
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTail-approach based design, synthesis, and cytotoxic evaluation of novel disubstituted and trisubstituted 1,3-thiazole benzenesulfonamide derivatives with suggested carbonic anhydrase IX inhibition mechanism†
Samir Bondock *a,
Tallah Albarqia,
Mohamed Abbouda,
Tamer Nasrbc,
Nada M. Mohamedc and
Moaz M. Abdoud
*a,
Tallah Albarqia,
Mohamed Abbouda,
Tamer Nasrbc,
Nada M. Mohamedc and
Moaz M. Abdoud
aChemistry Department, Faculty of Science, King Khalid University, 9004 Abha, Saudi Arabia. E-mail: bondock@mans.edu.eg
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Helwan University, 11795 Helwan, Cairo, Egypt
cPharmaceutical Chemistry Department, Faculty of Pharmacy, Modern University for Technology and Information, MTI, 12055 Cairo, Egypt
dEgyptian Petroleum Research Institute, Nasr City, 11727, Cairo, Egypt
First published on 10th August 2023
Abstract
A novel series of 2,4,5- and 2,3,4-trisubstituted thiazole hybrids with 1,3,4-thiadiazolylbenzenesulfonamide was designed following the tail approach as possible hCAIX inhibitors. The key intermediate 1 was condensed with thiosemicarbazide 2a to give 1,3,4-thiadiazolylthiosemicarbazone 3, which upon hetero-cyclization with substituted α-haloketones and esters afforded 2,4,5-trisubstituted thiazole-1,3,4-thiadiazole conjugates 4–8. Furthermore, the trisubstituted thiazole-1,3,4-thiadiazole hybrids 12a–d were synthesized via the regioselective cyclization of 4-substituted-1,3,4-thiadiazolylthiosemicarbazones with phenacyl bromide. The cyclized 2,4-disubstituted thiazole 4 enhanced cytotoxicity by nine, four and two times against HepG-2, Caco2, and MCF-7, respectively. Moreover, the simple methyl substitution on the thiosemicarbazone terminus 9a improved the parent derivative 3 cytotoxicity by nine, fourteen, and six times against HepG-2, Caco2, and MCF-7, respectively. This astonishing cytotoxicity was elaborated with hCAIX molecular docking simulation of 4, 9a, and 12d demonstrating binding to zinc and its catalytic His94. Furthermore, molecular dynamic simulation 9a revealed stable hydrogen bonding with hCAIX with interaction energy of −61.07 kcal mol−1 and ΔGbinding MM-PBSA of −9.6 kcal mol−1.
1. Introduction
There is a rising rate of research in medicinal chemistry to discover new effective and specific chemotherapeutic agents for cancer treatment.1 Although many available anticancer drugs exist, drug resistance is still a major problem for their effective use. The hybridization approach is considered one of the most effective methods in drug design to attain novel anticancer agents capable of limiting their proliferation.2 The synergistic effects of the hybrid structures can display pharmacological properties that are higher than those of individual structures, as well as decreased toxicity and fewer adverse effects. This trend in drug design is most obvious in thiazole chemistry,3 where a growing number of novel thiazole-derived hybrids have been described with a strong emphasis on their various enzyme-inhibitory properties.4 Among the several thiazole derivative platforms, promising and expanding applications with the sulfonamide system have been well exploited for producing carbonic anhydrase inhibitors.5 Furthermore, sulfonamide-substituted 1,3,4-thiadiazoles have been demonstrated to have potential carbonic anhydrase inhibitory characteristics with anticancer activity.6Carbonic anhydrases (hCAs, EC 4.2.1.1) are metalloenzymes that control the balance between carbon dioxide and bicarbonate ions through the adding-removing water mechanism. Their catalytic activity involved zinc(II) ions as a cofactor to regulate the cellular pH.7,8 To date, fifteen human isoforms have been isolated; 12 of them are catalytic isoforms with a zinc-binding site.9–11 Out of the 12 catalytic isoforms, the transmembrane hCAIX and hCAXII were strongly correlated with cancer progression, which assisted the cancerous cell in accommodating the hypoxia conditions promoting metastases.12 They were considered an interesting target for new anticancer agents' development, especially with their established protein structures. Despite their variable tissue distribution, the hCA isoforms show high similarity in their protein structure. The main chain structure of the tumor-associated hCAIX demonstrated RMSD 2.02, 2.05, and 1.87 Å with hCAI, hCAII, and hCAXII, respectively.13 Moreover, hCAIX shared 83 conserved amino acids with the other tumor-associated isoform hCAXII while sharing 79 and 75 conserved residues with hCAI and hCAII, respectively.13
Among the hCAIX inhibitors, thiadiazole-benzenesulfonamide derivatives had established appreciated activity, with the sulfonamide moiety acting as the zinc-binding group (ZBG). For instance, acetazolamide AAZ and methazolamide MAZ are clinically proven non-selective hCA inhibitors with thiadiazole sulfonamide core that possess the common structural features of classical inhibitors (Fig. 1).14,15 Merging the thiadiazole scaffold with the ZBG benzenesulfonamide was followed into Ia showed hCAXII Ki of 34.2 nM and Ib had hCAIX Ki of 86.4 nM.16 Several thiazole benzenesulfonamides had established valued hCA inhibition, such as II with Ki 0.4 and 0.11 μM of isoforms hCAI and hCAII, respectively.17 Similarly, the thiazole-benzenesulfonamide hybrids IIIa–c were reported with a potential inhibition of hCAIX, especially when increasing the lipophilicity of IIIa. This lipophilicity optimization improved their inhibitory potentials by three and 1.5 times against hCAIX and hCAXII, respectively, upon adding a phenyl IIIb or 4-chlorophenyl IIIc moieties to the C4-thiazole ring (Fig. 1).18
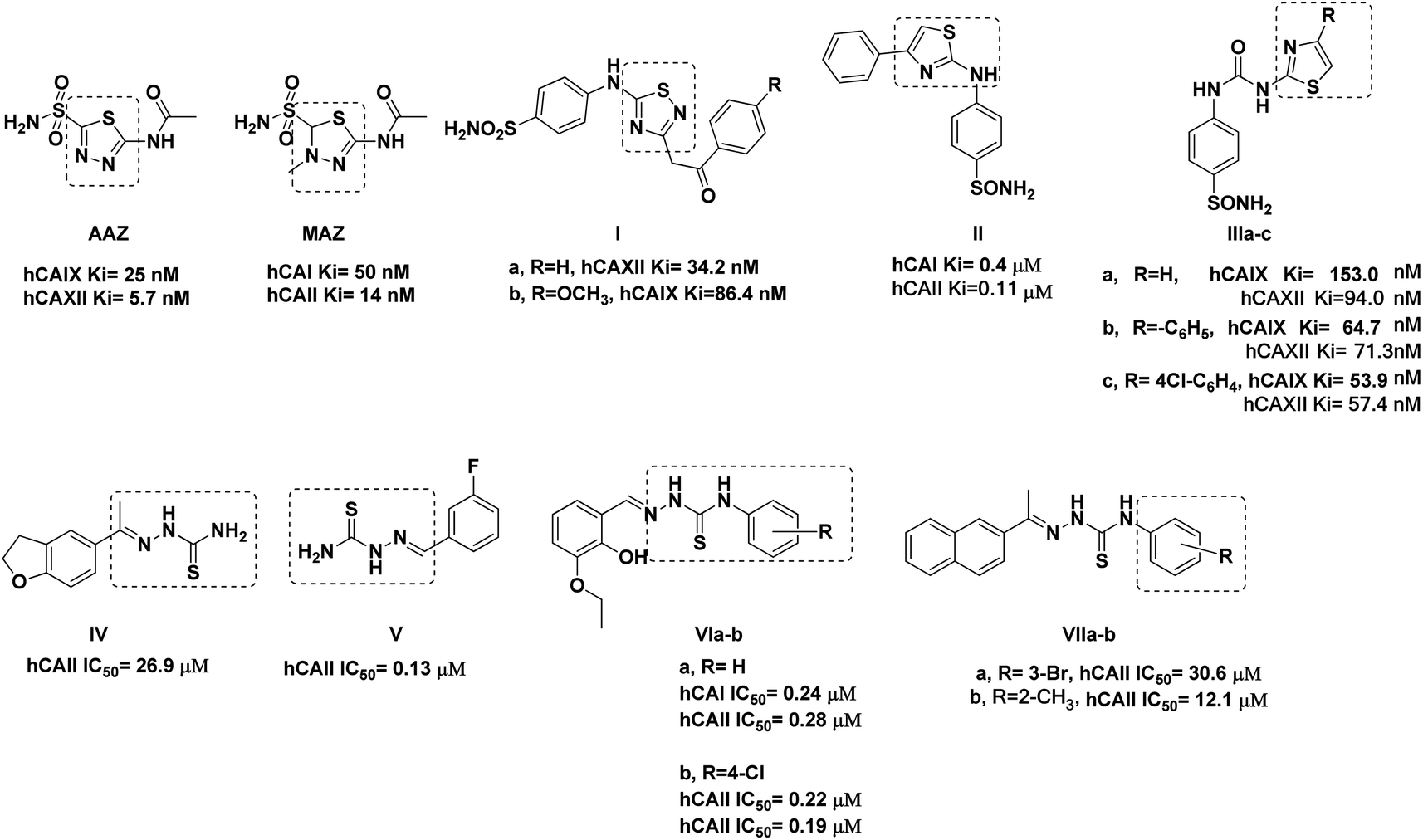 | ||
| Fig. 1 Reported thiadiazole, thiazole, and thiosemicarbazone derivatives with carbonic anhydrase inhibition and anticancer activity. | ||
On the other hand, several thiosemicarbazone derivatives were reported as hCA isoform inhibitors, such as IV–V, which showed hCAII IC50 of 26.9 and 0.13 μM, respectively.19,20 Moreover, the aromatic linked thiosemicarbazone proved appreciated inhibition against both hCA isoforms hCAI and hCAII with sub-micromolar IC50 values, including VIa and VIb with IC50 range 0.19–0.28 μM (Fig. 1).21 Similarly, the naphthalene–thiosemicarbazone VIIa and VIIb hybrids were reported to exhibit hCAII inhibition at the micromolar level (Fig. 1), with their molecular docking simulation highlighting the importance of the thiosemicarbazone sulfur and nitrogen groups to anchor zinc ion and the binding site residues Gln91, Thr198, and Thr199.20
Based on the information above, we designed novel hCAIX inhibitors by optimizing the structure of the non-selective hCA inhibitor AAZ (Fig. 2). In the target compounds, we kept the sulfonamide moiety as a zinc-binding group. Besides its ability to bind to the zinc in the active site, the sulfonamide moiety could bind to His94, Leu198, and/or Thr199 through H-bond formation. We then replaced the 1,3,4-thiadiazole spacer of AAZ with 5-imino-4,5-dihydro-4-(phenyl)-1,3,4-thiadiazole moiety. The main reason is the ability of the imino group at C5-thiadiazole to form a hydrogen bond with His64. It was proved that the role of His64 is shuttling the proton during hCAs catalytic activity.13
 | ||
| Fig. 2 Design the targeted 2,4,5- and 2,3,4-trisubstituted thiazole hybrids with 1,3,4-thiadiazolylbenzenesulfonaminde derivatives as hCAIX inhibitors. | ||
Moreover, the phenyl thiadiazole could form hydrophobic interactions with the amino acids within the active site of hCA, such as Leu198 and Leu135. Also, we replaced the small acetyl tail of AAZ with longer and flexible substituted thiosemicarbazide moieties. This moiety allows hydrogen bond formation with the amino acids Gln67, Asn62, Arg60, and/or Gln92 of the active site of hCA. On the other hand, the ring closure strategy was applied to introduce the thiazole moiety in the tail part by cyclization of the thiosemicarbazide moiety. Aromatic substitution at C4 of the thiazole ring might form hydrophobic interaction with the Val131 or Leu91. Besides, the aromatic group at C3-thiazole might form extra hydrophobic interaction with Gln92 and/or Val131. In a trial for finding the thiazole ring's best substitution pattern, we tried a set of substitutions to get 2,3,4- and 2,4,5-trisubstituted thiazole derivatives to adopt a conformation that might fill the unoccupied region of hCAIX active site.
2. Results and discussion
2.1. Chemistry
The synthetic strategy for constructing the target two series of disubstituted and trisubstituted 1,3-thiazole sulfonamide derivatives (Schemes 1 and 2). | ||
| Scheme 1 Synthesis of 1,3,4-thiadiazolylbenzenesulfonaminde tagged with 2,4-disubstituted and 2,4,5-trisubstituted thiazole derivatives 4–8. | ||
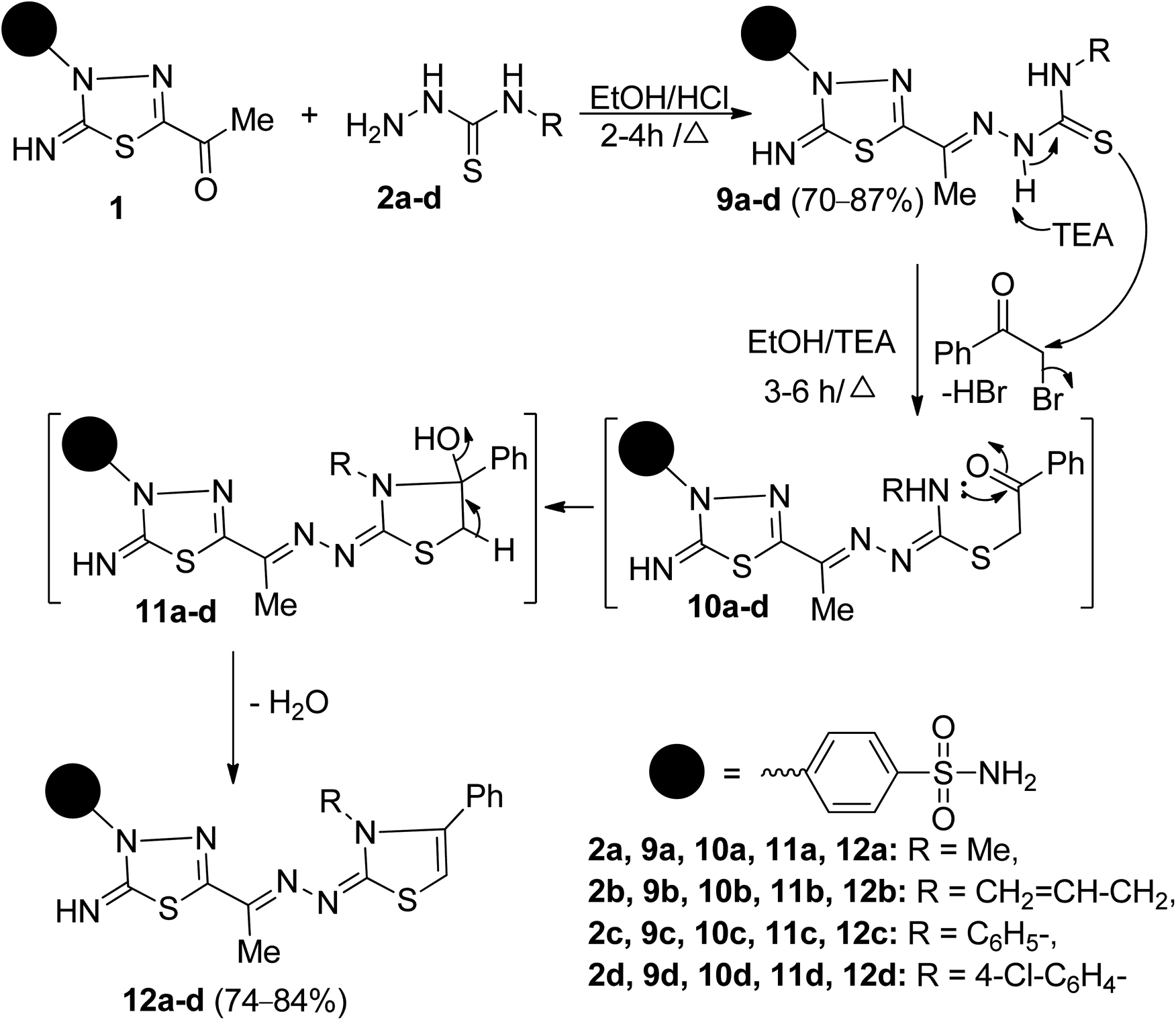 | ||
| Scheme 2 Synthesis of 1,3,4-thiadiazolylbenzenesulfonaminde tagged with 2,3,4-trisubstituted thiazole derivatives 12a–d. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, SO2 groups, and CS, respectively. Its 1H-NMR spectrum exhibits the expected signals from aromatic protons and six singlet signals at δ 2.32, 7.39, 7.64, 8.60, 9.41, and 10.83 ppm assigned to CH3, SO2NH2, 4N–Hb, 4N–Ha, NH and 2N–H protons, respectively. The 13C-NMR spectrum of 3 showed the existence of nine carbon signals which compatible with the suggested structure. The most significant carbon signals resonate at δ 12.50, 158.56, and 179.07 ppm, assignable to CH3, thiadiazole-C5, and CS carbon, respectively. The mass spectrum (MS) of 3 revealed a molecular ion peak (M+) at m/z = 371 confirming its molecular weight.
N, SO2 groups, and CS, respectively. Its 1H-NMR spectrum exhibits the expected signals from aromatic protons and six singlet signals at δ 2.32, 7.39, 7.64, 8.60, 9.41, and 10.83 ppm assigned to CH3, SO2NH2, 4N–Hb, 4N–Ha, NH and 2N–H protons, respectively. The 13C-NMR spectrum of 3 showed the existence of nine carbon signals which compatible with the suggested structure. The most significant carbon signals resonate at δ 12.50, 158.56, and 179.07 ppm, assignable to CH3, thiadiazole-C5, and CS carbon, respectively. The mass spectrum (MS) of 3 revealed a molecular ion peak (M+) at m/z = 371 confirming its molecular weight.It is worth mentioning that the 4N-unsubstituted thiosemicarbazone 3 can adopt syn conformation (NHCS) and 1E (CN) configuration (Scheme 1) based on previous studies.23 The suggested molecular configuration with the 4N–Hb cis to the CS group, leaving the second 4N–Ha disposed of intramolecular H-bond formation with 1NC. The proposed configuration of 3 supports the appearance of the germinal protons of the NH2 group in the 1H-NMR spectrum as magnetically nonequivalent protons.
Next, the reactivity of 3 with various α-chlorocarbonyl reagents was explored to prepare bioactive 1,3-thiazole-1,3,4-thiadiazole conjugates. Thus, the reaction of 3 with each of phenacyl bromide and chloroacetone in ethanol in the presence of sodium acetate under reflux afforded benzenesulfonamides 4 and 5, respectively (Scheme 1). The structures of 4 and 5 were confirmed based on microanalyses and spectral data. For example, the IR spectrum of 5 displayed the lack of NH2 and CS functions and the presence of two bands at 3142, 3101 cm−1 for two NH groups besides the azomethine (CN) band at 1602 cm−1. The 1H-NMR spectrum of 5 indicated six singlet signals at δ 2.13, 2.26, 6.30, 7.37, 9.29, and 11.79 ppm related to the two CH3 groups, thiazole-H5, NH2, imine NH, and NH, as well as to the expected two doublets for the four aromatic protons. Its 13C-NMR spectrum revealed two sp3 hybridized carbon atoms at δ 13.34 and 17.46 ppm due to two methyl carbons, besides ten carbon signals assigned to sp2 hybridized carbon atoms. The M+ peaks at m/z = 471 and 409 in the mass spectra of 4 and 5 are compatible with their molecular formula (C19H17N7O2S3) and (C14H15N7O2S3), respectively.
Furthermore, the mass fragmentation pattern of 5 is depicted in Scheme S1.† Compound 5 underwent cleavage to give two ion peaks at m/z = 128 and 281 due to the formation of 2-methylamino-4-methylthiazole and 5-cyano-2-imino-3-(4-sulfamoylphenyl)-1,3,4-thiadiazoline, respectively. The formed thiazole cleavages further to produce the base peak at m/z = 59 (100%), which is assigned to 1,2-thiazine cation radical. On the other hand, the thiadiazole fragment cleavages further to give rise to a sulfanilamide ion peak at m/z = 172.
Similarly, 2,4,5-trisubstituted thiazoles 6 and 7 were obtained in good yields by heating 3 with each of 3-chloro-2,4-pentanedione and ethyl 2-chloroacetoacetate in ethanol in the presence of sodium acetate. Heterocyclization of 3 with ethyl chloroacetate in ethanol under reflux in sodium acetate furnished the thiazolidine-4-one 8 (Scheme 1). Their correct elemental analyses and spectral data clarified the structures of 6, 7, and 8. Their IR spectra exhibited, in each case, a strong absorption band in the region 1713–1668 cm−1, characteristic of the carbonyl function. The 1H-NMR spectrum of 6, as a representative example, showed, along with aromatic proton signals, six singlet signals, three signals exchangeable with D2O, resonate downfield at δ 7.35, 9.25, 11.79 ppm corresponding to NH2, and two NH protons, respectively, and three signals appeared upfield at δ 2.19, 2.26, 2.39 ppm related to three methyl protons. Its 13C-NMR spectrum revealed the appearance of 14 carbon signals. The sp3-hybridized carbon atoms resonated at δ 12.98, 27.48, 30.87 ppm is assignable to the three methyl carbons. Furthermore, the characteristic signal of the CO resonates at δ 189.00 ppm. The mass spectrum of 6 showed a M+ peak at m/z 451, which is adaptable to a molecular formula (C16H17N7O3S3).
The structures 9a–d were proved based on their elemental analyses, spectral data, and chemical transformations. Their IR spectra exhibited characteristic absorption bands in the region 3339–3030 cm−1 attributed to NH2 and three NH groups and at 1635–1625 cm−1 characteristics for conjugated azomethine (CN) groups and 1247–1187 cm−1 due to thiocarbonyl groups, respectively. The mass spectra of 9a–d exhibited a M+ peak in each case, which was compatible with its molecular weight. The 1H-NMR spectrum of 9a, as an example of these series, revealed a doublet signal at δ 3.04 ppm due to methylamino protons, five singlet signals at δ 2.28, 7.66, 8.70, 11.04, and 11.43 ppm due to CH3, NH2, and three NH protons, respectively, and two doublet signals at δ 7.92 and 8.05 ppm due to four aromatic protons. Its 13C-NMR spectrum exhibited 10 carbon signals. The sp3 hybridized carbon signals resonate at δ 12.78 and 31.53 ppm, attributed to two methyl carbons. The two downfield carbon signals appear at δ 167.79 and 178.67 ppm characteristics for CNH and CS, respectively. Hence, NMR spectra for these analogues, 9a–d, showed only one set of peaks, demonstrating the E configuration's preferential formation. The ZCN configuration is not observed because of steric hindrance, and the non-planar conformation of CN–NH does not exist because it would violate the n–π conjugation.27–29 To get more evidence on the geometry of our molecular structure, we performed Quantum DFT-B3LYP/6-31G(d) calculations for each isomer of 9a as an example. Accordingly, the calculated energy for optimized geometry of the (E) isomer of −2194.475905 Hartree appears to be slightly lower than that of the (Z) isomer (−2194.465113 Hartree). Theoretically, the (E) isomer is more stable than the (Z) isomer, with an energy difference of 6.77 kcal mol−1 (ESI, Fig. S57†).
2,3,4-Trisubstituted thiazoles were prepared to investigate the effect of substituents inserted on the N-3 position for anticancer activity. Thus, cyclocondensation of 9a–d with phenacyl bromide furnished a sole product, in each case, identified as 2,3,4-trisubstituted thiazoles 12a–d (Scheme 2). Spectral data and microanalyses secured the structures of the later products. Their IR spectra were free of absorption bands in regions 3130–3100 cm−1 and 1247–1187 cm−1 related to two secondary NH groups and a thiocarbonyl group. For example, the 1H-NMR spectrum of 12b revealed four singlet signals at δ 2.27, 6.62, 7.46, 9.98 ppm due to CH3, thiazole-H5, NH2, and NH protons, respectively, and a doublet signal resonates at δ 4.47 ppm due NCH2 protons. It also exhibited two doublets of doublets, and one multiplet signal resonated around δ 5.12, 5.19, 5.82 ppm due to the vinyl protons (CH2CH), and two doublets resonate at δ 7.96 and 8.08 ppm, indicating the existence of 4-disubstituted benzene, besides the other expected signals for five aromatic ring residues. The 13C-NMR spectrum of 12b revealed eighteen carbon signals. The aliphatic carbons resonate at δ 12.49, 47.95 ppm, assignable for the carbons of CH3, and NCH2 groups, respectively. The olefinic carbons appear at δ 117.09, 132.07 ppm due to the carbons of the vinyl group (CH2CH). Further, the signals resonate at δ 102.35, 146.27, 155.51, 159.51, 171.48 ppm assignable to the carbons of thiazole-C5, CN, thiadiazole-C5, thiazole-C2, and thiadiazole-C2, respectively. The mass spectrum showed a M+ peak at m/z = 511, corresponding to a molecular formula C22H21N7O2S3. The mass spectrometry fragmentations are characteristic of the appearance of the fragment at m/z = 496 and 470 due to the loss of methyl and allyl groups from the parent ion peak, respectively. The peak showed at m/z = 102 (59%), assigned to the loss of phenylacetylene molecule [M+–C8H6] and the base peak appeared at m/z = 134 (100%) due to the formation of 2-phenylthirine cation radical [C8H6S]+˙ that confirms the formation of thiazole ring.
The formation of 12a–d, as depicted in Scheme 2, is expected to start with the SN2-displacement reaction of bromine atom by the thiolate anion of 9a–d, generated from deprotonation of more acidic N–H2 proton, to afford the non-isolable thioether intermediates 10a–d that subsequently undergoes tandem 5-exo-trig ring closure via nucleophilic addition of a secondary NH group to CO group to give intermediates 11a–d that loss water molecule to afford the target compounds 12a–d.Another structural feature of the synthesized thiazoles 12a–d is they can theoretically exist in EZ or/and EE diastereomers. The preference formation of the thermodynamically stable diastereomers was demonstrated for 12a-as example using B3LYP/6-31G(d) calculation. The results revealed that the optimized geometry for the diastereomer EZ has an energy of −2501.718252 Hartree, which is lower than that calculated for the EE-diastereomer (−2501.705865 Hartree) by about 7.77 kcal mol−1 (ESI, Fig. S58†).
2.2. Bio-evaluation
| Compound | hCAIX binding energy (kcal mol−1) | IC50 (μM ± SD) | |||
|---|---|---|---|---|---|
| HepG-2 | Caco2 | MCF-7 | WI-38 | ||
| a NA: not available. | |||||
| 1 | −10.13 | 59.60 ± 3.40 | 11.1 ± 0.60 | 14.90 ± 0.70 | 91.50 ± 4.70 |
| 3 | −10.64 | 4.57 ± 0.30 | 14.9 ± 0.70 | 6.38 ± 0.30 | 44.40 ± 2.30 |
| 4 | −11.61 | 0.50 ± 0.04 | 3.19 ± 0.20 | 3.52 ± 0.20 | 21.40 ± 1.10 |
| 6 | −11.65 | 32.90 ± 1.90 | 33.8 ± 1.70 | 21.20 ± 1.20 | 110.00 ± 5.70 |
| 7 | −11.96 | 19.60 ± 1.10 | 7.39 ± 0.40 | 12.60 ± 0.60 | 42.90 ± 2.20 |
| 8 | −11.39 | 11.30 ± 0.60 | 5.31 ± 0.30 | 27.20 ± 1.20 | 22.90 ± 1.20 |
| 9a | −11.47 | 0.53 ± 0.01 | 1.01 ± 0.10 | 1.12 ± 0.10 | 18.40 ± 1.70 |
| 9b | −11.21 | 149.00 ± 8.40 | 74.80 ± 3.70 | 73.00 ± 3.30 | 92.90 ± 4.80 |
| 9c | −11.51 | 37.10 ± 2.10 | 34.70 ± 1.70 | 21.10 ± 0.90 | 28.50 ± 1.50 |
| 9d | −11.52 | 153.00 ± 8.60 | 98.70 ± 4.90 | 42.60 ± 1.80 | 60.70 ± 3.10 |
| 12b | −11.13 | 17.50 ± 1.50 | 25.50 ± 1.30 | 15.10 ± 0.70 | 63.50 ± 3.90 |
| 12d | −12.04 | 4.26 ± 0.20 | 1.06 ± 0.10 | 3.79 ± 0.20 | 46.60 ± 2.40 |
| Staurosporine | NAa | 13.60 ± 0.80 | 8.18 ± 0.70 | 6.19 ± 0.30 | 25.20 ± 1.30 |
 | ||
| Fig. 3 Graphical presentation of the cytotoxic activity of derivatives 3, 4, 8, 9a and 12d against staurosporine as the positive control expressed as IC50 (μM). | ||
In the same context, derivatives 9a and 12d were more potent than staurosporine against the three tested cancer cell lines. Yet, the open chain thiosemicarbazone 9a showed the best growth inhibition potential among all the synthesized derivatives demonstrating IC50 0.53, 1.01, 1.12 μM against HepG-2, Caco2, and MCF-7, respectively. Nevertheless, it showed appreciable inhibition of the normal WI-38 giving IC50 18.4 μM. The most intriguing finding was that derivatives 1, 6, and 7 were less hazardous to the normal fibroblast cell line than staurosporine, as indicated by their IC50 values (Fig. 3). These findings indicated that these compounds could be considered safer than staurosporine. Similarly, IC50 of compounds 9c, 12b, and 12d on normal fibroblasts were higher than staurosporine, confirming their safety preferences. The remaining derivatives exhibited weak to moderate inhibitory activity against the three cancer cell lines with IC50 range of 34–153 μM.
On the other hand, preserving a small hydrophobic group like a methyl substitution at thiazole C4 with another H-bond forming moieties like ketone 6 or ester 7 at C5 did not add much to their cytotoxicity. As concluded, the di-substitution of the thiazole at C4 and C5 was not preferable but rather mono-substitution with a phenyl moiety at C4.
Nevertheless, N3 and C4 di-substitution of the thiazole ring was evaluated in series 12a–d. It was observed that incorporating the para-chlorophenyl group to N3 with another phenyl ring at C4 significantly improved the lipophilicity of 12d compared to the aliphatic allyl substitution at N3 of 12b. This modification led to 4–25 times improvement of cytotoxicity of 12d over 12b against the three tested cell lines, which might be due to the lipophilicity improvement to increase the drug's cellular permeability, as discussed earlier.
In the same context, increasing the lipophilicity of 3 was implemented by another strategy through adding lipophilic moieties such as methyl, allyl, and phenyl groups to the open chain thiosemicarbazone giving derivatives 9a–d. In this case, the best cytotoxic activity was achieved with the small methyl group substitution at 9a, showing three to nine times improvement against the three cell lines. Nonetheless, increasing the lipophilic moiety size by adding a phenyl group to the thiosemicarbazone terminus 9c showed drop-in cytotoxicity by 10 times. Further reduction in the cytotoxicity was observed upon substituting the thiosemicarbazone terminus with an allyl or para-chlorophenyl group of 9b and 9d, respectively. The size of the added substitution to the thiosemicarbazone terminus of 3 reduced the cytotoxicity probably by interfering with the possible interactions of the terminal sulfur of 3 with the active site residues of hCAIX, as discussed later in the molecular docking simulation (Fig. 5a). A summary of the effect of the structural modification of 3 on the activity is illustrated in Fig. 4.
 | ||
| Fig. 4 Schematic representation of the structure–activity relationship of the synthesized derivatives. | ||
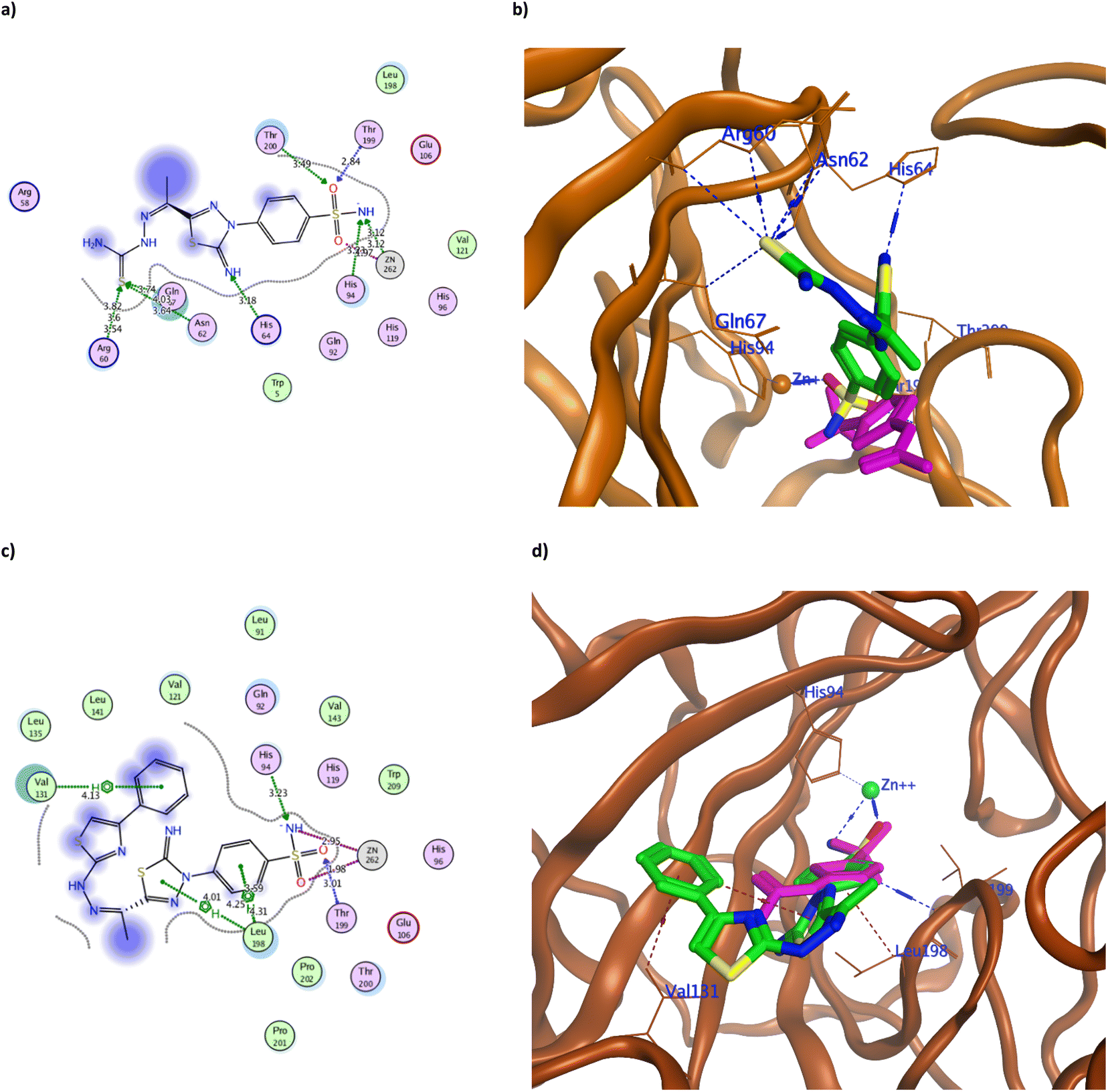 | ||
| Fig. 5 The 2D and 3D binding pattern of molecular docking simulation of 3 (a and b) and 4 (c and d) using hCAIX (PDB 3IAI, 2.20 Å). The tested derivatives were shown in the stick model in green relative to AAZ in magenta color, with the H-bonds and hydrophobic interaction appearing in blue and red dotted lines, respectively. | ||
The achieved molecular docking results (Tables 1 and 2, Fig. 5–7 and S60 and S61†) revealed better binding energy scores of all the tested benzenesulfonamide derivatives than AAZ ranging from −10.13 to −12.04 kcal mol−1 compared to −8.83 kcal mol−1 of AAZ. Moreover, all derivatives conserved the usual zinc coordination between the deprotonated nitrogen and one of the oxygen groups of the sulfonamide moiety (Table 2). It is well established that the sulfonamide nitrogen gets deprotonated in the physiological pH unveiling a negative charge that anchors the zinc ion.31,32 The zinc ion is an essential cofactor in maintaining the catalytic activity of the hCA isoforms.7,10,33 Therefore interfering with it would ultimately hinder hCA activity. The bi-dentate coordination of the derivatives sulfonamide moiety to zinc ion would replace the bound water/hydroxide ion from the zinc at the enzyme's active site as an establishing mechanism to initiate the catalytic inhibition.11,34 Moreover, all tested derivatives maintained the H-bond formation with the active site Thr199, similar to AAZ. Additionally, the most potent derivatives 3, 4, 8, 9a, 9c, and 12d managed to form H-bond with one of the hCAIX catalytic histidine triad His94 with an average distance of 3.30 Å that augmented with their biological activity (Fig. 5–7).10
| Compound | Ligand | hCAIX (PDB: 3IAI) | Interaction | |||||
|---|---|---|---|---|---|---|---|---|
| Interacting moiety | Interacting moiety | Interacting residue | Interaction type | Distance (Å) | Energy (kcal mol−1) | |||
| AAZ | N3 | 11 | OG1 | Thr | 200 | H-acceptor | 3.04 | −0.90 |
| O1 | 14 | N | Thr | 199 | H-acceptor | 2.98 | −4.40 | |
| O3 | 16 | NE2 | Gln | 92 | H-acceptor | 3.07 | −0.70 | |
| N1 | 8 | ZN | Zn | 262 | Ionic | 2.03 | −15.60 | |
| O2 | 15 | ZN | Zn | 262 | Ionic | 3.15 | −3.50 | |
| 5-ring | CD2 | Leu | 198 | Pi-H | 3.76 | −0.70 | ||
| 1 | O | 17 | N | Thr | 199 | H-acceptor | 2.97 | −4.70 |
| N | 16 | ZN | Zn | 262 | Ionic | 2.68 | −7.00 | |
| O | 18 | ZN | Zn | 262 | Ionic | 2.00 | −16.20 | |
| 6-ring | CD2 | Leu | 198 | Pi-H | 3.87 | −0.20 | ||
| 3 | N | 4 | ND1 | His | 64 | H-acceptor | 3.18 | −4.80 |
| S | 13 | CB | Arg | 60 | H-acceptor | 3.82 | −0.70 | |
| S | 13 | CD | Arg | 60 | H-acceptor | 3.60 | −1.00 | |
| S | 13 | NH1 | Arg | 60 | H-acceptor | 3.54 | −2.50 | |
| S | 13 | CB | Asn | 62 | H-acceptor | 4.03 | −0.80 | |
| S | 13 | ND2 | Asn | 62 | H-acceptor | 3.64 | −3.90 | |
| S | 13 | CB | Gln | 67 | H-acceptor | 3.74 | −0.70 | |
| O | 21 | N | Thr | 199 | H-acceptor | 2.84 | −3.00 | |
| O | 21 | OG1 | Thr | 200 | H-acceptor | 3.49 | −0.50 | |
| N | 23 | CE1 | His | 94 | H-acceptor | 3.23 | −0.30 | |
| O | 22 | ZN | Zn | 262 | Ionic | 1.97 | −16.80 | |
| N | 23 | ZN | Zn | 262 | Ionic | 3.12 | −3.70 | |
| 4 | N | 23 | CE1 | His | 94 | H-acceptor | 3.23 | −0.30 |
| O | 24 | N | Thr | 199 | H-acceptor | 3.01 | −4.50 | |
| N | 23 | ZN | Zn | 262 | Ionic | 2.95 | −4.80 | |
| O | 25 | ZN | Zn | 262 | Ionic | 1.98 | −16.60 | |
| 6-ring | CG2 | Val | 131 | Pi-H | 4.13 | −0.60 | ||
| 6-ring | CB | Leu | 198 | Pi-H | 4.31 | −0.50 | ||
| 5-ring | CD1 | Leu | 198 | Pi-H | 4.01 | −0.30 | ||
| 6-ring | CD1 | Leu | 198 | Pi-H | 4.25 | −0.40 | ||
| 6-ring | CD2 | Leu | 198 | Pi-H | 3.59 | −0.40 | ||
| 5 | O | 23 | N | Thr | 199 | H-acceptor | 3.02 | −4.40 |
| N | 22 | ZN | Zn | 262 | Ionic | 2.02 | −15.90 | |
| O | 24 | ZN | Zn | 262 | Ionic | 3.03 | −4.20 | |
| 6-ring | CD2 | Leu | 198 | Pi-H | 4.26 | −0.40 | ||
| 6 | O | 17 | ND1 | His | 64 | H-acceptor | 2.85 | −3.90 |
| O | 26 | N | Thr | 199 | H-acceptor | 2.92 | −4.90 | |
| N | 24 | ZN | Zn | 262 | Ionic | 2.08 | −14.70 | |
| O | 25 | ZN | Zn | 262 | Ionic | 2.78 | −6.20 | |
| 6-ring | CD2 | Leu | 198 | Pi-H | 3.54 | −0.40 | ||
| 7 | N | 10 | O | Pro | 201 | H-donor | 2.98 | −1.50 |
| S | 12 | O | Pro | 201 | H-donor | 3.63 | −2.90 | |
| O | 17 | ND1 | His | 64 | H-acceptor | 2.98 | −2.60 | |
| O | 25 | N | Thr | 199 | H-acceptor | 2.90 | −5.00 | |
| N | 24 | ZN | Zn | 262 | Ionic | 2.69 | −6.90 | |
| O | 26 | ZN | Zn | 262 | Ionic | 1.97 | −16.70 | |
| 5-ring | CD2 | Leu | 135 | Pi-H | 4.87 | −0.70 | ||
| 6-ring | CD2 | Leu | 198 | Pi-H | 3.59 | −0.60 | ||
| 8 | S | 12 | OE1 | Gln | 67 | H-donor | 3.23 | −1.80 |
| N | 8 | ND1 | His | 64 | H-acceptor | 3.38 | −2.60 | |
| O | 16 | CB | His | 64 | H-acceptor | 3.36 | −0.20 | |
| N | 23 | CA | Leu | 198 | H-acceptor | 3.43 | −0.90 | |
| N | 23 | N | Thr | 199 | H-acceptor | 3.33 | −5.90 | |
| O | 25 | CE1 | His | 94 | H-acceptor | 3.38 | −0.20 | |
| O | 24 | ZN | Zn | 262 | Ionic | 1.93 | −17.70 | |
| O | 25 | ZN | Zn | 262 | Ionic | 3.43 | −2.20 | |
| 6-ring | CD2 | Leu | 198 | Pi-H | 4.55 | −0.20 | ||
| 9a | N | 14 | NE2 | Gln | 92 | H-acceptor | 3.38 | 2.80 |
| O | 23 | CE1 | His | 94 | H-acceptor | 3.47 | −0.30 | |
| O | 24 | CA | Leu | 198 | H-acceptor | 3.18 | −0.40 | |
| O | 24 | N | Thr | 199 | H-acceptor | 3.03 | −4.00 | |
| N | 22 | ZN | Zn | 262 | Ionic | 2.00 | −16.30 | |
| 9b | N | 7 | NE2 | Gln | 92 | H-acceptor | 3.50 | −0.70 |
| O | 23 | N | Thr | 199 | H-acceptor | 2.96 | −4.80 | |
| N | 22 | ZN | Zn | 262 | Ionic | 2.67 | −7.10 | |
| O | 24 | ZN | Zn | 262 | Ionic | 1.98 | −16.60 | |
| 9c | N | 22 | CE1 | His | 94 | H-acceptor | 3.28 | −0.80 |
| O | 23 | N | Thr | 199 | H-acceptor | 3.05 | −4.00 | |
| N | 22 | ZN | Zn | 262 | Ionic | 3.35 | −2.50 | |
| O | 24 | ZN | Zn | 262 | Ionic | 1.94 | −17.40 | |
| 5-ring | CZ2 | Trp | 5 | Pi-H | 4.34 | −0.50 | ||
| 6-ring | CG1 | Val | 131 | Pi-H | 3.60 | −0.40 | ||
| 9d | O | 24 | N | Thr | 199 | H-acceptor | 2.92 | −4.90 |
| N | 22 | ZN | Zn | 262 | Ionic | 2.08 | −14.80 | |
| O | 23 | ZN | Zn | 262 | Ionic | 2.59 | −7.90 | |
| 6-ring | CB | Asp | 132 | Pi-H | 3.74 | −0.80 | ||
| 12a | O | 25 | N | Thr | 199 | H-acceptor | 2.88 | −5.10 |
| N | 24 | ZN | Zn | 262 | Ionic | 2.48 | −9.10 | |
| O | 26 | ZN | Zn | 262 | Ionic | 1.93 | −17.60 | |
| 12b | O | 25 | ZN | Zn | 262 | H-acceptor | 2.92 | −0.40 |
| O | 26 | N | Thr | 199 | H-acceptor | 2.96 | −4.70 | |
| N | 24 | ZN | Zn | 262 | Ionic | 2.06 | −15.10 | |
| O | 25 | ZN | Zn | 262 | Ionic | 2.92 | −5.00 | |
| 6-ring | CG2 | Val | 131 | Pi-H | 4.22 | −0.50 | ||
| 5-ring | CD1 | Leu | 198 | Pi-H | 4.12 | −0.30 | ||
| 6-ring | CD2 | Leu | 198 | Pi-H | 3.54 | −0.30 | ||
| 12c | O | 25 | N | Thr | 199 | H-acceptor | 2.95 | −4.80 |
| N | 24 | ZN | Zn | 262 | Ionic | 2.68 | −7.00 | |
| O | 26 | ZN | Zn | 262 | Ionic | 1.97 | −16.70 | |
| 6-ring | CD2 | Leu | 198 | Pi-H | 4.24 | −0.30 | ||
| 12d | N | 53 | CE1 | His | 94 | H-acceptor | 3.33 | −0.60 |
| O | 56 | CA | Leu | 198 | H-acceptor | 3.27 | −0.30 | |
| O | 56 | N | Thr | 199 | H-acceptor | 3.07 | −3.90 | |
| N | 53 | ZN | Zn | 262 | Ionic | 3.34 | −2.60 | |
| O | 55 | ZN | Zn | 262 | Ionic | 1.93 | −17.60 | |
| 6-ring | CD2 | Leu | 91 | Pi-H | 4.81 | −0.20 | ||
| 6-ring | NE2 | Gln | 92 | Pi-H | 4.55 | −0.80 | ||
| 6-ring | CG1 | Val | 131 | Pi-H | 4.61 | −0.30 | ||
| 6-ring | CG2 | Val | 131 | Pi-H | 4.27 | −0.30 | ||
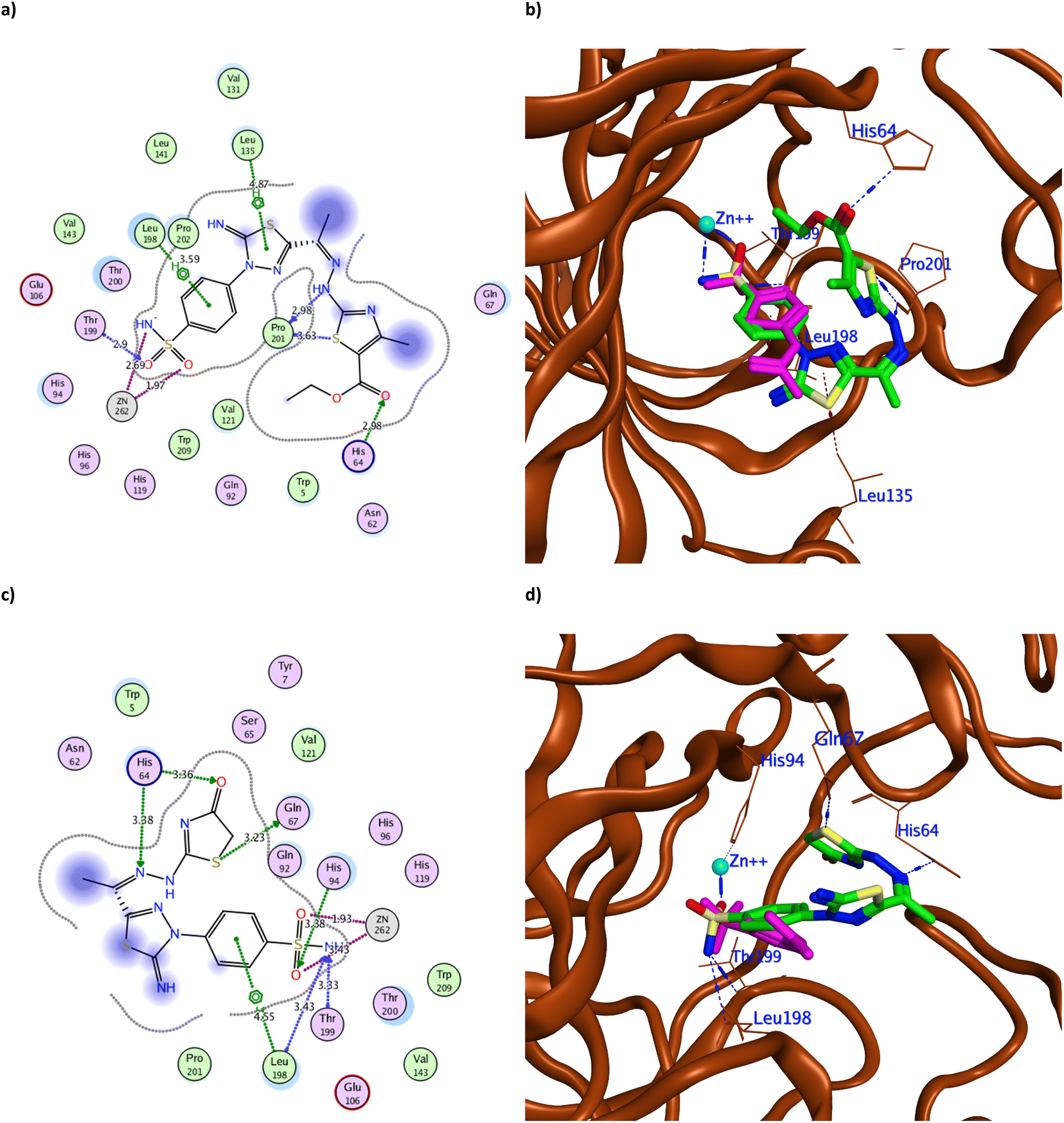 | ||
| Fig. 6 The 2D binding pattern of molecular docking simulation of 7 (a and b) and 8 (c and d) using hCAIX (PDB 3IAI, 2.20 Å) showing distance in Å. The tested derivatives were shown in the stick model in green relative to AAZ in magenta color, with the H-bonds and hydrophobic interaction appearing in blue and red dotted lines, respectively. | ||
 | ||
| Fig. 7 The 2D and 3D binding pattern of molecular docking simulation of 9a (a and b) and 12d (c and d) using hCAIX (PDB 3IAI, 2.20 Å). The tested derivatives were shown in the stick model in green relative to AAZ in magenta color, with the H-bonds and hydrophobic interaction appearing in blue and red dotted lines, respectively. | ||
It was observed that replacing the terminal acetyl moiety of 1 with thiourea in 3 allowed the derivative to interact with Gln67, Asn62, and Arg60 through H-bonds. In addition, it adopted a conformation that allowed the deprotonated nitrogen of 3 to approach His94 and Thr200, thus enhancing its cytotoxicity against HepG-2 and MCF-7 (Fig. 5a and b and S60a†). Conversely, cyclizing the terminal thiourea into a thiazole ring with phenyl group 4 preserved the H-bond with Thr199 and the catalytic His94 with extra hydrophobic interaction with Val131 (Fig. 5c and d). However, replacing the terminal phenyl moiety of 4 with an acetyl group, 6, greatly decreased its cytotoxic effect by losing the interaction with His94 and Val131 (Fig. S60c†). Changing the acetyl group with an ethyl ester 7 achieved a proper conformation to interact with His64, Leu135, Thr199, Leu198, and Pro201 (Fig. 6a) that was translated into double the cytotoxic effect on HepG-2 and MCF-7. Unlike the small molecule AAZ, this extended thiazole ring with ethyl ester of 7 adopted a conformation that filled the unoccupied active site region with the interaction with residues His64 and Pro201 (Fig. 6b). Similarly, altering the ester group with a carbonyl moiety 8 maintained the interaction with His64, and Gln67 at the unoccupied pocket of the active site (Fig. 6c and d). Moreover, it demonstrated further improvement in the cytotoxicity by regaining the H-bond with His94 and additional H-bond with the active site His64 (Fig. 6c). The role of His64 in shuttling the proton during hCAs catalytic activity was valued, especially with its proven inward and outward conformation.13
Among the aliphatic substituted thiourea derivatives 9a–d, derivatives 9a and 9c exhibited the best binding conformation inside hCAIX active site while maintaining the H-bond with His94 by their sulfonamide moiety (Fig. 7a and b and S60e†). Moreover, 9a showed other H-bonds with Gln92, Leu198, and Thr199 with an average distance of 3.20 Å (Fig. 7a). In a similar way; among the cyclized thiazole derivatives 12a–d, derivative 12d was the best oriented inside hCAIX with the lowest binding energy of −12.04 kcal mol−1. The deprotonated nitrogen of 12d conserved the H-bond with His94, while the terminal para chloro-substituted phenyl displayed additional hydrophobic interactions with Gln92 and Val131 (Fig. 7c and d). Furthermore, 12d showed multiple interactions with hCAIX active site residues Leu198, Thr199, and Leu91 that rationalized its good cytotoxicity.
Acknowledging the similarity between the CA isoenzymes, molecular docking simulation was performed using hCAI, hCAII, and hCAXII for the most potent derivatives 4, 9a, and 12d to assess their off-target potentials. Their molecular docking simulation used the reported PDB: 3W6H,35 3HS4,36 and 1JD0 (ref. 37) for hCAI, hCAII, and hCAXII, respectively. The attained data revealed the unfavorable binding pattern of the tested derivatives to the evaluated isoenzymes in contrast to hCAIX (Table 3 and ESI, Fig. S62–S64†). The deprotonated sulfonamide moiety of the three derivatives exhibited the classical coordination with zinc among the evaluated isoenzymes similar to hCAIX. Although 4 and 9a demonstrated H-bond formation with Thr199, this solely formed bond was not expected to be sufficient for appreciable enzyme inhibition. Moreover, 12d is only bound to Thr199 in the case of hCAII, not the other isoenzymes and showed single coordination with zinc of 3.27 Å length. In the same context, the predicted binding conformation and interaction of these promising derivatives with the tumor-associated hCAXII supported the assumption of exerting their anticancer activity through hCAIX inhibition, not hCAXII.
| Compound | hCAI (PDB: 3W6H) | hCAII (PDB: 3HS4) | hCAXII (PDB: 1JD0) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Binding energy (kcal mol−1) | Interacting residue | Interaction type | Distance (Å) | Binding energy (kcal mol−1) | Interacting residue | Interaction type | Distance (Å) | Binding energy (kcal mol−1) | Interacting residue | Interaction type | Distance (Å) | ||||
| 4 | −10.24 | Thr | 199 | H-acceptor | 3.21 | −10.59 | Thr | 199 | H-acceptor | 2.96 | −11.53 | Thr | 199 | H-acceptor | 3.01 |
| Zn | 302 | Ionic | 2.03 | Zn | 301 | Ionic | 2.76 | Zn | 901 | Ionic | 2.82 | ||||
| Zn | 302 | Ionic | 2.68 | Zn | 301 | Ionic | 2.05 | Zn | 901 | Ionic | 1.95 | ||||
| 9a | −11.13 | Asn | 69 | H-acceptor | 3.53 | −10.35 | Thr | 199 | H-acceptor | 3.10 | −10.93 | Lys | 67 | H-acceptor | 3.35 |
| Thr | 199 | H-acceptor | 2.93 | Zn | 301 | Ionic | 2.03 | Thr | 199 | H-acceptor | 3.01 | ||||
| Zn | 302 | Ionic | 2.01 | Zn | 301 | Ionic | 3.02 | Zn | 901 | Ionic | 2.85 | ||||
| Zn | 302 | Ionic | 3.10 | Zn | 901 | Ionic | 1.95 | ||||||||
| 12d | −7.43 | Gln | 92 | H-acceptor | 3.56 | −7.35 | Thr | 199 | H-acceptor | 2.95 | −8.42 | Zn | 262 | Ionic | 3.27 |
| Zn | 262 | Ionic | 3.23 | Zn | 262 | Ionic | 2.67 | ||||||||
| Val | 131 | Pi-H | 4.14 | Zn | 262 | Ionic | 1.99 | ||||||||
| Val | 131 | Pi-H | 4.21 | ||||||||||||
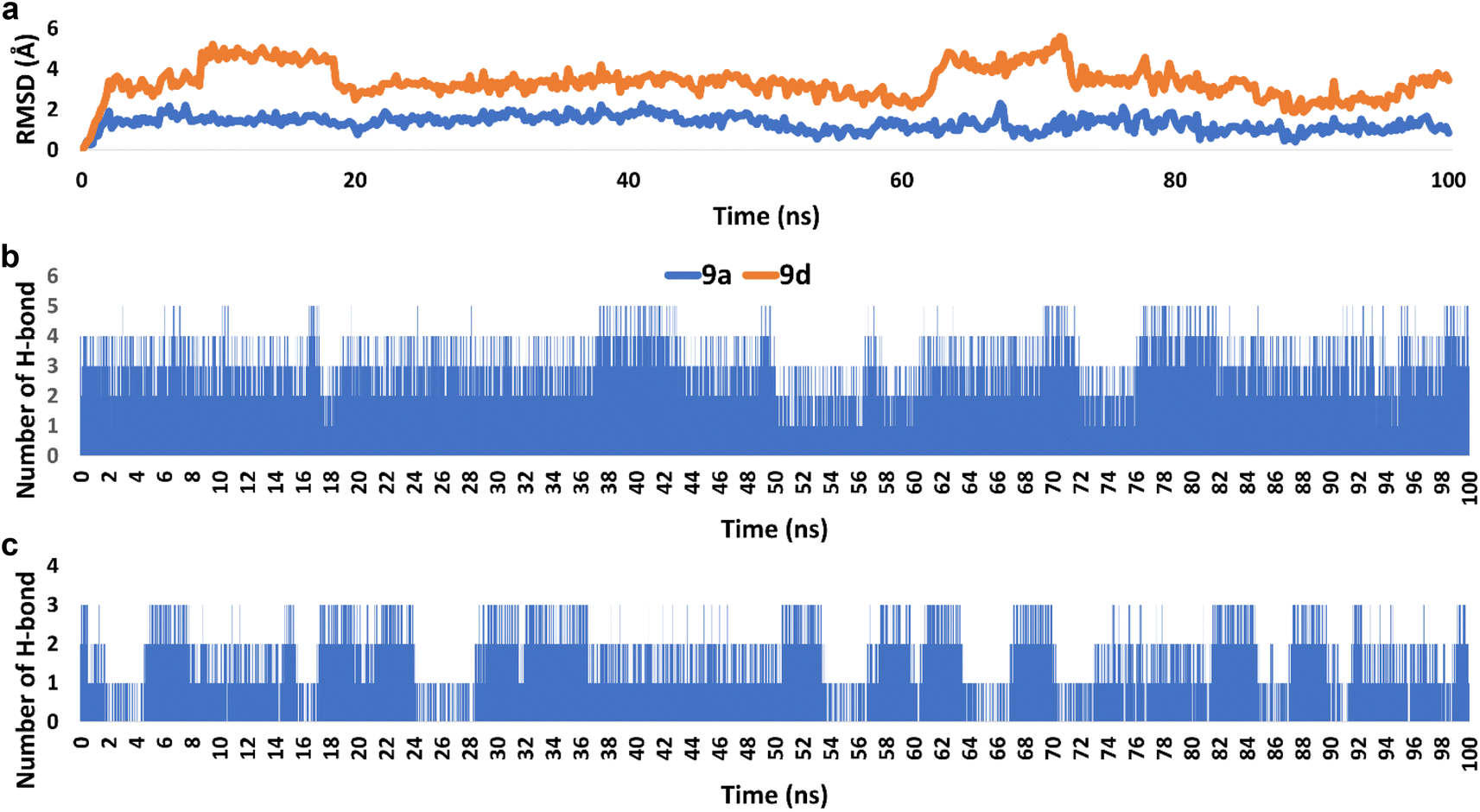 | ||
| Fig. 8 (a) RMSDs of derivatives 9a and 9d inside the active site of hCAIX (PDB ID: 3IAI) throughout 100 ns-long MD simulation. (b) and (c) The number of H-bonds detected for 9a and 9d, respectively inside the active site of hCAIX over the course of 100 ns-long MD simulation. The cut-off distance for H-bonds was set to be 2.5 Å. | ||
Accordingly, the overall interaction energies of both derivatives averaged around −61.07 and −22.58 kcal mol−1, respectively (Fig. 9). Furthermore, their calculated binding free energies (ΔGbinding) in terms of MM-PBSA were found to be −9.6 and −4.3 kcal mol−1, respectively showing a significant difference in affinities between the two structures towards the active site of hCAIX (Table 4). Accordingly, 9a was likely more active as an inhibitor of hCAIX in comparison to 9d.
 | ||
| Fig. 9 Interaction energies (i.e., electrostatic and van der Waals interaction energies) of 9a and 9d (A and B, respectively) inside the active site of hCAIX (PDB ID: 3IAI) for 100 ns-long MD simulation. | ||
| Energy component (kcal mol−1) | 9a | 9d |
|---|---|---|
| ΔGgas | −21.7654 | −14.1167 |
| ΔGsolv | 12.1875 | 9.8145 |
| ΔGtotal | −9.5779 | −4.3022 |
| Compounds | 3 | 4 | 7 | 8 | 9a | 12b | 12d |
|---|---|---|---|---|---|---|---|
| Physicochemical properties | |||||||
| Mol. weight in g mol−1 | 371.5 | 471.6 | 481.6 | 411.5 | 385.5 | 511.6 | 582.1 |
| #Heavy atoms | 23 | 31 | 31 | 26 | 24 | 34 | 38 |
| #Aromatic heavy atoms | 11 | 22 | 16 | 11 | 11 | 22 | 28 |
| Fraction Csp3 | 0.09 | 0.05 | 0.24 | 0.15 | 0.17 | 0.09 | 0.04 |
| #Rotatable bonds | 5 | 6 | 8 | 5 | 6 | 6 | 6 |
| #H-bond acceptors | 6 | 7 | 9 | 8 | 6 | 7 | 7 |
| #H-bond donors | 4 | 3 | 3 | 3 | 4 | 2 | 2 |
| MR | 90.5 | 122.4 | 118.0 | 102.3 | 95.4 | 135.9 | 151.0 |
| TPSA (Å2) | 221.0 | 204.0 | 230.3 | 217.6 | 207.0 | 196.3 | 196.3 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| Lipophilicity | |||||||
| iLOGP | 1.42 | 2.48 | 2.59 | 1.34 | 1.53 | 1.53 | 1.53 |
| XLOGP3 | 0.40 | 3.77 | 3.06 | 0.98 | 0.80 | 0.80 | 0.80 |
|
|||||||
| Water solubility | |||||||
| ESOL logS |
−2.42 | −5.27 | −4.61 | −2.99 | −2.68 | −2.68 | −2.68 |
| ESOL solubility (mg ml−1) | 1.42 × 100 | 2.54 × 10−3 | 1.19 × 10−2 | 4.19 × 10−1 | 8.11 × 10−1 | 8.11 × 10−1 | 8.11 × 10−1 |
| ESOL class | Soluble | Moderately soluble | Moderately soluble | Soluble | Soluble | Soluble | Soluble |
|
|||||||
| Pharmacokinetics | |||||||
| GI absorption | Low | Low | Low | Low | Low | Low | Low |
| BBB permeant | No | No | No | No | No | No | No |
| P-gp substrate | No | No | No | No | No | No | No |
| CYP1A2 inhibitor | No | No | No | No | No | No | No |
| CYP2C19 inhibitor | No | No | No | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No | No | No | No |
| CYP2D6 inhibitor | No | No | No | No | No | No | No |
| CYP3A4 inhibitor | No | No | Yes | No | No | No | No |
| Skin permeation logKp (cm s−1) |
−8.28 | −6.5 | −7.06 | −8.11 | −8.08 | −8.08 | −8.08 |
|
|||||||
| Drug-likeness | |||||||
| Lipinski #violations | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Ghose #violations | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Veber #violations | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Egan #violations | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Muegge #violations | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
On the other hand, all tested derivatives had acceptable drug-likeness properties according to Lipinski's rule, with one violation of molecular weight of more than 500 g mol−1 for 12b and 12d.41,42 However, their bioavailability scores revealed good physiological activity according to Abbott's bioavailability score.43
3. Experimental section
3.1. Synthesis and spectroscopic characterization
N), 1596 (CC), 1335, 1282 (SO2), 1248 (CS); 1H-NMR (DMSO-d6): δppm = 2.32 (s, 3H, CH3), 7.39 (s, 2H, NH2), 7.64 (s, 1H, 4N–Hb), 7.87 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.23 (d, J = 8.5 Hz, 2H, Ar–H3,5), 8.60 (s, 1H, 4N–Ha), 9.41 (s, 1H, CNH), 10.83 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 12.50 (CH3), 121.52 (2CH Ar–C2,6), 126.45 (2CH Ar–C3,5), 140.26 (Ar–C4), 141.38 (Ar–C1), 141.94 (CN), 148.62 (thiadiazole-C2), 158.56 (thiadiazole-C5), 179.07 (CS); MS m/z (%): 371 (M+, 1.3), 370 (1.9), 367 (1.8), 366 (2.0), 356 (1.7), 339 (2.1), 326 (1.3), 312 (1.6), 297 (5.1), 285 (2.3), 281 (1.7), 273 (1.5), 258 (1.6), 172 (1.5), 93 (12.7), 83 (58.1), 57 (100). Anal. calc. For C11H13N7O2S3 (371.45): C, 35.57; H, 3.53; N, 26.40%; found: C, 35.56; H, 3.54; N, 26.41%.3.1.2.1. 4-(2-Imino-5-(1-(2-(4-phenylthiazol-2-yl)hydrazineylidene)ethyl)-1,3,4-thiadiazol-3(2H)yl)benzenesulfonamide (4). The compound was obtained by heating 2-bromo-1-phenylethan-1-one (0.42 g, 0.002 mol) for 3 h under reflux. Brown powder; yield 84%; mp 279–281 °C; IR νmax/cm−1 = 3305, 3237 (NH2), 3197, 3109 (2NH), 1657 (C
N), 1548 (CC), 1328, 1267 (SO2); 1H-NMR (DMSO-d6): δppm = 2.33 (s, 3H, CH3), 7.49 (s, 2H, NH2), 7.55 (s, 1H, thiazole-C5), 7.32–8.22 (m, 9H, Ar–H), 9.45 (s, 1H, NH), 12.05 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 12.86 (CH3), 106.82 (thiazole-C5), 121.80 (2CHAr–C2,6), 126.01 (2CHAr–C2′,6′), 126.45 (1CHAr–C4′), 126.99 (2CHAr–C3′,5′), 127.32 (2CHAr–C3,5), 128.28 (Ar–C4), 129.19 (Ar–C1′), 140.30 (Ar–C1), 142.54 (CN), 152.82 (thiadiazole-C5), 158.83 (thiazole-C4), 162.82 (thiadiazole-C2), 168.83 (thiazole-C2); MS m/z (%): 471 (M+, 15.6), 470 (0.7), 464 (0.2), 456 (0.4), 392 (2.4), 382 (0.3), 356 (0.6), 259 (1.0), 217 (0.7), 191 (0.9), 176 (9.6), 134 (100), 92 (1.1), 66 (7.7), 54 (6.1). Anal. calc. For C19H17N7O2S3 (471.57): C, 48.39; H, 3.63; N, 20.79%; found: C, 48.38; H, 3.64; N, 20.78%.
3.1.2.2. 4-(2-Imino-5-(1-(2-(4-methylthiazol-2-yl)hydrazineylidene)ethyl)-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide (5). The compound was obtained by heating 1-chloropropan-2-one (0.19 g, 0.002 mol) for 7 h under reflux. Brown powder; yield 94%; mp 259–260 °C; IR νmax/cm−1 = 3291, 3186 (NH2), 3142, 3101 (NH), 1602 (C
N), 1558 (CC), 1320, 1304 (SO2); 1H-NMR (DMSO-d6): δppm = 2.13 (s, 3H, CH3), 2.26 (s, 3H, CH3), 6.30 (s, 1H, thiazole-C2), 7.37 (s, 2H, NH2), 7.87 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.24 (d, J = 8.5 Hz, 2H, Ar–H3,5), 9.29 (s, 1H, NH), 11.79 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 13.34 (CH3), 17.46 (CH3), 106.82 (thiazole-C5), 121.21 (2CHAr–C2,6), 127.42 (2CHAr–C3,5), 129.29 (Ar–C4), 140.01.42 (Ar–C1), 142.09 (CN), 149.77 (thiazole-C4), 158.89 (thiadiazole-C5), 162.82 (thiadiazole-C2), 168.83 (thiazole-C2); MS m/z (%): 409 (M+, 4.4), 367 (3.0), 313 (4.3), 299 (1.9), 269 (2.1), 262 (1.6), 207 (4.5), 199 (1,6), 177 (2.4), 153 (8.8), 117 (9.0), 99 (23.9), 76 (71.7), 65 (62.7), 59 (100), 51 (20.5); anal. calc. For C14H15N7O2S3 (409.50): C, 41.06; H, 3.69; N, 23.94%; found: C, 41.05; H, 3.68; N, 23.93%.
3.1.2.3. 4-(5-(1-(2-(5-Acetyl-4-methylthiazol-2-yl)hydrazineylidene)ethyl)-2-imino-1,3,4-thiadi azol-3(2H)-yl)benzenesulfonamide (6). The compound was obtained by heating 3-chloro-2,4-pentanedione (0.29 ml, 0.002 mol) for 5 h under reflux. Brown powder; yield 82%; mp 260–262 °C; IR νmax/cm−1 = 3300, 3237 (NH2), 3197, 3105 (2NH), 1668 (C
O), 1585 (CN), 1488 (CC), 1305, 1277 (SO2); 1H-NMR (DMSO-d6): δppm = 2.19 (s, 3H, CH3), 2.26 (s, 3H, CH3), 2.39 (s, 3H, CH3), 7.35 (s, 2H, NH2), 7.85 (d, J = 8.5 Hz, 2CH, Ar–H2,6), 8.22 (d, J = 8.5 Hz, 2CH, Ar–H3,5), 9.25 (s, 1H, NH), 11.79 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 12.98 (CH3), 27.48 (CH3), 30.87 (CH3), 113.48 (2CHAr–C2,6), 126.43 (2CHAr–C3,5), 126.54 (Ar–C4), 140.05 (thiazole-C5), 141.05 (Ar–C1), 143.25 (CN), 149.39 (thiadiazole-C5), 158.73 (thiazole-C4), 162.45 (thiadiazole-C2), 169.63 (thiazole-C2), 189.00 (CO); MS m/z (%): 451 (M+, 32.9), 450 (1.9), 393 (2.9), 326 (1.4), 305 (1.8), 414 (2.1), 296 (1.8), 253 (1.5), 209 (2.1), 171 (5.6), 118 (9.5), 92 (65.1), 77 (27.0), 67 (100), 56 (19.5); anal. calc. For C16H17N7O3S3 (451.54): C, 42.56; H, 3.80; N, 21.71%; found: C, 42.55; H, 3.81; N, 21.72%.
3.1.2.4. Ethyl-2-(2-(1-(5-imino-4-(4-sulfamoylphenyl)-4,5-dihydro-1,3,4-thiadiazol-2-yl)ethylidene)hydrazineyl)-4-methylthiazole-5-carboxylate (7). The compound was obtained by heating ethyl 2-chloro-3-oxobutanoate (0.35 ml, 0.002 mol) for 6 h under reflux. Brown powder; yield 80%; mp 100–102 °C; IR νmax/cm−1 = 3310, 3232 (NH2), 3190, 3115 (2NH), 1683 (C
O), 1654 (CN), 1540 (CC), 1320, 1268 (SO2); 1H-NMR (DMSO-d6): δppm = 1.23 (t, J = 6.8 Hz, 3H, CH3), 2.26 (s, 3H, CH3), 2.87 (s, 3H, CH3), 4.19 (q, J = 6.8 Hz, 2H, CH2), 7.40 (s, 2H, NH2), 7.89 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.15 (d, J = 8.5 Hz, 2H, Ar–H3,5), 9.53 (s, 1H, NH), 12.33 (s, 1H, NH–N); 13C-NMR (DMSO-d6): δppm = 12.52 (CH3), 16.31 (CH3), 30.86 (CH3), 60.58 (CH2), 113.48 (2CHAr–C2,6), 121.35 (thiazole-C5), 126.90 (2CHAr–C3,5), 127.35 (Ar–C4), 141.22 (Ar–C1), 142.15 (CN), 146.56 (thiadiazole-C5), 161.5 (thiazole-C4), 162.45 (thiadiazole-C2), 168.38 (thiazole-C2), 173.72 (CO); MS m/z (%): 481 (M+, 11.5); MS m/z (%): 481 (M+, 11.5), 437 (3.3), 401 (0.9), 382 (1.2), 373 (1.1), 302 (0.8), 294 (0.7), 255 (0.9), 222 (1.7), 199 (3.0), 134 (9.9), 127 (5.7), 96 (14.4), 67 (100), 56 (14.2); anal. calc. For C17H19N7O4S3 (481.56): C, 42.40; H, 3.98; N, 20.36%; found: C, 4.41; H, 3.97; N, 20.35%.
3.1.2.5. 4-(2-Imino-5-(1-(2-(4-oxo-4,5-dihydrothiazol-2-yl)hydrazineylidene)ethyl)-1,3,4-thiadi azol-3(2H)-yl)benzenesulfonamide (8). The compound was obtained by heating ethyl 2-chloroacetate (0.26 ml, 0.002 mol) for 4 h under reflux. Orange powder; yield 86%; mp 193–195 °C; IR νmax/cm−1 = 3303, 3278 (NH2), 3189, 3102 (2NH), 1713 (C
O), 1599 (CN), 1563 (CC), 1329, 1305 (SO2); 1H-NMR (DMSO-d6): δppm = 2.32 (s, 3H, CH3), 3.91 (s, 2H, CH2), 7.38 (s, 2H, NH2), 7.88 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.22 (d, J = 8.5 Hz, 2H, Ar–H3,5), 9.35 (s, 1H, NH), 12.24 (s, 1H, N–H); 13C-NMR (DMSO-d6): δppm = 12.80 (CH3), 33.15 (CH2), 120.79 (2CHAr–C2,6), 126.89 (2CHAr–C3,5), 141.92 (Ar–C4), 149.17 (Ar–C1), 153.85 (CN), 155.85 (thiadiazole-C5), 158.49 (thiadiazole-C2), 165.37 (thiazole-C2), 171.77 (CO); MS m/z (%): 411 (M+, 2.9), 410 (2.2), 367 (1.9), 340 (1.8), 313 (7.7), 297 (1.9), 242 (5.6), 208 (2.1), 197 (1.6), 177 (1.7), 153 (3.7), 111 (17.0), 90 (8.3), 77 (11.3), 61 (8.6), 55 (100); anal. calc. For C13H13N7O3S3 (411.47): C, 37.95; H, 3.18; N, 23.83%; found: C, 37.94; H, 3.19; N, 23.82%.
3.1.3.1. 2-(1-(5-Imino-4-(4-sulfamoylphenyl)-4,5-dihydro-1,3,4-thiadiazol-2-yl)ethylidene)-N-methylhydrazine-1-carbothioamide (9a). The compound was obtained by heating 2a (0.35 g, 0.003 mol) for 2 h under reflux. Orange powder; yield 78%; mp 251–253 °C; IR νmax/cm−1 = 3339, 3301 (NH2), 3199, 3115, 3109 (3NH), 1628 (C
N), 1559 (CC), 1327, 1292 (SO2), 1217 (CS); 1H-NMR (DMSO-d6): δppm = 2.28 (s, 3H, CH3), 3.04 (d, J = 5.1 Hz, 3H, NH–CH3), 7.66 (s, 2H, NH2), 7.92 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.05 (d, J = 8.5 Hz, 2H, Ar–H3,5), 8.70 (s, 1H, NH), 11.04 (s, 1H, NH), 11.43 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 12.78 (CH3), 31.53 (CH3), 127.20 (2CHAr–C2,6), 127.55 (2CHAr–C3,5), 137.68 (Ar–C4), 139.07 (Ar–C1), 145.91 (CN), 155.60 (thiadiazole-C2), 167.79 (thiadiazole-C5), 178.67 (CS); MS m/z (%): 385 (M+, 0.1), 384 (0.1), 379 (0.1), 375 (0.1), 372 (0.1), 369 (0.1), 364 (0.1), 352 (0.2), 346 (0.1), 265 (0.7), 259 (0.2), 245 (0.3), 194 (1.2), 165 (1.7), 95 (22.2), 57 (100). Anal. calc. For C12H15N7O2S3 (385.48): C, 37.39; H, 3.92; N, 25.44%; found: C, 37.38; H, 3.93; N, 25.45%.
3.1.3.2. N-Allyl-2-(1-(5-imino-4-(4-sulfamoylphenyl)-4,5-di-hydro-1,3,4-thiadiazol-2-yl)ethylid ene)hydrazine-1-carbothioamide (9b). The compound was obtained by heating 2b (0.44 g, 0.003 mol) for 4 h under reflux. Orange powder; yield 70%; mp 263–265 °C; IR νmax/cm−1 = 3303, 3268 (NH2), 3197, 3120, 3100 (3NH), 1630 (C
N), 1555 (CC), 1313, 1293 (SO2), 1187 (CS); 1H-NMR (DMSO-d6): δppm = 2.30 (s, 3H, CH3), 4.24 (t, J = 5.9 Hz, 2H, N–CH2), 5.11 (dd, J = 8.5, 1.5 Hz, 1H, CH), 5.16 (dd, J = 15.3, 1.5 Hz, 1H, CH), 5.88 (m, 1H, CH), 7.64 (s, 2H, NH2), 7.93 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.05 (d, J = 8.5 Hz, 2H, Ar–H3,5), 8.83 (s, 1H, NH), 9.48 (s, 1H, NH), 11.13 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 12.81 (CH3), 46.19 (CH2), 115.83 (2CHAr–C2,6), 127.03 (CH2), 127.52 (2CHAr–C3,5), 134.36 (Ar–C4), 137.74 (Ar–C1), 139.34 (CH), 145.80 (CN), 155.51 (thiadiazole-C2), 167.64 (thiadiazole-C5), 178.33 (CS); MS m/z (%): 411 (M+, 0.7), 410 (1.1), 396 (1.0), 384 (1.1), 369 (1.9), 354 (2.2), 290 (0.7), 264 (4.5), 126 (3.5), 112 (10.4), 100 (3.4), 57 (100); anal. calc. For C14H17N7O2S3 (411.52): C, 40.86; H, 4.16; N, 23.83%; found: C, 40.87; H, 4.17; N, 23.82%.
3.1.3.3. Synthesis of 2-(1-(5-imino-4-(4-sulfamoylphenyl)-4,5-di-hydro-1,3,4-thiadiazol-2-yl)ethylidene)-N-phenylhydrazine-1-carbothioamide (9c). This compound was obtained by heating 2c (0.56 g, 0.003 mol) for 4 h under reflux. Orange powder; yield 86%; mp 260–261 °C; IR νmax/cm−1 = 3295, 3257 (NH2), 3187, 3114, 3102 (3NH), 1625 (C
N), 1554 (CC), 1318, 1294 (SO2), 1237 (CS); 1H-NMR (DMSO-d6): δppm = 2.35 (s, 3H, CH3), 7.24–756 (m, 5H, Ar–H), 7.65 (s, 2H, NH2), 7.94 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.06 (d, J = 8.5 Hz, 2H, Ar–H3,5), 10.49 (s, 1H, NH), 11.11 (s, 1H, NH), 11.48 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 13.06 (CH3), 121.54 (2CHAr–C2,6), 125.34 (2CHAr–C2′,6′), 125.84 (1CHAr–C4′), 127.15 (2CHAr–C3′,5′), 127.59 (2CHAr–C3,5), 128.47 (Ar–C4), 137.73 (Ar–C1), 138.94 (Ar–C1), 140.04 (CN), 145.91 (thiadiazole-C2), 167.85 (thiadiazole-C5), 177.61 (CS); MS m/z (%): 447 (M+, 0.2), 423 (0.3), 412 (0.2), 400 (0.2), 382 (0.2), 368 (0.2), 208 (0.2), 179 (0.5), 167 (1.1), 152 (1.3), 137 (0.7), 93 (100), 78 (3.9), 66 (40). Anal. calc. For C17H17N7O2S3 (447.55): C, 45.62; H, 3.83; N, 21.91%; found: C, 45.61; H, 3.82; N, 21.92%.
3.1.3.4. N-(4-Chlorophenyl)-2-(1-(5-imino-4-(4-sulfamoylphenyl)-4,5-dihydro-1,3,4-thiadiazol-2-yl)-ethylidene)hydrazine-1-carbothioamide (9d). The compound was obtained by heating 2d (0.67 g, 0.003 mol) for 3 h under reflux. Orange powder, yield 87%; mp 262–264 °C; IR νmax/cm−1 = 3339, 3258 (NH2), 3165, 3130, 3104 (3NH), 1635 (C
N), 1553 (CC), 1363, 1292 (SO2), 1247 (CS); 1H-NMR (DMSO-d6): δppm = 2.37 (s, 3H, CH3), 7.47 (d, J = 8.5 Hz, 2H, Ar–H3′,5′), 7.61 (d, J = 8.5 Hz, 2H, Ar–H2′,6′), 7.67 (s, 2H, NH2), 7.96 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.07 (d, J = 8.5 Hz, 2H, Ar–H3,5), 10.51 (s, 1H, NH), 10.99 (s, 1H, HN), 11.56 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 13.50 (CH3), 127.59 (2CHAr–C2,6), 127.96 (2CHAr–C3′,5′), 128.78 (2CHAr–C3,5), 130.21 (Ar–C4), 138.18 (2CHAr–C2′,6′), 138.36 (Ar–C4′), 140.80 (Ar–C1′), 140.99 (Ar–C1), 143.32 (CN), 146.23 (thiadiazole-C2), 155.76 (thiadiazole-C5), 178.08 (CS); MS m/z (%): 481 (M+, 0.2), 480 (0.3),467 (0.3), 397 (0.3), 381 (0.2), 367 (0.2), 333 (0.2), 325 (0.2), 295 (0.2), 268 (0.2), 264 (0.4), 254 (0.2), 250 (0.5), 239 (0.5), 227 (0.2), 201 (0.2), 199 (0.2), 191 (0.3), 182 (0.2), 171 (1.4), 127 (100), 93 (4.5), 78 (3.03), 66 (8.9). Anal. calc. For C17H16ClN7O2S3 (481.99): C, 42.36; H, 3.35; N, 20.34%; found: C, 42.37; H, 3.35; N, 20.33%.
3.1.4.1. 4-(2-Imino-5-(1-((3-methyl-4-phenylthiazol-2(3H)-ylidene)hydrazono)ethyl)-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide (12a). Orange powder; yield 77%; mp 231–233 °C; IR νmax/cm−1 = 3334, 3245 (NH2), 3172 (NH), 1652 (C
N), 1582 (CC), 1324, 1297 (SO2); 1H-NMR (DMSO-d6): δppm = 2.33 (s, 3H, CH3), 2.5 (s, 3H, NCH3), 6.56 (s, H, thiazole-H5), 7.39 (s, 2H, NH2),7.51 (s, 5H, Ar–H), 7.88 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.25 (d, J = 8.5 Hz, 2H, Ar–H3,5), 9.33 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 12.50 (CH3), 33.78 (CH3), 101.48 (thiazole-C5), 120.58 (2CHAr–C2,6), 126.46 (1CHAr–C4′), 128.85 (2CHAr–C2′,6′), 128.87 (2CHAr–C3′,5′), 129.09 (2CHAr–C3,5), 129.41 (Ar–C1′), 130.11 (Ar–C4), 140.14 (Ar–C1), 142.01 (thiazole-C4), 146.83 (CN), 153.00 (thiadiazole-C5), 159.19 (thiazole-C2), 171.56 (thiadiazole-C2); MS m/z (%): 485 (M+, 25.4), 484 (1.1), 443 (0.1), 412 (0.1), 394 (0.1), 325 (0.1), 315 (0.1), 298 (0.1), 256 (12.0), 190 (2.8), 160 (1.2), 102 (100), 72 (4.1); anal. calc. For C20H19N7O2S3 (485.60): C, 49.47; H, 3.94; N, 20.19%; found: C, 49.48; H, 3.93; N, 20.18%.
3.1.4.2. 4-(5-(1-((3-Allyl-4-phenylthiazol-2(3H)-ylidene)hydrazono)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide (12b). Orange powder; yield 80%; mp 202–204 °C; IR νmax/cm−1 = 3303, 3237 (NH2), 3165 (NH), 1637 (C
N), 1584 (CC), 1326, 1287 (SO2); 1H-NMR (DMSO-d6): δppm = 2.27 (s, 3H, CH3), 4.47 (d, J = 4.2 Hz, 2H, CH2), 5.12 (dd, J = 91, 1.7 Hz, 1H, CH), 5.19 (dd, J = 16, 1.7 Hz, 1H, CH), 5.82 (m, 1H, CH), 6.62 (s, 1H, thiazole-H5), 7.46 (s, 2H, NH2), 7.47–7.49 (m, 5H, Ar–H), 7.96 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.08 (d, J = 8.5 Hz, 2H, Ar–H3,5), 9.98 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 12.49 (CH3), 47.95 (N–CH2), 102.35 (thiazole-C5), 117.09 (CH2), 121.19 (2CHAr–C2,6), 126.00 (1CHAr–C4′), 126.87 (2CHAr–C2′,6′), 127.00 (2CHAr–C3′,5′), 128.82 (2CHAr–C3,5), 129.00 (Ar–C1′), 129.61 (Ar–C4), 130.04 (Ar–C1), 132.07 (CH), 140.75 (thiazole-C4), 146.27 (CN), 155.51 (thiadiazole-C5), 159.51 (thiazole-C2), 171.48 (thiadiazole-C2); MS m/z (%): 511 (M+, 15.8), 509 (1.6), 497 (1.3), 454 (3.6), 437 (6.7), 419 (2.4), 391 (1.4), 368 (2.1), 277 (2.6), 256 (9.8), 177 (2.4), 134 (100); anal. calc. For C22H21N7O2S3 (511.64): C, 51.65; H, 4.14; N, 19.16%; found: C, 51.66; H, 4.15; N, 19.17%.
3.1.4.3. 4-(5-(1-((3,4-Diphenylthiazol-2(3H)-ylidene)hydrazono)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide (12c). Green powder; yield 74%; mp 210–214 °C; IR νmax/cm−1 = 3330, 3274 (2NH), 3177 (NH), 1653 (C
N), 1586 (CC), 1325, 1280 (SO2); 1H-NMR (DMSO-d6): δppm = 2.07 (s, 3H, CH3), 6.78 (s, 1H, thiazole-H5), 7.15–7.31 (m, 10H, Ar–H), 7.44 (s, 2H, NH2), 7.90 (d, J = 9.3 Hz, 2H, Ar–H2,6), 7.99 (d, J = 9.3 Hz, 2H, Ar–H3,5), 9.68 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 12.61 (CH3), 103.16 (thiazole-C5), 121.28 (2CHAr–C2,6), 126.78 (1CHAr–C4′′), 126.89 (2CHAr–C2′′,6′′), 127.31 (1CHAr–C4′), 128.32 (2CHAr–C2′,6′), 128.38 (2CHAr–C3′,5′), 128.48 (2CHAr–C3′,5′), 128.50 (2CHAr–C3,5), 128.68 (Ar–C1′), 128.76 (Ar–C4), 140.25 (Ar–C1′′), 140.40 (Ar–C1), 147.79 (C–N), 148.31 (CN), 152.73 (thiadiazole-C5), 162.45 (thiazole-C2), 172.46 (thiadiazole-C2); MS m/z (%): 547 (M+, 45.4), 546 (1.9), 523 (1.3), 520 (1.6), 488 (2.6), 487 (4.5), 479 (1.5), 468 (1.9), 443 (1.3), 435 (1.4), 407 (1.7), 393 (1.4), 368 (1.9), 252 (4.9), 239 (4.2), 163 (2.4), 107 (10.9), 92 (21.1), 77 (100), 65 (25.0). Anal. calc. For C25H21N7O2S3 (547.67): C, 54.83; H, 3.87; N, 17.90%; found: C, 54.82; H, 3.86; N, 17.91%.
3.1.4.4. 4-(5-(1-((3-(4-Chlorophenyl)-4-phenylthiazol-2(3H)-ylidene)hydrazono)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide (12d). Dark green powder; yield 84%; mp 227–230 °C; IR νmax/cm−1 = 3303, 3276 (NH2), 3155 (NH), 1654 (C
N), 1584 (CC), 1306, 1279 (SO2), 738 (C–Cl); 1H-NMR (DMSO-d6): δppm = 2.12 (s, 3H, CH3), 6.81 (s, 1H, thiazole-H5), 7.18 (d, J = 2.5 Hz, 2H, Ar–H2′,6′), 7.29 (d, J = 4.2 Hz, 2H, Ar–H3′,5′), 7.34–7.45 (m, 5H, Ar–H), 7.59 (d, J = 8.5 Hz, 2H, Ar–H2,6), 7.90 (d, J = 8.5 Hz, 2H, Ar–H3,5), 8.19 (s, 2H, NH2), 9.49 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 12.42 (CH3), 103.67 (thiazole-C5), 114.33 (2CHAr–C2,6), 121.69 (2CHAr–C2′′,6′′), 127.03 (1CHAr–C4′′), 127.30 (1CHAr–C4′), 128.69 (2CHAr–C2′,6′), 128.91 (2CHAr–C3′,5′), 129.37 (2CHAr–C3′′,5′′), 130.03 (2CHAr–C3,5), 130.54 (Ar–C4), 130.83 (Ar–C1′), 131.12 (Ar–C1′′), 133.01 (Ar–C1), 136.43 (thiazole-C4), 140.25 (CN), 148.87 (thiadiazole-C5), 158.73 (thiazole-C2), 162.45 (thiadiazole-C2); MS m/z (%): 582 (M+, 22.1) 584 (M2+, 8.1), 581 (28.5), 524 (1.9), 503 (1.6), 488 (1.9), 425 (1.6), 381 (2.6), 355 (2.6), 351 (1.9), 339 (2.1), 315 (1.7), 294 (1.7), 279 (1.8), 2.51 (2.7), 210 (2.4), 155 (4.1), 127 (7.6), 93 (15.2), 71 (52.1), 57 (100). Anal. calc. For C25H20ClN7O2S3 (582.11): C, 51.58; H, 3.46; N, 16.84%; found: C, 51.59; H, 3.47; N, 16.85%.
3.2. Biological evaluation
The evaluated human cancer cell lines were cultivated to ensure their presence in the log phase then 3 mL of the cultivation medium was introduced to 96-well microplate. The total cells count in the medium should not exceed 106 cells per cm2 and the medium did not contain phenol or serum. Afterward, an equivalent volume of MTT (M-5655) to 10% of the medium was added and incubated at room temperature for 2–4 hours. The deposited formazan crystals were dissolved in an equivalent MTT solubilizing solution (M-8910) in the culture medium. The intensity of the obtained solution was measured as absorbance at wavelength 570 nm against the blank solution. The blank solution was prepared following the same conditions without cells at every test.44,453.3. Molecular docking simulation
The protein structure of hCAIX was recovered from the Protein Data Bank using PDB: 3IAI.46 Its crystal structure was subjected to the preparation step by deleting the unnecessary chains, co-factors, and water molecules while keeping the zinc ion. The Molecular Operating Environment (MOE) software-embedded autocorrection tool was used to check the validity of the chain conformation then hydrogens were added at a cutoff of 15 Å using AMBER10:EHT forcefield. The molecular docking site was adjusted to a radius of 4.5 Å around the co-crystallized ligand. Afterward, the actual docking simulation was implemented using Triangle Matcher, London dG, and GBVI/WSA as the placement, and rescoring functions 1 and 2, respectively. On the other hand, the chemical structures of the derivatives were constructed using ChemAxon Marvin JS, then pasted as smiles to MOE builder, protonated, and energy minimized at the same forcefield before commencing the simulation protocol.3.4. Molecular dynamic simulation
The simulation was performed using NAMD 3.0.0, applying Charmm-36 as the forcefield. According to the literature,47,48 Protein systems were built, corrected and prepared using the QwikMD.48,49 The binding free energy was calculated according to Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) embedded in the MMPBSA.py module of AMBER18 (ref. 50) using the following equation:| ΔGbinding = ΔGcomplex − ΔGreceptor − ΔGinhibitor |
Each equation required calculating multiple energy components, including van der Waals energy, electrostatic energy, internal energy from molecular mechanics, and polar contribution to solvation energy.
4. Conclusion
Herein, the tail approach synthesized a bundle of different hybrids of disubstituted/trisubstituted 1,3-thiazoles with 1,3,4-thiadiazolylbenzenesulfonaminde started with 1. The best cytotoxicity was observed with 4 and 9a against HepG-2, Caco2, and MCF-7 compared to staurosporine, with 4 showing a better safety margin as declared from its WI38 IC50 21.4 μM and 18.4 μM, respectively. Similarly, the phenylthiazole trisubstituted analogue 12d showed good cytotoxicity against the three tested cancer cell lines with higher safety than both 4 and 9a, as concluded from its WI38 IC50 46.6 μM. Moreover, the molecular modeling simulation of 4, 9a, and 12d against hCAIX revealed the bidentate coordination with zinc and forming H-bond with the catalytic His94, which explained their astonishing cytotoxicity. The molecular dynamic simulation of the more potent 9a and the least potent 9d with hCAIX demonstrated appreciable support of the proposed mechanism of their anticancer activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Deanship of Scientific Research at King Khalid University for funding this work through Small group Research Project under grant number RGP1/152/44.References
- J. Liu, S.-Q. Li, J. Wang, N. Li, J. Zhou and H. Chen, Recent Pat. Anti-Cancer Drug Discovery, 2022, 18, 125–132 CrossRef PubMed.
- A. I. Ilovaisky, A. M. Scherbakov, V. M. Merkulova, E. I. Chernoburova, M. A. Shchetinina, O. V. Andreeva, D. I. Salnikova, I. V. Zavarzin and A. O. Terent’ev, J. Steroid Biochem. Mol. Biol., 2023, 228, 106245 CrossRef CAS PubMed.
- M. Gümüş, M. Yakan and İ. Koca, Future Med. Chem., 2019, 11, 1979–1998 CrossRef PubMed.
- M. Arshad, A. Alam, A. S. Alshammari, M. B. Alhazza, I. M. Alzimam, Md. A. Alam, G. Mustafa, M. S. Ansari, A. M. D. A. Alotaibi, A. Alotaibi, S. Kumar, S. M. B. Asdaq, M. Imran, P. K. Deb, K. N. Venugopala and S. Jomah, Molecules, 2022, 27, 3994 CrossRef CAS PubMed.
- P. C. Sharma, K. K. Bansal, A. Sharma, D. Sharma and A. Deep, Eur. J. Med. Chem., 2020, 188, 112016 CrossRef CAS PubMed.
- A. Jain, S. Sharma, A. Vaidya, R. Veerasamy and R. K. Agrawal, Chem. Biol. Drug Des., 2013, 81, 557–576 CrossRef CAS PubMed.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- Y. L. Duc, E. Licsandru, D. Vullo, M. Barboiu and C. T. Supuran, Bioorg. Med. Chem., 2017, 25, 1681–1686 CrossRef PubMed.
- C. T. Supuran, Expert Opin. Ther. Pat., 2018, 28, 729–740 CrossRef PubMed.
- C. Temperini, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem., 2010, 20, 474–478 CrossRef CAS PubMed.
- C. T. Supuran and A. Scozzafava, Bioorg. Med. Chem., 2007, 15, 4336–4350 CrossRef CAS PubMed.
- F. Cianchi, M. C. Vinci, C. T. Supuran, B. Peruzzi, P. De Giuli, G. Fasolis, G. Perigli, S. Pastorekova, L. Papucci, A. Pini, E. Masini and L. Puccetti, J. Pharmacol. Exp. Ther., 2010, 334, 710–719 CrossRef CAS PubMed.
- M. Aggarwal, C. G. Boone, B. Kondeti and R. McKenna, J. Enzyme Inhib. Med. Chem., 2012, 28, 267–277 CrossRef PubMed.
- C. T. Supuran, Expert Opin. Ther. Pat., 2018, 28, 709–712 CrossRef CAS PubMed.
- C. W. McMonnies, J. Optom., 2017, 10, 71–78 CrossRef PubMed.
- R. Kumar, A. Kumar, S. Ram, C. T. Supuran, A. Bonardi, P. Gratteri and P. K. Sharma, Arch. Pharm., 2022, 355, 2100241 CrossRef CAS PubMed.
- S. Kilicaslan, M. Arslan, Z. Ruya, C. Bilen, A. Ergün, N. Gençer and O. Arslan, J. Enzyme Inhib. Med. Chem., 2016, 31, 1300–1305 CrossRef CAS PubMed.
- A. A. Almehizia, W. M. Eldehna, C. T. Supuran, H. M. Ibrahim, S. Bua, H. A. Abdel-Aziz and S. M. Abou-Seri, Bioorg. Chem., 2019, 87, 794–802 CrossRef PubMed.
- P. A. Channar, R. D. Alharthy, S. A. Ejaz, A. Saeed and J. Iqbal, Molecules, 2022, 27, 8723 CrossRef CAS PubMed.
- M. Khan, S. A. Halim, R. Csuk, M. N. Islam, M. A. Shehzad, A. Ibrard, F. R. Khan, N. Marraiki, J. Uddin, A. Khan and A. Al-Harrasi, Curr. Pharm. Des., 2022, 28, 3010–3022 CrossRef CAS PubMed.
- M. Ishaq, P. Taslimi, R. Csuk, S. Khan, R. E. Salmas, M. M. Zangeneh, A. Saeed, A. Zangeneh, P. Taslimi, A. Asari and H. Mohamad, Bioorg. Chem., 2020, 100, 103924 CrossRef CAS PubMed.
- A. W. Erian, S. S. Sherif and H. M. Gaber, Molecules, 2003, 8, 793–865 CrossRef CAS.
- R. Venkatraman, H. Ameera, L. Sitole, E. D. Ellis, F. R. Fronczek and E. J. Valente, J. Chem. Crystallogr., 2009, 39, 711–718 CrossRef CAS.
- A. A. Hassan, S. K. Mohamed, N. K. Mohamed, K. M. El-Shaieb, A. T. Abdel-Aziz, J. T. Mague and M. Akkurt, J. Sulfur Chem., 2016, 37, 162–175 CrossRef CAS.
- H. Yakan, Res. Chem. Intermed., 2020, 46, 3979–3995 CrossRef CAS.
- M. A. Fouad, M. Y. Zaki, R. A. Lotfy and W. H. Mahmoud, Bioorg. Chem., 2021, 112, 104985 CrossRef CAS PubMed.
- V. V. Syakaev, S. N. Podyachev, B. I. Buzykin, S. K. Latypov, W. D. Habicher and A. I. Konovalov, J. Mol. Struct., 2006, 788, 55–62 CrossRef CAS.
- D. T. G. Gonzaga, F. De C Da Silva, V. F. Ferreira, J. L. Wardell and S. M. S. V. Wardell, J. Braz. Chem. Soc., 2016, 27, 2322–2333 CAS.
- E. Abdel-Latif, M. M. Fahad, A. El-Demerdash and M. A. Ismail, J. Heterocycl. Chem., 2020, 57, 3071–3081 CrossRef CAS.
- V. Alterio, M. Hilvo, A. Di Fiore, C. T. Supuran, P. Pan, S. Parkkila, A. Scaloni, J. Pastorek, S. Pastorekova, C. Pedone, A. Scozzafava, S. M. Monti and G. De Simone, Proc. Natl. Acad. Sci., 2009, 106, 16233–16238 CrossRef CAS PubMed.
- K. Kanamori and J. D. Roberts, Biochemistry, 1983, 22, 2658–2664 CrossRef CAS PubMed.
- N. Chiaramonte, M. N. Romanelli, E. Teodori and C. T. Supuran, Metabolites, 2018, 8, 36 CrossRef PubMed.
- C. T. Supuran, Expert Opin. Invest. Drugs, 2018, 27, 963–970 CrossRef CAS PubMed.
- S. Imran, M. Taha, N. H. Ismail, S. Fayyaz, K. M. Khan and M. I. Choudhary, Bioorg. Chem., 2015, 62, 83–93 CrossRef CAS PubMed.
- Y. Takaoka, Y. Kioi, A. Morito, J. Otani, K. Arita, E. Ashihara, M. Ariyoshi, H. Tochio, M. Shirakawa and I. Hamachi, Chem. Commun., 2013, 49, 2801–2803 RSC.
- K. H. Sippel, A. H. Robbins, J. F. Domsic, C. Genis, M. Agbandje-McKenna and R. McKenna, Acta Crystallogr., Sect. A: Found. Crystallogr., 2009, 65, 992–995 CAS.
- D. A. Whittington, A. Waheed, B. Ulmasov, G. N. Shah, J. H. Grubb, W. S. Sly and D. W. Christianson, Proc. Natl. Acad. Sci., 2001, 98, 9545–9550 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- A. T. Balaban, Theor. Chim. Acta, 1979, 53, 355–375 CrossRef CAS.
- J. Lin and M. Yamazaki, Clin. Pharmacokinet., 2003, 42, 59–98 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. W. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- Y. C. Martin, J. Med. Chem., 2005, 48, 3164–3170 CrossRef CAS PubMed.
- S. Bondock, T. Albarqi, T. Nasr, N. M. Mohamed and M. M. Abdou, Arabian J. Chem., 2023, 16, 104956 CrossRef CAS.
- S. Bondock, T. Albarqi, I. A. Shaaban and M. M. Abdou, RSC Adv., 2023, 13, 10353–10366 RSC.
- F. Briganti, S. Mangani, P. Orioli, A. Scozzafava, G. Vernaglione and C. T. Supuran, Biochemistry, 1997, 36, 10384–10392 CrossRef CAS PubMed.
- J. G. Phillips, R. Braun, W. Wang, J. C. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. V. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- J. Ribeiro, R. C. Bernardi, T. Rudack, K. Schulten and E. Tajkhorshid, Biophys. J., 2018, 114, 673a–674a CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- B. R. Miller, T. D. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02528d |
| This journal is © The Royal Society of Chemistry 2023 |