
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sustainable metal and base-free direct amidation of esters using water as a green solvent†
Nanthini Rajendran‡
a,
Kiruthigadevi Kamaraj‡a,
Saranya Janakiramana,
Mary Sarala,
Pierre H. Dixneuf *b and
Charles Beromeo Bheeter*a
*b and
Charles Beromeo Bheeter*a
aDepartment of Chemistry, School of Advanced Sciences, Vellore of Institute of Technology, Vellore-632014, TamilNadu, India. E-mail: charlesbheeter@vit.ac.in
bUniversity of Rennes, ISCR, UMR CNRS 6226, 35000 Rennes, France
First published on 16th May 2023
Abstract
Herein, we report a simple and efficient synthetic approach for direct amidation of esters via C(acyl)–O bond cleavage without any additional reagents or catalysts, using only water as a green solvent. Subsequently, the reaction byproduct is recovered and utilized for the next phase of ester synthesis. This method emphasized metal-free, additive-free, and base-free characteristics making it a new, sustainable, and eco-friendly way to realize direct amide bond formation. In addition, the synthesis of the drug molecule diethyltoluamide and the Gram-scale synthesis of a representative amide are demonstrated.
Amide bond formation is an essential step in pharmaceutical industries for drug designing, synthesis of polymers and material products since amide bonds are an indispensable constituent in peptide derivatives, natural products, and drugs.1 Ester derivatives can be powerful alternatives for amidation reactions rather than the commonly used organic acid halides due to their environment-friendly nature and ready availability. Oils, fatty acids, triesters of glycerol, and fruits are good sources of esters. Thus the methods of conversion of esters into amides are becoming significant.2 Traditionally, most pharmaceutical industries follow the acid-amine coupling method with an over-stoichiometric amount of coupling reagents and bases for amide bond formation.3
Wu and Beller et al. successfully developed a new catalytic method for the synthesis of amides using CO gas by amino carbonylation methodology.4 Another well-known approach in amide bond formation is the C(acyl)–O bond cleavage of esters via the catalytic pathway, using metal catalysts,5 (Scheme 1, Part A, B) strong base5,6 (Scheme 1, Part C), various additives,7 and often non-greener solvents.8 This method requires suitable substituents for activating esters9 and for effective conversion. In addition, aggressive organometallic reagents such as BuLi, AlMe3, Al(O-tBu) along with Ni(cod)5g catalyst or strong bases like LiHMDS5a and NaH10 are required for activating some esters. When highly activated esters are used, amidation is achieved using K2CO3 as a base with amines.11
 | ||
| Scheme 1 Different approaches towards C(acyl)–O bond cleavage of esters to produce amides. (A) Precious metal approach for amide bond formation from esters (i)5a, (ii)7a (B) base metal approach for amide bond18 (C) metal-free with a strong base approach for amide bond formation8a (D) this work. | ||
While the cleavage of esters yields carbonylative and decarbonylative12 products, the selectivity is controlled intermittently by the nature of the ligands.5b,12c For instance, Ni-(N-heterocyclic carbene) ligand gives carbonylative product13 while [Ni-bis-phosphine] ligand yields decarbonylative product.12e,14 Other non-noble metals also cleaved the C(acyl)–O bond of esters with hybrid ligands such as NHC with nitrogen donors in Mn(pincer)15 (Scheme 1, Part B) and La(OTf)3 as lewis acid catalyst.16 Besides, the amidation process is achieved through transition metal complexes such as Pd–NHC,17a,b Ni–NHC, Pd-Phosphine ligands, Mn pincer complex,15 and heterobimetallic lanthanide sodium alkaloids.18 In all these methods, the need for a catalyst, reagents, additives, and/or base, non-green solvents are prevalent.
Hence, an environmentally friendly methodology is necessary for direct amidation reactions, especially for the industrial synthesis of amides in large quantities. Herein, we are reporting amide bond formation via the coupling of inactivated esters with amines in the presence of water in an oil bath at 110 °C with the recovery of side product phenol, which is further used in the synthesis of ester moieties. Overall, the conversion of simple esters into amides under metal and base-free conditions using water as a green solvent leading to an atom-economical with reduced cost is expected to be excellent progress in the area of amide synthesis. To the best of our knowledge, a sustainable direct amidation of esters under metal-free, additive-free, and base-free conditions using water as a solvent has not been explored.
At the outset, we have outlined the model scheme for the amidation of esters using phenyl benzoate (Table 1, 1a) and benzylamine (Table 1, 2a) as the ester and amine sources, respectively. Initially, we preferred using heterogeneous catalysts over traditional ones as the former can be recycled and reused several times. Accordingly, the reaction was performed using silica material, namely KIT-6 (mesoporous silica nanoparticles 10–100 μm) and KIT-6 with metal precursor (Fe-KIT-6), which furnished an amidation product (Table 1, 3a) with 78% and 82% yield respectively (Table 1, entries 1–2). Then we moved to another silica material, SBA-15 (mesoporous silica nanoparticles < 150 μm), which yielded 60% of the product (3a). Subsequently, we tried the reaction with normal mesoporous silica 200–400 mesh (25 mg), which yielded 80% of the product (3a) using THF at 110 °C (Table 1, entries 3–4).
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yield (%) |
| a Conditions: phenyl benzoate 0.2525 mmol, benzylamine 0.5050 mmol, catalyst 25 mg, solvent 3 mL, 110 °C in oil bath, 12 h, isolated yields. | |||
| 1 | Fe KIT-6 | THF | 82 |
| 2 | KIT-6 | THF | 78 |
| 3 | SBA-15 | THF | 60 |
| 4 | Silica | THF | 80 |
| 5 | Catalyst-free | THF | 80 |
| 6 | Catalyst-free | CH3CN | 78 |
| 7 | Catalyst-free | Water | 95 |
| 8 | Catalyst-free | Ethanol | 45 |
| 9 | Catalyst-free | Methanol | 32 |
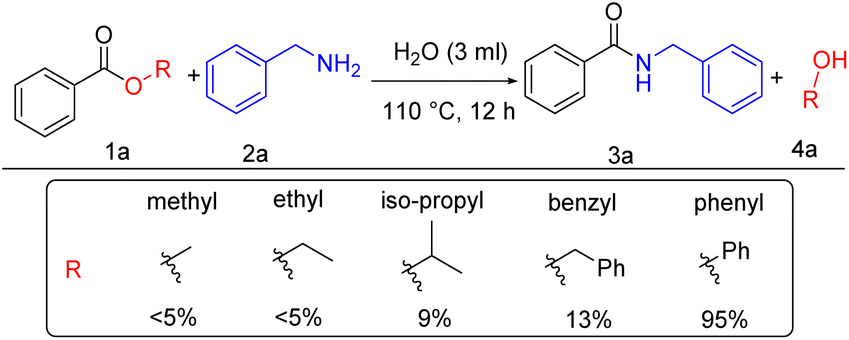
Interestingly, standard mesoporous silica produced a very good yield, better than SBA-15, KIT-6, and comparable to Fe-KIT-6. As the reaction proceeded well with ordinary mesoporous silica, we wanted to determine the catalyst's role in the reaction. Subsequently, we tested for the catalyst-free performance of the reaction with different solvents. Surprisingly, the reaction proceeded well without a catalyst. So, to study the catalyst-free reaction further, we selected two different types of solvents according to their nature. The polar aprotic solvents like tetrahydrofuran and acetonitrile yielded 80% and 78% products (Table 1, entries 5–6). On the other hand, polar protic solvents, ethanol, and methanol showed low yields of 45% and 32%, respectively (Table 1, entries 8–9). Interestingly, when we tested with a universal solvent, water, it was observed to form the desired product with an excellent yield of 95% after 12 h (Table 1, entry 7). Hence, we concluded that the amidation process of esters proceeds well without a catalyst using the universal solvent water. This approach is best as it is greener and more atom economical than all previous approaches known so far. After optimization, we screened different esters such as methyl benzoate, ethyl benzoate, iso-propyl-benzoate, benzyl benzoate, and phenyl benzoate. Under our standard reaction condition methyl and ethyl benzoate gave a yield of <5%, iso-propyl- and benzyl benzoate gave 9% and 13% yield respectively, whereas phenyl benzoate gave the desired product with a 95% yield (Scheme 2).
 | ||
| Scheme 2 Optimization of different esters. | ||
From this, we hypothesized that phenol could act as a good leaving group in phenylesters compared to others esters without coupling reagent and base. Also, the inherent nature of the phenoxy group in phenyl benzoate could be related to its ability to involve in hydrogen bonding interaction with water to facilitate the amidation reaction of esters in water.19 Accordingly, the affinity of the phenolic moiety of the phenyl esters in water plays an important role than in organic solvents.
In the first part of the substrate scope, we explored the activity of aromatic esters with aliphatic amines employing the novel methodology. Benzylamine-bearing electron-donating and electron-withdrawing substituents in the para position, such as 4-methyl benzylamine with phenyl ester, give 76% yield, 4-methoxy benzylamine afforded 89% yield and 4-chloro benzylamine with 84% yield (Table 2, 3b–3d). Then we introduced substituents in ester moieties such as 4-chloro-, 2-methyl-, 3-methyl- and 2-methoxy-phenyl benzoate, which afforded corresponding amides with 94%, 94%, 96%, and 73% of yields respectively (Table 2, 3e–3h). Later, we introduced benzylamine-bearing electron-withdrawing substituents in ortho positions, such as 2-fluoro benzylamine, which converted into respective amide with a moderate yield of 39% (Table 2, 3i). Next, we introduced the 2-methoxy (Table 2, 3j–3k), and 3-methoxy (Table 2, 3l–3m) substituents in ester moieties, and we correlated it with different substituents in benzyl amines such as 2-chloro-, 2-fluoro-, 4-fluoro- and 4-chloro-benzylamine which afforded 72% to 96% yield (Table 2, 3j–3m).
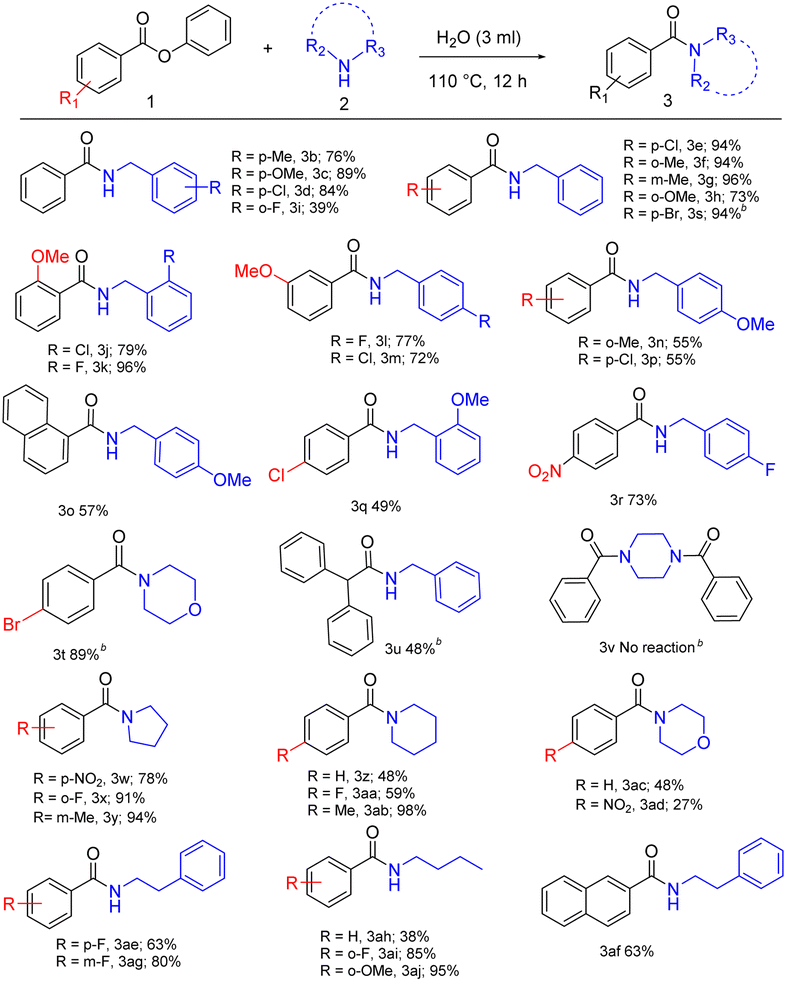
Also, 4-methoxy benzylamine with 2-methyl phenyl benzoate, 1-phenyl naphtholate, and 4-chloro phenyl benzoate as ester moieties yielded 55% to 57% of respective amides (Table 2, 3n–3p). In the case of 4-chloro phenyl benzoate with 2-methoxy benzylamine and 4-nitro phenyl benzoate with 4-fluoro benzylamine afforded yields of 49% and 73%, respectively (Table 2, 3q–3r). Likewise, 4-bromo phenyl benzoate afforded 94% yield with benzylamine (Table 2, 3s), and 4-bromo phenyl benzoate afforded 89% yield with morpholine (Table 2, 3t). The reaction between phenyl 2,2 diphenyl acetate with benzylamine afforded a 48% yield (Table 2, 3u). But under the same optimized conditions, n-Boc piperazine was completely inactive towards phenyl esters (Table 2, 3v). Aliphatic cyclic amines were also utilized under the optimized reaction conditions. Pyrrolidine underwent the amidation process with 4-nitro-, 2-fluoro-, and 3-methyl-phenyl benzoate affording 78%, 91% and 94% yield, respectively (Table 2, 3w–3y). The reaction of piperidine with phenyl benzoate, 4-fluoro-, and 4-methyl-phenyl benzoate provided 48%, 59%, and 98% of corresponding amides (Table 2, 3z, 3aa, 3ab). Morpholine reacted with phenyl benzoate to give a 48% yield while the reaction with 4-nitro phenyl benzoate afforded only a 27% yield (Table 2, 3ac, 3ad).
This may be due to the influence of the electronic nature of ester moieties in the amidation process. Particularly, when the alkyl chain of the amine was lengthened as in the reaction of phenethylamine with ester moieties such as 4-fluoro phenyl benzoate, 2 phenyl naphtholate and 3-fluoro phenyl benzoate giving corresponding amides, which gave a good yield of 63%, 63%, and 80% (Table 2, 3ae, 3af, 3ag). Phenyl benzoate, 2-fluoro- and 2-methoxy-phenyl benzoate coupled with n-butylamine provided the corresponding amides with moderate to good yields of 38%, 85%, and 95% (Table 2, 3ah, 3ai, 3aj).
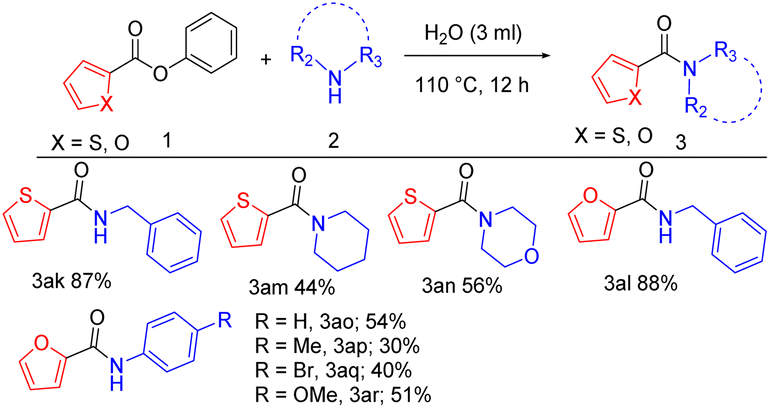
In the second part of the substrate scope, we have explored the activity of heteroaromatic esters with aliphatic and aromatic amines via new methodology. When benzylamine is reacted with phenyl thiophene-2-carboxylate and phenyl furan-2-carboxylate, 87%, and 88% yield respectively (Table 3, 3ak, 3al). Phenyl thiophene-2-carboxylate, when coupled with piperidine and morpholine gives 44%, and 56% yield (Table 3, 3am, 3an). To evaluate the scope of aromatic amines, a diverse range of heterocyclic esters was explored. Accordingly, phenyl furan-2-carboxylate coupled successfully with aniline, 4-toluidine, 4-bromo aniline, and 4-anisidine affording 54%, 30%, 40%, and 51% yield respectively (Table 3, 3ao, 3ap, 3aq, 3ar).
| a Reaction conditions: 1 esters (1 eq), 2 amines (1.2 eq), H2O (3 mL), 110 °C, 12 h. |
|---|
 |
In the third part of the substrate scope, we explored the activity of aliphatic esters, amino acid ester with aliphatic and aromatic amines. A selection of fused aliphatic esters was employed for the reaction as they are vital components of bioactive compounds and useful intermediates in drug synthesis. Phenyl cyclopropane carboxylate coupled with benzylamine yields 96% of the product (Table 4, 3as). Phenyl cyclopropane carboxylate with aniline, 4-methyl- and 4-chloro-aniline provide 42% to 81% of yield (Table 4, 3at, 3au, and 3av). Phenyl cyclobutane carboxylate reacts with aniline affording a corresponding yield of 74% (Table 4, 3aw), and the reaction with morpholine yields a trace amount of product (Table 4, 3ax).
| a Conditions: ester (1 eq), amine (1.2 eq), solvent 3 mL, 110 °C, 12 h. |
|---|
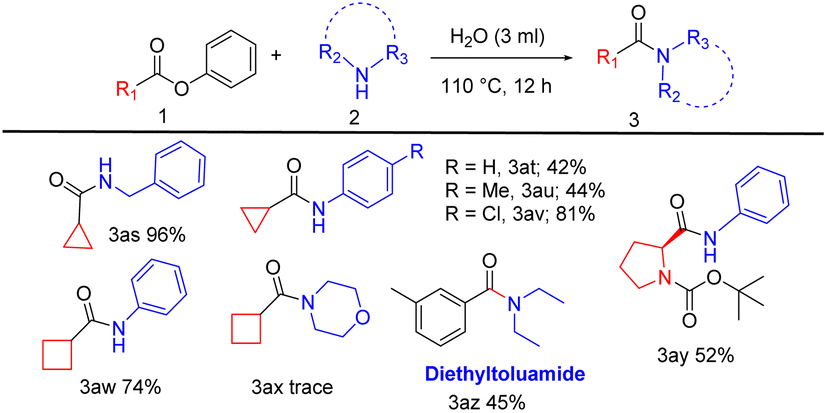 |
Also, this reaction was influential in the context of the direct synthesis of biologically relevant compounds like tert-butyl(S)-2-(phenyl carbamoyl)pyrrolidine-1-carboxylate (Table 4, 3ay) from n-boc L-proline an amino acid with 52% yield. Significantly, we prepared the drug, diethyltoluamide (Table 4, 3az), an insect repellent,20 by aminolysis of meta-methyl phenyl benzoate with diethylamine to give a moderate yield of 45%.
To prove the proposed methodology's environment-friendly nature, the side product phenol (Scheme 3) was recovered as it could be used further in the preparation of ester derivatives (see the ESI† for more details).
 | ||
| Scheme 3 Recycling of side product. | ||
Having demonstrated that water is an effective solvent for a range of direct amidation products employing the present methodology, we sought to exemplify the method on a Gram scale. We assessed the Gram scale synthesis (Scheme 4) with a mixture of phenyl benzoate 1a (1g, one equiv), benzylamine 2a (810 mg, 1.5 equiv), which was stirred in the presence of H2O at 110 °C for 12 h to afford the formation of 953 mg of benzyl benzamide 3a with 90% yield.
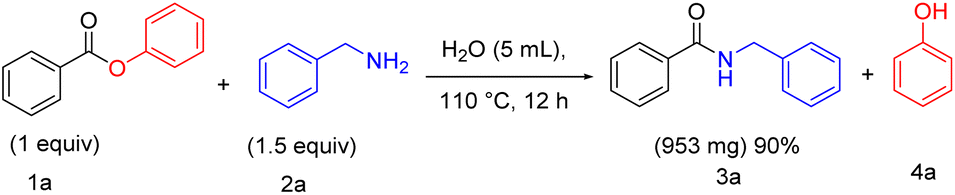 | ||
| Scheme 4 Gram scale synthesis. | ||
We have found a metal-free, base/additive-free amidation protocol under aqueous conditions. Based on the substrate scope and Gram scale experiment, we show that the aromatic, aliphatic, and amino acid ester derivatives with aliphatic and aromatic amines exhibit good activity to give the corresponding amidation products. Furthermore, successful recovery of side product for further use in ester derivative synthesis is done for making the discussed approach sustainable. This is the first report on ester acyl(C–O) bond cleavage for amide formation with water as an eco-friendly benign solvent.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
NR and KK acknowledge VIT for providing a fellowship. Authors acknowledge VIT for seed grants (SG20210184), infrastructure and instrumentation facilities, and SERB (CRG/2021/07286) for research funding.Notes and references
- (a) M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS; (b) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (d) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC; (e) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- (a) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC; (b) M. Ishikawa, Y. Warita, E. Takahisa and Y. Ohkubo, in Comprehensive Natural Products II, ed. H.-W. Liu and L. Mander, Elsevier, Oxford, 2010, pp. 595–629, DOI: DOI:10.1016/B978-008045382-8.00107-6; (c) A. J. Meléndez-Martínez, P. Mapelli-Brahm, D. Hornero-Méndez and I. M. Vicario, in Carotenoid Esters in Foods: Physical, Chemical and Biological Properties, The Royal Society of Chemistry, 2019, pp. 1–50, DOI: 10.1039/9781788015851-00001.
- (a) N. G. Anderson, in Practical Process Research and Development, ed. N. G. Anderson, Academic Press, Oxford, 2nd edn, 2012, pp. 89–120, DOI: DOI:10.1016/B978-0-12-386537-3.00004-6; (b) R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed; (c) F. Albericio and A. El-Faham, Org. Process Res. Dev., 2018, 22, 760–772 CrossRef CAS.
- L. Wu, X. Fang, Q. Liu, R. Jackstell, M. Beller and X.-F. Wu, ACS Catal., 2014, 4, 2977–2989 CrossRef CAS.
- (a) T. Zhou, G. Li, S. P. Nolan and M. Szostak, Org. Lett., 2019, 21, 3304–3309 CrossRef CAS PubMed; (b) T. Ben Halima, J. K. Vandavasi, M. Shkoor and S. G. Newman, ACS Catal., 2017, 7, 2176–2180 CrossRef CAS; (c) A. Mondal, M. Subaramanian, A. Nandakumar and E. Balaraman, Org. Lett., 2018, 20, 3381–3384 CrossRef CAS; (d) M. B. Kurosawa, R. Isshiki, K. Muto and J. Yamaguchi, J. Am. Chem. Soc., 2020, 142, 7386–7392 CrossRef CAS PubMed; (e) A. H. Dardir, P. R. Melvin, R. M. Davis, N. Hazari and M. Mohadjer Beromi, J. Org. Chem., 2018, 83, 469–477 CrossRef CAS PubMed; (f) K. Kim and S. H. Hong, J. Org. Chem., 2015, 80, 4152–4156 CrossRef CAS PubMed; (g) L. Hie, N. F. Fine Nathel, X. Hong, Y.-F. Yang, K. N. Houk and N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 2810–2814 CrossRef CAS PubMed; (h) Y.-L. Zheng and S. Newman, ACS Catal., 2019, 9, 4426–4433 CrossRef CAS; (i) W. B. Wang and E. J. Roskamp, J. Org. Chem., 1992, 57, 6101–6103 CrossRef CAS; (j) G. Li and M. Szostak, Nat. Commun., 2018, 9, 4165 CrossRef PubMed; (k) J. L. Vrijdag, F. Delgado, N. Alonso, W. M. De Borggraeve, N. Pérez-Macias and J. Alcázar, Chem. Commun., 2014, 50, 15094–15097 RSC.
- (a) L. H.-G. Kim BoRam, K. Seung-Beom, G. H. Sung, J.-J. Kim, J. K. Park, L. Sang-Gyeong and Y. Y. Jin, Synthesis, 2012, 44, 42–50 CrossRef; (b) R. Zhang, W.-Z. Yao, L. Qian, W. Sang, Y. Yuan, M.-C. Du, H. Cheng, C. Chen and X. Qin, Green Chem., 2021, 23, 3972–3982 RSC; (c) W. I. Nicholson, F. Barreteau, J. A. Leitch, R. Payne, I. Priestley, E. Godineau, C. Battilocchio and D. L. Browne, Angew. Chem., Int. Ed., 2021, 60, 21868–21874 CrossRef CAS PubMed; (d) T. Ohshima, Y. Hayashi, K. Agura, Y. Fujii, A. Yoshiyama and K. Mashima, Chem. Commun., 2012, 48, 5434–5436 RSC.
- (a) G. Li and M. Szostak, Synthesis, 2020, 52, 2579–2599 CrossRef CAS; (b) N. Caldwell, C. Jamieson, I. Simpson and A. J. B. Watson, Chem. Commun., 2015, 51, 9495–9498 RSC; (c) A. Ramirez, B. Mudryk, L. Rossano and S. Tummala, J. Org. Chem., 2012, 77, 775–779 CrossRef CAS PubMed; (d) C. G. McPherson, N. Caldwell, C. Jamieson, I. Simpson and A. J. B. Watson, Org. Biomol. Chem., 2017, 15, 3507–3518 RSC; (e) D. A. Rankic, C. M. Stiff, C. W. am Ende and J. M. Humphrey, J. Org. Chem., 2017, 82, 12791–12797 CrossRef CAS PubMed; (f) K. E. Price, C. Larrivée-Aboussafy, B. M. Lillie, R. W. McLaughlin, J. Mustakis, K. W. Hettenbach, J. M. Hawkins and R. Vaidyanathan, Org. Lett., 2009, 11, 2003–2006 CrossRef CAS PubMed.
- (a) Y. Liu, S. Shi, M. Achtenhagen, R. Liu and M. Szostak, Org. Lett., 2017, 19, 1614–1617 CrossRef CAS PubMed; (b) M. M. Rahman, G. Li and M. Szostak, J. Org. Chem., 2019, 84, 12091–12100 CrossRef CAS PubMed; (c) P. V. Ramachandran, H. J. Hamann and S. Choudhary, Org. Lett., 2020, 22, 8593–8597 CrossRef CAS PubMed.
- (a) L. Ling, C. Chen, M. Luo and X. Zeng, Org. Lett., 2019, 21, 1912–1916 CrossRef CAS PubMed; (b) C. W. Cheung, N. Shen, S.-P. Wang, A. Ullah, X. Hu and J.-A. Ma, Org. Chem. Front., 2019, 6, 756–761 RSC; (c) L.-Y. Chen and M.-F. Wu, Synthesis, 2019, 51, 1595–1602 CrossRef CAS; (d) T. Ooi, E. Tayama, M. Yamada and K. J. S. Maruoka, Synlett, 1999, 6, 729–730 CrossRef; (e) X. Yang and V. B. Birman, Org. Lett., 2009, 11, 1499–1502 CrossRef CAS PubMed; (f) Y. Dongqiang and X. Jiaxi, Curr. Microw. Chem., 2020, 7, 74–82 CrossRef.
- S. A. Rzhevskiy, A. A. Ageshina, G. A. Chesnokov, P. S. Gribanov, M. A. Topchiy, M. S. Nechaev and A. F. Asachenko, RSC Adv., 2019, 9, 1536–1540 RSC.
- S. Karthik, R. Sreedharan and T. Gandhi, ChemistrySelect, 2019, 4, 175–180 CrossRef CAS.
- (a) K. Amaike, K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 13573–13576 CrossRef CAS PubMed; (b) K. Muto, J. Yamaguchi, D. G. Musaev and K. Itami, Nat. Commun., 2015, 6, 7508 CrossRef PubMed; (c) A. Chatupheeraphat, H.-H. Liao, W. Srimontree, L. Guo, Y. Minenkov, A. Poater, L. Cavallo and M. Rueping, J. Am. Chem. Soc., 2018, 140, 3724–3735 CrossRef CAS PubMed; (d) T. B. Boit, A. S. Bulger, J. E. Dander and N. K. Garg, ACS Catal., 2020, 10, 12109–12126 CrossRef CAS PubMed; (e) L. Guo and M. Rueping, Acc. Chem. Res., 2018, 51, 1185–1195 CrossRef CAS PubMed.
- T. Ben Halima, J. Masson-Makdissi and S. G. Newman, Angew. Chem., Int. Ed., 2018, 57, 12925–12929 CrossRef CAS PubMed.
- C. A. Malapit, M. Borrell, M. W. Milbauer, C. E. Brigham and M. S. Sanford, J. Am. Chem. Soc., 2020, 142, 5918–5923 CrossRef CAS PubMed.
- Z. Fu, X. Wang, S. Tao, Q. Bu, D. Wei and N. Liu, J. Org. Chem., 2021, 86, 2339–2358 CrossRef CAS PubMed.
- H. Morimoto, R. Fujiwara, Y. Shimizu, K. Morisaki and T. Ohshima, Org. Lett., 2014, 16, 2018–2021 CrossRef CAS PubMed.
- S. Shi and M. Szostak, ChemComm, 2017, 53, 10584–10587 RSC.
- Z. Li, C. Wang, Y. Wang, D. Yuan and Y. Yao, Asian J. Org. Chem., 2018, 7, 810–814 CrossRef CAS.
- R. Parthasarathi, V. Subramanian and N. Sathyamurthy, J. Phys. Chem., 2005, 109, 843–850 CrossRef CAS PubMed.
- D. L. Sudakin and T. Osimitz, in Hayes' Handbook of Pesticide Toxicology, ed. R. Krieger, Academic Press, New York, 3rd edn, 2010, pp. 2111–2125, DOI: DOI:10.1016/B978-0-12-374367-1.00098-7.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02637j |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |