg-1::GFP) in an automated manner. However, similar to the imaging and analysis pipeline we presented in this work, the identities of the worms were lost in the analysis stage and therefore it was not possible to track and monitor the worms over an extended period of time. The latest microfluidic device for host–microbiota interactions allowed in vivo fluorescence imaging of C. elegans worms immobilized via traps at a moderate resolution for a duration of 30 hours while maintaining the identities of the worms.28 However, the immobilization is not sufficient to allow high-resolution z-stack fluorescence imaging. Indeed, in this study, a widefield microscope was used to perform time-lapse fluorescence images and no z-stack imaging was performed. However, widefield microscopes are inherently limited in resolution as the light signals from out-of-focus planes blur acquired images. This, in particular, is noticeable when bacterial cells are present in the intestine at high densities, which is typically the case in older worms. As a result, intact fluorescent bacteria in densely populated bacterial clusters will appear as diffusive fluorescent background when imaged using a widefield microscope and consequently, will be mistakenly interpreted as disrupted bacteria. This shows the necessity of utilizing a SDCM to accurately image and analyze the bacterial colonies in the intestine and thus the superior imaging resolution and accuracy afforded by our imaging and analysis pipeline.
As an outlook, a simple way to further improve the functionality of our chip would be to include traps for aligning the worms before performing fixing. This would allow us to not only to identify the bacterial spots but also to systematically characterize their positions within the gut and discover any preferential place for colonization. Our group has previously designed a microfluidic device that enables in vivo extraction of phenotypes such as length, area, motility and first egg release time in an automated fashion.21 The imaging analysis involved for the determination of these phenotypes could be modified such they can be used in the new device. Additionally, our group has also reported microfluidic devices for in vivo studying of bacterial load dynamics and absorption (e.g. measurement of the pharyngeal pumping rate, and the time evolution of bacterial load in the worm gut).28 The functions exhibited by the latter device could be complemented with image analysis techniques of the former device and combined with the ideas expressed in this work (high-resolution fluorescent z-stack imaging, fixing the worms on chip, shifting assays using fluorescent and non-fluorescent bacteria) to result in a more complete picture of bacterial proliferation and/or pathogenesis within the worm.
Conclusions
We have performed high-resolution z-stack fluorescence imaging of the gut bacteria in C. elegans. We were able to automatically construct a 3D representation of the bacteria in the intestine using IMARIS and characterized the bacterial load of the worm (the sizes and the fluorescence intensities of the bacterial spots) during early adulthood. Our analysis showed that the conventional sampling and averaging method for determining the number of bacteria in the gut is not always representative for all the worms and in some cases can be misleading. Our ideas on high-resolution imaging, on-chip fixing and shifting assays can complement currently existing microfluidic solutions providing a step forward towards a complete description of bacterial pathogenesis in C. elegans.Conflicts of interest
Authors declare no conflicts of interests.Acknowledgements
This work is supported by the Swiss National Foundation (Grant No. 205321-179021). Microfluidic chips were fabricated at the EPFL Center of MicroNanoTechnology (EPFL-CMi). High-resolution imaging was carried out at Bioimaging and Optics Platform (BIOP) at EPFL. We thank Dr Vittorio Viri and Dr Huseyin B. Atakan for their kind suggestions and feedbacks. We also thank Dr Yuan Zhao and Prof. David Gems (UCL, C. elegans Ageing Laboratory) for providing the RFP-labelled E. coli OP50 strain.References
- D. D. Shaye and I. Greenwald, PLoS One, 2011, 6(5), e20085 CrossRef CAS PubMed.
- K. V. Christensen, M. G. Morch, T. H. Morthorst, S. Lykkemark and A. Olsen, in Ageing: Lessons from C. elegans, ed. A. Olsen and M. S. Gill, Springer International Publishing, Cham, 2017, pp. 411–429 Search PubMed.
- L. S. Yilmaz and A. J. M. Walhout, Trends Genet., 2014, 30, 496–503 CrossRef CAS PubMed.
- F. Cabreiro and D. Gems, EMBO Mol. Med., 2013, 5, 1300–1310 CrossRef CAS PubMed.
- C. D. Sifri, J. Begun and F. M. Ausubel, Trends Microbiol., 2005, 13, 119–127 CrossRef CAS PubMed.
- P. Dirksen, S. A. Marsh, I. Braker, N. Heitland, S. Wagner, R. Nakad, S. Mader, C. Petersen, V. Kowallik, P. Rosenstiel, M.-A. Félix and H. Schulenburg, BMC Biol., 2016, 14, 1–16 CrossRef PubMed.
- F. Zhang, M. Berg, K. Dierking, M.-A. Félix, M. Shapira, B. S. Samuel and H. Schulenburg, Front. Microbiol., 2017, 8 DOI:10.3389/fmicb.2017.00485.
- A. Aballay, P. Yorgey and F. M. Ausubel, Curr. Biol., 2000, 10, 1539–1542 CrossRef CAS PubMed.
- M.-W. Tan, S. Mahajan-Miklos and F. M. Ausubel, Proc. Natl. Acad. Sci., 1999, 96, 715–720 CrossRef CAS PubMed.
- C. L. Kurz, S. Chauvet, E. Andrès, M. Aurouze, I. Vallet, G. P. F. Michel, M. Uh, J. Celli, A. Filloux, S. de Bentzmann, I. Steinmetz, J. A. Hoffmann, B. B. Finlay, J.-P. Gorvel, D. Ferrandon and J. J. Ewbank, EMBO J., 2003, 22, 1451–1460 CrossRef CAS PubMed.
- C. Portal-Celhay and M. J. Blaser, Infect. Immun., 2012, 80, 1288–1299 CrossRef CAS PubMed.
- A. San-Miguel and H. Lu, Microfluidics as a tool for C. elegans research, WormBook, 2018 Search PubMed.
- S. X. Guo, F. Bourgeois, T. Chokshi, N. J. Durr, M. A. Hilliard, N. Chronis and A. Ben-Yakar, Nat. Methods, 2008, 5, 531–533 CrossRef CAS PubMed.
- S. K. Gokce, S. X. Guo, N. Ghorashian, W. N. Everett, T. Jarrell, A. Kottek, A. C. Bovik and A. Ben-Yakar, PLoS One, 2014, 9, e113917 CrossRef PubMed.
- S. K. Gokce, E. M. Hegarty, S. Mondal, P. Zhao, N. Ghorashian, M. A. Hilliard and A. Ben-Yakar, Sci. Rep., 2017, 7, 9837 CrossRef PubMed.
- B. P. Gupta and P. Rezai, Micromachines, 2016, 7, 123 CrossRef PubMed.
- S. E. Hulme, S. S. Shevkoplyas, A. P. McGuigan, J. Apfeld, W. Fontana and G. M. Whitesides, Lab Chip, 2010, 10, 589–597 RSC.
- R. B. Kopito and E. Levine, Lab Chip, 2014, 14, 764–770 RSC.
- S. Berger, E. Lattmann, T. Aegerter-Wilmsen, M. Hengartner, A. Hajnal, A. deMello and X. Casadevall i Solvas, Lab Chip, 2018, 18, 1359–1368 RSC.
- W. Keil, L. M. Kutscher, S. Shaham and E. D. Siggia, Dev. Cell, 2017, 40, 202–214 CrossRef CAS PubMed.
- H. B. Atakan, M. Cornaglia, L. Mouchiroud, J. Auwerx and M. A. M. Gijs, Lab Chip, 2019, 19, 120–135 RSC.
- M. Cornaglia, T. Lehnert and M. A. M. Gijs, Lab Chip, 2017, 17, 3736–3759 RSC.
- D. Midkiff and A. San-Miguel, Molecules, 2019, 24(23), 4292 CrossRef CAS PubMed.
- S. Mondal, E. Hegarty, C. Martin, S. K. Gökçe, N. Ghorashian and A. Ben-Yakar, Nat. Commun., 2016, 7, 13023 CrossRef CAS PubMed.
- J. Yang, Z. Chen, P. Ching, Q. Shi and X. Li, Lab Chip, 2013, 13, 3373–3382 RSC.
- K. S. Lee, L. E. Lee and E. Levine, Sci. Rep., 2016, 6, 1–9 CrossRef PubMed.
- V. Viri, M. Cornaglia, H. B. Atakan, T. Lehnert and M. A. M. Gijs, Lab Chip, 2020, 20, 2696–2708 RSC.
- V. Viri, M. Arveiler, T. Lehnert and M. A. M. Gijs, Micromachines, 2021, 12, 832 CrossRef PubMed.
- Y. Zhao, A. F. Gilliat, M. Ziehm, M. Turmaine, H. Wang, M. Ezcurra, C. Yang, G. Phillips, D. McBay, W. B. Zhang, L. Partridge, Z. Pincus and D. Gems, Nat. Commun., 2017, 8, 15458 CrossRef CAS PubMed.
- Y. Zhao, H. Wang, R. J. Poole and D. Gems, Nat. Commun., 2019, 10, 5087 CrossRef PubMed.
- Y. Zhao, B. Zhang, I. Marcu, F. Athar, H. Wang, E. R. Galimov, H. Chapman and D. Gems, Aging Cell, 2021, 20, e13324 CAS.
- G. Ball, R. M. Parton, R. S. Hamilton and I. Davis, in Methods in Enzymology, ed. P. M. conn, Academic Press, 2012, vol. 504, pp. 29–55 Search PubMed.
- Z. Pincus, T. C. Mazer and F. J. Slack, Aging, 2016, 8, 889–898 CrossRef CAS PubMed.
- C. Portal-Celhay, E. R. Bradley and M. J. Blaser, BMC Microbiol., 2012, 12, 49 CrossRef CAS PubMed.
- M. J. Youngman, Z. N. Rogers and D. H. Kim, PLoS Genet., 2011, 7, e1002082 CrossRef CAS PubMed.
- J. Collins, C. Huang, S. Hughes and K. Kornfeld, WormBook, 2008, 1–21, DOI:10.1895/wormbook.1.137.1.
- S.-K. Ooi, T.-Y. Lim, S.-H. Lee and S. Nathan, Virulence, 2012, 3, 485–496 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02934d |
| This journal is © The Royal Society of Chemistry 2023 |