
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophilic ionic liquid assisted hydrothermal synthesis of ZnO nanostructures with controllable morphology†
Mousumi Aktera,
Md. Arif Faisala,
Ajaya Kumar Singh b and
Md. Abu Bin Hasan Susan*ac
b and
Md. Abu Bin Hasan Susan*ac
aDepartment of Chemistry, University of Dhaka, Dhaka 1000, Bangladesh. E-mail: susan@du.ac.bd; Fax: +880 255167810; Fax: +880 9666911463; Tel: ext. 7162
bDepartment of Chemistry, Govt. V. Y. T. PG. Autonomous, College, Durg, Chhattisgarh 491001, India
cDhaka University Nanotechnology Center (DUNC), University of Dhaka, Dhaka 1000, Bangladesh
First published on 13th June 2023
Abstract
Nanostructured ZnO with controllable morphology was prepared by a hydrothermal method in the presence of three different hydrophilic ionic liquids (ILs), 1-ethyl-3-methylimidazolium methylsulfate, ([C2mim]CH3SO4), 1-butyl-3-methylimidazolium methylsulfate, ([C4mim]CH3SO4) and 1-ethyl-3-methylimidazolium ethylsulfate, ([C2mim]C2H5SO4) as soft templates. The formation of ZnO nanoparticles (NPs) with and without IL was verified using FT-IR and UV-visible spectroscopy. X-ray diffraction (XRD) and selected area electron diffraction (SAED) patterns indicated the formation of pure crystalline ZnO with a hexagonal wurtzite phase. Field emission scanning electron microscopic (FESEM) and high-resolution transmission electron microscopic (HRTEM) images confirmed the formation of rod-shaped ZnO nanostructures without using IL, whereas the morphology varied widely following addition of ILs. With increasing concentrations of [C2mim]CH3SO4, the rod-shaped ZnO nanostructures transformed into flower-shaped nanostructures whereas with rising concentrations of [C4mim]CH3SO4 and [C2mim]C2H5SO4 the morphology changed into petal- and flake-like nanostructures, respectively. The selective adsorption effect of the ILs could protect certain facets during the formation of ZnO rods and promote the growth in directions other than [0001] to yield petal- or flake-like architectures. The morphology of ZnO nanostructures was, therefore, tunable by the controlled addition of hydrophilic ILs of different structures. The size of the nanostructures was widely distributed and the Z-average diameter, evaluated from dynamic light scattering measurements, increased as the concentration of the IL increased and passed through a maximum before decreasing again. The optical band gap energy of the ZnO nanostructures decreased when IL was added during the synthesis which is consistent with the morphology of the ZnO nanostructures. Thus, the hydrophilic ILs serve as self-directing agents and soft templates for the synthesis of ZnO nanostructures and the morphology and optical properties of ZnO nanostructures are tunable by changing the structure of the ILs as well as systematic variation of the concentration of ILs during synthesis.
1. Introduction
It is truly the tiny things that make the giant world so incredible. Nanochemistry has made this saying stronger by presenting a unique approach to building devices with molecular scale precision.1–6 In particular, due to their specific characteristics that depend on shape and size, metal oxides in the nanodimension have recently received a surge of interest. Among the nanostructured metal oxides, ZnO has been one of the most auspicious materials for its vast utilization in different sectors.7–13 ZnO is a semiconductor with a significant free exciton binding energy of about 60 MeV and a wide band gap of about 3.37 eV.14–17 It is a significant functional material that has very wide applications in electronics, photo-electronics, optics, sensors, and catalysis. ZnO nanoparticles (NPs) are an effective adsorbent for the elimination of numerous hazardous compounds as well as dyes, and they have shown promise for short wavelength optoelectronic applications.18–21 For storage and processing information in magnetic devices, ZnO NPs are required.21–23 High reactivity associated with a greater surface to volume ratio and different morphology help ZnO NPs play a vital role in suppressing bacterial growth in aqueous and solid media.24–26Nanostructured ZnO has been found useful in sensors, nanogenerators, and photovoltaics. Interestingly, ZnO can be prepared in a large variety of conceivable nanostructures. The large number of attainable morphologies combined with the wide range of physical characteristics of nanostructured ZnO serves up intriguing prospects for tailoring ZnO nanostructures to construct more intelligent electronics. Physical properties may be modulated via control of morphology by implementing various conditions under which the nanostructured materials are synthesized. Major nanostructures so far observed are nanospheres, nanowires, tetrapods, nanorods, nanobelts, nanoflowers, nanoplates, nanopyramids, nanotubes, quantum dots, etc.27–34 Involvement of elevated temperature or prolonged treatment is a requisite and can be achieved by hydrothermal,35,36 thermal decomposition,37–39 microemulsion,40 sol–gel,41,42 electrochemical–thermal,43 chemical vapour deposition,44 and precipitation techniques.45 However, the hydrothermal method offers several advantages, such as a comparatively mild reaction condition, environmental friendliness, a one-step synthetic route, and a wide temperature and pressure range. Specifically, when the task is to control the morphology of nanomaterials, the hydrothermal method provides better tuning of temperature and pressure so that control over nanostructures can be easily achieved. Therefore, the composition and morphology can be well tuned through a liquid phase or multiphase chemical reactions in the hydrothermal route. Consequently, the hydrothermal method has been preferred by researchers over the other synthetic procedures for the synthesis of ZnO NPs with controllable morphologies.
It is still difficult to synthesize ZnO in nanodimension with variable morphology on large-scale using a simple yet effective and practical manner. So far, capping agents, including starch, ligands, poly(vinyl pyrrolidone), poly(vinyl alcohol), and molecular solvents have been used.46,47 Often polymeric materials are found to be difficult to be removed from the surface of the synthesized ZnO NPs. It also causes ruptures in the shape of the nanostructures which damages the target NPs. Soft templates were thus critically sought for and ionic liquids (ILs) were introduced for the synthesis of ZnO NPs.48–59 ILs, salts comprising only ions, remain as liquid below 100 °C or even at room temperature. The unique properties that suggest template synthesis are good dissolving ability, good thermal stability, low vapor pressure, highly ordered structure in the liquid state, and recyclability of ILs. ILs serve as a better template or capping agent for the synthesis of ZnO NPs with novel structures, special shapes, and improved properties, which can be further controlled by adjusting the concentration of the IL. The cationic or anionic components of ILs, also otherwise called “designer salts”, may be changed to adjust their hydrophilicity or hydrophobicity. Research so far attempts to establish a mechanism based on the effect of hydrophobic ILs on the morphology of ZnO NPs.60–66
According to Duan and coworkers, ILs are a very effective medium for producing various kinds of metal oxides.67 Oprea et al. produced spherical and polyhedral ZnO NPs using a straightforward single-step solution method under low-temperature conditions in mixtures of ILs and organic solvents.61 The development of a methodology by exploiting the hydrophilicity of ILs to regulate the morphology, in particular, the size of metal oxide NPs, specifically ZnO NPs, is still in the rudimentary stage. The preliminary success has been by the use of hydrophilic ILs, 1-butyl-3-methylimidazolium tetrafluoroborate ([C4mim]BF4), 1-ethyl-3-methylimidazolium tetrafluoroborate ([C2mim]BF4) and 1-hexyl-3-methylimidazolium tetrafluoroborate ([C6mim]BF4).68 Gandhi and coworkers found that the PF6− anion of [C4mim]PF6, which is hydrophobic in nature, has weaker hydrogen bonding strength and stronger coordination, and in comparison to hydrophilic ILs, the interaction between the ZnO nuclei and the anion is too weak to be beneficial for nucleation and growth.69 In our previous article, we reported the successful synthesis of ZnO in nanodimension with variation in shapes by chemical precipitation method with systematic addition of different concentrations of 1-ethyl-3-methyl imidazolium methylsulfate, ([C2mim]CH3SO4) as a templating and self-directing material.70
ZnO NPs with controllable size and morphology have continued to be the aim of researchers since properties for task-specific applications may be tuned by their appropriate control. It appears intriguing to exploit the templating properties of ILs during the synthesis of nanostructured ZnO. The unique properties and structural variation of ILs are advantageous for the preparation of ZnO with desirable morphology. We, therefore, aimed at using a convenient hydrothermal method through the use of ILs with varying cationic and anionic structures to prepare ZnO NPs. The ultimate goal has been to acquire a systematic comprehension of the role of ILs in regulating the size and shape of ZnO nanostructures.
2. Experimental
2.1 Materials
ZnO NPs were synthesized from zinc acetate dihydrate (Zn(OAc)2·2H2O, >98%; Merck) and NaOH (>98%; Merck). Isopropanol (>99.5%; Sigma-Aldrich) was used as a dispersant and reference medium. ILs used were 1-ethyl-3-methylimidazolium methylsulfate, [C2mim]CH3SO4, 1-butyl-3-methylimidazolium methylsulfate, [C4mim]CH3SO4, and 1-ethyl-3-methylimidazolium ethylsulfate, [C2mim]C2H5SO4 (Sigma-Aldrich). Water was passed through an ion exchange column and the deionized water was distilled by a water distiller. It was further purified by a water purifier and the collected water was HPLC grade (conductivity at 25.0 °C = 0.055 μS m−1; BOECO pure, Model-BOE8704000, Germany).2.2 Method of preparation
A zinc acetate solution was prepared by dissolving 0.6 g of Zn(OAc)2·2H2O in 30.0 mL of HPLC-grade water. 10.0 mL of a freshly prepared 4.0 M NaOH solution was added to zinc acetate solution. The calculated volume of IL was added dropwise to have a final IL concentration of 0.1–2.5% (v/v). The mixture was vigorously stirred for 10 min and transferred to an autoclave (model: H1clave HV-25). The autoclave was sealed and maintained at 121 °C for 2 h to ensure the completeness of the reaction.2.3 Characterizations
The prepared samples were characterized by a Fourier transform-infrared/near-infrared (FT-IR/NIR) spectrometer (Frontier, PerkinElmer) in the wavenumber range of 400 to 4000 cm−1. Small amounts of solid materials and KBr were mixed to prepare pellets to record the spectrum. X-ray diffraction (XRD) patterns were recorded by Philips PW-1724 with XDC-700 Guinier Hagg focusing camera using Cu Kα radiation (λ = 1.540598 Å). A double-beam UV-visible spectrophotometer (Labomed UVD-500) was used for the spectroscopic analysis. Isopropanol was used as the reference for recording a spectrum of ZnO NPs synthesized without IL, while for ZnO NPs prepared in the presence of IL, the reference used had the IL added to isopropanol at a concentration identical to that employed during synthesis. The hydrodynamic diameter (Dh) and size distribution of ZnO NPs were determined by dynamic light scattering measurements by a particle size analyser (Zeta sizer Nano ZS90, ZEN3690, Malvern Instruments). The dispersions were sonicated before measurement and isopropanol was used as a dispersant.The morphology of ZnO samples was analysed using a scanning electron microscope (SEM, JSM-6490LA), a field emission scanning electron microscope (FESEM, JSM-7600F), and a high resolution transmission electron microscope (HRTEM, JEOL JEM 2100 HRTEM). Selected area electron diffraction (SAED) pattern was also simultaneously examined during HRTEM analysis. The optical band gap energy (Eg) of the samples was evaluated from specular reflectance spectra recorded using an integrating sphere attachment DRA-CA-30I of a double beam UV-visible spectrophotometer (UV-1800, Shimadzu).
3. Results and discussion
3.1 Analysis of size distribution
Fig. 1 shows the distributions of particle size of ZnO NPs prepared in the presence of ILs at different concentrations. The Z-average diameter of NPs synthesized without any template is 400 nm. On the other hand, with the addition of 0.5% (v/v) [C2mim]CH3SO4, the diameter decreased to 244 nm (Fig. 1a). With the further addition of 1.0% (v/v) IL, the average particle size increased to a maximum value of 596 nm. When there was no IL present in the reaction medium, there was no agent to control the growth. Thus, the size of ZnO NPs obtained without IL was found to be greater. With the introduction of 0.5% (v/v) [C2mim]CH3SO4, the diameter decreased since the IL acted as a soft template to control the growth of the ZnO clusters. However, at 1.0% (v/v) IL concentration, the diameter increased, which is even greater than the size obtained without IL. This might have happened because of the greater concentration of the IL which could cause weaker arrangement of the ions in the reaction medium due to steric hindrance. Consequently, at 1.0% (v/v) concentration, [C2mim]CH3SO4 could not serve as the size controlling agent and 0.5% (v/v) is the optimum concentration of [C2mim]CH3SO4 for the production of particles of smaller size. | ||
| Fig. 1 Distribution of particle size of ZnO NPs synthesized in presence of (a) [C2mim]CH3SO4, (b) [C4mim]CH3SO4, and (c) [C2mim]C2H5SO4 at different concentrations and (d) correlation of size of the NPs with varying concentration of ILs. | ||
Sort of a similar trend is followed when [C4mim]CH3SO4 was added to the reaction media (Fig. 1b). The Z-average size of the NPs decreased at 0.5% (v/v) [C4mim]CH3SO4 addition and increased at 1.0% (v/v) IL addition. Although further addition of [C4mim]CH3SO4 resulted in a decrease in size (526 nm) again. Moreover, 0.5% (v/v) is the optimum concentration of [C4mim]CH3SO4 for the production of smaller hydrodynamic diameters of ZnO NPs. In case of [C2mim]C2H5SO4, a rather opposite trend is observed in size distribution (Fig. 1c). With the addition of 0.5% (v/v) [C2mim]C2H5SO4, the Z-average size was maximum (934 nm), but when 1.0% (v/v) IL was added to the system, the average particle size decreased to 448 nm. Again, with further addition of 2.5% (v/v) IL, the average size increased to 534 nm. It is apparent from Fig. 1d that both [C2mim]CH3SO4 and [C4mim]CH3SO4 increased the size by their presence in the reaction media, while [C2mim]C2H5SO4 decreased the size with a higher concentration. 1.0% (v/v) is the optimum concentration for [C2mim]C2H5SO4, while this concentration did not serve well for the other two ILs with the common anion. This might be due to the alteration of the anion into a bigger one, which caused a better array of the [C2mim]C2H5SO4 template at a higher concentration. On the contrary, small anion (CH3SO4−) formed a better array in the system at lower concentrations due to less steric hindrance and a suitable compacting environment. Nevertheless, C2H5SO4− anion caused difficulties for the IL to form a suitable arrangement of ions at lower concentrations, resulting in the weakest control over the growth of the ZnO clusters. Hence, the highest diameter was obtained for 0.5% (v/v) [C2mim]C2H5SO4 and the lowest diameter was seen at 0.5% (v/v) [C2mim]CH3SO4.
Also, change in the cation of the ILs didn't affect the trend of change in size of ZnO NPs significantly with increasing concentration, but with change in the anion size, a noticeable change was observed with increasing concentration. Anions of the ILs thus regulate the size distribution of ZnO nanostructures. This might be caused as the bigger anions at higher concentrations can prevent the agglomeration of the particles or induce a preferential growth direction of the NPs and thus can implement a variation of size of the nanostructures. Thus, the anion effect of the ILs causes the size of the NPs to be altered with certain trends. As the particle size is dictated by the rate of nucleation and product growth, ILs can play a dual role here by protecting the growth of the particles through a certain direction and by promoting a suitable facet of the particle to agglomerate, and thus acting as a soft template. Therefore, increased concentration of the IL with larger anion causes difficulty for the agglomeration of the NPs through certain directions and consequently results in a decrease in particle size. Moreover, it also indicates the change in the shape of the NPs with varying concentrations of different ILs.
3.2 Morphological analysis
FT-IR spectra (Fig. S2 ESI†) confirm that ZnO was successfully synthesized both in presence and absence of ILs. XRD patterns helped identifying the crystalline phases and measuring the crystallite size of ZnO NPs. XRD patterns of ZnO NPs synthesized without and with IL as template are shown in Fig. 2a. All of the XRD patterns show several peaks at certain Bragg's positions, indicating that the structure of the produced powders is wurtzite and they are polycrystalline in nature. The JCPDS (No. 36-1451) data were used to match the peak positions of each diffraction pattern.61 The Zn(OH)2 synthesized in presence of distinct ILs transforms entirely into ZnO with the characteristic hexagonal wurtzite phase. No other intense peaks could be marked corresponding to other ZnO phases or any other impurities. The diffraction peaks were sharp to indicate the highly crystalline nature of the nanostructure.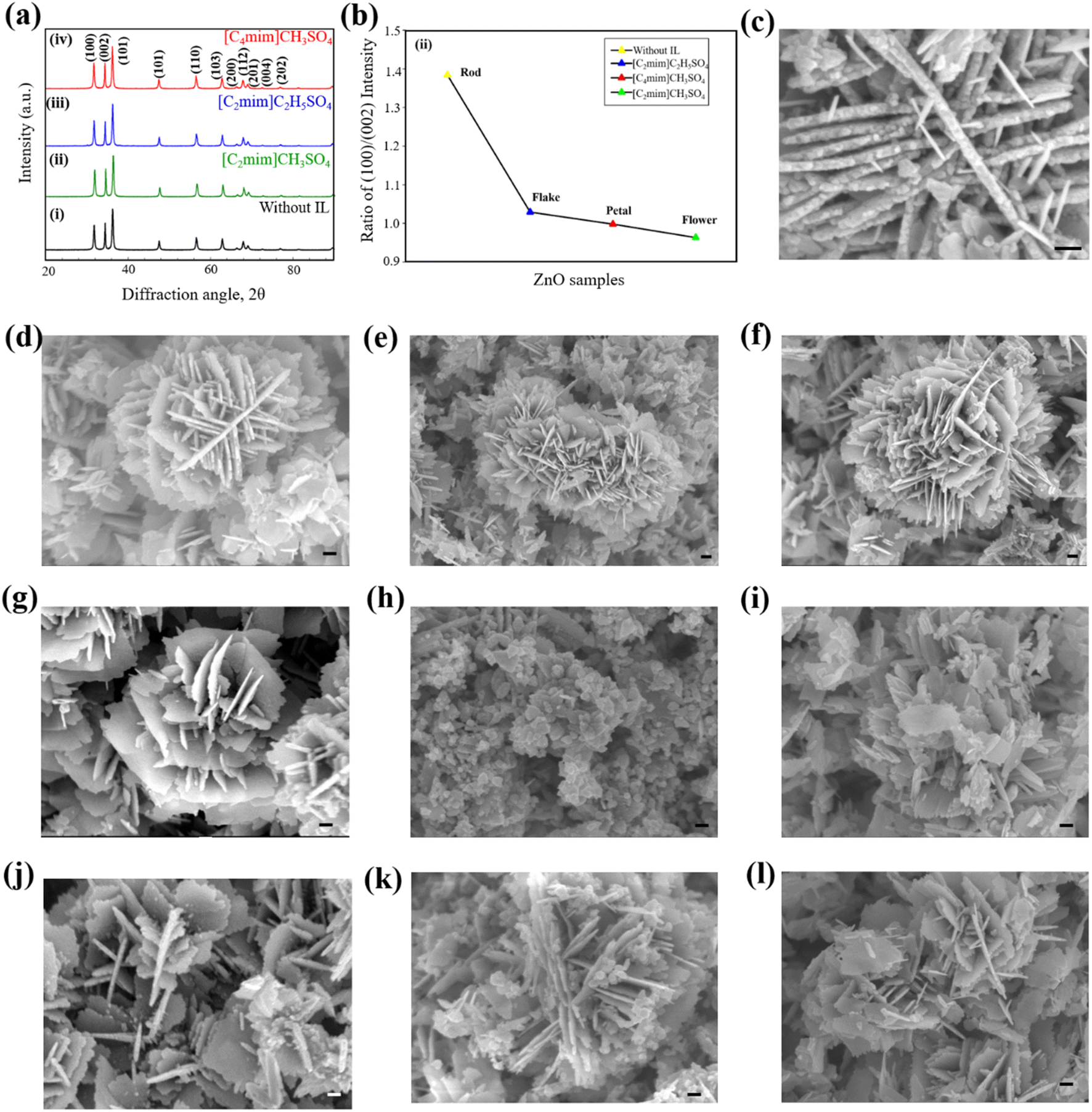 | ||
| Fig. 2 (a) XRD patterns of ZnO nanostructures with and without ILs and (b) variation of ratio of peak intensities of (100)/(002). FESEM images of ZnO nanostructures synthesized (c) without IL and with addition of (d) 0.5% [C2mim]CH3SO4, (e) 1.0% [C2mim]CH3SO4, (f) 2.5% [C2mim]CH3SO4, (g) 0.5% [C4mim]CH3SO4, (h) 1.0% [C4mim]CH3SO4, (i) 2.5% [C4mim]CH3SO4, (j) 0.5% [C2mim]C2H5SO4, (k) 1.0% [C2mim]C2H5SO4, and (l) 2.5% [C2mim]C2H5SO4. The scale bar is presented in the inset of the FESEM images and the length of the scale bar is 100 nm. | ||
The average crystallite size shows an apparent decrease from 29 nm to 23 nm (Table S1 ESI†) once a hydrophilic IL is added to the system. Therefore, with the addition of IL, the crystallite size of ZnO NPs decreases along with the change in the shape of the nanostructures. Different ILs act as different templates or typical capping agents providing a certain growth direction. This is evident from the XRD pattern; the (002) peak in pattern (ii) has a relative intensity higher than that of the pattern (i), while the intensities of (002) peaks in patterns (iii) and (iv) are similar to suggest the variation of growth of ZnO nanostructures.71 ZnO samples prepared using various ILs show differences in the relative intensities of their diffraction peaks, indicating that each IL has unique nanostructures in particular directions of the growing material.
With changes in the IL, the ratio of planes with Miller indices (100) and (002) varies from 1.384 to 0.963 (Fig. 2b and Table S2 ESI†). The surface energy is the lowest for the [001] direction rendering it to be the most likely growth direction for ZnO. The ratios are all less than that of the product obtained without any IL, so the ILs must be acting as definite templates or directing agents for the growing NPs. In the case of [C2mim]CH3SO4, the increased (002) peak intensity corresponds to increased growth of the nanostructures along with the [001] direction. But as the ratios increase for [C4mim]CH3SO4 and [C2mim]C2H5SO4, it may be concluded that these two ILs caused less growth along the [001] direction than [C2mim]CH3SO4. Again, almost similar (002) peak intensities are observed for [C4mim]CH3SO4 and [C2mim]C2H5SO4 but they differ to a greater extent from the peak obtained from [C2mim]CH3SO4. Thus, the change in growth direction might be caused by both the anion and cation effect of the IL structure. When the cation changed to a shorter chain ([C4mim] > [C2mim]) and the anion to a shorter size (C2H5SO4− > CH3SO4−), the growth increased along the [001] direction. The short chain cation and shorter anion might be suitable for a better arrangement of the template to provide a better growth in a certain direction.
FESEM images of ZnO nanostructures prepared using an IL and without IL at different concentrations are shown in Fig. 2c–l. ZnO synthesized without IL shows a well-defined structure composed of nanosized, irregular, and non-uniform rod-shaped particles (Fig. 2c) with an average diameter of ca. 50 nm. Upon addition of 0.5% (v/v) [C2mim]CH3SO4, the structure appears to be very unique star-like one (Fig. 2d), containing small flakes with denticulate edges which are connected in the center with other similar flakes. The lateral flake serves as a surface on top of which other flakes can develop and form a structure like a star. With the addition of 1.0% (v/v) [C2mim]CH3SO4, the morphology of ZnO changed from a star-like structure to a uniform 3D flower-like structure. According to Fig. 2e, the flower-like structures comprised several intertwined nanoflakes that are 20–30 nm thick and as compared to other samples their flakes have the largest diameter. Fig. 2f shows that with the addition of 2.5% (v/v) [C2mim]CH3SO4, the morphology of ZnO NPs has remained the same as a uniform 3D flower-like structure. ZnO structures that resemble flowers develop through the processes of (i) the nucleation and growth of ZnO NPs, (ii) the directed attachment of ZnO NPs to form nanoflakes, and (iii) the self-assembly of nanoflakes into three-dimensional flower-like structures. The overall growth process is strongly dependent on the concentration of Zn2+ and OH− ions.
Flower-shaped ZnO NPs are formed with the addition of 0.5% (v/v) [C4mim]CH3SO4 in the reaction medium (Fig. 2g). Such structures were formed by irregular nanoflakes and nanosheets. Further addition of [C4mim]CH3SO4 in the system caused the flower-shaped nanostructures to transform into petal-like ZnO nanostructures in agglomerated form with a consequent decrease in the average diameter. With the increasing concentration of [C4mim]CH3SO4 in the system, petal-shaped ZnO restructured into flakes-shaped nanostructures (Fig. 2i). These ZnO flakes were randomly and uniformly distributed with agglomeration. The diameter of ZnO flakes was lower compared to the petal-shaped ZnO nanostructures. While the peripheral surfaces are faceted and display a hexagonal pattern, the main surface seems to be relatively smooth. These characteristics suggest that each ZnO flake is most likely a single crystal with a flat [0001] surface.
A mixture of star-shaped and flakes-shaped ZnO nanostructures was found with the addition of 0.5% (v/v) [C2mim]C2H5SO4 as a template. According to Fig. 2j, these particles emerged from intersecting nanosheets that were, on average, 25 nm thick and predominantly interlaced and overlapped at an angle of around 60° to produce a cross-like morphology. Fig. 2k shows FESEM images of uniform needle-shaped ZnO nanostructures synthesized using 1.0% (v/v) [C2mim]C2H5SO4 to the reaction media. The needle-shaped ZnO NPs show no alignment and are distributed in a dispersive way, but their diameters and lengths are rather uniform. Needles with diameters of approximately 100–200 nm and lengths of 200–600 nm have made up the nanostructures. Flakes-shaped ZnO nanostructures are formed with the addition of 2.5% (v/v) [C2mim]C2H5SO4. Needle-shaped ZnO NPs are also slightly present in the sample (Fig. 2l). The average diameter of flakes-shaped ZnO is around 50–60 nm.
These are consistent with XRD results where [C2mim]CH3SO4 as a template showed higher (002) peak intensity than [C4mim]CH3SO4 and [C2mim]C2H5SO4. As a consequence, with [C2mim]CH3SO4, the product nanostructures had a preferential growth direction and thus a 3D flower like structure could be found whereas the flat petal like surfaces and nanosheets were observed in the case of [C4mim]CH3SO4 and [C2mim]C2H5SO4, respectively.
The HRTEM image of ZnO nanostructures without any template during synthesis is shown in part (a) of Fig. 3. This consists of nanorods that vary in length from 300 to 400 nm and diameter from 30 to 40 nm. The interplanar spacing of the lattice stripes in the HRTEM image of ZnO nanostructures taken from the edge of nanorods is around 0.25 nm, which corresponds to the distance between neighbouring two (0002) planes of hexagonal ZnO. This suggests the preferred growth orientation of ZnO nanorods along the [0001] crystal axis. The SAED pattern on the edge of a single nanorod reveals that ZnO nanorods are polycrystalline.
 | ||
| Fig. 3 HRTEM images of ZnO nanostructures synthesized (a) without IL and with (b) 0.5% and (c) 1.0% [C2mim]CH3SO4 and (d) 0.5% and (e) 2.5% [C4mim]CH3SO4, and (f) 0.5% and (g) 2.5% [C2mim]C2H5SO4. Inset shows the SAED pattern. | ||
The introduction of [C2mim]CH3SO4 in the reaction media caused significant changes in the morphology and crystallinity of ZnO NPs. According to Fig. 3b, the diameters of ZnO nanostructures produced with 0.5% (v/v) [C2mim]CH3SO4 varied between 20 and 40 nm, whereas those made with 1.0% had diameters between 40 and 50 nm. ZnO nanostructures synthesized using 0.5% (v/v) [C2mim]CH3SO4 are polycrystalline in nature (see SAED pattern), but with increasing concentration of [C2mim]CH3SO4, single crystalline ZnO was obtained. Since the interplanar spacing between two adjacent (0002) lattice planes of ZnO NPs is approximately 0.25 nm, it can be inferred from the inset of Fig. 3b that ZnO nanostructures are prone to growing along the [0001] crystal axis. The interplanar spacing of the crystalline stripes (0.5 nm), which is approximate twice the distance between two adjacent (0002) lattice planes of ZnO, is shown in the inset of Fig. 3c and probably explains the single crystalline nature of the nanostructures.
Both HRTEM and FESEM images gave consistent results regarding morphologies of ZnO nanostructures. The diameters of ZnO nanostructures formed using 0.5% (v/v) [C4mim]CH3SO4 increased to 80–100 nm with increasing [C4mim]CH3SO4 concentration, as shown in Fig. 3d. Thus, an increase in the diameter of the particle is observed due to change in the cationic structure of the IL. The diameter of the nanostructures increased as the length of the cation chain increased. The cation chain might be the driving architect here to create a template that produces ZnO NPs with higher diameters. However, this is only consistent with the DLS results of [C2mim]CH3SO4 and [C4mim]CH3SO4 (Fig. 1d). At the optimum concentration of these two ILs, hydrodynamic diameter increased with the increase of the cation chain length. But, this is inconsistent with the result of [C2mim]C2H5SO4, probably because of the dominant role of the larger anion in controlling the size of ZnO NPs. SAED patterns confirm that ZnO NPs synthesized using 0.5% and 1.0% (v/v) [C4mim]CH3SO4 are polycrystalline in nature which agrees with XRD results. The separation between the two adjacent (0002) lattice planes of hexagonal ZnO is close to the interplanar spacing of the crystalline stripes, which is ca. 0.50 nm (inset of Fig. 3d), suggesting that the ZnO nanostructures have a preferential growth direction along the [0001] crystal axis. But the interplanar spacing between two adjacent (0002) lattice planes of ZnO nanostructures is around 0.25 nm (inset of Fig. 3e) indicating that the [0001] crystal axis is not the preferred growth direction for ZnO. This is why the flower-like shape was transformed into a petal-like structure with less growth in directions compared to the nanostructures formed with [C2mim]CH3SO4.
Fig. 3f and g show HRTEM images of ZnO nanostructures synthesized using [C2mim]C2H5SO4 at different concentrations. Here, the diameters of ZnO prepared using 0.5% (v/v) [C2mim]C2H5SO4 are ca. 50–60 nm; however, when the concentration of [C2mim]C2H5SO4 increased, the diameter shrank to 30–40 nm. With longer chain anion, the IL creates an environment where the diameter of the nanostructures decreases to obtain a different morphology. SAED patterns gave evidence that ZnO nanostructures synthesized using 0.5% and 2.5% (v/v) [C2mim]C2H5SO4 are polycrystalline in nature which agrees with XRD results. The interplanar spacing between two adjacent (0002) lattice planes of ZnO nanostructures is around 0.75 nm (inset of Fig. 3f), which is about three times the separation of two adjacent (0002) lattice planes of ZnO. However, the crystalline stripes have an interplanar spacing of about 1.0 nm (inset of Fig. 3g). So, higher interplanar spacing was noted in the case of [C4mim]CH3SO4 and [C2mim]C2H5SO4 compared to [C2mim]CH3SO4. These support the XRD and FESEM results (vide infra). Higher interplanar spacing means lesser preferential growth in a certain direction causing the petal-like flat surfaces found in the products, which were prepared with the templates, [C4mim]CH3SO4 and [C2mim]C2H5SO4. But as the spacing is close to 0.25 nm in the case of [C2mim]CH3SO4, the 3D flower-like structure emerged along the [0001] direction via preferential growth.
3.3 Measurement of band gap energy from specular reflectance spectroscopy
The Eg for ZnO nanostructures was evaluated using the modified Kubelka–Munk method (Fig. 4 and Table S3 ESI†). The Eg obtained from the extrapolation of the curve is found to be approximately 3.17 eV, which is smaller than that of the bulk photonic wurtzite n-type semiconductor, ZnO (direct band gap of 3.37 eV) probably due to the presence of defects in the crystal lattice. When [C2mim]CH3SO4 is added to the reaction medium, the Eg of ZnO nanostructures changes. With the addition of 0.5% and 1.0% (v/v) [C2mim]CH3SO4, the Eg values of ZnO were 2.63 and 2.90 eV, respectively. These variations correspond to the increase in particle size and shape of ZnO NPs upon the addition of [C2mim]CH3SO4 in the system and may be explained in terms of quantum size effects.72,73 However, this variation of the bandgap is not supported by the DLS results, maybe due to the limitation of DLS as it assumes every shape of nanostructures as a sphere. Therefore, the anomaly might originate from the variation in the shape of the NPs in the presence of [C2mim]CH3SO4. | ||
| Fig. 4 Determination of band gap energy of ZnO NPs synthesized using (a) [C2mim]CH3SO4, (b) [C4mim]CH3SO4, and (c) [C2mim]C2H5SO4 as the template. | ||
When [C4mim]CH3SO4 was added during synthesis, the Eg decreases. With 1.0% and 2.5% (v/v) [C4mim]CH3SO4, the Eg values of ZnO NPs were 2.97 and 3.20 eV, respectively, while with 1.0% and 2.5% (v/v) [C2mim]C2H5SO4, the Eg values were 3.05 and 3.00 eV, respectively. In these two cases, the variation of the Eg values with different concentrations of ILs is consistent with the size distribution analysis discussed in section 3.1. However, it should be noted that DLS analysis gave the size distribution of the produced NPs in different IL templates, but it cannot provide the exact size of the NPs. Thus, the anomalies observed in the Eg values and the diameter obtained from DLS, indicate that the variation of the Eg values is due to the cumulative effect of the change of size and shape of the nanostructures. Therefore, the bandgap energy analysis also supports the shape variation of the ZnO NPs obtained with different IL templates. Moreover, the defects present in the crystal lattice of various nanostructures could vary the bandgap. These defects can be important to enhance the potential of the products for various applications.
The density of point/surface defects in semiconductor crystals increases with decreasing particle size below a certain threshold. Because of a slight delocalization of molecular orbitals on the surface, defects in the bulk semiconductor can result in deep and shallow traps close to the band edge of its electronic state. This results in a reduction in the band gap, which causes a redshift in the absorption spectrum. There are five intrinsic defects in the ZnO lattice such as oxygen vacancy VO, zinc vacancy VZn, interstitial oxygen Oi, interstitial zinc Zni and antisite oxygen OZn. The size quantization (Q-size) effect manifests when the spatial confinement of charge carriers causes the size of the semiconductor particle to decrease from its bulk to that of the Bohr radius, for example, in the initial excitation state. As a result, electrons and holes in the quantum-sized semiconductor are contained in a potential well and do not experience the delocalization that occurs in the bulk phase. In light of this, the band gap expands as particle size drops when the size of an ultra-fine semiconductor particle is below the band gap minimum. It is possible to exploit the size- and shape-dependencies of such optoelectronic features for a wide range of applications.
3.4 Interaction between ZnO nuclei and hydrophilic IL
ZnO with fascinating morphologies such as vermicelli, nanorods, nanoflakes, nanoflowers, and star-shaped nanostructures were obtained with varying concentration of hydrophilic ILs such as [C2mim]CH3SO4, [C4mim]CH3SO4, and [C2mim]C2H5SO4 by hydrothermal method. The growth unit for ZnO crystal is considered to be Zn(OH)42−.74| Zn(CH3CO2)2 + 2NaOH → Zn(OH)2 + 2CH3COONa + 2H2O |
| Zn(OH)2 + 2H2O → Zn(OH)42− + 2H+ |

The IL has a significant impact on the size and shape of the nanostructures by acting as a size and shape directing agent. The IL serves as a soft template based on electrostatic attraction, π–π* stacking interactions, hydrogen bonding, self-assembled mechanism, and so on.75,76 When added at low concentration to the reaction medium, the IL may dissipate into constituent ions. Due to its ability to withdraw electrons, which is aided by an electron pair sharing between hydrogen and carbon at position 2 of the imidazole ring, the cation of the IL may interact with the bulk. A portion of the forming material is constrained since the IL is adsorbed on the surface of the growing ZnO crystals and thereby, brings about anisotropy. Accordingly, it makes sense that the IL induces the ZnO material to develop certain nanostructures using a template comprised of a mechanism of hydrogen bonding-co-π–π* stacking. In the hydrothermal method, growth rate is higher than the nucleation rate. Under hydrothermal conditions, Zn(OH)2 precipitate dissolves to generate Zn2+ and OH− ions; ZnO crystals precipitate out of the solution once the product of [Zn2+] and [OH−] exceeds a critical value (which is necessary for the formation of ZnO crystals). Under hydrothermal circumstances, ZnO has a substantially lower solubility than that of Zn(OH)2, and consequently, the precipitate of Zn(OH)2 has a strong tendency to be converted into ZnO crystals via the following reactions:
| Zn(OH)2 ⇌ Zn2+ + 2OH− | (1) |
| Zn2+ + 2OH− ⇌ ZnO + H2O | (2) |
Initially, the concentrations of Zn2+ and OH− were significantly increased, resulting in considerable growth of crystal in different directions. The nucleation and crystal formation of ZnO commence once the concentration of Zn2+ and OH− reaches the supersaturation level of ZnO.
The 4-fold tetrahedral coordination of O2− and Zn2+ ions are stacked alternately along the c axis to form alternating planes of the wurtzite-structured ZnO crystal. This results in Zn2+-terminated (0001) top and O2−-terminated (0001) bottom surfaces. Other common side surfaces of ZnO include those that are O2−-terminated (1011), (1010). When compared to other crystal surfaces, the O2−-terminated one shows slower growth behaviour since by electrostatic force, the cations are easily adsorbed on the O2−-terminated surface. Additionally, the hydrogen bond that forms between the oxygen atoms of O–Zn and the hydrogen atom at position 2 of the imidazole ring may act as a useful bridge to link the developed metal oxide and the cation of ILs. As a result, the Zn2+-terminated (0001) face, which had the largest initial growth rates, experiences preferential growth.
Again, a shorter alkyl group (C2mim–C4mim) in the cation of the IL limits the steric effect and allows the NPs to grow longer. So, with the small alkyl chain length at the subsidiary chain of the imidazole cation in the IL, the size of NPs becomes larger. But with the large alkyl group of the anion in ILs, the size of the NPs also grows larger. A smaller anion might assist ILs to self-assemble in the aqueous medium and serve as a soft template for the preferential growth direction of the ZnO NPs. Therefore, ZnO nanostructures of different well-defined shapes were formed from the hydrothermal process using different ILs at varying concentrations.
The probable mechanism of formation of the ZnO nanostructures is illustrated in Fig. 5A. The zinc acetate hydrate molecule breaks down into ultrafine ZnO NPs, which are unstable due to their high chemical potential and surface energy. Therefore, the ZnO rod is formed by deciding on the [0001] growth direction as a result of the oriented coarsening process. However, the addition of the ILs as templates caused the protection along the (0001) facet and produced different nanostructures such as flower, petal, and intercrossed flakes.
 | ||
| Fig. 5 Schematic illustration of (A) the process of formation of ZnO nanostructures and (B) the effect of concentration of the ILs on the shapes and corresponding FESEM images. | ||
The size and shape of ZnO crystals are mostly influenced by the concentration of the ILs (Fig. 5B). For [C2mim]CH3SO4, the shape transforms from star to flower with increasing concentration. While [C4mim]CH3SO4 causes flowers to turn into petals with increasing concentration. But [C2mim]C2H5SO4 changes the star-like structure to intercrossed flakes. These findings support the impact of selective adsorption of ILs in promoting growth in directions other than the [0001] direction to form petal- or flake-like structures by protecting certain facets during the process of formation of ZnO rods.
4. Conclusions
It was possible to effectively synthesize ZnO nanostructures with controllable morphology using a hydrothermal method in the presence of three different hydrophilic ILs, [C2mim]CH3SO4, [C4mim]CH3SO4 and [C2mim]C2H5SO4 at various concentrations. Polycrystalline ZnO was formed with a high phase purity. The size of the nanostructures varied interestingly with different ILs used during preparation and it also exhibited a significant change with varying concentrations of the ILs. The band gap energy varied with the change in size of the ZnO nanostructures due to the concentration effect of ILs. Depending on the structure of the hydrophilic ILs, different morphologies could be obtained. The flower-like structure was observed with [C2mim]CH3SO4 and changing the cation caused the formation of petal-shaped nanostructures with [C4mim]CH3SO4. A mixture of star- and flake-shaped nanostructures were obtained in the presence of [C2mim]C2H5SO4 where the anion is bulkier. Hydrophilic ILs thus work well as a self-directing agent and template for the successful synthesis of ZnO nanostructures. Systematic changes in the concentration of ILs cause changes in the size, shape, and optical properties of ZnO nanostructures.Author contributions
Mousumi Akter: writing – original draft preparation, data curation, formal analysis, methodology, reviewing and editing, Md. Arif Faisal: writing – original draft preparation, formal analysis, reviewing and editing, Ajaya Kumar Singh: HRTEM and SAED data analysis and Md. Abu Bin Hasan Susan: conceptualization, writing – reviewing and editing, supervision, funding acquisition, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MAF acknowledges the Semiconductor Technology Research Centre of the University of Dhaka, Bangladesh for a research fellowship. The authors also express their gratitude for permission to use the Multi Scale Characterization Lab of the Department of Glass and Ceramic Engineering of BUET for FESEM and EDX and the Sophisticated Test and Instrumentation Centre, Kochi University, India for HRTEM and SAED facilities.Notes and references
- G. Oskam, J. Sol-Gel Sci. Technol., 2006, 37, 161–164 CrossRef CAS.
- A. Tavakoli, M. Sohrabi and A. Kargari, Chem. Pap., 2007, 61, 151–170 CAS.
- W. T. Liu, J. Biosci. Bioeng., 2006, 102, 1–7 CrossRef CAS PubMed.
- C. S. C. Santos, B. Gabriel, M. Blanchy, O. Menes, D. García, M. Blanco, N. Arconada and V. Neto, Mater. Today: Proc., 2015, 2, 456–465 Search PubMed.
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 1–13 RSC.
- Y. Ju-Nam and J. R. Lead, Sci. Total Environ., 2008, 400, 396–414 CrossRef CAS PubMed.
- G. Xiong, U. Pal, J. G. Serrano, K. B. Ucer and R. T. Williams, Phys. Status Solidi C, 2006, 142, 3577–3581 CrossRef.
- M. I. Khalil, M. M. Al-Qunaibit, A. M. Al-Zahem and J. P. Labis, Arabian J. Chem., 2014, 7, 1178–1184 CrossRef CAS.
- G. C. Yi, C. Wang and W. Park, Semicond. Sci. Technol., 2005, 20, 22–34 CrossRef.
- W. J. E. Beek, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2004, 16, 1009–1013 CrossRef CAS.
- S. Syeeda, M. S. Miran, M. Y. A. Mollah and M. M. Rahman, J. Bangladesh Chem. Soc., 2008, 21, 29 Search PubMed.
- P. Ahmed, M. S. Miran, M. A. B. H. Susan and M. Y. A. Mollah, J. Bangladesh Chem. Soc., 2013, 26, 20–29 CrossRef.
- S. S. Satter, M. Hoque, M. M. Rahman, M. Y. A. Mollah and M. A. B. H. Susan, RSC Adv., 2014, 4, 20612–20615 RSC.
- D. G. Thomas, J. Phys. Chem. Solids, 1960, 15, 86–96 CrossRef CAS.
- M. Dvorak, S. Wei and Z. Wu, Phys. Rev. Lett., 2013, 110, 016402 CrossRef PubMed.
- Y. Q. Su, Y. Zhu, D. Yong, M. Chen, L. Su, A. Chen, Y. Wu, B. Pan and Z. Tang, J. Phys. Chem. Lett., 2016, 7, 1484–1489 CrossRef CAS PubMed.
- B. Subash, B. Krishnakumar, M. Swaminathan and M. Shanthi, Ind. Eng. Chem. Res., 2014, 53, 12953–12963 CrossRef CAS.
- C. Mondal, J. Pal, M. Ganguly, A. K. Sinha, J. Jana and T. Pal, New J. Chem., 2014, 38, 2999–3005 RSC.
- K. K. Chattopadhyay, S. Maiti and S. Pal, CrystEngComm, 2015, 17, 9264–9295 RSC.
- E. Kowsari and B. Bazri, Appl. Catal., A, 2014, 475, 325–334 CrossRef CAS.
- J. M. Hancock, W. M. Rankin, T. M. Hammad, J. S. Salem, K. Chesnel and R. G. Harrison, J. Nanosci. Nanotechnol., 2015, 15, 3809–3815 CrossRef CAS PubMed.
- M. A. Garcia, J. M. Merino, E. F. Pinel, A. Quesada, J. de la Venta, M. L. R. Gonžalez, G. R. Castro, P. Crespo, J. Llopis, J. M. Gonžalez-Calbet and A. Hernando, Nano Lett., 2007, 7, 1489–1494 CrossRef CAS PubMed.
- H. Xu, Q. Zhao, H. Yang and Y. Chn, J. Nanopart. Res., 2009, 11, 615 CrossRef CAS.
- A. Sirelkhatim, S. Mahmud, A. Seeni, N. H. M. Kaus, L. C. Ann, S. K. M. Bakhori, H. Hasan and D. Mohamad, Nano-Micro Lett., 2015, 7, 219–242 CrossRef CAS PubMed.
- Z. Emami-Karvani and P. Chehrazi, Afr. J. Microbiol. Res., 2011, 5(12), 1368–1373 CAS.
- Y. Xie, Y. He, P. L. Irwin, T. Jin and X. Shi, Appl. Environ. Microbiol., 2011, 77(7), 2325–2331 CrossRef CAS PubMed.
- P. Gao, J. Lee and Z. Wang, J. Phys. Chem. C, 2007, 11, 13763–13769 CrossRef.
- B. Liu and H. Zeng, J. Am. Chem. Soc., 2003, 125, 4430–4431 CrossRef CAS PubMed.
- Z. Li, Y. Xiong and Y. Xie, Inorg. Chem., 2003, 42, 8105–8109 CrossRef CAS PubMed.
- J. Xie, P. Li, Y. Wang and Y. Wei, J. Phys. Chem. Solids, 2009, 70, 112–116 CrossRef CAS.
- A. Wei, X. Sun and C. Xu, Nanotechnology, 2006, 17, 1740–1744 CrossRef CAS PubMed.
- Z. Gui, J. Liu and Z. Wang, J. Phys. Chem. B, 2005, 109, 1113–1117 CrossRef CAS PubMed.
- Y. H. Ni, X. W. Wei, X. Ma and J. M. Hong, J. Cryst. Growth, 2005, 283, 48–56 CrossRef CAS.
- H. Zhao, X. Su, F. Xiao, J. Wang and J. Jian, Mater. Sci. Eng., B, 2011, 176, 611–615 CrossRef CAS.
- H. Zhang, D. Yang, Y. Ji, X. Y. Ma, J. Xu and D. L. Que, J. Phys. Chem. B, 2004, 18, 3955–3958 CrossRef.
- D. Ramimoghadam, M. Z. B. Hussein and Y. H. Taufiq-Yap, J. Phys. Chem. C, 2007, 111, 13763–13769 CrossRef.
- S. Baskoutas, P. Giabouranis, S. N. Yannopoulos, V. Dracopoulos, L. Toth, A. Chrissanthopoulos and N. Bouropoulos, Thin Solid Films, 2007, 515, 8461–8464 CrossRef CAS.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, 829 CrossRef.
- B. Hutera, A. Kmita, E. Olejnik and T. Tokarski, Arch. Metall. Mater., 2011, 58, 489–491 Search PubMed.
- O. A. Yıldırım and C. Durucan, J. Alloys Compd., 2010, 506, 944–949 CrossRef.
- M. Ristiac, S. Musiac, M. Ivanda and S. Popoviac, J. Alloys Compd., 2005, 397, 1–4 CrossRef.
- H. Y. Yue, W. D. Fei, Z. J. Li and L. D. Wang, J. Sol-Gel Sci. Technol., 2007, 44, 259–262 CrossRef CAS.
- K. G. Chandrappa and T. V. Venkatesha, Nano-Micro Lett., 2012, 4, 14 CrossRef CAS.
- M. Purica, E. Budianu, E. Rusu and R. Gavrila, Thin Solid Films, 2002, 404, 485–488 CrossRef.
- A. H. Moharram, S. A. Mansour, M. A. Hussein and M. Rashad, J. Nanomater., 2014, 716210 Search PubMed.
- D. Ramimoghadam, M. Z. B. Hussein and Y. H. Taufiq-Yap, Chem. Cent. J., 2013, 7, 136 CrossRef PubMed.
- L. Guo, S. Yang, C. Yang, P. Yu, J. Wang and W. Ge, Appl. Phys. Lett., 2000, 76, 2901–2903 CrossRef CAS.
- J. Dupont, R. F. Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS PubMed.
- M. Freemantle, An introduction to ionic liquids, RSC Publishing, Cambridge, 2010 Search PubMed.
- N. Papageorgiou, Y. Athanassov, M. Armand, P. Bonhôte, H. Pettersson, A. Azam and M. Grätzel, J. Electrochem. Soc., 1996, 143, 3099 CrossRef CAS.
- R. Kawano, H. Matsui, C. Matsuyama, A. Sato, M. A. B. H. Susan, N. Tanabe and M. Watanabe, J. Photochem. Photobiol., A, 2004, 164, 87 CrossRef CAS.
- M. Doyle, S. K. Choi and G. Proulx, J. Electrochem. Soc., 2000, 147, 34 CrossRef CAS.
- A. Noda, M. A. B. H. Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2003, 107, 4024 CrossRef CAS.
- M. A. B. H. Susan, A. Noda, S. Mitsushima and M. Watanabe, Chem. Commun., 2003, 8, 938–939 RSC.
- W. Lu, A. G. Fadeev, B. Qi, E. Smela, B. R. Mattes, J. Ding, G. M. Spinks, J. Mazurkiewicz, D. Zhou, G. G. Wallace, D. R. MacFarlane, S. A. Forsyth and M. Forsyth, Science, 2002, 297, 983 CrossRef CAS PubMed.
- M. A. B. H. Susan, T. Kaneko, A. Noda and M. Watanabe, J. Am. Chem. Soc., 2005, 127, 4976 CrossRef CAS PubMed.
- M. A. B. H. Susan, S. Saha, S. Ahmed, F. Begum, M. M. Rahman and M. Y. A. Mollah, Mater. Res. Innovations, 2012, 16, 345 CrossRef CAS.
- M. S. Miran, H. Kinoshita, T. Yasuda, M. A. B. H. Susan and M. Watanabe, Phys. Chem. Chem. Phys., 2012, 14, 5178 RSC.
- M. S. Miran, T. Yasuda, M. A. B. H. Susan, K. Dokko and M. Watanabe, RSC Adv., 2013, 3, 4141 RSC.
- S. Rabieh and M. Bagheri, Mater. Lett., 2014, 122, 190–192 CrossRef CAS.
- O. Oprea, O. Ciocirlan, A. Badanoiu and E. Vasile, Cent. Eur. J. Chem., 2014, 12, 749–756 CAS.
- K. Qi, J. Yang, J. Fu, G. Wang, L. Zhu, G. Liua and W. Zheng, CrystEngComm, 2013, 15, 6729–6735 RSC.
- Z. He and P. Alexandridis, Phys. Chem. Chem. Phys., 2015, 35, 20 Search PubMed.
- M. Movahedi, E. Kowsari, A. R. Mahjoub and I. Yavari, Mater. Lett., 2008, 62, 3856–3858 CrossRef CAS.
- H. Zou, Z. Li, Y. Luan, T. Mu, Q. Wang, L. Li, J. Ge and G. Chen, Curr. Opin. Solid State Mater. Sci., 2010, 14, 75–82 CrossRef CAS.
- T. Singh, T. J. Trivedi and A. Kumar, Nanomater. Energy, 2012, 1, 207–215 CrossRef CAS.
- X. Duan, J. Ma, J. Lian and W. Zheng, CrystEngComm, 2014, 16, 2550 RSC.
- A. Wittmar, D. Gautam, C. Schilling, U. Dorfler, W. Mayer-Zaika, M. Winterer and M. Ulbricht, J. Nanopart. Res., 2014, 16, 2341 CrossRef.
- R. Gandhi, S. Gowri, J. Suresh and M. Sundrarajan, J. Mater. Sci. Technol., 2013, 29, 533 CrossRef CAS.
- M. Akter, S. S. Satter, A. K. Singh, M. M. Rahman, M. Y. A. Mollah and M. A. B. H. Susan, RSC Adv., 2016, 6, 92040 RSC.
- F. Jakuphanoglu, S. Ilican, M. Caglar and Y. Caglar, J. Optoelectron. Adv. Mater., 2007, 9, 2180 Search PubMed.
- R. Koole, E. Groeneveld, D. Vanmaekelbergh, A. Meijerink and C. de Mello Donegá, Nanoparticles, 2014, 13–51 CrossRef PubMed.
- H. Lin, C. P. Huang, W. Li, C. Ni, S. I. Shah and Y. H. Tseng, Appl. Catal., B, 2006, 68, 1–11 CrossRef CAS.
- W. J. Li, E. W. Shi, W. Z. Zhong and Z. W. Yin, J. Cryst. Growth, 1999, 203, 186–196 CrossRef CAS.
- M. Sabbaghan, A. S. Shahvelayati and S. E. Bashtani, Solid State Sci., 2012, 14, 1191–1195 CrossRef CAS.
- M. Sabbaghan, J. Beheshtian and S. A. M. Mirsaeidi, Ceram. Int., 2013, 12, 119 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02681g |
| This journal is © The Royal Society of Chemistry 2023 |