
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring novel aryl/heteroaryl-isosteres of phenylthiazole against multidrug-resistant bacteria†
Mariam Omaraa,
Mohamed Hagras *b,
Mohamed M. Elsebaieb,
Nader S. Abutalebcd,
Hanzada T. Nour El-Dine,
Maria O. Mekhailf,
Ahmed S. Attiaeg,
Mohamed N. Seleemch,
Marwa T. Sarga and
Abdelrahman S. Mayhoubbi
*b,
Mohamed M. Elsebaieb,
Nader S. Abutalebcd,
Hanzada T. Nour El-Dine,
Maria O. Mekhailf,
Ahmed S. Attiaeg,
Mohamed N. Seleemch,
Marwa T. Sarga and
Abdelrahman S. Mayhoubbi
aDepartment of Pharmaceutical Organic Chemistry, College of Pharmacy (Girls), Al-Azhar University, Cairo, Egypt
bDepartment of Pharmaceutical Organic Chemistry, College of Pharmacy (Boys), Al-Azhar University, Cairo 11884, Egypt. E-mail: m.hagrs@azhar.edu.eg
cDepartment of Biomedical Sciences and Pathobiology, Virginia-Maryland College of Veterinary Medicine, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, USA
dDepartment of Microbiology and Immunology, Faculty of Pharmacy, Zagazig University, Zagazig 44519, Egypt
eDepartment of Microbiology and Immunology, Faculty of Pharmacy, Cairo University, Cairo 11562, Egypt
fPharmD-Clinical Pharmacy Undergraduate Program, Faculty of Pharmacy, Cairo University, Cairo 11562, Egypt
gDepartment of Microbiology and Immunology, School of Pharmacy, Newgiza University, Giza, Egypt
hCenter for One Health Research, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, USA
iNanoscience Program, University of Science and Technology, Zewail City of Science and Technology, Giza, Egypt
First published on 6th July 2023
Abstract
Antimicrobial resistance has become a concern as a worldwide threat. A novel scaffold of phenylthiazoles was recently evaluated against multidrug-resistant Staphylococci to control the emergence and spread of antimicrobial resistance, showing good results. Several structural modifications are needed based on the structure–activity relationships (SARs) of this new antibiotic class. Previous studies revealed the existence of two key structural features essential for the antibacterial activity, the guanidine head and lipophilic tail. In this study, a new series of twenty-three phenylthiazole derivatives were synthesized utilizing the Suzuki coupling reaction to explore the lipophilic part. The in vitro antibacterial activity was evaluated against a range of clinical isolates. The three most promising compounds, 7d, 15d and 17d, with potent MIC values against MRSA USA300 were selected for further antimicrobial evaluation. The tested compounds exhibited potent results against the tested MSSA, MRSA, and VRSA strains (concentration: 0.5 to 4 μg mL−1). Compound 15d inhibited MRSA USA400 at a concentration of 0.5 μg mL−1 (one-fold more potent than vancomycin) and showed low MIC values against ten clinical isolates, including linezolid-resistant strain MRSA NRS119 and three vancomycin-resistant isolates VRSA 9/10/12. Moreover, compound 15d retained its potent antibacterial activity using the in vivo model by the burden reduction of MRSA USA300 in skin-infected mice. The tested compounds also showed good toxicity profiles and were found to be highly tolerable to Caco-2 cells at concentrations of up to 16 μg mL−1, with 100% of the cells remaining viable.
1. Introduction
Antimicrobial resistance (AMR) has recently become one of the most significant worldwide threats and has acquired an obvious priority status in global public health.1 Mainly, the combination of antibiotics overuse and the slow discovery of new antibiotics has led to the increase in the incidence of AMR via stimulation of bacterial natural defense mechanisms, such as point mutations, bacterial evolution, and horizontal resistance gene transfer.2 In the United States, annual reports have referred to millions of resistant infections with more than 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths.3 In addition, the pandemic COVID-19 is expected to increase AMR development as 70% of COVID-19 patients use antibiotics.4,5 The adverse effects of AMR are not limited to health problems, as it clearly has an influence on the global economy where billions of USD have been spent for the sake of treating resistant bacterial infections.6 According to the World Health Organization (WHO) priority pathogen list, both methicillin-resistant and vancomycin-intermediate or – resistant Staphylococcus aureus (MRSA, VISA and VRSA, respectively) are categorized as high priorities.7 Methicillin-resistant S. aureus (MRSA) is a major cause of nosocomial and community-acquired infections worldwide.8 MRSA is resistant to most traditional antibiotics, such as β-lactams, fluoroquinolones, aminoglycosides, and tetracyclines.9–13 Furthermore, MRSA has developed resistance against vancomycin and linezolid, the drugs of choice for treatment against it. Thus, there is an urgent need to develop new effective antimicrobial agents.14–16
000 deaths.3 In addition, the pandemic COVID-19 is expected to increase AMR development as 70% of COVID-19 patients use antibiotics.4,5 The adverse effects of AMR are not limited to health problems, as it clearly has an influence on the global economy where billions of USD have been spent for the sake of treating resistant bacterial infections.6 According to the World Health Organization (WHO) priority pathogen list, both methicillin-resistant and vancomycin-intermediate or – resistant Staphylococcus aureus (MRSA, VISA and VRSA, respectively) are categorized as high priorities.7 Methicillin-resistant S. aureus (MRSA) is a major cause of nosocomial and community-acquired infections worldwide.8 MRSA is resistant to most traditional antibiotics, such as β-lactams, fluoroquinolones, aminoglycosides, and tetracyclines.9–13 Furthermore, MRSA has developed resistance against vancomycin and linezolid, the drugs of choice for treatment against it. Thus, there is an urgent need to develop new effective antimicrobial agents.14–16
Previously, a novel antibacterial scaffold was discovered by our research group containing n-butylphenylthiazole (I) (Fig. 1). It showed a MIC value of 4.8 μg mL−1 against MRSA and was considered as a lead compound for further development. The lead I has two essential features: a lipophilic moiety (blue color, Fig. 1) and a cationic moiety (red color, Fig. 1). During the optimization of the lead compound, the replacement of the n-butyl group with a phenyl ring (II) decreased the MIC value to 2.4 μg mL−1 (Fig. 1).17 Meanwhile, rigidification by acetylene linked to a heteroaromatic system (III) afforded an improvement of the antibacterial activity (MIC = 2 μg mL−1).18 In this study, we aim to further advance the lead developments based on the previous results by utilizing two new lead optimization strategies: scaffold simplification and bioisosteric replacement techniques to develop novel and potent derivatives. Furthermore, we aim to investigate the structure–activity relationship of novel phenylthiazoles using the Suzuki–Miyaura coupling reaction (Fig. 1).19–24
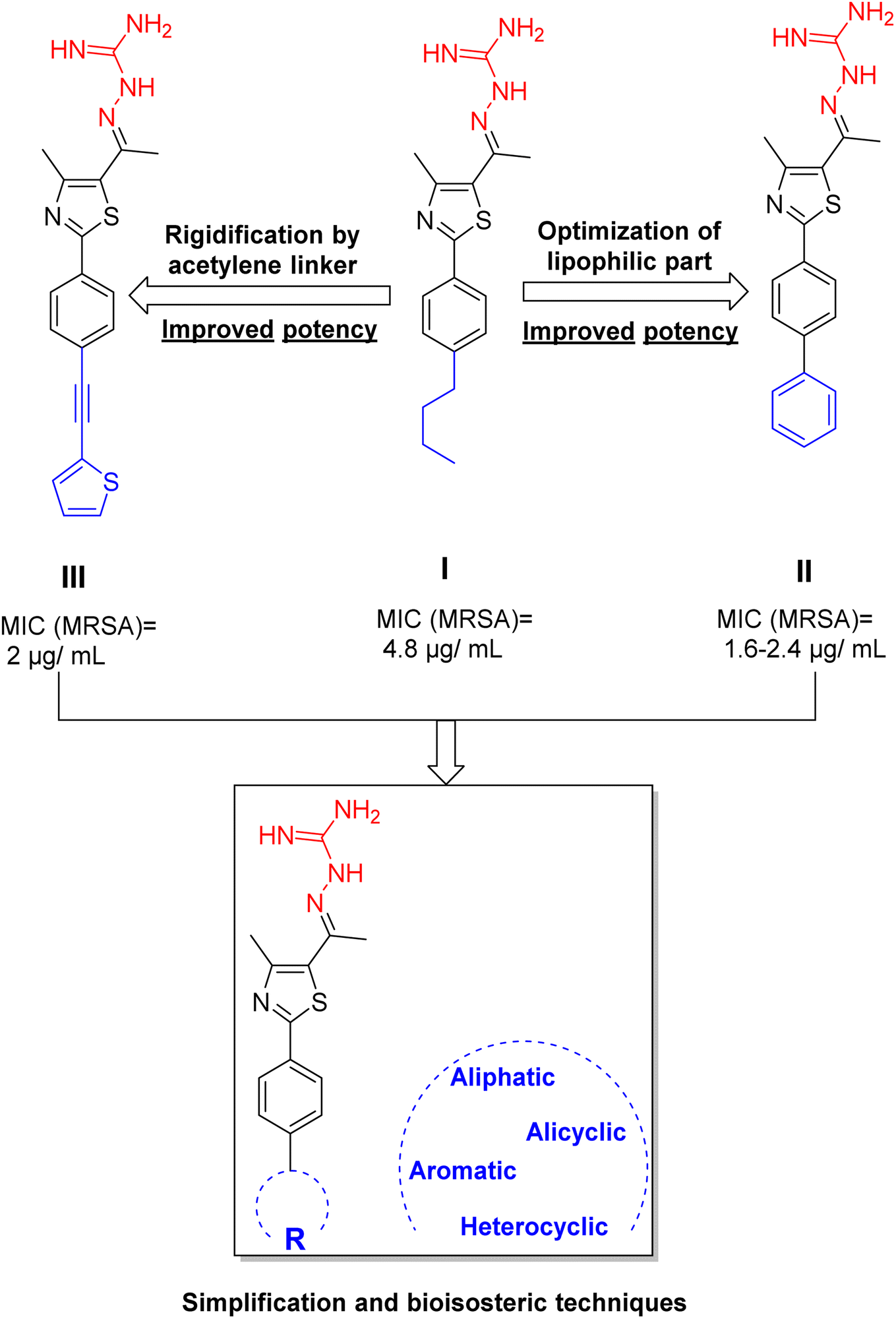 | ||
| Fig. 1 The design of the current study based on the previous study. | ||
2. Results and discussion
2.1. Chemistry
Hantzsch thiazole synthesis between 4-halobenzothioamide (1a,b) and α-chloroacetylacetone afforded the key starting material phenylthiazole (2a,b).18,25 The alkyl and aryl derivatives were introduced using typical Suzuki cross-coupling conditions, as mentioned in Scheme 1, via two synthetic routes. The first one involved the direct reaction of boronic acid derivatives with p-iodophenylthiazole in one step.22 The second method is an in situ borylation of p-chlorophenylthiazole, followed by reaction with the second aryl halide, which was suitable for a wide range of derivatives (Table 1).26 Thiophene sulfonamide derivatives 19–25 were synthesized via nucleophilic substitution reaction of the corresponding sulfonyl chloride with proper amines in the presence of triethylamine.27 Finally, aminoguanidine derivatives (3–25d) were obtained through condensation between 3–25c with aminoguanidine HCl in the presence of a catalytic amount of hydrochloric acid (Scheme 1).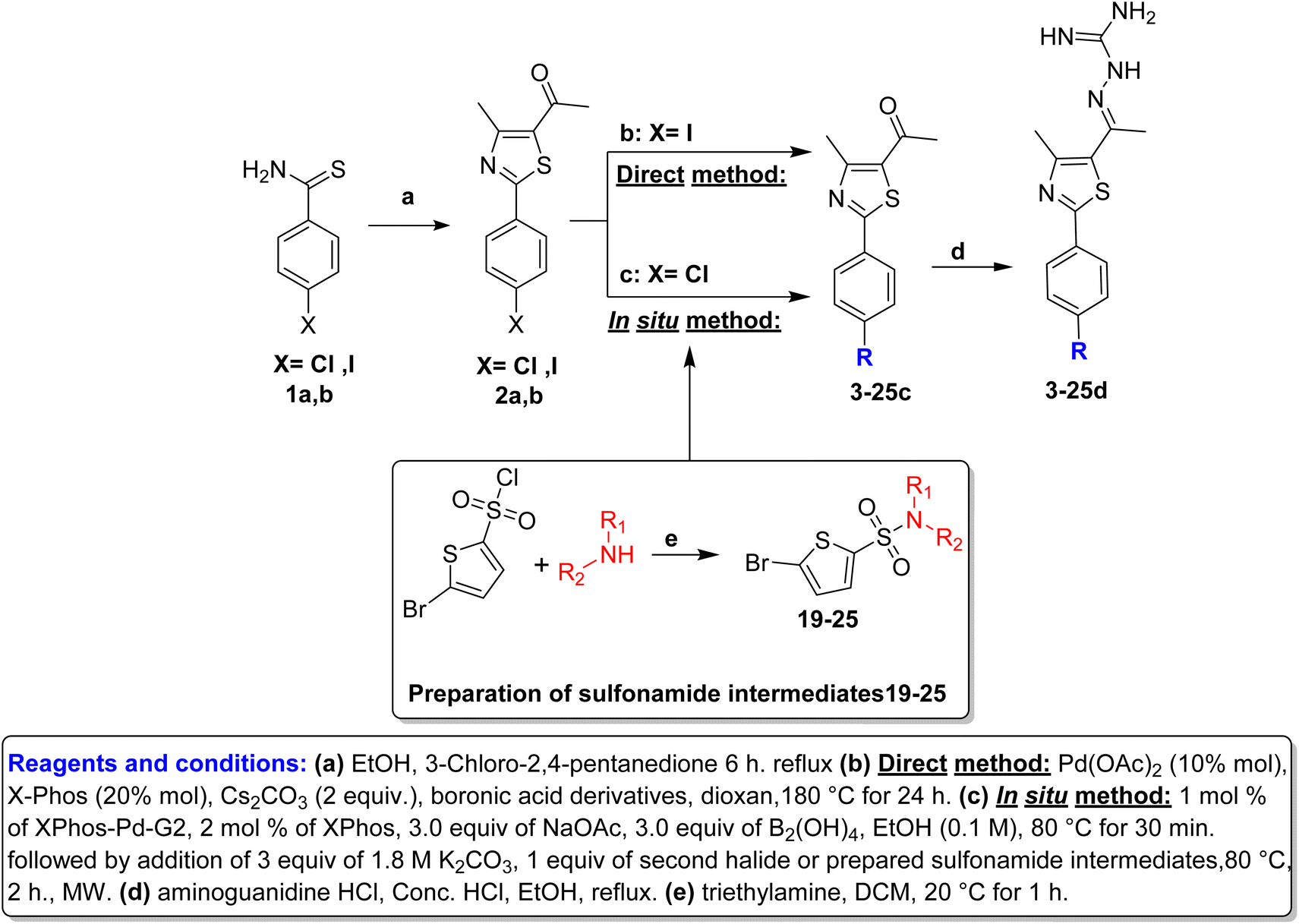 | ||
| Scheme 1 | ||










2.2. Biological evaluation
| Compound number | R | MIC against MRSA USA 300 | Compound number | R | MIC against MRSA USA 300 |
|---|---|---|---|---|---|
| 3d |  |
8 | 15d |  |
1 |
| 4d |  |
32 | 16d | 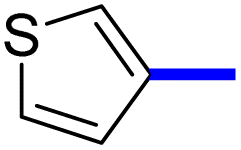 |
4 |
| 5d | 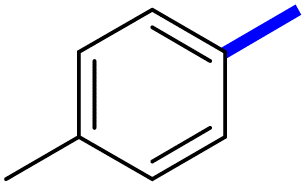 |
4 | 17d |  |
2 |
| 6d | 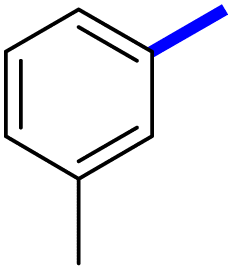 |
8 | 18d |  |
4 |
| 7d |  |
2 | 19d | 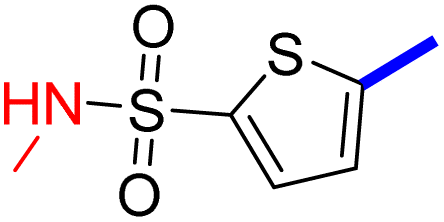 |
32 |
| 8d | 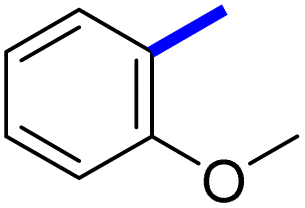 |
2 | 20d | 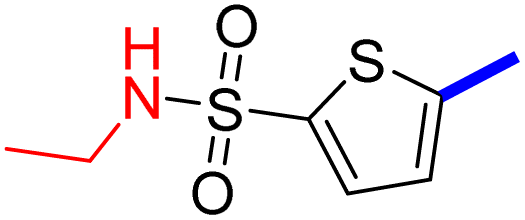 |
16 |
| 9d |  |
8 | 21d |  |
32 |
| 10d | 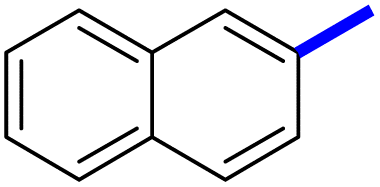 |
4 | 22d | 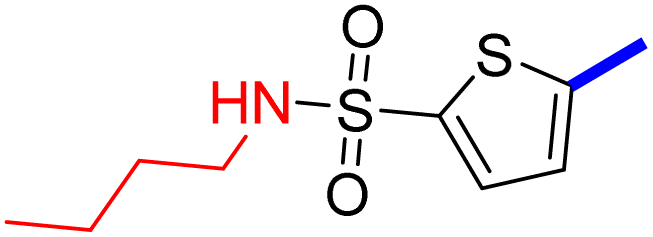 |
>32 |
| 11d | 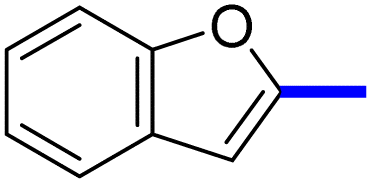 |
32 | 23d | 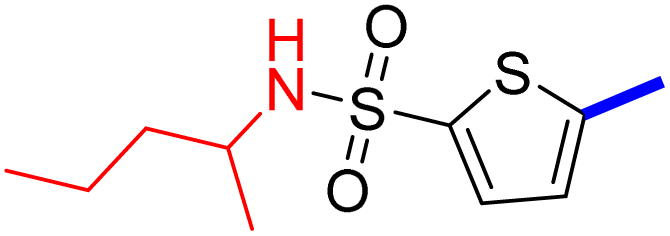 |
16 |
| 12d | 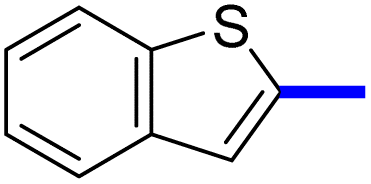 |
32 | 24d | 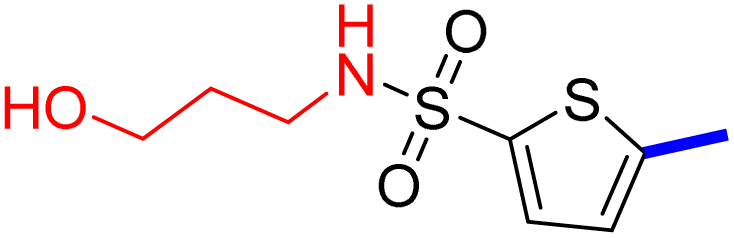 |
>32 |
| 13d | 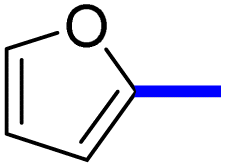 |
64 | 25d |  |
>32 |
| 14d |  |
2 | 26d |  |
16 |
| Vancomycin | 1 | ||||
Our compounds exhibited potent antibacterial activity against the tested MSSA, MRSA, and VRSA strains, inhibiting their growth at concentrations ranging from 0.5 to 4 μg mL−1. Notably, they maintained their potency against linezolid-resistant and vancomycin-resistant staphylococcal strains. Compound 15d displayed the most potent activity against the tested strains, inhibiting their growth at concentrations ranging from 0.5 to 1 μg mL−1 (Table 3). Interestingly, the compounds maintained their potency against linezolid-resistant and vancomycin-resistant staphylococcal strains, suggesting that they do not share the same resistance mechanism with linezolid or vancomycin.
| Bacterial isolates | Compounds/control antibiotics | ||||
|---|---|---|---|---|---|
| 7d | 15d | 17d | Linezolid | Vancomycin | |
| MSSA ATCC 6538 | 2 | 1 | 4 | 1 | 0.5 |
| MSSA NRS 107 | 4 | 1 | 4 | 0.5 | 1 |
| MRSA NRS 119 | 4 | 1 | 4 | 64 | 2 |
| MRSA USA 400 | 2 | 0.5 | 2 | 2 | 1 |
| MRSA USA 500 | 4 | 1 | 4 | 1 | 2 |
| MRSA USA 700 | 2 | 1 | 4 | 1 | 1 |
| VRSA 9 | 2 | 0.5 | 4 | 1 | >64 |
| VRSA 10 | 2 | 1 | 4 | 2 | 64 |
| VRSA 12 | 4 | 1 | 4 | 1 | >64 |
| S. epidermidis NRS 101 | 2 | 0.5 | 2 | 1 | 1 |
| Bacterial isolates | Compounds/control antibiotics | ||||
|---|---|---|---|---|---|
| 7d | 15d | 17d | Linezolid | Vancomycin | |
| S. pneumoniae ATCC 51916 | 4 | 1 | 4 | 1 | 2 |
| S. pneumoniae ATCC 700677 | 4 | 1 | 4 | 1 | 1 |
| E. faecalis ATCC 51299 | 4 | 2 | 4 | 1 | 64 |
| E. faecium ATCC 700221 | 2 | 1 | 2 | 1 | >64 |
| L. monocytogenes ATCC 19111 | 4 | 1 | 8 | 1 | 1 |
| C. difficile ATCC BAA 1870 | 4 | 2 | 2 | NT | 1 |
Among the tested compounds, compound 15d displayed the most potent activity among the tested compounds with MIC values ranging from 1 μg mL−1 to 2 μg mL−1. Compounds 7d and 17d also exhibited strong activity, inhibiting the bacterial strains at MICs ranging from 2 to 8 μg mL−1. Importantly, our compounds maintained their potency against vancomycin-resistant enterococcal strains, suggesting that they are not subjected to the same resistance mechanism as vancomycin.
 | ||
| Fig. 2 Analyzing the toxicity of compounds 7d, 15d and 17d (tested in triplicates at 8, 16 and 32 μg mL−1) against human colorectal cells (Caco-2) using the MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium assay. Results are presented as the percentage of viable cells relative to DMSO (negative control) to determine a baseline measure for the cytotoxic impact of each compound. The absorbance values represent an average of three samples analyzed for each compound. Error bars represent the standard deviation values. Data were analyzed via a two-way ANOVA with post hoc Dunnett's test for multiple comparisons. (*) denotes a statistical difference (p < 0.05) between values obtained for the compounds and the DMSO. | ||
log10 – reduction after 24 hours, while compounds 15d and 17d generated 2.0 and 1.7log10 – reduction in bacterial CFU, respectively. In contrast, linezolid resulted in only a 1.1log10 -reduction in bacterial burden after 24 hours (see Fig. 3).
 | ||
| Fig. 3 Killing kinetics of compounds (tested in triplicates at 5 × MIC) against methicillin-resistant Staphylococcus aureus NRS123 over a 24 hours incubation period at 37 °C. DMSO (solvent for the compounds) served as a negative control, and linezolid and vancomycin served as control antibiotics. The error bars represent standard deviation values obtained from triplicate samples used for each compound/antibiotic studied. Data were analyzed via a two-way ANOVA with post hoc Dunnett's test for multiple comparisons. An asterisk (*) denotes a statistical difference (p < 0.05) between the values obtained for each test agent as compared to DMSO. | ||
000-fold increase in MIC by the end of the experiment. These results indicate that MRSA was unable to develop rapid resistance to any of the tested phenylthiazole compounds, but could rapidly develop resistance to rifampicin (Fig. 4).
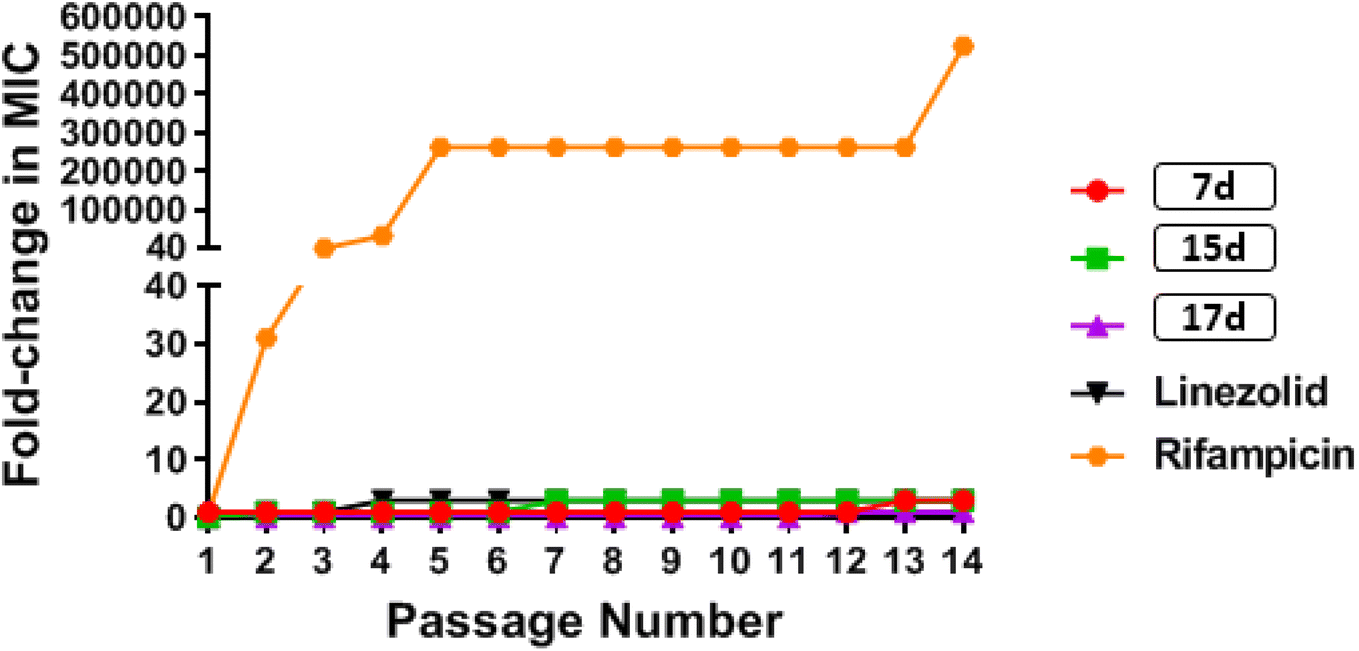 | ||
| Fig. 4 Multi-step resistance selection of compounds against methicillin-resistant S. aureus USA 400. Bacteria were serially passaged over a 14 days period, and the broth microdilution assay was used to determine the minimum inhibitory concentration of each compound against MRSA after each successive passage. A four-fold shift in MIC would be indicative of bacterial resistance to the test agent. | ||
 | ||
| Fig. 5 In vivo anti-MRSA activity in a murine model of skin infection. Seventy-two hours post-infection with S. aureus USA300, a visible lesion developed at the site of infection. Twice a day topical application of petroleum jelly (PJ), commercial 2% fusidic acid ointment (FA) or 2% 15d in PJ (15d) was carried out for 4 consecutive days. (A) Photographs of representative mice from the three groups on the sacrifice day. Box plots of the bacterial burden recovered from the skin lesions (B) and the spleens (C) of the infected mice of the three groups. In both plots, the whiskers span the difference between the minimum and maximum readings, the horizontal bar represents the median, and the (+) sign represents the mean of the log10CFU mL−1. Statistical analysis was done using the ordinary one-way ANOVA, followed by Tukey's multiple comparisons test. The * indicates a statistical difference (p ≤ 0.05). The charts were generated using GraphPad Prism (version 9.0). | ||
Bacterial burdens in the skin lesions were estimated, and it was observed that compound 15d resulted in a decrease in the MRSA burden in the mice skin lesions by almost one log10 cycle, which was not significant compared to the FA treatment that generated around three log10 reduction as compared to the vehicle-treated mice (Fig. 5B). However, compound 15d demonstrated weaker activity than FA in controlling the systemic dissemination of S. aureus, as evidenced by the low reduction in the bacterial burden detected in the spleens of infected mice compared to the vehicle-treated group (Fig. 5C). These results suggest that while compound 15d shows promising potential in controlling S. aureus skin infection, its efficacy in controlling systemic dissemination is weaker than that of FA.
3. Conclusion
The present study aimed to enhance the antibacterial activity of phenylthiazoles against multidrug-resistant Staphylococci by exploring the lipophilic part of the compounds via Suzuki coupling reaction. First, twenty-three compounds were synthesized, and their efficacy was evaluated against a range of clinical isolates, including MRSA USA400, MRSA NRS119, and vancomycin-resistant isolates VRSA 9/10/12. Compound 15d was found to be particularly potent, exhibiting an inhibitory concentration of 0.5 μg mL−1 against MRSA USA400, which is one-fold more potent than vancomycin. Additionally, compound 15d maintained its efficacy against ten clinical isolates, including MRSA NRS119 and three VRSA strains. In vivo experiments on skin-infected mice showed that compound 15d was able to reduce the bacterial burden of MRSA USA300. These results provide evidence that exploring the lipophilic part of phenylthiazoles is promising, and can lead to the development of more potent antibiotics against multidrug-resistant Staphylococci.4. Experimental
4.1. Chemistry
General: 1H NMR spectra were run at 400 MHz and 13C NMR spectra were determined at 100 MHz in dimethyl sulfoxide (DMSO-d6) on a Bruker VX-400 NMR spectrometer. Chemical shifts are given in parts per million (ppm) on the delta (δ) scale. Chemical shifts were calibrated relative to those of the solvents. Flash chromatography was performed on 230–400 mesh silica. The progress of reactions was monitored with Merck silica gel IB2-F plates (0.25 mm thickness). The infrared spectra were recorded in potassium bromide disks on pye Unicam SP 3300 and Shimadzu FT IR 8101 PC infrared spectrophotometer. Mass spectra were recorded at 70 eV. High-resolution mass spectra for all ionization techniques were obtained from a FinniganMAT XL95. Melting points were determined using capillary tubes with a Stuart SMP30 apparatus and are uncorrected. All yields reported refer to isolated yields.:ethyl acetate = 4:1) to afford compounds 19–25 as white solids as reported.33–354.1.2.1 General procedure. A. Direct method: To a dioxan
:water (9:1 mL) mixture in a 75 mL sealed tube, compound 2b (350 mg, 1.02 mmol), palladium diacetate (24 mg, 10% mol), 2-dicyclohexylphosphino-2′,4′,6′-triiso-propylbiphenyl (X-phos) (146 mg, 0.3 mmol) and Cesium carbonate (833 mg, 2.56 mmol) were dissolved. After the reaction mixture was purged with dry nitrogen gas for 10 min, appropriate bronic acid derivatives (1.61 mmol) were added. The sealed tube was then placed in an oil bath and stirred at 100 °C for 24 h. After cooling to room temperature, the reaction mixture was passed through Celite, followed by ethyl acetate (2 × 50 mL), and dried over anhydrous magnesium sulphate. The organic materials were then concentrated under reduced pressure. The crude materials were purified via silica gel flash column chromatography using hexane–ethyl acetate (7:3).B. In situ: In a 75 mL sealed tube with 15 mL EtOH, compound 2a (350 mg, 1.39 mmol, 1 eq.), tetrahydroxydiboron (373.94 mg, 4.17 mmol, 3 eq.), XPhos-PdG2 (11 mg, 14 μmol, 0.01 eq.), XPhos (13.23 mg, 28 μmol, 0.02 eq.), and NaOAc (342 mg, 4.17 mmol, 3 eq.) were added respectively under N2 flushing. The reaction mixture was then heated to 80 °C until the solution changed into a red color, and a precipitation was formed, indicating that the boronic acid derivative was formed and confirmed by TLC. Then, a solution of K2CO3 (577 mg, 4.17 mmol, 3 eq.) in 5 mL distilled water was added to the reaction mixture, followed by the addition of the second halide (2.8 mmol, 2 eq.). The reaction mixture was further heated to 80 °C for 15 h. After cooling to room temperature, the reaction mixture was passed through Celite, followed by ethyl acetate (2 × 50 mL) and dried over anhydrous magnesium sulphate. The organic materials were then concentrated under reduced pressure. The crude materials were purified via silica gel flash column chromatography using hexane–ethyl acetate (7:3).
1-(2-(4-Isobutylphenyl)-4-methylthiazol-5-yl)ethan-1-one (3c): Following the general procedure (method A), compound 3c was obtained as a light-brown oil (240 mg, 86%). 1H NMR (DMSO-d6) δ: 8.01 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 2.71 (s, 3H), 2.62 (s, 3H), 2.52 (d, J = 7.2 Hz, 2H), 1.93–1.83 (m, 1H), 0.90 (d, J = 8.2 Hz, 6H); 13C NMR (DMSO-d6); δ 200.6, 148.5, 142.3, 137.0, 135.2, 129.7, 126.6, 117.2, 44.8, 30.1, 25.2, 22.6, 17.5; MS (m/z); 273.
1-(2-(4-Cyclopropylphenyl)-4-methylthiazol-5-yl)ethan-1-one (4c): Following the general procedure (method A), compound 4c was obtained as a light-brown oil (226 mg, 86%). 1H NMR (DMSO-d6) δ: 7.98 (d, J = 8.2 Hz, 2H), 7.18 (d, J = 8.2 Hz, 2H), 2.64 (s, 3H), 2.23 (s, 3H), 1.99–194 (m, 1H), 1.01–0.97 (m, 2H), 0.75–0.72 (m, 2H); 13C NMR (DMSO-d6); δ 196.0, 153.4, 146.0, 137.0, 136.1, 135.1, 126.03, 117.0, 25.2, 17.5, 15.4, 10.1; MS (m/z); 257.
1-(4-Methyl-2-(4′-methyl-[1,1′-biphenyl]-4-yl)-thiazol-5-yl)ethan-1-one (5c): Following the general procedure (method A), compound 5c was obtained as a yellow oil (293 mg, 93%). 1H NMR (DMSO-d6) δ: 7.97 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.2 Hz, 2H), 2.61 (s, 3H), 2.36 (s, 3H), 2.33 (s, 3H); MS (m/z); 307.
1-(4-Methyl-2-(3′-methyl-[1,1′-biphenyl]-4-yl)thiazol-5-yl)ethan-1-one (6c): Following the general procedure (method A), compound 6c was obtained as a light brown oil (246 mg, 78%). 1H NMR (DMSO-d6) δ: 8.20 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.4 Hz, 2H), 7.56 (s, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.40 (t, J = 8.2 Hz, 1H), 7.22 (d, J = 8.2 Hz, 1H), 2.67 (s, 3H), 2.40 (s, 3H), 2.26 (s, 3H); MS (m/z); 307.
1-(2-(3′-Methoxy-[1,1′-biphenyl]-4-yl)-4-methylthiazol-5-yl)ethan-1-one (7c): Following the general procedure (method B), compound 7c was obtained as a yellow oil (193 mg, 43%). 1H NMR (DMSO-d6) δ: 8.08 (d, J = 8 Hz, 2H), 7.85 (d, J = 8 Hz, 2H), 7.74 (t, J = 8 Hz, 1H), 7.32 (d, J = 8 Hz, 1H), 7.27 (s, 1H), 7.01 (d, J = 8 Hz, 1H), 3.85 (s, 3H), 2.73 (s, 3H), 2.58 (s, 3H); 13C NMR (DMSO-d6); δ: 192.1, 168.5, 160.3, 158.7, 143.2, 140.8, 132.6, 131.7, 130.6, 128.1, 127.5, 119.5, 114.3, 112.6, 55.6, 30.6, 18.6; MS (m/z); 323.
1-(2-(2′-Methoxy-[1,1′-biphenyl]-4-yl)-4-methylthiazol-5-yl)ethan-1-one (8c): Following the general procedure (method B), compound 8c was obtained as a buff oil (188 mg, 42%). 1H NMR (DMSO-d6) δ: 8.03 (d, J = 8 Hz, 2H), 7.65 (d, J = 8 Hz, 2H), 7.41–7.36 (m, 2H), 7.16 (t, J = 8 Hz, 1H), 7.08 (t, J = 8 Hz, 1H), 3.79 (s, 3H), 2.72 (s, 3H), 2.57 (s, 3H); 13C NMR (DMSO-d6); δ: 191.8, 168.8, 158.6, 156.6, 141.7, 132.5, 131.0, 130.7, 130.6, 130.0, 129.0, 126.7, 121.3, 112.3, 55.0, 30.8, 18.6; MS (m/z); 323.
Methyl 4′-(5-acetyl-4-methylthiazol-2-yl)-[1,1′-biphenyl]-2-carboxylate (9c): Following the general procedure (method B), compound 9c was obtained as a yellow oil (293 mg, 60%). 1H NMR (DMSO-d6) δ: 8.05 (d, J = 8 Hz, 2H), 7.82 (d, J = 8 Hz, 1H), 7.69 (t, J = 8 Hz, 1H), 7.56 (t, J = 8 Hz, 1H), 7.50 (d, J = 8 Hz, 1H), 7.45 (d, J = 8 Hz, 2H), 3.64 (s, 3H), 2.73 (s, 3H), 2.58 (s, 3H); 13C NMR (DMSO-d6); δ: 191.1, 168.6, 158.7, 144.0, 140.7, 132.6, 131.5, 130.9, 130.0, 129.6, 128.5, 126.9, 128.5, 126.9, 52.5, 30.9, 18.6; MS (m/z); 351.
1-(4-Methyl-2-(4-(naphthalen-2-yl)phenyl)-thiazol-5-yl)ethan-1-one (10c): Following the general procedure (method A), compound 10c was obtained as a yellow oil (302 mg, 86%). 1H NMR (DMSO-d6) δ: 8.30 (s, 1H), 8.25 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 8.4 Hz, 2H), 7.90 (s, 1H), 7.80 (t, J = 8.4 Hz, 1H), 7.58 (t, J = 8.4 Hz, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.40 (d, J = 8.2 Hz, 1H), 2.69 (s, 3H), 2.26 (s, 3H); MS (m/z); 343.
1-(2-(4-Benzofuran-2-yl)phenyl)-4-methylthiazol-5-yl)ethan-1-one (11c): Following the general procedure (method A), compound 14c was obtained as a yellow oil (280 mg, 82%). 1H NMR (DMSO-d6) δ: 8.26 (d, J = 8.4 Hz, 2H), 8.04 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.68 (s, 1H), 7.57 (t, J = 8.4 Hz, 1H), 7.35 (t, J = 8.2 Hz, 1H), 2.67 (s, 3H), 2.25 (s, 3H); MS (m/z); 333.
1-(2-(4-Benzo[b]thiophen-2-yl)phenyl)-4-methylthiazol-5-yl)ethan-1-one (12c): Following the general procedure (method A), compound 12c was obtained as a yellow oil (290 mg, 81%). 1H NMR (DMSO-d6) δ: 8.23 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.4 Hz, 2H), 7.89 (d, J = 8.4 Hz, 1H), 7.86 (s, 1H), 7.82 (t, J = 8.4 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.57 (t, J = 8.2 Hz, 1H), 2.67 (s, 3H), 2.25 (s, 3H); MS (m/z); 349.
1-(2-(4-Furan-2-yl)phenyl)-5-methylthiazol-5-yl)ethan-1-one (13c): Following the general procedure (method A), compound 13c was obtained as a yellow oil (250 mg, 86%). 1H NMR (DMSO-d6) δ: 8.01 (d, J = 8.2 Hz, 2H), 7.80 (d, J = 8.2 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 7.20 (t, J = 8.4 Hz, 1H), 2.71 (s, 3H), 2.56 (s, 3H); 13C NMR (DMSO-d6); δ 191.0, 168.3, 158.7, 142.9, 136.8, 132.4, 131.3, 129.3, 127.7, 127.5, 126.4, 125.4, 30.8, 18.6; MS (m/z); 283.
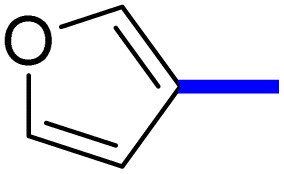
1-(2-(4-Furan-3-yl)phenyl)-5-methylthiazol-5-yl)ethan-1-one (14c): Following the general procedure (method A), compound 14c was obtained as a pale-yellow oil (244 mg, 84%). 1H NMR (DMSO-d6) δ: 8.29 (s, 1H), 7.91 (d, J = 8.2 Hz, 2H), 7.78 (d, J = 8.2 Hz, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 6.4 Hz, 1H), 2.59 (s, 3H), 2.32 (s, 3H); 13C NMR (DMSO-d6); δ 191.0, 168.6, 158.7, 140.7, 138.2, 132.3, 131.5, 129.8, 127.9, 127.5, 125.5, 122.9, 30.8, 18.6; MS (m/z); 283.
1-(4-Methyl-2-(4-(thiophen-2-yl)phenyl)thiazol-5-yl)ethan-1-one (15c): Following the general procedure (method B), compound 15c was obtained as a yellow oil (263 mg, 63%). 1H NMR (DMSO-d6) δ: 8.01 (d, J = 8 Hz, 2H), 7.70 (d, J = 8 Hz, 2H), 7.66–7.64 (m, 2H), 7.20–7.18 (m, 1H), 2.70 (s, 3H), 2.56 (s, 3H); 13C NMR (DMSO-d6); δ 191.0, 168.3, 158.7, 142.5, 136.8, 132.4, 131.3, 129.3, 127.7, 127.5, 126.4, 125.4, 30.8, 18.6; MS (m/z); 299.
1-(4-Methyl-2-(4-(thiophen-3-yl)phenyl)thiazol-5-yl)ethan-1-one (16c): Following the general procedure (method B), compound 16c was obtained as a yellow oil (210 mg, 50%). 1H NMR (DMSO-d6) δ: 8.02 (d, J = 8 Hz, 2H), 7.98 (s, 1H), 7.86 (d, J = 8 Hz, 2H), 7.68 (d, J = 8 Hz, 1H), 7.63 (d, J = 8 Hz, 1H), 2.7 (s, 3H), 2.55 (s, 3H); 13C NMR (DMSO-d6); δ 191.0, 168.4, 158.5, 140.7, 136.2, 132.5, 132.3, 131.1, 127.9, 127.2, 126.5, 122.9, 30.8, 18.6; MS (m/z); 299.
1-(4-Methyl-2-(4-(5-methylthiophen-3-yl)phenyl)thiazol-5-yl)ethan-1-one (17c): Following the general procedure (method B), compound 17c was obtained as a brown oil (180 mg, 41%). 1H NMR (DMSO-d6) δ: 8.01 (d, J = 8 Hz, 2H), 7.82 (d, J = 8 Hz, 2H), 7.77 (s, 1H), 7.33 (s, 1H), 2.71 (s, 3H), 2.57 (s, 3H), 2.39 (s, 3H); 13C NMR (DMSO-d6); δ 191.0, 168.6, 158.7, 141.0, 140.3, 138.4, 132.3, 130.9, 127.5, 126.9, 124.8, 120.8, 30.9, 18.6, 15.5; MS (m/z); 313.
1-(2-(4-(4,5-Dimethylthiophen-3-yl)phenyl)-4-methylthiazol-5-yl)ethan-1-one (18c): Following the general procedure (method B), compound 18c was obtained as a yellow oil (305 mg, 67%). 1H NMR (DMSO-d6) δ: 8.03 (d, J = 8 Hz, 2H), 7.52 (d, J = 8 Hz, 2H), 7.32 (s, 1H), 2.72 (s, 3H), 2.57 (s, 3H), 2.38 (s, 3H), 2.12 (s, 3H); 13C NMR (DMSO-d6); δ 191.0, 168.4, 158.5, 142.3, 140.5, 134.3, 132.5, 131.4, 131.0, 129.5, 127.0, 120.4, 30.8, 18.5, 13.9, 13.4; MS (m/z); 327.
5-[4-(5-Acetyl-4-methylthiazol-2-yl)phenyl]-N-methylthiophene-2-sulfonamide (19c): Following the general procedure (method B), compound 19c was obtained as a yellow oil (130 mg, 23%); 1H NMR (DMSO-d6) δ: 8.06 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.4 Hz, 2H), 7.79 (brs, 1H), 7.72 (d, J = 4 Hz, 1H), 7.62 (d, J = 4 Hz, 1H), 2.71 (s, 3H), 2.57 (s, 3H), 2.56 (s, 3H); 13C NMR (DMSO-d6) δ: 191.1, 167.9, 158.7, 148.0, 140.1, 135.2, 133.2, 132.9, 132.7, 127.8, 127.1, 125.6, 30.9, 29.3, 18.6; MS (m/z) 392.
5-[4-(5-Acetyl-4-methylthiazol-2-yl)phenyl]-N-ethylthiophene-2-sulfonamide (20c): Following the general procedure (method B), compound 20c was obtained as a yellow oil (150 mg, 27%); 1H NMR (DMSO-d6) δ: 8.06 (d, J = 8.4 Hz, 2H), 7.90 (brs, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 4 Hz, 1H), 7.61 (d, J = 4 Hz, 1H), 2.94 (m, 2H), 2.73 (s, 3H), 2.58 (s, 3H), 1.07 (t, J = 7.2 Hz, 3H); 13C NMR (DMSO-d6) δ: 191.1, 167.9, 158.7, 147.8, 141.5, 135.2, 132.96, 132.90, 132.6, 127.8, 127.1, 125.6, 38.3, 30.9, 18.6, 15.1; MS (m/z) 406.
5-[4-(5-Acetyl-4-methylthiazol-2-yl)phenyl]-N-isopropylthiophene-2-sulfonamide (21c): Following the general procedure (method B), compound 21c was obtained as a yellow oil (160 mg, 28%); 1H NMR (DMSO-d6) δ: 8.07 (d, J = 8.4 Hz, 2H), 7.95 (brs, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 4 Hz, 1H), 7.61 (d, J = 4 Hz, 1H), 3.46 (m, 1H), 2.72 (s, 3H), 2.58 (s, 3H), 1.07 (m, 6H); 13C NMR (DMSO-d6) δ: 191.1, 167.9, 158.7, 147.6, 142.9, 135.3, 132.89, 132.80, 132.6, 127.8, 127.0, 125.5, 46.2, 30.9, 23.6, 19.0; MS (m/z) 421.
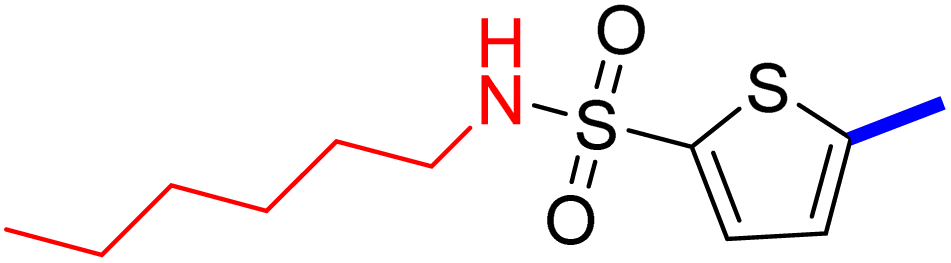
5-(4-(5-Acetyl-4-methylthiazol-2-yl)phenyl)-N-butylthiophene-2-sulfonamide (22c): Following the general procedure (method B), compound 22c was obtained as a yellow oil (175 g, 30%); 1H NMR (DMSO-d6) δ: 8.07 (d, J = 8.4 Hz, 2H), 7.92 (brs, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 4 Hz, 1H), 7.61 (d, J = 4 Hz, 1H), 2.92 (m, 2H), 2.73 (s, 3H), 2.58 (s, 3H), 1.43 (m, 2H), 1.31 (m, 2H), 0.84 (t, J = 7.2 Hz, 3H); 13C NMR (DMSO-d6) δ: 191.1, 167.9, 158.7, 147.8, 141.5, 135.2, 132.96, 132.90, 132.6, 127.8, 127.1, 125.6, 42.9, 31.4, 30.9, 19.7, 18.6, 13.9; MS (m/z) 434.
5-[4-(5-Acetyl-4-methylthiazol-2-yl)phenyl]-N-(pentan-2-yl)thiophene-2-sulfonamide (23c): Following the general procedure (method B), compound 23c was obtained as a yellow oil (134 mg, 22%); 1H NMR (DMSO-d6) δ: 8.08 (d, J = 8.4 Hz, 2H), 7.89 (brs, 1H), 7.87 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 4 Hz, 1H), 7.61 (d, J = 4 Hz, 1H), 3.30–3.20 (m, 1H), 2.72 (s, 3H), 2.58 (s, 3H), 1.33–1.21 (m, 4H), 0.99–0.97 (m, 3H), 0.79–0.77 (m, 3H); 13C NMR (DMSO-d6) δ: 191.3, 167.9, 158.7, 147.5, 143.2, 135.3, 132.9, 132.7, 132.6, 127.9, 127.0, 125.5, 49.8, 30.9, 21.5, 18.8, 18.6, 14.1; MS (m/z) 448.6.
5-[4-(5-Acetyl-4-methylthiazol-2-yl)phenyl]-N-hexylthiophene-2-sulfonamide (24c): Following the general procedure (method B), compound 24c was obtained as a yellow oil (144 mg, 22%); 1H NMR (DMSO-d6) δ: 8.06 (d, J = 8.4 Hz, 2H), 7.92 (brs, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 4 Hz, 1H), 7.60 (d, J = 4 Hz, 1H), 2.90 (q, J = 6.8 Hz, 2H), 2.72 (s, 3H), 2.58 (s, 3H), 1.45–1.38 (m, 2H), 1.28–1.16 (m, 6H), 0.83–0.80 (t, J = 6.4 Hz, 3H); 13C NMR (DMSO-d6) δ: 191.0, 167.9, 158.7, 147.8, 141.6, 135.2, 132.9, 132.6, 132.2, 127.8, 127.0, 125.5, 43.2, 31.2, 30.9, 29.2, 26.1, 22.4, 18.6, 14.3; MS (m/z) 462.11.
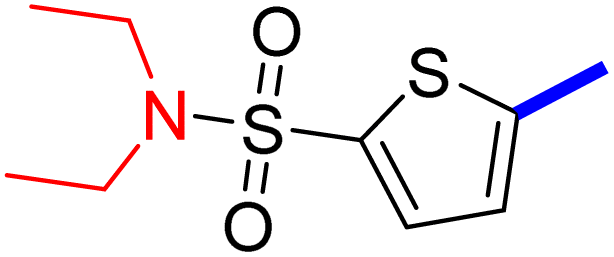
5-[4-(5-Acetyl-4-methylthiazol-2-yl)phenyl]-N,N-diethylthiophene-2-sulfonamide (25c): Following the general procedure (method B), compound 25c was obtained as a light yellow oil (280 mg, 46%); 1H NMR (DMSO-d6) δ: 8.06 (d, J = 8.4 Hz, 2H), 7.9 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 4 Hz, 1H), 7.69 (d, J = 4 Hz, 1H), 3.25 (q, J = 7.2 Hz, 4H), 2.72 (s, 3H), 2.58 (s, 3H), 1.14 (t, J = 7.2 Hz, 6H); 13C NMR (DMSO-d6) δ: 192.2, 167.9, 158.7, 148.2, 139.6, 135.1, 133.3, 132.9, 132.7, 127.8, 127.1, 125.8, 42.9, 30.9, 18.6, 14.6; MS (m/z) 434.
4.1.3.1 General procedure. Acetyl derivatives 3–25c (0.375 mmol) was dissolved in absolute ethanol (15 mL), then concentrated hydrochloric acid (0.5 mL) and aminoguanidine hydrochloride (0.75 mmol) were added. The reaction mixture was heated at reflux for 4 h. The solvent was concentrated under reduced pressure, then poured into crushed ice and neutralized with sodium carbonate to pH 7–8. The formed precipitated solid was collected by filtration, and washed with a copious amount of water. Crystallization from dichloromethane afforded the desired products.
2-(1-(2-(4-Isobutylphenyl)-4-methylthiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (3d): Light-brown solid (90 mg, 73%); mp 117–119 °C; 1H NMR (DMSO-d6) δ: 8.01 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 5.70 (brs, 2H), 5.50 (brs, 2H), 2.63 (s, 3H), 2.52 (d, J = 8.4 Hz, 2H), 2.23 (s, 3H), 1.90–1.85 (m, 1H), 0.91 (d, J = 8.4 Hz, 6H); 13C NMR (DMSO-d6); δ 160.2, 155.4, 153.4, 148.5, 137.0, 135.2, 129.7, 126.6, 117.2, 44.8, 30.1, 25.28, 22.6, 17.5; HPLC purity 95.1% (acetonitrile-3% TEA, 1:4).
2-(1-(2-(4-Cyclopropylphenyl)-4-methylthiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (4d): Light-brown solid (87 mg, 74%); mp 121–123 °C; 1H NMR (DMSO-d6) δ: 7.98 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 5.71 (brs, 2H), 5.50 (brs, 2H), 2.62 (s, 3H), 2.23 (s, 3H), 1.99–1.94 (m, 1H), 1.01–0.97 (t, J = 8.4 Hz, 2H), 0.74–0.72 (t, J = 8.4 Hz, 2H); 13C NMR (DMSO-d6); δ 160.2, 155.4, 153.4, 148.6, 145.0, 137.0, 136.1, 126.7, 126.0, 25.2, 17.5, 15.4, 10.1; HPLC purity 99% (acetonitrile-3% TEA, 1:4).
2-(1-(4-Methyl-2-(4′-methyl-[1,1′-biphenyl]-4-yl)thiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (5d): Beige solid (90 mg, 76%); mp 133–135 °C; 1H NMR (DMSO-d6) δ: 7.97 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.2 Hz, 2H), 5.78 (brs, 2H), 5.71 (brs, 2H), 2.61 (s, 3H), 2.36 (s, 3H), 2.33 (s, 3H); 13C NMR (DMSO-d6); δ 160.3, 155.6, 152.9, 148.5, 140.7, 137.7, 137.1, 137.0, 135.4, 130.0, 127.3, 126.8, 117.4, 25.3, 21.1, 17.5; HPLC purity 100% (acetonitrile-3% TEA, 1:4).
2-(1-(4-Methyl-2-(3′-methyl-[1,1′-biphenyl]-4-yl)thiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (6d): Beige solid (97 mg, 71%); mp 137–139 °C; 1H NMR (DMSO-d6) δ: 8.20 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.4 Hz, 2H), 7.56 (s, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.40 (t, J = 8.2 Hz, 1H), 7.22 (d, J = 8.2 Hz, 1H), 5.82 (brs, 2H), 5.71 (brs, 2H), 2.67 (s, 3H), 2.51 (s, 3H), 2.26 (s, 3H); 13C NMR (DMSO-d6); δ 160.1, 155.6, 153.0, 148.7, 140.9, 140.0, 138.1, 137.9, 137.1, 135.4, 129.3, 128.7, 127.7, 124.2, 117.4, 25.3, 21.6, 17.5; HPLC purity 99.5% (acetonitrile-3% TEA, 1:4).
2-(1-(2-(3′-Methoxy-[1,1′-biphenyl]-4-yl)-4-methylthiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (7d): Yellow solid (80 mg, 68%) mp = 125–127 °C; 1H NMR (DMSO-d6) δ: 7.98 (d, J = 8 Hz, 2H), 7.80 (d, J = 8 Hz, 2H), 7.41 (t, J = 8 Hz, 1H), 7.31 (d, J = 8 Hz, 1H), 7.26 (s, 1H), 6.99 (d, J = 8 Hz, 1H), 5.78 (brs, 2H), 5.67 (brs, 2H), 3.84 (s, 3H), 2.61 (s, 3H), 2.33 (s, 3H); 13C NMR (DMSO-d6) δ: 162.7, 160.2, 160.1, 140.4, 143.1, 141.5, 141.1, 135.9, 132.8, 130.5, 127.8, 126.6, 119.4, 114.0, 112.5, 55.6, 18.7, 16.5; MS (m/z) 379; HPLC purity 93% (acetonitrile-3% TEA, 1:4).
2-(1-(2-(2′-Methoxy-[1,1′-biphenyl]-4-yl)-4-methylthiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (8d): Yellow solid (77 mg, 65%) mp = 130–132 °C; 1H NMR (DMSO-d6) δ: 7.94 (d, J = 8 Hz, 2H), 7.60 (d, J = 8 Hz, 2H), 7.40–7.38 (m, 2H), 7.15 (d, J = 8 Hz, 1H), 7.08 (t, J = 8 Hz, 1H), 5.98 (brs, 4H), 3.79 (s, 3H), 2.61 (s, 3H), 2.33 (s, 3H); 13C NMR (DMSO-d6) δ: 162.7, 159.7, 156.6, 148.8, 143.6, 140.1, 135.2, 132.1, 130.6, 130.4, 129.8, 129.3, 125.8, 121.3, 112.3, 56.0, 18.6, 16.7; MS (m/z) 379; HPLC purity 90.9% (acetonitrile-3% TEA, 1:4).
Methyl-4′-(5-(1-(2-carbamimidoylhydrazono)ethyl)-4-methylthiazol-2-yl)-[1,1′-biphenyl]-2-carboxylate (9d): Yellow solid (75 mg, 64%) mp = 136 °C; 1H NMR (DMSO-d6) δ: 7.96 (d, J = 8 Hz, 2H), 7.80 (d, J = 8 Hz, 1H), 7.68 (t, J = 8 Hz, 1H), 7.54–7.51 (m, 2H), 7.41 (d, J = 8 Hz, 2H), 6.22 (brs, 4H), 3.63 (s, 3H), 2.61 (s, 3H), 2.35 (s, 3H); 13C NMR (DMSO-d6) δ: 173.1, 168.8, 162.6, 159.5, 149.2, 143.8, 142.4, 140.9, 132.5, 132.1, 131.1, 130.8, 129.9, 129.4, 126.0, 52.4, 18.6, 16.9; MS (m/z) 407; HPLC purity 95.7% (acetonitrile-3% TEA, 1:4).
2-(1-(4-Methyl-2-(4-(naphthalen-2-yl)phenyl)thiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (10d): Yellow solid (100 mg, 67%); mp 141–143 °C; 1H NMR (DMSO-d6) δ: 8.30 (s, 1H), 8.25 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 8.4 Hz, 2H), 7.83 (t, J = 8.4 Hz, 2H), 7.78 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 5.84 (brs, 2H), 5.69 (brs, 2H), 2.69 (s, 3H), 2.26 (s, 3H); 13C NMR (DMSO-d6); δ 160.2, 155.7, 152.9, 148.7, 140.5, 138.1, 137.3, 135.5, 133.8, 132.8, 129.0, 128.7, 127.9, 126.7, 125.4, 117.5, 25.3, 17.5; HPLC purity 100% (acetonitrile-3% TEA, 1:4).
2-(1-(2-(4-(Benzofuran-2-yl)phenyl)-4-methyl-thiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (11d): Yellow solid (90 mg, 62%); mp 136–138 °C; 1H NMR (DMSO-d6) δ: 8.26 (d, J = 8.4 Hz, 2H), 8.04 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.68 (s, 1H), 7.57 (t, J = 8.4 Hz, 1H), 7.35 (t, J = 8.2 Hz, 1H), 5.81 (brs, 2H), 5.62 (brs, 2H), 2.67 (s, 3H), 2.25 (s, 3H); 13C NMR (DMSO-d6); δ 160.2, 155.3, 154.8, 152.5, 139.1, 137.1, 136.1, 135.7, 130.3, 129.3, 127.4, 125.2, 123.8, 121.7, 117.6, 111.6, 103.0, 25.3, 17.5; HPLC purity 98.8% (acetonitrile-3% TEA, 1:4).
2-(1-(2-(4-(Benzo[b]thiophen-2-yl)phenyl)-4-methylthiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (12d): Yellow solid (96 mg, 63%); mp 132–134 °C; 1H NMR (DMSO-d6) δ: 8.21 (s, 1H), 8.01–7.78 (m, 8H), 5.77 (brs, 2H), 5.60 (brs, 2H), 2.63 (s, 3H), 2.35 (s, 3H); 13C NMR (DMSO-d6); δ 160.2, 155.7, 152.5, 148.5, 143.2, 140.9, 139.1, 137.1, 135.7, 134.1, 127.5, 126.8, 125.3, 124.3, 120.8, 117.5, 25.3, 17.5; HPLC purity 97.6% (acetonitrile-3% TEA, 1:4).
2-(1-(2-(4-(Furan-2-yl)phenyl)-4-methylthiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (13d): Beige solid (90 mg, 71%); mp 122–124 °C; 1H NMR (DMSO-d6) δ: 7.94 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 7.19 (t, J = 6.2 Hz, 1H), 5.78 (brs, 2H), 5.67 (brs, 2H), 2.61 (s, 3H), 2.33 (s, 3H); 13C NMR (DMSO-d6); δ 162.3, 160.1, 148.3, 145.0, 143.1, 140.5, 135.6, 133.8, 132.1, 126.6, 126.5, 125.7, 109.0, 18.6, 16.5; HPLC purity 96.5% (acetonitrile-3% TEA, 1:4).
2-(1-(2-(4-(Furan-3-yl)phenyl)-4-methylthiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (14d): Beige solid (84 mg, 66%); mp 120–122 °C; 1H NMR (DMSO-d6) δ: 8.30 (s, 1H), 7.92 (d, J = 8.2 Hz, 2H), 7.78 (d, J = 8.2 Hz, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 6.4 Hz, 1H), 5.77 (brs, 2H), 5.66 (brs, 2H), 2.60 (s, 3H), 2.32 (s, 3H); 13C NMR (DMSO-d6); δ 162.3, 160.1, 148.3, 145.0, 143.1, 140.5, 135.6, 133.8, 132.1, 126.6, 126.5, 125.7, 109.0, 18.6, 16.5; HPLC purity 97% (acetonitrile-3% TEA, 1:4).
2-(1-(4-Methyl-2-(4-(thiophen-2-yl)phenyl)thiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (15d): Yellow solid (90 mg, 76%) mp = 120–122 °C; 1H NMR (DMSO-d6) δ: 7.94 (d, J = 8 Hz, 2H), 7.76 (d, J = 8 Hz, 2H), 7.63–7.60 (m, 2H), 7.19–7.17 (m, 1H), 5.79 (brs, 2H), 5.67 (brs, 2H), 2.60 (s, 3H), 2.32 (s, 3H); 13C NMR (DMSO-d6) δ: 160.2, 158.1, 148.4, 142.9, 135.9, 133.6, 131.9, 129.2, 126.8, 126.3, 124.9, 18.6, 16.5; MS (m/z) 355; HPLC purity 96.2% (acetonitrile-3% TEA, 1:4).
2-(1-(4-Methyl-2-(4-(thiophen-3-yl)phenyl)thiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (16d): Yellow solid (102 mg, 88%) mp = 123–125 °C; 1H NMR (DMSO-d6) δ: 7.97 (s, 1H), 7.93 (d, J = 8 Hz, 2H), 7.83 (d, J = 8 Hz, 2H), 7.67 (d, J = 8 Hz, 1H), 7.63 (d, J = 8 Hz, 1H), 5.79 (brs, 2H), 5.67 (brs, 2H), 2.60 (s, 3H), 2.32 (s, 3H); 13C NMR (DMSO-d6) δ: 162.2, 160.1, 148.3, 143.1, 141.0, 136.7, 135.7, 132.2, 127.8, 127.0, 126.6, 126.5, 122.2, 18.6, 16.5; MS (m/z) 355; HPLC purity 94.7% (acetonitrile-3% TEA, 1:4).
(1-(4-Methyl-2-(4-(5-methylthiophen-3-yl)phenyl)thiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (17d): Yellow solid (105 mg, 89%) mp = 120–122 °C; 1H NMR (DMSO-d6) δ: 7.91 (d, J = 8 Hz, 2H), 7.78 (d, J = 8 Hz, 2H), 7.71 (s, 1H), 7.32 (s, 1H), 5.79 (brs, 2H), 5.68 (brs, 2H), 2.60 (s, 3H), 2.42 (s, 3H), 2.32 (s, 3H); 13C NMR (DMSO-d6) δ: 162.2, 160.2, 148.3, 143.0, 140.8, 140.6, 136.9, 135.7, 132.1, 126.8, 126.6, 124.8, 120.0, 18.6, 16.5, 15.5; MS (m/z) 369; HPLC purity 97% (acetonitrile-3% TEA, 1:4).
2-(1-(2-(4-(4,5-Dimethylthiophen-3-yl)phenyl)-4-methylthiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (18d): Yellow solid (106 mg, 90%) mp = 123–125 °C; 1H NMR (DMSO-d6) δ: 7.94 (d, J = 8 Hz, 2H), 7.49 (d, J = 8 Hz, 2H), 7.29 (s, 1H), 5.78 (brs, 2H), 5.69 (brs, 2H), 2.60 (s, 3H), 2.38 (s, 3H), 2.32 (s, 3H), 2.12 (s, 3H); 13C NMR (DMSO-d6) δ: 162.3, 160.1, 148.4, 143.1, 142.6, 138.9, 135.7, 134.1, 132.1, 131.4, 129.4, 126.2, 119.9, 18.6, 16.5, 13.9, 13.5; MS (m/z) 383; HPLC purity 98.9% (acetonitrile-3% TEA, 1:4).
2-{1-[4-Methyl-2-(4-(5-(N-methylsulfamoyl)thiophen-2-yl)phenyl)thiazol-5-yl]ethylidene}hydrazine-1-carboximidamide (19d): Yellow solid (80 mg, 70%) mp = 129–131 °C; 1H NMR (DMSO-d6) δ: 7.97 (d, J = 8.4 Hz, 2H), 7.84 (d, J = 8.4 Hz, 2H), 7.71 (brs, 1H), 7.68 (d, J = 4 Hz, 1H), 7.60 (d, J = 4 Hz, 1H), 5.78 (brs, 2H), 5.67 (brs, 2H), 2.60 (s, 3H), 2.56 (s, 3H), 2.32 (s, 3H); 13C NMR (DMSO-d6) δ: 161.5, 160.2, 148.5, 148.4, 142.9, 139.6, 136.4, 133.9, 133.6, 133.2, 126.98, 126.94, 125.0, 29.3, 18.6, 16.4; MS (m/z) 448.5; HPLC purity 95.9% (acetonitrile-3% TEA, 1:4).
2-(1-(2-(4-(5-(N-Ethylsulfamoyl)thiophen-2-yl)phenyl)-4-methylthiazol-5-yl)ethylidene)hydrazine-1-carboximidamide (20d): Yellow solid (83 mg, 73%) mp = 130–132 °C; 1H NMR (DMSO-d6) δ: 7.97 (d, J = 8.4 Hz, 2H), 7.90 (brs, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 4 Hz, 1H), 7.60 (d, J = 4 Hz, 1H), 5.89 (brs, 4H), 2.98 (q, J = 6.8 Hz, 2H), 2.60 (s, 3H), 2.33 (s, 3H), 1.06 (t, J = 6.8 Hz, 3H); 13C NMR (DMSO-d6) δ: 161.7, 159.9, 148.9, 148.3, 143.3, 140.9, 136.0, 133.8, 133.7, 132.9, 126.9, 126.6, 125.0, 38.3, 18.6, 16.6, 15.1; MS (m/z) 462.6; HPLC purity 98.9% (acetonitrile-3% TEA, 1:4).
2-{1-[2-(4-(5-(N-Isopropylsulfamoyl)thiophen-2-yl)phenyl)-4-methylthiazol-5-yl]ethylidene}hydrazine-1-carboximidamide (21d): Yellow solid (77 mg, 68%) mp = 133–135 °C; 1H NMR (DMSO-d6) δ: 7.97 (d, J = 8.4 Hz, 2H), 7.91 (brs, 1H), 7.84 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 4 Hz, 1H), 7.60 (d, J = 4 Hz, 1H), 5.84 (brs, 2H), 5.73 (brs, 2H), 3.43–3.39 (m, 1H), 2.60 (s, 3H), 2.31 (s, 3H), 1.04 (d, J = 6.4 Hz, 6H); 13C NMR (DMSO-d6) δ: 161.5, 160.3, 148.5, 148.1, 142.8, 142.3, 136.4, 133.8, 133.7, 132.8, 127.2, 126.9, 124.9, 46.1, 23.6, 18.6, 16.4; MS (m/z) 476.6; HPLC purity 91% (acetonitrile-3% TEA, 1:4).
2-{1-[2-(4-(5-(N-Butylsulfamoyl)thiophen-2-yl)phenyl)-4-methylthiazol-5-yl]ethylidene}hydrazine-1-carboximidamide (22d): Yellow solid (79 mg, 70%) mp = 136–138 °C; 1H NMR (DMSO-d6) δ: 7.97 (d, J = 8.4 Hz, 2H), 7.90 (brs, 1H), 7.84 (d, J = 8.4 Hz, 2H), 7.67 (d, J = 4 Hz, 1H), 7.60 (d, J = 4 Hz, 1H), 5.86 (brs, 4H), 2.90 (t, J = 6.4 Hz, 2H), 2.60 (s, 3H), 2.33 (s, 3H), 1.43 (m, 2H), 1.31 (m, 2H), 0.85 (t, J = 7.2 Hz, 3H); 13C NMR (DMSO-d6) δ: 161.6, 159.9, 148.8, 148.2, 143.2, 141.0, 136.1, 133.8, 133.7, 132.9, 127.2, 126.9, 125.4, 42.9, 31.4, 19.7, 18.6, 16.6, 13.9; MS (m/z) 490; HPLC purity 95.4% (acetonitrile-3% TEA, 1:4).
2-{1-[4-Methyl-2-(4-(5-(N-(pentan-2-yl)sulfamoyl)thiophen-2-yl)phenyl)thiazol-5-yl]ethylidene}hydrazine-1-carboximidamide (23d): Yellow solid (83 mg, 73%) mp = 136–138 °C; 1H NMR (DMSO-d6) δ: 7.97 (d, J = 8.4 Hz, 2H), 7.89 (brs, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 4 Hz, 1H), 7.59 (d, J = 4 Hz, 1H), 5.83 (brs, 4H), 3.37–3.28 (m, 1H), 2.60 (s, 3H), 2.32 (s, 3H), 1.33–1.21 (m, 4H), 0.98–0.97 (m, 3H), 0.79–0.76 (m, 3H); 13C NMR (DMSO-d6) δ: 161.6, 160.0, 148.7, 148.0, 143.2, 142.6, 136.2, 133.8, 133.7, 132.7, 126.95, 126.92, 124.9, 49.8, 42.6, 21.5, 18.8, 18.6, 16.5, 14.1; MS (m/z) 504.6; HPLC purity 98.13% (acetonitrile-3% TEA, 1:4).
2-{1-[2-(4-(5-(N-Hexylsulfamoyl)thiophen-2-yl)phenyl)-4-methylthiazol-5-yl]ethylidene}hydrazine-1-carboximidamide (24d): Yellow solid (87 mg, 77%) mp = 139–141 °C; 1H NMR (DMSO-d6) δ: 7.97 (d, J = 8.4 Hz, 2H), 7.88 (brs, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 4 Hz, 1H), 7.59 (d, J = 4 Hz, 1H), 5.79 (brs, 2H), 5.71 (brs, 2H), 2.89 (t, J = 8 Hz, 2H), 2.60 (s, 3H), 2.32 (s, 3H), 1.43–1.37 (m, 2H), 1.28–1.16 (m, 6H), 0.83–0.80 (m, 3H); 13C NMR (DMSO-d6) δ: 161.5, 160.2, 148.5, 148.2, 143.0, 141.0, 136.3, 133.8, 133.7, 132.9, 126.98, 126.94, 124.9, 43.2, 31.2, 29.2, 26.1, 22.4, 18.6, 16.5, 14.3; MS (m/z) 518.7; HPLC purity 97.7% (acetonitrile-3% TEA, 1:4).
2-{1-[2-(4-(5-(N,N-Diethylsulfamoyl)thiophen-2-yl)phenyl)-4-methylthiazol-5-yl]ethylidene}hydrazine-1-carboximidamide (25d): Yellow solid (80 mg, 70%) mp = 135–137 °C; 1H NMR (DMSO-d6) δ: 7.97 (d, J = 8.4 Hz, 2H), 7.85 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 4 Hz, 1H), 7.67 (d, J = 4 Hz, 1H), 5.78 (brs, 2H), 5.7 (brs, 2H), 3.25 (q, J = 7.2 Hz, 4H), 2.60 (s, 3H), 2.32 (s, 3H), 1.39 (t, J = 7.2 Hz, 6H); 13C NMR (DMSO-d6) δ: 161.4, 160.2, 148.7, 148.5, 143.0, 139.0, 136.4, 133.9, 133.5, 133.3, 127.0, 126.9, 125.2, 42.9, 18.6, 16.5, 14.6; MS (m/z) 490.6; HPLC purity 95.7% (acetonitrile-3% TEA, 1:4).
4.2. Biological assays
Conflicts of interest
All authors declare that they have no conflict of interest.Acknowledgements
This paper is based on work supported by Science, Technology & Innovation Funding Authority (STDF) under grant number (43229) young researcher.References
- G. M. Knight, R. E. Glover, C. F. McQuaid, I. D. Olaru, K. Gallandat, Q. J. Leclerc, N. M. Fuller, S. J. Willcocks, R. Hasan and E. van Kleef, Elife, 2021, 10, e64139 CrossRef CAS PubMed.
- C. L. Ventola, Pharm. Ther., 2015, 40, 277 Search PubMed.
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao and E. Wool, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- P. Mazumder, A. Kalamdhad, G. T. Chaminda and M. Kumar, Case Stud. Chem. Environ. Eng., 2021, 3, 100093 CrossRef CAS.
- B. J. Langford, M. So, V. Leung, S. Raybardhan, J. Lo, T. Kan, F. Leung, D. Westwood, N. Daneman and D. R. MacFadden, Clin. Microbiol. Infect., 2022, 28, 491–501 CrossRef CAS PubMed.
- R. J. Fair and Y. Tor, Perspect. Med. Chem., 2014, 6, S14459 CrossRef PubMed.
- B. Tornimbene, S. Eremin, M. Escher, J. Griskeviciene, S. Manglani and C. L. Pessoa-Silva, Lancet Infect. Dis., 2018, 18, 241–242 CrossRef PubMed.
- E. F. Kong, J. K. Johnson and M. A. Jabra-Rizk, PLoS Pathog., 2016, 12, e1005837 CrossRef PubMed.
- P. Rodríguez-López, V. Filipello, P. A. Di Ciccio, A. Pitozzi, S. Ghidini, F. Scali, A. Ianieri, E. Zanardi, M. N. Losio and A. C. Simon, Foods, 2020, 9, 1141 CrossRef PubMed.
- R. H. Deurenberg, C. Vink, S. Kalenic, A. Friedrich, C. Bruggeman and E. Stobberingh, Clin. Microbiol. Infect., 2007, 13, 222–235 CrossRef CAS PubMed.
- G. J. Moran, A. Krishnadasan, R. J. Gorwitz, G. E. Fosheim, L. K. McDougal, R. B. Carey and D. A. Talan, N. Engl. J. Med., 2006, 355, 666–674 CrossRef CAS PubMed.
- B. W. Frazee, J. Lynn, E. D. Charlebois, L. Lambert, D. Lowery and F. Perdreau-Remington, Ann. Emerg. Med., 2005, 45, 311–320 CrossRef PubMed.
- S. K. Fridkin, J. C. Hageman, M. Morrison, L. T. Sanza, K. Como-Sabetti, J. A. Jernigan, K. Harriman, L. H. Harrison, R. Lynfield and M. M. Farley, N. Engl. J. Med., 2005, 352, 1436–1444 CrossRef CAS PubMed.
- S. Kishore, D. Verma and M. Siddique, J. Clin. Diagn. Res., 2014, 8, DC12 CAS.
- H. T. Nour El-Din, A. S. Yassin, Y. M. Ragab and A. M. Hashem, Infect. Drug Resist., 2021, 1557–1571 CrossRef PubMed.
- A. W. Karchmer and A. S. Bayer, Clin. Infect. Dis., 2008, 46, S342–S343 CrossRef PubMed.
- H. Mohammad, A. S. Mayhoub, A. Ghafoor, M. Soofi, R. A. Alajlouni, M. Cushman and M. N. Seleem, J. Med. Chem., 2014, 57, 1609–1615 CrossRef CAS PubMed.
- M. M. Elsebaei, H. Mohammad, A. Samir, N. S. Abutaleb, A. B. Norvil, A. R. Michie, M. M. Moustafa, H. Samy, H. Gowher and M. N. Seleem, Eur. J. Med. Chem., 2019, 175, 49–62 CrossRef CAS PubMed.
- N. Brown, Mol. Inf., 2014, 33, 458–462 CrossRef CAS PubMed.
- K. Okamoto, A. Ishikawa, R. Okawa, K. Yamamoto, T. Sato, S.-i. Yokota, K. Chiba and S. Ichikawa, Bioorg. Med. Chem., 2022, 55, 116556 CrossRef CAS PubMed.
- M. M. Elsebaie, H. T. N. El-Din, N. S. Abutaleb, A. A. Abuelkhir, H.-W. Liang, A. S. Attia, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2022, 234, 114204 CrossRef CAS PubMed.
- A. M. Sayed, N. S. Abutaleb, A. Kotb, H. G. Ezzat, M. N. Seleem, A. S. Mayhoub and M. M. Elsebaie, J. Heterocycl. Chem., 2023, 60, 134–144 CrossRef CAS.
- M. A. Seleem, A. M. Disouky, H. Mohammad, T. M. Abdelghany, A. S. Mancy, S. A. Bayoumi, A. Elshafeey, A. El-Morsy, M. N. Seleem and A. S. Mayhoub, J. Med. Chem., 2016, 59, 4900–4912 CrossRef CAS PubMed.
- H. Mohammad, P. N. Reddy, D. Monteleone, A. S. Mayhoub, M. Cushman and M. N. Seleem, Eur. J. Med. Chem., 2015, 94, 306–316 CrossRef CAS PubMed.
- M. M. Elsebaei, N. S. Abutaleb, A. A. Mahgoub, D. Li, M. Hagras, H. Mohammad, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2019, 182, 111593 CrossRef PubMed.
- G. A. Molander, S. L. Trice and S. M. Kennedy, J. Org. Chem., 2012, 77, 8678–8688 CrossRef CAS PubMed.
- J. Yu, A. Ciancetta, S. Dudas, S. Duca, J. Lottermoser and K. A. Jacobson, J. Med. Chem., 2018, 61, 4860–4882 CrossRef CAS PubMed.
- M. Carrel, E. N. Perencevich and M. Z. David, Emerging Infect. Dis., 2015, 21, 1973 CrossRef PubMed.
- G. R. Golding, P. N. Levett, R. R. McDonald, J. Irvine, B. Quinn, M. Nsungu, S. Woods, M. Khan, M. Ofner-Agostini and M. R. Mulvey, Emerging Infect. Dis., 2011, 17, 722 CrossRef PubMed.
- U. Seybold, E. V. Kourbatova, J. G. Johnson, S. J. Halvosa, Y. F. Wang, M. D. King, S. M. Ray and H. M. Blumberg, Clin. Infect. Dis., 2006, 42, 647–656 CrossRef CAS PubMed.
- B. A. Diep, H. A. Carleton, R. F. Chang, G. F. Sensabaugh and F. Perdreau-Remington, J. Infect. Dis., 2006, 193, 1495–1503 CrossRef CAS PubMed.
- P. Peters, J. Brooks, B. Limbago, H. Lowery, S. McAllister, R. Mindley, G. Fosheim, R. Gorwitz, J. Guest and J. Hageman, Epidemiol. Infect., 2011, 139, 998–1008 CrossRef CAS PubMed.
- G. Zhen, G. Zeng, K. Jiang, F. Wang, X. Cao and B. Yin, Chem.–Eur. J., 2023, 29, e202203217 CrossRef CAS PubMed.
- A. U. Meyer, A. L. Berger and B. König, Chem. Commun., 2016, 52, 10918–10921 RSC.
- L. Zhou, T. Kawate, X. Liu, Y. B. Kim, Y. Zhao, G. Feng, J. Banerji, H. Nash, C. Whitehurst and S. Jindal, Bioorg. Med. Chem., 2012, 20, 750–758 CrossRef CAS PubMed.
- M. A. Wikler, CLSI (NCCLS), 2006, 26, pp. M7–A7 Search PubMed.
- N. S. Abutaleb, A. Elkashif, D. P. Flaherty and M. N. Seleem, Antimicrob. Agents Chemother., 2021, 65, e01715–e01720 CrossRef CAS PubMed.
- N. S. Abutaleb and M. N. Seleem, Antimicrob. Agents Chemother., 2020, 64, e02115–e02119 CrossRef PubMed.
- N. S. Abutaleb and M. N. Seleem, Sci. Rep., 2020, 10, 1–8 CrossRef PubMed.
- N. S. Abutaleb, A. E. Elhassanny, D. P. Flaherty and M. N. Seleem, PeerJ, 2021, 9, e11059 CrossRef PubMed.
- N. S. Abutaleb and M. N. Seleem, Int. J. Antimicrob. Agents, 2020, 55, 105828 CrossRef CAS PubMed.
- N. S. Abutaleb, A. E. Elhassanny, A. Nocentini, C. S. Hewitt, A. Elkashif, B. R. Cooper, C. T. Supuran, M. N. Seleem and D. P. Flaherty, J. Enzyme Inhib. Med. Chem., 2022, 37, 51–61 CrossRef CAS PubMed.
- H. Mohammad, W. Younis, H. G. Ezzat, C. E. Peters, A. AbdelKhalek, B. Cooper, K. Pogliano, J. Pogliano, A. S. Mayhoub and M. N. Seleem, PLoS One, 2017, 12, e0182821 CrossRef PubMed.
- C. W. Tseng, M. Sanchez-Martinez, A. Arruda and G. Y. Liu, J. Vis. Exp., 2011, 48, 2528 Search PubMed.
- M. Hagras, N. S. Abutaleb, N. M. Elhosseiny, T. M. Abdelghany, M. Omara, M. M. Elsebaei, M. Alhashimi, A. B. Norvil, M. I. Gutay and H. Gowher, ACS Infect. Dis., 2020, 6, 2887–2900 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02778c |
| This journal is © The Royal Society of Chemistry 2023 |