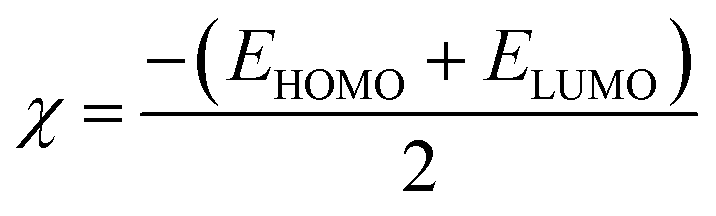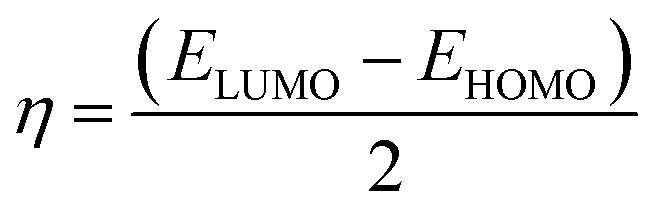DOI:
10.1039/D3RA02393A
(Paper)
RSC Adv., 2023,
13, 14580-14593
Antioxidant activity of novel nitrogen scaffold with docking investigation and correlation of DFT stimulation†
Received
11th April 2023
, Accepted 2nd May 2023
First published on 15th May 2023
Abstract
Heterocyclic scaffolds are frequently employed in drug development to treat a variety of conditions, including cancers. These substances have the ability to engage covalently or non-covalently with particular residues in the target proteins, inhibiting them. In this study, the formation of N-, S-, and O-containing heterocycles by the interaction of chalcone with nitrogen-containing nucleophiles such as hydrazine, hydroxyl amine, guanidine, urea, and aminothiourea was explored. FT-IR, UV-visible, NMR, and mass spectrometric studies were used to confirm the heterocyclic compounds that were produced. These substances were tested for their antioxidant activity by their capacity to scavenge the artificial radicals 2,2-diphenyl-1-picrylhydrazyl (DPPH). The strongest antioxidant activity was demonstrated by compound 3 (IC50 = 93.4 μM), whereas compound 8 (IC50 = 448.70 μM) had the lowest activity when compared to vitamin C (IC50 141.9 μM). Also, the experimental findings and the docking estimation of these heterocyclic compounds with PDBID:3RP8 were in agreement. Additionally, the compounds' global reactivity characteristics, such as HOMO–LUMO gaps, electronic hardness, chemical potential, electrophilicity index, and Mulliken charges, were identified using DFT/B3LYP/6-31G(d,p) basis sets. The two chemicals that displayed the best antioxidant activity also had their molecular electrostatic potential (MEP) ascertained using DFT simulations.
1. Introduction
Heterocycles are extremely important in organic chemistry because they have so many uses in the biological, chemical, and medicinal sciences.1–4 Organic heterocycle refers to cyclic organic compounds containing one or more hetero-atoms, including nitrogen, oxygen, sulfur, etc.5–8 Different heterocyclic substances are essential to nature, medicine, and innovation. Moreover, these ring structures serve as the fundamental structural elements of a large number of biological molecules, including haemoglobin, DNA, RNA, vitamins, hormones, etc. Many medications that the FDA has approved for the treatment of various disorders also contain this structure.9–12 A number of N-containing heterocycles with distinct properties and uses have recently gained popularity in the fields of chemical, pharmaceutical, and medical chemistry.13–16 The heterocyclic nitrogen compounds such as pyrazole, pyrimidine, pyrazoline, and isoxazoline have shown high biological evaluation as Epirizole used as anti-inflammatory drug,17,18 Dasatinib used to treat leukemia, 18and Trimethoprim used as antibiotics.19 Pyrazoline serves as a vital backbone in the development of new anticancer treatment agents, with several already being utilized clinically and others in ongoing research studies. By combining pyrazoline with alternative heterocyclic structures, there is potential to decrease adverse effects and combat drug resistance, making it an essential component in the design of efficient and secure cancer treatments.20 Moreover, pyridines are important building blocks in pharmaceuticals due to their analgesic, anti-inflammatory, antitumor, and antiviral properties.21
Likewise, the O-containing heterocycles display a variety of pharmacological properties, including antibacterial, anti-HIV, anti-malaria, anti-cancer, anti-tubercular, and diabetic properties.22–26 The iso-oxazolines exhibit antibacterial, antifungal, anticancer, and anti-inflammatory properties.27 Furthermore, S-containing heterocyclic compounds are used in dyes, medicines, and agrochemicals as well as being key components of primary (cystine and methionine amino acids) and secondary metabolites (biotin and thiamine).28,29 Benzothiazepines are recognized for their impact on cardiovascular functioning by acting as calcium channel inhibitors, and they have also been found to exhibit anticonvulsant and anticancer properties.30 Antioxidants, either enzymatic or non-enzymatic, can prevent or reduce cellular damage caused by free radicals, unstable molecules generated by the body as a response to environmental or other pressures.31,32 The radical scavengers are molecules that play an important role in chemistry, biology, and material science. For example, they are used in food storage, pharmaceuticals, cosmetics, petroleum products, oil, rubber, and electronic device application.33–37 Recently, it has been increasingly important to develop new radical scavengers with applications for industry and pharmaceuticals.38–40 Microwave (MW) irradiation is a very useful tool in organic synthesis, where the required reaction times for the formation of the target compounds in synthetic organic chemistry are shorter compared to the conventional heating method.41
In this elucidation we synthesized different N-, S-, and O-containing heterocycles from the 2′-hydroxychalcone compound and analyzed them through spectral analysis such as FT-IR, mass, and NMR. The synthesized compounds were examined through scavenger radical inhibition as antioxidant activity using DPPH. Docking analysis of these compounds were evaluated through PDBID:3RP8 and it was found that they exhibited a strong correlation interaction with hydrogen bond interaction. Furthermore, the DFT analysis of these heterocyclic compounds showed computability of their physical descriptors and its HOMO–LUMO analysis.
2. Results and discussion
2.1. Chemistry
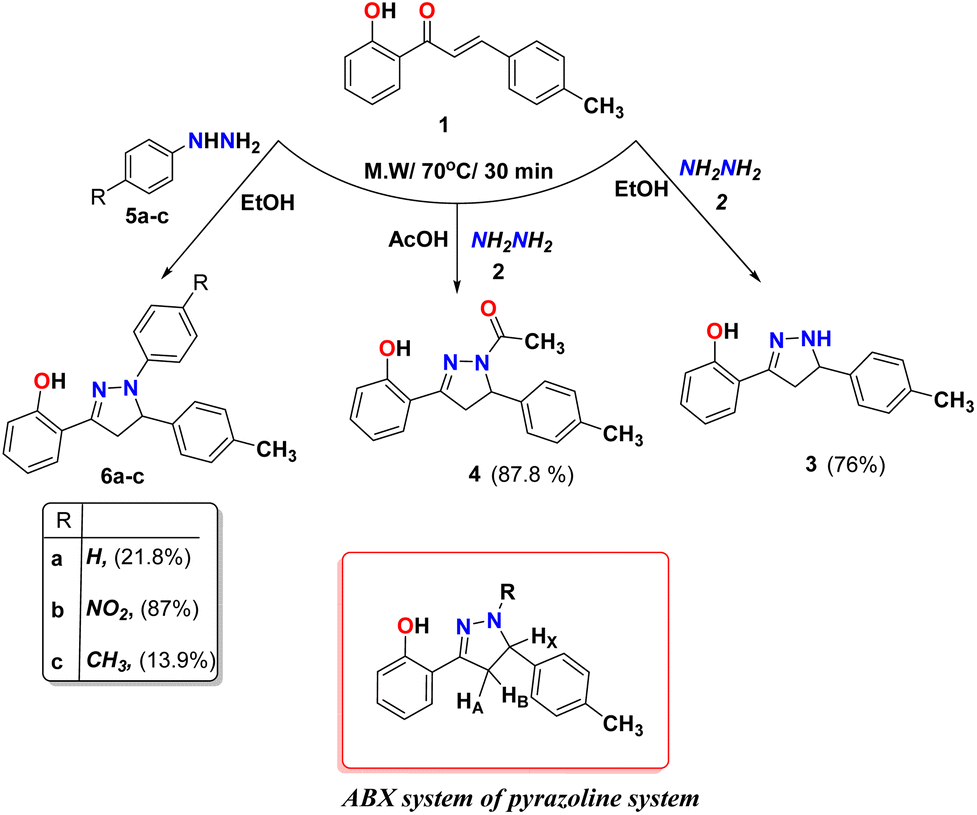
We examined the reactivity of chalcone 1 towards aza-Michael addition reactions using different nitrogen-attacking nucleophiles (Michael donors) such as hydrazine hydrate, phenyl hydrazine derivatives, hydroxylamine, guanidine, urea, thiourea, and heterocyclic amines to form different heterocyclic active compounds. Different pyrazolines 3 and 4 were formed under microwave radiation from the reaction of chalcone 1 with hydrazine hydrate 2 in different solvents such as ethanol and acetic acid as the reported method42,43 (Scheme 1). The IR spectrum of compound 3 showed characteristic stretching bands at 3339, 3006, and 1619 cm−1 due to NH, aromatic C–H, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N groups, respectively. Whereas compound 4, the IR spectrum showed bands at 3032, 1615, and 1648 cm−1 due to aromatic C–H, and CN, and CO group, respectively. The two compounds 3 and 4 were supported the ABX system of pyrazoline ring protons and showed in 1H-NMR at δ 2.94 and 3.21 ppm for HA resonated as a pair of doublets of doublets, while for HB appeared at δ 3.56 and 3.90 ppm as a pair of doublets of doublets, respectively. The HX in the pyrazoline ring for compounds 3 and 4 appeared as a doublet of triplets or doublet of doublets at δ 4.79 and 5.44 ppm, respectively. The D2O exchangeable singlet peak of OH appeared at 12.38 and 10.20 ppm for compounds 3 and 4, respectively, while the NH proton for compound 3 appeared at 7.80 ppm. All the other aromatic protons were observed with their expected chemical shifts. Moreover, the formations of pyrazolines 6a–c were synthesized via condensation reaction of chalcone 1 with different phenyl hydrazine derivatives 5a–c as displayed in Scheme 1. The IR spectrum of pyrazoline rings of 6a–c showed a characteristic bands at 1616, 1620, and 1615 cm−1 due to CN group, also the 1HNMR of ABX structure of pyrazoline ring protons for compounds 6a–c. The signals for compounds 6a–c appeared, respectively, at δ 3.21, 3.33, and 3.19 ppm for HA resonated as a pair of doublets of doublets, while for HB appeared as a pair of doublets of doublets at δ 4.01, 4.08, and 3.98 ppm, respectively. Also, The HX in the pyrazoline ring for compounds 6a–c appeared as a doublet of doublets at δ 5.40, 5.65, and 5.35 ppm, respectively. The D2O exchangeable singlet peak of OH appeared at 10.55, 10.22 and 10.58 ppm for compounds 6a–c, respectively. All the remaining aromatic protons were observed with their expected chemical shifts.
N groups, respectively. Whereas compound 4, the IR spectrum showed bands at 3032, 1615, and 1648 cm−1 due to aromatic C–H, and CN, and CO group, respectively. The two compounds 3 and 4 were supported the ABX system of pyrazoline ring protons and showed in 1H-NMR at δ 2.94 and 3.21 ppm for HA resonated as a pair of doublets of doublets, while for HB appeared at δ 3.56 and 3.90 ppm as a pair of doublets of doublets, respectively. The HX in the pyrazoline ring for compounds 3 and 4 appeared as a doublet of triplets or doublet of doublets at δ 4.79 and 5.44 ppm, respectively. The D2O exchangeable singlet peak of OH appeared at 12.38 and 10.20 ppm for compounds 3 and 4, respectively, while the NH proton for compound 3 appeared at 7.80 ppm. All the other aromatic protons were observed with their expected chemical shifts. Moreover, the formations of pyrazolines 6a–c were synthesized via condensation reaction of chalcone 1 with different phenyl hydrazine derivatives 5a–c as displayed in Scheme 1. The IR spectrum of pyrazoline rings of 6a–c showed a characteristic bands at 1616, 1620, and 1615 cm−1 due to CN group, also the 1HNMR of ABX structure of pyrazoline ring protons for compounds 6a–c. The signals for compounds 6a–c appeared, respectively, at δ 3.21, 3.33, and 3.19 ppm for HA resonated as a pair of doublets of doublets, while for HB appeared as a pair of doublets of doublets at δ 4.01, 4.08, and 3.98 ppm, respectively. Also, The HX in the pyrazoline ring for compounds 6a–c appeared as a doublet of doublets at δ 5.40, 5.65, and 5.35 ppm, respectively. The D2O exchangeable singlet peak of OH appeared at 10.55, 10.22 and 10.58 ppm for compounds 6a–c, respectively. All the remaining aromatic protons were observed with their expected chemical shifts.
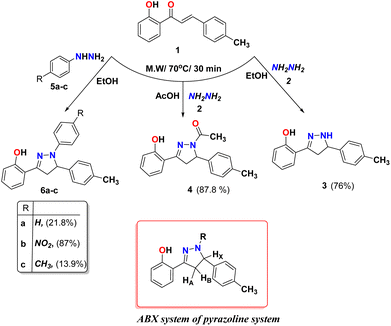 |
| | Scheme 1 Reaction of chalcone 1 with hydrazine hydrate in different condition and chalcone 1 with phenyl hydrazine derivatives. | |
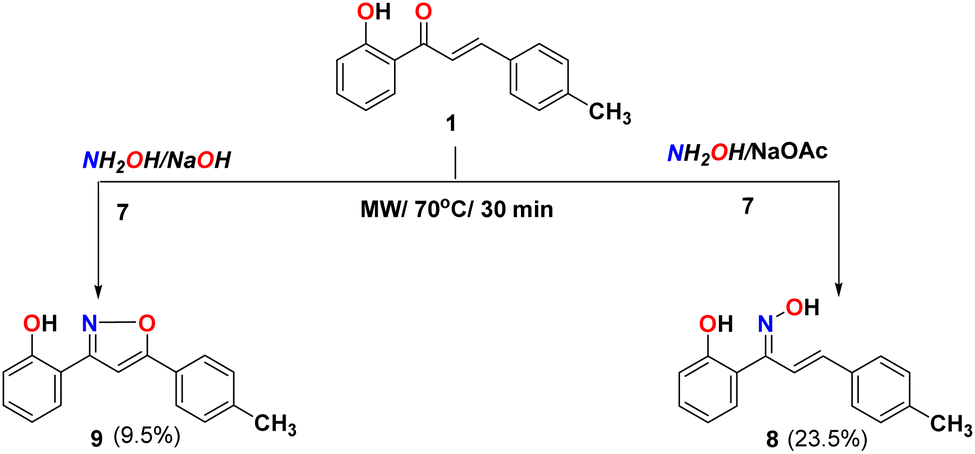
Furthermore, different varieties of condition reactions of chalcone 1 with hydroxylamine hydrochloride 7 were examined either with sodium acetate in ethanol, or sodium hydroxide as a base medium instead of using pyridine as previously reported44 (Scheme 2). The reaction of chalcone 1 with hydroxylamine hydrochloride and sodium acetate in ethanol formed oxime 8 and didn't form the isoxazoline 9. The method with sodium acetate using microwave afforded oxime 8 as the only product for the reaction. The IR spectra of compound 8 showed bands at 3321, 3028, and 1635 cm−1 due to OH, aromatic C–H, and CN groups, respectively. The 1H NMR supported the structure identity due to the appearance of the double bond peaks (J = 16.8 Hz) at 6.42 and 7.50 ppm revealed the pure geometrical with trans-configuration in addition to the D2O exchangeable singlet peak of two OH at 9.65 and 11.35 ppm. While the reaction in presence of NaOH as a base afforded the isoxazoline 9. Their IR spectra showed bands at 3281, 3028, and 1635 cm−1 due to OH, aromatic C–H, and CN groups, respectively. The NMR supported the structure identity (cf. experimental part).
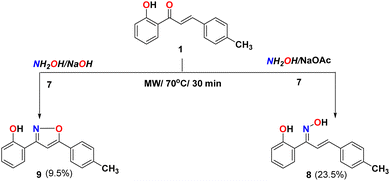 |
| | Scheme 2 Reactivity of chalcone 1 with NH2OH in different basic condition. | |
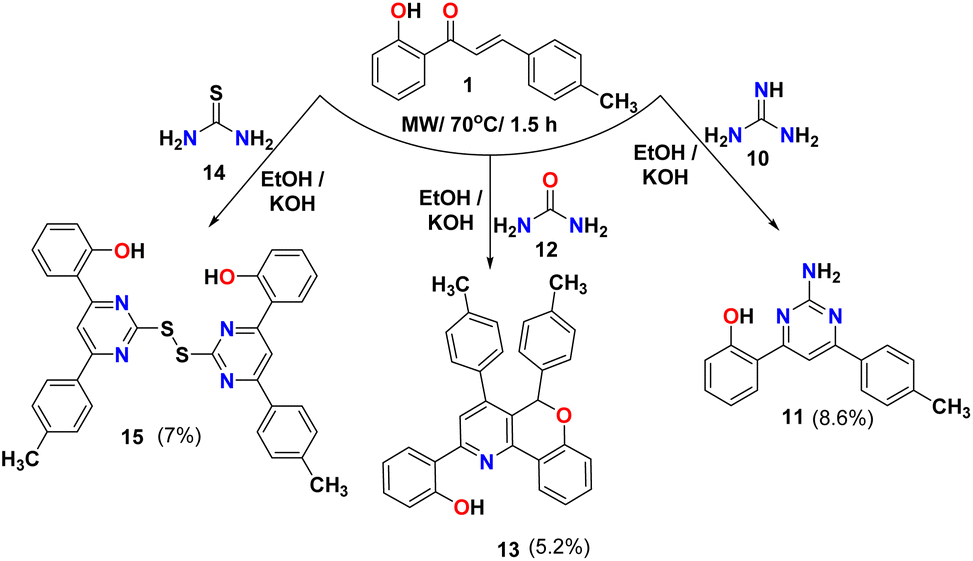
The reaction of chalcone 1 with guanidine 10 in the presence of KOH as a base gave the amino pyrimidine 11, as shown in Scheme 3. This reaction was reported using t-BuOK as a base and 2-propanol as a solvent.45 The IR spectra of amino pyrimidine 11 showed bands at 3356, 3259, 3089, and 1672 cm−1 due to OH, NH, aromatic C–H, and CN groups, respectively. The 1H NMR for compound 11 showed singlet peaks at 7.17, 7.80, and 14.03 ppm due to NH, CH pyrimidine ring, and OH, respectively. While the reaction of chalcone 1 with urea did not produce the expected compound pyrimidine and gave triaryl-5H-chromeno[4,3-b]pyridine 1346 and confirmed via spectral analysis such as FT-IR spectra that shows bands at 3053 and 1600 cm−1 due to aromatic C–H, and CN groups, respectively. The 1HNMR of compound 13 showed three singlet peaks appeared at 6.44, 8.10, and 13.80 ppm due to CH of the chromene ring, CH of the pyridine ring, and OH, respectively. Also, condensation of chalcone 1 with thiourea 14 gave the dimer product thiopyrimidine compound 15, which was detected with the IR that shows bands at 3019 and 1613 cm−1 due to aromatic C–H, and CN groups; respectively. The 1H NMR for thiopyrimidine 15 showed one singlet peak at 8.52 ppm due to the CH of pyrimidine ring as showed in Scheme 3.
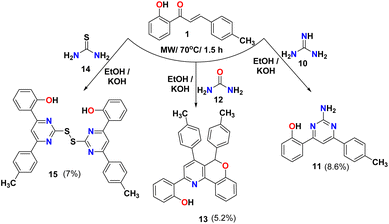 |
| | Scheme 3 Reaction of chalcone 1 with guanidine, urea, and thiourea. | |
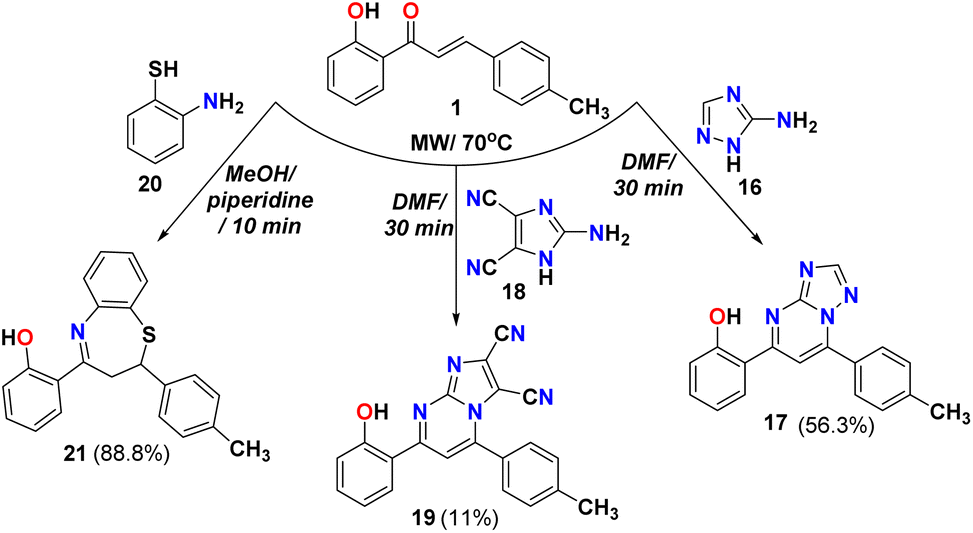
The cyclocondensation of chalcone 1 with 1,2,4-triazol-3-amine 16, as shown in Scheme 4 in DMF gave the 2-(7-(p-tolyl)-[1,2,4]triazolo[1,5-a]pyrimidin-5-yl)phenol 17. The IR for compound 17 showed bands at 3496 and 1687 cm−1 due to OH and CN groups and the 1H NMR showed singlet peaks at 8.72, and 12.38 ppm due to CH pyrimidine ring and OH, respectively. While the reaction of chalcone 1 with 2-amino-1H-imidazole-4,5-dicarbonitrile 18 in DMF as solvent afforded the corresponding 7-(2-hydroxyphenyl)-5-(p-tolyl)imidazo [1,2-a]pyrimidine-2,3-dicarbonitrile 19 and confirmed through spectral analysis such as FT-IR spectra that showed bands at 3493, 2246 and 1614 cm−1 due to OH, CN, and CN groups; respectively. The 1H NMR showed three singlet peaks appeared at 8.14, and 11.68 ppm due to CH of the pyrimidine ring, and OH, respectively. Furthermore, the Michael addition reaction of o-aminothiophenol 20 with chalcone 1 followed by cyclocondensation to give the desired 1,5-benzothiazepine 21. The same reaction was reported using N-methylimidazolium nitrate as a catalyst.47 The IR spectra showed characteristic bands at 3428, 3051, 1598 cm−1 due to OH, aromatic C–H, and CN double bond in the seven-membered ring heterocycle. The 1H NMR of benzothiazepine 21 supported the ABX system where it showed a triplet at δ 2.90 ppm for HA and two doublets of doublet at δ 3.50 and 5.22 ppm for the HB and HX; respectively. The mass spectrum exhibited a peak at m/z 345 attributable to the molecular ion as displayed in Scheme 4.
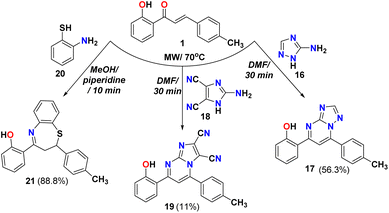 |
| | Scheme 4 Reactivity of chalcone 1 with amino-heterocyclic derivatives. | |
2.2. Antioxidant activities
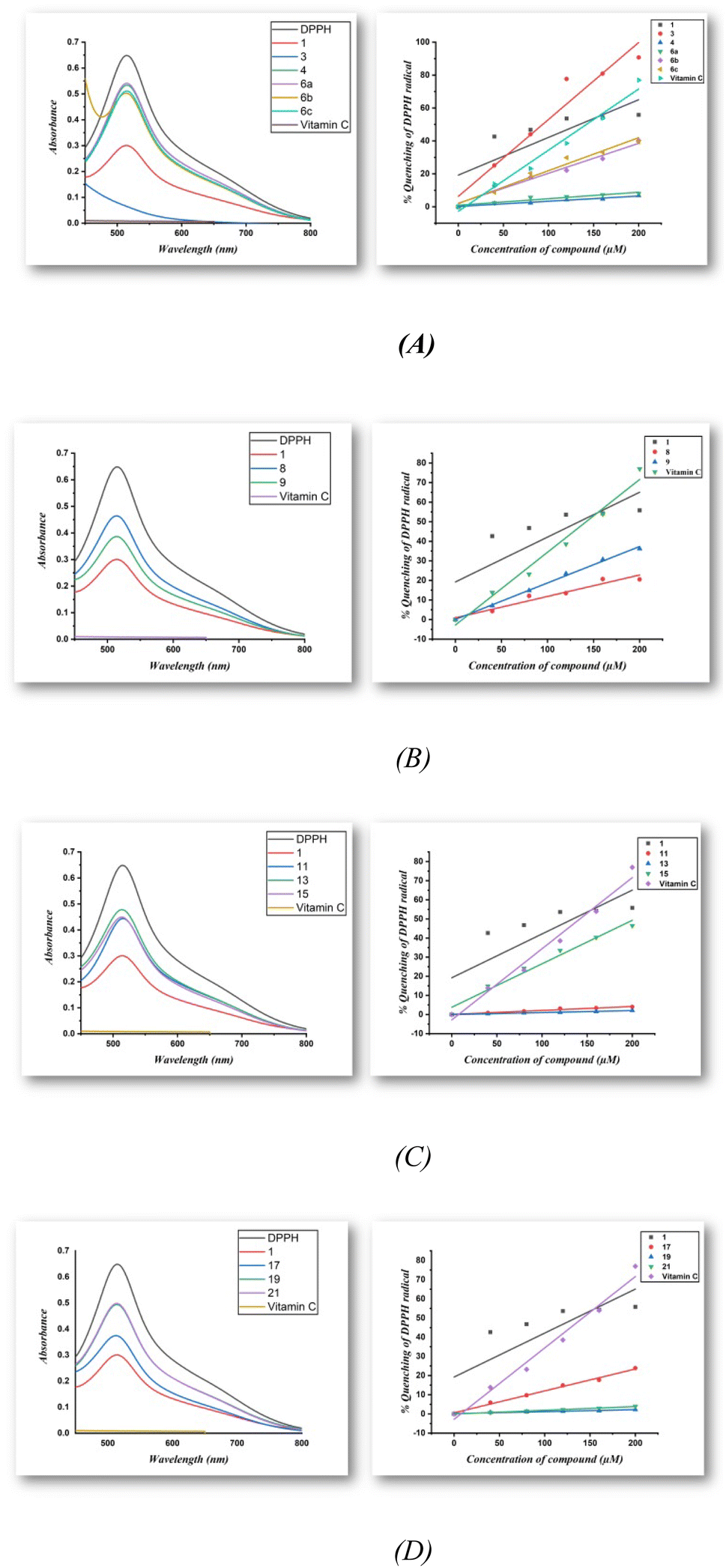
The radical scavenging properties of compounds 3, 4, 6a–c, 8, 9, 11, 13, 15, 17, 19, and 21 were studied using a DPPH assay in methanol, whereby DPPH free radicals change their color from purple to yellow upon the quenching of the radical in the presence of an antioxidant. In general, the DPPH radical is monitored at 517 nm (Fig. 1 (left)), at which the absorption decreases in the presence of antioxidants, and Fig. 1 (right) shows the percentage inhibition plotted against antioxidant concentration. From Table 1, it can be seen that all compounds, except for 4, 6a, 11, 13, 19, and 21, possess antioxidant properties. In particular, compound 3 showed the strongest antioxidant properties among all synthesized compounds with an IC50 of approximately 93.4 μM and comparable to those of vitamin C (IC50 ∼ 141.9 μM). The IC50 values of the remaining compounds are decrease to approach the value of vitamin C in the order of 1 > 15 > 6c > 6b > 9 >17 > 8. Upon replacing the hydrogen with acetyl (i.e., compound 4) or with phenyl group (i.e., compound 6a) in the pyrazoline ring in compound 3, no antioxidant activity is observed in both compounds. On the other hand, in the presence of an electron-donating or electron-withdrawing substituents like methyl and nitro in the para position for the phenyl group attached to the pyrazoline ring, the antioxidant activity of the pyrazoline compound 6c and 6b are increased compared with compound 6a. Also, the antioxidant activity of the oxime compound 8 (IC50: 448.7 μM) is decreased compared with the isoxazoline 9 (IC50: 267.9 μM). By comparing the amino pyrimidine 11 with the dimer product thiopyrimidine compound 15, no antioxidant activity is observed for compound 11 while compound 15 exhibits good antioxidant properties. Furthermore, triazolo[1,5-a]pyrimidine compound 17 exhibits good antioxidant properties (IC50: 432.90 μM) compared with imidazo[1,2-a]pyrimidine compound 19 which does not show any antioxidant activity. Accordingly, compounds 1 and 3 exhibit greater DPPH radical scavenging capacity than vitamin C as a reference compound, indicating that they are useful as antioxidants.
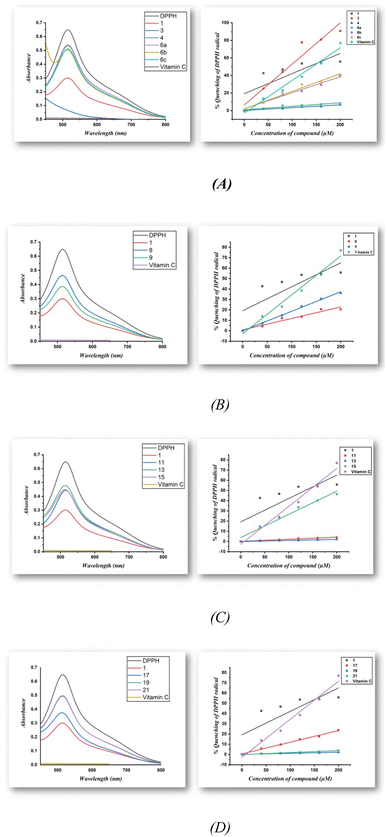 |
| | Fig. 1 (A–D) Typical absorption spectra of 100 μM of the DPPH radical alone and in presence of a 200 μM concentration of compounds and vitamin C (left) along with a diagram of the percentage of quenching of the DPPH radical against the concentration of the antioxidants compounds and vitamin C (right). | |
Table 1 IC50 values of afforded heterocyclic compounds (1–21) for the DPPH radical in methanol
| Compounds |
IC50 (μM) |
| 1 |
134.26 |
| 3 |
93.40 |
| 4 |
— |
| 6a |
— |
| 6b |
263.01 |
| 6c |
240.07 |
| 8 |
448.70 |
| 9 |
267.90 |
| 11 |
— |
| 13 |
— |
| 15 |
203.08 |
| 17 |
432.90 |
| 19 |
— |
| 21 |
— |
| Vitamin C |
141.90 |
2.3. Docking analysis
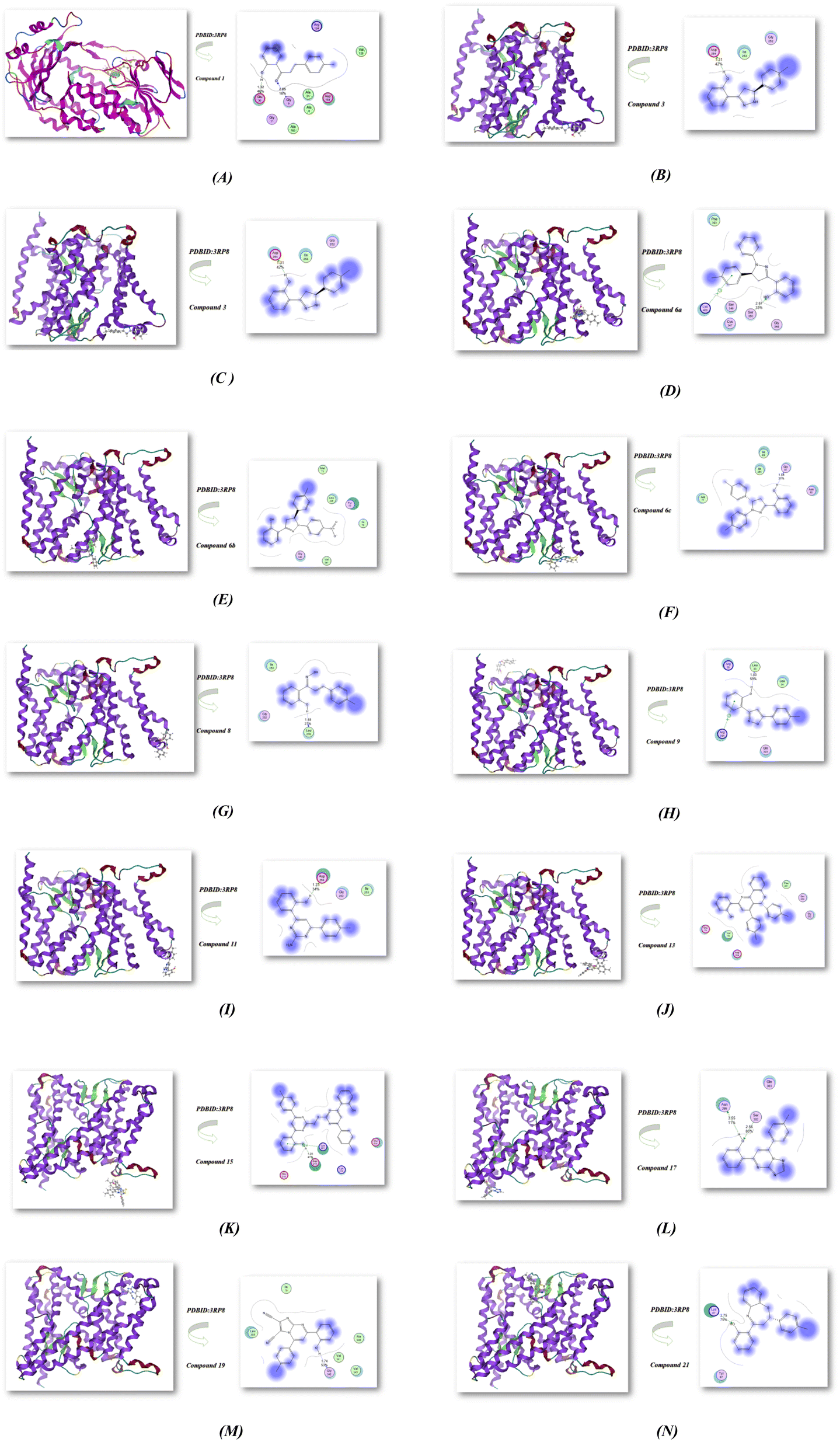
The complex docking molecular analysis was enhanced with bond lengths in Å units using Moe software.48 The minimization energies were then employed to maintain the geometrical optimization and systematic investigations with an RMS gradient of 0.01 Å. Also, the crystal structure of Klebsiella pneumoniae R204Q HpxO complexed with FAD (PDBID:3RP8),49 as shown in Fig. 2 and Table 2. Chalcone 1 exhibited the highest binding affinity to the protein, with an energy affinity of −13.521 kcal mol−1 and the shortest distance ranging from 1.32–2.85 Å. This strong binding resulted from the hydrogen bond interactions between the OH group and the CO group of chalcone, enabling a compact connection with the protein's amino group, specifically Glu 30 and Gly 9. Compound 3 also demonstrated strong compatibility with the protein 3RP8, having an energy affinity of −13.782 kcal mol−1 with its OH group, and binding to Asp254 at a distance of 1.31 Å. In contrast, compounds 4 and 6a displayed reduced affinities, with energy levels of −12.624 and −10.625 kcal mol−1, respectively, and increased interaction distances in the range of 2.52–2.62 Å. While compound 6a did bind to Ser 350 through its OH group, compounds 6b and 6c showcased remarkable binding affinities with values of −12.524 and −12.841 kcal mol−1, respectively. This was attributed to the presence of withdrawing (NO2) and electron-donating (CH3) groups that influenced their binding capabilities with proteins and enhanced hydrogen bond interactions. Furthermore, the reaction of chalcone with hydroxyl amine in different conditions showed different binding interactions in energy −10.025 kcal mol−1 and −11.254 kcal mol−1 for compounds 8 and 9, respectively. Both compounds showed binding with OH of the phenyl ring with short bond lengths 1.48–1.43 Å with different amino acids (Leu 249, Gly 252, Ile 253) and (Leu 33, Leu 34, Arg 35, Arg 304, Gln 303); respectively. Moreover, the reactivity of chalcone with guanidine, urea and thiourea afford different heterocyclic compounds 11, 13 and 15. These compounds showed also least binding affinity with PDBID:3RP8 with −10.521, −11.762 and −12.400 kcal mol−1 and length ranges (1.23–2.05 Å). The interactions mainly occurred with the OH group of the phenyl ring but compound 15 showed compatibility with one OH group due it is a bis compound, leading to its increased stability when interacting with proteins such as (Lys 221, Asp 219, Glu 218, Lys 213, Glu 214). Also, the reactivity of chalcone with various nitrogen heterocyclic produced compounds 17, 19, and 21. These compounds showed least binding affinities with values of −9.621, −8.0696, and −8.0052 kcal mol−1 and distance with range 1.74–3.55 Å. The binding mostly occurred with OH and amino group in amino acids with (Asn 299, Gly 342 and Lys 67; respectively). We concluded that the antioxidant activity was exhibited from hydrogen bonding interaction between OH group of the phenyl ring with DPPH radical scavenger which is be confirmed through docking investigation as displayed in Fig. 2(A–N).50–53
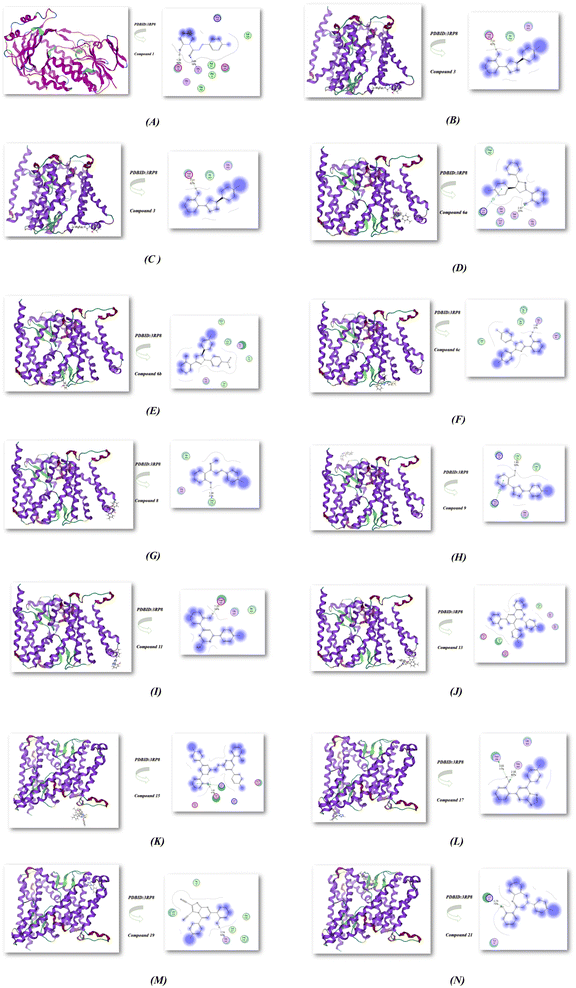 |
| | Fig. 2 (A–N) Molecular docking investigation of nitrogen heterocycles utilized PDBID:3RP8 protein. | |
Table 2 The molecular docking of synthesized compounds with PDBID:3RP8
| (PDB:3RP8) |
| |
Affinity of energy (kcal mol−1) |
Distance (Å) |
Amino acids |
|
Affinity of energy (kcal mol−1) |
Distance (Å) |
Amino acids |
| 1 |
−13.521 |
1.32, 2.85 Å |
Glu 30, Gly 9, Gly 7, Ala8, Ala 31, Ala 153, Asp 154, Val 125 |
9 |
−11.254 |
1.43 Å |
Leu 33, Leu 34, Arg 35, Arg 304, Gln 303 |
| 3 |
−13.782 |
1.31Å |
Asp 254, Ile 253, Gly 252 |
11 |
−10.521 |
1.23 Å |
Asp 254, Gly 252, Ile 253 |
| 4 |
−12.624 |
2.52 Å |
Arg 125, Gly 189, Ser 302, Gly 187, Cys 188 |
13 |
−11.762 |
2.05 Å |
Phe 251, Ser 250, Gly 252, Glu 75, Val 76, Asp 254 |
| 6a |
−10.6257 |
2.67 Å |
Ser 350, Cys 347,Gly 348, Ser 349, Lys 400 |
15 |
−12.400 |
1.28 Å |
Lys 221, Asp 219, Glu 218, Lys 213, Glu 214 |
| 6b |
−12.542 |
2.74 Å |
Gly 342, Val 341, Leu 338, Tyr 77, ile 79 |
17 |
−9.6211 |
2.56,3.55 Å |
Asn 299, Ser 302 |
| 6c |
−12.841 |
1.54 Å |
Gly 81, Asn 80, Ile 83, Ile 354 |
19 |
−8.0696 |
1.74 Å |
Gly 342, Val 341, Val 345, Ala 346, Leu 338, Ile 79 |
| 8 |
−10.025 |
1.48 Å |
Leu 249, Gly 252, Ile 253 |
21 |
−8.0052 |
2.75 Å |
Lys 67, Tyr 87 |
2.4. DFT study
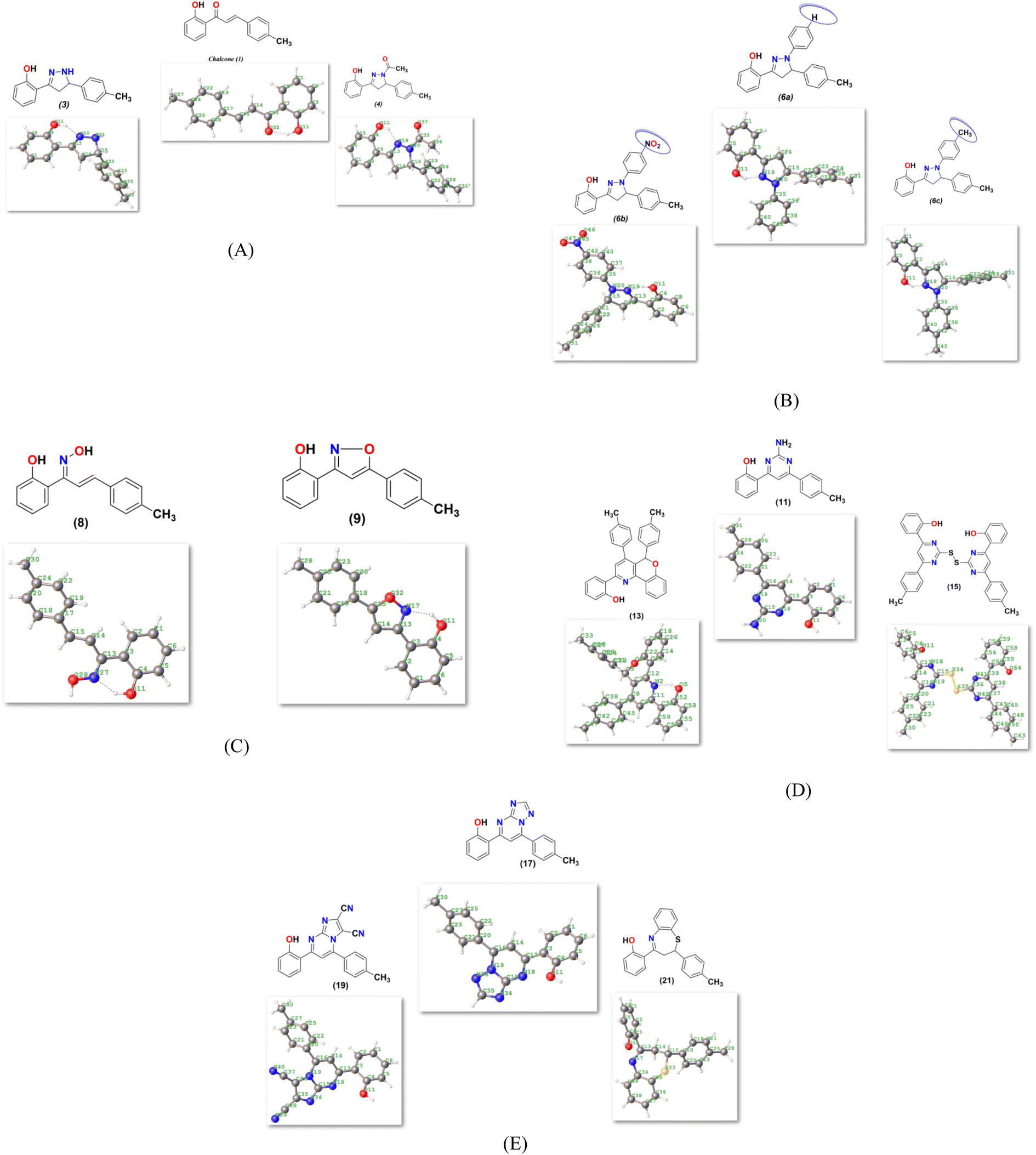
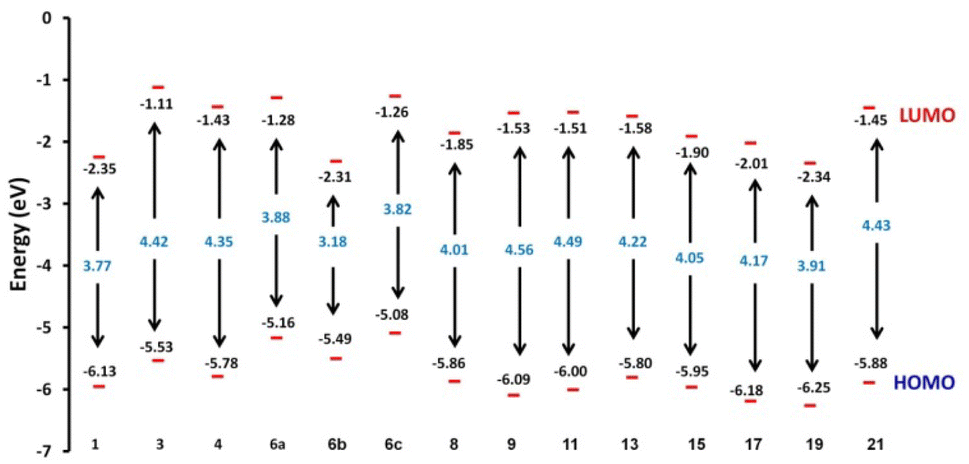
2.4.1. Molecular orbital analysis and chemical reactivity. Full optimization was carried out for all produced compounds along with chalcone 1 using the DFT/B3LYP/6-31G(d,p) basis set,54–56 as displayed in Fig. 3. A frequency calculation was done to guarantee that there were no imaginary frequencies. From eqn (1)–(8) estimated from B3LYP/6-31G(d,p), Table 3 lists the physical properties used in the optimization of molecular structures of all compounds concerning (σ) absolute softness, (χ) electronegativity, (ΔNmax) electronic charge, (η) absolute hardness, (ω) global electrophilicity, (S) global softness, and (Pi) chemical potential. Compared to the starting chalcone 1, all synthesized compounds 3–21 showed higher energy stability, as well as thiopyrimidine 15, which showed greater stability with energy (−2477.18 au) (−67407.34 e V) because of the presence of N, O, and S in its structure and increased electronegativity. Additionally, compound 19 has a very high dipole moment compared to other compounds. The difference in dipole moment between compound 19 and the chalcone 1 is 5.8 D, which is caused by the presence of two CN groups, which facilitates the separation of charges and increases reactivity.3 A molecule's kinetic stability and chemical hardness-softness can be evaluated using the energy gap (ΔE) between its highest occupied molecular orbital (HOMO) and its lowest unoccupied molecular orbital (LUMO).57–59 Consequently, the hardness and softness of the molecule are controlled by its ΔE, where large ΔE values indicate high kinetic stability, while low values indicate high chemical reactivity.60 In addition, the hardness of a chemical system is a measure of its resistance to deformation of the electron cloud by small perturbations encountered during the chemical process. Therefore, the electron density of soft molecules can be easily altered unlike that of hard molecules. The ability of an electrophile to acquire electronic charge is described by electrophilicity, besides its resistance to exchanging electronic charge with its surroundings. Furthermore, it provides information about electron transfer (chemical potential) and stability (hardness), as well as a better understanding of global chemical reactivity.
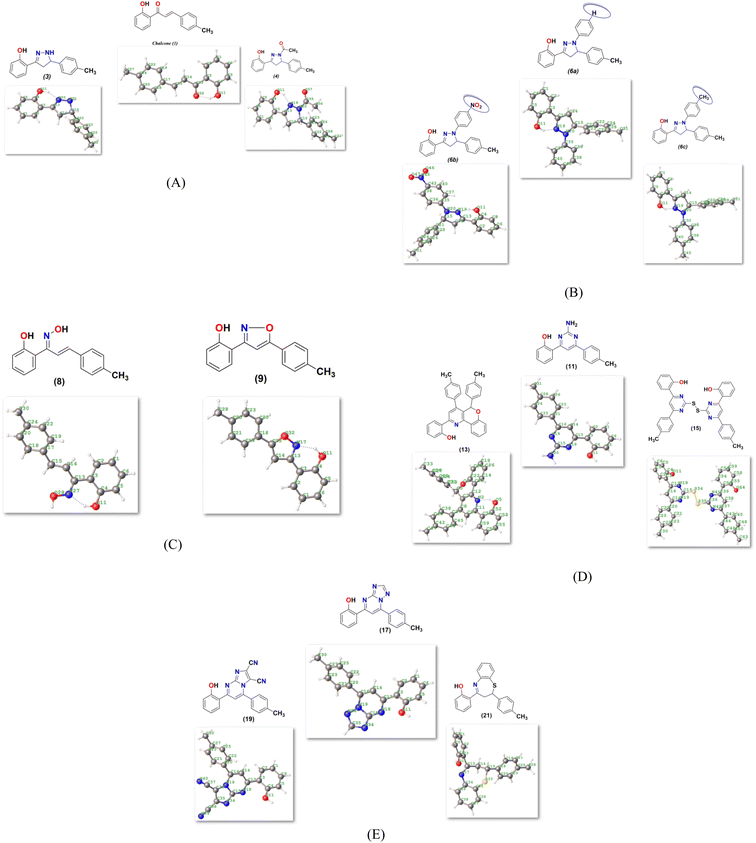 |
| | Fig. 3 (A–E) Chemical and the optimized structure of compounds 1–21 utilized DFT/631(G) basis set. | |
Table 3 Ground state energies of compounds utilizing DFT/B3LYP/6-31G(d,p) and their physical parameters
| Physical descriptors |
| Compound |
ET (au) |
EHOMO (eV) |
ELUMO (e V) |
Eg (eV) |
μ (D) |
χ (eV) |
η (eV) |
σ (eV) |
Pi (eV) |
S (eV) |
ω (eV) |
ΔN max |
| 1 |
−768.61 |
−6.13 |
−2.35 |
3.77 |
4.78 |
4.24 |
1.88 |
0.52 |
−4.24 |
0.264 |
4.763 |
2.246 |
| 3 |
−804.07 |
−5.53 |
−1.11 |
4.42 |
4.01 |
3.33 |
2.21 |
0.45 |
−3.33 |
0.226 |
2.507 |
1.507 |
| 4 |
−956.74 |
−5.78 |
−1.43 |
4.35 |
6.63 |
3.61 |
2.17 |
0.46 |
−3.61 |
0.229 |
2.991 |
1.657 |
| 6a |
−1035.13 |
−5.16 |
−1.28 |
3.88 |
3.91 |
3.22 |
1.94 |
0.51 |
−3.22 |
0.257 |
2.681 |
1.662 |
| 6b |
−1239.64 |
−5.49 |
−2.31 |
3.18 |
9.10 |
3.90 |
1.59 |
0.62 |
−3.90 |
0.313 |
4.783 |
2.450 |
| 6c |
−1074.45 |
−5.08 |
−1.26 |
3.82 |
3.76 |
3.17 |
1.91 |
0.52 |
−3.17 |
0.261 |
2.633 |
1.659 |
| 8 |
−823.90 |
−5.86 |
−1.85 |
4.01 |
3.40 |
3.86 |
2.00 |
0.49 |
−3.86 |
0.249 |
3.720 |
1.927 |
| 9 |
−822.72 |
−6.09 |
−1.53 |
4.56 |
4.76 |
3.81 |
2.27 |
0.43 |
−3.81 |
0.219 |
3.186 |
1.672 |
| 11 |
−896.37 |
−6.00 |
−1.51 |
4.49 |
1.71 |
3.76 |
2.24 |
0.44 |
−3.76 |
0.222 |
3.155 |
1.677 |
| 13 |
−1440.04 |
−5.80 |
−1.58 |
4.21 |
5.03 |
3.69 |
2.10 |
0.47 |
−3.69 |
0.237 |
3.233 |
1.751 |
| 15 |
−2477.18 |
−5.95 |
−1.90 |
4.05 |
2.06 |
3.93 |
2.02 |
0.49 |
−3.93 |
0.246 |
3.817 |
1.941 |
| 17 |
−988.59 |
−6.18 |
−2.01 |
4.17 |
5.07 |
4.10 |
2.08 |
0.48 |
−4.10 |
0.240 |
4.038 |
1.969 |
| 19 |
−1157.03 |
−6.25 |
−2.34 |
3.91 |
10.59 |
4.30 |
1.95 |
0.51 |
−4.30 |
0.256 |
4.730 |
2.199 |
| 21 |
−1377.96 |
−5.88 |
−1.45 |
4.43 |
2.52 |
3.66 |
2.21 |
0.45 |
−3.66 |
0.225 |
3.033 |
1.653 |
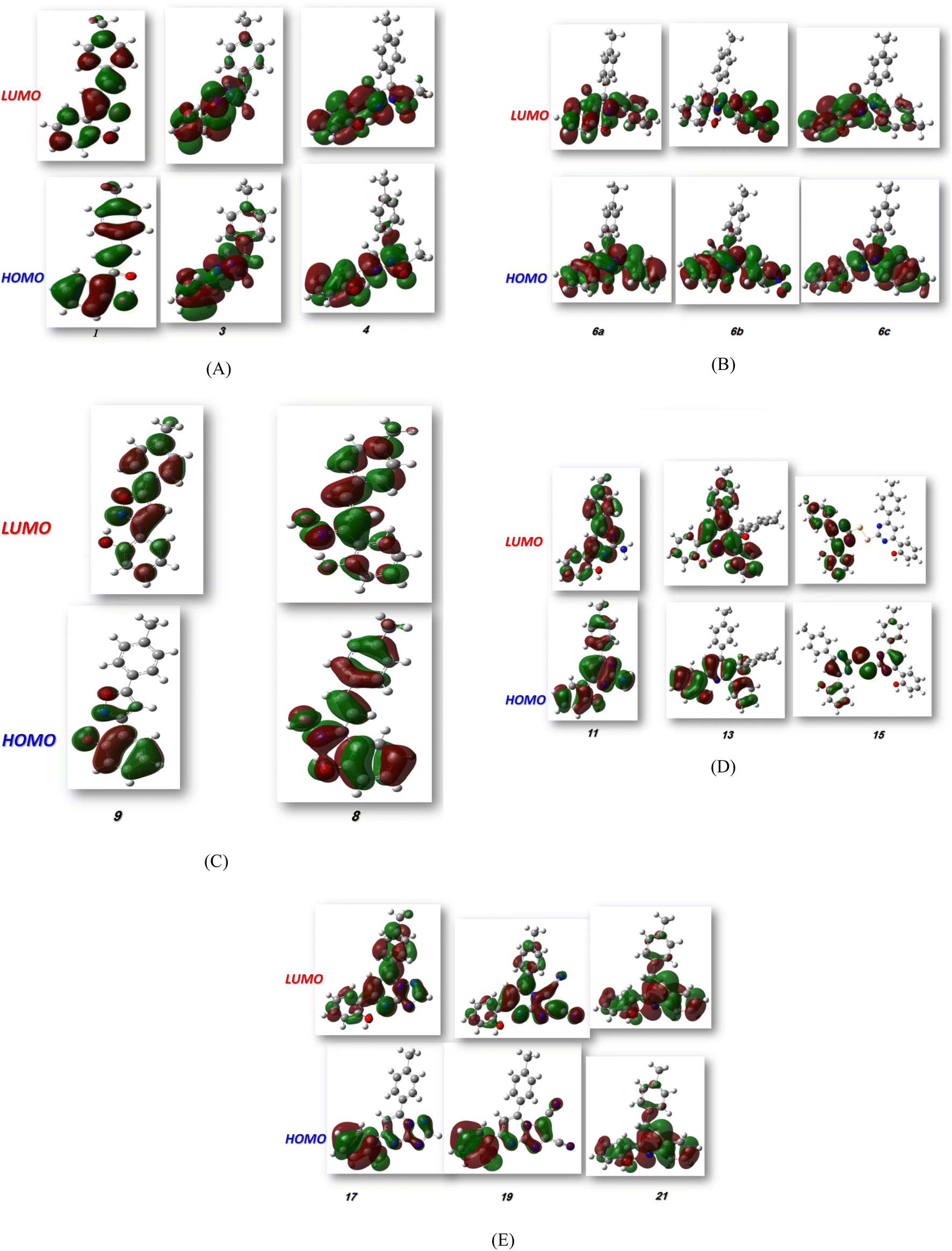
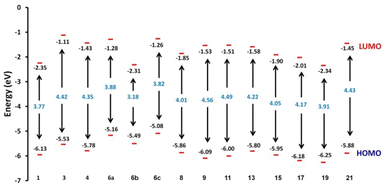
Hard molecules have a huge HOMO–LUMO gap, whereas soft molecules have a small HOMO–LUMO gap. Furthermore, hard molecules tend to have high kinetic stability and low chemical reactivity, whereas soft molecules have the opposite properties. According to Table 3, the higher hardness values (2.27 eV) and low softness (0.43 eV) display lower intramolecular charge transfer present in compound 9. Also, compound 9 has the highest ΔE among all compounds reflecting its kinetic stability, whereas compound 6b has the lowest ΔE reflecting its high chemical reactivity. Fig. 4 illustrates the energy levels of HOMO–LUMO orbitals, while Fig. 5 shows their orbital analyses of LUMO and HOMO calculated at the B3LYP/6-31G(d, p) level for all compounds 1–21. Positive and negative phases were characterized by red and green colors, respectively. For the start compound 1, the electron clouds in the HOMO distribute over the π-electron of the two phenyl rings together with the double bond of the CHCH group, the oxygen atom of the OH group, and the methyl group (i.e. the whole molecule except for the carbonyl group), while its LUMO distributes charges over the molecule except for the methyl group. For the produced heterocyclic compounds, the electron clouds in both HOMO and LUMO of compound 3 distribute charges over the entire molecule except for the methyl group. Similar charge distribution was found in the HOMO of compound 4 except for the methyl group in both acetyl and tolyl group, while the electron clouds LUMO distribute charges over the entire molecule except for the tolyl group and the attached CH group in the pyrazolyl ring. The electronic clouds in compounds 6a, 6b, and 6c behave similarly in a HOMO, where they are mainly distributed in the molecule except for the tolyl groups. Their LUMO differs based on the group that is attached to the second phenyl ring, where the H atom and the donating methyl group in compounds 6a and 6c distribute charges toward the hydroxyphenyl ring, whereas in compound 6b, the electron withdrawing nitro group causes charges to be distributed towards the nitrophenyl ring, as shown in Fig. 5. Oxime 8 has different HOMO distribution charges than isoxazoline 9, where the electron density is located on the entire molecule in compound 8, whereas it is populated in the hydroxyphenyl ring and the isoxazoline ring in compound 9. On the contrary, their LUMO electron clouds distribute the charges over the entire molecule of both compounds. The HOMO of the aminoprymidine 11 is localized on the entire molecule, while the HOMO of the dimer of thiopyrimidine 15 is mainly localized on the center unit of disulfide bis-pyrimidine and part of charges distributed over one hydroxy phenyl ring and one benzene ring in the tolyl group. Their LUMO amost behave the same for both, where the LUMO of compound 11 is localized on the entire molecule except the amino group (i.e. pyrimidine, hydroxyphenyl, and tolyl group), while for compound 15 it is localized on the half unit of the dimer except sulfer (i.e. pyrimidine, hydroxyphenyl, and tolyl group). The HOMO for compound 13 is mostly localized on the 5H-chromeno[4,3-b]pyridine and phenol rings, while the LUMO is localized on the 5H-chromeno[4, 3-b]pyridine ring as reported.46 The HOMO for both compounds 17 and 19 is localized on the whole molecule except the tolyl group, while their LUMO is localized on the whole molecule except the methyl group. The HOMO and LUMO for compound 21 are localized over the entire molecule except for the methyl group and little charge is distributed on the benzene ring of the tolyl group. Further, Mulliken atomic charges and electronic populations were determined using the B3LYP/6-31G(d, p) basis sets. Fig. S32 and S33† shows the atomic charges of all compounds 1–21, and Fig. 3 shows their optimized structures with atomic numbers. Delocalization of negative charges in atoms with a lower electronegative results from electron attraction from atoms with a higher electronegative. Fig. S32 and S33† show that carbon atoms have both positive and negative charges. The heteroatoms (N and O) have negative charges and a high electron density. The carbon atoms that bonded to the S atom have negative charges. The most acidic hydrogens are the phenolic hydrogen in all compounds.2,6
| |
 | (2) |
| |
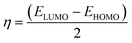 | (3) |
 |
| | Fig. 4 Schematic diagrams of HOMO and LUMO energy levels of compounds 1–21 obtained from the DFT calculation with B3LYP/6-31G(d,p). | |
 |
| | Fig. 5 (A–E) FMO profiles of synthesized heterocyclic compounds. | |
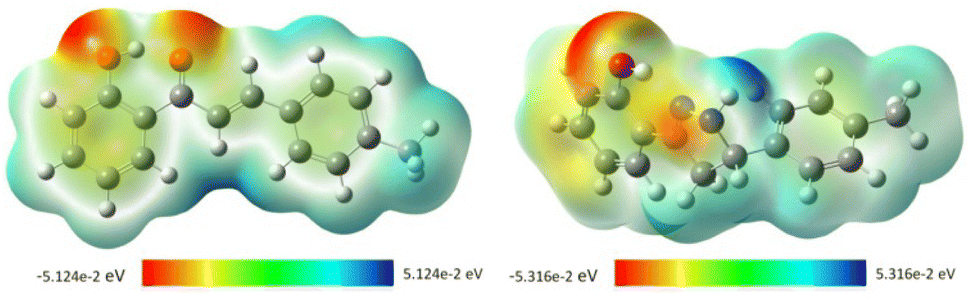
2.4.2. Molecular electrostatic potential (MEP). MEP mapping, a tool for analyzing and predicting molecular behavior, was used to identify electrophilic and nucleophilic active sites and hydrogen bonding interactions.46,61 This tool allows visualization of the charge distribution and helps comprehend the relative polarity of a molecule and predict their reactive sites. Also, this method is extremely helpful for determining relationships between physicochemical properties and molecular structure. In MEP mapping, red represents electron-rich, nucleophilic regions, while blue represents electron-poor, electrophilic regions. Different colors describe the electrostatic potential on the surface, decreasing from blue to green to yellow to orange to red. Based on full-geometry optimization at DFT/B3LYP/6-31G (d,p) level, MEP was calculated for the two most active antioxidant compounds (compound 1 and compound 3). A detailed analysis of the MEP maps (Fig. 6) indicates that the highly negative electrostatic potential regions (red) are centered on the oxygen atoms of the OH group and the ketone group in compound 1 with values of −5.124 × 10−2 eV (at an isovalue of 0.001 electrons per Å3), while the highly positive regions (blue color) are concentrated near the olefin proton. For compound 3, the negative values are mainly located over the oxygen atom of the hydroxyl group with a value of −5.316 × 10−2 eV, indicating a potential electrophilic attack site, while the most positive regions were localized on the H19 of the NH group with a value of −5.316 × 10−2 eV, indicating a potential nucleophile attack site. Based on these findings, we are able to predict the probable sites for electrophilic attacks, which would clarify the docking results in regard to the binding of each molecule to the amino acid residues of the studied protein's active site.62,63
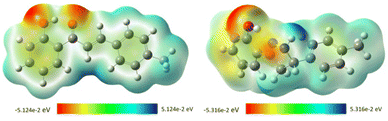 |
| | Fig. 6 Molecular electrostatic potential (MEP) maps of chalcone 1 (left) and compound 3 (right). | |
3. Experimental section
3.1. General procedure
The Gallenkamp melting point instrument was used to measure the melting points. Thin-layer chromatography (TLC) was conducted on Polygram SIL G/UV254 TLC plates, and the results were visualized with ultraviolet light at 254 nm and 350 nm. In this study, the CEM Discover LabMate microwave apparatus (300 W, ChemDriver software; Matthews, NC) was used for microwave experiments. In closed vessels under pressure, microwave irradiated Pyrex tubes with PCS caps were used to conduct the reactions. A Bruker DPX 400 superconducting NMR spectrometer was used to record the 1H and 13C nuclear magnetic resonance (NMR) spectra, and the IR spectra were measured with a Jasco Fourier transform/IR-6300 FT-IR spectrometer. The Elementar Vario MICRO Cube was used for the elemental analysis. Electron impact (EI) was used to determine mass analyses on a Thermo double-focusing sector (DFS) mass spectrometer. Varian Cary 5 spectrometer and a Shimadzu UV2600 spectrophotometer were used in UV-vis studies. Utilizing X-ray microdiffraction and single-crystal X-ray diffractometers, Rigaku D/MAX Rapid II and Bruker X8 Prospector were used to determine the X-ray crystal structure.
3.2. Materials and reagents
1-(2-Hydroxyphenyl)ethan-1-one, 4-methylbenzaldehyde were purchased from Aldrich Chemical CO that was used in the synthesis of chalcone 1 as reported in the literature.46,64 In this study, all solvents were obtained from Aldrich.
3.2.1. Synthesis of 2-(5-(p-tolyl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (3)42. A mixture of chalcone 146 (1 g, 4.2 mmol) and hydrazine hydrate (10 equivalent) in ethanol (8 mL) was mixture was irradiated utilizing microwave at 70 °C for 30 min. White crystals was formed after cooling and filtered. Yield (0.8 g, 76%), M p 106 °C (literature:65 105–106 °C). FT-IR (νmax, cm−1): 3339 (NH), 3006 (C–H Ar), 1619 (CN). 1H NMR (DMSO-d6, δ ppm): 2.28 (s, 3H, H3C), 2.94–2.98 (dd, 1H, J = 10.8 Hz, H-4A), 3.56–3.60 (dd, 1H, J = 10.8 Hz, H-4B), 4.79–4.83 (dt, 1H, J = 10.8 Hz, H-5X), 6.86–6.91 (m, 2H, ArH), 7.15 (d, 2H, J = 7.8 Hz, ArH), 7.21–7.29 (m, 4H, ArH), 7.80 (d,1H, HN), 11.16 (s, 1H, HO), 13C NMR (DMSO-d6):δ 20.6 (CH3), 40.78 (CH), 61.84 (CH), 115.69 (C), 116.76 (CH), 119.10 (CH), 126.59 (CH), 127.72 (CH), 128.98 (CH), 129.67 (C), 136.43 (C), 139.22 (C), 152.46 (C), 156.71 (C). MS (m/z): 252 (M+, 100.0%), anal. calcd. for C16H16N2O (252.13): C, 76.16; H, 6.39; N, 11.10. Found: C, 76.00; H, 6.41; N, 11.08.
3.2.2. Synthesis of 1-(3-(2-hydroxyphenyl)-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (4). A mixture of 1 (1 g, 4.2 mmol) and hydrazine hydrate (10 equivalent) in glacial acetic acid (5 mL) was irradiated utilizing microwave at 70 °C for 30 min. The reaction mixture was cooled and poured onto crushed ice. The obtained precipitate was filtered, washed with H2O, dried, and recrystallized from ethanol to afford 7. Yield (1.08 g, 87.8%). Mp 131 °C (literature:65 131–134 °C). FT-IR (νmax, cm−1): 3032 (C–H Ar), 1648 (CO), 1615 (CN). 1H NMR (DMSO-d6, δ ppm): 2.26 (s, 6H, 2 H3C), 3.21 (dd, 1H, J = 18.6 Hz, H-4A), 3.90 (dd, 1H, J = 18.6 Hz, H–4B), 5.44 (dd, 1H, J = 12 Hz, H-5X), 6.90 (t, 1H, J = 8.4 Hz, ArH), 6.96 (d, 1H, J = 7.2 Hz, ArH), 7.08–7.18 (m, 4H, ArH), 7.33 (t,1H, J = 8.4 Hz, ArH), 7.55 (d,1H, J = 7.8 Hz, ArH), 10.20 (s, 1H, HO), 13C NMR (DMSO-d6):δ 20.6 (CH3), 21.7 (CH3), 43.58 (CH2), 58.16 (CH), 116.23 (C), 119.54 (CH), 125.39 (CH), 129.01 (CH), 131.79 (CH), 136.36 (C), 139.26 (C), 155.65 (C), 156.69 (C), 166.88 (CO). MS (m/z): 294 (M+, 100.0%), anal. calcd. for C18H18N2O2 (294.14): C, 73.45; H, 6.16; N, 9.52. Found: C, 73.40; H, 6.20; N, 9.49.
3.2.3. Synthesis of 2-(1-phenyl-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (6a). A mixture of chalcone 1 (1 g, 4.2 mmol), phenylhydrazine (0.45 g, 4.2 mmol) in dry EtOH (8 mL) was irradiated utilizing microwave at 70 °C for 30 min. The reaction mixture poured into ice water. Yield (0.3 g, 21.8%). M p 133–134 °C. FT-IR (νmax, cm−1): 3155 (OH), 3093 (C–H Ar), 1616 (CN). 1H NMR (DMSO-d6, δ ppm): 2.25 (s, 3H, H3C), 3.21 (dd, 1H, J = 17.4 Hz, H-4A), 4.01 (dd, 1H, J = 17.4 Hz, H-4B), 5.40 (dd, 1H, J = 12 Hz, 5X), 6.75 (t, 1H, J = 7.2 Hz, ArH), 6.91–6.93 (m, 3H, ArH), 6.97–6.99 (dd, 1H, J = 8.4 Hz, ArH), 7.14 (d, 2H, J = 7.8 Hz, ArH), 7.17–7.21 (m, 4H, ArH), 7.26–7.29 (m, 1H, ArH), 7.40 (d, 1H, J = 18.6 Hz, ArH),10.55 (s, 1H, HO), 13C NMR (DMSO-d6): δ 20.62 (CH3), 43.84 (CH2), 61.97 (CH), 112.92 (C), 116.04 (CH), 116.58 (CH), 119.14 (CH), 125.88 (CH), 128.04 (CH), 129.05 (CH), 129.65 (CH), 130.37 (CH), 136.71 (C), 139.10 (C), 143.65 (C), 149.98 (C), 156.19 (C). MS (m/z): 328 (M+, 100.0%), anal. calcd. for C22H20N2O (328.16): C, 80.46; H, 6.14; N, 8.53. Found: C, 80.39; H, 6.20; N, 8.48.
3.2.4. Synthesis of 2-(1-(4-nitrophenyl)-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (6b). A mixture of chalcone 1 (1 g, 4.2 mmol), 4-nitrophenylhydrazine hydrochloride (0.79 g, 4.2 mmol) in dry EtOH (8 mL) was irradiated utilizing microwave at 70 °C for 30 min. The red precipitate which formed was filtered. Yield (1.36 g, 87%). M p 208–210 °C. FT-IR (νmax, cm−1): 3329 (OH), 3093 (C–H Ar), 1620 (CN). 1H NMR (DMSO-d6, δ ppm): 2.24 (s, 3H, H3C), 3.33–3.39 (m, 1H, H-4A), 4.08 (dd, 1H, J = 18 Hz, H-4B), 5.65 (dd, 1H, J = 11.4 Hz, H-5X), 6.94–7.06 (m, 4H, ArH), 7.15 (1, 4H, J = 19.8 Hz, ArH), 7.32 (dt, 1H, J = 3.6 Hz, ArH), 7.62 (dd, J = 7.8 Hz, 1H, ArH), 8.06 (d, 2H, J = 9.6 Hz, ArH), 10.22 (s, 1H, HO), 13C NMR (DMSO-d6): δ 20.61 (CH3), 44.87 (CH2), 61.10 (CH), 111.86 (C), 116.49 (CH), 119.62 (CH), 125.57 (CH), 125.81 (CH), 128.76 (CH), 128.97 (CH), 129.76 (CH), 131.50 (C), 137.06 (C), 137.99 (C), 147.67 (C), 154.00 (C), 156.32 (C). MS (m/z): 373 (M+, 55.0%), anal. calcd. for C22H19N3O3 (373.14): C, 70.76; H, 5.13; N, 11.25. Found: C, 70.69; H, 5.20; N, 11.20.
3.2.5. Synthesis of 2-(1,5-di-p-tolyl-4,5-dihydro-1H-pyrazol-3-yl)phenol (6c). A mixture of chalcone 1 (1 g, 4.2 mmol), p-tolylhydrazine hydrochloride (0.66 g, 4.2 mmol) in dry EtOH (8 mL) was irradiated utilizing microwave at 70 °C for 30 min. The reaction mixture poured into ice water and the beige precipitate which formed was filtered. Yield (0.2 g, 13.9%). M p 144–145 °C. FT-IR (νmax, cm−1): 3555 (OH), 3095 (C–H Ar), 1615 (CN). 1H NMR (DMSO-d6, δ ppm): 2.16 (s, 3H, H3C), 2.25 (s, 3H, H3C), 3.19 (dd, 1H, J = 18 Hz, H-4A), 3.98 (dd, 1H, J = 16.8 Hz, H-4B), 5.35 (dd, 1H, J = 12 Hz, H-5X), 6.81 (d, 2H, J = 8.4 Hz, H–ArH), 6.89 (t, 1H, J = 7.8 Hz, ArH), 6.96–7.00 (m, 3H, ArH), 7.13 (d, 2H, J = 7.8 Hz, ArH), 7.18 (d, J = 8.4 Hz, 2H, ArH), 7.25–7.28 (dt, J = 8.4 Hz, 1H, ArH), 7.38 (dd, 1H, J = 7.8 Hz, ArH), 10.58 (s, 1H, HO), 13C NMR (DMSO-d6):δ 20.05 (CH3), 20.61 (CH3), 43.70 (CH2), 62.32 (CH), 113.16 (C), 115.98 (CH), 116.61 (CH), 119.50 (CH), 125.95 (CH), 127.91 (CH), 129.46 (CH), 130.20 (C), 136.65 (C), 139.09 (C), 141.60 (C), 149.57 (C), 156.15 (C). MS (m/z): 342 (M+, 100.0%), anal. calcd. for C22H19N3O3 (342.17): C, 80.67; H, 6.48; N, 8.18. Found: C, 80.63; H, 6.40; N, 8.20.
3.2.6. Synthesis of 1-(2-hydroxyphenyl)-3-(p-tolyl)prop-2-en-1-one oxime (8). A mixture of chalcone 1 (1 g, 4.2 mmol), hydroxylamine hydrochloride (0.43 g, 4.2 mmol) and NaOAc (0.68 g, 8.4 mmol) in ethanol (5 mL) was irradiated at 70 °C for 30 min using microwave. The reaction mixture was poured into ice water to give a white precipitate (0.3 g, 23.5%). M p 174–176 °C (literature:66 174–176 °C). FT-IR (νmax, cm−1): 3321 (OH), 3028 (C–H Ar), 1635 (CN). 1H NMR (DMSO-d6, δ ppm): 2.29 (s, 3H, H3C), 6.42 (d, 1H, J = 16.8 Hz, CHCH), 6.85 (dt, 1H, J = 7.8 Hz, ArH), 6.89 (dd, 1H, J = 7.8 Hz, ArH), 7.14 (m, 2H, ArH), 7.23 (t, 2H, J = 8.4 Hz, ArH), 7.37 (d, 2H, J = 8.4 Hz, ArH), 7.50 (d, 1H, J = 16.8 Hz, CHCH), 9.65 (s, 1H, N–HO), 11.35 (s, 1H, HO), 13C NMR (DMSO-d6): δ 20.72 (CH3), 115.83 (CH), 116.26 (C), 118.74 (CH), 121.92 (CH), 127.03 (CH), 129.42 (CH), 129.76 (CH), 130.83 (CH), 133.37 (CH), 135.94 (C), 138.37 (C), 154.56 (C), 155.69 (C). MS (m/z): 253 (M+, 60.0%), anal. calcd. for C16H15NO2 (253.11): C, 75.87; H, 5.97; N, 5.53. Found: C, 75.82; H, 5.93; N, 5.50.
3.2.7. Synthesis of 2-(5-(p-tolyl)isoxazol-3-yl)phenol (9)67. A mixture of chalcone 1 (1 g, 4.2 mmol), hydroxylamine hydrochloride (0.43 g, 4.2 mmol) and NaOH (0.25 g, 6.25 mmol) in ethanol (8 mL) was irradiated at 70 °C for 30 min using microwave. The reaction mixture was poured into ice water to give a beige precipitate (0.1 g, 9.5%). M p 142–143 °C (literature:66 142–143 °C). FT-IR (νmax, cm−1): 3281 (OH), 3028 (C–H Ar), 1607 (CN). 1H NMR (DMSO-d6, δ ppm): 2.32 (s, 3H, H3C), 7.38 (d, 1H, J = 12 Hz, ArH), 7.45 (t, 1H, J = 12 Hz, ArH), 7.75–7.83 (m, 3H, ArH), 8.11 (m, 4H, ArH), 9.53 (s, 1H, HO), 13C NMR (DMSO-d6): δ 21.02 (CH3), 118.36 (CH), 121.29 (C), 124.50 (CH), 124.75 (CH), 127.56 (CH), 128.49 (C), 129.12 (CH), 133.62 (CH), 138.73 (CH), 139.78 (C), 145.4 (C), 154.50 (C), 172.84 (C). MS (m/z): 252 ((M + H)+, 100.0%), anal. calcd. for C16H13NO2 (251.09): C, 76.48; H, 5.21; N, 5.57. Found: C, 76.52; H, 5.29; N, 5.50.
3.2.8. Synthesis of 2-(2-amino-6-(p-tolyl)pyrimidin-4-yl)phenol (11). A mixture of compound 1 (1.0 g, 4.2 mmol), guanidine (0.4 g, 4.2 mmol) and KOH (0.47 g, 8.4 mmol) was dissolved in EtOH (8 mL) and irradiated at 70 °C for 1.5 h using microwave. After the accomplishment of reaction (as examined through TLC), the entire mixture was poured on to crushed ice to give a yellow precipitate which was filtered, washed successively with water and dried. Yield (0.1 g, 8.6%). M p 214 °C (literature:68 191–192 °C). FT-IR (νmax, cm−1): 3356 (OH), 3259 (NH), 3089 (C–H Ar), 1672 (CN). 1H NMR (DMSO-d6, δ ppm): 2.39 (s, 3H, H3C), 6.90 (m, 2H, H-3′, 5′), 7.17 (s, 2H, H2N), 7.33–7.37 (m, 3H, ArH), 7.79 (s, 1H, ArH), 8.15 (d, 2H, J = 12.6 Hz, ArH), 8.21 (d, 1H, J = 12.6 Hz, ArH), 14.03 (s, 1H, HO), 13C NMR (DMSO-d6): δ 20.97 (CH3), 99.49 (CH), 117.55 (CH), 118.05 (CH), 118.62 (C), 127.10 (CH), 128.01 (CH), 129.24 (CH), 132.58 (CH), 134.20 (C), 140.68 (C), 160.34 (C), 165.07 (C), 165.23 (C). MS (m/z): 277 (M+, 100.0%), anal. calcd. for C17H15N3O (277.12): C, 73.63; H, 5.45; N, 15.15. Found: C, 73.67; H, 5.52; N, 15.10.
3.2.9. Synthesis of 2-(4,5-di-p-tolyl-5H-chromeno[4,3-b]pyridin-2-yl)phenol (13)46. A mixture of compound 1 (1.0 g, 4.2 mmol), urea (0.25 g, 4.2 mmol) and KOH (0.47 g, 8.4 mmol) was dissolved in EtOH (8 mL) and irradiated at 70 °C for 1.5 h using microwave. After the accomplishment of reaction (as examined through TLC), the entire mixture was poured on to crushed ice to give a yellow precipitate which was filtered, washed successively with water and dried. Yield (0.1 g, 5.2%). M p 86 °C. FT-IR (νmax, cm−1): 3053 (C–H Ar), 1600 (CN). 1H NMR (DMSO-d6, δ ppm): 2.20 (s, 3H, H3C), 2.35 (s, 3H, H3C), 6.44 (s, 1H, H-5), 6.93–7.07 (m, 7H, ArH), 7.15 (t, 1H, J = 10.8 Hz, ArH), 7.22–7.27 (m, 4H, ArH), 7.33–7.40 (m, 2H, ArH), 7.99 (d, 1H, J = 10.2 Hz, ArH), 8.10 (s, 1H, ArH), 8.18 (d, 1H, J = 10.8 Hz, ArH), 13.80 (s, 1H, HO), 13C NMR (DMSO-d6): δ 20.59 (CH3), 20.73 (CH3), 99.49 (CH), 117.72 (C), 118.17 (CH), 119.17 (CH), 119.27 (CH), 120.56 (C), 121.85 (C), 122.50 (CH), 122.85 (CH), 123.55 (CH), 127.65 (CH), 127.95 (CH), 128.27 (CH), 129.12 (CH), 129.23 (CH), 131.65 (CH), 132.28 (CH), 133.58 (C), 135.60 (C),137.99 (C), 138.41 (C), 145.33 (C), 149.40 (C), 153.71 (C), 156.36 (C), 158.76 (C). MS (m/z): 455 (M+, 70.0%), anal. calcd. for C32H25NO2 (455.19): C, 84.37; H, 5.53; N, 3.07. Found: C, 84.36; H, 5.53; N, 3.05.
3.2.10. Synthesis of 2,2'-(disulfanediylbis(6-(p-tolyl)pyrimidine-2,4-diyl))diphenol (15). A mixture of compound 1 (1.0 g, 4.2 mmol), thiourea (0.319 g, 4.2 mmol) and KOH (0.47 g, 8.4 mmol) was dissolved in EtOH (8 mL) and irradiated at 70 °C for 1.5 h using microwave. After the accomplishment of reaction (as examined through TLC), the entire mixture was poured on to crushed ice to give a yellow precipitate which was filtered, washed successively with water and dried. Yield (0.17 g, 7%). M p 248–250 °C. FT-IR (νmax, cm−1): 3019 (C–H Ar), 1613 (CN). 1H NMR (DMSO-d6, δ ppm): 2.39 (s, 3H, H3C), 6.88–6.93 (m, 4H, ArH), 7.34 (d, 6H, J = 12 Hz, ArH), 8.19 (d, 6H, J = 16.2 Hz, ArH), 8.52 (s, 2H, ArH), 13C NMR (DMSO-d6): δ 21.05 (CH3), 109.70 (CH), 117.95 (CH), 119.42 (CH), 127.50 (C), 129.04 (CH′), 129.72 (CH), 132.46 (CH), 133.61 (CH), 142.17 (C), 159.10 (C), 164.71 (C), 165.05, (C), 165.31 (C), 166.63 (C). MS (m/z): 585 ((M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −H)+, 97.0%), anal. calcd. for C34H26N4O2S2 (586.15): C, 69.60; H, 4.47; N, 9.55. Found: C, 69.56; H, 4.50; N, 9.49.
−H)+, 97.0%), anal. calcd. for C34H26N4O2S2 (586.15): C, 69.60; H, 4.47; N, 9.55. Found: C, 69.56; H, 4.50; N, 9.49.
3.2.11. Synthesis of 2-(7-(p-tolyl)-[1,2,4]triazolo[1,5-a]pyrimidin-5-yl)phenol (17). A solution of chalcone 1 (1 g, 4.2 mmol) and 3-amino-1,2,4-triazole (0.35 g, 4.2 mmol) in DMF (5 mL) was irradiated at 70 °C for 30 min using microwave. The reaction mixture was cooled to room temperature, diluted with water (50 mL) and stirred sufficiently. The resulting mixture was filtered and washed with water to give the crude product, which was further purified by recrystallization from ethanol. Yellow precipitate, yield (0.71 g, 56.3%). Mp > 300 °C. FT-IR (νmax, cm−1): 3336 (OH), 3089 (C–H Ar), 1687 (CN). 1H NMR (DMSO-d6, δ ppm): 2.46 (s, 3H, H3C), 7.02–7.07 (m, 2H, ArH), 7.45–7.49 (m, 3H, ArH), 8.18 (d, 2H, J = 12.6 Hz, ArH), 8.22 (s, 1H, ArH), 8.28 (d, 1H, J = 12 Hz, ArH), 8.72 (s, 1H, ArH), 12.38 (s, 1H, HO), 13C NMR (DMSO-d6): δ 21.12 (CH3), 107.07 (CH), 117.80 (CH), 119.44 (CH), 119.55 (C), 126.90 (CH), 129.20 (CH), 129.73 (CH), 129.84 (C), 133.34 (CH), 142.05 (C), 147.66 (C), 154.21 (C-3a), 155.76 (CH), 158.79 (C), 161.28 (C). MS (m/z): 302 (M+, 100.0%), anal. calcd. for C18H14N4O (302.12): C, 71.51; H, 4.67; N, 18.53. Found: C, 71.48; H, 4.65; N, 18.50.
3.2.12. Synthesis of 7-(2-hydroxyphenyl)-5-(p-tolyl)imidazo[1,2-a]pyrimidine-2,3-dicarbonitrile (19). A solution of chalcone 1 (1 g, 4.2 mmol) and 2-amino-1H-imidazole-4,5-dicarbonitrile (0.55 g, 4.2 mmol) in DMF (5 mL) was irradiated at 70 °C for 30 min using microwave. The reaction mixture was cooled to room temperature, filtered and washed with water to give the crude product. Yellow precipitate, yield (0.16 g, 11%). Mp 247–250 °C. FT-IR (νmax, cm−1): 3189 (C–H Ar), 2246, 2228 (CN), 1614 (CN). 1H NMR (DMSO-d6, δ ppm): 2.32 (s, 3H, H3C), 7.02 (t, 1H, J = 7.2 Hz, ArH), 7.06 (d, 1H, J = 9 Hz, ArH), 7.45–7.49 (m, 3H, ArH), 7.67 (d, 2H, J = 8.4 Hz, ArH), 8.14 (s, 1H, ArH), 8.18 (d, 1H, J = 7.8 Hz, ArH), 11.68 (s, 1H, HO), 13C NMR (DMSO-d6): δ 21.14 (CH3), 102.64 (C-3), 108.23 (CH), 112.43 (CN), 112.55 (CN), 117.7 (CH), 119.78 (CH), 126.91 (C), 127.24 (CH), 129.04 (CH), 129.22 (CH), 129.42 (C), 130.19 (C), 134.00 (CH), 141.87 (C), 148.38 (C), 149.03 (C-8a), 158.50 (C), 162.07 (C). MS (m/z): 351 (M+, 100.0%), anal. calcd. for C21H13N5O (351.11): C, 71.79; H, 3.73; N, 19.93. Found: C, 71.71; H, 3.77; N, 19.89.
3.2.13. Synthesis of 2-(2-(p-tolyl)-2,3-dihydrobenzo[b][1,4]thiazepin-4-yl)phenol (21)47. To a flask containing an equimolar mixture (4.2 mmol) of chalcone 1 (1 g) and 2-aminothiophenol (0.5 g) in methanol (8 mL), drops of piperidine was added, and the mixture was irradiated at 70 °C for 10 min using microwave. Filter the formed yellow precipitate. Yield (1.28, 88.8%). Mp 152–154 °C (literature:47 156–158 °C). FT-IR (νmax, cm−1): 3428 (OH), 3051 (C–H Ar), 1598 (CN). 1H NMR (DMSO-d6, δ ppm): 2.28 (s, 3H, H3C), 2.90 (t, 1H, J = 13.2 Hz, H-1A), 3.50 (dd, 1H, J = 13.2 Hz, H–1B), 5.22 (dd, 1H, J = 12.6 Hz, H-2X), 6.95 (t, 1H, J = 8.4 Hz, ArH), 6.99 (d, 1H, J = 7.8 Hz, ArH), 7.12 (d, 2H, J = 7.8 Hz, ArH), 7.23 (d, 2H, J = 8.4 Hz, ArH), 7.27 (dt, 1H, J = 7.8 Hz, ArH), 7.37 (dd, 1H, J = 7.8 Hz, ArH), 7.45 (dt, 1H, J = 7.2 Hz, ArH), 7.56–7.60 (m, 2H, ArH), 7.86 (d, 1H, J = 7.8 Hz, ArH), 13C NMR (DMSO-d6): δ 20.65 (CH3), 36.44 (CH2), 58.86 (CH), 117.67 (CH), 118.06 (CH), 118.86 (C), 123.55 (CH), 125.35 (CH), 125.92 (CH), 126.61 (C-9a), 129.05 (CH), 129.79 (CH), 130.29 (CH), 133.79 (CH), 136.02 (CH), 136.94 (C), 140.84 (C), 148.35 (C-5a), 161.68 (C), 174.11 (C). MS (m/z): 345 (M+, 20.0%), anal. calcd. for C22H19NOS (345.12): C, 76.49; H, 5.54; N, 4.05. Found: C, 76.41; H, 5.49; N, 3.93.
3.3. Antioxidant activities
DPPH (2,2-diphenyl-1-picrylhydrazyl) radical scavenging activity. The scavenging activity of different heterocyclic compounds were determined using the free radical DPPH (2,2-diphenyl-1-picrylhydrazyl). Equal volumes of 100 μM DPPH chemical solution was mixed in methanol and added to different concentrations of the test compounds (0–200 μM mL−1) in methanol and mixed well. The reaction mixture was incubated for 30 min at room temperature in the dark and was then measured at 520 nm. Plotting the percentage DPPH˙ scavenging against concentration gave the standard curve and the percentage scavenging was calculated from the following equation:
| % scavenging = [(Absorbance of blank − Absorbance of test)/Absorbance of blank] × 100 |
IC50 was obtained from a plot between concentration of test compounds and % scavenging. Ascorbic acid (vitamine C) was used as standard for comparison.
3.4. Molecular docking simulation
The complex docking molecular analysis was enhanced with bond lengths in Å units using Moe software.48,69 Afterwards, with an RMS gradient of 0.01, the minimization energies were put into practise to sustain the geometrical optimisation and systematic studies, also Crystal Structure of Klebsiella pneumoniae R204Q HpxO complexed with FAD (PDBID:3RP8)49 The authorization described by E-conformation, overall statistics, and connected to amino acids around the protein's binding compact.70
3.5. Computational studies
Molecular geometry was directly taken from the experimental outcomes of X-ray diffraction without any constraints. Density functional theory including Becke's three-parameter hybrid functional using the LYP correlation functional (B3LYP) with the 6-31G(d, p) basis set via the Berny method54,55 were proceeded with the Gaussian 09W program71 In order to investigate the reactive sites for compound 1 and 3, the molecular electrostatic potential was calculated using the B3LYP/6-31G(d, p) method. Furthermore, the Mulliken atomic charges of all heterocyclic compounds were calculated as well.
4. Conclusions
In the present work, the reaction of chalcone with different nitrogen-containing investigated to form N-, S-, and O-containing heterocycles. The resulting heterocyclic compounds were confirmed by spectroscopic analyses, including FTIR, UV-visible, NMR, and mass spectrometry. The antioxidant activity of these compounds was studied for their efficacy as antioxidants through their ability to scavenge the synthetic radicals 2,2-diphenyl-1-picrylhydrazyl (DPPH). Compound 3 (IC50 = 93.4 μM) and compound 1 (IC50 = 134.26 μM) showed the highest antioxidant activity, indicating their potential as antioxidants. On the other hand, compound 8 (IC50 = 448.70 μM) exhibited the lowest activity when compared to vitamin C (IC50 ∼ 141.9 μM). In addition, the docking estimations were compatible with experimental results for these heterocyclic compounds evaluated with PDBID:3RP8. Furthermore, DFT/B3LYP/6-31G(d,p) basis sets were used to optimize the compounds and identify their Global reactivity descriptors, including HOMO–LUMO gaps, electronic hardness, chemical potential, electrophilicity index, and Mulliken charges. In comparison to the original chalcone 1, all synthesized compounds 3–21 exhibited higher energy stability. Among them, compound 19 demonstrated a particularly high dipole moment, promoting the separation of charges and increasing reactivity. Meanwhile, compound 9 displayed higher hardness and lower softness values, displaying lower intramolecular charge transfer. Furthermore, compound 9 had the highest ΔE value, indicating greater kinetic stability, while compound 6b had the lowest ΔE, indicating high chemical reactivity. The DFT calculations were also used to determine the molecular electrostatic potential (MEP) of the two compounds that demonstrated the highest antioxidant activity.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The Authors acknowledge the National research centre and Ain Shams University for the facilities provided.
References
- A. Mermer, T. Keles and Y. Sirin, Bioorg. Chem., 2021, 114, 105076 CrossRef CAS PubMed.
- A. M. Fahim and M. A. Shalaby, J. Mol. Struct., 2019, 1176, 408–421 CrossRef CAS.
- A. I. Khodair, S. E. Kassab, N. A. Kheder and A. M. Fahim, Carbohydr. Res., 2022, 514, 108546 CrossRef CAS PubMed.
- A. M. Farag and A. M. Fahim, J. Mol. Struct., 2019, 1179, 304–314 CrossRef CAS.
- R. J. Obaid, N. Naeem, E. U. Mughal, M. M. Al-Rooqi, A. Sadiq, R. S. Jassas, Z. Moussa and S. A. Ahmed, RSC Adv., 2022, 12, 19764–19855 RSC.
- A. M. Fahim, M. A. Shalaby and M. A. Ibrahim, J. Mol. Struct., 2019, 1194, 211–226 CrossRef CAS.
- S. A. Rizk, S. S. Abdelwahab and H. A. Sallam, J. Heterocycl. Chem., 2018, 55, 1604–1614 CrossRef CAS.
- M. A. Fahim, S. M. Elshikh and M. N. Darwish, Curr. Comput.-Aided Drug Des., 2020, 16, 486–499 CrossRef PubMed.
- S. Wang, X.-H. Yuan, S.-Q. Wang, W. Zhao, X.-B. Chen and B. Yu, Eur. J. Med. Chem., 2021, 214, 113218 CrossRef CAS PubMed.
- P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, J. Med. Chem., 2021, 64, 2339–2381 CrossRef CAS PubMed.
- P. Das, M. D. Delost, M. H. Qureshi, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2018, 62, 4265–4311 CrossRef PubMed.
- A. M. Fahim and E. H. Ismael, Egypt. J. Chem., 2019, 62, 1427–1440 Search PubMed.
- S. A. Rizk and S. Shaban, J. Heterocycl. Chem., 2019, 56(9), 2379–2388 CrossRef CAS.
- K. N. M. Halim, S. A. Rizk, M. A. El-Hashash and S. K. Ramadan, J. Heterocycl. Chem., 2021, 58, 636–645 CrossRef CAS.
- Y. U. Cebeci, H. Bayrak, Ş. A. Karaoğlu and A. M. Fahim, J. Mol. Struct., 2022, 1260, 132810 CrossRef CAS.
- S. A. Rizk, A. A. El-Sayed and M. M. Mounier, J. Heterocycl. Chem., 2017, 54, 3358–3371 CrossRef CAS.
- H. ur Rashid, M. A. U. Martines, A. P. Duarte, J. Jorge, S. Rasool, R. Muhammad, N. Ahmad and M. N. Umar, RSC Adv., 2021, 11, 6060–6098 RSC.
- B. Colom-Fernández, A. Kreutzman, A. Marcos-Jiménez, V. García-Gutiérrez, C. Cuesta-Mateos, I. Portero-Sainz, Y. Pérez-García, L. F. Casado, F. Sánchez-Guijo and J. Martínez-López, Front. Pharmacol., 2019, 10, 1340 CrossRef PubMed.
- F. C. K. Dolk, K. B. Pouwels, D. R. Smith, J. V. Robotham and T. Smieszek, J. Antimicrob. Chemother., 2018, 73, ii2–ii10 CrossRef CAS PubMed.
- K. Haider, M. Shafeeque, S. Yahya and M. S. Yar, J. Med. Chem. Rep., 2022, 100042 CAS.
- A. Kumar, A. K. Singh, H. Singh, V. Vijayan, D. Kumar, J. Naik, S. Thareja, J. P. Yadav, P. Pathak and M. J. P. Grishina, Pharmaceuticals, 2023, 16, 299 CrossRef CAS.
- A. Aboelnaga, E. Mansour, A. M. Fahim and G. H. Elsayed, J. Mol. Struct., 2022, 1251, 131945 CrossRef CAS.
- A. M. Fahim, A. M. Farag, A. Mermer, H. Bayrak and Y. Şirin, J. Mol. Struct., 2021, 1233, 130092 CrossRef CAS.
- J. Cossy and A. Guerinot, in Advances in Heterocyclic Chemistry, Elsevier, 2016, vol. 119, pp. 107–142 Search PubMed.
- A. M. Fahim, H. E. M. Tolan and H. Awad, J. Iran. Chem. Soc., 2021, 18, 2965–2981 CrossRef CAS.
- S. Rizk, Egypt. J. Chem., 2008, 51, 611–621 Search PubMed.
- X. Wang, Q. Hu, H. Tang and X. Pan, Pharmaceuticals, 2023, 16, 228 CrossRef CAS.
- M. Ashfaq, M. N. Tahir, K. S. Munawar, R. Behjatmanesh-Ardakani and H. Kargar, J. Mol. Struct., 2022, 1261, 132952 CrossRef CAS.
- B. Meyer, Chem. Rev., 1976, 76, 367–388 CrossRef CAS.
- R. Mehmood, E. U. Mughal, E. B. Elkaeed, R. J. Obaid, Y. Nazir, H. A. Al-Ghulikah, N. Naeem, M. M. Al-Rooqi, S. A. Ahmed and S. W. A. Shah, ACS omega, 2022, 7, 30215–30232 CrossRef CAS PubMed.
- S. B. Nimse and D. Pal, RSC Adv., 2015, 5, 27986–28006 RSC.
- A. Mermer and S. Alyar, Chem.-Biol. Interact., 2022, 351, 109742 CrossRef CAS PubMed.
- A. Gusain, N. Kumar, J. Kumar, G. Pandey and P. K. Hota, J. Fluoresc., 2021, 31, 51–61 CrossRef CAS PubMed.
- W. A. Yehye, N. A. Rahman, A. Ariffin, S. B. Abd Hamid, A. A. Alhadi, F. A. Kadir and M. Yaeghoobi, Eur. J. Med. Chem., 2015, 101, 295–312 CrossRef CAS PubMed.
- T. Jaehnert, M. D. Hager and U. S. Schubert, J. Mater. Chem. A, 2014, 2, 15234–15251 RSC.
- M. Carocho and I. C. Ferreira, Food Chem. Toxicol., 2013, 51, 15–25 CrossRef CAS PubMed.
- S. Schmidt and J. Pokorný, Czech J. Food Sci., 2005, 23, 93–102 CrossRef CAS.
- J. Hu, L. Yang, P. Yang, S. Jiang, X. Liu and Y. Li, Biomater. Sci., 2020, 8, 4940–4950 RSC.
- C. Nopo-Olazabal, J. Hubstenberger, L. Nopo-Olazabal and F. Medina-Bolivar, J. Agric. Food Chem., 2013, 61, 11744–11758 CrossRef CAS PubMed.
- D. Charles, Antioxidant Properties of Spices, Herbs and Other Sources, Springer, New York, NY, 2012 Search PubMed.
- S. K. Ramadan and S. A. Rizk, J. Iran. Chem. Soc., 2022, 19, 187–201 CrossRef CAS.
- S. Upadhyay, A. C. Tripathi, S. Paliwal and S. K. Saraf, Pharm. Chem. J., 2017, 51, 564–575 CrossRef CAS.
- M. Shyam, H. Verma, G. Bhattacharje, P. Mukherjee, S. Singh, S. Kamilya, P. Jalani, S. Das, A. Dasgupta, A. Mondal, A. K. Das, A. Singh, F. Brucoli, C. Bagnéris, R. Dickman, V. N. Basavanakatti, P. Naresh Babu, V. Sankaran, A. Dev, B. N. Sinha, S. Bhakta and V. Jayaprakash, J. Med. Chem., 2022, 65, 234–256 CrossRef CAS PubMed.
- A. M Fahim, E. H. I. Ismael, G. H. Elsayed and A. M. Farag, J. Biomol. Struct. Dyn., 2022, 40(19), 9177–9193 CrossRef CAS PubMed.
- A. Nie, J. Wang and Z. Huang, J. Comb. Chem., 2006, 8, 646–648 CrossRef CAS PubMed.
- M. A. Shalaby, H. M. Al-Matar, A. M. Fahim and S. A. Rizk, J. Phys. Chem. Solids, 2022, 170, 110933 CrossRef CAS.
- H. Loghmani-Khouzani, P. Tamjidi, I. Mohammadpoor-Baltork, M. Yaeghoobi, N. A. Rahman, A. R. Khosropour, M. Moghadam, S. Tangestaninejad, V. Mirkhani, M. H. Habibi, A. Kashima and T. Suzuki, J. Heterocycl. Chem., 2014, 51, 138–150 CrossRef CAS.
- S. Vilar, G. Cozza and S. Moro, Curr. Top. Med. Chem., 2008, 8, 1555–1572 CrossRef CAS PubMed.
- K. A. Hicks, S. E. O'Leary, T. P. Begley and S. E. Ealick, Biochemistry, 2013, 52, 477–487 CrossRef CAS PubMed.
- G. H. Elsayed, S. Dacrory and A. M. Fahim, Int. J. Biol. Macromol., 2022, 222, 3077–3099 CrossRef CAS PubMed.
- P. Pyykkö, Chem. Soc. Rev., 2008, 37, 1967–1997 RSC.
- M. Spiegel, T. Marino, M. Prejanò and N. Russo, Phys. Chem. Chem. Phys., 2022, 24, 16353–16359 RSC.
- M. Spiegel, T. Marino, M. Prejanò and N. Russo, J. Mol. Liq., 2022, 366, 120343 CrossRef CAS.
- C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49–56 CrossRef CAS.
- H. B. Schlegel, J. Comput. Chem., 1982, 3, 214–218 CrossRef CAS.
- A. Mohamed, A. M. Fahim and M. A. Ibrahim, J. Mol. Model., 2020, 26, 354 CrossRef CAS PubMed.
- G. Zhang and C. B. Musgrave, J. Phys. Chem. A, 2007, 111, 1554–1561 CrossRef CAS PubMed.
- V. Choudhary, A. Bhatt, D. Dash and N. Sharma, J. Comput. Chem., 2019, 40, 2354–2363 CrossRef CAS PubMed.
- A. Nekrouf, K. Toubal, Y. Megrouss, N. E. H. Belkafouf, A. Djafri, N. Khelloul, J.-C. Daran, A. Djafri and A. Chouaih, J. Mol. Struct., 2022, 1262, 133002 CrossRef CAS.
- Z. Demircioğlu, G. Kaştaş, Ç. A. Kaştaş and R. Frank, J. Mol. Struct., 2019, 1191, 129–137 CrossRef.
- J. Pandey, P. Prajapati, A. Srivastava, P. Tandon, K. Sinha, A. P. Ayala and A. K. Bansal, Spectrochim. Acta, Part A, 2018, 203, 1–12 CrossRef CAS PubMed.
- A. M. Fahim, H. S. Magar and N. H. Mahmoud, Appl. Organomet. Chem., 2023, 37(4), e7023 CrossRef CAS.
- A. Attia, A. Aboelnaga and A. M. Fahim, Egypt. J. Chem., 2022 DOI:10.21608/ejchem.2022.166142.7046.
- B. M. Ivković, K. Nikolic, B. B. Ilić, Ž. S. Žižak, R. B. Novaković, O. A. Čudina and S. M. Vladimirov, Eur. J. Med. Chem., 2013, 63, 239–255 CrossRef PubMed.
- M. Shyam, H. Verma, G. Bhattacharje, P. Mukherjee, S. Singh, S. Kamilya, P. Jalani, S. Das, A. Dasgupta and A. Mondal, J. Med. Chem., 2022, 65, 234–256 CrossRef CAS PubMed.
- A. M. A. Algohary, A. M. Hassan, A. Y. Alzahrani and S. A. Rizk, J. Heterocycl. Chem., 2023, 1 DOI:10.1002/jhet.4647.
- M. A. Shalaby, A. M. Fahim and S. A. Rizk, Sci. Rep., 2023, 13, 4999 CrossRef CAS PubMed.
- R. Ramajayam, R. Giridhar, M. R. Yadav, H. Djaballah, D. Shum and C. Radu, J. Enzyme Inhib. Med. Chem., 2007, 22(6), 716–721 CrossRef CAS PubMed.
- A. M. Fahim, J. Mol. Struct., 2023, 1277, 134871 CrossRef CAS.
- K. M. Elokely and R. J. Doerksen, J. Chem. Inf. Model., 2013, 53, 1934–1945 CrossRef CAS PubMed.
- J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. Hratchian and A. Izmaylov, Gaussian 09, Revision A.02, Inc., Wallingford, 2009 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Sameh A. Rizka
*b and
Sameh A. Rizka