
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of natural polyprenols for the production of biological prenylquinones and tocochromanols†
Jerzy Kruk *a and
Renata Szymańskab
*a and
Renata Szymańskab
aDepartment of Plant Physiology and Biochemistry, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Gronostajowa 7, 30-387 Kraków, Poland. E-mail: jerzy.kruk@uj.edu.pl; Tel: +48 126646361
bFaculty of Physics and Applied Computer Science, AGH University of Science and Technology, Reymonta 19, 30-059 Kraków, Poland
First published on 31st July 2023
Abstract
We elaborate the chemical synthesis of polyprenols by chain lengthening, which is considerably less time-consuming than the other previously described methods. Our method eliminates critical steps requiring low temperature and toxic chemicals, which are difficult to perform in ordinary laboratories. The critical step of acetylene addition in liquid ammonia was replaced by a new approach, namely, the use of sodium acetylide in dimethoxyethane at room temperature, where the reaction is completed within one hour. This method is of general significance as it can also be applied to the synthesis of any other acetylides. Our method provides reasonable yields and can be scaled depending on the requirements. All the reactions were followed by high-performance liquid chromatography, allowing the formation of undesired isomers and other side-products to be controlled. The resulting polyprenols were further used in the synthesis of plastoquinones, although a variety of biological prenylquinones can be synthesized this way. Moreover, we found a new method for the direct formation of tocochromanols (plastochromanols, tocochromanols) from polyprenols and aromatic head groups.
1. Introduction
Polyprenols fulfil a variety of important functions in all organisms. They constitute a side chain of such prenylquinones as ubiquinones, menaquinones and plastoquinones engaged in respiratory and photosynthetic electron transport systems.1 In the reduced form, these prenyllipids also have an antioxidant function.1–4 The number of isoprenoid units in the side chains of prenylquinones is especially variable in microorganisms.1 The configuration of double bonds is always all-trans (all-E) in these compounds. Naturally, polyprenols are synthesized by specific enzymes (isoprenyl diphosphate synthases) that are specialized in the synthesis of polyprenols with a fixed number of isoprenoid units.5 The enzymes engaged in the synthesis of side-chains of plasto-, mena- and ubiquinones have been characterized6,7 and the molecular mechanism of determination of the side-chain length has been found.5 Phytol, a partially saturated polyprenol, is known as a side-chain of chlorophylls, tocopherols and vitamin K1.8 Besides, there are known less widespread, free polyprenols, such as dolichols and plant polyprenols of the Z/E configuration,9–15 which are characterized by a wide range of the hydrocarbon chain length (up to C500), and have a variety of biological functions and potential pharmacological applications.16 Their biosynthesis has been recently recognized.17,18As polyprenols are constituents of vitamin E (tocopherols), ubiquinones (coenzyme Q) and menaquinones (vitamin K2), there is a need for efficient and economic synthesis of polyprenols of different chain lengths, not only for commercial but also for basic research, especially those with a number of isoprenoid units between 5 and 8 and longer than 9, which are not available commercially.
The chemical synthesis of polyprenols has been the subject of numerous studies for a long time.12,19–26 However, most if not all, of these methods are multistep, very time-consuming and frequently require low and high temperatures or toxic chemicals. This makes synthesis, especially in a non-specialized laboratory, difficult and discouraging. One of the first of these methods is that of Rüegg et al.19 which relied on subsequent addition of isoprenoid units (Fig. S1†). However, in the critical step it required work with metallic sodium and toxic liquid ammonia under elevated pressure, yielded undesired isomers and took nearly 3 days of continuous work to synthesize one isoprenoid unit.
In our work, we focused on modifying this method, to make it as short as possible, avoiding critical steps, where one isoprenoid unit can be synthesized within 4–5 hours. Moreover, all the synthetic steps were monitored using high-performance liquid chromatography (HPLC) analysis, which allowed control of the formation of undesired isomers and other by-products.
2. Experimental
2.1. Chemicals and materials
All the chemicals and solvents came from Sigma-Aldrich (Poznań, Poland) unless otherwise stated. Anhydrous diethyl ether (ethanol-free) was stabilized with BHT (butylhydroxytoluene). Ag2O and AgNO3 were purchased from POCH (Gliwice, Poland). Natural all-E geranylgeraniol (GG-OH) (98% purity) came from Chemos GmbH & Co KG (Regenstauf, Germany), while solanesol (90% purity) was purchased from MolPort (Riga, Latvia). All-E pentaprenol and all-E decaprenol were a kind gift of Dr Thomas Netscher (Research and Development, DSM Nutritional Products, Basel, Switzerland). Compressed hydrogen was purchased from Messer (Gliwice, Poland) (5.0 purity, >99.999%). Alumina N (activity grade I) came from MP Biomedicals (Eschwege, Germany). Alumina N grade III, IV or V, used for column chromatography, was prepared from grade I Alumina by the addition of 6, 10 or 15% water (w/w), respectively and careful mixing of the chromatography bed. After cooling to room temperature (RT), the bed was ready to use. AgNO3-impregnated Alumina N, deactivated to the desired grade, was prepared by dissolving an appropriate amount of AgNO3 in water used for deactivation. All operations with AgNO3-impregnated Alumina N were performed under dim light.2.2. Synthetic pathway of polyprenols
The following procedure is described for the synthesis of pentaprenol (prenol-5) from GG-OH but it can be applied to the synthesis of polyprenols with any chain length. Similarly, the scale of the synthesis can be changed depending on the needs, keeping the reagents in appropriate proportions.(a) Bromination – 10 mmol (2.9 g) GG-OH (compound I, Fig. 1) was dissolved in 25 ml of diethyl ether (Et2O) and 0.435 ml (4.35 mmol) of PBr3 was added during magnetic stirring. The reaction was run for 30 min at RT under continuous stirring. Afterwards, the reaction mixture was transferred in approximately equal proportions to two 50 ml Falcon tubes, 20 ml of hexane and 15 ml of water were added to each tube, followed by vigorous shaking and centrifugation at 5500g × 2 min. Then, the upper organic layers were transferred with a pipette to round-bottom evaporator flask. The extraction of the lower phase with hexane was repeated, the extracts were combined and the solvent was evaporated on a rotary evaporator (BUCHI Rotavapor R-300, Mainz, Germany), resulting in II.
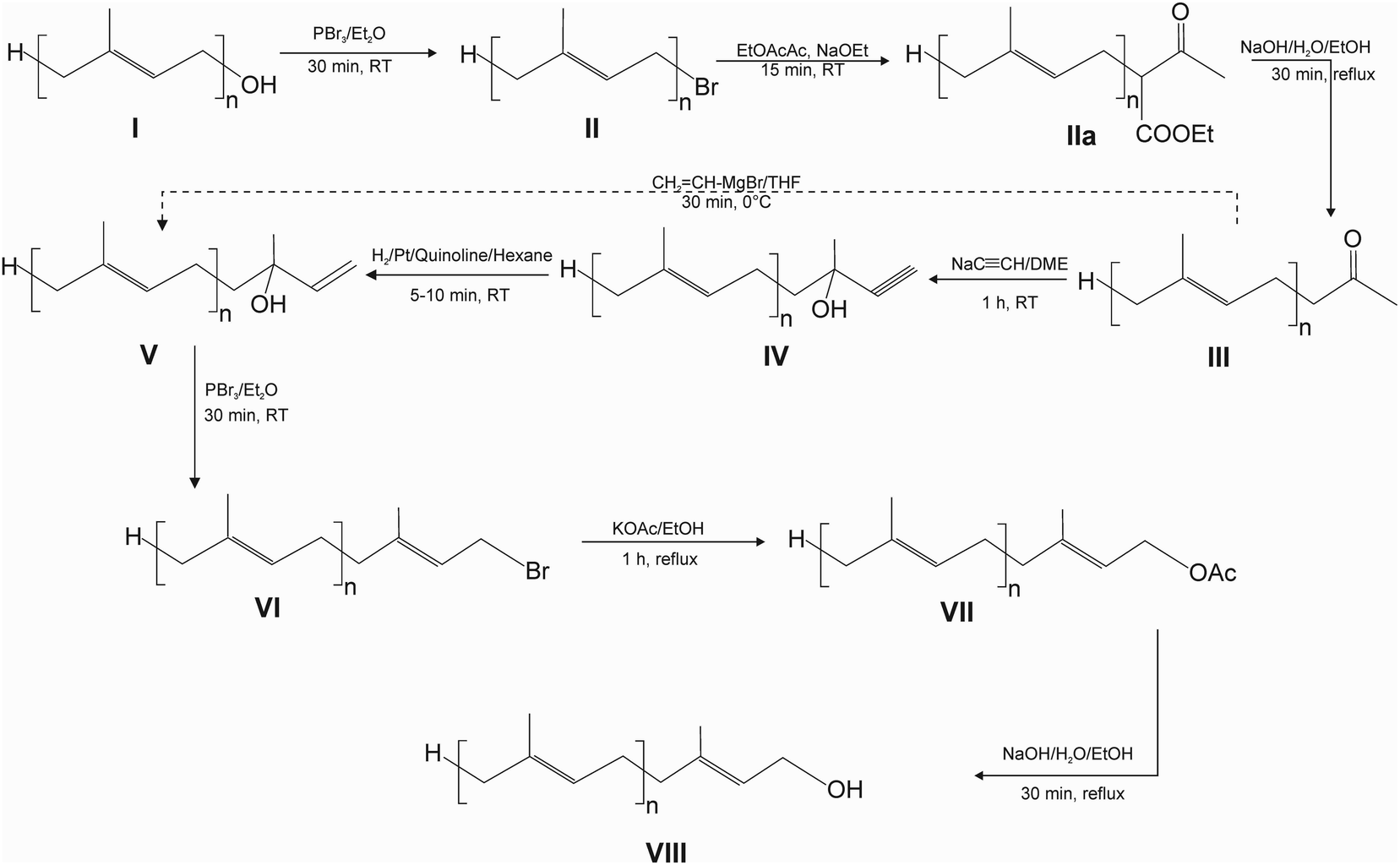 | ||
| Fig. 1 Modified synthesis of polyprenols via chain lengthening. | ||
(b) Acetoacetic ester synthesis – compound II was dissolved in 5.7 ml of ethyl acetoacetate (EtOAcAc) and 3.5 ml of sodium ethoxide (NaOEt) solution (21% wt. in ethanol) was added during magnetic stirring and the mixture was stirred for another 15 min at RT resulting in IIa. Afterwards, the reaction mixture was treated as in ‘a’ with additional washing of the organic phase with water in Falcon tubes (15 ml of H2O/30 ml of the organic phase) to remove unreacted EtOAcAc from the organic phase.
(c) Hydrolysis – compound IIa was dissolved in 20 ml of absolute ethanol (EtOH), followed by the addition of 2 g NaOH and 1 ml of water. The mixture was heated under reflux for 30 min resulting in III. Afterwards, the reaction mixture was treated as in ‘a’. If the organic phase was cloudy (emulsion), 2 ml of 3 N HCl per Falcon was added and the extraction repeated.
(d) Acetylide addition – compound III was dissolved in 15 ml of 1,2-dimethoxyethane (DME) (anhydrous, inhibitor-free) and 4.2 ml of sodium acetylide suspension (18% wt. slurry in xylene: light mineral oil) was added to the stirred solution. The mixture was stirred for 1 h at RT resulting in IV. Afterwards, the reaction mixture was treated as in ‘a’.
(e) Hydrogenation – compound IV was dissolved in 24 ml of hexane, followed by the addition of 0.238 g of Lindlar's catalyst and 0.1 ml of quinoline. The mixture was bubbled with hydrogen, using a stainless-steel HPLC inlet filter, for 5–10 min at RT at a hydrogen flow of ca. 50 ml min−1. After the reaction, the mixture was transferred to a round-bottom evaporator flask and evaporated on a rotary evaporator, resulting in V.
(f) Bromination – compound V was dissolved in 25 ml of Et2O and 0.435 ml (4.35 mmol) of PBr3 was added during magnetic stirring. The reaction was run for 30 min at RT under continuous stirring resulting in VI. Afterwards, the reaction mixture was treated as in ‘a’.
(g) Substitution – compound VI was dissolved in 10 ml EtOH and 2.1 g anhydrous potassium acetate (KOAc) was added. The mixture was heated for 1 h under reflux with magnetic stirring, resulting in VII. Afterwards, the reaction mixture was treated as in ‘a’.
(h) Hydrolysis – compound VII was dissolved in 21 ml of EtOH, followed by the addition of 1 g NaOH and 1 ml of water. The mixture was heated under reflux for 30 min, resulting in VIII. Afterwards, the reaction mixture was treated as in ‘a’.
Alternatively, steps ‘d’ and ‘e’ can be replaced by direct vinylation of III, taking advantage of the Grignard reaction: compound III, obtained from 5 mmol of GG-OH, was dissolved in 6 ml of tetrahydrofuran (THF) and 12.5 ml of 1 M vinyl-Mg-Br in THF was added at 0 °C and the solution was stirred for 30 min at 0 °C.
2.3. Chromatographic purification of polyprenols
After synthesis, the crude polyprenols were first purified on Alumina N, followed by chromatography on AgNO3-impregnated Alumina N to separate Z/E isomers. During chromatography, the 50 or 100 ml fractions were analyzed using HPLC. Routinely, 140 g of Alumina N were used for the single purification step (3 cm i.d. of the column).Pentaprenol was purified on Alumina N (grade III) column in 20–40% Et2O/hexane to obtain a mixture of Z/E isomers, which were eluted with 35–40% Et2O/hexane, followed by chromatography on Alumina N (grade IV) – 5% AgNO3 column in 20–50% Et2O/hexane. The all-E isomer was eluted with 30–50% Et2O/hexane.
Hexaprenol was first purified on Alumina N (grade III) column in 15–40% Et2O/hexane to obtain a mixture of Z/E isomers, which were eluted with 25–35% Et2O/hexane and then on Alumina N (grade IV) – 5% AgNO3 column in 15–50% Et2O/hexane. All-E prenol-6 was eluted with 20–50% Et2O/hexane.
Heptaprenol was purified on Alumina N (grade III) column in 20–30% Et2O/hexane. Z isomer was eluted with 20%, while all-E prenol-7 was eluted with 20–25% Et2O/hexane.
Octaprenol was first purified on Alumina N (grade III) column in 20% Et2O/hexane to obtain a mixture of Z/E isomers and then on Alumina N (grade V) – 5% AgNO3 column in 15–60% Et2O/hexane. Z isomer was eluted with 40%, while all-E prenol-8 was eluted with 40–50% Et2O/hexane.
Decaprenol was first purified on Alumina N (grade III) column in 15% Et2O/hexane to obtain a mixture of Z/E isomers and then on Alumina N (grade V) – 5% AgNO3 column in 15–50% Et2O/hexane. All-E prenol-10 was eluted with 20–50% Et2O/hexane.
2.4. Synthesis of prenylquinones and tocochromanols
For the synthesis of prenylquinones, 5 mmol (0.7 g) of 2,3-dimethylhydroquinone (DMHQ) and 2 mmol of prenol-n were dissolved in 6 ml of dioxane, followed by the addition of 0.8 ml of BF3 diethyl etherate (≥46.5% BF3 basis) and the solution was stirred for 15 min at RT. The progress of the reaction was followed by HPLC, using fluorescence detection (290/330 nm, excitation/emission) in acetonitrile/methanol/water (ACN/MeOH/H2O) (72/8/1, v/v) for plastoquinone-4 (PQH2-4) and PQH2-5, MeOH/hexane (340/20, v/v) for PQH2-6, MeOH/hexane (340/50, v/v) for PQH2-7 and MeOH/hexane (340/85, v/v) for PQH2-8 and PQH2-9. Afterwards, the reaction mixture was partitioned between hexane/water as described in 2.2.a and the organic phase was evaporated on a rotary evaporator. The resulting phenylquinone was oxidized with 2 g of Ag2O in 50 ml of Et2O for 1 h at RT, according to Mayer and Isler27 Afterwards, the solution was centrifuged (5500g × 2 min) and the supernatant was evaporated on a rotary evaporator to obtain the prenylquinone.For the synthesis of tocochromanols, 5 mmol (0.7 g) of DMHQ and 2 mmol of prenol-n were dissolved in 6 ml of anhydrous THF, followed by the addition of 0.8 ml BF3 diethyl etherate (≥46.5% BF3 basis) and the solution was left for 48 h at RT (22–23 °C). The progress of the reaction was followed by HPLC using fluorescence detection (290/330 nm, excitation/emission) in methanol/hexane (340/85, v/v). Afterwards, the reaction mixture was partitioned between hexane/water as described in 2.2.a and the organic phase was evaporated on a rotary evaporator. The crude tocochromanol was purified using column chromatography on Lichroprep RP-18 (43–60 μm) (13 × 2.2 cm i.d.). In the case of plastochromanol-8 (PC-8), the column was first developed in MeOH/hexane (340/20, v/v) to elute DMHQ, afterwards in MeOH/hexane (340/37) to elute PQH2-9 and finally in MeOH/hexane (340/60) to elute PC-8. During chromatography, the 50 ml-fractions collected were analyzed using HPLC.
2.5. HPLC chromatography
During all steps of the synthesis and chromatographic purification of the compounds, HPLC analysis was used. HPLC was performed using a Nucleosil 100 C18 reverse-phase column (MZ Analysentechnik, Germany, 250 × 4 mm, 5 μm) at a solvent flow rate of 1.5 ml min−1, unless otherwise stated. The HPLC setup included a Jasco PU-2080 Plus pump (Jasco, Tokyo, Japan), a Jasco UV-VIS detector (Jasco, Tokyo, Japan) and Shimadzu RF-10 AXL fluorescence detector (MD, USA) (290/330 nm, excitation/emission). The loop was 100 μl. The absorption detection was at 210 nm for polyprenols or 255 nm for plastoquinones. The solvents used are given in the legends of the chromatograms. During synthesis of the polyprenols, the reaction mixtures were routinely diluted 1000-fold for the HPLC analysis.2.6. NMR measurements
1H spectra were recorded at 600.26 MHz in CD3Cl. Residual chloroform (δ = 7.26 ppm and 77.16 ppm for proton and carbon respectively) was used as internal references for proton spectra measured. Proton spectra were recorded with Bruker Avance III 600 MHz (USA).2.7. Absorption measurements
Absorption spectra were collected using a Jasco UV-VIS V-650 spectrophotometer (Jasco, Tokyo, Japan).2.8. Mass spectrometry
High-resolution mass spectrometry (HRMS) analyses were performed by direct injection of methanol solutions of isolated fractions using electrospray ion source in a positive mode and high-resolution tandem mass spectrometer Bruker Impact II (Bremen, Germany), equipped with an and quadrupole time-of-flight mass analyzer (ESI-QTOF). The obtained m/z data were fitted to molecular formula using ChemCalc software (https://www.chemcalc.org) and the following atom composition range C0-100 H0-200 O0-20 Na1.3. Results
3.1. Synthesis of polyprenols
Alkyl bromides are indispensable substrates in the synthesis of polyprenols and are mostly obtained by the action of PBr3 on the corresponding primary alcohols. In the first step of our synthesis (Fig. 1), we found that besides the expected all-E GG-Br, Z and other isomers are also formed during this reaction (Fig. S2†). We tested the bromination reaction with PBr3 in various conditions, such as a different temperature, solvent system, reaction time, the addition of pyridine (Pyr), but without any significant influence on the composition of the isomers. Next, we tried several other halogenation systems described in the literature, such as N-bromosuccinimide (NBS),29 α,α-dibromo-β-dicarbonyl compounds30 and CBr4 (ref. 31) in combination with triphenylphosphine and tetramethyl-α-chlorphenamine (TMCE)32 or halogenation via mesylates. Even though these systems are frequently considered mild and selective, all of them resulted in the formation of isomers other than all-E GG-Br ones (data not shown), at proportions similar to those in the case of PBr3 treatment. Therefore, in our method we used the reaction with PBr3 in Et2O but with a shorter reaction time at RT (30 min) which under these conditions led to complete conversion of the substrate (Fig. S4†). The Z isomer of GG-Br, together with its all-E isomer, underwent all the subsequent reactions that can be seen in HPLC chromatograms (Fig. S4†). Interestingly, when bromination with PBr3 was performed on the other all-E prenols, such as phytol, farnesol, pentaprenol and higher homologues, only all-E products were observed.The next stage, acetoacetic ester synthesis, is a two step reaction (Fig. S3†) that yields ketone III. We found that the first reaction is completed in as little as 15 min at RT, and hydrolysis after 30 min of heating (Fig. 1).
In the original procedure,19 ketone III is reacted with sodium acetylide (prepared from metallic sodium and acetylene) in liquid NH3 at elevated pressure (Fig. S1†). To replace this critical, time-consuming reaction, we searched unsuccessfully in the literature for another sodium acetylide solvent. Therefore, we tested various solvents for this purpose and found that the reaction of ketone III with acetylide proceeds successfully in anhydrous DME at RT within one hour. The progress of the reaction can be followed visually by darkening the solution (Fig. S5†). This indicates that sodium acetylide is at least partially soluble in DME.
The acetylene bond was hydrogenated similarly as in the original procedure, in the presence of Lindlar's catalyst, taking advantage of compressed hydrogen, using the setup shown in Fig. S6.† The reaction was usually complete within 5–10 min. It was important not to extend the reaction time, which resulted in the formation of compounds with reduced double bonds, even though the catalyst had been poisoned by quinoline. These derivatives are seen in the chromatograms with the longer retention time (Fig. S4,† chromatogram of compound V). Under the conditions applied, the content of these derivatives did not exceed 10–15% of compound V.
Alternatively, acetylide addition and hydrogenation can be replaced by direct vinylation of III, taking advantage of the Grignard reaction (Fig. 1). However, this reaction turned out to be less efficient than the two-step procedure.
In the next, bromination step of the allelic alcohol V with PBr3, formation of the Z-isomer was observed, which was tested separately using geranyllinalool (Fig. S2†). The use of the other, previously described methods of bromination, did not result in an improvement in the E/Z isomers ratio. In this case, the reaction time of 30 min at RT was also sufficient to complete the reaction.
Substitution of bromide by the acetyl group was found to proceed considerably faster using EtOH instead of acetone, allowing the reaction time to be shortened to one hour (Fig. 1). This was mainly due to considerably better solubility of KOAc in EtOH than in acetone. The subsequent hydrolysis was already completed within 30 min resulting in the final product VIII.
The main by-product of the synthesis was Z-isomer which is formed during two steps of bromination and is separated from the all-E isomer using column chromatography.
As in the case of the synthesis of pentaprenol from GG-OH discussed above (Fig. 1), higher polyprenols homologues were also synthesized (hexaprenol, heptaprenol, octaprenol), whose progress of synthesis was followed by HPLC (Fig. S7–S9,† respectively). When the homologues are synthesized one after another, the bromide VI could be directly used as II for the next synthesis (Fig. 1). However, it is recommended to proceed via the purified prenol to remove the Z-isomer before the next round of reactions.
3.2. Chromatographic purification of polyprenols
After synthesis, the crude polyprenols were first purified on Alumina N, followed by chromatography on AgNO3-impregnated Alumina N to separate the Z/E isomers. During chromatography, the fractions were analyzed using HPLC.The pentaprenol was purified on Alumina N (grade III) column in 20–40% Et2O/hexane to obtain a mixture of Z/E isomers, which were eluted with 35–40% Et2O/hexane, followed by chromatography on Alumina N (grade IV) – 5% AgNO3 column in 20–50% Et2O/hexane. The all-E isomer was eluted with 30–50% Et2O/hexane. The yield of Z/E isomers in relation to the starting amount of GG-OH was 25–30%.
The hexaprenol was first purified on Alumina N (grade III) column in 15–40% Et2O/hexane to obtain mixture of Z/E isomers, which were eluted with 25–35% Et2O/hexane, and then on Alumina N (grade IV) – 5% AgNO3 column in 15–50% Et2O/hexane. All-E prenol-6 was eluted with 20–50% Et2O/hexane. The yield of Z/E isomers, after the first column, in relation to the starting amount of pentaprenol was 33–39%.
The heptaprenol was purified on Alumina N (grade III) column in 20–30% Et2O/hexane. The Z isomer was eluted with 20%, and all-E prenol-7 with 20–25% Et2O/hexane. The yield of Z/E isomers, after the first column, in relation to the starting amount of hexaprenol was 25%.
The octaprenol was first purified on Alumina N (grade III) column in 20% Et2O/hexane to obtain a mixture of Z/E isomers and then on Alumina N (grade V) – 5% AgNO3 column in 15–60% Et2O/hexane. Z isomer was eluted with 40%, and all-E prenol-8 with 40–50% Et2O/hexane. The yield of Z/E isomers, after the first column, in relation to the starting amount of heptaprenol was 20–25%.
The decaprenol was first purified on Alumina N (grade III) column in 15% Et2O/hexane to obtain a mixture of Z/E isomers and then on Alumina N (grade V) – 5% AgNO3 column in 15–50% Et2O/hexane. All-E prenol-10 was eluted with 20–50% Et2O/hexane.
The purified all-E polyprenols, besides octaprenol, showed one peak in the HPLC chromatogram (Fig. 2), indicating high purity of the polyprenols obtained. All-E octaprenol, which showed the presence of Z and other minor isomers, could be further purified on an Alumina-AgNO3-impregnated column as described above.
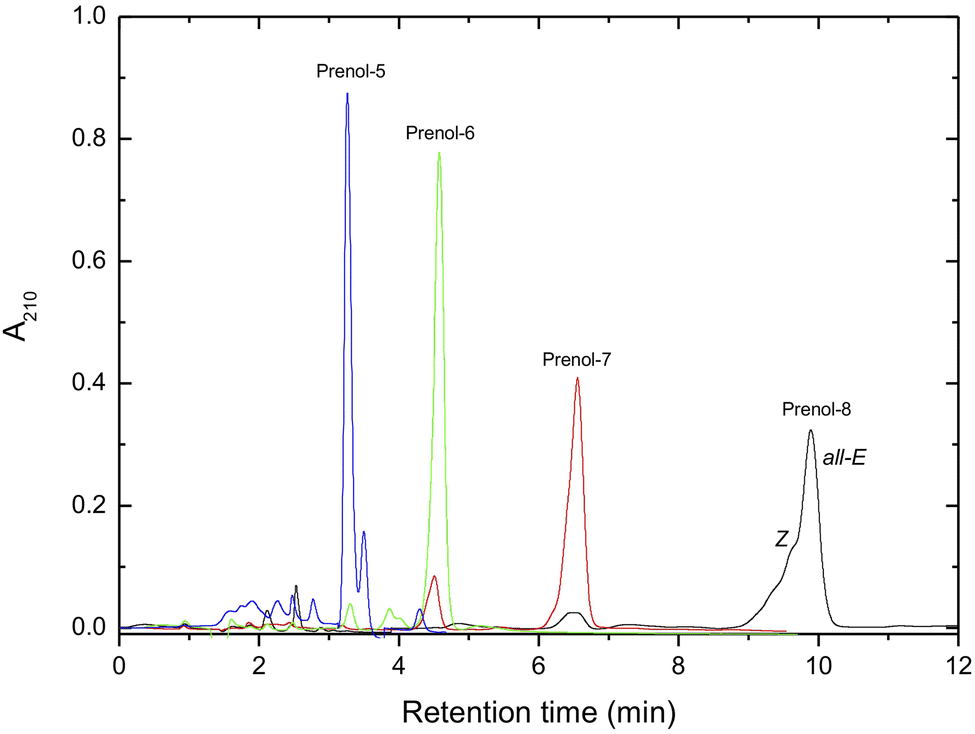 | ||
| Fig. 2 HPLC chromatogram of purified polyprenols (Nucleosil 100 C18 column, methanol/hexane 340/20, v/v). Further details are given in the Experimental section. | ||
3.3. Synthesis of prenylquinones and tocochromanols
In order to couple the polyprenols with the aromatic ring, the widely used method that takes advantage of BF3 was used. In the literature, various conditions are applied in this method.33 To optimize the conditions, we tested a variety of solvents, temperature and reaction times. We found that elevated temperature and extended time resulted in the formation of by-products. The optimal conditions were 15 min at RT in dioxane (Fig. 3). After oxidation with Ag2O, the prenylquinones were obtained with the following yields: 35–45% for plastoquinones (PQ): PQ-4, 30% for PQ-5, 45% for PQ-6, 36% for PQ-7, 44% for PQ-8 and 30% for PQ-9. All the plastoquinones showed absorption maxima at 255 nm, shoulder at 262 nm and 230 nm minimum in absolute ethanol, which is consistent with the literature data.34 The HPLC chromatogram of the synthesized plastoquinones is shown in Fig. 4. | ||
| Fig. 3 Chemical synthesis of plastoquinones and plastochromanols with a different side-chain length. | ||
 | ||
| Fig. 4 HPLC chromatograms of the synthesized plastoquinones and plastochromanols with a different side-chain length (Nucleosil 100 C18 column, methanol/hexane, 340/50, v/v). Absorption detection was at 255 nm (upper panel) for plastoquinones and fluorescence detection at 290/330 nm ex./em. (lower panel) for plastochromanols. Further details are given in the Experimental section. | ||
We found that during the coupling reaction, an elevated temperature and extended time resulted in the formation of tocochromanols (plastochromanols). After testing various reaction conditions, the optimal ones were 48 h at RT in THF (Fig. 3, S10†). Under these conditions, the yield of PC-8 was 15–20%. It could be increased to 40–60% using an elevated temperature and a shorter reaction time (e.g., 60 °C × 30 min), but under these conditions, nonspecific PC-8 isomers were formed in increased amounts. Using this procedure, all the homologues from PC-4 to PC-8 were obtained from the corresponding polyprenols (Fig. 4). The plastochromanols demonstrated absorption and fluorescence maxima consistent with the data in the literature.35
3.4. Spectral data
4. Disscussion
In this study, we develop a method for the chemical synthesis of polyprenols by chain lengthening which is considerably shorter than other methods and eliminates critical steps, requiring low temperatures and toxic chemicals, that are difficult to perform in ordinary laboratories. The reactions are performed at room temperature or under the reflux temperature of ethanol. Our method provides reasonable yields and can be scaled depending on requirements. Introducing short reaction times and simple product isolation between the reaction steps with two phase extraction and centrifugation allowed for one-day polyprenol lengthening by one isoprene unit and column chromatography purification of the product the same or next day. All the reactions can be followed using HPLC, allowing the formation of undesired isomers and other by-products to be controlled.The critical step of acetylene addition in liquid ammonia was replaced by the use of sodium acetylide in DME at room temperature, where the reaction is completed within one hour. To our knowledge, DME is the first solvent in which reaction of acetylene addition can be performed efficiently at room temperature. This method is of general importance as it can also be used in the synthesis of any other acetylides.
The main problems of the synthetic pathway remain the bromination steps of allylic prenols, resulting in the formation of Z isomers that lower the yield of the reaction and makes purification of the final all-E product difficult, even using AgNO3-based column chromatography. Bromination of the primary prenyl alcohols is a reaction frequently used in many, if not all synthesis procedures of polyprenols,19–23 but the formation of Z-isomers is not considered during the bromination step in these methods, which lowers the yield and makes it difficult to separate the isomers that have formed when pure all-E polyprenol is desired. Even though we tried different bromination methods, considered mild and specific, we could not obtain an improved E/Z ratio of the formed bromides. On the other hand, allylic rearrangement resulting in Z/E isomerisation during the bromination reaction of tertiary allylic alcohols is known,36–40 but there are no effective methods described in the literature to eliminate this problem. It is worth mentioning here that the use of metal catalysts41 or a certain combination of solvents42 improves the E/Z ration while tertiary alcohols are being coupled to aromatic nucleus.
The resulting polyprenols could then be used in the synthesis of a variety of biological prenylquinones and tocochromanols. We found reaction conditions, resulting in the direct formation of tocochromanols, which can be used for example to synthesize plastochromanol-8, an efficient natural antioxidant.43,44 The method for the synthesis of this compound described in the literature27 relied on heating PQ-9 in Pyr, followed by a reduction of the chromenol formed, using metallic sodium in ethanol. This procedure is in fact ineffective and the unreacted chromenol is difficult to separate from plastochromanol.
In summary, our method can be applied to the synthesis of polyprenols of any chain-length and thereafter to the synthesis of various biological prenylquinones, such as plastoquinones, ubiquinones or menaquinones, as well as to the corresponding tocochromanols (plastochromanols, tocotrienols, tocochromanols or tocochromanols).
5. Conclusions
We have presented a new, chemical synthesis method for polyprenols by chain lengthening which is considerably less time-consuming than other methods. It eliminates critical steps requiring low temperature and toxic chemicals. The addition reaction of acetylene in liquid ammonia was replaced by a new approach, the use of sodium acetylide in dimethoxyethane at room temperature. Our method is of general significance as it can also be used to synthesize any other acetylides. All the reactions were followed by high-performance liquid chromatography, allowing the formation of undesired isomers and other by-products to be controlled. The resulting polyprenols can further be used in the synthesis of a variety of biological prenylquinones. Moreover, we found a new method for the synthesis of tocochromanols (plastochromanols, tocochromanols) from polyprenols and aromatic head group directly.Author contributions
J. K. designed the study, performed the experiments, and wrote the text. R. S. wrote the text and drew the figures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are indebted to Dr Thomas Netscher (Research and Development, DSM Nutritional Products, Basel, Switzerland) for the kind gift of all-E pentaprenol and all-E decaprenol. This research was supported by scientific subsidy N19/DBS/000009 and MPK: 19000882 obtained from the Ministry of Science and Higher Education of Poland.References
- B. Nowicka and J. Kruk, Biochim. Biophys. Acta – Bioenerg., 2010, 1797, 1587–1605 CrossRef CAS.
- J. Kruk, K. Strzałka and G. H. Schmid, Free Radical Res., 1994, 21, 409–416 CrossRef CAS.
- J. Kruk, M. Jemioła-Rzemińska and K. Strzałka, Chem. Phys. Lipids, 1997, 87, 73–80 CrossRef CAS.
- J. Gruszka, A. Pawlak and J. Kruk, Free Radical Biol. Med., 2008, 45, 920–928 CrossRef CAS PubMed.
- K. Wang and S. Ohnuma, Trends Biochem. Sci., 1999, 24, 445–451 CrossRef CAS PubMed.
- M. O. Jones, L. Perez-Fons, F. P. Robertson, P. M. Bramley and P. D. Fraser, Biochem. J., 2013, 449, 729–740 CrossRef CAS PubMed.
- A. Block, R. Fristedt, S. Rogers, J. Kumar, B. Barnes, J. Barnes, C. G. Elowsky, Y. Wamboldt, S. A. Mackenzie, K. Redding, S. S. Merchant and G. J. Basset, J. Biol. Chem., 2013, 288, 27594–27606 CrossRef CAS.
- F. Bouvier, A. Rahier and B. Camara, Prog. Lipid Res., 2005, 44, 357–429 CrossRef CAS.
- T. Chojnacki and G. Dallner, Biochem. J., 1988, 251, 1–9 CrossRef CAS PubMed.
- L. Surmacz and E. Świeżewska, Biochem. Biophys. Res. Commun., 2011, 407, 627–632 CrossRef CAS PubMed.
- Q. Zhang, L. Huang, C. Zhang, P. Xie, Y. Zhang, S. Ding and F. Xu, Fitoterapia, 2015, 106, 184–193 CrossRef CAS PubMed.
- I. D. Boateng, Food Chem., 2023, 418, 136006 CrossRef CAS PubMed.
- J. W. Rip, C. A. Rupar, K. Ravi and K. K. Carroll, Prog. Lipid Res., 1985, 24, 269–309 CrossRef CAS PubMed.
- W. J. Jankowski, E. Swiezewska, W. Sasak and T. Chojnacki, J. Plant Physiol., 1994, 143, 448–452 CrossRef CAS.
- H. Sagami, E. Swiezewska and Y. Shidojic, Biosci., Biotechnol., Biochem., 2018, 82, 947–955 CrossRef CAS PubMed.
- A. A. Antipina, V. S. Popov and V. Y. Balabaniyan, Farmatsiya (Moscow), 2021, 70, 15–21 Search PubMed.
- T. A. Akhtar, P. Surowiecki, H. Siekierska, M. Kania, K. Van Gelder, K. A. Rea, L. K. A. Virta, M. Vatta, K. Gawarecka, J. Wojcik, W. Danikiewicz, D. Buszewicz, E. Swiezewska and L. Surmacz, Plant Cell, 2017, 29, 1709–1725 CrossRef CAS.
- A. Lipko, C. Pączkowski, L. Perez-Fons, P. D. Fraser, M. Kania, M. Hoffman-Sommer, W. Danikiewicz, M. Rohmer, J. Poznanski and E. Swiezewska, Biochem. J., 2023, 480, 495–520 CrossRef CAS PubMed.
- R. Rüegg, U. Gloor, R. N. Goel, G. Ryser, O. Wiss and O. Isler, Helv. Chim. Acta, 1959, 42, 2616–2621 CrossRef.
- Y. Naruta, J. Org. Chem., 1980, 45, 4097–4104 CrossRef CAS.
- C. P. Casey and D. F. Marten, Synth. Commun., 1973, 3, 321–324 CrossRef CAS.
- S. Sato, S. Inoue, A. Onishi, N. Uchida and N. Minowa, J. Chem. Soc., Perkin Trans., 1981, 1, 761–769 RSC.
- A. Baj, P. Wałejko, A. Kutner, Ł. Kaczmarek, J. W. Morzycki and S. Witkowski, Org. Process Res. Dev., 2016, 20, 1026–1033 CrossRef CAS.
- K. Sato and S. Inoue, J. Synth. Org. Chem., Jpn., 1971, 29, 237–253 CrossRef CAS.
- Y. Totsuka, Y. Yasuno and T. Shinada, Chem. Lett., 2019, 48, 491–494 CrossRef CAS.
- T. Netscher, W. Bonrath, I. Bendik, P. J. Zimmermann, F. Weber and A. Rüttimann, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH and Co. KGaA, Weinheim, 2020, pp. 1–25 Search PubMed.
- H. Mayer and O. Isler, Methods Enzymol., 1971, 38, 241–348 Search PubMed.
- J. Kruk, Biophys. Chem., 1998, 30, 143–149 CrossRef PubMed.
- M. M. Ponpipom and S. Hanessian, Carbohydr. Res., 1971, 18, 342–344 CrossRef CAS.
- X.-M. Cui, Y.-H. Guan, N. Li, H. Lv, L.-A. Fu, K. Guo and X. Fan, Tetrahedron Lett., 2014, 55, 90–93 CrossRef CAS.
- E. H. Axelrod, G. M. Milne and E. E. van Tamelen, J. Am. Chem. Soc., 1970, 92, 2139–2141 CrossRef CAS.
- W. Gati, F. Munyemana, A. Colens, A. Srour, M. Dufour, K. H. V. Reddy, B. Techy, G. Rosse, E. Schweiger, Q. Qiao and L. Ghosez, Tetrahedron, 2020, 76, 131441 CrossRef CAS.
- H. Mayer and O. Isler, Methods Enzymol., 1971, 38, 491–547 Search PubMed.
- R. Barr and F. L. Crane, Methods Enzymol., 1971, 23, 372–408 CAS.
- K. J. Whittle, P. J. Dunphy and J. F. Pennock, Biochem. J., 1965, 96, 17c–19c CrossRef CAS PubMed.
- Y. Yuasa and Y. Yuasa, Synth. Commun., 2006, 36, 1671–1677 CrossRef CAS.
- J. H. Babler, J. Org. Chem., 1976, 41, 1262–1264 CrossRef CAS.
- P. J. R. Nederlof, M. J. Moolenaar, E. R. De Waard and H. O. Huisman, Tetrahedron, 1977, 33, 579–580 CAS.
- A. K. Bakkestuen, L.-L. Gundersen, D. Petersen, B. T. Utenova and A. Vik, Org. Biomol. Chem., 2005, 3, 1025–1033 RSC.
- R. Rüegg, U. Gloor, A. Langemann, M. Kofler, C. von Planta, G. Ryser and O. Isler, Helv. Chim. Acta, 1960, 43, 1745–1751 CrossRef.
- B. Wüstenberg, M. Stettler and W. Bonrath, 41 Jahrestreffen Deutscher Katalytiker, Weimar, 2008, pp. 27–29 Search PubMed.
- H. Eto and C. Eguchi, Chem. Lett., 1988, 17, 1597–1600 CrossRef.
- J. Kruk, R. Szymańska, J. Cela and S. Munne-Bosch, Phytochemistry, 2014, 108, 9–16 CrossRef CAS.
- B. Nowicka, J. Gruszka and J. Kruk, Biochim. Biophys. Acta – Biomembr., 2013, 1828, 233–240 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02872k |
| This journal is © The Royal Society of Chemistry 2023 |