
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, and characterization of non-hemolytic antimicrobial peptides related to human cathelicidin LL-37†
Rajavenkatesh Krishnamoorthy *ab,
Priyanka Adhikaric and
Parthiban Anaikuttic
*ab,
Priyanka Adhikaric and
Parthiban Anaikuttic
aOrganic and Bioorganic Chemistry Laboratory, CSIR-CLRI, Adyar, Chennai-600020, Tamil Nadu, India
bDepartment of Chemistry, Sethu Institute of Technology, Kariapatti, Virudunagar-626115, Tamil Nadu, India. E-mail: krajavenkateshkcmp5@gmail.com; rajavenkateshk2@sethu.ac.in
cCentre for GMP Extraction Facility (Dept. of Biotechnology), National Institute of Pharmaceutical Education and Research, Guwahati-781101, Assam, India
First published on 23rd May 2023
Abstract
We designed and synthesised the N-terminally labeled cationic and hydrophobic peptides, i.e., FFKKSKEKIGKEFKKIVQKI (P1) and FRRSRERIGREFRRIVQRI (P2) related to the human cathelicidin LL-37 peptide. The integrity and molecular weight of the peptides were confirmed by mass spectrometry. The purity and homogeneity of peptides P1 and P2 were determined by comparing LCMS or analytical HPLC chromatograms. The circular dichroism spectroscopy reveals the conformational transitions upon interaction with membranes. Predictably, peptides P1 and P2 showed a random coil structure in the buffer and formed α-helix secondary structure in TFE and SDS micelles. This assessment was further confirmed by 2D NMR spectroscopic methods. The analytical HPLC binding assay measurements revealed that peptides P1 and P2 display preferential interactions with the anionic lipid bilayer (POPC:POPG) moderately than zwitterionic (POPC). The efficacies of the peptides were tested against Gram-positive and Gram-negative bacteria. It is imperative to note here that the arginine-rich P2 exerted higher activity against all the test organisms as compared with that shown by the lysine-rich peptide P1. To test the toxicity of these peptides, a hemolytic assay was performed. P1 and P2 showed very little to no toxicity for a hemolytic assay, which is significant for P1 and P2 to be used as potential therapeutic agents in practical applications. Both peptides P1 and P2 were non-hemolytic and appeared to be more promising as they demonstrated wide-spectrum antimicrobial activity.
Introduction
A series of structurally different AMPs called cathelicidin is found at the carboxyl end of a highly conserved 15–18 kDa cathepsin-L-inhibitor (cathelin)-like domain. Cathelin is an abbreviation for cathepsin L inhibitor. LL-37 is an antimicrobial peptide in the cathelicidin family, and it is important to fully comprehend how this peptide works.1,2 LL-37, in contrast to most other defensins, maintains a broad-spectrum bactericidal activity at physiological or high salt concentrations, which is a distinguishing advantage for prospective therapeutic purposes.3 It lacks disulfide bridges, making chemical synthesis simpler and less expensive.4 Its rapid microbial destruction should be advantageous for topical applications, allowing bacterial death before the peptide is mechanically removed or rendered inactive, while other natural antimicrobial peptides are very sensitive to enzymatic degradation. It was also demonstrated that LL-37 inhibits Lipopolysaccharide (LPS) action.5,6Due to LL-37's rapid rate of microbial killing, topical treatments should benefit from the ability to destroy germs before the peptide is mechanically removed or rendered inactive. In contrast to other natural antimicrobial peptides, LL37 appears to form dimeric and trimeric aggregates in solution under experimental conditions7 as well as in lipid bilayer, as demonstrated by 14N solid-state NMR studies8 and is strongly shielded from proteolytic destruction. E. coli's LL37 has been purified and biologically expressed, as described.9,10 The high-resolution structure of detergent micelles in previous NMR investigations on LL37 (ref. 11 and 12) and the mechanism of membrane rupture were both documented.13,14 Recent biophysical studies have demonstrated membrane interactions and the development of LL37 fibrils15–18 and fibril formation of LL37.19
Nonetheless, it has been demonstrated that LL-37 and a subset of peptides matching various segments of LL-37 exhibit powerful lytic activity against microorganisms, cancer cells, and red blood cells.20 The identification of a 21-residue peptide segment corresponding to residues 7–27 of LL-37, or LL-37(7–27), to demonstrate membrane selective activity has been made as a result of the search for peptides with increased or acceptable antibacterial activity and cell selectivity.21 The investigation of the hemolytic and antimicrobial activities of LL37(7–27) and its ability to disrupt the outer membrane of E. coli and to interact with model membranes mimicking bacterial and mammalian membranes has provided valuable information that is helpful in fine-tuning the activities of this potentially interesting peptide. To obtain information on the membrane selectivity, the binding energy using isothermal titration calorimetry (ITC) and the lipid induced conformational transitions using circular dichroism (CD) experiments have been determined. Specific lipid–peptide interactions have been observed following the phase transition behavior of multilamellar lipid vesicles (MLVs) formed in the dimyristoylphosphatidylcholine (DMPC) and 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dimyristoylphosphatidyl choline (DMPC):dimyristoylphosphatidylglycerol (DMPG) mixture. The peptide's ability to permeabilize anionic model membranes mimicking the bacterial inner membrane has also been studied using 31P-NMR experiments.22 The data from this study suggested that the peptide's miscibility with anionic membrane and lipid-induced conformational transitions form the basis for the observed antimicrobial activity of LL-37(7–27).
:1 dimyristoylphosphatidyl choline (DMPC):dimyristoylphosphatidylglycerol (DMPG) mixture. The peptide's ability to permeabilize anionic model membranes mimicking the bacterial inner membrane has also been studied using 31P-NMR experiments.22 The data from this study suggested that the peptide's miscibility with anionic membrane and lipid-induced conformational transitions form the basis for the observed antimicrobial activity of LL-37(7–27).
The present study deals with the design and synthesis of non-hemolytic antimicrobial peptides from LL-37(5–24). The overall charge on the control peptide LL-37(5–24) and the designed peptides P1 and P2 remains the same. The retention times from HPLC analysis of these peptides P1 and P2 are correlated with the aliphatic content of the peptides. The role of hydrophobicity in the membrane selective association of peptides is presented. A possible structure activity relationship deduced from CD and NMR analyses of the peptides and the HPLC membrane binding assay results shows that both peptides P1 and P2 preferentially bind with an anionic lipid membrane (POPC:POPG) rather than zwitterionic lipid membrane (POPOC). Since LL-37 is hemolytic because of its high hydrophobicity and amphiphilicity, short peptides with potent antimicrobial properties would be useful for therapeutic purposes. The hemolytic and antimicrobial activities of these peptides were also examined. The designed peptides P1 and P2 are expected to selectively target bacterial cells.
Materials and methods
Chemical and reagents
Rink amide-methylbenzhydrylaminehydrochloride salt (MBHA) resin, 9-fluorenyl-methoxycarbonyl (Fmoc) protected amino acids (Merck India Pvt. Ltd), diisopropylethylamine (DIPEA), 1-hydroxy benzotriazole (HOBT), trifluoroacetic acid (TFA), thioanisole, ethanedithiol, and the peptide synthesis vessel were purchased from Sigma Aldrich-Merck Chemicals Pvt. Ltd (Bangalore, India). Both Gram-positive Staphylococcus aureus (ATCC 9144) & Bacillus subtilis and Gram-negative Escherichia coli (ATCC 25922) & Pseudomonas aeruginosa (ATCC 1688) bacteria were obtained from Sigma Aldrich Chemicals Pvt. Ltd (Bangalore, India). The lipids like 2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (POPC) and 2-oleoyl-1-palmitoyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt (POPG) used in the study were purchased from Sigma Aldrich Chemical Inc. Lipids (Alabaster, AL). Solvents like acetonitrile, water, dichloromethane, dimethylformamide, chloroform, and methanol were procured from Spectrochem Pvt. Ltd. (Mumbai, India). Buffers were prepared using water obtained from NANO pure A filtration system. All the chemicals were used without any further purification. The Fmoc-Rink amide MBHA resin and the Fmoc-protected amino acids were purchased from Novabiochem, Merck, Germany. Lipids were purchased from Avanti Polar Lipids Inc., Alabaster. All chemicals utilised in this project were from Merck and used as received. All of the chemicals were used without further purification.Peptides synthesis
The peptides were prepared using the Rink amide resin support and the Fmoc-chemistry solid phase peptide synthesis protocol. First, a 5% solution of DIPEA was used to neutralise rink amide resin. All amino acids were coupled as HBTU with one equivalent of HOBT and 2.5 equivalents of DIPEA for the synthesis of all peptides. The Fmoc protective group was eliminated after the synthesis was complete. The HBTU active ester of peptides P1 and P2 was applied to the resins overnight. Before being dried in a vacuum, the resin was additionally carefully washed with DMF, DCM, acetic acid, and diethyl ether. Using a combination of TFA, thioanisole, and ethanedithiol (9.0:0.5:0.5, v/v) at room temperature for 2 h, the peptide was separated from the resin. Filtration was used to collect the cleavage mixture, which was then concentrated and given diethyl ether treatment to precipitate the crude peptide. Centrifugation was used to separate the crude peptide, which was then dried with dry air and kept at 4 °C. Therefore, an LC-MS with an ESI source was used to calculate the molecular weight of the synthesised peptides.
Reversed-phase high-performance liquid chromatography (RP-HPLC)
The crude peptides were purified using a semi-preparative column, YMC-Pack, ODS-A, (250 × 6.0) mm, 15μ (YMC Co., Ltd. Japan.) stainless steel column filled with octadecyl bonded on a porous silica surface. 0.1% TFA in Milli-Q water was used as mobile phase-A, and 0.1% TFA in acetonitrile was used as mobile phase-B. The purification was carried out using the flow rate of 3.0 mL min−1 and gradient elution program of 0 min/22% B, 13 min/30% B, and 14 min/22% B for P1. The column was equilibrated 3 min before each analysis. The purification was carried out using the flow rate of 3.0 mL min−1 and gradient elution program of 0 min/22% B, 11 min/29% B, 14 min/60% B, and 15 min/22% B for P2. The column was equilibrated 3 min before each analysis.The purity of semi preparative HPLC purified peptides was checked using an analytical column, Xterra RP-18, (150 × 4.6) mm, 5μ. Waters Alliance HPLC system (Waters Corporation, USA) has gradient elution capability. 0.1% TFA in Milli-Q water was used as mobile phase-A, and 0.1% TFA in acetonitrile was used as mobile phase-B. The wavelength of detection was performed at 215 nm using Waters 2487 dual absorbance UV detector. Flow rate was set at 1.0 mL min−1 and gradient compositions of 0 min/20% B, 20 min/30% B, 30 min/50% B, and 35 min/20% B. The column was equilibrated for 10 min before each analysis for both P1 and P2. The peptide was purified using various gradient compositions of organic modifier. The collected fractions were pooled together and solidified using the chiller at −70 °C and lyophilized using a lyophilizer. The percentage recovery of pure peptide was about 55% with respect to the weight of crude peptide.
Secondary structure of peptides studied by circular dichroism
Circular dichroism (CD) spectra were recorded on a JASCO J-715 spectropolarimeter, which was calibrated with 10-(+) camphor sulfonic acid. About 20 μM of each peptide in different solutions were prepared individually using various solvents like 20% TFE, 10 mM sodium phosphate buffer (pH 7.4), and 30 mM SDS solutions. Measurements were made using a 1 mm cell in the range from 270 to 190 nm at 1 nm intervals. An average of four scans of each control sample without peptide was subtracted from the corresponding peptide sample, and then the spectra were normalized for concentration and path length.Secondary structure of peptides studied by NMR spectroscopy
NMR samples were prepared by dissolving ∼2 mg of peptide in 0.5 mL of water (90% H2O, 10% D2O, pH 5.0) and 0.5 mL of 80 mM SLS-d25, which is well above the critical micelle concentration (8 mM) for SLS. The SLS-to-peptide ratio of the NMR sample was ∼40:1. All NMR spectra in SLS and buffer were recorded on Bruker DMX 400 spectrometer, operating at 400 MHz proton resonance frequency. The data were processed by the Topspin 1.3 program on an HP workstation. All two-dimensional spectra were acquired in the phase-sensitive mode. The NOESY and TOCSY spectra were recorded with 32 scans, a relaxation delay of 1.5 s, a spectral width of 5020 Hz in both dimensions, 512 increments in t1, and 2K data points in t2. The NOESY spectra were recorded with mixing times of 200, 300, and 400 ms (different mixing times were used to evaluate the linear build-up of NOE and to find the mixing time appropriate to the two-spin approximation).
Preparation of multilamellar lipid vesicles
The lipid vesicles were prepared by the vortex mixing method. From the stock solutions, 3 mg lipid [1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine] (POPC) or 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol (POPG)] in chloroform was transferred into a vertex tube and the solvent was removed using a stream of nitrogen gas. The lipids were rehydrated to a concentration of 3 mg mL−1 using PBS buffer and vigorously mixed using a vortex mixer. The multilamellar vesicles thus obtained were used for binding experiments without delay.Peptide–lipid binding assay23
In the first attempt, an analytical column, Xterra RP-18, 150 mm × 4.6 mm dimension; 5μ particle size; Waters Alliance HPLC system (Waters Corporation, USA) having gradient elution capability was used for determining the free peptide. About 10 μM peptide (1.0 mL) was titrated against different amounts of POPC MLVs. After incubation and centrifugation (×g), 100 μL of the supernatant was injected into the analytical column. However, the binding kinetics could not be followed owing to the sensitivity of the instrument (limit of detection). However, a reversed phase bio-analytical column [YMC-Pack, ODS-A 100 mm × 2.0 mm dimension; 5μ particle size] was used. In the case of P1, a flow rate of 0.5 mL min−1 was set using the at gradient compositions: 0 min/10% B, 7 min/30% B, 13 min/50% B, 16 min/90% B, 25 min/90% B, and 28 min/10% B. For P2, the gradient composition was set as follows: 0 min/15% B, 7 min/30% B, 13 min/50% B, 16 min/90% B, 25 min/90% B, and 28 min/10% B. The column was equilibrated for 3 min before each analysis. The absorbance of peptide at 215 nm was measured using a Waters 2487 dual absorbance UV detector.In order to determine the relative binding affinities of the peptides for zwitterionic and anionic lipid membranes, a dilute solution of each peptide (P1 and P2) in PBS buffer was used. A constant amount of each peptide (∼10 5 μM, 1.0 3 mL) was mixed with increasing amounts of lipid vesicles (MLVs). The contents were incubated for 30 min. and centrifuged at 14000 rpm. The supernatant liquid was withdrawn and examined by HPLC using 0.1% TFA in water as mobile phase-A and 0.1% TFA in acetonitrile as mobile phase-B.
In vitro assay for evaluation of antibacterial activity
Results and discussion
Peptide design and synthesis
The peptides were prepared using the Rink amide resin support and the Fmoc-chemistry solid phase peptide synthesis.25 The central segment of LL-37 comprising residues 15–31 has a high potential to fold into the α-helix.20 Under appropriate conditions, LL-37 forms a regular amphipathic helix due to the segregation of polar and non-polar residues along the α-helix. The amphipathic nature of the helix and the hydrophobicity of the peptide facilitate the insertion of the peptide into bio-membranes and the lysis of the cell. For the purpose of designing non-hemolytic antimicrobial peptides, the segment comprising residues LL-37(5–24) was considered after omitting the hydrophobic C- and N-terminal segments.The peptide sequence thus obtained LL-37(5–24) showed an aliphatic index value of 73 and a net positive charge of 7. This sequence contains 3 arginine and 5 lysine amino acid residues. Peptides P1 and P2 were synthesized using Fmoc-chemistry protocols on Rink amide MBHA resin and the identity of the peptides was established by biophysical methods. Also, studies on the biological activity and binding affinities of P1 and P2 were carried out to explore the role of subtle variations in hydrophobicity, α-helix propensity, and ionic interactions that arise due to specific amino acid (lysine or arginine) substitution. The schematic line structure of peptides P1 and P2 is given in Fig. 1. Without altering the charge characteristics and aliphatic index,26 we modified the sequence of LL-37(5–24) to P1, in which 3 arginine residues were substituted by lysine. Similarly, P2 was made by replacing 5 lysine residues with arginine. The human antimicrobial peptide LL-37 is highly hydrophobic and cationic that it is active against a wide spectrum of Gram-positive and Gram-negative bacteria and transforms human cells as well as human erythrocytes. The hemolytic activity of LL-37 makes it not suitable for therapeutic purposes in spite of its wide spectrum activity. The toxicity of LL-37 is partly attributed to the presence of a high degree of hydrophobic moieties in the peptide. Moreover, the high cost involved in the synthesis of such a long peptide is also a factor in drug development. In this work, attempts have been made to design a potentially antimicrobial peptide that is not toxic to human erythrocytes. A reduction in the length of the peptide to reduce the cost factor is also accomplished. Two 20-residue peptides have been synthesized, allowing the direct comparison of the role of side chains in the membrane selective activity of antimicrobial peptides.
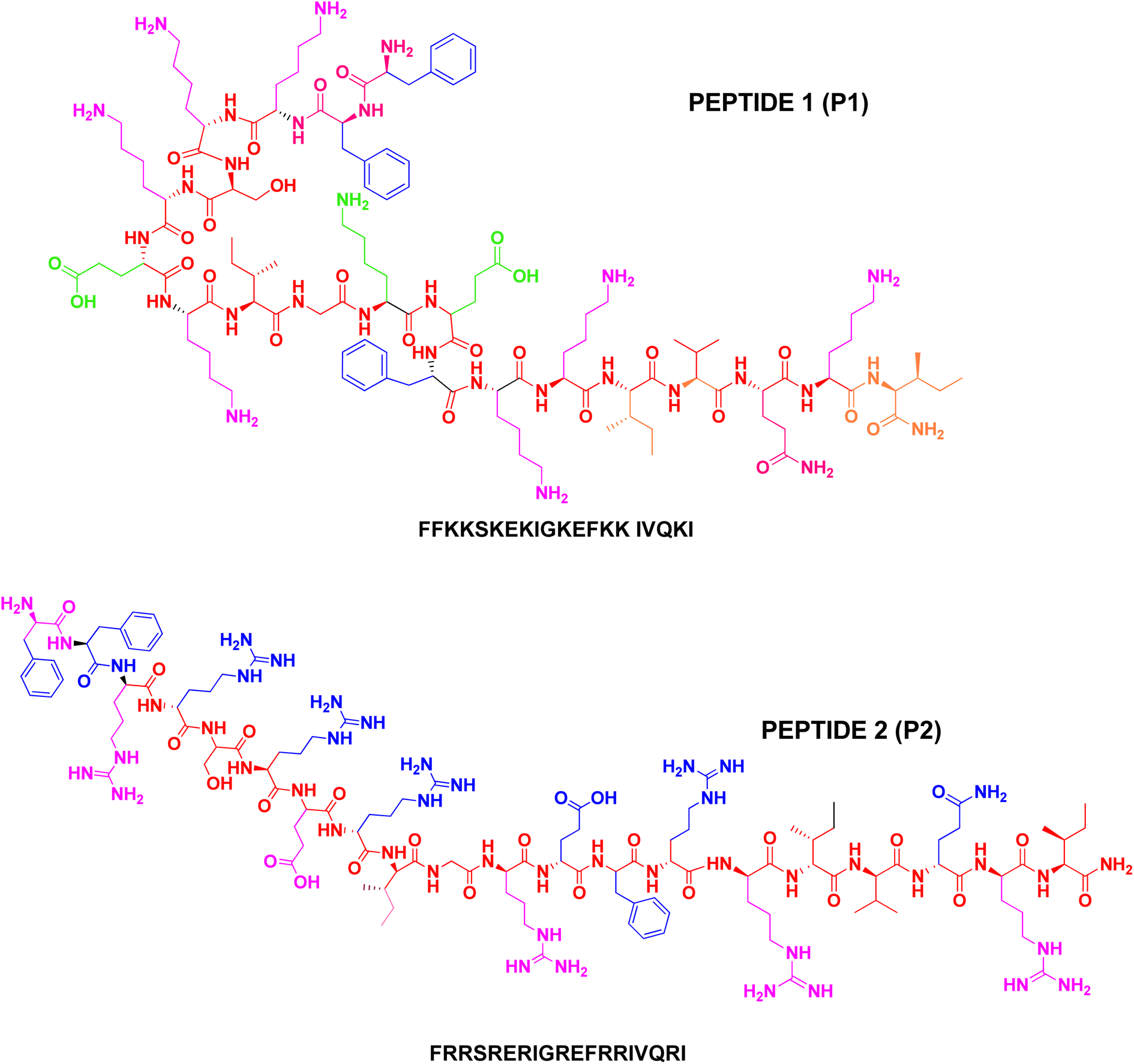 | ||
| Fig. 1 Schematic line structure of P1 and P2. | ||
Peptide hydrophobicity
RP-HPLC retention behaviour is a particularly good method to represent apparent peptide hydrophobicity, and the retention times of peptides are highly sensitive to the conformational states of peptides upon interaction with the hydrophobic environment of the column matrix. The non-polar face of an amphipathic α-helical peptide represents a preferred domain for binding the hydrophobic matrix of a reversed-phase column. Both peptides P1 and P2 have a reduced helix propensity and hydrophobicity as compared with LL-37 (Table 1). The peptides differ from each other only in the grand average hydrophobicity (GRAVY).27 In this study, the observed hydrophobicity of the peptides (as expressed by RP-HPLC retention time) is in the order P1 < P2 (Table 2). As predicted, P2 is more hydrophobic than P1. As shown in Table 2, the number of hydrophobic interactions on the non-polar face of the peptide analog appears to correlate with the observed hydrophobicity27 values of the peptides with comparable homology.| Name | Peptide sequence | Net charge | GRAVY |
|---|---|---|---|
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES-OH Aliphatic index:89.46 | 9 | −0.724 |
| LL-37(5–24) | FFRKSKEKIGKEFKRIVQRI-CONH2 Aliphatic index:73.00 | 7 | −0.930 |
| P1 | FFKKSKEKIGKEFKKIVQKI-CONH2 Aliphatic index:73.00 | 7 | −0.840 |
| P2 | FFRRSRERIGREFRRIVQRI-CONH2 Aliphatic index:73.00 | 7 | −1.080 |
| Peptidea | Hydrophobicityb | Buffercr | 80% TFEc | SDSc | |||||
|---|---|---|---|---|---|---|---|---|---|
| S. no | Denotation | tR | ΔtR(X-P1) | [θ]222 | % Helixd | [θ]222 | % Helixd | [θ]222 | % Helixd |
| a Peptide sequences are shown in Table 1.b The hydrophobicity order of peptides was deduced from observed tR values on a RP-HPLC at room temperature and at pH 2.c The mean [θ]222 values (in deg cm2 dmol−1) at wavelength 222 nm were measured at RT under benign conditions (100 mM KCl, 50 mM PO42−, pH 7.4), in benign buffer containing 50% TFE and in SDS micelles using circular dichroism spectropolarimeter.d The helical content (helix percentage) of a peptide relative to the molar ellipticity value of peptide P1/P2 in benign conditions (100 mM KCl, 50 mM PO42−, pH 7.4) in benign buffer containing 50% TFE and in SDS micelles. | |||||||||
| 1 | P1 | 12.13′ | 0.00′ | −100 | — | −7600 | 25 | −7900 | 26 |
| 2 | P2 | 15.58′ | 3.45′ | −700 | — | −7700 | 26 | −8300 | 28 |
Experimental measurement of helicity using circular dichroism of peptides has been used in conjunction with the prediction programs for the helicity of peptides. AGADIR28 is an algorithm that is based on helix-coil theory but modified to incorporate experimentally derived parameters. The algorithm attempts to obtain an energetic description of the system by splitting the conformational energy of the peptide into a sum of energies: intrinsic helical tendencies of each residue, main chain–main chain hydrogen bonding, side chain–side chain interactions, helical dipole effects, and effects of non-helical residues. The algorithm is useful for predicting the secondary structure of polypeptides consisting of less than 30 residues.
Under suitable conditions, the peptides can fold into a maximum of six helical turns that is sufficient to span the transmembrane space, in which case, the peptides would be membrane active. The low GRAVY values of the peptides may not permit the peptides to cause extensive membrane disruption and leakage of cellular contents. From Table 1, the aliphatic index of LL-37 is 89.46, and its grand average hydrophobicity (GRAVY) value is −0.724. Two peptides related to LL-37 were identified with a reduced hydrophobicity index value of 73.0. Of these two, P1 showed a lower GRAVY value of −0.840, while peptide P2 had a value of −1.080. The net positive charge 7 should be sufficient to establish lipid membrane selective binding and favour the localization of peptide at the membrane–water interface as opposed to the transmembrane orientation of LL-37. At the membrane–water interface, the peptides must be able to compromise the membrane barrier function and inhibit the growth of microorganisms. Because of the low hydrophobicity, the peptides may not bind zwitterionic cells such as the red blood cells.
Peptide homogeneity
The presence of impurities in any multi-step synthesis is a common occurrence. However, the level of impurities is drastically reduced in solid-phase step-wise synthesis. The HPLC chromatogram of the crude peptide P1 shown below is a manifestation of the advantage of solid-phase synthesis. The peptides P1 and P2 were purified to homogeneity and used for other experiments and NMR data collection. An analysis of the chromatograms shows that peptides P1 and P2 are about 69% and 71% pure, respectively. The yield and homogeneity of the peptides were checked using reversed-phase analytical HPLC methods. However, large-scale purification was done on a semi-preparative HPLC column. On both analytical and semi-preparative elution conditions (Fig. 2A–E), peptide P2 eluted later than P1, suggesting the hydrophobic nature of the arginine-rich sequence.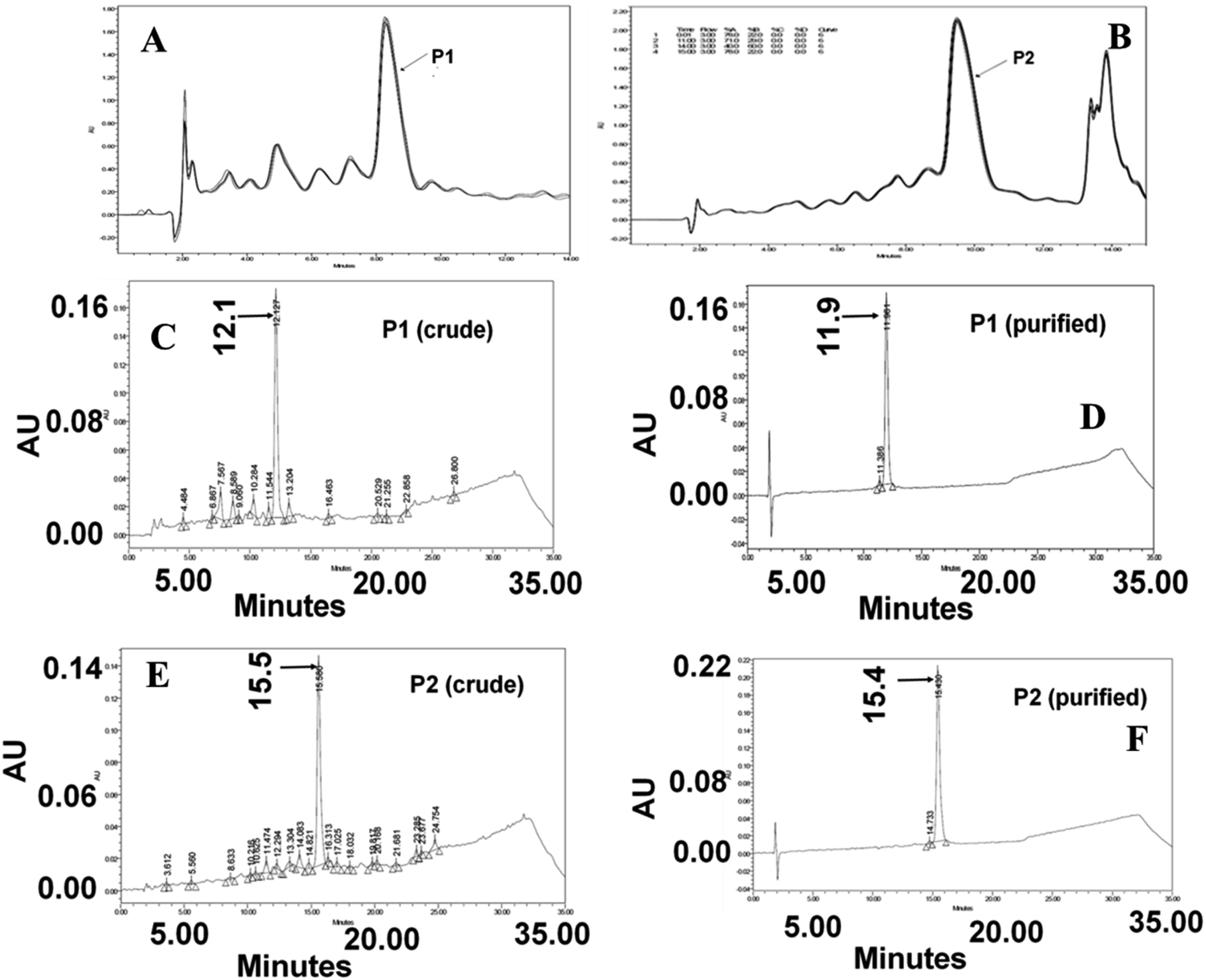 | ||
| Fig. 2 An overlay of semi preparative HPLC chromatograms. (A) Crude P1 (B) crude P2. (C and D) Analytical HPLC chromatograms of crude and purified P1. (E and F) Analytical HPLC chromatograms of crude and purified P2. | ||
Even though the peptides were collected as a single ensemble (peak), it was imperative to verify the integrity of the peptides. To alleviate the element of doubt on the integrity of the peptides along with side chain modification during synthesis and work up, ESI mass analysis was carried out at various ionising conditions. The multiple charged ions produced during ionization were analyzed to obtain the monoisotopic mass of P1 (m/z = 2451.5) and P2 (m/z = 2675.6). The calculated mass of these peptides matched very well with the observed monoisotopic mass values.29 The absence of mass peaks corresponding to more than one monoisotopic mass of any given peptide established the integrity of the peptide.
The mass of the purified peptides was determined by the ESI-Quad Mass spectrometer. Observation of molecular mass is critical to establish the structural integrity of the peptides. Mass spectrometer also provides information related to fragment ions, the ability to form adducts with ligands and metal ions, as well as peptide aggregation (dimer, trimer etc.). The ESI mass of spectra of peptides P1 and P2 are presented in Fig. S1 and S2,† respectively.
Secondary structures of peptides
Circular dichroism is a convenient technique to study the conformational transitions of proteins and peptides in media of different polarities. The CD spectra of P1 and P2 in buffer pH 7.0, SDS micelles, and TFE are given in Fig. 3. The spectrum in an aqueous solution shows the peptide in a random coil structure, having a weak maximum at ∼217 nm and a strong minimum at ∼198 nm. The addition of the structure-inducing TFE or SDS micelles induced a shift toward helical conformation as the spectra had a minimum at 222 nm (helical ππ* transition) and a second minimum between 206 and 208 nm (overlapping helical and random coil ππ* transition at 208 nm and 200 nm), respectively.30–32 CD data were also analyzed using two parameters, R1 and R2, which are independent of inaccuracies in the determined peptide concentration as well as those caused by small shifts in wavelength.34 Here, R1 is the ratio of the intensity of the maximum between 190 and 195 nm and the intensity of the minimum between 200 and 210 nm, and R2 is the ratio of the intensity of the minimum near 222 nm and the intensity of minimum between 200 and 210 nm. For a random structure, R1 is positive, and R2 is close to zero. On the other hand, in a highly helical state, R1 will be close to −2, and R2 will approach the value 1.0.33,34 These parameters calculated for P1 in buffer, TFE, and SDS show an induction of the helical structure on the addition of TFE (R1 = −1.7, R2 = 0.61) and SDS (R1 = −1.9, R2 = 0.69). The parameters calculated for P2 in the buffer, TFE, and SDS show an induction of the helical structure on the addition of TFE (R1 = −0.91, R2 = 0.62) and SDS (R1 = −0.85, R2 = 0.67). The spectra showing the transition from a random coil (buffer) to α-helix (in SDS micelles or TFE) are presented in Fig. 3 as a function of mean residue ellipticity vs. wavelength.35 Regardless of the difference in secondary structures of the peptides in the buffer, a highly helical structure could be induced by the non-polar environment of TFE. TFE is known to promote and stabilize the α-helix structure of polypeptides. The excellent hydrophobic environment provided by TFE in aqueous mixtures is often compared to the hydrophobic environment of bio-membranes as well as lipid membranes. Thus, the observed folding behaviour of peptides in TFE and SDS micelles is likely to be reflected in the membrane environment where only the folded peptides are expected to bind and elicit the activity on the membranes.36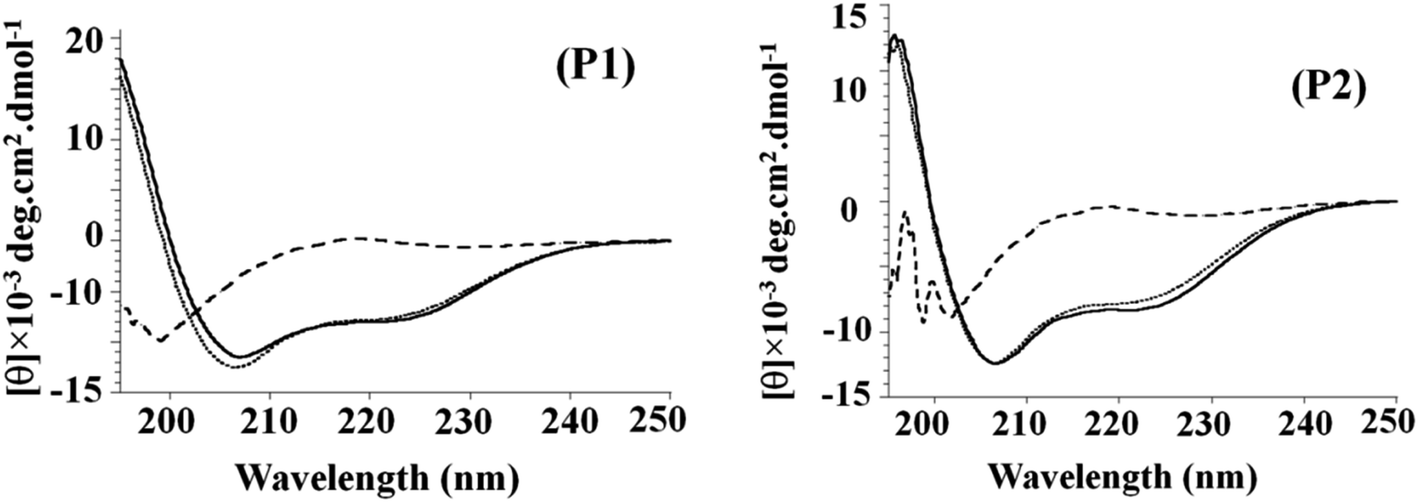 | ||
| Fig. 3 CD spectra of P1 and P2 in buffer (dashed line), TFE (dotted line) and SDS (solid line). | ||
Binding affinity of peptides for zwitterionic and anionic lipid membranes
Almost all spectroscopic and separation methods have been used for the determination of the binding affinities of a ligand to a given substrate. HPLC is an excellent method for the quantitative determination of residual ligands in a binding assay.37 Using the YMC-Pack bio-analytical column and appropriate elution conditions, the amount of free peptide isolated from the peptide-MLV mixture could be determined quantitatively. The amount of peptide bound to MLVs was calculated indirectly. Both peptides P1 and P2 bind only marginally to MLVs made of zwitterionic POPC lipids, as seen in Fig. 4. However, they bind significantly to POPC-POPG (7:3) MLVs (Fig. 4). This remarkable enhancement in binding can only be attributed to the presence of anionic POPG lipids in the MLVs. The charged interaction between the anionic phosphate group of POPG and the cationic amino group at biological pH appears to play a determining role in the membrane selective binding of P1 and P2. A quantitative description of the binding characteristics of P1 and P2 with regard to the anionic POPC-POPG (7:3) membrane is presented in Fig. 5. Interestingly, the arginine-rich P2 appears to be more hydrophobic and anionic membrane selective than the lysine-rich P1 (Fig. 4 and 5). The binding profiles of each peptide with POPC as well as POPC-POPG (7:3) MLVs are presented, and the decrease in the area of the peptide.s as a function of lipid concentration is presented in Fig. 4 and 5. Considering the very weak interaction of P2 with zwitterionic POPC vesicles and its very strong association with anionic POPC/POPG (7:3) lipid vesicles, P2 could be a promising candidate for the future designing of cell-selective antimicrobial peptides. It is apparent from the binding data that both peptides show little preference for zwitterionic POPC membrane. As shown in Fig. 5, the affinity for anionic membrane turns out to be higher in the case of P2 as compared with peptide P1. However, when a reversed-phase bio-analytical column [5μ particle size] was used, the eluted unbound peptides isolated from peptide-MLVs mixtures showed sharp peaks (Fig. S6†), facilitating the binding analysis.
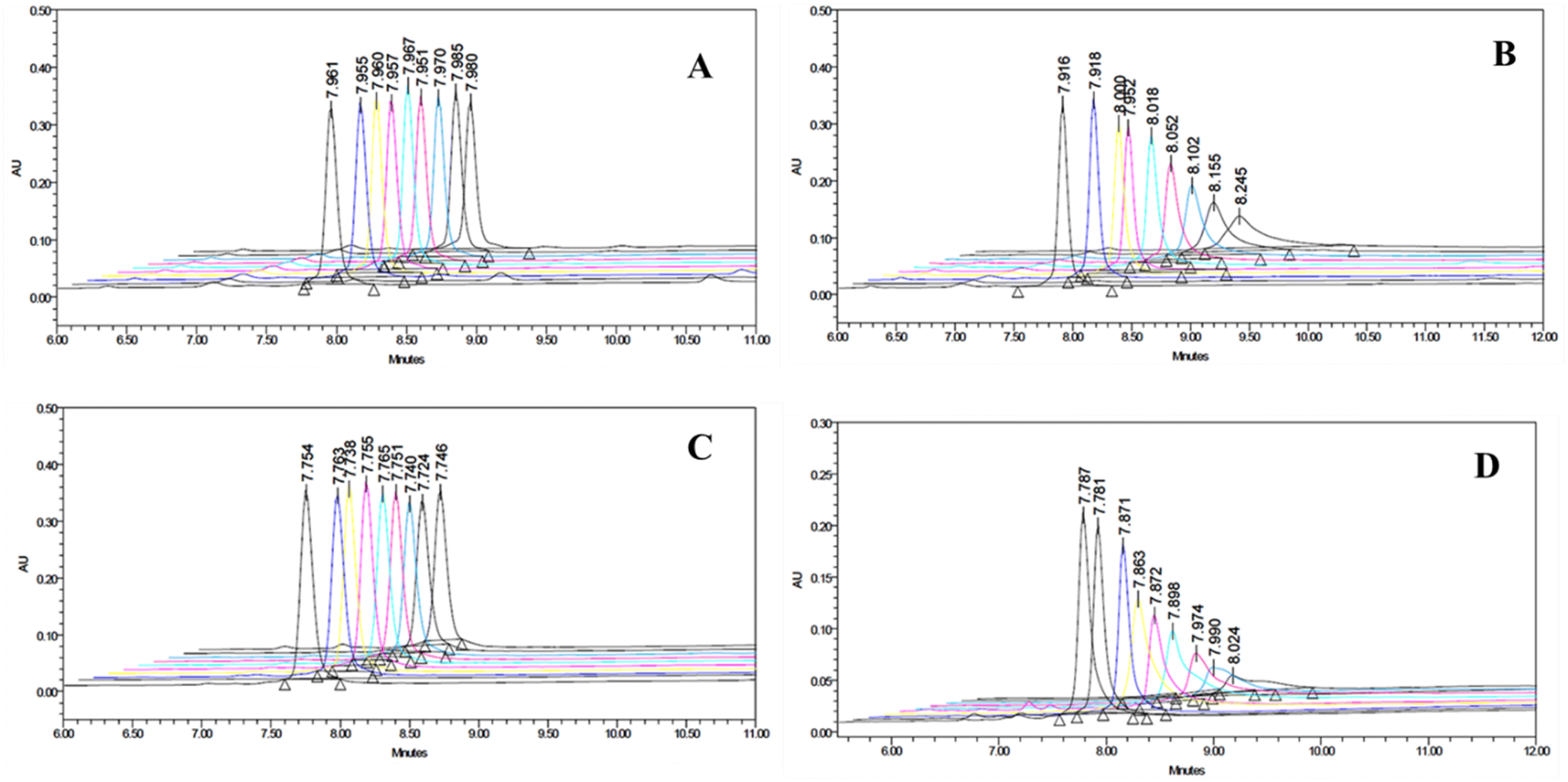 | ||
| Fig. 4 Overlay of HPLC chromatograms. (A) P1 as a function of lipid concentration (POPC MLVs). (B) P1 as a function of lipid concentration [POPC-POPG (7:3) MLVs]. (C) P2 as a function of lipid concentration (POPC MLVs). (D) P2 as a function of lipid concentration [POPC-POPG (7:3) MLVs]. Note: from right to left (A) free peptide eluted retention time = 7.980 min, (C) free peptide eluted retention time = 7.740 min. | ||
 | ||
| Fig. 5 Plot of area under the curve showing the relative binding affinities of the peptides. (A) Free P1 concentration versus average lipid concentration of MLVs. (B) Free P2 concentration versus average lipid concentration of MLVs. (C) Bound peptide (P1 and P2) versus average lipid concentration of MLVs. | ||
Peptide P1 and P2 exhibit a broad spectrum of antimicrobial activities
The antimicrobial activity of the control LL-37(5–24) and synthetic peptides P1 and P2 was tested against four test organisms following standard protocols. The zone of inhibition of the peptides indicates that P1 and P2 are active against both Gram-positive and Gram-negative bacteria. It was also stated that the cationic and amphipathic properties of antimicrobial peptides contribute to their antibacterial activity. Between 12 to 100 amino acids in length, they have a wide range of structural variations, and they have the ability to attach and damage microbial membranes. They can prevent the biosynthesis of cell walls, nucleic acids, and proteins.38 Moreover, control peptide LL-37(5–24) has a higher zone of inhibition and lower MIC values than P1 and P2 against Gram-positive, i.e., B. subtilis (10 μg mL−1), S. aureus (0.6 μg mL−1), and Gram-negative, namely E. Coli (0.8 μg mL−1), and P. aeruginosa (25 μg mL−1), bacteria. Results in the similar line against the tested bacterial strain were also reported in a previous study by Thennarasu et al.39 Both the peptides P1 and P2 have inhibited the growth of all the tested bacterial strains, but they have higher MIC values in comparison to the control parent peptide LL37(5–24). For S. aureus and E. coli, P1 has upright antibacterial activity with MIC 25 μg mL−1 and 20 μg mL−1, respectively. Likewise, against B. subtilis and P. aeruginosa, P2 had higher antibacterial activity with MIC 30 μg mL−1. The results are shown in Fig. 6 and Table S1.† | ||
| Fig. 6 Antimicrobial activity of peptide 1 (P1), peptide 2 (P2), and control = LL37(5–24). MIC (minimum inhibitory concentration). | ||
The slightly lower activity of synthetic peptides could be ascribed to the shorter length of the peptides that promote solubility and thereby retard membrane binding. Since the peptides are relatively hydrophilic, lipid-induced peptide aggregation could not be considered as a probable cause for the slightly reduced activity of peptides P1 and P2. Another possible contributing factor is the high degree of cationic content in a short stretch of peptide that could favour a tight binding of the peptide to the bacterial anionic membrane.21 As a result, more than the required number of peptide molecules bind each cell wall, and the free peptide concentration required to elicit activity on other remaining bacterial cells is dramatically reduced. Moreover, peptide aggregation on the membrane surface can lead to the formation of a peptide-rich domain. Such membrane domains may fall out of the membrane as it happens during the endocytosis of cell membranes. One of the two forms of free peptides isolated (Fig. 4D) from the mixture of POPC-POPG (7:3) MLVs and P2 could be the peptide–lipid complex fallen out of MLVs. This could form the basis for the observed antimicrobial activity of P2.
Structured P1 and P2 selectively bind anionic membranes
The difference in the membrane composition of bacterial and mammalian cell membranes has been thought to play a major role in the selectivity of antimicrobial peptides. For example, the presence of anionic lipids in bacterial membranes enables the selective interaction of cationic AMPs.37 For this study, the role of anionic lipid on the induction of secondary structures in P1 and P2 was studied using circular dichroism spectroscopy. Both P1 and P2 adopt a random coil structure in an aqueous buffer at biological pH. However, they fold into a regular α-helix in the helix-promoting solvent TFE (Fig. 3). This shows the intrinsic ability of the peptide to form the helix structure on the membrane surface. TFE is considered as a solvent to mimic the lipid membrane environment. Interestingly, both peptides form α-helix structures in an anionic micellar medium. This observation reinforces the notion that cationic antimicrobial peptides fold into α-helix structures when they come in contact with anionic lipid membranes such as the bacterial membrane.The formation of the α-helix structure was further confirmed by 1H NMR and NOESY methods (Fig. S3–S5†). In an aqueous buffer at pH 7.0, both peptides P1 and P2 are unordered coils, as seen from the cluster of amide NH proton resonance peaks (Fig. S3 and S4†). However, the spread of amide NH proton resonance peaks observed in the presence of anionic micelles suggests the induction of secondary structures, presumably, α-helix formation. The formation of helix structure is also seen in the NOESY spectra (Fig. S5†) of peptides in SLS-d26. Even though the assignment of NOESY was proved to be difficult due to the presence of a large number of lysine or arginine residues that impeded the unambiguous assignment of cross peaks, the presence of a substantial number of NH–NH connectivity (Fig. S5†) supports the conclusions drawn based on CD and 1H NMR studies. Together, these results suggest that the electrostatic interactions between the cationic peptides (P1 and P2) and anionic bacterial membranes play a very critical role in cell-selective activity and the absence of hemolytic activity. This study confirms our hypothesis that α-helix structure is not a hallmark of antimicrobial activity.35 NOESY spectrum with a mixing time of 300 ms was recorded for obtaining the NH–NH cross peaks. As seen in Fig. S3–S5,† the absence of NH–NH cross peaks in the buffer suggests a random conformation while the spectra of both peptides in SDS micelles indicate the formation a regular helix.40
Carpet-type mechanism of membrane disruption by P1 and P2
Based on the results of the present investigation, the possibility of a transmembrane orientation41 of membrane-bound peptide and any barrel-stave42 like ion-channel mechanism of membrane disruption cannot be ruled out. However, the two forms of free peptides (Fig. 4) clearly suggest that P2 and possibly P1 should have strong interactions with anionic lipids in the bilayer. Such strong binding of peptides often results in the formation of membrane domains.21,37 Membrane domain formation is one of the steps in the proposed carpet mechanism of membrane disruption. Therefore, peptides P1 and P2 are also likely to function via a carpet-type43 mechanism as in the case of the peptide LL-37(7–27) that may lead to either toroidal-pore formation and/or micellization of the membrane at higher concentrations of the peptide. Both peptides were considered non-hemolytic as neither a significant amount of absorbance at 540 nm nor release of hemoglobin (naked eye detection) was observed. Only a small value of absorbance (5–8%) with respect to 100% hemolysis was observed from individual samples. This basal level absorbance could be due to residual blood plasma and RBC membrane proteins present in test samples.Conclusion
Using a 20-residue membrane active fragment and refining the sequence, two non-hemolytic (not active on mammalian cells) antimicrobial peptides were designed. Both peptides P1 and P2 were non-hemolytic and appeared to be more promising as it displayed wide spectrum antimicrobial activity. Mass spectrometry confirmed that the calculated molecular weight of the peptide is correlated with the observed molecular weight. The RP-HPLC chromatograms reveal the purity of peptides P1 and P2. The binding affinity of the peptides for zwitterionic and acidic lipid membranes was determined using HPLC-based binding assays. Both P1 and P2 showed about 20% binding as inferred from the amount of free peptide isolated from the peptide–lipid complex. This binding may be ascribed to the non-specific binding of peptides to lipid vesicles or a limitation of the binding assay itself. Interestingly, the binding profiles of P1 and P2 to anionic lipid vesicles clearly established the membrane-selective binding of these cationic peptides. A plot of bound peptide versus anionic lipid concentration allowed a direct comparison of the binding affinity of P1 and P2 for anionic membranes and explains the bacterial membrane selective activity of these peptides. The exceptional affinity for POPC/POPG membrane shown by P2 signified the role of guanidine moiety membrane selective activity.The conformational preferences of the peptides in media of different polarities were studied using circular dichroism spectroscopy to assess the conformational transitions upon interaction with membranes. Predictably, both peptides showed a completely random structure in an aqueous buffer and a regularly folded α-helix in TFE and SDS micelles. This observation was further confirmed by 2D NMR methods. Amphipathic α-helical cationic peptides are known to bind bacterial membranes with a certain degree of specificity and induce membrane permeabilization. The membrane permeabilization process is thought to involve the formation of membrane defects and/or pore structures that allow the leakage of cytoplasmic contents. Most of the antimicrobial peptides that form transmembrane pores on bacterial membranes also form pore structures on mammalian cell membranes and behave like lytic peptides. The results obtained in this study suggest that peptides P1 and P2 are non-lytic, and therefore, could be considered for therapeutic purposes.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors heartily thank Dr S. Thennarasu, Chief Scientist, CSIR-CLRI, Adyar, Chennai, Tamilnadu, India, for providing research support. There has been no external research funding for carrying out this work.References
- M. Zanetti, R. Gennaro and R. Circo, Cathelicidin peptides as candidates for a novel class of antimicrobials, Curr. Pharm. Des., 2002, 8, 779–793 CrossRef CAS PubMed.
- M. G. Scott, D. J. Davidson and R. E. Hancock, The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses, J. Immunol., 2002, 169, 3883–3891 CrossRef CAS PubMed.
- U. H. N. Durr, U. S. Sudheendra and A. Ramamoorthy, LL-37, the only human member of the cathelicidin family of antimicrobial peptides, Biochim. Biophys. Acta, 2006, 1758, 1408–1425 CrossRef PubMed.
- A. Nijnik and R. E. W. Hancock, The roles of cathelicidin LL-37 in immune defences and novel clinical applications, Curr. Opin. Hematol., 2009, 16, 41–47 CrossRef CAS PubMed.
- N. Mookherjee and R. E. W. Hancock, Cationic host defence peptides: innate immune regulatory peptides as a novel approach for treating infections, Cell. Mol. Life Sci., 2007, 64, 922–933 CrossRef CAS PubMed.
- Y. Rosenfeld, N. Papo and Y. Shai, Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. Peptide properties and plausible modes of action, J. Biol. Chem., 2005, 281, 1636–1643 CrossRef PubMed.
- Z. Oren, J. C. Lerman and Y. Shai, Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity, Biochem. J., 1999, 341, 501–513 CrossRef CAS PubMed.
- A. Ramamoorthy, D. K. Lee and K. A. Henzler-Wildman, Nitrogen-14 solid-state NMR spectroscopy of aligned phospholipid bilayers to probe peptide-lipid interaction and oligomerization of membrane associated peptides, J. Am. Chem. Soc., 2008, 130, 11023–11029 CrossRef CAS PubMed.
- J. Y. Moon, K. A. Henzler-Wildman and A. Ramamoorthy, Expression and purification of a recombinant LL-37 from Escherichia coli, Biochim. Biophys. Acta, 2006, 1758, 1351–1358 CrossRef CAS PubMed.
- Y. Li, X. Li, H. Li, O. Lockridge and G. A. Wang, novel method for purifying recombinant human host defensecathelicidin LL-37 by utilizing its inherent property of aggregation, Protein Expr. Purif., 2007, 54, 157–165 CrossRef CAS PubMed.
- F. Porcelli, R. Verardi and G. Veglia, NMR structure of the cathelicidin- derived human antimicrobial peptide LL-37 in dodecylphosphocholinemicelles, Biochemistry, 2008, 47, 5565–5572 CrossRef CAS PubMed.
- G. Wang, Structures of human host defensecathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles, J. Biol. Chem., 2008, 283, 32637–32643 CrossRef CAS PubMed.
- K. A. Henzler Wildman, D. K. Lee and A. Ramamoorthy, Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37, Biochemistry, 2003, 42, 6545–6558 CrossRef CAS PubMed.
- K. A. Henzler-Wildman, G. V. Martinez and A. Ramamoorthy, Perturbation of the hydrophobic core of lipid bilayers by the human antimicrobial peptide LL-37, Biochemistry, 2004, 43, 8459–8469 CrossRef CAS PubMed.
- F. Neville, M. Cahuzac and D. Gidalevitz, The interaction of antimicrobial peptide LL-37 with artificial biomembranes: epifluorescence and impedance spectroscopy approach, J. Phys. Condens. Matter., 2004, 16, S2413–S2420 CrossRef CAS.
- F. Neville, M. Cahuzac and D. Gidalevitz, Lipid head group discrimination by antimicrobial peptide LL-37: insight into mechanism of action, Biophys. J., 2006, 90, 1275–1287 CrossRef CAS PubMed.
- E. Sevcsik, G. Pabst and K. Lohner, Interaction of LL-37 with model membrane systems of different complexity: influence of the lipid matrix, Biophys. J., 2008, 94, 4688–4699 CrossRef CAS PubMed.
- R. Sood and P. K. Kinnunen, Cholesterol, lanosterol, and ergosterol attenuate the membrane association of LL-37(W27F) and temporinL, Biochim. Biophys. Acta, 2008, 1778, 1460–1466 CrossRef CAS PubMed.
- R. Sood, Y. Domanov and P. K. Kinnunen, Binding of LL-37 to model biomembranes: insight into target vs host cell recognition, Biochim. Biophys. Acta, 2008, 1778, 983–996 CrossRef CAS PubMed.
- X. Li, Y. Li and G. Wang, Solution structures of human LL-37 fragments and NMR-based identification of a minimal membranetargeting antimicrobial and anticancer region, J. Am. Chem. Soc., 2006, 128, 5776–5785 CrossRef CAS PubMed.
- S. Thennarasu, A. Tan, R. Penumatchu, C. E. Shelburne, D. L. Heyl and A. Ramamoorthy, Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide LL37, Biophys. J., 2010, 98, 248–257 CrossRef CAS PubMed.
- A. Ramamoorthy, S. Thennarasu, D.-K. Lee, A. Tan and M. Lee, Solid-State NMR Investigation of the Membrane-Disrupting Mechanism of Antimicrobial Peptides MSI-78 and MSI-594 Derived from Magainin 2 and Melittin, Biophys. J., 2006, 91(1), 206–216, DOI:10.1529/biophysj.105.073890.
- G. Saberwal and R. Nagaraj, Interaction of hydrophobic peptides with model membranes: slow binding to membranes and not subtle variations in pore structure is responsible for the gradual release of entrapped solutes, Biochim. Biophys. Acta, Biomembr., 1993, 1151, 43–50 CrossRef CAS PubMed.
- P. Adhikari, A. Pandey, V. Agnihotri and V. Pandey, Selection of solvent and extraction method for determination of antimicrobial potential of Taxus wallichiana Zucc., Res. Pharm., 2018, 8, 1–9 Search PubMed.
- C. A. Roque-Borda, B. A. Antunes, A. B. Toledo Borgues, J. T. Costa de Pontes, A. B. Meneguin, M. Chorilli, E. Trovatti, S. R. Teixeira, F. R. Pavan and E. F. Vicente, Conjugation of Ctx(Ile21)-Ha Antimicrobial Peptides to Chitosan Ultrathin Films by N-Acetylcysteine Improves Peptide Physicochemical Properties and Enhances Biological Activity, ACS Omega, 2022, 7, 28238–28247, DOI:10.1021/acsomega.2c02570.
- A. J. Ikai, Thermostability and aliphatic index of globular proteins, J. Biochem., 1980, 88, 1895–1898 CAS.
- J. Kyte and R. F. Doolittle, A simple method for displaying the hydropathic character of a protein, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS PubMed.
- E. Lacroix, A. R. Viguera and L. Serrano, Elucidating the folding problem of α-helices: local motifs, long-range electrostatics, ionic-strength dependence and prediction of NMR parameters, J. Mol. Biol., 1998, 284, 173–191 CrossRef CAS PubMed.
- S. J. Gaskell, Electrospray: Principles and Practice, J. Mass Spectrom., 1997, 32, 677–688 CrossRef CAS.
- G. Hojzwarth and P. Doty, The ultraviolet circular dichroism of polypeptides, J. Am. Chem. Soc., 1965, 87, 218–228 CrossRef PubMed.
- A. J. Alder, N. J. Greenfield and G. D. Fasman, Circular dichroism and optical rotatory dispersion of proteins and polypeptides, Methods Enzymol., 1973, 27, 675–735 Search PubMed.
- T. C. Chang, C. S. Wu and J. T. Yung, Circular dichroic analysis of protein conformation: inclusion of the beta-turns, Anal. Biochem., 1978, 91, 13–31 CrossRef PubMed.
- M. D. Bruch, M. M. Dhingra and L. M. Gierasch, Side Chain–Backbone Hydrogen Bonding Contributes to Helix Stability in Peptides Derived from an α–Helical Region of Carboxypeptidase A, Proteins Struct. Funct. Genet., 1991, 10, 130–139 CrossRef CAS PubMed.
- J. Rizo, F. J. Blanco, B. Kobe, M. D. Bruch and L. M. Gierasch, Conformational Behavior of E. coli OmpA Signal Peptides in Membrane Mimetic Environments, Biochemistry, 1993, 32, 4881–4894 CrossRef CAS PubMed.
- S. Thennarasu and R. Nagaraj, Solution conformations of peptides representing the sequence of the toxin pardaxin and analogues in trifluoroethanol–water mixtures: analysis of CD spectra, Biopolymers, 1997, 41, 635–645 CrossRef CAS PubMed.
- E. F. Vicente, L. G. M. Basso and E. Crusca Junior, et al., Biophysical Studies of TOAC Analogs of the Ctx(Ile21)-Ha Antimicrobial Peptide Using Liposomes, Braz. J. Phys., 2022, 52, 71, DOI:10.1007/s13538-022-01077-9.
- A. Ramamoorthy, S. Thennarasu, A. Tan, K. Gottipati, S. Sreekumar, D. L. Heyl, F. Y. An and C. E. Shelburne, Deletion of all cysteines in tachyplesin I abolishes hemolytic activity and retains antimicrobial activity and lipopolysaccharide selective binding, Biochemistry, 2006, 45, 6529–6540 CrossRef CAS PubMed.
- A. J. Duplantier and M. L. Van Hoek, The human cathelicidin antimicrobial peptide LL-37 as a potential treatment for polymicrobial infected wounds, Front. Immunol., 2013, 4, 1–14 CAS.
- S. Thennarasu, A. Tan, R. Penumatchu, C. E. Shelburne, D. L. Heyl and A. Ramamoorthy, Antimicrobial and Membrane Disrupting Activities of a Peptide Derived from the Human Cathelicidin Antimicrobial Peptide LL37, Biophys. J., 2010, 98, 248–257 CrossRef CAS PubMed.
- S. Thennarasu, R. Huang, D.-K. Lee, P. Tang, L. Maloy, Z. Chen and R. Ramamoorthy, Limiting an Antimicrobial Peptide to the Lipid–Water Interface Enhances Its Bacterial Membrane Selectivity: A Case Study of MSI-367, Biochemistry, 2010, 49, 10595–10605 CrossRef CAS PubMed.
- C. C. Lee, Y. Sun, S. Qian and H. W. Huang, Transmembrane pores formed by human antimicrobial peptide LL-37, Biophys. J., 2011, 100, 1688–1696 CrossRef CAS PubMed.
- L. Yang, T. A. Harroun, T. M. Weiss, L. Ding and H. W. Huang, Barrel-stave model or toroidal model? A case study on melittin pores, Biophys. J., 2001, 81, 1475–1485 CrossRef CAS PubMed.
- Y. Shai and Z. Oren, From "carpet" mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides, Peptides, 2001, 22, 1629–1641 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02473c |
| This journal is © The Royal Society of Chemistry 2023 |