DOI:
10.1039/D3RA02646A
(Paper)
RSC Adv., 2023,
13, 17587-17594
Isolation, structural elucidation, and cytotoxic activity investigation of novel styryl-lactone derivatives from Goniothalamus elegans: in vitro and in silico studies†
Received
21st April 2023
, Accepted 4th June 2023
First published on 12th June 2023
Abstract
Two styryl-lactone derivatives (1 and 2) were isolated from the aerial parts of Goniothalamus elegans. Compound 1 is a newly discovered natural product, and compound 2 is reported in this plant for the first time. The absolute configuration of 1 was determined based on the ECD spectrum. The two styryl-lactone derivatives were tested for cytotoxicity activity against five cancer cell lines and human embryonic kidney cells. The newly discovered compound demonstrated potent cytotoxicity, with IC50 values ranging from 2.05 to 3.96 μM. Computational methods were also applied to investigate the mechanism of the cytotoxic activity of the two compounds. Density functional theory and molecular mechanisms were used to assess the interaction between protein targets to compound 1 and 2, respectively, through the EGF/EGFR signaling pathway. The results indicated that 1 showed a strong binding affinity for two proteins EGFR and HER-2. Finally, ADMET predictions were used to validate the pharmacokinetics and toxicity of these compounds. The results showed that both compounds are likely to be absorbed in the gastrointestinal tract and penetrate the blood–brain barrier. Based on our findings, these compounds may have potential for further studies to be developed into active ingredients for cancer treatment.
1. Introduction
The American Cancer Society reported that there were about two million new cancer cases and 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cancer-related deaths in the United States in 2021. The majority of these deaths were attributed to prostate, breast, colorectal, and lung cancers.1 Recently, a revolution in cancer therapies has been seen due to early cancer identification, as well as the discovery of novel treatments through improved technology and pharmaceuticals. Furthermore, funding for cancer research is increasing significantly, facilitating these improvements.
000 cancer-related deaths in the United States in 2021. The majority of these deaths were attributed to prostate, breast, colorectal, and lung cancers.1 Recently, a revolution in cancer therapies has been seen due to early cancer identification, as well as the discovery of novel treatments through improved technology and pharmaceuticals. Furthermore, funding for cancer research is increasing significantly, facilitating these improvements.
The epidermal growth factor receptor (EGFR) signaling pathway is one of the most essential pathways in mammalian cells. This pathway regulates several crucial functions during cell development, including intercellular communication, proliferation, migration, and cell death.2,3 As the mutation of the specific elements in this pathway results in human cancer, it has been regarded as an ideal therapeutic target for cancer studies. A typical outcome of the activation of the EGFR-mediated downstream pathways in cancer cells is abnormal gene activity, which promotes uncontrolled tumor growth and death. EGFRs, commonly referred to as ErbB receptors, are members of the receptor tyrosine kinase protein family. Recently, the discovery of small-molecule tyrosine kinase inhibitors (TKIs) has revolutionized cancer therapy because of the correlation between EGFR and malignancies.3,4 Interestingly, significant clinical trials are currently being conducted to investigate the effects of suppressing EGFR and ErbB2 signaling in malignancies. For example, the US Food and Drug Administration (FDA) has approved lapatinib and neratinib, which target the ErbB2 receptor, for the treatment of breast cancer, while afatinib, dacomitinib, erlotinib, gefitinib, and osimertinib, which are EGFR/ErbB1 inhibitors, are used to treat non-small cell lung cancer.5,6
Drug development is an expensive and complex process. This process begins with target selection and then continues through in vitro and in vivo experiments and pre-clinical studies. Virtual screening techniques have enhanced the ability to use computational approaches for the discovery of pharmacological candidates.7,8 In fact, it is well acknowledged that combining computational and experimental methods can increase the success rate of drug development.9,10 Recently, the development of herbal products has been increasing in popularity.11–13 The 160 species of the diverse plant genus Goniothalamus, a member of the Annonaceae family, are primarily native to Southern Asia's tropical climates.14 Several Goniothalamus species are used in traditional medicine to treat a range of symptoms, including fever, stomachaches, typhoid, and skin conditions.14 Previous studies have also shown that specific extracts and chemicals derived from Goniothalamus species offer beneficial biological features, such as antibacterial, antimalarial, antioxidant, and particularly cytotoxic properties.15,16 Vietnam and other Southeast Asian countries are the main locations of Goniothalamus elegans. This species is frequently used to treat cardiac conditions and hemorrhagic diarrhea in traditional Vietnamese medicine.14 However, publications on the phytochemical properties of the species—including anticancer activity—are limited. Notably, our previous research revealed that isolated chemicals, fractions, and extracts from Vietnamese G. elegans were cytotoxic to human breast cancer cell lines.9,10 Based on these findings, we continued to isolate and evaluate the anticancer activity of new chemical components from G. elegans. In this study, one new and one known styryl-lactone were isolated from G. elegans. The cytotoxic properties of these compounds were validated on several human cancer cell lines. Subsequently, a molecular docking method was used to investigate the potential ability of these compounds to inhibit EGFR. The stabilities of the ligand–protein complexes were evaluated with molecular dynamics simulations. Theoretical calculation of electronic circular dichroism (ECD), density functional theory (DFT), and molecular docking were used to study molecular mechanism between these styryl-lactone derivatives and proteins ErbB1 and ErbB2. Finally, absorption, distribution, metabolism, excretion, and toxicity (ADMET) prediction were used to assess drug-likeness of two styryl-lactone derivatives.
2. Experimental
2.1. General experimental procedures
Optical rotations were obtained on a JASCO-2000 Polarimeter (Hachioji, Japan). UV spectra were recorded on a Shimadzu UV-1800 spectrophotometer (Shimadzu, Kyoto, Japan). The NMR spectra were recorded on a Bruker Avance Neo 600 spectrometer operating at 600 MHz for 1H and 150 MHz for 13C. The HRESIMS data were recorded on SCIEX X500 QTOF (SCIEX, MA, USA) mass spectrometer. Silica gel (60 N, spherical, neutral, 40–50 mm, Kanto Chemical Co., Inc., Tokyo, Japan), YMC RP-18 (Fuji Silysia Chemical Ltd, Kasugai, Aichi, Japan), Sephadex LH-20 (DowexVR 50WX2-100, Sigma-Aldrich, USA) were used to remove the pigment and for coarse separation. Precoated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm) and precoated RP-18 F254s plates (Merck) were used for analytical thin-layer chromatography analyses. Five human cancer cell lines, including breast carcinoma (MCF-7), hepatocellular carcinoma (HepG2), lung carcinoma (LU-1), gastric adenocarcinoma (AGS), colon adenocarcinoma (SW-480), and human embryonic kidney cells (HEK-293A) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA).
2.2. Plant material
During July 2020, the aerial parts of G. elegans were collected from Quang Tri province in Vietnam (N16°44′38.9′′ E107°14′51.1′′). The authenticity of the samples was confirmed by the Vietnam National Museum of Nature, VAST in Vietnam. A sample of the plant (GE01) was also preserved at the Faculty of Pharmacy, Hue University of Medicine and Pharmacy, Hue University in Vietnam.
2.3. Extraction and isolation
The dried powder of G. elegans (4.5 kg) was extracted with MeOH (3 times, 10.0 L each) at room temperature and then concentrated under reduced pressure to yield 375 g of a dark solid extract. This extract was then suspended in water and successively partitioned with n-hexane, CH2Cl2, and EtOAc (3 times, 5.0 L each) to obtain the n-hexane (117.6 g), CH2Cl2 (91.2 g), EtOAc (83.3 g), and water (74.7 g) fractions after removal of the solvents in vacuo (Fig. 1). The EtOAc extract was partitioned on a silica gel column eluted with a gradient of n-hexane–EtOAc (100:0, 95:5, 90:10, 50:10, 10:10, and 0:100, v/v, 1.0 L each) to afford 6 fractions (E1–E6). Fraction E2 (11.4 g) was chromatographed on a silica gel column using n-hexane–CH2Cl2–MeOH (3:1:0.1, v/v/v) as the eluent to give 6 sub-fractions (E2A–E2F). Fraction E2B (1.4 g) was partitioned on a Sephadex LH-20 column using CH2Cl2–MeOH (1:1, v/v) as the eluent to obtain 5 additional fractions (E2B1–E2B5). Fraction E2B3 (280 mg) was submitted to silica gel chromatography column eluting with CH2Cl2–EtOAc (20:1, v/v) to afford 6 fractions (E2B3A–E2B3F). Fraction E2B3B (58 mg) was chromatographed on a YMC RP-18 column eluted with acetone–H2O (5:1, v/v) to give compound 1 (5.5 mg). Fraction E2D (1.7 g) was partitioned on a YMC RP-18 column eluted with acetone–H2O (3:1, v/v) to obtain 5 additional fractions (E2D1–E2D5). Fraction E2D3 (230 mg) was chromatographed on a Sephadex LH-20 column eluted with MeOH to furnish compound 2 (11.5 mg).
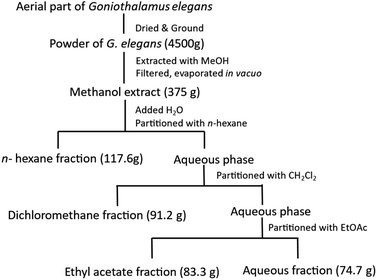 |
| | Fig. 1 Extraction and fractions of G. elegans. | |
Compound 1: white amorphous powder, α25D −188.1 (c 0.13, CH3OH), UV 256, 282 nm; IR spectrum 2956, 2923, 1737, and 1708 cm−1; ECD λmax (Δε) 210 (−11.32), 249 (6.73), 276 (−11.88) nm; HR-ESI-MS m/z 347.1278 [M + H]+ (calcd for C22H19O4+, 347.1278). 1H-(CDCl3) and 13C-NMR (CDCl3) data are listed in Table 1.
Table 1 1H- (600 MHz) and 13C- (150 MHz) NMR data of 1a
| Position |
1Hb (J in Hz) |
13Cb |
| Measured in CDCl3. Chemical shift may be overlapped signals which were confirmed by HSQC, and HMBC experiments. |
| 2 |
— |
162.5 |
| 3 |
6.30 d (9.7) |
124.9 |
| 4 |
7.08 dd (9.7, 5.5) |
140.9 |
| 5 |
5.51 dd (5.5, 3.1) |
63.9 |
| 6 |
5.26 ddd (6.6, 3.1, 1.3) |
79.4 |
| 7 |
6.28 dd (16.0, 6.6) |
121.1 |
| 8 |
6.87 d (16.0) |
135.2 |
| 9 |
— |
135.7 |
| 10 |
7.39 m |
126.9 |
| 11 |
7.31 t (7.3) |
128.7 |
| 12 |
7.28 tt (7.3, 1.3) |
128.5 |
| 13 |
7.31 t (7.3) |
128.7 |
| 14 |
7.39 m |
126.9 |
| 15 |
— |
165.8 |
| 16 |
6.42 d (16.0) |
116.4 |
| 17 |
7.70 d (16.0) |
146.8 |
| 18 |
— |
133.9 |
| 19 |
7.50 m |
128.3 |
| 20 |
7.39 m |
130.8 |
| 21 |
7.39 m |
129.0 |
| 22 |
7.39 m |
130.8 |
| 23 |
7.50 m |
128.3 |
2.4. Theoretical calculation of the ECD spectrum of compound 1
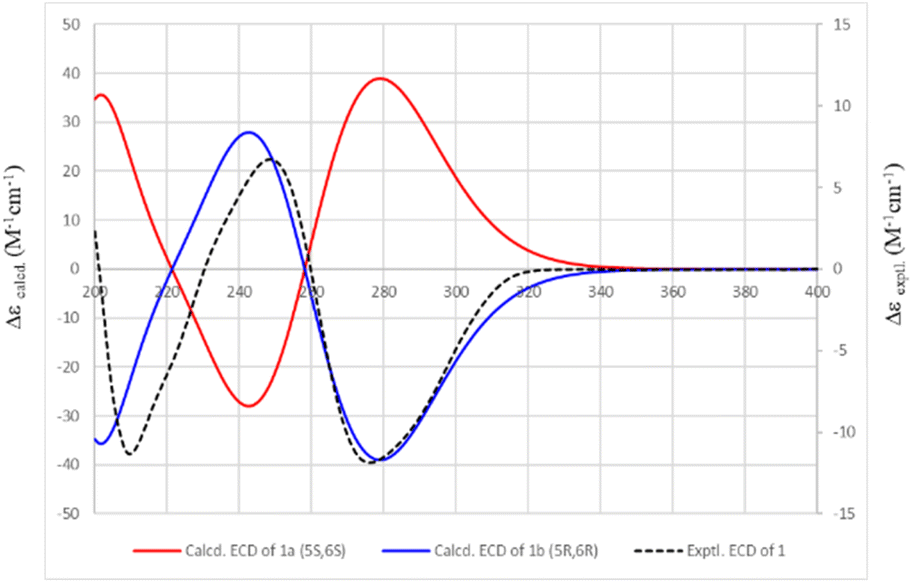
Conformational analyses were performed on the Spartan 14 program. Main conformers were selected, optimized, and subjected to TDDFT calculation using the Gaussian 09 program. The calculated ECD spectra were composed, based on the Boltzmann distribution of the stable conformers using SpecDis v1.71 software. Compound 1a (5S,6S form) was submitted to conformational searches at the MMFF set using the Spartan 14. Thirteen conformers with Boltzmann distribution larger than 1% were then DFT optimized by the Gaussian 09 program at the B3LYP/6-31G(d,p) basic set and polarizable continuum model (PCM) of methanol. Optimized conformers were then subjected to TDDFT calculations using the Gaussian 09. TDDFT parameters were set at the CAM-B3LYP/6-31G(d,p) level and PCM of methanol. The ECD spectra at 30 excited states for each conformer were collected and summed to obtain theoretical ECD spectra using SpecDis v1.71. The half-band was taken at ζ = 0.40 eV. The calculated ECD spectrum of the 1b (5R,6R form) was composed as the mirror image of that of 1a.
2.5. Quantum chemical calculation
After the structure of compound 1 was determined based on the ECD spectrum, the molecular structure of compound 1 and compound 2 are optimized by density functional theory (DFT) using 6-311++G(d,p) basis sets by the new local density functional (M052X) method.17–19 Natural bond orbital (NBO) analysis was executed using NBO 5.1 in order to frontier orbital analysis at the level of theory M052X/def2TZVPP.19 Kooman's theorem was applied to calculate the quantum chemical characteristics including the ionization potential (IP), electron affinity (EA), electronegativity (χ), chemical hardness (η), chemical potential (μ), chemical softness (S), and electrophilicity index (ω). GaussView 6 was used in the representation of the results.20 All the DFT calculations mentioned above were using the Gaussian 09 package.21
2.6. Cytotoxic assays
The cytotoxicity test was performed according to the protocol of previously publication.10 The cytotoxicity of two selected compounds from the plant was evaluated using the sulforhodamine B (SRB) colorimetric assay on five human cancer cell lines: breast carcinoma (MCF-7), hepatocellular carcinoma (HepG2), lung carcinoma (LU-1), gastric adenocarcinoma (AGS), and colon adenocarcinoma (SW-480) and human embryonic kidney cells (HEK-293A). Stock cultures of cells were grown in T-75 flasks containing Dulbecco's modified Eagle's medium (DMEM) supplemented with L-glutamine, sodium bicarbonate, and fetal bovine serum (FBS). The media were changed every 48 hours. The cells were cultured with a specific mix of media and sub-cultured every 3 to 5 days in a controlled environment. The cytotoxicity was determined by adding the cells to 96-well plates and incubating them for 72 hours. The viability of the cells was then measured using the SRB assay. The cells were fixed and treated with SRB solution, and the optical density was recorded on an ELISA reader at 515 nm. The study tested the cytotoxicity of the compounds at doses of 100, 20, 4, and 0.8 μg mL−1 and compared the results with a negative control (DMSO) and a positive control (Ellipticine). The half-maximal IC50 was calculated using TableCurve Version 4.0 software. The experiment was repeated three times.
2.7. Protein and ligand preparation
Because the 3D crystal structure of EGFR on the published RCSB library has missing residues. To solve this problem, the 3D structure of EGFR was downloaded from the AlphaFold Protein Structure Database. Then, the binding domain of EGFR from residue Asn700 to Ile1018 and binding domain of HER2 from residue Met706 to Asp993 were selected for molecular docking. Pairwise structural alignment between the predicted structure and crystal protein of EGFR (PDB ID: 6Z4B, the resolution is 2.50 Å) and HER2 (PDB ID: 3PP0, the resolution is 2.25 Å) were used to validate the accuracy of modeling. The result showed in (ESI, Fig. S10†). Then, energy minimization performed by standard optimization parameter of Swiss PDB Viewer.22 Next, polar hydrogen molecules were added to two protein structures using the AutoDock Tool 1.5.6 and were exported into a dockable pdbqt format in preparation for molecular docking.23 The 3D structures of two styryl-lactone derivatives were prepared using MarvinSketch software (ChemAxon, USA). Later, the torsion bonds were automatically set for all ligands and converted to the pdbqt format apted for screening.
2.8. Molecular docking simulation
The molecular docking process was carried out using the program AutoDock Vina 1.1.2.24 The grid box covering the active site of the protein was determined using the position of crystal ligand. Docking scores were reported in kcal mol−1. Results of interactions between compounds and protein targets were visualized by software Discovery Studio Visualizer 2020.
2.9. In silico drug-likeness and toxicity
Prediction drug-likeness of compounds was predicted based on an already established concept by Lipinski rule. SwissADME server was used to calculate the ADME properties of ligands which provided information about absorption, distribution, metabolism, excretion (ADME) properties of the ligands.25 The acute oral toxicity (T) was predicted using the DL-AOT prediction server.26
2.10. Statistical analysis
Quantitative data were obtained by triplicated experiments and analysis was carried out with one-way ANOVA followed by Tukey Post Hoc test in Excel 2020.
3. Results
3.1. Chemical structure elucidation
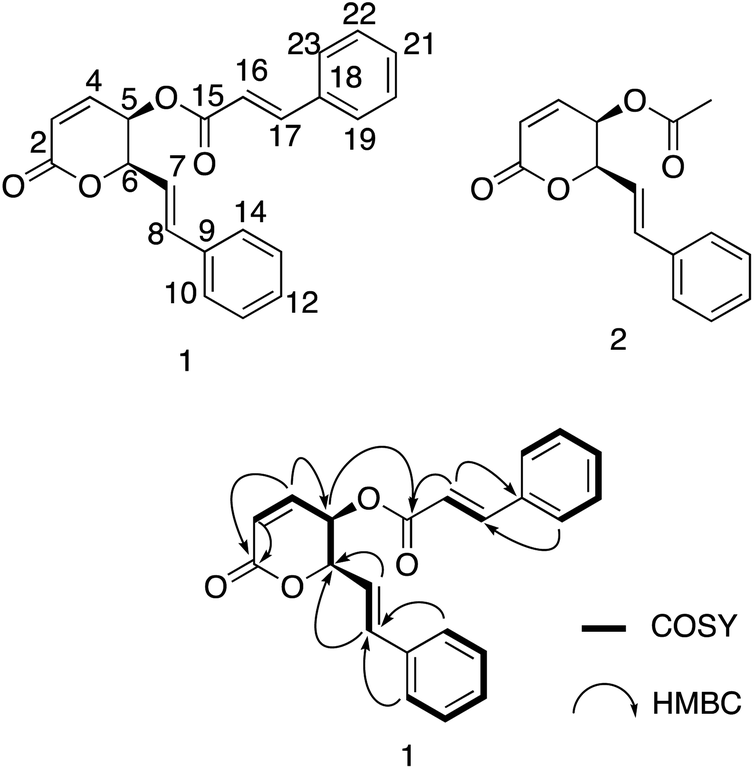
Compound 1 was isolated as a white amorphous powder with an optical rotation [α]25D −188.1 (c 0.13, CH3OH). The molecular formula of 1 was deduced to be C22H18O4 based on the quasi-molecular ion peak at m/z 347.1278 [M + H]+ (calcd for C22H19O4+, 347.1278) in the HRESIMS spectrum. Its IR spectrum showed vibrational bands of double bonds at 2956 and 2923 cm−1, and carbonyl groups at 1737 and 1708 cm−1. The 1H-NMR spectrum displayed two olefinic proton signals at δH 6.30 (d, J = 9.7 Hz, H-3) and 7.08 (dd, J = 9.7, 5.5 Hz, H-4), and two methine protons at δH 5.51 (dd, J = 5.5, 3.1 Hz, H-5) and 5.26 (ddd, J = 6.6, 3.1, 1.3 Hz, H-6). These protons showed HSQC correlations to four methine carbons at δC 124.9 (C-3), 140.9 (C-4), 63.9 (C-5), and 79.4 (C-6), respectively. In the 13C-NMR spectrum, a ketone carbon is present at δC 162.5 (C-2). This carbon exhibited HMBC correlations with H-3 and H-4 (Fig. 2). The COSY cross-peaks indicated the presence of a linear spin system, C-3(H)/C-4(H)/C-5(H)/C-6(H) in 1. The above NMR characteristics were assignable to a 5,6-dihydro-2H-pyran-2-one group. In addition, the 1H-NMR spectrum of 1 showed a set of five aromatic protons resonating at δH 7.39 (2H, m, H-10 and H-14), 7.31 (2H, t, J = 7.3 Hz, H-11 and H-13), and 7.28 (1H, tt, J = 7.3, 1.3 Hz, H-12) assigned to a phenyl group. The two olefinic proton signals were observed at δH 6.28 (1H, dd, J = 16.0, 6.6 Hz, H-7), and 6.87 (1H, d, J = 16.0 Hz, H-8), which characterized the trans configuration of the double bond Δ7 (Table 1). This evidence—together with the carbon signals at δC 121.1 (C-7), 135.7 (C-8), and 135.7 (C-9), and the set of five methine carbons at δC 126.9 (C-10 and C-14), 128.7 (C-11 and C-13), and 128.5 (C-12)—was indicative of the presence of a styryl group. Interestingly, the 1D-NMR spectra of 1 showed the resonance signal of one more aromatic ring at δH 7.50 (2H, m, H-19 and H-23), 7.39 (3H, m, H-20, H-22, and 21), and δC 133.9 (C-18), 128.3 (C-19, C-23), 130.8 (C-20, C-22), and 129.0 (C-21). Another two paired olefin proton signals were observed at δH 6.42 (1H, d, J = 16.0 Hz, H-16) and 7.70 (1H, d, J = 16.0 Hz, H-17), and characterized as the trans configuration of the double bond Δ16. Taken together, a signal observed at δC 165.8 (C-15) was assigned to a carbonyl carbon. The above resonance signals suggested the presence of a cinnamoyl moiety in the structure of compound 1. Examination of the 13C-NMR and HSQC spectra confirmed the presence of 22 carbon atoms, which exhibited three parts, including 5 signals to a pyran ring, 8 to a styryl, and 9 to cinnamoyl moieties. This evidence also suggested that 1 possessed a styryl-lactone skeleton—the main phytochemical constituent of Goniothalamus sp. The above NMR data were comparable to those of styryl lactones, goniothalamin, and 5-acetoxygoniothalamin (compound 2), which were isolated from Goniothalamus uvaroides.27 However, the difference between 1 and 2 was the appearance of a cinnamoyl residue in 2 instead of an acetyl as in 1. Next, the HMBC correlations from H-5 (δH 5.51) to C-15 (δC 165.8) suggested the attachment of cinnamoyloxy at C-5. Similarly, the cross-peaks between H-6 (δH 5.26) and δC 121.1 (C-7), and between H-7 (δH 6.28)/H-8 (δH 6.87) and C-6 (δC 79.4), also supported the presence of a styryl group at C-6. The coupling constants, J4,5 5.5 Hz, J5,6 3.1 Hz, and J6,7 6.6 Hz, were consistent with those of (+)-5β-hydroxygoniothalamin (5.4, 2.9, 6.4 Hz, respectively), but very different from those of (+)-5α-hydroxygoniothalamin (2.0, 8.0, 7.3 Hz, respectively).28,29 Therefore, the cis configuration was unambigiously assigned for 1. This was supported by NOESY correlation between H-5 and H-6 (ESI†). In addition, the absolute configuration at the C-5 and C-6 chiral centers of 1 was determined based on the ECD calculation (Fig. 3). The experimental ECD spectrum of 1 (in methanol) was compared with the TDDFT-calculated ECD spectra of 1a (5S,6S) and 1b (5R,6R) isomers (ESI†). Because the experimental ECD of 1 agreed with that of 1b, the 5R,6R configuration was assigned for 1. Therefore, the chemical structure of 1 was fully elucidated as (5R,6R)-2-oxo-6-((E)-styryl)-5,6-dihydro-2H-pyran-5-yl cinnamate (trivial name: 5-cinnamoyloxygoniothalamin).
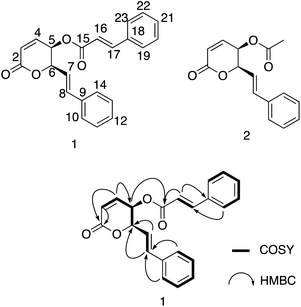 |
| | Fig. 2 Chemical structure of 1, 2 and selected COSY and HMBC of 1. | |
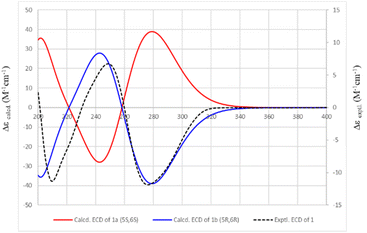 |
| | Fig. 3 Experimental and calculated ECD spectra of 1. | |
3.2. DFT studies
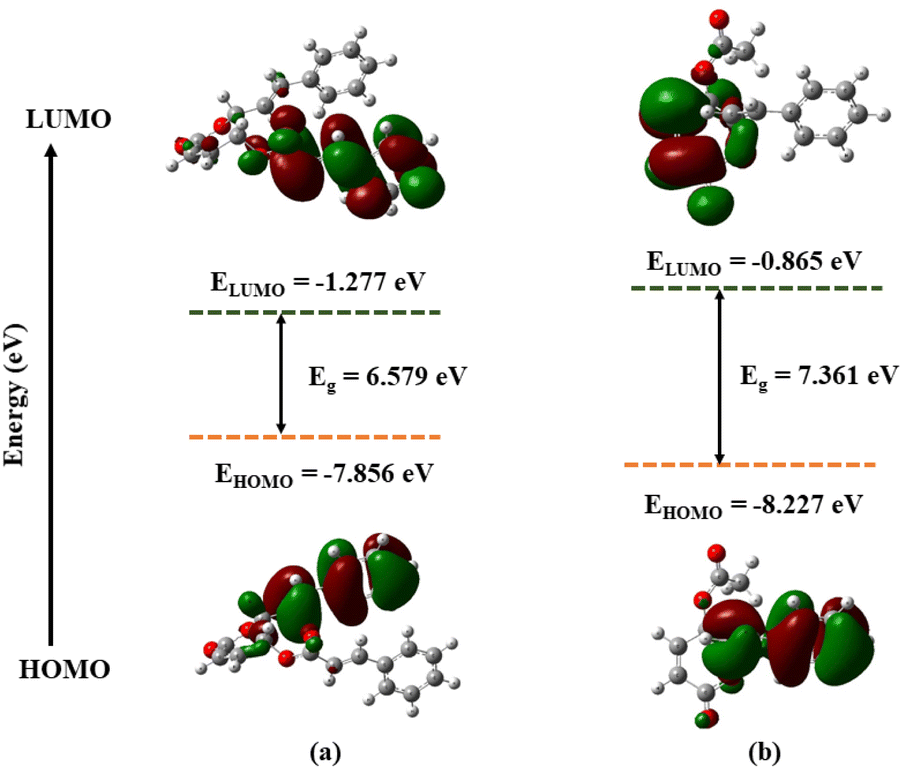
The optimized geometries of 1 and 2, with the numbering scheme of atoms obtained from DFT calculations, are shown in the ESI.† The optimized structural parameters were determined using the M05-2X method with 6-311++G(d,p) basis sets and tabulated in the ESI.† The results showed 1 exhibited high dipole moments and polarizability—with values of 5.04 Debye and 266.65 a.u.—compared to 2, with values of 2.47 Debye and 178.83 a.u., respectively. In theory, high dipole moment values suggest better bonding or complex formation between the ligand and the target protein.30 Therefore, 1 had a better binding interaction with the target protein than 2. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are important quantum chemical parameters for determining the reactivity descriptors.31 The DFT-M05-2X/def2TZVPP level of theory was used to calculate the quantum descriptors, and data was listed in the ESI.† Fig. 4 displays the HOMO and LUMO orbitals and related energy for the substances under investigation. The energy gap values (Eg) between HOMO and LUMO were 6.579 and 7.361 eV for 1 and 2, respectively. A smaller Eg leads to lower kinetic stability and higher chemical reactivity; the opposite is also true. Moreover, the calculated chemical hardness (η) and chemical softness (S) values of 3.290 and 0.152 eV for compound 1, and 3.681 and 0.136 eV for compound 2 also confirmed compound 1 to be more reactive compared to compound 2.
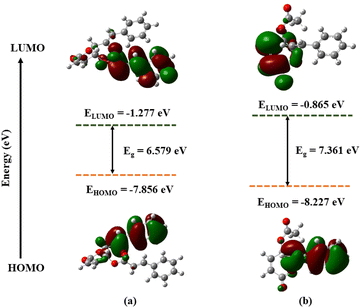 |
| | Fig. 4 Frontier molecular orbitals (HOMO, LUMO) and related energy compound 1 (a), compound 2 (b) calculated by DFT(M052X/def2TZVPP). | |
3.3. Cytotoxic assay
Cytotoxicity studies of two styryl-lactone derivatives were tested on five human cancer cell lines—MCF-7, HepG2, LU-1, AGS, and SW-480—and the HEK-293A human embryonic kidney cells (Table 2). Both styryl-lactone derivatives 1 and 2 displayed notable cytotoxic activities in vitro against the tested cell lines, with IC50 values ranging from 2.05 to 3.96 μM, and from 3.33 to 7.00 μM, respectively. At the same time, ellipticine, the positive control, showed strong cytotoxicity, with IC50 values ranging from 1.42 to 1.83 μM. Moreover, the cytotoxicity of 1 was stronger than 2 in all tested cell lines. The current findings provide a basis for further research and development of G. elegans as a new source of natural anticancer drugs.
Table 2 Cytotoxic activities of 1 and 2
| Cell lines |
IC50 (μM)a |
| 1 |
2 |
Ellipticineb |
| IC50 concentration that inhibits 50% of cell growth. Positive control. |
| MCF-7 |
2.66 ± 0.06 |
5.89 ± 0.43 |
1.83 ± 0.20 |
| HepG2 |
3.03 ± 0.03 |
4.38 ± 0.35 |
1.50 ± 0.16 |
| LU-1 |
2.19 ± 0.09 |
4.76 ± 0.58 |
1.75 ± 0.08 |
| SW480 |
3.96 ± 0.43 |
7.00 ± 0.77 |
1.75 ± 0.08 |
| AGS |
2.08 ± 0.06 |
6.58 ± 0.54 |
1.46 ± 0.12 |
| HEK-293A |
2.05 ± 0.12 |
3.33 ± 0.19 |
1.42 ± 0.12 |
3.4. Molecular docking analysis
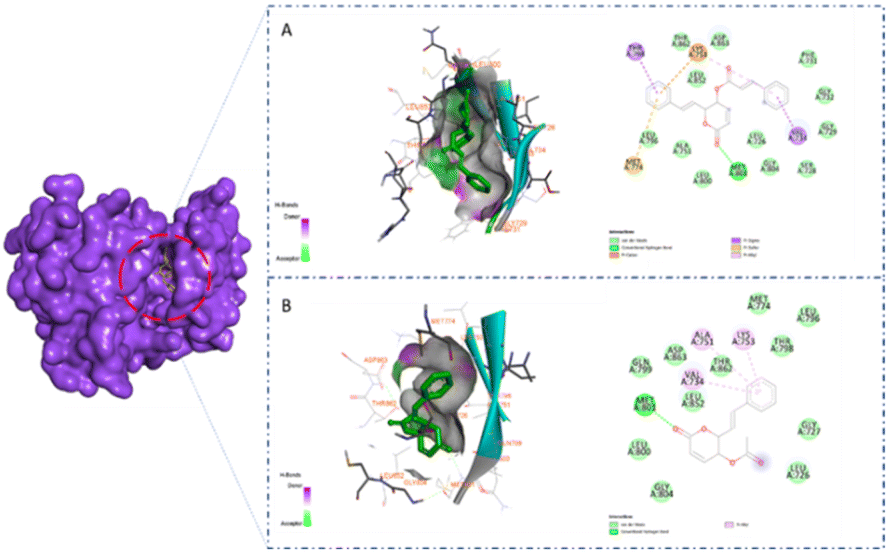
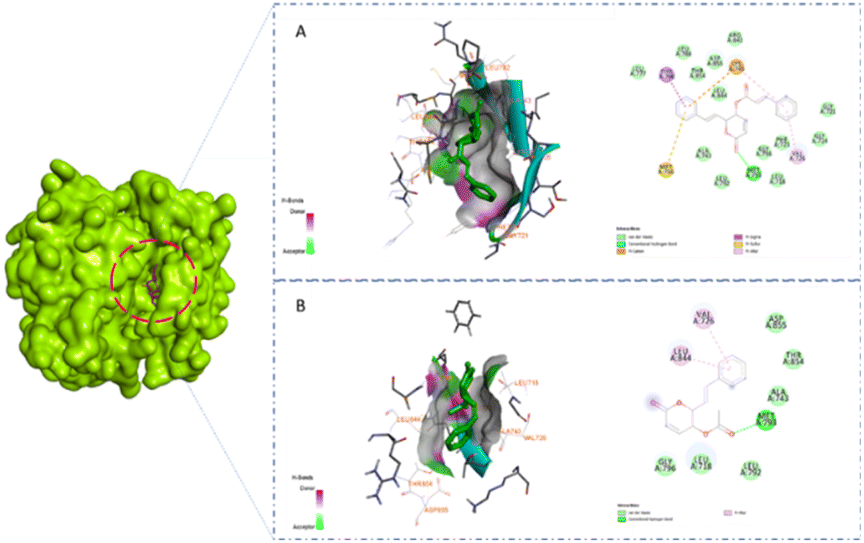
A previous study showed that Osimertinib, a commercial anticancer drug, binds to EGFR and HER-2.6 Therefore, we selected osimertinib as a reference with which to compare the binding affinities of the candidates. The molecular docking results are shown in Table 3. It is highlighted that compound 1 showed stronger binding affinity with EGFR (−8.6 kcal mol−1) and HER-2 (−8.7 kcal mol−1) compared to osimertinib. Regarding EGFR, compound 1 formed one hydrogen bond at amino acid Met793 and five hydrophobic interactions at amino acids Thr790, Met766, Lys745, and Val 726 (Fig. 5A). Meanwhile, compound 2 was found to interact with one hydrogen bond at Met793 and two pi–alkyl interactions at amino acids Leu844 and Val726 (Fig. 5B). A similar result could be seen in the interaction between the two styryl-lactone derivatives and HER-2: compound 1 showed one hydrogen bond at amino acid Met801 and five hydrophobic interactions at amino acids Thr798, Lys753, Met774, and Val734, while compound 2 interacted with HER-2 with one hydrogen bond at Met801 and three pi–alkyl interactions at Val734, Ala751, and Lys753 (Fig. 6).
Table 3 Binding energy of compounds towards EGFR and HER2
| Compound |
EGFR (kcal mol−1) |
HER2 (kcal mol−1) |
| 1 |
−8.6 |
−8.7 |
| 2 |
−6.4 |
−6.9 |
| Osimertinib |
−8.1 |
−7.9 |
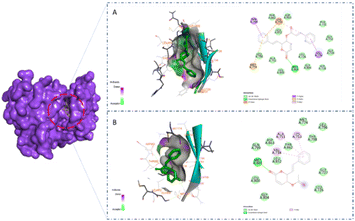 |
| | Fig. 5 Surface and 2D interaction between ligand and EGFR (A) compound 1 (B) compound 2. | |
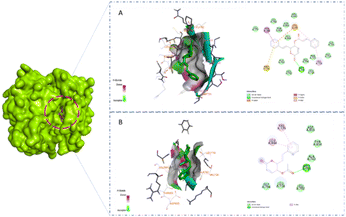 |
| | Fig. 6 Surface and 2D interaction between ligand and HER-2 (A) compound 1 (B) compound 2. | |
Structurally, the benzene ring in the styryl-lactone framework can form hydrophobic interactions with protein residues. In addition, the cyclic ketone group and heterocyclic oxy can also form strong hydrogen bonds. This is consistent with the growth potential of styryl-lactone derivatives in cancer cell lines. Compared to 2, 1 contained one phenyl group at the position of the carboxyl group. Therefore, 1 can form additional hydrophobic interactions, which explains its greater binding affinity. In addition, the interaction site of EGFR and HER-2 is a long groove on the protein surface. Interestingly, the three rings in the structure of 1 are linked together by rotatable bonds; this makes the molecule flexible, enabling it to change its conformation to bind to these proteins.
3.5. ADMET studies
Compounds 1 and 2 acquired acceptable drug-like properties and other drug-likeness parameters following the standard rule.32 The logS values of 1 and 2 were −4.71 and −2.87, respectively, which suggests moderate water solubility. The TPSA values of both also indicated potential for good intestinal absorption. Additionally, the nRotB value of less than 10 suggests that the drug molecules have a favorable stereochemical property contributing to their potential for oral bioavailability. The computational prediction of the pharmacokinetics of 1 and 2 is displayed in the ESI.† The results showed that both compounds are likely to be absorbed in the gastrointestinal tract and are also expected to penetrate the blood–brain barrier. According to the SwissADME prediction, 1 was found to inhibit CYP1A2, CYP2C19, and CYP2C9, while 2 was observed to inhibit only CYP2C19. The expected LD50 values of 1 and 2 were 3.72 mg kg−1 and 3.61 mg kg−1, respectively. The classification of these compounds under “Caution” indicates that high doses of them may be toxic to the human body (Tables 4 and 5).
Table 4 Drug properties of isolated compounds analyzed with SwissADMEa
| Compound |
1 |
2 |
| MW: molecular weight; logP: log of octanol/water partition coefficient; nHBA: number of hydrogen bond acceptor(s); nHBD: number of hydrogen bond donor(s); MR: molar refractivity; nRotB: number of rotatable bonds; logS: log of solubility; TPSA: total polar surface area. |
| MW (g mol−1) |
346.38 |
258.27 |
| LogP |
3.62 |
2.17 |
| nHBD |
0 |
0 |
| nHBA |
4 |
4 |
| TPSA |
52.6 |
52.6 |
| MR |
99.78 |
70.16 |
| Lipinski violation |
0 |
0 |
| LogS |
−4.71 |
−2.87 |
| nRotB |
6 |
4 |
Table 5 ADME predictions of isolated compounds computed by SwissADMEa
| Compound |
1 |
2 |
| LogKp: log of skin permeability; GI abs: gastro-intestinal absorption; BBB per: blood brain barrier permeability; P-gp: P-glycoprotein; CYP: cytochrome-P. |
| GI abs |
High |
High |
| BBB per |
Yes |
Yes |
| P-gp substrate |
No |
No |
| CYP1A2 inhibitor |
Yes |
No |
| CYP2C19 inhibitor |
Yes |
Yes |
| CYP2C9 inhibitor |
Yes |
No |
| CYP2D6 inhibitor |
No |
No |
| CYP3A4 inhibitor |
No |
No |
4. Discussion
A previous study showed that extracts from Goniothalamus species contain compounds with potent cytotoxic effects against cancer cells, including lung, breast, and liver cancer.33 Furthermore, our previous study demonstrated that the crude extract from G. elegans had significant cytotoxic activity on cancer cell lines SW-480, AGS, and SK-LU1.9 In this study, we isolated two styryl-lactone derivatives from G. elegans, including (5R,6R)-2-oxo-6-((E)-styryl)-5,6-dihydro-2H-pyran-5-yl cinnamate, and (−)-5-acetylgoniothalamin. Styryl-lactone derivatives have been extensively studied for their potential anticancer activity. These compounds belong to a class of natural or synthetic compounds that have been found to have significant cytotoxic effects against various cancer cell lines. A previous study showed that styryl-lactones induce apoptosis in cancer cells by disrupting the balance between proapoptotic and antiapoptotic signaling pathways.33 In addition, styryl-lactones have been found to exhibit anti-restenosis activity, which involves abnormal proliferation of vascular smooth muscle cells.34 Our study indicated that both 1 and 2 exhibit strong inhibitory capacity on the growth of cancer cell lines. It is noted that 1 exhibited stronger cytotoxic activity activity than 2. This result is considered to be consistent with theoretical calculation-based chemical structure and molecular modeling results. However, experimental experiments are still needed to evaluate the in silico results.
Although the cytotoxic potential of styryl-lactone derivatives has been demonstrated in many publications, the drawback is that these compounds are not selective on cancer cell lines. Therefore, our study provides evidence based on simulation experiments to further investigate the mechanism of action of these compounds on cancer cell lines. This could be the basis for targeted drug development for further studies in the future. Styryl-lactone derivatives have been found to have antiproliferative and proapoptotic effects on cancer cells, especially those with altered EGFR signaling. Studies have shown that styryl-lactone derivatives can bind to EGFR35,36 and interfere with its activation, leading to the suppression of downstream signaling pathways and the induction of apoptosis in cancer cells.37 Some styryl-lactones also showed cytotoxic and anti-inflammatory effects through the NF-κB pathway via TNF-α inhibition.38 Taken together, this evidence makes styryl-lactones attractive candidates for the development of novel cancer therapeutics. In our study, a molecular docking method was used to validate the binding affinities of 1 and 2 to EGFR and HER-2 proteins through EGF/EGFR signalling pathway. The results indicated 1 had strong binding affinity for both EGFR and HER-2. Additionally, 1 also showed cytotoxic activity on various cancer cell lines. ADMET studies showed 1 acquired acceptable drug-like properties. Although further experiments are needed to elucidate the molecular mechanism of compound 1 on cancer cell lines, this evidence suggests that compound 1 may be considered as a hit compound for the development of cancer drugs through the EGF/EGFR pathway.
5. Conclusions
In conclusion, our study successfully isolated two styryl-lactone derivatives from the aerial parts of G. elegans, one of which is a novel compound and one of which is reported for the first time in this plant. The absolute configuration of the novel compound, (5R,6R)-2-oxo-6-((E)-styryl)-5,6-dihydro-2H-pyran-5-yl cinnamate, was determined through ECD spectrum analysis. The cytotoxicity activity of the two styryl-lactone derivatives was tested against various cancer cell lines and human embryonic kidney cells, with the new compound demonstrating the strongest activity. Molecular docking analysis revealed the potential molecular mechanisms of the compounds in interacting with the target proteins of EGF/EGFR signaling. ADMET predictions further validated the pharmacokinetics and toxicity of these compounds. Overall, this study provides important insights into the potential therapeutic applications of styryl-lactone derivatives from G. elegans.
Author contributions
Tan Khanh Nguyen: methodology, investigation, writing – original draft preparation, writing – reviewing and editing. Linh Thuy Thi Tran: methodology, investigation, writing – original draft preparation, writing – reviewing and editing. Truong Tan Trung: methodology, writing – original draft preparation, writing – reviewing and editing. Phu Tran Vinh Pham: investigation, writing – original draft preparation, writing – reviewing and editing. Linh Thuy Khanh Nguyen: investigation, writing – reviewing and editing. Hoai Thi Nguyen: investigation, writing – original draft preparation, writing – reviewing and editing. Duc Viet Ho: resources, formal analysis, data curation, supervision. Manh Hung Tran: investigation, writing – reviewing and editing.
Conflicts of interest
There is no conflict to declare.
Acknowledgements
This work was supported by Hue University under the Core Research Program, Grant No. NCM.DHH.2023.02.
References
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Ca-Cancer J. Clin., 2021, 71, 7–33 CrossRef PubMed.
- S. Cohen, Biosci. Rep., 1986, 6, 1017–1028 CrossRef CAS PubMed.
- K. D. Brown, Eur. J. Gastroenterol. Hepatol., 1995, 7, 914–922 CrossRef CAS PubMed.
- R. Roskoski, Pharmacol. Res., 2014, 79, 34–74 CrossRef CAS PubMed.
- R. Roskoski, Pharmacol. Res., 2014, 87, 42–59 CrossRef CAS PubMed.
- R. Roskoski, Pharmacol. Res., 2019, 139, 395–411 CrossRef CAS PubMed.
- T. T. T. Nguyen, T. T. T. Nguyen, H. D. Nguyen, T. K. Nguyen, P. T. V. Pham, L. T. Tran, L. T. T. Tran and M. H. Tran, Nat. Prod. Commun., 2023, 18, 1934578X2311672 CrossRef.
- T. K. Nguyen, T. N. Le Nguyen, K. Nguyen, H. V. T. Nguyen, L. T. T. Tran, T. X. T. Ngo, P. T. V. Pham and M. H. Tran, J. Mol. Struct., 2022, 133627 CrossRef CAS.
- L. T. T. Tran, L. H. D. Pham, N. Y. T. Dang, N. T. Nguyen Le, H. B. Nguyen and T. K. Nguyen, Nat. Prod. Commun., 2022, 17(11) DOI:10.1177/1934578X221138435.
- L. T. T. Tran, N. Y. T. Dang, N. T. Nguyen Le, H. T. Nguyen, D. V. Ho, T. T. Do, M. H. Tran, T. K. Nguyen and P. T. V. Pham, Nat. Prod. Commun., 2022, 17(3) DOI:10.1177/1934578X221088110.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS.
- C. Van Vo, Dictionary of Vietnamese medicinal plants [in Vietnamese], Medicine Publishing House, Ha Noi, Vietnam, 2000 Search PubMed.
- N. Suchaichit, K. Kanokmedhakul, N. Panthama, K. Poopasit, P. Moosophon and S. Kanokmedhakul, Fitoterapia, 2015, 103, 206–212 CrossRef CAS.
- R. Lekphrom, S. Kanokmedhakul and K. Kanokmedhakul, J. Ethnopharmacol., 2009, 125, 47–50 CrossRef CAS.
- W. J. Hehre, K. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- T. Van Mourik, M. Bühl and M. P. Gaigeot, Philos. Trans. R. Soc., A, 2014 DOI:10.1098/RSTA.2012.0488.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- GaussView 6.0.16 Free Download - Getintopc, https://webintopc.com/gaussview-6-0-16-free-download/, accessed 13 April 2023.
- K. Mori, N. Matsumoto, S. Nomoto and K. Tsuruta, Open J. Compos. Mater., 2017, 7, 179–184 CrossRef CAS.
- W. Kaplan and T. G. Littlejohn, Briefings Bioinf., 2001, 2, 195–197 CrossRef CAS PubMed.
- G. M. Morris, H. Ruth, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31(2), 455–461 CAS.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- Y. Xu, J. Pei and L. Lai, J. Chem. Inf. Model., 2017, 57, 2672–2685 CrossRef CAS PubMed.
- F. B. Ahmad, W. A. Tukol, S. Omar and A. M. Sharif, Phytochemistry, 1991, 30, 2430–2431 CrossRef CAS.
- T. C. W. Mak, G. C. L. Ee and C. H. Chuah, Nat. Prod. Lett., 1995, 5, 255–259 CrossRef.
- T. K. Kotammagari, S. Paul and A. K. Bhattacharya, ACS Omega, 2019, 4, 22549–22556 CrossRef CAS PubMed.
- N. Van Hue, T. D. Cuong, P. T. Quy, T. Q. Bui, N. T. T. Hai, N. T. Triet, D. D. Thanh, N. T. T. Nhi, N. M. Thai, T. Van Chen and N. T. A. Nhung, ChemistrySelect, 2022, 7(17), e202200680 CrossRef CAS.
- S. M. A. Kawsar, M. A. Hosen, S. Ahmad, Y. El Bakri, H. Laaroussi, T. Ben Hadda, F. A. Almalki, Y. Ozeki and S. Goumri-Said, PLoS One, 2022, 17(11), e0273256 CrossRef.
- M. S. Alam, J. U. Ahmed and D. U. Lee, Appl. Biol. Chem., 2016, 59, 181–192 CrossRef CAS.
- C. Wiart, J. Evidence-Based Complementary Altern. Med., 2007, 4, 299–311 CrossRef.
- K. M. Chan, N. F. Rajab, M. H. A. Ishak, A. M. Ali, K. Yusoff, L. B. Din and S. H. Inayat-Hussain, Chem. Biol. Interface, 2006, 159, 129–140 CrossRef CAS PubMed.
- L. Sun, R. Zhao, X. Lan, R. Chen, S. Wang and G. Du, Molecules, 2014, 19, 19501 CrossRef.
- N. V. Bihud, N. E. Rasol, S. Imran, K. Awang, F. B. Ahmad, C. W. Mai, C. O. Leong, G. A. Cordell and N. H. Ismail, J. Nat. Prod., 2019, 82, 2430–2442 CrossRef CAS.
- P. Khaw-On, W. Pompimon and R. Banjerdpongchai, BioMed Res. Int., 2018, 2018, 7049053 Search PubMed.
- B. Orlikova, M. Schumacher, T. Juncker, C. C. Yan, S. H. Inayat-Hussain, S. Hajjouli, C. Cerella, M. Dicato and M. Diederich, Food Chem. Toxicol., 2013, 59, 572–578 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Linh Thuy Thi Tran‡
a,
Linh Thuy Thi Tran‡