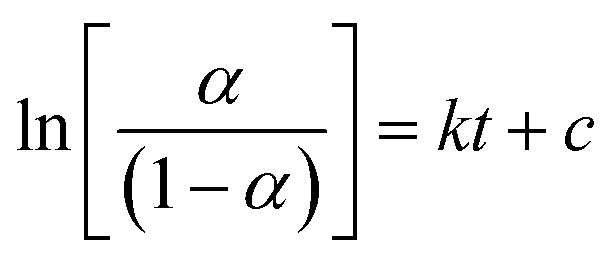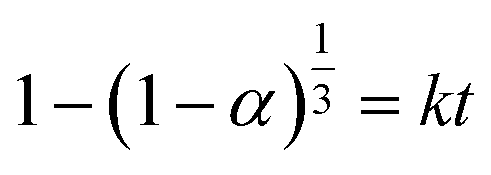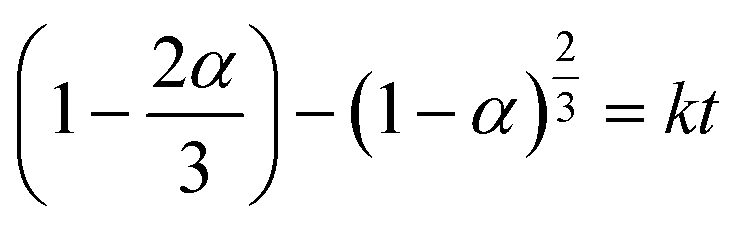DOI:
10.1039/D3RA02521G
(Paper)
RSC Adv., 2023,
13, 22172-22184
Solubility determination, dissolution properties and solid transformation of resmetirom (form A) in heptane and seven alcohols†
Received
16th April 2023
, Accepted 17th July 2023
First published on 27th July 2023
Abstract
In this work, the solubility of resmetirom (form A) was initially measured in heptane and seven alcohol solvents by gravimetric methods. Then, the transformation temperature between form A and ethanol solvate was determined at 333.76 K. Subsequently, some commonly used models were applied to fit the solubility data, and it was found that the modified Apelblat equation and the Jouyban–Acree–van't Hoff (J–A–V) model achieved the highest correlation accuracy for those in mono-solvents and heptane + propanol, respectively. And the average relative deviation (ARD) values of models were less than 0.5%, indicating a good agreement with the experimental results. Additionally, through density functional theory calculation and the analysis of solvent parameters, it was observed that hydrogen-bonding played primary roles in the dissolution process of resmetirom. The multiple factors such as the polarity of solvent, active site interaction, the molecular size and free volume all affect the solubility of resmetirom. Furthermore, by comparing the experimental and simulated infrared spectra of form A and two alcohol solvates, five characteristic bands were selected for quantification. Partial least squares regression (PLSR), a multivariate statistical analysis method, was used to extract quantitative information. The quantitative analysis model was established based on specific wavelength intervals, which were associated with inter-molecular interactions. Combined with PLSR, a new high-precision quantitative method was established to study the solid transformation process between form A and solvates. From 303.15 to 323.15 K, the rate of transformation from form A to methanol solvate or ethanol solvate was decreased with increasing temperature, revealing that the transformation process was driven by the solubility difference between form A and solvates under the studied conditions. This research will definitely afford necessary solubility data and solvent selection for the design of the crystallization process of resmetirom (form A) in industry, and provide basic data for the production of resmetirom (form A) in the pharmaceutical industry.
Introduction
Active pharmaceutical ingredients (APIs) have been proven to commonly possess various solid forms, in which more than half of the APIs are able to form solvates.1,2 Because of different conformations and arrangements of active molecules as well as disparate binding types of solvent molecules in solid structures, different solid forms of drug molecules often show various physicochemical properties, processing performance, solubility and bioavailability, leading further to different therapeutic effect and safety. Hence, research on the different solid forms of APIs is crucial in the drug development process.3–5
Various solvents are inevitably used in drug production, and the transformation of certain crystalline forms turning to solvates are common.6 Therefore, the study of solid transformation is vital for drug production. The solid–solid transformation kinetics model is typically obtained through online or offline detection techniques such as differential scanning calorimetry (DSC), powder X-ray diffraction (PXRD), Fourier transform infrared (FTIR), etc.7,8 Among them, FTIR is an ideal method for the quantitative analysis to study the conversion between different solid forms because of its easy operation, high sensitivity and rich spectral information. The vibrational frequency of the molecule is characterized by the atomic structure, in which functional groups can be easily detected on spectra with the wavenumber from 4000 to 400 cm−1 and 3600 spectral points (i.e., variables) were produced. However, low root mean square errors in prediction have been pursued blindly in current selection methods for infrared spectra, which lacks the interpretability of variables. Thereby, when requirements of quantitative analysis accuracy are satisfied, improvement on interpretability of variable selection is one of the development directions of variable selection methods.9,10
The concept of “nonalcoholic steatohepatitis (NASH)” was first introduced by Ludwig in 1980,11 and in 1986, Schaffner introduced “nonalcoholic steatohepatic disease (NAFLD)”.12 NASH is considered the most severe histological lesion within NAFLD due to its higher propensity for hepatocellular carcinoma development. NASH patients exhibit a fibrosis progression rate of 40.76% and a mean annual progression rate.13 Recent estimates suggest that NASH will lead to a 30% increase in indirect and direct healthcare costs over the next 5 years due to rising rates of overweight and obesity. Consequently, the development of therapeutic drugs for NASH is urgently needed. Resmetirom is a highly selective THR-β agonist with the ability to regulate lipid metabolism, and Phase III clinical trials of its therapy for NASH have been finished.14,15 The molecular structure of resmetirom is depicted in Fig. 1, which includes pyridazine ring (R1), dichlorobenzene ring (R2) and triazine ring (R3) from left to right. The Hansen solubility parameters of resmetirom in variously chosen solvents and the single-crystal structure of the solvates were acquired by Tian et al.16 However, solubility and solid transformation in organic solvents remains unreported.
 |
| | Fig. 1 Molecular structure of resmetirom. | |
Building upon the work of Tian et al.,16 the solubility of resmetirom (form A) in heptane and seven alcohol solvents was firstly determined in this study, which were correlated using four commonly used models. Then, the dissolution process of resmetirom could be analyzed based on the binding energy calculation by density functional theory (DFT). Subsequently, combined with DFT calculations, a vibrational frequency range was chosen, which was sensitive to the frequency change caused by solvent molecules in the lattice.17,18 Finally, based on variable selection method, a high-precision quantitative analysis was built up by partial least squares regression (PLSR), which was adopted to reveal the impact on temperatures to transformation processes between form A and two solvates.
Materials and method
Materials
The resmetirom (form A), methanol, ethanol, propanol, isopropyl alcohol, butanol, isobutyl alcohol, 3-methyl-1-butanol and heptane were supplied by Aladdin Company (Shanghai, China). Details of these materials were given in Table 1.
Table 1 Detailed information of materials used in this work
| Material |
Molecular formula |
Molar volume (cm3 mol−1) |
Molar mass (g mol−1) |
Mass fraction purity |
Analytical method |
| High performance liquid chromatography Gas chromatographic method. |
| Resmetirom |
C17H13Cl2N5O6 |
263.77 |
435.22 |
≥0.990 |
HPLCa |
| Methanol |
CH4O |
42.50 |
32.00 |
≥0.995 |
GCb |
| Ethanol |
C2H5O |
58.39 |
46.07 |
≥0.995 |
| Propanol |
C3H8O |
75.50 |
60.10 |
≥0.995 |
| Isopropyl alcohol |
C3H8O |
75.90 |
60.10 |
≥0.995 |
| Butanol |
C4H10O |
92.00 |
74.12 |
≥0.995 |
| Isobutyl alcohol |
C4H10O |
92.40 |
74.12 |
≥0.995 |
| 3-Methyl-1-butanol |
C5H12O |
108.90 |
88.15 |
≥0.995 |
| Heptane |
C7H16 |
144.00 |
100.20 |
≥0.995 |
Characterization
The solid-state characterization methods used in this work included DSC, PXRD and FTIR. Based on the principle of power-compensated heat flow differences, the DSC data were obtained by an STA 499 F1 differential scanning calorimeter (Netzsch, Bavaria, Germany) armed with a mainframe, a workstation and a gas controller. The PXRD spectrum were obtained by a multifunctional Fringe EV X-ray diffractometer (LANScientific, Jiangsu, China), Cu-Kα radiation (λ = 1.54 Å) with a tube voltage of 40 kV and a tube current of 35 mA was used to analyze samples over 2θ range of 5–45°, and imaged by MDI Jade 6.5 software. The FTIR data were collected on a Shimadzu IRTracer-100 spectrometer (Shimadzu, Tokyo, Japan) and imaged on Omnic software.
Solubility measurement
From 273.15 to 333.15 K, the solubility of form A was determined in seven mono-solvents as well as mixed solvent of heptane + propanol using a gravimetric method. Firstly, the appropriate organic solvent was added to a crystallizer, and excess samples were added at desired temperatures. Then, the commixture was stirred utilizing magnetic stirrers for 12 h, and stood for another 12 h until the suspension reached solid–liquid equilibrium. Subsequently, the supernatants were withdrawn using preheated syringes, followed by being transferred to a pre-weighed sample vial. After that the filtrate and vial were weighted by an FA1004T analytical balance (Supo, Zhejiang, China), followed by being dried in a constant-temperature WGL-65B oven (Taisite, Tianjin, China) until the mass kept unchanged. Finally, the crystalline forms of residual solids were characterized. The reported experimental data were averaged of three experiments. The molar fraction solubility of form A (xi) in mono-solvent and binary solvent system were calculated using eqn (1):19–21| |
 | (1) |
where M1, M2, M3 were the molecular mass of form A, alcohols as well as heptane, respectively. m1 and m2 were the mass of form A and solvent, respectively. ω was the mass fraction of propanol in mixed solvents.
Solid transformation determination
The solid transformation experiments for form A were carried out in 50 mL crystallizer. Firstly, in light of solubility data above, initial suspend solutions of form A at 303.15, 313.15, and 323.15 K were prepared in 10 g methanol/ethanol. Then the suspension was extracted and filtered in an interval time, and residual solids were dried at corresponding temperature for 30 min. Finally, a high-precision calibration of FTIR for quantitative analysis was used in this study, and the purity of the methanol solvate (R-MT) and ethanol solvate (R-ET) was determined.
Establishment of quantitative analysis methods
Firstly, form A and the two solvates were weighed accurately. Then, characterization of the solids was detected on PXRD to confirm that no polymorphic transition occurred during the measurements. After that, fifteen samples were prepared in the 0–100% concentration range, and all of which were mixed in 5 mL vials followed by being shaken for 15 min. Subsequently, the small samples were further ground with 200 mg KBr powder. The powder was mixed thoroughly in an agate mortar and tableted into transparent discs with a diameter in approximately 1 cm by an FW-5A tablet press machine (Shengda, Tianjin, China). Multiple measurements on the samples to decrease the error of quantitative analysis. Finally, the full processes of quantitative analysis were completed of the PLSR method on Matlab 2020 software.22,23
All quantum calculations were used in the Gaussian16 software with the B3LYP exchange–correlation functionals and the 6-311G (d, p) basis set, combined with the BJ-damping function proposed by Grimme to account for long-range effects.24 In order to establish a more precise quantitative analysis model, geometry optimization and vibrational analysis were carried out for both form A and solvates, meanwhile, FTIR spectra were simulated. The correction factor (0.961) was also applied to calibrate all calculated vibrational frequencies. The simulated FTIR spectra showed no imaginary frequencies, indicating that the optimized molecule corresponded to the minimum of the potential energy surface. For improvement of model performance, PLSR, a multivariate statistical analysis method, was used to extract quantitative information to overcome the nonlinear relationship between spectral response signals and concentrations. The quantitative analysis model was established based on specific wavelength intervals, which were associated with inter-molecular interactions. The coefficient of determination, R2, which was worked as an indicator of assessing model accuracy, was calculated as follows:
| |
 | (2) |
here,
n represented the sample size.
yP,
yi and
ym were denoted as the predicted, actual and mean values of the sample, respectively.
Quantum mechanics calculation
The simulations pertaining to resmetirom molecules were performed using DFT in the Gaussian16 software. Firstly, the structures of resmetirom molecules and various solvent molecules were optimized and verified by frequency calculations, ensuring that the potential energy surface on their structures was a local minimum. Then, the optimized molecular structures were plotted separately for the electrostatic distribution on the molecular surface. Based on the principle of potential attraction, the possible interaction sites of solvent–solute molecules were predicted. Subsequently, the solvent and solute molecules were placed at suitable positions as the initial conformation according to the predicted sites, and the optimization of structure, position, and frequency calculation were performed again. The optimized solute–solvent units were analyzed by interaction region indicator (IRI) to calculate the intermolecular interaction forces. The Multiwfn software calculated the molecular electrostatic potential (MEP) maps and IRI analysis maps, which were presented in the VMD 1.9.4 program.25,26 Finally, binding energies (ΔE) corrected by basis set superposition error (BSSE) between resmetirom and various solvents was calculated using DFT to quantify the binding abilities of the solute with different solvents at the B3LYP-D3 (BJ)/6-311G (d,p) level. ΔE was expressed as:27–29| | |
ΔE = Eunit − (EA + EB) + EBSSE
| (3) |
here, Eunit, EA, EB and EBSSE represented the energy of solute–solvent unit, solute molecules, solvent molecules and BSSE, respectively.
Results and discussion
Characterization
The solids are dried for 30 min to remove surface solvent, and they are subsequently performed using PXRD. The PXRD patterns of the solid residues and resmetirom (form A) are depicted in Fig. 2. When methanol and ethanol are solvents, the PXRD patterns of residual solids are different to form A, indicating that two solvates are formed. The peaks of residual solids in other solvents are basically similar except in methanol and ethanol, indicating that solvates are not formed. The characteristic peaks of form A are identified at 2θ = 8.19 ± 0.1°, 11.16 ± 0.1°, 18.32 ± 0.1°, 18.67 ± 0.1°, 22.23 ± 0.1° and 32.20 ± 0.1°. The characteristic peaks for R-MT appear at 2θ = 5.71 ± 0.1°, 10.46 ± 0.1°, 17.65 ± 0.1°, 22.59 ± 0.1°, 24.61 ± 0.1°. The characteristic peaks for R-ET appear at 2θ = 5.71 ± 0.1°, 10.52 ± 0.1°, 11.41 ± 0.1°, 21.19 ± 0.1°, 23.19 ± 0.1°, 26.24 ± 0.1°.
 |
| | Fig. 2 PXRD diagrams of form A as well as residual solids for solubility measurements. | |
Form A, R-MT, and R-ET forms of DSC and TGA curves are illustrated in Fig. 3(a)–(c), respectively. At 606.30 K, the crystals start to decompose and melt with an enthalpy of decomposition of 120.32 J g−1. The DSC curves of the solvates show a clear endothermic peak before the decomposition temperature. The heat absorption enthalpy of the methanol solvate is 95.97 J g−1, while that of the ethanol solvate is 113.31 J g−1. This is a consequence of solvent molecules evaporating from the lattice during heating. By the mass loss on TGA curves, the stoichiometric ratio between the solute and solvent molecules can be obtained for two solvates, which are both 1. The theoretical and observed mass loss rates for the R-MT are 6.45% and 6.56%, respectively, while those for the R-ET are 9.57% and 9.51%, indicating the data of experimental mass loss exhibit a satisfactory agreement with the theoretical data.
 |
| | Fig. 3 DSC/TGA curves of form A (a), R-MT (b) and R-ET (c). | |
The original FTIR spectra of form A, R-MT and R-ET are shown in Fig. 4, which reveal molecular vibration information is related to the crystal structure. The main difference between form A and the two solvates is reflected in the range of the 3600–3000 cm−1, which is obviously due to the O–H stretching vibrations in hydroxyl and N–H stretching vibrations in amide groups. Additionally, the intensity of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N peak also varies significantly, which is attributed to the intramolecular hydrogen bonding between resmetirom and the solvent molecules. The characteristic peaks in the range of 1800–1700 cm−1 could be ascribed to the stretching vibration of carbonyl groups. The carbonyl stretching vibrational peaks on the R3 and R1 rings in form A appear at 1736 and 1719 cm−1, respectively, while the corresponding vibrations for the solvates appear at 1760 and 1727 cm−1. In the range of 1700–1600 cm−1, a single peak occurs in the spectrum of form A, while three small peaks occur in that of solvates (as shown in the embedded figure), which may be related to the solvent molecules in the lattice. The FTIR spectra of the two solvates are very similar, but there is a bathochromic shift in the peaks in the spectrum of R-MT compared to that of R-ET, probably due to hydrogen bonding. The main distinction of peaks between R-MT and R-ET occurs within the spectral interval of 820–700 cm−1, where the O–H vibrations in hydroxyl group in the solvent molecule is caused by a N atom in the R3 ring. The spectra of solvates exhibit the difference with that of form A resulting from intermolecular interactions, which lays a foundation for quantification by FTIR spectra.
N peak also varies significantly, which is attributed to the intramolecular hydrogen bonding between resmetirom and the solvent molecules. The characteristic peaks in the range of 1800–1700 cm−1 could be ascribed to the stretching vibration of carbonyl groups. The carbonyl stretching vibrational peaks on the R3 and R1 rings in form A appear at 1736 and 1719 cm−1, respectively, while the corresponding vibrations for the solvates appear at 1760 and 1727 cm−1. In the range of 1700–1600 cm−1, a single peak occurs in the spectrum of form A, while three small peaks occur in that of solvates (as shown in the embedded figure), which may be related to the solvent molecules in the lattice. The FTIR spectra of the two solvates are very similar, but there is a bathochromic shift in the peaks in the spectrum of R-MT compared to that of R-ET, probably due to hydrogen bonding. The main distinction of peaks between R-MT and R-ET occurs within the spectral interval of 820–700 cm−1, where the O–H vibrations in hydroxyl group in the solvent molecule is caused by a N atom in the R3 ring. The spectra of solvates exhibit the difference with that of form A resulting from intermolecular interactions, which lays a foundation for quantification by FTIR spectra.
 |
| | Fig. 4 FTIR spectra of form A, R-MT and R-ET. The embedded figure is a schematic diagram of some bands. | |
Solubility
The stability analysis of polymorph is an essential part of pharmaceutical research. The transformation temperatures between form A and R-MT or R-ET are illustrated in Fig. 5(a). The solubility of resmetirom in ethanol vary with temperature in two temperature regions, in which the breakpoint is 333.76 K. While transformation temperature between form A and R-MT is above the boiling temperature of methanol under experimental conditions. In both temperature regions, the correlation of temperature to solubility is linear in two temperature regions, in which the slope of AB curve in low-temperature region is steeper than that of BC curve in high-temperature region. The intersection temperature point B of the two curves corresponds to 333.76 K, which is the solid transformation temperature of solvated and unsolvated forms. The solubility of the unsolvated form in the low temperature range and the solvate in the high temperature range are indicated by the two dashed lines BD and BE in the figure, respectively. According to the stability rule of crystal polymorphs, the crystal form with lower solubility has thermodynamic stability at the same temperature.30 Therefore, when the temperature is lower than 333.76 K, the solvated crystalline form of resmetirom is stable, otherwise the form A is stable. The trend of polymorphic transformation can be explained by the difference in two forms of Gibbs free energy ΔG as follows:31,32| |
ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(xi/xR-ET) ln(xi/xR-ET)
| (4) |
where xi and xR-ET are the solubilities of form A and R-ET at corresponding temperature, respectively. The relationship between ΔG and temperature for two forms in ethanol from 273.15 to 348.15 K is depicted in Fig. 5(b). The data from eqn (4) reveal temperature exhibits a linear relationship with ΔG. Furthermore, with a transformation point at 333.76 K, ΔG is negative at lower temperatures and positive at higher temperatures, which is congruent with the results obtained in Fig. 5(a).
 |
| | Fig. 5 Solubility of unsolvated and solvated resmetirom in methanol and ethanol (a); ΔG between form A and R-ET (b). | |
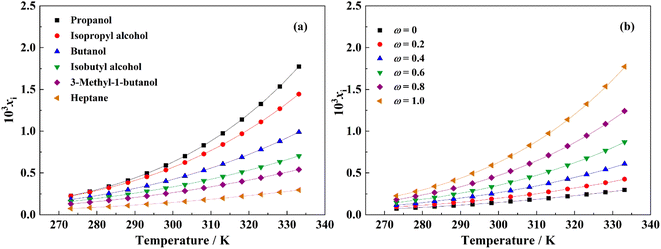
The solubility data of form A in various mono-solvents from 273.15 to 333.15 K are collected in Table 2. The solubility data of form A in binary mixtures of propanol and heptane are shown in Table 3. The correlation results by the modified Apelblat equation for the experimental solubility of form A in pure solvents and those in propanol + heptane by the J–A–V model are presented in Fig. 6. The results demonstrate that the solubility of form A show an upward trend as temperature increases throughout all experimental measurements, indicating that this is an endothermic process.
Table 2 Molar fraction solubility (xi/mol mol−1) of resmetirom (form A) in mono-solventsa (p = 0.1 MPa)
| T (K) |
Propanol |
Isopropyl alcohol |
Butanol |
| 103xexp |
103xcal |
103xexp |
103xcal |
103xexp |
103xcal |
| Apelblat |
λh |
Wilson |
Apelblat |
λh |
Wilson |
Apelblat |
λh |
Wilson |
| The standard uncertainties u of pressure and temperature are 0.5 kPa and 0.01 K, respectively. The relative standard uncertainty of the solubility measurement is ur (xi) = 0.06. |
| 273.15 |
0.227 |
0.226 |
0.227 |
0.224 |
0.226 |
0.225 |
0.226 |
0.218 |
0.183 |
0.183 |
0.184 |
0.171 |
| 278.15 |
0.278 |
0.278 |
0.279 |
0.276 |
0.271 |
0.270 |
0.272 |
0.265 |
0.216 |
0.217 |
0.217 |
0.207 |
| 283.15 |
0.340 |
0.339 |
0.339 |
0.338 |
0.324 |
0.323 |
0.325 |
0.319 |
0.255 |
0.255 |
0.255 |
0.247 |
| 288.15 |
0.411 |
0.410 |
0.410 |
0.410 |
0.385 |
0.385 |
0.385 |
0.381 |
0.298 |
0.298 |
0.298 |
0.292 |
| 293.15 |
0.494 |
0.493 |
0.493 |
0.493 |
0.455 |
0.454 |
0.455 |
0.453 |
0.347 |
0.347 |
0.346 |
0.343 |
| 298.15 |
0.591 |
0.590 |
0.589 |
0.590 |
0.535 |
0.534 |
0.534 |
0.534 |
0.401 |
0.402 |
0.401 |
0.399 |
| 303.15 |
0.702 |
0.701 |
0.700 |
0.702 |
0.625 |
0.624 |
0.624 |
0.625 |
0.463 |
0.463 |
0.461 |
0.462 |
| 308.15 |
0.829 |
0.828 |
0.827 |
0.830 |
0.727 |
0.726 |
0.725 |
0.728 |
0.531 |
0.531 |
0.529 |
0.532 |
| 313.15 |
0.975 |
0.974 |
0.972 |
0.976 |
0.841 |
0.840 |
0.839 |
0.844 |
0.606 |
0.607 |
0.604 |
0.609 |
| 318.15 |
1.140 |
1.139 |
1.137 |
1.142 |
0.969 |
0.968 |
0.967 |
0.973 |
0.689 |
0.690 |
0.687 |
0.694 |
| 323.15 |
1.327 |
1.325 |
1.323 |
1.329 |
1.112 |
1.110 |
1.110 |
1.117 |
0.781 |
0.781 |
0.779 |
0.787 |
| 328.15 |
1.537 |
1.535 |
1.534 |
1.540 |
1.270 |
1.268 |
1.269 |
1.276 |
0.881 |
0.882 |
0.880 |
0.889 |
| 333.15 |
1.773 |
1.772 |
1.770 |
1.776 |
1.445 |
1.443 |
1.445 |
1.452 |
0.991 |
0.992 |
0.991 |
1.000 |
| T (K) |
Isobutyl alcohol |
3-Methyl-1-butanol |
Heptane |
| 103xexp |
103xcal |
103xexp |
103xcal |
103xexp |
103xcal |
| Apelblat |
λh |
Wilson |
Apelblat |
λh |
Wilson |
Apelblat |
λh |
Wilson |
| 273.15 |
0.160 |
0.160 |
0.162 |
0.149 |
0.131 |
0.131 |
0.132 |
0.119 |
0.075 |
0.075 |
0.076 |
0.062 |
| 278.15 |
0.186 |
0.186 |
0.187 |
0.176 |
0.151 |
0.151 |
0.152 |
0.141 |
0.086 |
0.086 |
0.087 |
0.075 |
| 283.15 |
0.214 |
0.215 |
0.215 |
0.206 |
0.173 |
0.173 |
0.174 |
0.165 |
0.098 |
0.098 |
0.099 |
0.089 |
| 288.15 |
0.246 |
0.246 |
0.246 |
0.240 |
0.198 |
0.198 |
0.197 |
0.191 |
0.112 |
0.112 |
0.112 |
0.105 |
| 293.15 |
0.281 |
0.281 |
0.280 |
0.277 |
0.225 |
0.225 |
0.224 |
0.220 |
0.126 |
0.127 |
0.126 |
0.121 |
| 298.15 |
0.319 |
0.320 |
0.318 |
0.317 |
0.254 |
0.254 |
0.253 |
0.251 |
0.142 |
0.143 |
0.142 |
0.139 |
| 303.15 |
0.361 |
0.362 |
0.360 |
0.361 |
0.286 |
0.287 |
0.285 |
0.285 |
0.159 |
0.160 |
0.159 |
0.159 |
| 308.15 |
0.408 |
0.409 |
0.406 |
0.409 |
0.321 |
0.322 |
0.319 |
0.322 |
0.179 |
0.179 |
0.178 |
0.180 |
| 313.15 |
0.458 |
0.459 |
0.456 |
0.461 |
0.359 |
0.359 |
0.357 |
0.362 |
0.199 |
0.200 |
0.198 |
0.202 |
| 318.15 |
0.512 |
0.514 |
0.511 |
0.517 |
0.399 |
0.400 |
0.398 |
0.404 |
0.221 |
0.222 |
0.221 |
0.226 |
| 323.15 |
0.572 |
0.573 |
0.570 |
0.578 |
0.444 |
0.444 |
0.442 |
0.450 |
0.245 |
0.245 |
0.244 |
0.251 |
| 328.15 |
0.635 |
0.636 |
0.635 |
0.643 |
0.491 |
0.492 |
0.490 |
0.499 |
0.270 |
0.271 |
0.270 |
0.279 |
| 333.15 |
0.704 |
0.705 |
0.705 |
0.714 |
0.542 |
0.542 |
0.542 |
0.552 |
0.297 |
0.298 |
0.298 |
0.308 |
Table 3 Molar fraction solubility (xi/mol mol−1) of resmetirom (form A) in propanol + heptanea (p = 0.1 MPa)
| T (K) |
ω = 0.2 |
ω = 0.4 |
| 103xexp |
103xcal |
103xexp |
103xcal |
| Apelblat |
λh |
J–A–V |
Apelblat |
λh |
J–A–V |
| The standard uncertainties u of pressure and temperature are 0.5 kPa and 0.01 K, respectively. The relative standard uncertainty of the solubility measurement is ur (xi) = 0.06. |
| 273.15 |
0.094 |
0.095 |
0.094 |
0.097 |
0.117 |
0.116 |
0.117 |
0.116 |
| 278.15 |
0.109 |
0.108 |
0.109 |
0.109 |
0.138 |
0.137 |
0.137 |
0.137 |
| 283.15 |
0.126 |
0.125 |
0.126 |
0.125 |
0.162 |
0.163 |
0.160 |
0.163 |
| 288.15 |
0.145 |
0.144 |
0.145 |
0.144 |
0.189 |
0.191 |
0.187 |
0.191 |
| 293.15 |
0.166 |
0.165 |
0.166 |
0.164 |
0.219 |
0.220 |
0.216 |
0.220 |
| 298.15 |
0.190 |
0.193 |
0.189 |
0.192 |
0.252 |
0.250 |
0.249 |
0.251 |
| 303.15 |
0.215 |
0.215 |
0.214 |
0.214 |
0.290 |
0.292 |
0.286 |
0.291 |
| 308.15 |
0.243 |
0.242 |
0.242 |
0.241 |
0.331 |
0.330 |
0.327 |
0.330 |
| 313.15 |
0.274 |
0.274 |
0.273 |
0.273 |
0.377 |
0.375 |
0.373 |
0.377 |
| 318.15 |
0.308 |
0.310 |
0.307 |
0.309 |
0.428 |
0.429 |
0.423 |
0.429 |
| 323.15 |
0.344 |
0.344 |
0.343 |
0.344 |
0.483 |
0.484 |
0.478 |
0.483 |
| 328.15 |
0.383 |
0.382 |
0.383 |
0.382 |
0.543 |
0.543 |
0.538 |
0.543 |
| 333.15 |
0.426 |
0.426 |
0.426 |
0.427 |
0.609 |
0.608 |
0.605 |
0.609 |
| T (K) |
ω = 0.6 |
ω = 0.8 |
| 103xexp |
103xcal |
103xexp |
103xcal |
| Apelblat |
λh |
J–A–V |
Apelblat |
λh |
J–A–V |
| 273.15 |
0.146 |
0.145 |
0.148 |
0.145 |
0.182 |
0.183 |
0.184 |
0.181 |
| 278.15 |
0.175 |
0.177 |
0.176 |
0.177 |
0.221 |
0.223 |
0.222 |
0.223 |
| 283.15 |
0.207 |
0.205 |
0.209 |
0.205 |
0.266 |
0.264 |
0.267 |
0.268 |
| 288.15 |
0.245 |
0.246 |
0.246 |
0.246 |
0.317 |
0.317 |
0.319 |
0.316 |
| 293.15 |
0.287 |
0.286 |
0.288 |
0.286 |
0.377 |
0.379 |
0.378 |
0.377 |
| 298.15 |
0.335 |
0.334 |
0.336 |
0.334 |
0.445 |
0.449 |
0.446 |
0.444 |
| 303.15 |
0.390 |
0.391 |
0.390 |
0.391 |
0.523 |
0.522 |
0.524 |
0.523 |
| 308.15 |
0.450 |
0.449 |
0.451 |
0.449 |
0.612 |
0.613 |
0.612 |
0.613 |
| 313.15 |
0.518 |
0.518 |
0.519 |
0.518 |
0.711 |
0.712 |
0.712 |
0.711 |
| 318.15 |
0.594 |
0.595 |
0.594 |
0.595 |
0.823 |
0.827 |
0.824 |
0.823 |
| 323.15 |
0.677 |
0.678 |
0.678 |
0.677 |
0.948 |
0.951 |
0.950 |
0.948 |
| 328.15 |
0.769 |
0.770 |
0.771 |
0.769 |
1.088 |
1.083 |
1.090 |
1.088 |
| 333.15 |
0.871 |
0.873 |
0.874 |
0.872 |
1.243 |
1.246 |
1.247 |
1.243 |
 |
| | Fig. 6 The experimental (points) and correlated (lines) solubility of form A in pure solvents (a) and propanol + heptane (b), respectively. | |
In mono-solvents, the solubility of resmetirom (form A) in alcohols is greater than in alkanes, meanwhile, the solubility of form A in the binary mixed solvent decreases with increasing proportion of heptane, indicating that heptane can be used as an anti-solvent for resmetirom crystal preparation. Obviously, the solubility of form A in various solvents is ranked as: propanol > isopropyl alcohol > butanol > isobutyl alcohol > 3-methyl-1-butanol > heptane.
Model evaluation
Solid–liquid equilibrium is the foundation of separation processes and a prerequisite for chemical production. Drawing from thermodynamic theory, solid–liquid models are established by correlating model parameters with temperature based on experimental solubility. The modified Apelblat equation, λh equation and Wilson model are employed to analyze the correlation of the solubility data of resmetirom (form A) in pure solvents, and J–A–V model are utilized for correlation of solubility data of form A in propanol + heptane, as outlined in Table 4.33,34 The applicability as well as the accuracy of these four equations for determining the solubility of resmetirom are assessed using the average relative deviation (ARD%) as shown below:35| |
 | (5) |
where, xiexp and xical refer to the experimental and calculated solubility data of resmetirom (form A), respectively. N is the number of data.
Table 4 Details of the thermodynamic equations used in this work
| Model |
Equations |
Parameters |
| Modified Apelblat equation |
 |
A, B, C |
| λh equation |
 |
λ, h |
 |
| Wilson model |
 |
δ12, δ21 |
 |
 |
| J–A–V model |
 |
A1, A2, B1, B2, Ji |
The results indicate that the predicted solubility from the equations were in excellent conformity with the measured data. Based on the values of ARD% in Tables S1–S4,† the modified Apelblat equation in mono-solvent exhibits the optimal regression performance for the chosen system, while the Wilson model shows large deviations which is not suitable for this system. For the mixed solvent, the J–A–V model exhibits the optimal regression performance for the chosen system. This means that the modified Apelblat equation can predict the solubility of the selected mono-solvent at higher or lower temperatures, and the J–A–V model can predict the solubility of the selected mixed solvent.36
Thermodynamic analysis of dissolution processes
Utilizing experimental solubility data of the solubility of solids in solvents, the apparent thermodynamic properties of form A in mono-solvents and mixed solvents are calculated in terms of energy. This includes the standard dissolution enthalpy change (ΔdisH°), standard entropy change (ΔdisS°) and Gibbs free energy change (ΔdisG°) of resmetirom (form A) dissolved in different solvents, as illustrated in the following eqn (6)–(9):37| |
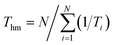 | (6) |
| | |
ΔdisS° = (ΔdisH° − ΔdisG°)/Thm
| (9) |
here, Thm denote average harmonic temperature, which is calculated as 301.99 K. k and d are the slope and intercept of the lnx versus 1/Ti − 1/Thm curve, respectively.
As shown in Tables 5 and 6, within the range of experimental temperatures, ΔdisH° values are positive for both mono-solvent and binary solvent mixtures, indicating that it is an endothermic process. The heat absorption effect during the dissolution process may be due to the fact that the interaction of solute–solvent molecule is weaker than the solvent–solvent. Furthermore, in mono-solvent systems, the ΔdisS° values that describe the randomness of the dissolution behavior are positive in propanol, isopropyl alcohol, and butanol. In the mixed solvent of propanol and heptane, the ΔdisS° values are also positive at ω = 0.2, 0.4 and 0.6, suggesting that the dissolution process is entropy-favored. When the dissolution process occurred of resmetirom molecules, the arrangement of solvent molecules is thrown into disarray, resulting the entropy of system is increasing.38 At the same time, ΔdisG° values for the dissolution behavior of form A are also positive in all chosen solvent systems. The ΔdisG° during dissolution determines the ease of dissolution, the smaller the ΔdisG° values, the lower the energy barrier to be overcome in the dissolution behavior and the higher the solubility of the solute.39
Table 5 Thermodynamic parameters of resmetirom (form A) in mono-solvents
| Solvent |
ΔdisH° (kJ mol−1) |
ΔdisG° (kJ mol−1) |
ΔdisS° (J mol−1 K−1) |
| Propanol |
25.931 |
17.932 |
26.490 |
| Isopropyl alcohol |
23.421 |
18.209 |
17.257 |
| Butanol |
21.317 |
18.942 |
7.865 |
| Isobutyl alcohol |
18.648 |
19.537 |
−2.944 |
| 3-Methyl-1-butanol |
17.867 |
20.110 |
−7.428 |
| Heptane |
17.368 |
21.540 |
−13.815 |
Table 6 Thermodynamic parameters of resmetirom (form A) in mixed solvents
| ω |
ΔdisH° (kJ mol−1) |
ΔdisG° (kJ mol−1) |
ΔdisS° (J mol−1 K−1) |
| 0.2 |
24.177 |
18.644 |
18.322 |
| 0.4 |
22.531 |
19.369 |
10.471 |
| 0.6 |
20.818 |
20.086 |
2.425 |
| 0.8 |
18.956 |
20.813 |
−6.150 |
Binding energy analysis based on DFT
According to Hossain, the dissolution of solutes in certain solvents is essentially governed by a two-stage process.40 Firstly, a relatively large pore or cavity is generated in the solvent phase to accommodate the solute molecule, and then the solute molecule enters the pore and interacts with the surrounding solvent molecules. Based on above, the solubility of a solute is closely related not only to the properties of the chosen solvent but also to the solute–solvent interactions. Therefore, DFT simulations were used to calculate the interaction forces and ΔE between form A and the selected solvents.
MEPs are based on the attraction between positive and negative potentials, which reveals the possible binding sites between solute and solvent molecules and facilitates the generation of initial configuration. The MEPs of resmetirom and various solvent molecules are depicted in Fig. 7. Blue zone is positive potential and red zone is negative potential. Furthermore, the yellow and cyan balls correspond to the points of maximum and minimum electrostatic potentials, respectively. The magnitude of the electrostatic potential values reveals that the H atom in the R3 ring of the resmetirom molecule corresponds to the extreme value of the surface electrostatic potential (61.07 kcal mol−1), while in the solvent molecules of alcohols, the O atom on the hydroxyl group corresponds to the extreme value of the surface electrostatic potential. Above sites are predicted as bonding sites between resmetirom and solvent molecules for subsequent configuration.
 |
| | Fig. 7 MEPs of resmetirom (a), methanol (b), ethanol (c), propanol (d), isopropyl alcohol (e), butanol (f), isobutyl alcohol (g), 3-methyl-1-butanol (h), heptane (i). | |
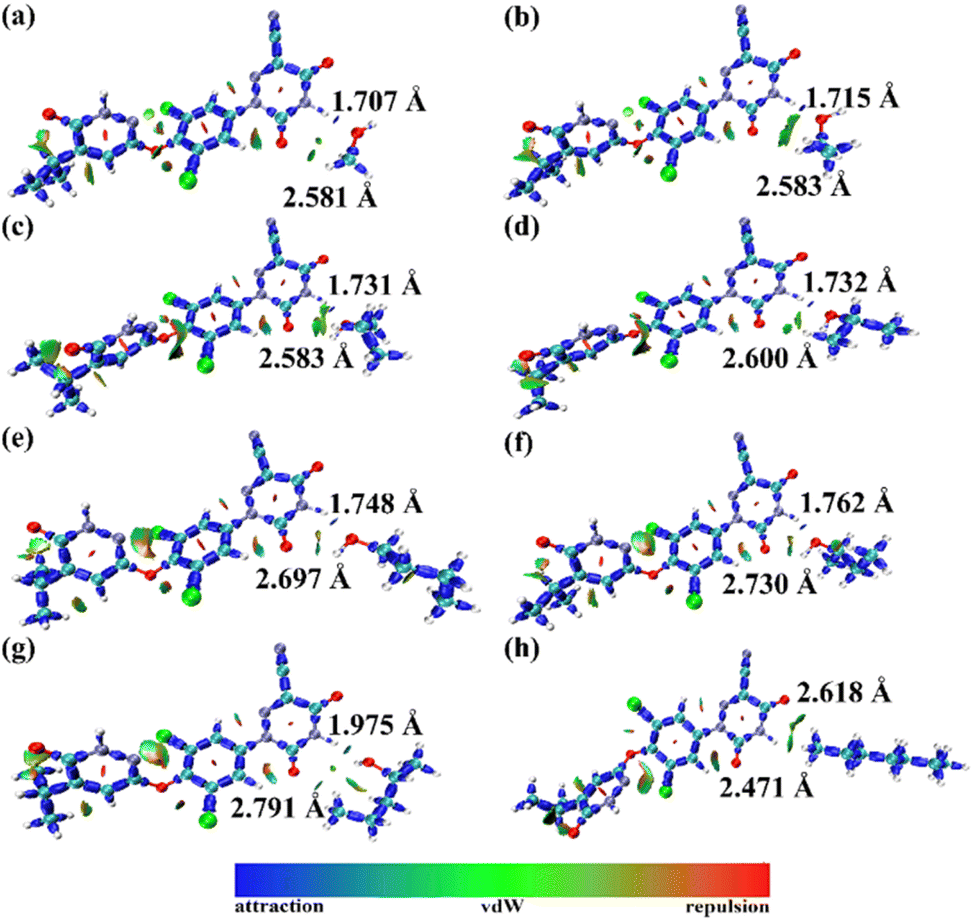
The optimized schematic of IRI resmetirom–solvent unit is shown in Fig. 8. The isosurface diagrams exhibit two isosurface domains of hydrogen bonds (blue) as well as van der Waals force (green), respectively. The main physicochemical properties and related calculated energy of the selected solvents are shown in Table 7, indicate that the solubility decreases with decreasing solvent polarity. Hydrogen bond acceptor and donor capabilities are both presented in alcohol solvents, while the R3 ring of resmetirom also contains an O atom and an H atom, so two hydrogen bonds will appear in unit, represented by the blue isosurface area. However, due to the difference in solvent polarity, the interaction force is varied, and the magnitude of the energy can be judged from the marked-out distance and the color of the isosurface. For example, the distances between the hydrogen bonds are the shortest in the unit of resmetirom + methanol, at 1.707 Å and 2.581 Å, respectively. While hydrogen bond acceptor and donor are lacked in the molecular structure of heptane, a large number of green isosurface area is performed, indicating there is only dispersion force rather than hydrogen-bond interaction. Therefore, it can be concluded that hydrogen bonds play primary roles in the dissolution of resmetirom in the studied solvents.
 |
| | Fig. 8 Isosurface diagrams of IRI analysis of solute with methanol (a), ethanol (b), propanol (c), isopropyl alcohol (d), butanol (e), isobutyl alcohol (f), 3-methyl-1-butanol (g) and heptane (h). | |
Table 7 Main physicochemical properties of the solvents and ΔE between the corresponding units
| Solvent |
πa |
∑αb |
∑βc |
EA + EBd |
Eunite |
ΔEf (kcal mol−1) |
| The polarity and dipole moment of solvent.41 Hydrogen bond donor ability.42 Hydrogen bond acceptor ability.42 Energy of two monomers in their respective basis sets. Total energy after correction. Binding energy between two molecules. |
| Methanol |
0.60 |
0.43 |
0.47 |
−2436.351 |
−2436.378 |
−17.25 |
| Ethanol |
0.54 |
0.37 |
0.48 |
−2359.950 |
−2359.966 |
−9.75 |
| Propanol |
0.48 |
0.37 |
0.48 |
−2399.271 |
−2399.286 |
−9.02 |
| Isopropyl alcohol |
0.48 |
0.33 |
0.56 |
−2399.279 |
−2399.293 |
−8.92 |
| Butanol |
0.47 |
0.37 |
0.48 |
−2438.600 |
−2438.614 |
−8.83 |
| Isobutyl alcohol |
0.40 |
0.37 |
0.48 |
−2438.594 |
−2438.606 |
−7.87 |
| 3-Methyl-1-butanol |
0.40 |
0.37 |
0.48 |
−2477.915 |
−2477.927 |
−7.48 |
| Heptane |
−0.08 |
0.00 |
0.00 |
−2481.346 |
−2481.353 |
−4.07 |
The ΔE between resmetirom (form A) and solvents are collected in Table 7. Specifically, the ΔE of these solvent–solute is as follows: methanol (−17.25) > ethanol (−9.75) > propanol (−9.02) > isopropyl alcohol (−8.92) > butanol (−8.83) > isoamyl alcohol (−7.87) > 3-methyl-1-butanol (−7.48) > heptane (−4.07). The ΔE between solute and solvent is always decreased accompanied by an increase in c-chain length, and the lower value of ΔE, the higher the solubility, which is consistent with the experimental results.
Validation of quantitative analysis methods
The standard curve is established by FTIR to quantitatively analyze the polymorphic composition of resmetirom during the transformation from form A to solvates, and the kinetics is also studied. Since the dynamic view of the calculated vibration frequencies are allowed to be viewed through the Gaussian View 6.0 software, and the peaks that reflected the interaction force between resmetirom and solvents are chosen and used in a quantitative model. The infrared spectra of R-ET and R-MT are very similar and therefore the same bands can be selected for analysis. In Fig. 9, the comparison of theoretical spectra of three solids are compared. In addition, the selected ranges and relevant vibrations are shown in Table 8.
 |
| | Fig. 9 The theoretical spectra of form A, R-MT and R-ET. | |
Table 8 Variable selection range and corresponding relative vibration mode
| Spectral range (cm−1) |
Related vibrations |
| 3720–3180 |
The stretching vibration of N–H in amide and the O–H stretching vibrations in hydroxyl of intermolecular hydrogen bonding |
| 1800–1600 |
The N–H in-plane bending vibration in R1 ring; the stretching vibration of carbonyl group in amide |
| 1275–1150 |
The N–H in-plane bending vibration caused by the C–N and N–N stretching vibration of R1 ring |
| 980–850 |
The in-plane bending vibration of N–H of R1 ring; the out-of-plane rocking vibration of N–H of R3 ring |
| 645–600 |
The out-of-plane rocking vibration of N–H of R1 ring |
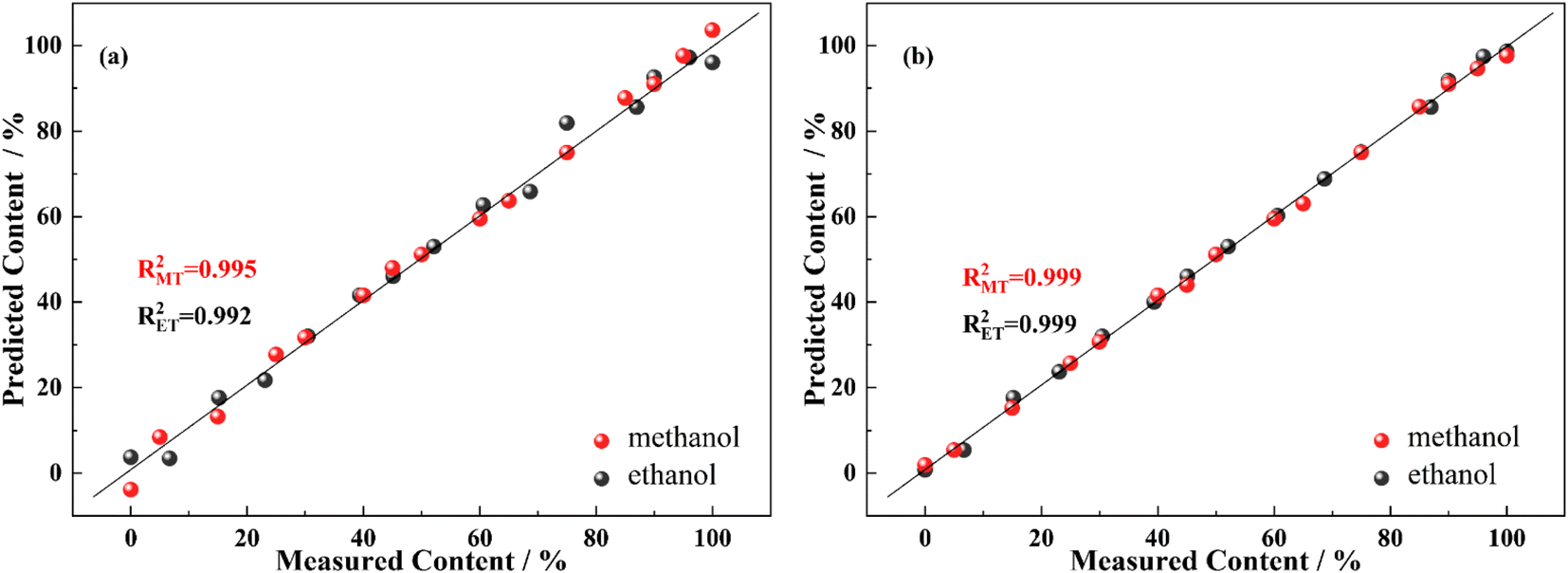
In Fig. 10, the actual and predicted concentration of solvents in the binary mixture based on PLSR are compared. The model correlation coefficients are obtained from the quantitative analysis using the full spectral bands when the solvent compound is in the 0–100% concentration range depicted in Fig. 10(a). In addition, the accuracy of the model is improved significantly with the model correlation coefficient rising to 0.999 by combining with the specific bands selected by DFT and PLSR, as shown in Fig. 10(b). The results suggest that the combination of variable selection and DFT calculations is feasible for the analysis of solvent compound concentration in the complex range.
 |
| | Fig. 10 The standard curves for quantitative analysis with a full spectrum (a) and with selected specific bands (b), respectively. | |
Process of solid transformation
Based on the high-precision quantitative analysis method, the solid transformation of form A in methanol and ethanol has been studied from 303.15 to 323.15 K with the same suspension density as well as stirring. The solid transformation of form A to its solvates is graphically shown in Fig. 11. Generally, the transformation rate from the form A to the solvates decreases with increasing temperature, which is probably because the supersaturation of solvates (i.e. the difference between the concentration of solvates and their solubility) at lower temperature is larger than that at higher temperature, and the larger supersaturation of solvates is benefit for the formation of solvate nuclei.43 Based on the rate of transition in Fig. 11, it can be seen that three stages of solid transformation. Initially, the number of nuclei formed by crystallization is small and therefore the crystallization rate is relatively slow. As the number of nuclei in the solution increases, the crystallization rate gradually increases, driven by the degree of supersaturation. Finally, as the crystals precipitate, resulting in a decrease in the crystallization rate after reaching its maximum value. That is, the transformation process is driven by the solubility difference between form A and solvates.44 Besides, it is obvious that the transformation time of form A to R-MT is faster than that of form A to R-ET, which is probably because the binding capacity between resmetirom and methanol is stronger than that between resmetirom and ethanol.
 |
| | Fig. 11 Transformation profiles at different temperatures in methanol (a) and ethanol (b). | |
Solid–solid transformation models are theoretical and mathematical description of experiment results, which are usually based on mechanical assumptions or experience. The goal of kinetic research is always to acquire rate constants k, which can be utilized for characterizing the reaction mechanism.45 Similarly, the study of transformation processes involves the same objective. Various solid–solid kinetics models are employed to evaluate the time-dependent functions of the solid transformation rate at different temperatures. And α represents the content of the predicted solid transformation with FTIR spectra, k denotes the reaction rate constant, t stands for the solid transformation time. Nine classic solid–solid kinetics models are used to verify the process from form A to solvates, and the kinetic equation and relevant coefficients are shown in Table 9.46–48
Table 9 Details of the kinetic equations and the correlation coefficient of the models for resmetirom (form A) to solvate
| Model name |
Equation |
Parameters |
| M1: Prout–Tompkins |
 |
k, c |
| M2: Avrami–Erofeev (n = 3) |
 |
| M3: first order |
−ln(1 − α) = kt |
| M4: two dimensional phase boundary |
 |
| M5: three dimensional phase boundary |
 |
| M6: 1D diffusion |
α2 = kt |
| M7: 2D diffusion |
(1 − α)ln(1 − α) + α = kt |
| M8: 3D diffusion-Jander equation |
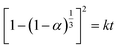 |
| M9: 3D diffusion-Ginstling–Brounshtein equation |
 |
Based on the equations and correlation coefficients in Table 10, optimal results for the transformation of form A to R-MT and R-ET at different temperatures are obtained from M1. M1 is a self-catalytic model in which the solvate plays a catalytic role along with production. To determine the activation energy, k is always assumed follows the Arrhenius equation:49
| |
 | (10) |
where
Ea and A represent the activation energy and the pre-exponential factor of the process, respectively. Regression analysis is based on the
eqn (10) to calculate the activation energy of the solid transformation of form A in methanol (
Ea = −17.79 kJ mol
−1) and ethanol (
Ea = −15.24 kJ mol
−1), respectively.
Table 10 Correlation coefficient (R2) of the model of resmetirom (form A) to solvates
| |
Methanol |
Ethanol |
| 308.15 K |
318.15 K |
328.15 K |
308.15 K |
318.15 K |
328.15 K |
| M1 |
0.997 |
0.999 |
0.998 |
0.996 |
0.998 |
0.998 |
| M2 |
0.912 |
0.982 |
0.907 |
0.984 |
0.998 |
0.991 |
| M3 |
0.995 |
0.995 |
0.992 |
0.989 |
0.997 |
0.997 |
| M4 |
0.922 |
0.936 |
0.999 |
0.987 |
0.938 |
0.984 |
| M5 |
0.925 |
0.993 |
0.995 |
0.990 |
0.962 |
0.978 |
| M5 |
0.913 |
0.917 |
0.984 |
0.972 |
0.982 |
0.982 |
| M7 |
0.980 |
0.982 |
0.977 |
0.978 |
0.960 |
0.968 |
| M8 |
0.907 |
0.960 |
0.957 |
0.873 |
0.961 |
0.950 |
| M9 |
0.956 |
0.982 |
0.988 |
0.967 |
0.989 |
0.989 |
Conclusions
The solubility of resmetirom (form A) in various solvents is ranked as: propanol > isopropanol > butanol > isobutyl alcohol > 3-methyl-1-butanol > heptane, in which propanol and heptane can be chosen as the good and the poor solvent for its anti-solvent crystallization, respectively. The modified Apelblat equation and the J–A–V model achieve the highest correlation accuracy for the experimental solubility data in mono-solvents and heptane + propanol, respectively. DFT calculation and solvent parameters analyses indicate that hydrogen-bonding plays primary roles in the dissolution process of resmetirom (form A) in the studied solvents. Furthermore, a high-precision quantitative method for probing transformation process is firstly and successfully established. Using this efficient and convenient approach, it is found that the rate of transformation from form A to methanol solvate or ethanol solvate is decreased with increasing temperature from 303.15 to 323.15 K, with activation energies of −17.79 and −15.24 kJ mol−1, respectively. In sum, this work shall definitely afford necessary solubility data and solvent selection for the design of the crystallization process of resmetirom (form A) in industry.
Author contributions
Chang Liu: data collection, writing original draft. Yue Xu: data analysis. Haikuan Yuan: supervision, validation. Guangxin Tian: methodology. Xiaolan Qin: editing. Boxuan Lou: validation. Xijian Liu: reviewing. Lijuan Zhang: conceptualization. Jie Lu: supervision, resources.
Conflicts of interest
The authors declared that they have no conflicts of interest to this work.
Acknowledgements
We appreciated the National Natural Science Foundation of China (No. 21978165, 92156020, 22081340412 and 22078191) to support this work.
Notes and references
- D. Singhal and W. Curatolo, Adv. Drug Delivery Rev., 2004, 56, 335–347 CrossRef CAS PubMed.
- K. M. Lutker, R. Quiñones, J. Xu, A. Ramamoorthy and A. J. Matzger, J. Pharm. Sci., 2011, 100, 949–963 CrossRef CAS PubMed.
- P. Klitou, C. M. Pask, L. Onoufriadi, I. Rosbottom and E. Simone, Cryst. Growth Des., 2020, 20, 6573–6584 CrossRef CAS.
- D. Braun and U. Griesser, CrystEngComm, 2017, 19, 3566–3572 RSC.
- R. Chang, Q. Fu, P. Yu, L. Wang, Y. Li, W. Du, C. Chang and A. Zeng, RSC Adv., 2016, 6, 85063–85073 RSC.
- X. Xiong, Q. Du, X. Zeng, J. He, H. Yang and H. Li, RSC Adv., 2017, 7, 23279–23286 RSC.
- G. Wang, Y. Ma, Y. Wang, H. Hao and Y. Jiang, Org. Process Res. Dev., 2015, 19, 1820–1825 CrossRef CAS.
- V. Suitchmezian, I. Jeß and C. Näther, J. Pharm. Sci., 2008, 97, 4516–4527 CrossRef CAS PubMed.
- Y.-H. Yun, Y.-Z. Liang, G.-X. Xie, H.-D. Li, D.-S. Cao and Q.-S. Xu, Analyst, 2013, 138, 6412–6421 RSC.
- J. Zhang, H. Yan, Y. Xiong, Q. Li and S. Min, RSC Adv., 2019, 9, 6708–6716 RSC.
- J. Ludwig, Mayo Clin. Proc., 1980, 55, 434–438 CAS.
- F. Schaffner and H. Thaler, Prog. Liver Dis., 1986, 8, 283 CAS.
- Z. M. Younossi, A. B. Koenig, D. Abdelatif, Y. Fazel, L. Henry and M. Wymer, Hepatology, 2016, 64, 73–84 CrossRef PubMed.
- A. Kannt, P. Wohlfart, A. N. Madsen, S. S. Veidal, M. Feigh and D. Schmoll, Br. J. Pharmacol., 2021, 178, 2412–2423 CrossRef CAS PubMed.
- S. Harrison, A. Mangia, P. Lasierra-Resa, M. S. Cubberley, R. Taub, G. Neff, N. Alkhouri and M. Bashir, Dig. Liver Dis., 2022, 54, S48 CrossRef.
- G. X. Tian, Y. Luo, B. X. Lou, J. Sui, X. Qin, Y. Shen, X. Zhu and J. Lu, J. Mol. Liq., 2023, 369, 120857 CrossRef CAS.
- P. Manjusha, S. Muthu and B. Raajaraman, J. Mol. Struct., 2020, 1203, 127394 CrossRef CAS.
- F. Jacinto, L. Siqueira and W. A. Alves, J. Raman Spectrosc., 2010, 40, 1585–1590 CrossRef.
- R. Xu, B. Hou, N. Wang, Y. Lou, Y. Li, J. Huang and H. Hao, J. Mol. Liq., 2018, 252, 194–202 CrossRef CAS.
- S. Liu, E. G. Macaringue, X. Li, L. Jia, Y. Liu and J. Gong, J. Mol. Liq., 2017, 238, 411–422 CrossRef CAS.
- Y. Cheng, D. Wang, Z. Zhang and Z. Wang, RSC Adv., 2015, 5, 80548–80552 RSC.
- V. Consonni, G. Baccolo, F. Gosetti, R. Todeschini and D. Ballabio, Chemom. Intell. Lab. Syst., 2021, 213, 104313 CrossRef CAS.
- S. Karimi, J. Feizy, F. Mehrjo and M. Farrokhnia, RSC Adv., 2016, 6, 23085–23093 RSC.
- T. Topal, Y. Zorlu and N. Karapınar, J. Mol. Struct., 2021, 1239, 130514 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and Q. Chen, Chem.: Methods, 2021, 1, 231–239 CAS.
- L. Bondesson, E. Rudberg, Y. Luo and P. Sałek, J. Comput. Chem., 2008, 29, 1725–1732 CrossRef CAS PubMed.
- Z. Ren, Y. Zeng, Y. Hua, Y. Cheng and Z. Guo, J. Chem. Eng. Data, 2014, 59, 2517–2522 CrossRef CAS.
- M. Singh, J. Cryst. Growth, 2014, 396, 14–23 CrossRef CAS.
- R. Van Santen, J. Phys. Chem., 1984, 88, 5768–5769 CrossRef CAS.
- Z. Fang, L. Zhang, S. Mao, S. Rohani, J. Ulrich and J. Lu, J. Chem. Thermodyn., 2015, 90, 71–78 CrossRef CAS.
- M. Mirmehrabi, S. Rohani, K. Murthy and B. Radatus, Int. J. Pharm., 2004, 282, 73–85 CrossRef CAS PubMed.
- L. Huang, X. Li, R. Han, Y. Li, L. Xu, Z. Zeng and K. Wu, J. Mol. Liq., 2022, 363, 119847 CrossRef CAS.
- M. Xue, D.-z. Huang, K.-x. Yang, L.-z. Chen, Z.-h. Zheng, Y. Xiang, Q.-w. Huang and J.-l. Wang, J. Mol. Liq., 2021, 330, 115639 CrossRef CAS.
- S. N. Mirheydari, M. Barzegar-Jalali, F. Martinez and A. Jouyban, Phys. Chem. Liq., 2020, 58, 769–781 CrossRef CAS.
- A. Jouyban, Pharmazie, 2007, 62, 365–367 CAS.
- C. Zhang, A. Jouyban, H. Zhao, A. Farajtabar and W. E. Acree Jr, J. Mol. Liq., 2021, 326, 115219 CrossRef CAS.
- H. Zhang, Y. Yang, M. Zhu, S. Jiang, S. Xu, S. Du, B. Yu and J. Gong, J. Mol. Liq., 2017, 240, 162–171 CrossRef CAS.
- C. Li, H. Wang, W. Huang, T. Wen, J. Xu, J. Ouyang and C. Zhang, J. Mol. Liq., 2021, 339, 116733 CrossRef CAS.
- S. Hossain, A. Kabedev, A. Parrow, C. A. Bergström and P. Larsson, Eur. J. Pharm. Biopharm., 2019, 137, 46–55 CrossRef CAS PubMed.
- C.-H. Gu, H. Li, R. B. Gandhi and K. Raghavan, Int. J. Pharm., 2004, 283, 117–125 CrossRef CAS PubMed.
- M. H. Abraham, Chem. Soc. Rev., 1993, 22, 73–83 RSC.
- L. Jia, Q. Yin, L. Zhou, X. Zhang, C. Wang, W. Du and L. Zhou, RSC Adv., 2018, 8, 9697–9706 RSC.
- F. Tian, H. Qu, A. Zimmermann, T. Munk, A. C. Jørgensen and J. Rantanen, J. Pharm. Pharmacol., 2010, 62, 1534–1546 CrossRef CAS PubMed.
- A. Khawam and D. R. Flanagan, J. Phys. Chem. B, 2006, 110, 17315–17328 CrossRef CAS PubMed.
- L. Zhou, Q. Yin, S. Du, H. Hao, Y. Li, M. Liu and B. Hou, RSC Adv., 2016, 6, 51037–51045 RSC.
- T. Liu, Y. Ran, B. Wang, W. Dong, W. U. Songgu and J. Gong, Front. Chem. Sci. Eng., 2014, 8, 55–63 CrossRef CAS.
- K. A. Alkhamis, M. S. Salem and R. M. Obaidat, J. Pharm. Sci., 2006, 95, 859–870 CrossRef CAS.
- L. E. O'Brien, P. Timmins, A. C. Williams and P. York, J. Pharm. Biomed. Anal., 2004, 36, 335–340 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Parameters of models. See DOI: https://doi.org/10.1039/d3ra02521g |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Guangxin Tian,
Xiaolan Qin,
Boxuan Lou,
Xijian Liu
*,
Guangxin Tian,
Xiaolan Qin,
Boxuan Lou,
Xijian Liu