
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gauging the importance of structural parameters for hyperfine coupling constants in organic radicals†
Conrad Szczuka *a,
Rüdiger-A. Eichelab and
Josef Granwehrac
*a,
Rüdiger-A. Eichelab and
Josef Granwehrac
aInstitute of Energy and Climate Research (IEK-9), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: co.szczuka@fz-juelich.de
bInstitute of Physical Chemistry, RWTH Aachen University, 52056 Aachen, Germany
cInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, 52056 Aachen, Germany
First published on 12th May 2023
Abstract
The identification of fundamental relationships between atomic configuration and electronic structure typically requires experimental empiricism or systematic theoretical studies. Here, we provide an alternative statistical approach to gauge the importance of structure parameters, i.e., bond lengths, bond angles, and dihedral angles, for hyperfine coupling constants in organic radicals. Hyperfine coupling constants describe electron–nuclear interactions defined by the electronic structure and are experimentally measurable, for example, by electron paramagnetic resonance spectroscopy. Importance quantifiers are computed with the machine learning algorithm neighborhood components analysis using molecular dynamics trajectory snapshots. Atomic–electronic structure relationships are visualized in matrices correlating structure parameters with coupling constants of all magnetic nuclei. Qualitatively, the results reproduce common hyperfine coupling models. Tools to use the presented procedure for other radicals/paramagnetic species or other atomic structure-dependent parameters are provided.
1 Introduction
Machine learning techniques are increasingly applied in chemical sciences.1 Applications can be roughly classified into predictions of properties based on atomic/electronic structure2 and (statistical) evaluation of experimental results.1 In the subfield of electron magnetic resonance, applications focus on the interpretation of acquired spectra, e.g., from continuous-wave electron paramagnetic resonance (EPR),3 double electron–electron resonance (DEER),4 electron–nuclear double resonance (ENDOR),5 or hyperfine sublevel correlation spectroscopy (HYSCORE).6Most machine learning techniques are prediction models that can predict a numerical output value by regression or classification based on input values (features). To improve model stability and accuracy, the dimensionality of feature sets can be reduced through preprocessing, e.g., by transformation into a new basis or selecting features through ranking their importance in determining the output. Neighborhood component analysis (NCA) is one example for a non-paramagnetic feature selection algorithm introduced by Goldberger et al.7 It was improved through introduction of a regularization term by Yang et al.,8 and validated in real-world applications.9,10 This algorithm can be used for feature selection but also simply to provide a gauge for importance of a feature for a particular response, as will be exploited herein.
Frequently targeted spectroscopic parameters of radicals and paramagnetic centers are hyperfine coupling constants, resulting from electron–nuclear interactions, that can be used to evaluate the underlying atomic and electronic structure. Hyperfine couplings arise from a non-zero spin density distribution evoked by spin delocalization and polarization. In small systems, some origins of hyperfine interactions based on, e.g., exchange interactions or hyperconjugation, were identified.11–14 These mechanisms are often described locally for simplicity; however, the entirety of the electronic structure dictated by the type and configuration of all atoms defines the spin density distribution and, thereby, hyperfine coupling magnitudes.
Hyperfine couplings are described by an anisotropic rank 2 tensor A with three principal components Ax,y,z and tr(A) = Aiso.15 Since interpretation of experimental Ax,y,z values is non-trivial, a comparison with calculated values from ab initio models is usually performed.16–19 Sufficiently accurate calculations are often possible, but require incorporation of electron correlation defined by the used electronic theory, solvation effects defined by the structural model, and dynamic contributions. Dynamics can be included by averaging computed hyperfine coupling constants from molecular dynamics snapshots,20,21 since molecular motion on timescales of fs is fast compared to the experimental time scale of ps to ns for EPR. Trajectory analyses generally show a non-Gaussian coupling constant distribution in histograms.21,22 In principle, these trajectories and the computed coupling constants contain statistical information on structure–hyperfine relations that are usually removed through the averaging, but will be exploited herein.
In this work, molecular dynamics trajectories are used to derive correlations between molecular structure and hyperfine coupling constants of common organic radicals using machine learning. Correlations are quantified by feature weights calculated using the NCA algorithm. Features involve all bonds, angles, and dihedrals extracted from the trajectory's coordinates, and targeted responses are Ax,y,z,iso. First, adequate simulation parameters are derived using the principle of magnetic equivalence. Secondly, example radicals are analyzed regarding structure–hyperfine relations extracted by the algorithm. The discussion is complemented by considering the structure, spin density distribution, hyperfine tensor orientations/magnitudes, and links to relations known in literature.
2 Methods
2.1 Density functional theory calculations
Density functional theory (DFT) calculations were conducted using ORCA 5.0.1.23 Organic radicals were geometry optimized using the hybrid functional B3LYP24,25 and def2-TZVP26 basis sets. XYZ files are accessible in the data repository found at https://doi.org/10.26165/JUELICH-DATA/UH0LM2. The ORCA-MD module27 was used to calculate ab initio molecular dynamics (MD) trajectories, with velocities initialized assuming a temperature of 900 K or, for temperature comparison, in a range of 150 to 2000 K. Initialized temperatures were maintained using a Nosé–Hoover chains28–30 (NHC) thermostat with a time constant of 20 fs. Time steps of 0.5 fs were applied to address high-frequency hydrogen motion. For MD snapshots in random steps of 0.5–40 fs along the trajectory, hyperfine coupling tensors for 1H, 13C, and 17O were calculated using B3LYP and EPR-III (ref. 31) basis sets, disregarding contributions from spin–orbit coupling. Hyperfine tensors are referenced to the g-tensor frame. Example ORCA input files are given in the ESI, Section F.†2.2 Data processing workflow
Further data processing and analysis was performed in MATLAB R2021b using the Statistics and Machine Learning Toolbox and Deep Learning Toolbox, involving (a) conversion of xyz coordinates into position-independent structure parameters comprising bonds, angles, and dihedrals, and (b) NCA to identify structure parameter importance for changes in hyperfine coupling constants. The MATLAB scripts are available online (https://doi.org/10.26165/JUELICH-DATA/UH0LM2).3 Results and discussion
3.1 Workflow
The workflow is schematized in Fig. 1. First, organic radicals (Table 1) are initialized as xyz structures, pre-optimized using force-fields, and ultimately optimized with density functional theory. These structures serve as input for ab initio molecular dynamics (MD) simulations using a simulation temperature of 900 K, chosen based on results presented in the section Parameter choice below. Up to 30 ps MD simulation trajectories were computed to provide sufficient statistics for the machine learning algorithm. To account for the non-linear increase of structure parameters with the number of atoms in a molecule, the MD step number lies between 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (≙10 ps) for the methyl and 60000 (≙30 ps) for the tryptophan-type radical, compromising between necessary statistics and computation resources. MD trajectories represent the computationally most demanding step, using more than 93% of the computational resources (Table S1†). Along MD trajectories, snapshots were chosen for hyperfine coupling tensor calculations. We deliberately chose not to calculate hyperfine tensors for every step to reduce structural similarity between single data points. Because structure parameters oscillate predominantly according to a superposition of regular sine waves,33 choosing snapshots in regular intervals will significantly decrease specific structure parameter variations if the interval corresponds to the frequency of a normal mode. Thus, we use an evenly distributed randomized step interval of 1–80 for hyperfine coupling calculations.
000 (≙10 ps) for the methyl and 60000 (≙30 ps) for the tryptophan-type radical, compromising between necessary statistics and computation resources. MD trajectories represent the computationally most demanding step, using more than 93% of the computational resources (Table S1†). Along MD trajectories, snapshots were chosen for hyperfine coupling tensor calculations. We deliberately chose not to calculate hyperfine tensors for every step to reduce structural similarity between single data points. Because structure parameters oscillate predominantly according to a superposition of regular sine waves,33 choosing snapshots in regular intervals will significantly decrease specific structure parameter variations if the interval corresponds to the frequency of a normal mode. Thus, we use an evenly distributed randomized step interval of 1–80 for hyperfine coupling calculations.
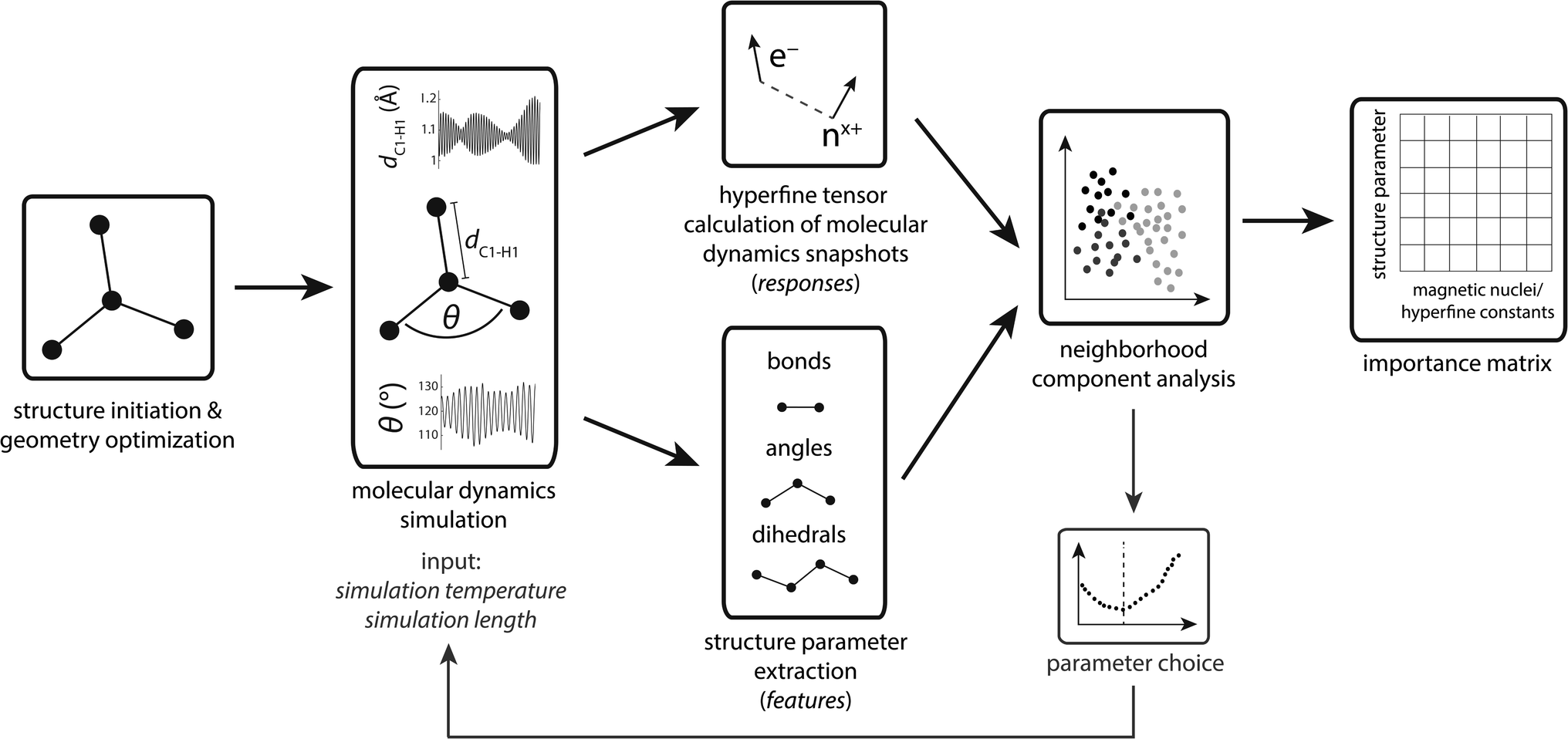 | ||
| Fig. 1 Workflow chart of the approach taken in this work. | ||
| Radical | # of MD steps | # of hyperfine calculations | # of bonds | # of angles | # of dihedrals |
|---|---|---|---|---|---|
| Methyl | 20000 |
505 | 3 | 3 | 0 |
| Ethyl | 50000 |
1250 | 6 | 9 | 1 |
| Methyl peroxy | 40000 |
969 | 5 | 7 | 1 |
| Semiquinone | 60000 |
1496 | 13 | 19 | 7 |
| Tyrosyl (see ESI) | 70000 |
1715 | 15 | 24 | 7 |
| Tryptophan-type (see ESI) | 60000 |
1466 | 19 | 31 | 11 |
3.2 Neighborhood components analysis
The hyperfine coupling constants can be described as output (response variable) given a defined set of intrinsic variables including distances (chemical bonds), angles, and dihedrals. These structure parameters can serve as features for the machine learning algorithm NCA.7,8 NCA mathematically maps the input features to the outputs by generating weights for each feature. The feature weights determine the numerical importance of each feature in this mapping. Intuitively, features with higher weights contribute more to the output than features with lower weights. For stochastically fluctuating features, regularization is a strategy to prevent the model from overaccurately tracing these fluctuations, which could lead to unphysical results that would be referred to as overfitting. Regularization penalizes feature weights of excessive value by adding a term to the error estimation between the real outputs and the model prediction. In NCA,8 the penalty term consists of the squared weights multiplied with a regularization parameter λ. Herein, λ = 0.05 is chosen based on results from the section Parameter choice below.To improve the accuracy of machine learning algorithms, choice and quality of features are crucial.34 Herein, features are selected as follows. In contrast to bond lengths being continuous variables, dihedrals are circular variables exhibiting a discontinuity, but can be converted to continuous sine and cosine values. Bond angles are not converted likewise since they are defined in the interval [0°, 180°] and thus, are pseudo-continuous. Another possibility to reduce the number of features is to consider overlapping dihedrals. Dihedrals are described by four chemically connected atomic positions. For a standard descriptive example like 1,2-dichloroethane, typically a single dihedral is used, assuming that the other atomic positions can be inferred by symmetry. However, for randomly moving atoms in molecular dynamics, all existing sets of four connected atoms need to be considered. Yet, we expect the additional dihedrals to be highly correlated. Hence, only one dihedral per two defining central atoms is kept as a feature. Before feeding the NCA algorithm, each feature is individually standardized to have zero mean and unit standard deviation, avoiding artefacts from different unit scales and variation magnitudes. This strategy for standardization is ideally suited for continuous and Gaussian-like distributed features, which are often observed in MD trajectories, particularly for bonds and angles.21,22,35,36 Other standardization strategies might be superior depending on the conformational energy landscape.
On the basis of the created feature set, NCA can be performed for an arbitrary response variable that depends on the features. Herein, we include hyperfine coupling constants Ax,y,z,iso for all magnetic nuclei in a given organic radical. A simple example is the methyl radical with atomic labels, spin density, and hyperfine tensor plot depicted in Fig. 2a–c, respectively. The importance matrix regarding the central 13C and all three 1H nuclei shows clear non-zero entries (Fig. 2d and e). Aiso correlations are also evident from visible inspection of the raw data plotted in Fig. 2f, confirming NCA-derived correlations. For the response Aiso(H1), positive correlation with the angle H2C1H3 opposite of H1 and negative correlations with the bond length H1C1 and the sum of all bond angles are inferred. For Aiso(C1), positive correlation with all bond lengths and negative correlation with the sum of all angles is inferred. Without discussing chemical and physical origins at this point, a set of symmetry arguments are evident for the matrix in Fig. 2d. Due to magnetic equivalence of H1, H2, and H3, the upper left 3 × 3 matrix characterizing bonds and the adjacent lower 3 × 3 matrix characterizing angles should ideally be centrosymmetric. Similarly, all hydrogen atoms should be equally important for Aiso(C1), so importance values for bonds and angles should be identical. Deviations from these symmetry considerations, like the non-centrosymmetric entries with importance values below one, are assumed to stem from insufficient statistics along the MD trajectory.
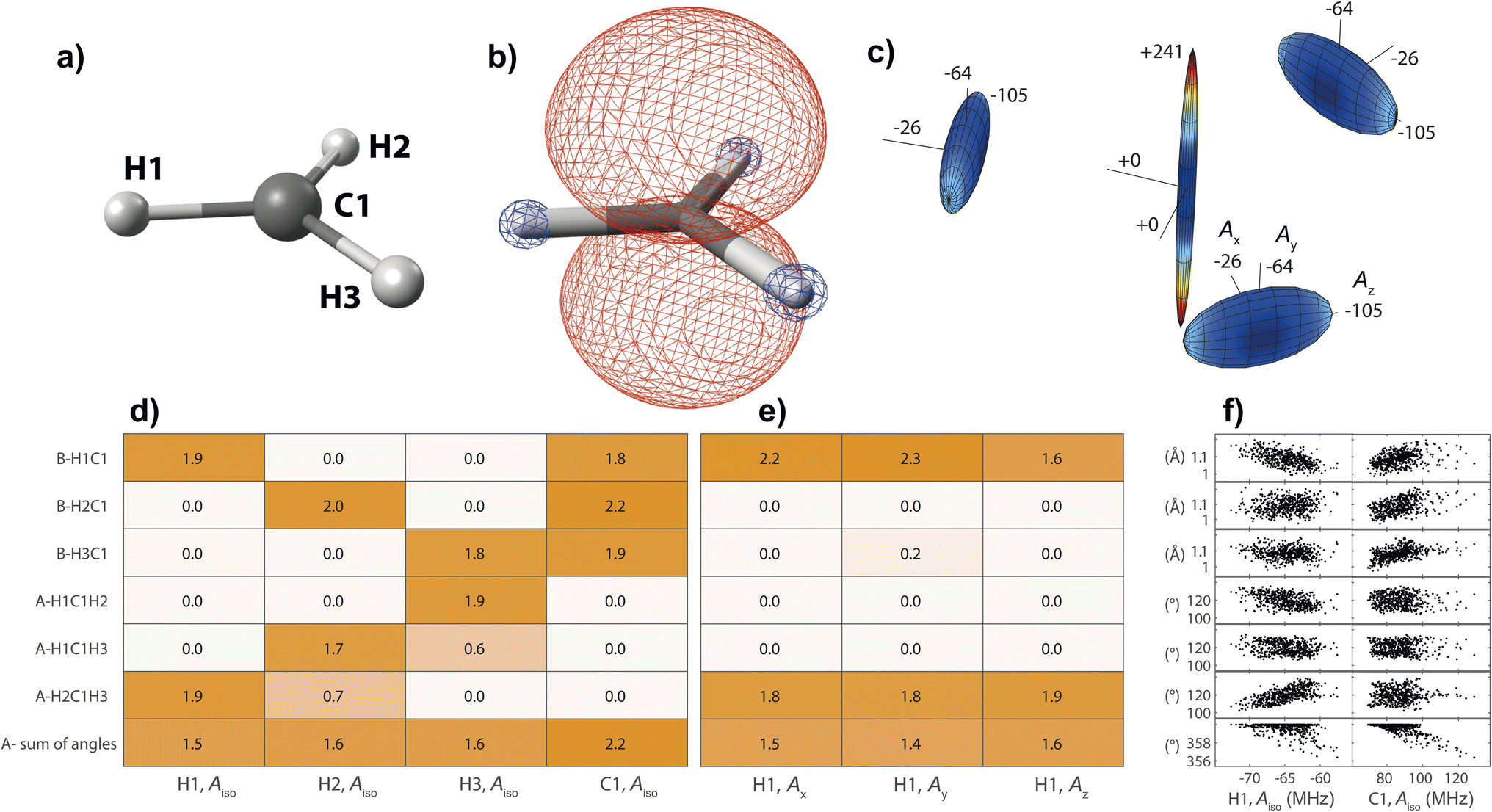 | ||
| Fig. 2 Structure–hyperfine relationship for the methyl radical with atomic labels given in (a). (b) For the geometry optimized structure, positive (red) and negative (blue) spin density is displayed at an iso-value of 0.007/a03, where a0 is the Bohr radius. (c) Hyperfine coupling tensors are visualized for each magnetic nucleus, normalized to the largest overall coupling constant. Importance matrices from neighborhood components analysis using λ = 0.05 are given for Aiso of all nuclei (d), and for all hyperfine tensor principal components of H1 (e). Structure parameters are classified as bond lengths (B) and angles (A). (f) Raw data plots including all 505 hyperfine calculations. | ||
3.3 Parameter choice
The chosen MD parameters have a large influence on the dataset quality for NCA. Since the importance matrix elements are unknown a priori, symmetry arguments as introduced in the last section are exploited to gauge parameter suitability. More specifically, for H atom symmetry, the first three matrix columns from Fig. 2d are permuted to meet the symmetry arguments, i.e., the first entry of nucleus HX, with X ∈ {1, 2, 3}, corresponds to the bond HXC1 and the fourth entry corresponds to the opposite angle not involving HX. Permutations are performed for Ax,y,z, and the cumulative mean square error comparing each pair of the {H1, H2, H3} group is calculated. For C atom symmetry, the standard deviation of the first three bond-associated entries and the following three angle-associated entries are calculated for Ax,y,z, and the overall mean standard deviation is computed.Using these symmetry-based descriptors, the MD temperature TMD is investigated (Fig. 3, central panel, see also Fig. S1†). For 0.03 ≤ λ ≤ 0.12, a window for minimum error is evident from roughly 700–1200 K, both for H and C symmetry gauges. By increasing TMD, the amplitude of structure parameter variations increases concomitantly. For example, the maximum amplitude of bond stretching/compression is 0.08 Å at 150 K and 0.26 Å at 2000 K. Maximum amplitude of angle stretching/compression is 7° at 150 K and 32° at 2000 K. Results suggest that in the low-to-intermediate TMD regime, an increase in TMD benefits the accuracy of the model, because the numerical spread of features/responses and sampling of the conformational space is enhanced. However, in the intermediate-to-high TMD regime, the trend is reversed. Extreme structure parameters might lead to unforeseen high-temperature effects on the electronic structure, e.g. relatively strong hydrogen 1s orbital overlap for angles <90° observed for TMD = 2000 K, resulting in alterations or discontinuities of the structure–hyperfine relationship. Deviations from realistic chemical structures using TMD = 1500 K were also reported37 and above 2000 K pyrolysis of hydrocarbons occurs.38 We note that importance values may be slightly dependent on TMD, which is however most likely of minor significance for the semi-quantitative findings presented herein (Fig. S1†).
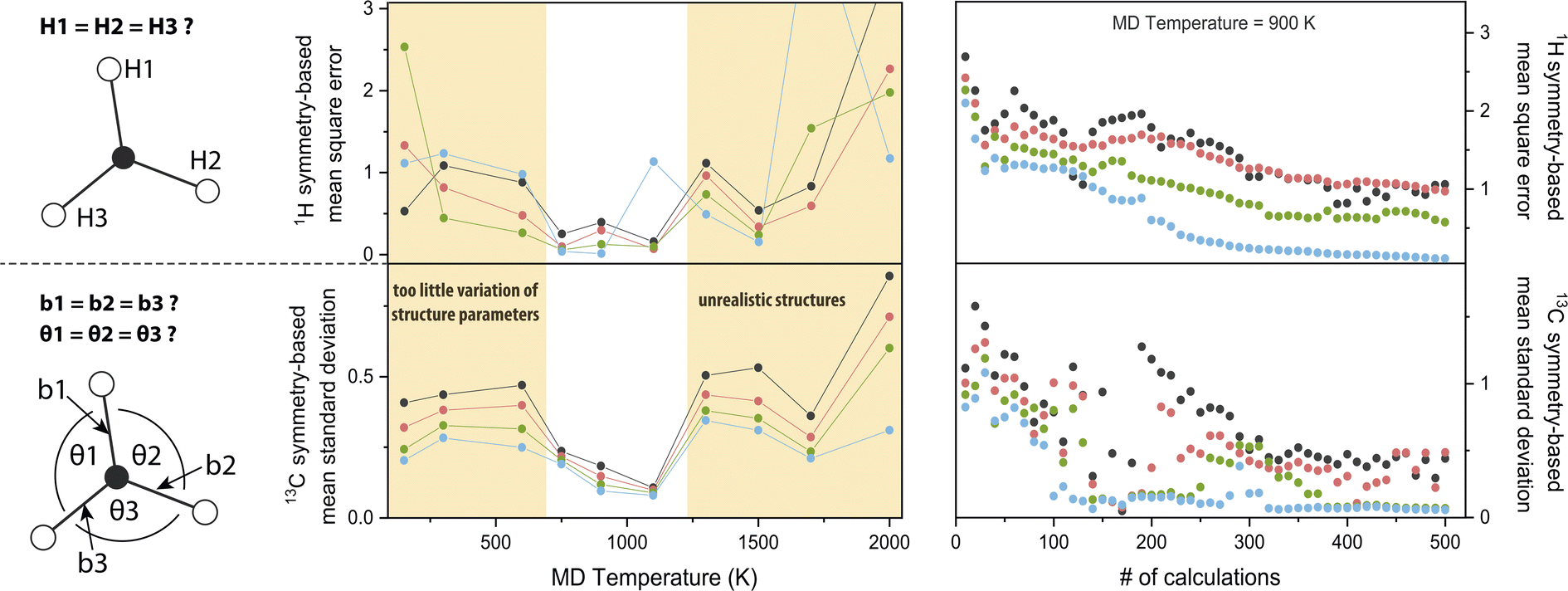 | ||
| Fig. 3 Symmetry-based analysis of molecular dynamics parameters determining the quality of methyl radical importance matrices. Symmetry arguments for 1H and 13C hyperfine couplings are visualized on the left. Design of symmetry-based descriptors is described in the section Parameter choice and plotted for λ = 0.03 (black), 0.05 (red), 0.08 (green), and 0.12 (blue). In the middle panel, the molecular dynamics temperature is optimized. In the right panel, error reduction with dataset size is analyzed. | ||
In Fig. 3 (right panel), the symmetry-based descriptors are used to investigate the dataset size N. For increasing N, statistics become better and correlations between atomic movements are averaged out. For high λ values, errors are generally smaller because weakly correlated features are forced to an importance value of zero. Equilibration of the descriptors for this trajectory is achieved with around N ≈ 350. Although minimization of the error is the ideal performance indicator, the importance matrix is already qualitatively reproduced with far smaller N; the underlying symmetry is indicated already for N = 25 (Fig. S1†) where the absolute importance values still deviate significantly from N = 505 (Fig. 2), but correlated or uncorrelated classification is already satisfying. Using rough extrapolation, dataset sizes were increased for increasing amounts of magnetic nuclei in a radical (Table 1). To compromise with available computational resources, the N-to-feature ratio decreases from 84 for the methyl radical to 24 for the tryptophan-type radical.
Lastly, the regularization parameter λ needs to be chosen appropriately, with feature weights approaching zero for λ → ∞, but a too small λ over-interprets the data (overfitting). λ can be tuned by optimizing the accuracy of the NCA model using cross-validation, where parts of the data are not used to train the model but to later validate its accuracy. Prediction accuracy optimization (Fig. S2†) for the methyl, ethyl, and methyl peroxy radicals was performed for each coupling constant individually, resulting in 0.01 ≤ λ ≤ 0.26 with an average of 0.07 and a median of 0.03 (Table S2†). The optimized λ values are very similar for magnetically equivalent nuclei and vary for different coupling magnitudes and nucleus type. Based on these test sets and to ensure better comparability in importance matrices, λ was chosen to be 0.05, in between the average and median for all figures in the main text. Additional figures with λ = 0.025 and 0.100 for all radicals are provided in the ESI, Section E.†
3.4 Chemical implications
In this section, organic radicals (Table 1) are investigated as test cases for the introduced feature design and NCA algorithm. In analogy to the methyl radical, importance matrices can be cross-checked by symmetry arguments. On that basis, the relationship of chemical information with atomic and electronic structure will be extracted, since hyperfine coupling constants directly result from the spin density distribution that is a function of the molecular orbital structure. The spin–orbit coupling contribution is disregarded in this work, as is common for organic radicals,16 saving computational resources and removing selection biases since spin–orbit coupling is significantly DFT-functional dependent.39,40 To reduce complexity, we will largely base the discussion on Aiso values.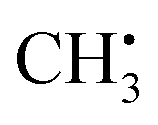 is an experimentally41 and theoretically42,43 well-characterized organic radical with a carbon 2pz orbital centered free electron (Fig. 2b), largely defining the anisotropy and magnitude of the 13C coupling tensor (Fig. 2c). In addition, spin density can spread over the molecule via spin polarization. For
is an experimentally41 and theoretically42,43 well-characterized organic radical with a carbon 2pz orbital centered free electron (Fig. 2b), largely defining the anisotropy and magnitude of the 13C coupling tensor (Fig. 2c). In addition, spin density can spread over the molecule via spin polarization. For 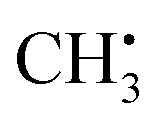 , exchange interactions between the (π)1 and (σCH)2 orbital result in negative spin density around H atoms through different potentials felt by electrons with parallel and anti-parallel spin (McConnell11 exchange), indicated by a H Mulliken spin population of −0.03 for the relaxed structure. The anisotropy of the 1H hyperfine tensors is affected by through-space contributions; particularly from positive spin density around the central 13C,44 which explains less negative Ax,y that is pointing toward the C atom and parallel to the 13C hyperfine z-axis, respectively.
, exchange interactions between the (π)1 and (σCH)2 orbital result in negative spin density around H atoms through different potentials felt by electrons with parallel and anti-parallel spin (McConnell11 exchange), indicated by a H Mulliken spin population of −0.03 for the relaxed structure. The anisotropy of the 1H hyperfine tensors is affected by through-space contributions; particularly from positive spin density around the central 13C,44 which explains less negative Ax,y that is pointing toward the C atom and parallel to the 13C hyperfine z-axis, respectively.The importance matrix shows that Aiso(HX) is correlated with the respective HXC1 bond length, likely increasing negative spin density at the nucleus through growing overlap between (π)1 and (σCH)2, and thus increasing the exchange interaction. Analogously, Aiso(C1) is affected by contributions from all bonds simultaneously. Regarding bond angles, Aiso(HX) correlation with the opposite-lying angle is observed, e.g. H2C1H3 for Aiso(H1), verified by symmetry arguments. Angles deviating from 120° likely alter the orbital configuration with respect to hybridization and inter-(σC1HX)2 overlap, where larger H–H distances were found to result in more negative Aiso.45 Consequently, also the two remaining angles should affect Aiso. However, the opposite-lying angle is the only angle where both other H atoms either move further away or get closer concurrently to a reference 1H nucleus. In contrast, if H1C1H2 got larger, H1C1H3 could get larger as well, amplifying the effect on Aiso(H1), or it could get smaller, compensating the effect. For 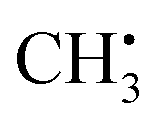 , the most analyzed structural deformation in literature is a trigonal pyramidal distortion, characterized by the angle of the H atom's positions and the relaxed structure's plane.43,46 To gauge this distortion, the sum of all angles was added as a feature herein and shows likewise correlation with all Aiso(HX), evoked by moving 1H away from the nodal plane toward the 2pz orbital lobes. Thus, Aiso(HX) becomes less negative with growing pyramidal distortion (Fig. 2f). Concurrently, Aiso(C1) becomes more positive because the pz-centered free electron transforms into a more sp3 hybridized-type electron, in analogy to ground-state pyramidal
, the most analyzed structural deformation in literature is a trigonal pyramidal distortion, characterized by the angle of the H atom's positions and the relaxed structure's plane.43,46 To gauge this distortion, the sum of all angles was added as a feature herein and shows likewise correlation with all Aiso(HX), evoked by moving 1H away from the nodal plane toward the 2pz orbital lobes. Thus, Aiso(HX) becomes less negative with growing pyramidal distortion (Fig. 2f). Concurrently, Aiso(C1) becomes more positive because the pz-centered free electron transforms into a more sp3 hybridized-type electron, in analogy to ground-state pyramidal  .47 Regarding importance matrices of the full hyperfine tensor, the H1C1 bond length has a greater effect on Ax,y(H1) compared to Az(H1). All principal components are affected by the change in spin population at C and H atoms, but through-space contributions affect Ax,y more significantly, as reflected above. For C1, Ax,y,z importance values are not significantly distinguishable (Fig. S3†).
.47 Regarding importance matrices of the full hyperfine tensor, the H1C1 bond length has a greater effect on Ax,y(H1) compared to Az(H1). All principal components are affected by the change in spin population at C and H atoms, but through-space contributions affect Ax,y more significantly, as reflected above. For C1, Ax,y,z importance values are not significantly distinguishable (Fig. S3†).
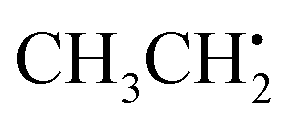 can be analyzed based on
can be analyzed based on 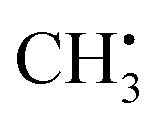 with one H atom exchanged for a CH3 group. The CH2 fragment behaves analogous to
with one H atom exchanged for a CH3 group. The CH2 fragment behaves analogous to 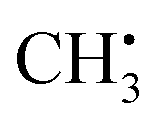 , exhibiting maximum spin population in the C1 pz orbital and negative spin density around H1 and H2 (Fig. 4a). Accordingly, non-zero importance values of H1, H2, and C1 are equivalent to the
, exhibiting maximum spin population in the C1 pz orbital and negative spin density around H1 and H2 (Fig. 4a). Accordingly, non-zero importance values of H1, H2, and C1 are equivalent to the 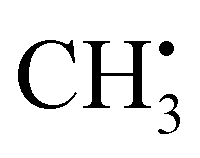 case, taking into account that all C1-centered angles are now correlated, because C3 rotational symmetry is absent and, therefore, each angle matters individually. Exchanging the H residue with CH3 stabilizes the ethyl radical via hyperconjugation,13,48 describing electron delocalization via adjacent σβ-CH bonds lying above or below the H1C1H2 plane. This is manifested in large-magnitude 1H hyperfine tensors of H4 and H5 and insignificant coupling for H3 lying in the nodal plane in the relaxed structure. However, low-energy CH3 rotation renders these atoms (β-H) magnetically equivalent along the MD trajectory. The involved large changes of Aiso (β-H) depending on the dihedral angle can be identified by a strong correlation in the importance matrix, representing Karplus-like14 behavior. Furthermore, non-zero angle importance values define the tetrahedral-to-pyramidal distortion, altering the β-H distance to the pz orbital lobes. Interestingly, the C2C1 bond length is also significant for the hyperfine coupling of β-H. This can be attributed to negative spin density around C2 (Mulliken spin population of −0.08), possibly evoked by similar exchange interactions along the C2C1 bond as for H1C1. Further exchange interactions can result in spin polarization of β-H orbitals, as pointed out by Geoffroy and Lucken,49 challenging the sole attribution of β-H hyperfine values to McConnell-type11,13,44 hyperconjugation. Accordingly, Aiso(C2) depends strongly on the C2C1 bond but also on the C1C2β-H angles, which affects spin delocalization via hyperconjugation toward β-H, thereby offering another path for exchange interactions between β-H and C2.
case, taking into account that all C1-centered angles are now correlated, because C3 rotational symmetry is absent and, therefore, each angle matters individually. Exchanging the H residue with CH3 stabilizes the ethyl radical via hyperconjugation,13,48 describing electron delocalization via adjacent σβ-CH bonds lying above or below the H1C1H2 plane. This is manifested in large-magnitude 1H hyperfine tensors of H4 and H5 and insignificant coupling for H3 lying in the nodal plane in the relaxed structure. However, low-energy CH3 rotation renders these atoms (β-H) magnetically equivalent along the MD trajectory. The involved large changes of Aiso (β-H) depending on the dihedral angle can be identified by a strong correlation in the importance matrix, representing Karplus-like14 behavior. Furthermore, non-zero angle importance values define the tetrahedral-to-pyramidal distortion, altering the β-H distance to the pz orbital lobes. Interestingly, the C2C1 bond length is also significant for the hyperfine coupling of β-H. This can be attributed to negative spin density around C2 (Mulliken spin population of −0.08), possibly evoked by similar exchange interactions along the C2C1 bond as for H1C1. Further exchange interactions can result in spin polarization of β-H orbitals, as pointed out by Geoffroy and Lucken,49 challenging the sole attribution of β-H hyperfine values to McConnell-type11,13,44 hyperconjugation. Accordingly, Aiso(C2) depends strongly on the C2C1 bond but also on the C1C2β-H angles, which affects spin delocalization via hyperconjugation toward β-H, thereby offering another path for exchange interactions between β-H and C2.
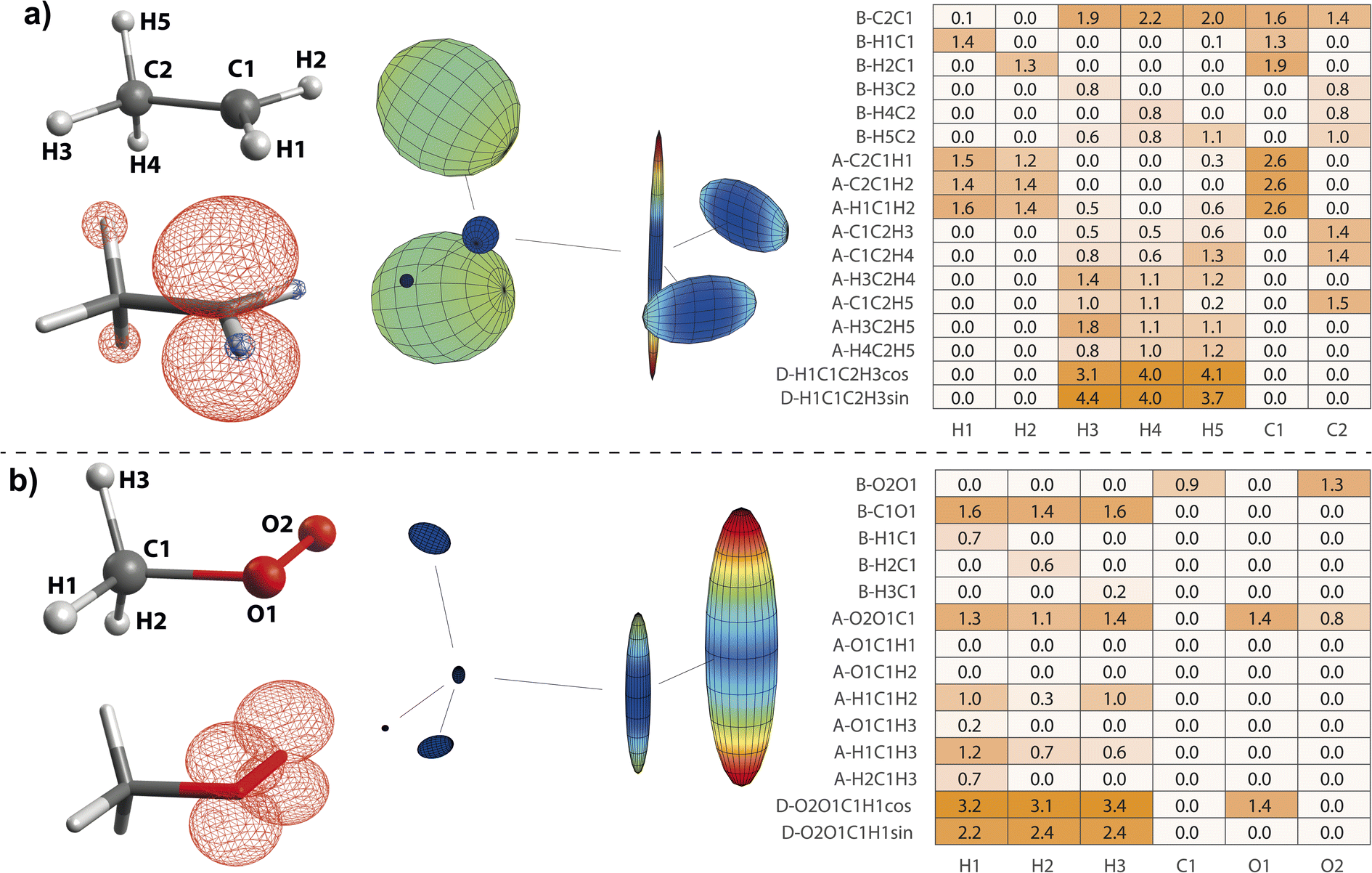 | ||
| Fig. 4 Structure–hyperfine relationships for the ethyl (a) and methyl peroxy (b) radicals. Atomic labels (left top), spin densities (left bottom), and hyperfine coupling tensor visualizations (center) are plotted in analogy to Fig. 2. Importance matrices (right) are given for Aiso values correlated with bonds (B), angles (A), and dihedrals (D). For dihedrals, importance values for cosine (cos) and sine (sin) values are given. | ||
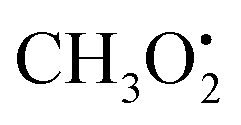 in particular is relevant for gas-phase atmospheric chemistry as an intermediate in the oxidative decomposition of methane.50 Maximum spin density is located in oxygen π-bonded pz orbitals with Mulliken spin population of +0.69 and +0.29 for O2 and O1, respectively (Fig. 4b). The 17O hyperfine tensors exhibit consistent anisotropy and magnitude. Similar to
in particular is relevant for gas-phase atmospheric chemistry as an intermediate in the oxidative decomposition of methane.50 Maximum spin density is located in oxygen π-bonded pz orbitals with Mulliken spin population of +0.69 and +0.29 for O2 and O1, respectively (Fig. 4b). The 17O hyperfine tensors exhibit consistent anisotropy and magnitude. Similar to 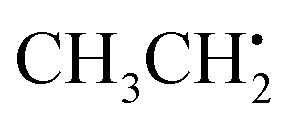 , the π network is further stabilized by σCH hyperconjugation and Geoffroy/Lucken49 exchange (vide supra), confirmed by large 1H importance values regarding the dihedral angle and the C1O1 bond, respectively. In contrast to
, the π network is further stabilized by σCH hyperconjugation and Geoffroy/Lucken49 exchange (vide supra), confirmed by large 1H importance values regarding the dihedral angle and the C1O1 bond, respectively. In contrast to 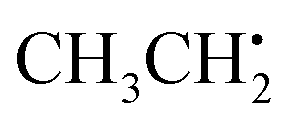 , 1H hyperfine coupling changes will not follow approximate C2 symmetry with the dihedral, reflecting pz symmetry, but are more complex due to increasing (dipolar) interactions when the dihedral approaches ecliptic HX and O2. In this position, the O2O1C1 angle dominates the distance of 1H to maximum spin density affecting Aiso(HX), as deducible from the importance matrix. Also, both 17O coupling tensors strongly correlate with O2O1C1, indicating a significant change of electronic structure, possibly involving facilitated hyperconjugation if O2O1C1 is small and/or changes in the π network. Aiso(C1) is comparably small and dictated by the O2O1 bond. This finding can be explained by a cascading effect; if O2O1 is short, π-bonding becomes more efficient, hence the spin population in the O1 pz orbital increases, and thus exchange interactions between the O1 pz and the C1O1 σ-bond are larger. Although there is no correlation of Aiso(O1) with O2O1 in the displayed importance matrix, an importance value of 1.6 is extracted for Az(O1) (Fig. S4†), which is consistent with the interpretation.
, 1H hyperfine coupling changes will not follow approximate C2 symmetry with the dihedral, reflecting pz symmetry, but are more complex due to increasing (dipolar) interactions when the dihedral approaches ecliptic HX and O2. In this position, the O2O1C1 angle dominates the distance of 1H to maximum spin density affecting Aiso(HX), as deducible from the importance matrix. Also, both 17O coupling tensors strongly correlate with O2O1C1, indicating a significant change of electronic structure, possibly involving facilitated hyperconjugation if O2O1C1 is small and/or changes in the π network. Aiso(C1) is comparably small and dictated by the O2O1 bond. This finding can be explained by a cascading effect; if O2O1 is short, π-bonding becomes more efficient, hence the spin population in the O1 pz orbital increases, and thus exchange interactions between the O1 pz and the C1O1 σ-bond are larger. Although there is no correlation of Aiso(O1) with O2O1 in the displayed importance matrix, an importance value of 1.6 is extracted for Az(O1) (Fig. S4†), which is consistent with the interpretation.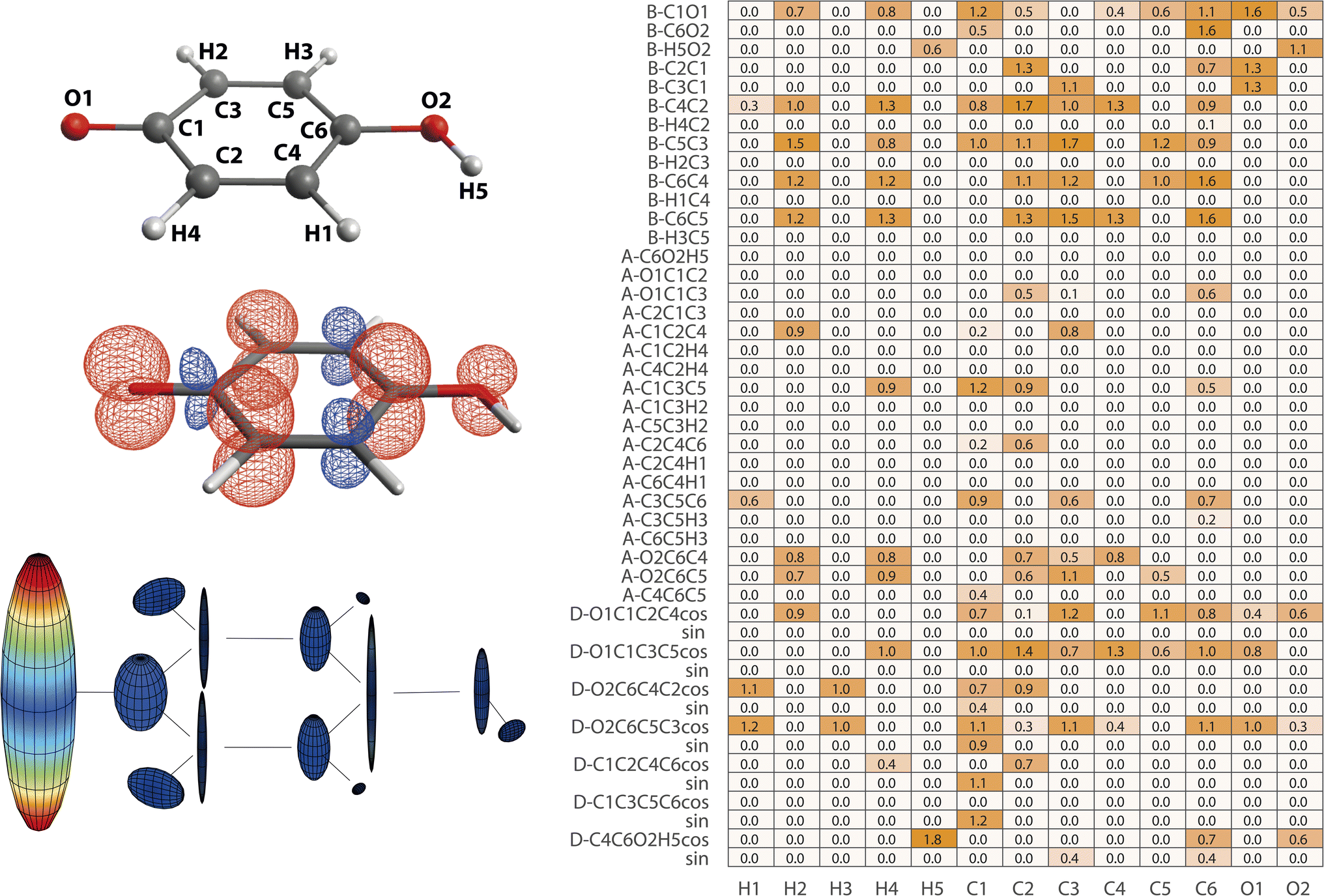 | ||
| Fig. 5 Structure–hyperfine relationships for p-benzosemiquinone. (left) Atomic labels, spin densities, and hyperfine coupling tensor visualizations are plotted in analogy to Fig. 2. (right) Importance matrix is given for Aiso values correlated with bonds (B), angles (A), and dihedrals (D). For dihedrals, importance values for cosine (cos) and sine (sin) values are given. | ||
Largest Aiso is observed for O1 and the importance matrix correlations suggest that its magnitude is predominantly affected by its inclusion into the aromatic ring system; via the next-nearest bonds C1O1, C2C1, and C3C1 and via its atomic positioning relative to the aromatic plane characterized by the dihedrals O1C1C3C5 and O1C1C2C4. Other deformations within the ring seem to be less impactful, in agreement with findings that the degree of aromaticity remains fairly resistant against medium deformations therein.53,54 Aiso values of ring carbons are more complex to define; the multitude of significant structure parameters results in some uncertainty of the algorithm indicated by deviations from C2/C3 and C4/C5 equivalence. Nonetheless, more general statements can be made. In accordance with Aiso(O1), the dihedrals involving O1 position relative to the ring also impact Aiso(CX) by alteration of the degree of delocalization into the ring. For that, also the C1O1 bond seems to be significant, especially for Aiso(C1). Within the ring, C–C bond lengths dominate Aiso(CX) via proposed sensitivity in orbital overlap and exchange, evoking alternating spin density signs around the ring. Inter-ring and ring-hydrogen angles are considered less impacting. Notably, the position of O2 defined by the angle O2C6C4 (and O2C6C5) seems to consistently affect Aiso of ortho- and meta-carbons, possibly due to symmetry cancelation along C1C3C5C6 versus C1C2C4C6 leading to altered delocalization pathways. A clearer picture arises from ring-hydrogens where symmetry of H1/H3 and H2/H4 is largely intact. It is known that spin density at ring-hydrogens is predominantly caused by McConnell11 exchange where Aiso is proportional to the spin density at the adjacent C ring-atom.12,52 Therefore, it is no surprise that C–C bond lengths also largely impact Aiso of ring-hydrogens, which is also evident for H1 and H3 when using a smaller λ (ESI, Section E†). Nonetheless, ring-hydrogens are more affected by their position above or below the nodal plane characterized by the correlation with appropriate dihedral angles.
The para-positioned OH group is the last fragment to be analyzed. O2 again is highly affected by the C1O1 bond, mediating the degree of delocalization originating from O1. Efficient delocalization is further depending on positioning above or below the aromatic plane, either on the spin density ‘sending’ (O1C1C2C4) or ‘receiving’ side (O2C6C5C3). H5 is positionally more flexible with regard to the aromatic plane (C4C6O2H5), which has a large impact on Aiso(H5). It is dominated by McConnell11 exchange mediated by the H5O2 bond when within the plane and by participation in the π network above or below the plane.
In addition to the radicals discussed thus far, we also analyzed the tyrosyl radical, structurally similar to semiquinone, and tryptophan-type radical. The corresponding importance matrices are shown in Fig. S5 and S6,† respectively.
3.5 Suggestions for application
On a qualitative basis, the introduced importance matrices reveal a multitude of atomic–electronic structure relationships, which typically necessitate elaborate analyses of individual structural changes and their effect(s).11–13,42–45,47,49,51,52,55 For small organic radicals, spin density distributions are often conceptually simple to understand and partly transferrable. In contrast, interdependencies in more complicated radicals or paramagnetic species often need to be specifically investigated. For example, systematic computational studies were recently conducted to evaluate the electronic structure of transition metal phosphoroxy compounds,35,56 which in part motivated this work. Finding relevant structure relations requires sound chemical intuition and time-consuming variational testing, where the holistic importance matrix approach could speed up the identification of structural relationships and prevent overlooked dependencies. Furthermore, molecular dynamics have often been used to precisely predict Aiso by averaging a conformational ensemble.21,22,36,46,57,58 Apart from mimicking experimental conditions and computing averaged values, MD trajectories could also be concurrently used for the importance matrix approach, given that the needed inputs are already available. The presented procedure can be used for any system, provided hyperfine coupling calculations can be performed with appropriate accuracy.From a more practical point of view, importance matrices can help answer two types of questions for experimentalists that aid the structure identification of a paramagnetic molecule: (1) which hyperfine coupling constants do we need to measure to estimate a specific structure parameter? With pre-experimental awareness of these correlations, EPR experiments can be planned using a design of experiments (DoE) approach. This might involve a decision on whether to investigate liquids or solids, which hyperfine spectroscopy techniques are needed, and which atoms need to be isotope-labeled (e.g. 13C, 14,15N, 17O). (2) Which structure parameters could be responsible for a detected change in a specific hyperfine coupling constant? Changes in structure parameters can have a multitude of reasons and are, therefore, frequently targeted with EPR spectroscopy. In (frozen) solutions, the solvent (or other interacting molecules) is often responsible for structural changes, either implicitly through its dielectric continuum or explicitly, e.g., via hydrogen bonding.59,60 Temperature was also identified to affect hyperfine couplings,61 although again through indirect solvent effects such as viscosity changes. Hyperfine couplings can also be sensitive to structural changes rather remote to the actual radical center (e.g., dihedral-dependent side chain hyperconjugation in semiquinones57,58), which might be disregarded in an analysis to reduce complexity/save computational resources or overlooked in the first place.
4 Conclusions
We have described an application of the NCA feature selection algorithm to identify relations between atomic configuration and electronic structure in paramagnetic species. The statistical analysis is based on structure parameters and hyperfine tensors of molecular dynamics snapshots, calculated by density functional theory. While conducting MD simulations at experimentally accessible temperatures would be desirable, results suggest that the semi-quantitative information of an importance matrix can be retained at elevated temperatures around 700–1200 K. Adjusting the temperature helps balancing computational costs, and it may also be helpful for sampling a larger configurational space within a particular MD trajectory, as long as the structure of a molecule is not qualitatively altered. Assigning importance quantifiers to structural parameters for Ax,y,z,iso creates a new perspective to look at, analyze, and understand organic radicals. For the well-characterized methyl and ethyl radicals, importance values reflect known (π)1 and (σCH)2 exchange interactions and hyperconjugation with β–σCH bonds. For larger radicals, more subtle correlations are identified, for example that Aiso(C) in the methyl peroxy radical is predominantly dictated by the oxygen–oxygen bond length, or that either exchange or π-delocalization dictates Aiso of the hydroxy-hydrogen in p-benzosemiquinone, depending on the dihedral angle.The computed importance matrices can help experimentalists in selecting the most sensitive hyperfine couplings for the assessment whether a specific structure parameter is changing. For example, changes can occur via H-bonding, which is known to lengthen the C–O bond in p-benzosemiquinone.57 A sensitive hyperfine coupling constant can also help in identifying confined conformational distributions, as shown for biological tyrosyl and tryptophan radicals.62,63 To incorporate all relevant effects, the analysis should be repeated including the explicit molecular environment, e.g., including solvent molecules. We encourage the reader to use and modify the implemented procedure written for ORCA and MATLAB and note that, in principle, also other structure-dependent atomic constants such as quadrupole couplings, chemical shift tensors, etc. or isotope variations are suitable for the presented procedure.
Data availability
The codes used for ORCA calculations are included in the ESI, Section F.† Matlab scripts/functions and trajectory data of the organic radicals investigated herein can be found at https://doi.org/10.26165/JUELICH-DATA/UH0LM2 or at https://github.com/conradsz/StructureHyperfineRelations.Author contributions
C. S.: conceptualization, methodology, software, formal analysis, visualization, writing – original draft. R.-A. E.: writing – review & editing, supervision. J. G.: conceptualization, writing – review & editing, supervision.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Valuable discussions with Davis Thomas Daniel and Prof. Mario Chiesa are acknowledged. We thank Prof. Stefan Stoll for allowing us to use and modify his hyperfine coupling tensor illustration tool. Computations were performed with computing resources granted by RWTH Aachen University under project rwth0701.References
- H. J. Kulik, T. Hammerschmidt, J. Schmidt, S. Botti, M. A. L. Marques, M. Boley, M. Scheffler, M. Todorović, P. Rinke, C. Oses, A. Smolyanyuk, S. Curtarolo, A. Tkatchenko, A. P. Bartók, S. Manzhos, M. Ihara, T. Carrington, J. Behler, O. Isayev, M. Veit, A. Grisafi, J. Nigam, M. Ceriotti, K. T. Schütt, J. Westermayr, M. Gastegger, R. J. Maurer, B. Kalita, K. Burke, R. Nagai, R. Akashi, O. Sugino, J. Hermann, F. Noé, S. Pilati, C. Draxl, M. Kuban, S. Rigamonti, M. Scheidgen, M. Esters, D. Hicks, C. Toher, P. v. Balachandran, I. Tamblyn, S. Whitelam, C. Bellinger and L. M. Ghiringhelli, Electron. Struct., 2022, 4, 023004 CrossRef.
- D. Dai, Q. Liu, R. Hu, X. Wei, G. Ding, B. Xu, T. Xu, J. Zhang, Y. Xu and H. Zhang, Mater. Des., 2020, 196, 109194 CrossRef CAS.
- A. Khailov, A. Ivannikov, K. Zhumadilov, V. Stepanenko, A. Kaprin, P. Shegay and S. Ivanov, Radiat. Meas., 2020, 137, 106435 CrossRef CAS.
- S. G. Worswick, J. A. Spencer, G. Jeschke and I. Kuprov, Sci. Adv., 2018, 4, 5218 CrossRef PubMed.
- Y. Pokern, B. Eltzner, S. F. Huckemann, C. Beeken, J. A. Stubbe, I. Tkach, M. Bennati and M. Hiller, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023615118 CrossRef CAS PubMed.
- A. T. Taguchi, E. D. Evans, S. A. Dikanov and R. G. Griffin, J. Phys. Chem. Lett., 2019, 10, 1115–1119 CrossRef CAS PubMed.
- J. Goldberger, S. Roweis, G. Hinton and R. Salakhutdinov, Adv. Neural Inf. Process. Syst., 2004, 513–520 Search PubMed.
- W. Yang, K. Wang and W. Zuo, J. Comput., 2012, 7, 162–168 Search PubMed.
- N. S. Malan and S. Sharma, Comput. Biol. Med., 2019, 107, 118–126 CrossRef PubMed.
- J. Nie, X. Wen, X. Niu, Y. Chu, F. Chen, W. Wang, D. Zhang, Z. Hu, J. Xiao and L. Guo, Polym. Test., 2022, 112, 107624 CrossRef CAS.
- H. M. McConnell and D. B. Chesnut, J. Chem. Phys., 1958, 28, 107–117 CrossRef CAS.
- H. M. McConnell, J. Chem. Phys., 1956, 24, 764–766 CrossRef CAS.
- A. D. McLachlan, Mol. Phys., 1958, 1, 233–240 CrossRef CAS.
- M. Karplus, J. Chem. Phys., 1959, 30, 11–15 CrossRef CAS.
- M. Bennati, in eMagRes, John Wiley & Sons, Ltd, Chichester, UK, 2017, pp. 271–282 Search PubMed.
- F. Neese, eMagRes, 2017, 6, 1–22 CAS.
- F. Neese and M. L. Munzarová, in Calculation of NMR and EPR Parameters, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2004, pp. 21–32 Search PubMed.
- M. L. Munzarova, P. Kubacek and M. Kaupp, J. Am. Chem. Soc., 2000, 122, 11900–11913 CrossRef CAS.
- P. J. Carl, S. L. Isley and S. C. Larsen, J. Phys. Chem. A, 2001, 105, 4563–4573 CrossRef CAS.
- R. Improta and V. Barone, Chem. Rev., 2004, 104, 1231–1253 CrossRef CAS PubMed.
- S. Vogler, J. C. B. Dietschreit, L. D. M. Peters and C. Ochsenfeld, Mol. Phys., 2020, 118(19–20), e1772515 CrossRef.
- A. A. Auer, V. A. Tran, B. Sharma, G. L. Stoychev, D. Marx and F. Neese, Mol. Phys., 2020, 118(19–20), e1797916 CrossRef.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Brehm, ORCA-MD module, https://brehm-research.de/orcamd Search PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- V. Barone and P. Cimino, Chem. Phys. Lett., 2008, 454, 139–143 CrossRef CAS.
- D. C. Liu and J. Nocedal, Math. Program., 1989, 45, 503–528 CrossRef.
- S. Wang, ACS Omega, 2019, 4, 9271–9283 CrossRef CAS PubMed.
- S. Khalid, T. Khalil and S. Nasreen, Proceedings of 2014 Science and Information Conference, SAI 2014, 2014, pp. 372–378 Search PubMed.
- C. Szczuka, P. Jakes, R.-A. Eichel and J. Granwehr, Adv. Energy Sustainability Res., 2021, 2, 2100121 CrossRef CAS.
- B. Sharma, V. A. Tran, T. Pongratz, L. Galazzo, I. Zhurko, E. Bordignon, S. M. Kast, F. Neese and D. Marx, J. Chem. Theory Comput., 2021, 17, 6366–6386 CrossRef CAS PubMed.
- R. E. Bruccoleri and M. Karplus, Biopolymers, 1990, 29, 1847–1862 CrossRef CAS PubMed.
- Y. Wang, S. Gong, H. Wang, L. Li and G. Liu, J. Anal. Appl. Pyrolysis, 2021, 155, 105045 CrossRef CAS.
- F. Neese, J. Chem. Phys., 2005, 122, 034107 CrossRef PubMed.
- S. Kossmann, B. Kirchner and F. Neese, Mol. Phys., 2007, 105, 2049–2071 CrossRef CAS.
- Y. A. Dmitriev, Phys. B, 2004, 352, 383–389 CrossRef CAS.
- M. Barfield, A. S. Babaqi, D. M. Doddrell and H. P. W. Gottlieb, Mol. Phys., 1981, 42, 153–164 CrossRef CAS.
- M. Karplus, J. Chem. Phys., 1959, 30, 15–18 CrossRef CAS.
- H. M. McConnell and J. Strathdee, Mol. Phys., 1959, 2, 129–138 CrossRef CAS.
- J. Higuchi, J. Chem. Phys., 1963, 39, 3455–3462 CrossRef CAS.
- H. Tachikawa, M. Igarashi and T. Ishibashi, Chem. Phys. Lett., 2002, 352, 113–119 CrossRef CAS.
- O. Edlund, A. Lund, M. Shiotani, J. Sohma and K. Thuomas, Mol. Phys., 1976, 32, 49–69 CrossRef CAS.
- H. Zipse, Top. Curr. Chem., 2006, 263, 163–189 CrossRef CAS.
- M. Geoffroy and E. A. C. Lucken, Mol. Phys., 1981, 43, 887–895 CrossRef CAS.
- P. D. Lightfoot, R. A. Cox, J. N. Crowley, M. Destriau, G. D. Hayman, M. E. Jenkin, G. K. Moortgat and F. Zabel, Atmos. Environ., Part A, 1992, 26, 1805–1961 CrossRef.
- T. H. Brown, D. H. Anderson and H. S. Gutowsky, J. Chem. Phys., 1960, 33, 720–726 CrossRef CAS.
- A. D. McLachlan, H. H. Dearman and R. Lefebvre, J. Chem. Phys., 1960, 33, 65–70 CrossRef CAS.
- J. Aihara, Bull. Chem. Soc. Jpn., 1990, 63, 1956–1960 CrossRef CAS.
- C. Fei Shen, Z. Zhong Liu, H. Xia Liu and H. Qing Zhang, Spectrochim. Acta, Part A, 2018, 201, 392–398 CrossRef PubMed.
- J. R. Bolton, A. Carrington and J. dos Santos-Veiga, Mol. Phys., 1962, 5, 465–473 CrossRef CAS.
- N.-A. Stamos, E. Ferentinos, M. Chrysina, C. P. Raptopoulou, V. Psycharis, Y. Sanakis, D. A. Pantazis, P. Kyritsis and G. Mitrikas, Inorg. Chem., 2020, 59, 3666–3676 CrossRef CAS PubMed.
- J. R. Asher and M. Kaupp, ChemPhysChem, 2007, 8, 69–79 CrossRef CAS PubMed.
- J. R. Asher and M. Kaupp, Theor. Chem. Acc., 2008, 119, 477–487 Search PubMed.
- T. Abe, S. Tero-Kubota and Y. Ikegami, J. Phys. Chem., 1982, 86, 1358–1365 CrossRef CAS.
- R. Owenius, M. Engström, M. Lindgren and M. Huber, J. Phys. Chem. A, 2001, 105, 10967–10977 CrossRef CAS.
- B. L. Bales, E. Wajnberg and O. R. Nascimento, J. Magn. Reson., Ser. A, 1996, 118, 227–233 CrossRef CAS.
- F. Lendzian, Biochim. Biophys. Acta, Bioenerg., 2005, 1707, 67–90 CrossRef CAS PubMed.
- M. Hiller, I. Tkach, H. Wiechers, B. Eltzner, S. Huckemann, Y. Pokern and M. Bennati, Appl. Magn. Reson., 2022, 53, 1015–1030 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02476h |
| This journal is © The Royal Society of Chemistry 2023 |