
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient and stereoselective synthesis of sugar fused pyrano[3,2-c]pyranones as anticancer agents†
Sandeep Kumar *ab,
Ram Krishna Sahuc,
Priti Kumaria,
Jyotirmoy Maityd,
Binayak Kumarc,
Rajni Johar Chhatwale,
Brajendra K. Singh*a and
Ashok K. Prasad‡
a
*ab,
Ram Krishna Sahuc,
Priti Kumaria,
Jyotirmoy Maityd,
Binayak Kumarc,
Rajni Johar Chhatwale,
Brajendra K. Singh*a and
Ashok K. Prasad‡
a
aBioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi-110007, India. E-mail: skbanger388@gmail.com
bDepartment of Chemistry, Ramjas College, University of Delhi, Delhi-110007, India
cNational Institute of Cancer Prevention & Research, Noida, Uttar Pradesh, India
dDepartment of Chemistry, St. Stephen College, University of Delhi, Delhi-110007, India
eDepartment of Chemistry, Maitreyi College, University of Delhi, Delhi, India
First published on 18th August 2023
Abstract
A highly stereoselective, efficient and facile route was achieved for the synthesis of novel and biochemically potent sugar fused pyrano[3,2-c]pyranone derivatives starting from inexpensive, naturally occurring D-galactose and D-glucose. First, β-C-glycopyranosyl aldehydes were synthesized from these D-hexose sugars in six steps, with overall yields 41–55%. Next, two different 1-C-formyl glycals were synthesized from these β-C-glycopyranosyl aldehydes by treatment in basic conditions. The optimization of reaction conditions was carried out following reactions between 1-C-formyl galactal and 4-hydroxycoumarin. Next, 1-C-formyl galactal and 1-C-formyl glucal were treated with nine substituted 4-hydroxy coumarins at room temperature (25 °C) in ethyl acetate for ∼1–2 h in the presence of L-proline to obtain exclusively single diastereomers of pyrano[3,2-c]pyranone derivatives in excellent yields. Four compounds were found to be active for the MCF-7 cancer cell line. The MTT assay, apoptosis assay and migration analysis showed significant death of the cancer cells induced by the synthesized compounds.
Introduction
Cancer is one of the most horrifying human diseases and prominent causes of death worldwide.1 Studies estimate that one in five people before hitting 75 years of age will suffer from cancer.2 Also, according to the World Health Organization (WHO) report, around 13 million people will die due to cancer in 2030.3,4 So there has been continuous efforts for the development of drugs to fight against cancer. But due to the cytotoxicity and drug-resistance problems encountered with many available treatments, it has become important to discover and develop highly effective drugs to treat cancer.1,5 Over 60% of conventional anticancer drugs have originated from natural sources.6 Sugars, being natural are an important class of biomolecules which act as building blocks for numerous biologically active compounds and they have been proven as an untapped source of new drug molecules, offering new therapeutic opportunities.7,8 On the other hand, oxygen containing heterocyclic moieties were found to be a key structural motif in many biological important molecules.9 So, fusion or combination of these two kinds of biologically significant structural moieties would have potential to give rise to new drug-like bioactive compounds, which would be able to interact with enzymes or receptors and inflect their functions.10 This inventiveness has encouraged synthetic chemists to take up challenges to produce these potent molecules in an efficient and facile way.Pyrano[3,2-c]pyranones constitute an important class of oxygen containing heterocyclic compounds and are found to play important roles in many biologically active drugs. In the past few decades, this motif has drawn attention in medicinal chemistry due to its biological and pharmaceutical activities such as anti-cancer, antifungal, anti-HIV, anti-inflammatory, antioxidant and antibacterial etc.11–15 Some of the pyrano[3,2-c]pyranones were used for the treatment of skin and anti-neurodegenerative disorders such as Alzheimer's and Parkinson's diseases.16,17 Moreover, this structure constituted building blocks for many natural products found in plants,18–20 where some of them showed high potency towards biological activities.21 For instance, triptiliospinocoumarin (A),22 vismiaguian A (B),23 pyripyropene T (C),24 were found naturally while synthetic compound D25 was cytotoxic to neuroblastoma and melanoma cells. The tricyclic E 26–F 17 might be utilised for the prevention and treatment of neurodegenerative diseases, such as Alzheimer's disease. Compounds G–J were strong inhibitors of acetylcholinesterase activity, DNA synthesis, and leukemic cell growth.21,27 Recently, sugar fused pyrano-pyranone derivative K28,29 has been proved to be an antiproliferative agent i.e. against breast and liver cancer cell (Fig. 1).
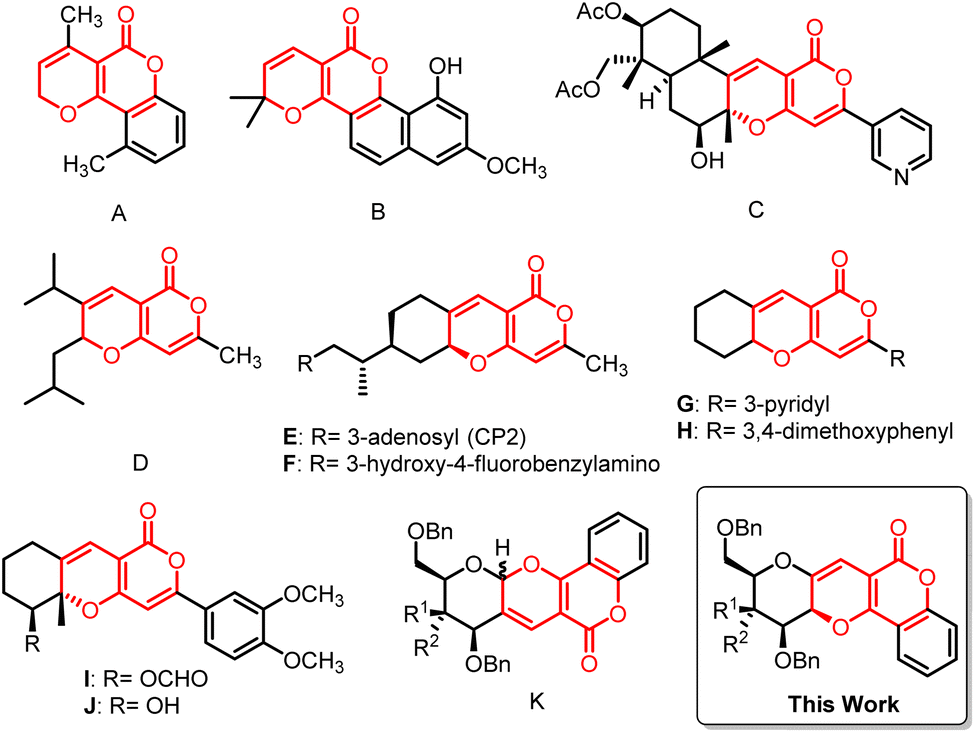 | ||
| Fig. 1 Significant pyrano[3,2-c]pyranone derivatives. | ||
The generation of enantiopure sugar fused molecules from readily available sugars is of much practical value. The values become more concerned when these individual reactions employ simple reagents, proceed smoothly, avoid complex separation in work-up procedures and enable favourable overall yields. Herein, we report a small library of two series of sugar fused pyrano[3,2-c]pyranone derivatives, which were afforded stereo-specifically in excellent yields starting from natural sugars i.e. D-galactose and D-glucose. No requirement of high temperature, short reaction time for the synthesis of these privileged compounds in diastereomeric pure form are the key factors of present synthesis.
Results and discussion
Chemistry
Sugar fused derivatives of pyrano[3,2-c]pyranone were synthesized earlier by Sagar28,29 and co-workers in 2009 and further explored for anti-cancer activity in 2018. This synthetic methodology involved 2-C-formyl glycals and 4-hydroxy coumarins as starting materials which were reacted in toluene–AcOH solvent mixture, using pyrrolidine as catalyst, when a diastereomeric mixture (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) was afforded for 2-C-formyl glucal.
:50) was afforded for 2-C-formyl glucal.
However, 2-C-formyl galactal showed a better diasteroselectivity and the mixture was obtained in 99:1 ratio (Scheme 1).28,29 It was also observed that elevated temperature helped the reaction to achieve the desired products in high yields.
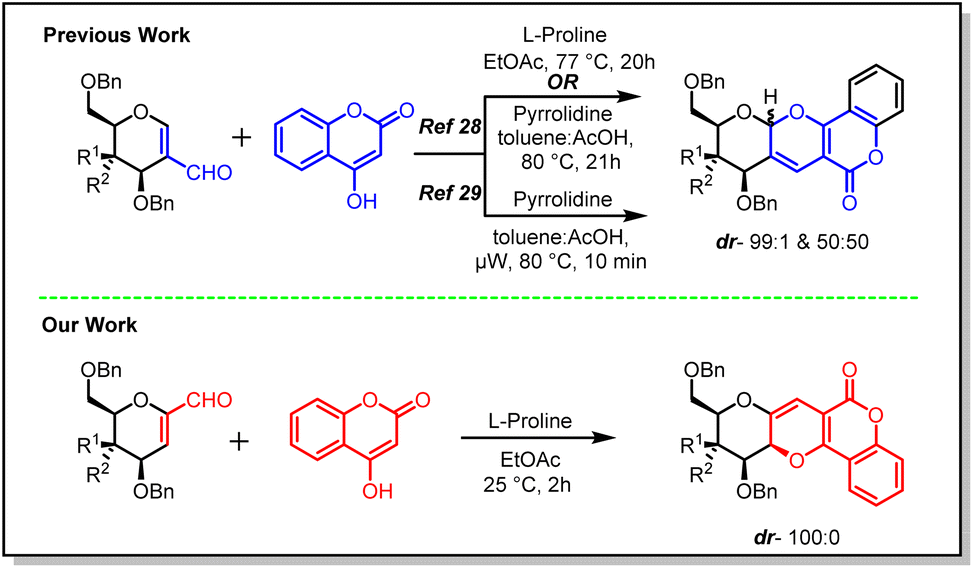 | ||
| Scheme 1 Synthetic protocol of two different sugar fused pyrano[3,2-c]pyranone derivatives. | ||
On this occasion, we undertook a similar reaction on 1-C-formyl glycals. The primary objective of this investigation was to eliminate the diastereoselectivity in the resulting product and additionally assess the biological activity of a single isomer of these sugar-fused pyrano[3,2-c]pyranones. Hence, our reaction methodology consisted of reaction between 1-C-formyl glycal and 4-hydroxy coumarins in ethyl acetate solvent system, where L-proline was used as catalyst. Under this reaction conditions, stereoselective single isomer formation was observed in case of both gluco- and galacto-derivatives of 1-C-formyl glycal (Scheme 1). Herein, the reaction proceeded well at room temperature and there was no requirement of higher temperature. In fact, the progress of the reaction was noted even at 5 °C. Comparatively, a better yield was afforded by this procedure in comparison to the previously mentioned literature procedures (Scheme 1). The sugar fused pyrano[3,2-c]pyranone derivatives synthesized following this method had C–C bond at anomeric position of sugar moiety, instead of C–O bond as was observed in the previous case, which guided us to categorise these molecules as C-glycosides. The structural alteration resulting from transformation of anomeric centre from C–O acetal to strong C–C bond in C-glycosides would stimulate the resistance of these molecules towards chemical/enzymatic hydrolysis and/or metabolic processes, which might enable them to be potent biologically active candidate.30
It was envisioned that the reaction of 1-C-formylglycal with 4-hydroxy coumarin could generate the desired pyrano[3,2-c]pyranone derivatives through C-1,2 nucleophilic addition followed by 6π-electrocyclization reaction. The pioneer reaction was carried out by Link and his co-workers,31,32 in the year 1944. Later, this reaction condition was explored by many research groups in attempt to synthesize variety of natural products and biologically active molecules.33–36 It was noted that the researchers faced challenges in terms of stereoselectivity, reaction time, temperature, isolated yield and substrate scope etc. while carrying out this reaction. Therefore, there was a constant need to develop an efficient and stereo-controlled method for the synthesis of pyrano[3,2-c]pyranones.
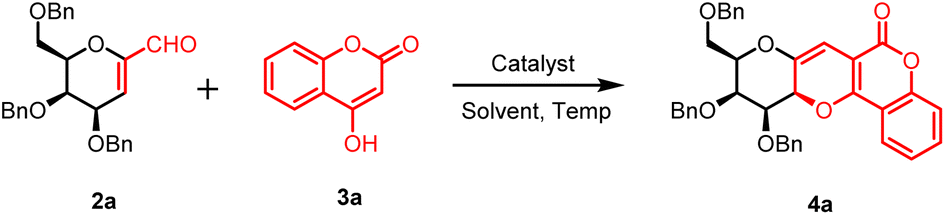
In our research work, we started the synthetic methodology with cheap and naturally available sugar molecules i.e. D-galactose and D-glucose. These natural sugars were transformed into C-glycopyranosyl aldehydes 1a–b following a reaction methodology consisting six steps, which afforded the compounds in 41–55% overall yields.37 Next, these sugar aldehydes 1a–b were treated with sodium methoxide in methanol at room temperature to afford 1-C-formyl glycals 2a–b in 90–92% yields. Next, the reaction conditions for the model reaction between 1-C-formyl galactal 2a with 4-hydroxy coumarin 3a was optimised with respect to catalyst, solvent and temperature (Table 1). Thus, starting materials 1-C-formyl galactal 2a and 4-hydroxy coumarin 3a in presence of catalyst L-proline in toluene–acetic acid solvent system at 25 °C afforded desired sugar fused pyrano[3,2-c]pyranone 4a in 68% yield (entry 1, Table 1). The crude product was purified by column chromatography using ethyl acetate and petroleum ether as the solvent system to obtain the pure compound as a solid material. 1H and 13C NMR spectra of crude product indicated the presence of only one isomer, inferring that the reaction was absolutely stereoselective. However, as the isolated yield was only 68%, there was need to improve the isolated yield. Thereafter, the catalyst and solvent system were kept same and optimization was done at higher temperatures i.e. 80 °C, 120 °C and 150 °C, which afforded the desired product in 55, 42 and 30% yields, respectively (entries 2–4, Table 1). It implicated that, with the raise in temperature, the yield of product was decreasing. It was concluded that the room temperate i.e. 25 °C would be optimum temperature to obtain higher yield. Next, catalyst was changed i.e. pyrrolidine was used which also did not show any promising effect on product yield (entry 5, Table 1). It was also observed that there was a slight increment in obtained yield as the temperature went down to 25 °C. So, catalyst L-proline was reconsidered as the chosen catalyst at temperature 25 °C, which afforded a higher yield (78%) (entry 5, Table 1). Further, solvent system was changed to ethyl acetate while retaining L-proline as catalyst at 25 °C, which produced the desired product in 91% yield (entry 6, Table 1). When the reaction was carried out without the catalyst L-proline, only trace of product was observed (entry 7, Table 1). Next, we used the polar solvents like DMSO, DMF, MeOH, EtOH and found that the former two solvents were inefficient for the product formation and later two solvents afforded the desired product with very poor yield (entries 8–11, Table 1). All other non-polar solvents like benzene, THF, MeCN, DCM, dioxane, acetone were also proved less effective for the conversion of starting material into product (entries 12–19, Table 1). Furthermore, an alternative greener solvent, iso-propyl acetate, was employed instead of ethyl acetate. However, the resulting yield was slightly lower compared to the yield obtained with ethyl acetate (entry 20, Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (20 mol%) | Solvent | Temp (°C) | Time | Yield (%) |
| a Condition: 3,4,6-tri-O-benzyl-1-formyl galactal (2a, 1.0 mmol), 4-hydroxy coumarin (3a, 1.2 mmol) and L-proline (0.2 mmol), ethyl acetate (5 mL), 25 °C, 1.5 h.b Alternative organocatalysts such as pyrrolidine, piperidine, and morpholine were also employed in addition to L-proline. Nevertheless, the optimal yield was achieved when L-proline was utilized instead of the other catalysts.c The reaction did not reach completion within the specified time frame. | |||||
| 1 | L-Proline | Toluene–AcOH | 25 | 2 h | 68 |
| 2 | L-Proline | Toluene–AcOH | 80 | 1 h 50 min | 55 |
| 3 | L-Proline | Toluene–AcOH | 120 | 1 h 10 min | 42 |
| 4 | L-Proline | Toluene–AcOH | 150 | 40 min | 30 |
| 5 | Pyrrolidine | Toluene–AcOH | 25 | 2 h | 58 |
| 6b | L-Proline | Ethyl acetate | 25 | 1 h 30 min | 91 |
| 7c | — | Ethyl acetate | 25 | 24 h | Trace |
| 8c | L-Proline | DMSO | 25 | 24 h | NR |
| 9c | L-Proline | DMF | 25 | 24 h | NR |
| 10 | L-Proline | MeOH | 25 | 24 h | 22 |
| 11 | L-Proline | EtOH | 25 | 24 h | 24 |
| 12 | L-Proline | Benzene | 25 | 2 h | 45 |
| 13 | L-Proline | THF | 25 | 2 h | 48 |
| 14 | L-Proline | CH3CN | 25 | 2 h | 35 |
| 15 | L-Proline | Toluene | 25 | 2 h | 57 |
| 16 | L-Proline | 1,4-Dioxane | 25 | 24 h | 25 |
| 17 | L-Proline | CHCl3 | 25 | 2 h | 58 |
| 18 | L-Proline | DCM | 25 | 24 h | 20 |
| 19c | L-Proline | Acetone | 25 | 24 h | Trace |
| 20 | L-Proline | iso-Propyl acetate | 25 | 2 h 30 min | 82 |
The optimised reaction conditions i.e. L-proline as catalyst in ethyl acetate at 25 °C was used for the reaction of 2a with nine other freshly prepared differently substituted 4-hydroxy coumarins 3a–i38 to afford sugar fused pyrano[3,2-c]pyranones 4a–i in excellent yields (Scheme 2).
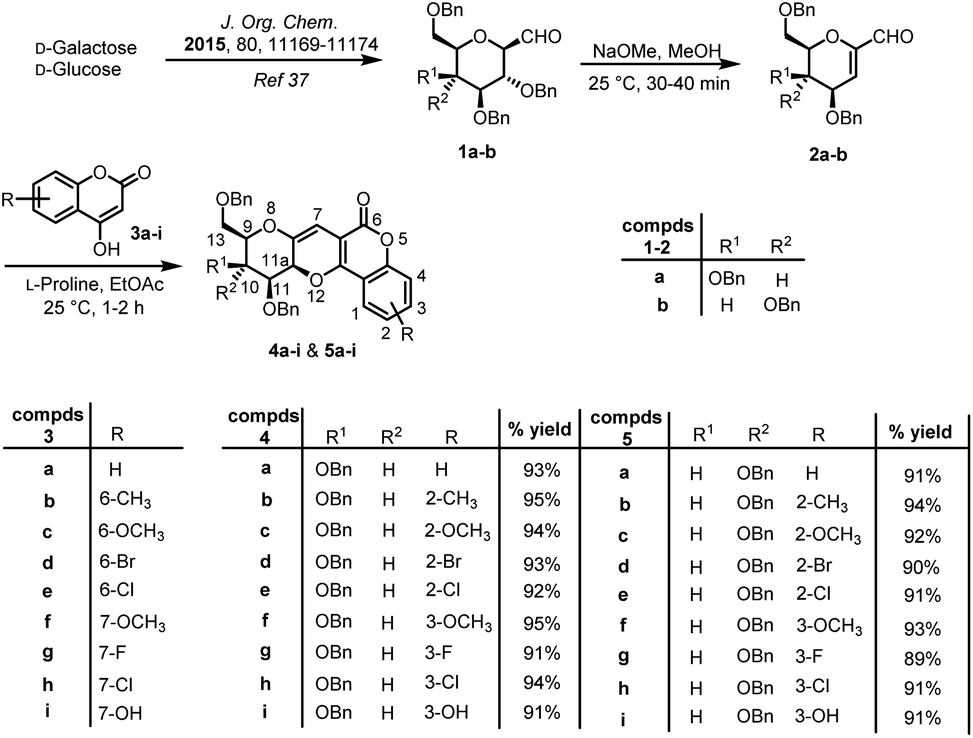 | ||
| Scheme 2 Synthesis of sugar fused pyrano[3,2-c]pyranone derivatives 4a–i and 5a–i. | ||
Similarly, the designed protocol was also applied on 1-C-formyl glucal 2b and interestingly, here also only one stereoisomer was observed on TLC, which was finally confirmed by 1H and 13C NMR spectra of crude product. Thus, taking optimised conditions in hand, further reactions of 1-C-formyl glucal 2b with substituted 4-hydroxy coumarins 3a–i were carried out to furnish sugar fused pyrano[3,2-c]pyranones 5a–i in excellent yields. Thus, a small library of eighteen potent biologically active sugar fused pyrano[3,2-c]pyranones were prepared (Scheme 2). It was noticed that the reaction for production of pyrano[3,2-c]pyranone derivatives proceeded smoothly for 1-C-formyl galactal 2a as compared to 1-C-formyl glucal 2b with respect to percentage yield and time taken for the completion of the reaction.
Stereochemistry
The stereochemistry at C-11a position of these compounds found to be “S” based on NOESY spectrum of compound 4a and 5a. In the NOESY spectrum of compound 4a, cross peaks at δ 4.08 ppm (for C-11H) and 5.52 ppm (for C-11aH) were observed. Similarly, cross peaks at δ 4.11 ppm (for C-11H) and 5.19 ppm (for C-11aH) were observed in the NOESY spectrum of compound 5a. So, both these spectra confirmed the stereochemistry as “S” at C-11a position of the compounds (Fig. 2). Similar trend was followed to assign the stereochemistry of all the synthesized compounds 4a–i and 5a–i.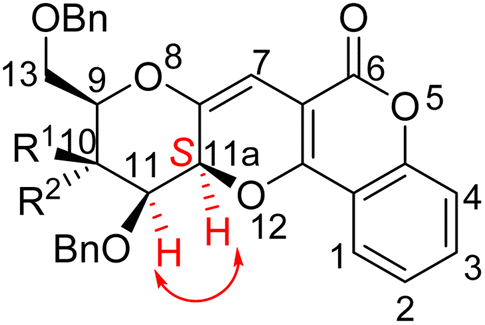 | ||
| Fig. 2 Compound 4a and 5a showing NOESY interaction. | ||
Plausible mechanism
The mechanism for the stereoselective synthesis of sugar fused pyrano[3,2-c]pyranone can be proposed as illustrated in Fig. 3. Attack of L-proline on 1-C-formyl glycals 2a–b produced intermediate I which underwent dehydration to form iminium ion II. Now 1,2-nucleophilic addition of 4-hydroxy coumarin on iminium ion II furnished intermediate III which on rearrangement produced intermediate IV. β-Elimination of intermediate IV furnished 6π electron system V which finally electro cyclized to form desired sugar fused pyrano[3,2-c]pyranone derivatives 4a–i and 5a–i. | ||
| Fig. 3 Plausible mechanism for the synthesis of sugar fused pyrano[3,2-c]pyranones. | ||
The structures of all the synthesized compounds 1a–c, 2a–b, 3a–i, 4a–i and 5a–i were unambiguously established on the basis of their spectral (IR, 1H-, 13C-NMR, 1H-1H COSY NMR, 1H-13C HETCOR NMR, NOESY NMR and HRMS) data analysis. The structure of known compounds 1a–c, 2a–b and 3a–i were further confirmed by the comparison of their physical and spectral data with those reported in literature.38,39
Biology
Anticancer potential of synthesized sugar fused pyrano[3,2-c]pyranones (n = 18) along with different substituted 4-hydroxycoumarin precursors (n = 9) at 100 μM & 120 μM was investigated by MTT assay in MCF-7 and MDA-MB-231 breast cancer cell lines. MCF-7 cells represent estrogen receptor (ER), progesterone receptor (PR) positive and Her-2 (HER-2) negative expression subtype of breast cancer whereas MDA-MB-231 is all three negative expression subtype called triple −ve breast cancer. Results showed four compounds have significant growth inhibition in MCF-7 cells whereas, none of the compound have significant growth inhibition in MDA-MB-231 cell line when treated with compound up to 120 μM concentrations. Results are indicating specificity of these compounds for ER +ve, PR +ve and Her-2 −ve sub-category of breast cancer but unfortunately it does not work for triple-negative breast cancer subtype (Table 2).| Compound | Cell viabilitya (%) ± SD | |||
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | |||
| 100 μM | 120a μM | 120 μM | 100a μM | |
| a Concentration of epirubicin is 125 μM instead of 120 μM. | ||||
| 4a | 94 ± 0.16 | 99 ± 0.17 | 99 ± 2.41 | 92 ± 0.42 |
| 4b | 98 ± 0.43 | 92 ± 0.17 | 92 ± 3.45 | 96 ± 0.09 |
| 4c | 93 ± 0.42 | 71 ± 0.09 | 89 ± 2.80 | 82 ± 0.80 |
| 4d | 93 ± 0.36 | 96 ± 0.17 | 98 ± 2.41 | 85 ± 2.22 |
| 4e | 86 ± 0.67 | 83 ± 0.28 | 89 ± 3.00 | 88 ± 0.98 |
| 4f | 99 ± 0.51 | 95 ± 0.27 | 91 ± 2.10 | 89 ± 0.08 |
| 4g | 83 ± 0.15 | 78 ± 0.07 | 92 ± 1.60 | 87 ± 1.45 |
| 4h | 80 ± 0.18 | 69 ± 0.20 | 84 ± 2.39 | 99 ± 2.38 |
| 4i | 93 ± 0.91 | 97 ± 0.12 | 99 ± 3.14 | 97 ± 0.05 |
| 5a | 92 ± 0.12 | 99 ± 0.23 | 93 ± 2.67 | 98 ± 2.35 |
| 5b | 96 ± 0.08 | 99 ± 0.09 | 96 ± 1.34 | 83 ± 1.28 |
| 5c | 56 ± 3.60 | 50 ± 0.80 | 86 ± 2.27 | 81 ± 2.30 |
| 5d | 98 ± 0.23 | 99 ± 0.29 | 93 ± 2.34 | 84 ± 0.94 |
| 5e | 99 ± 0.17 | 98 ± 0.13 | 97 ± 3.87 | 88 ± 0.89 |
| 5f | 98 ± 0.11 | 96 ± 0.07 | 82 ± 1.33 | 89 ± 0.45 |
| 5g | 50 ± 0.74 | 41 ± 2.71 | 96 ± 2.80 | 99 ± 2.80 |
| 5h | 50 ± 0.90 | 45 ± 2.00 | 80 ± 3.31 | 78 ± 3.21 |
| 5i | 59 ± 2.94 | 50 ± 0.71 | 88 ± 1.92 | 96 ± 1.45 |
| Epirubicin (positive control) | 49 ± 12.04 | 44 ± 11.93 | 79 ± 11.45 | 65 ± 15.05 |
From the study, it has been shown that glucal fused pyrano[3,2-c]pyranones compounds 5c, 5g, 5h and 5i exhibited moderate inhibitory effect against MCF-7 cells among all derivatives with an IC50 of 120, 100, 100 and 120 μM respectively (Fig. 4). While compounds of galactal series found to be inactive for both cancer cell lines.
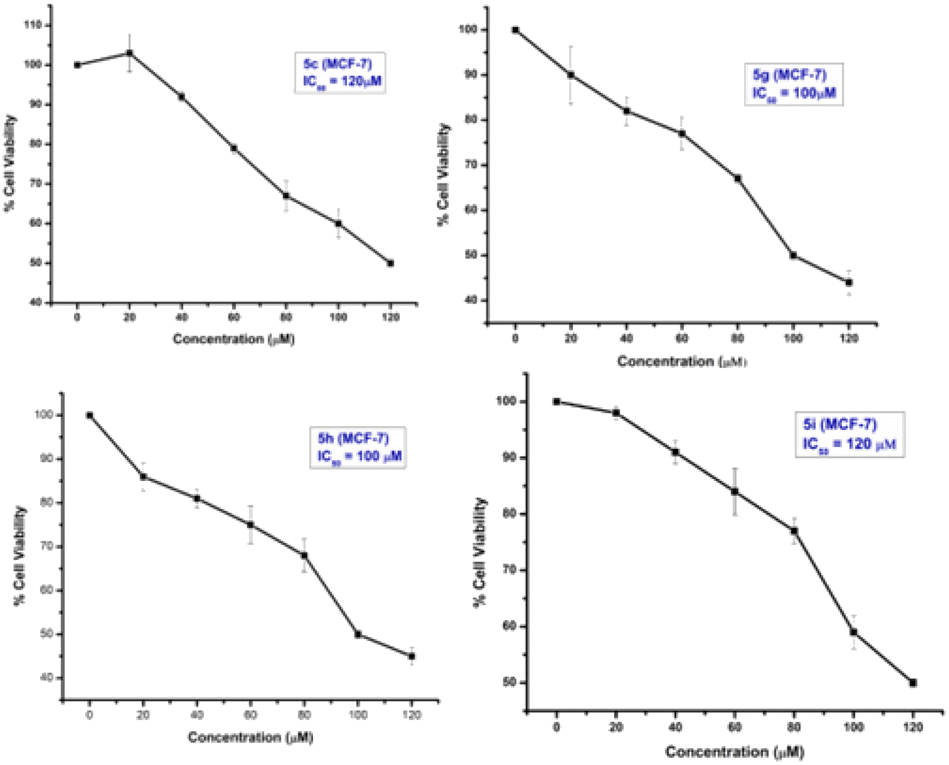 | ||
| Fig. 4 Anti-proliferative activity of sugar fused pyrano[3,2-c]pyranones. Anticancer potential of compounds were determined by conducting MTT-assay and IC50 were calculated for 5c, 5g, 5h and 5i in MCF-7 and MDA-MB231 cell lines after 24 h of treatment. IC50 values were determined by calculating mean value absorbance at 570 nm wavelength and percent cell viability against each concentration. The experiments were repeated thrice, ±SD-value was calculated for each value. | ||
Epirubicin is a chemotherapeutic drug currently being used against both subtypes of breast cancer. It was used as positive control for these experiments. The MTT assay of epirubicin at different concentration was observed for both cancer cell lines MCF-7 and MDA-MB-231 (ESI†). We further investigated if this anti-proliferative activity was the result of apoptotic mechanisms. MCF-7 cells treated with all the four compounds 5c, 5g, 5h and 5i at 20 to 120 μM showed altered morphology, cellular shrinkage, chromatin condensation and cell death which are the characteristics of apoptosis (Fig. 5 & ESI†).
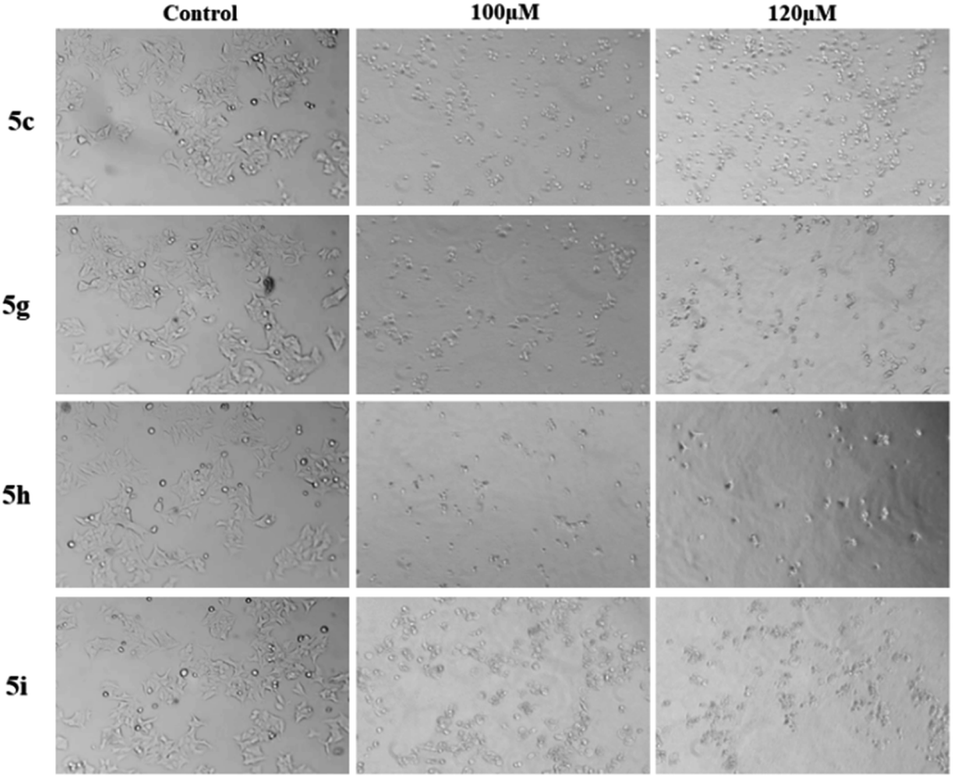 | ||
| Fig. 5 Sugar fused pyrano[3,2-c]pyranones induced morphological changes. MCF-7 cells treated with 5c, 5g, 5h and 5i for 24 hours and observed compound induced cell morphology changes. Images have been taken in inverted microscope at 10× magnification. | ||
Apoptosis is initiated with morphological changes. The characteristic of the apoptotic cell include chromatin condensation and nuclear fragmentation, plasma membrane blabbing and cell shrinkage. Ultimately the cells breaks into small fragments (apoptotic bodies).40 Herein for apoptosis analysis, MCF-7 & MDA-MB-231 cells were seeded into 96 well plate, incubated for up to 70–90% confluency then treated with different concentrations of compounds (5c, 5g, 5h, and 5i) for 24 hours. After treatment cells were fixed in 4% PFA for 15 min at 4 °C followed by staining with 1 mg mL−1 AO/Etbr solution for 10 min and washed with 1× PBS twice. Images were taken in blue and green monochromatic light by inverted fluorescence microscope at 10× magnification. Result showed gradually increased no. of apoptotic cells from 20 μM to 120 μM compound concentration in MCF-7 breast cancer cells and it is highly significant at 100 μM and 120 μM in 5c (P < 0.0001), 5g (P < 0.0011, P < 0.0012), 5h (P < 0.0001) and 5i (P < 0.0004, P < 0.0010) treated MCF-7 cells (Fig. 6). However, there is no significant apoptotic effects were observed in MDA-MB-231 triple negative breast cancer cell (results are shown in ESI†) line when treated with different concentration of above-mentioned compounds. The c-Myc gene is overexpressed in cancer cells, and this gene directly transactivates the GLUT1 gene.41 In contrast to glucose media, cells expressing c-myc that are cultivated in galactose medium undergo oxidative phosphorylation and survive and proliferate.42 According to Sheu et al.43 high glucose uptake induces apoptosis in cells via PI3K/AKT signalling, NF-B-COX2-Caspase3/BCL-2 pathway, ROS production, etc. In this study we have performed apoptosis analysis of glucal fused pyrano[3,2-c]pyranones compounds 5c, 5g, 5h and 5i on MCF-7 and MDA-MB-231 cell lines and found that these compound showed cytotoxic effects on MCF-7 cells as compare to MDA-MB-231 breast cancer cells. It might be possible that as MCF-7 cell overexpress c-myc gene compare to MDA-MB-231 cells which up-regulate the GLUT1 glucose transporter gene causes uptake of these glucose derivative compounds and induces apoptosis by above stated pathways in MCF-7 breast cancer cells.
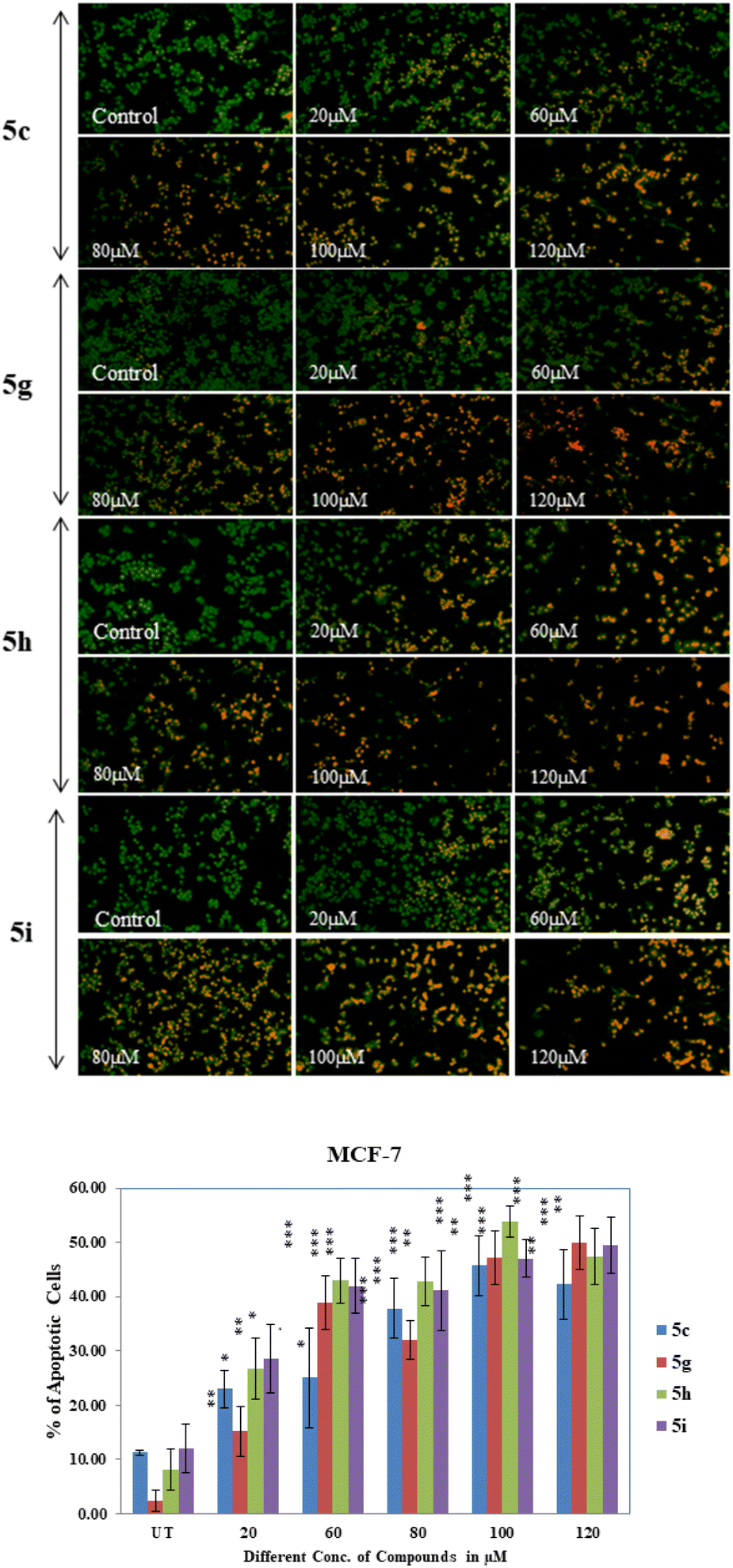 | ||
| Fig. 6 [A] Sugar fused pyrano[3,2-c]pyranones showing apoptotic changes: MCF-7 cells treated with 5c, 5g, 5h & 5i compounds for 24 hours, cells stained with acridine orange and ethidium bromide staining, drug intake by cells indicate imitation of cell death were observed in inverted fluorescence microscope at 10× magnification. [B] Quantification of apoptotic cell no. calculated by using ImageJ software and histogram was plotted as % of apoptotic cell vs. compound concentration. | ||
Cells migration is essential event for invasion and metastasis of all types of solid tumor including breast cancer during disease progression.44 Cell migration is evaluated in MCF-7 & MDA-MB-231 cells. MCF-7 and MDA-MB-231 cells were seeded into 6 well plate and the migration assay was performed by the scratch method. Cells were treated with different concentrations of compounds (5c, 5g, 5h, and 5i) for 24 hours and wound healing image was captured in 0 and 24 hours after Scratch using inverted microscope. The gap due to scratch was measured by ImageJ software. Due to cell migration gap is filled in the control whereas in the compound treated cells migration is inhibited. Cells migration was gradually reduced from 20 μM to 60 μM treatment as compared to control cells, and significant at the 60 μM (P < 0.0027) treated cells after 24 hours treatment of 5c, (P < 0.0001) in 5g and 5h treated MCF-7 cells, whereas P < 0.0092 in 5i treated MCF-7 cells (Fig. 7). However, there is no significant migration inhibition was observed on MDA-MB-231 triple negative breast cancer cell line (results are attached in ESI†). One of the most significant somatically mutated oncogenes in human cancer is the MYC transcription factor. MYC somatic amplification and overexpression are exceedingly frequent in human tumours and are linked to poor prognosis in a variety of human tumour types. Deregulation of the MYC pathway is one of the most frequent characteristics of human carcinogenesis since the majority of transforming oncogenes eventually drive MYC expression either directly or indirectly. The MYC oncoprotein gives cancer cells a selective advantage in a variety of situations by encouraging cell survival, proliferation, differentiation blockage, epithelial to mesenchymal transition, and genetic instability, all of which may indirectly aid metastasis.42,45 In this study we have used glucal fused pyrano[3,2-c]pyranones compounds 5c, 5g, 5h and 5i for the migration analysis. It might be possible that these compounds regulated the myc gene expression which in turn inhibited the migration of MCF-7 cells.
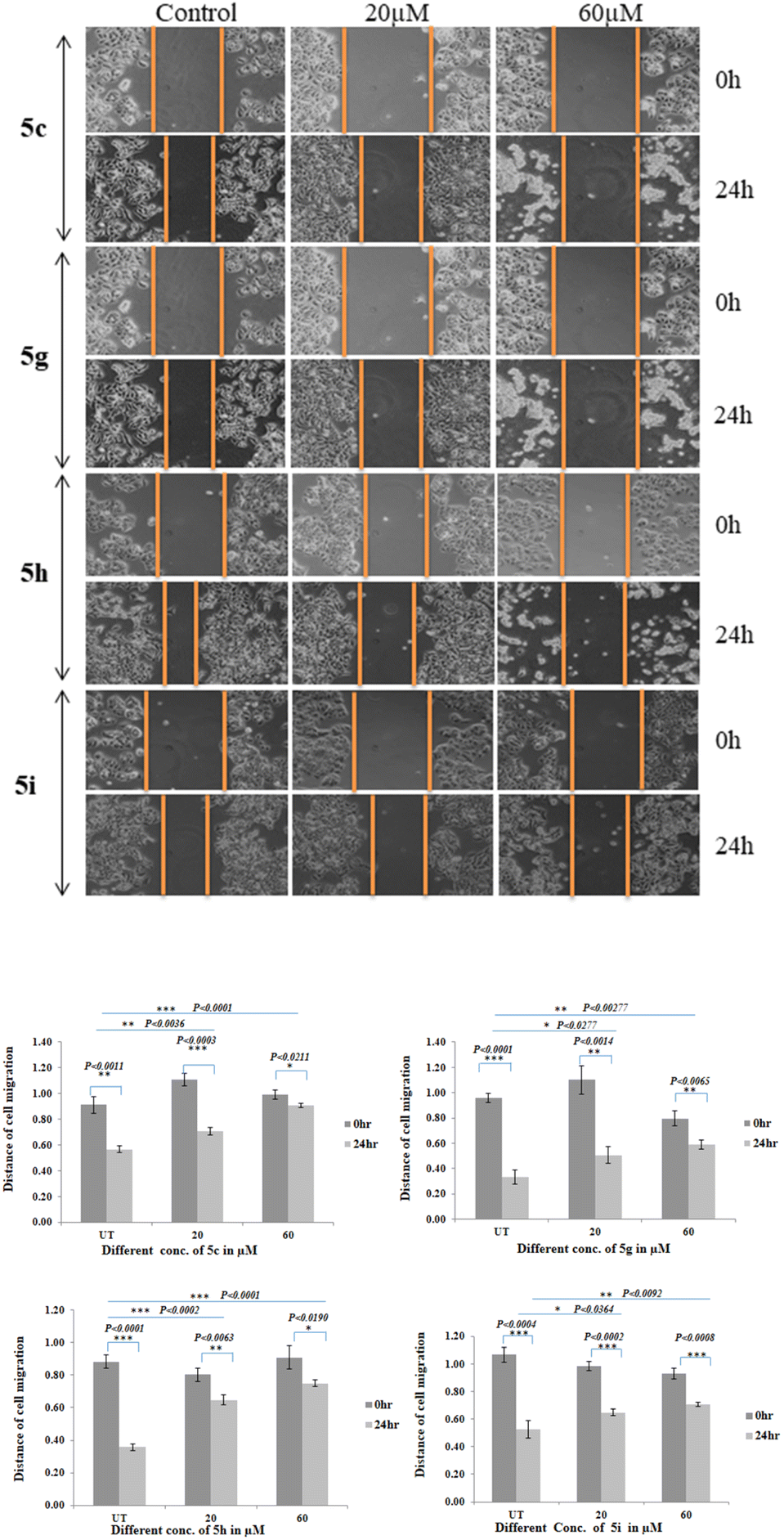 | ||
| Fig. 7 Compounds 5c, 5g, 5h and 5i regulate breast cancer cell migration. [A] The cell migration was analysed by wound healing assay. Confluent monolayer of MCF 7 cells was scratched using pipette tip and the cells were incubated with indicated concentrations of these compounds. Images were taken at 0 and 24 hours. In untreated control gap was filled after 24 hours, whereas compounds 5c, 5g, 5h and 5i treated cells, migration was inhibited and gap is still remained. [B] The images are representative of three assays. Bar graph (down) represents significant cell migration inhibition by these compounds at 20 and 60 μM as compared to untreated control. | ||
Structure–activity relationship
The relation between structure and activity concluded that only glucose series of carbohydrate moiety was found to be active against MCF-7 cell line. Structural correlation revealed that among four active compounds, three compounds had substituent at C-3 position and one had substituent at C-2 position. Compounds with substituents F, Cl, OH & OCH3 showed prominent biological activity. Out of these four potential compounds, F- and Cl-substituted compounds showed IC50 value at 100 μM and other two compounds with OH and OCH3 substitutions showed IC50 value at 120 μM (Fig. 8). | ||
| Fig. 8 The structure activity relation between gluco- and galacto fused pyrano[3,2-c]pyranones. | ||
Experimental
All chemicals & reagents were purchased from Alfa-Aesar by Thermo Fischer Scientific, India Pvt. Limited and from local commercial sources and were used without any further purification unless otherwise specified. Solvents were distilled prior to use for column chromatography. Silica gel (100–200 mesh) was used while performing column chromatography. Melting points were determined on Buchi M-560 instrument and are uncorrected. The IR spectra of compounds were recorded on PerkinElmer model 2000 FT-IR spectrometer and are expressed as wavenumber (cm−1). HRMS analysis was carried out using Agilent G6530AA LC Q-TOF mass spectrometer using ESI method. Specific rotation was measured on Rudolph Autopol II polarimeter. Rf values of compounds are reported for analytical TLC using the specified solvents and 0.25 mm silica gel 60 F254 plates that were visualized by UV irradiation or by charring with 5% alcoholic sulfuric acid solution. The 1H, 13C-NMR spectra were recorded on Jeol alpha-400 & Bruker-Avance Neo 400 FT-NMR spectrometers at 400 MHz, 100.6 MHz, respectively by using tetramethylsilane (TMS) as internal standard. The chemical shift values are on δ scale and the coupling constant (J) are in Hz. The cancer cell lines (MCF-7 and MDA-MB-231) used for this study were purchased from National Centre for Cell Science, Pune, Maharashtra, India.General procedure for the synthesis of 3,4,6-tri-O-benzyl-1-formylglycal (2a–b)
To a solution of β-C-glycopyranosyl aldehydes (1a–b, 1.0 mmol) in methanol (20 mL), NaOMe (1.2 mmol) was added with continuous stirring at 25 °C and further stirred for 30–40 min. The progress of the reaction was monitored by TLC. After completion, the solvent was evaporated on rota-vapour at high pressure. The desired compound was extracted with ethyl acetate (3 × 40 mL), brine solution (2 × 40 mL) and dried over anhydrous sodium sulphate. Excess of solvent was reduced over rotary evaporator to get the crude product. The residue thus obtained was purified by column chromatography using ethyl acetate in petroleum ether as gradient solvent system to afford the desired product 2a–b in 90–92% yield. All spectral data are identical as reported in the literature.39General procedure for the synthesis of (9R,10S,11S,11aS)-10,11-bis(benzyloxy)-9-((benzyloxy)methyl)-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one and (9R,10R,11S,11aS)-10,11-bis(benzyloxy)-9-((benzyloxy)methyl)-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one derivatives
A mixture of 3,4,6-tri-O-benzyl-1-formylglycal (2a–b, 1.0 mmol) and 4-hydroxy coumarin (3a–i, 1.2 mmol) and L-proline (0.2 mmol) in ethyl acetate (5 mL) was stirred at 25 °C for 1–2 h and the progress of the reaction was monitored by TLC. After completion, the desired compound was extracted with ethyl acetate (3 × 20 mL), brine solution (2 × 20 mL) and dried over anhydrous sodium sulphate. Excess of solvent was reduced over rotary evaporator to get the crude product. The residue thus obtained was purified by column chromatography using ethyl acetate in petroleum ether as gradient solvent system to afford the desired product 4a–i & 5a–i in 89–96% yield.(9R,10S,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (4a)
It was obtained as light-yellow viscous in 93% yield. Rf = 0.44 (15% ethyl acetate in petroleum ether); [α]24D = +45.65 (c 0.1, MeOH); IR (KBr, cm−1): 3034, 2965, 2843, 1708, 1539, 1435, 1389, 1058, 865, 768, 724, 565; 1H NMR (CDCl3, 400 MHz): δ 7.64 (1H, dd, J = 7.9, 1.5 Hz, ArH), 7.45–7.49 (3H, m, ArH), 7.27–7.40 (15H, m, ArH), 6.05 (1H, d, J = 2.5 Hz, C-7H), 5.52 (1H, dd, J = 9.4, 2.5 Hz, C-11aH), 4.96 (1H, d, J = 11.3 Hz, OCH2Ph), 4.89 (2H, q, J = 12.1 Hz, OCH2Ph), 4.65 (1H, d, J = 11.3 Hz, OCH2Ph), 4.49 (2H, q, J = 11.7 Hz, OCH2Ph), 4.16 (1H, d, J = 1.7 Hz, C-10H), 4.08 (1H, dd, J = 9.4, 2.6 Hz, C-11H), 3.94 (1H, t, J = 6.9 Hz, C-9H), 3.65–3.72 (2H, m, C-13H); 13C NMR (CDCl3, 100.6 MHz) δ 160.4 (CO), 155.5, 152.3, 145.4, 138.0, 137.9, 137.6, 131.4, 128.7, 128.6, 128.4, 128.3, 128.1, 128.0, 127.8, 127.7, 127.0, 124.1, 122.3, 116.7, 114.9 and 103.0 (ArC), 98.0 (C-7), 80.6 (C-10), 78.1 (C-9), 77.3 (C-11a), 75.0 (OCH2Ph), 73.7 (OCH2Ph), 73.6 (C-11), 72.9 (OCH2Ph), 68.1 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H33O7 [M + H]+: 589.2221; found: 589.2240.(9R,10S,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-2-methyl-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (4b)
It was obtained as light-yellow viscous in 95% yield. Rf = 0.45 (15% ethyl acetate in petroleum ether); [α]24D = +108.06 (c 0.1, MeOH); IR (KBr, cm−1): 3102, 2940, 2850, 1731, 1451, 1340, 1043, 942, 850, 730, 704, 660, 540; 1H NMR (CDCl3, 400 MHz): δ 7.50 (2H, d, J = 6.9 Hz, ArH), 7.30–7.44 (15H, m, ArH), 7.17 (1H, d, J = 8.4 Hz, ArH), 6.04 (1H, d, J = 2.5 Hz, C-7H), 5.51 (1H, dd, J = 9.4, 2.5 Hz, C-11aH), 4.97 (1H, d, J = 11.3 Hz, OCH2Ph), 4.88 (2H, s, OCH2Ph), 4.66 (1H, d, J = 11.3 Hz, OCH2Ph), 4.50 (2H, q, J = 11.7 Hz, OCH2Ph), 4.18 (1H, d, J = 1.6 Hz, C-10H), 4.08 (1H, dd, J = 9.4, 2.6 Hz, C-11H), 3.95 (1H, t, J = 6.6 Hz, C-9H), 3.69 (2H, dd, J = 6.6, 1.9 Hz, C-13H), 2.38 (3H, s, CH3); 13C NMR (CDCl3, 100.6 MHz): δ 160.6 (CO), 155.5, 150.5, 145.3, 138.0, 137.9, 137.6, 133.8, 132.5, 128.7, 128.6, 128.4, 128.3, 128.1, 128.0, 127.7, 122.0, 116.5, 114.5 and 102.8 (ArC), 98.1 (C-7), 80.8 (C-10), 78.1 (C-9), 75.0 (OCH2Ph), 73.7 (OCH2Ph), 73.4 (C-11), 72.8 (OCH2Ph), 68.1 (C-13), 21.1 (CH3); HR-ESI-TOF-MS: m/z cal. for C38H35O7 [M + H]+: 603.2377; found: 603.2382.(9R,10S,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-2-methoxy-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (4c)
It was obtained as light-yellow viscous in 94% yield. Rf = 0.41 (15% ethyl acetate in petroleum ether); [α]24D = +87.34 (c 0.1, MeOH); IR (KBr, cm−1): 3023, 2954, 2908, 2835, 1742, 1424, 1334, 1032, 887, 738, 720, 511; 1H NMR (CDCl3, 400 MHz): δ 7.47 (2H, d, J = 6.9 Hz, ArH), 7.28–7.40 (13H, m, ArH), 7.21 (1H, d, J = 9.0 Hz, ArH), 7.10 (1H, d, J = 2.9 Hz, ArH), 7.04 (1H, dd, J = 9.0, 3.0 Hz, ArH), 6.05 (1H, d, J = 2.5 Hz, C-7H), 5.54 (1H, dd, J = 9.4, 2.5 Hz, C-11aH), 4.96 (1H, d, J = 11.3 Hz, OCH2Ph), 4.89 (2H, q, J = 11.9 Hz, OCH2Ph), 4.66 (1H, d, J = 11.3 Hz, OCH2Ph), 4.50 (2H, q, J = 11.7 Hz, OCH2Ph), 4.19 (1H, d, J = 1.6 Hz, C-10H), 4.10 (1H, dd, J = 9.4, 2.6 Hz, C-11H), 3.96 (1H, t, J = 6.6 Hz, C-9H), 3.74 (3H, s, OCH3), 3.69 (2H, dd, J = 6.6, 2.6 Hz, C-13H); 13C NMR (CDCl3, 100.6 MHz): δ 160.6 (CO), 156.0, 155.2, 146.9, 145.5, 137.9, 137.8, 137.6, 128.7, 128.6, 128.4, 128.3, 128.1, 128.0, 127.5, 119.8, 117.9, 115.2, 103.8 and 103.1 (ArC), 98.1 (C-7), 80.7 (C-10), 78.1 (C-9), 75.0 (OCH2Ph), 73.8 (OCH2Ph), 73.4 (C-11), 72.8 (OCH2Ph), 68.1 (C-13), 55.6 (OCH3); HR-ESI-TOF-MS: m/z cal. for C38H35O8 [M + H]+: 619.2326; found: 619.2339.(9R,10S,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-2-bromo-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (4d)
It was obtained as light-yellow viscous in 93% yield. Rf = 0.49 (15% ethyl acetate in petroleum ether); [α]24D = +127.90 (c 0.1, MeOH); IR (KBr, cm−1): 3132, 3021, 2956, 2843, 1744, 1423, 1371, 1212, 1191, 1051, 980, 790, 712, 509; 1H NMR (CDCl3, 400 MHz): δ 7.78 (1H, d, J = 2.3 Hz, ArH), 7.54 (1H, dd, J = 8.8, 2.4 Hz, ArH), 7.43–7.50 (4H, m, ArH), 7.30–7.36 (11H, m, ArH), 7.14 (1H, d, J = 8.8 Hz, ArH), 6.01 (1H, d, J = 2.5 Hz, C-7H), 5.49 (1H, dd, J = 9.5, 2.5 Hz, C-11aH), 4.95 (1H, d, J = 11.3 Hz, OCH2Ph), 4.82 (2H, q, J = 12.0 Hz, OCH2Ph), 4.65 (1H, d, J = 11.3 Hz, OCH2Ph), 4.51 (2H, q, J = 11.7 Hz, OCH2Ph), 4.20 (1H, d, J = 1.6 Hz, C-10H), 4.06 (1H, dd, J = 9.5, 2.6 Hz, C-11H), 3.96 (1H, t, J = 6.9 Hz, C-9H), 3.68–3.70 (2H, m, C-13H); 13C NMR (CDCl3, 100.6 MHz): δ 159.8 (CO), 154.1, 151.0, 146.0, 137.9, 137.6, 137.5, 134.1, 128.8, 128.6, 128.5, 128.3, 128.1, 128.0, 127.9, 124.9, 118.4, 117.0, 116.5 and 104.0 (ArC), 97.7 (C-7), 80.4 (C-10), 78.1 (C-9), 75.0 (OCH2Ph), 73.8 (OCH2Ph), 73.1 (C-11), 72.6 (OCH2Ph), 68.0 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H32BrO7 [M + H]+: 667.1326; found: 667.1339.(9R,10S,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-2-chloro-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (4e)
It was obtained as light-yellow viscous in 92% yield. Rf = 0.50 (15% ethyl acetate in petroleum ether); [α]24D = +108.24 (c 0.1, MeOH); IR (KBr, cm−1): 3201, 3176, 3047, 2950, 2839, 1740, 1445, 1383, 1043, 865, 743, 734, 521; 1H NMR (CDCl3, 400 MHz): δ 7.62 (1H, d, J = 2.5 Hz, ArH), 7.31–7.48 (16H, m, ArH), 7.21 (1H, d, J = 8.8 Hz, ArH), 6.02 (1H, d, J = 2.5 Hz, C-7H), 5.49 (1H, dd, J = 9.4, 2.5 Hz, C-11aH), 4.96 (1H, d, J = 11.3 Hz, OCH2Ph), 4.86 (2H, q, J = 12.0 Hz, OCH2Ph), 4.66 (1H, d, J = 11.3 Hz, OCH2Ph), 4.51 (2H, q, J = 11.7 Hz, OCH2Ph), 4.19 (1H, d, J = 1.7 Hz, C-10H), 4.06 (1H, dd, J = 9.4, 2.6 Hz, C-11H), 3.96 (1H, t, J = 6.7 Hz, C-9H), 3.68–3.70 (2H, m, C-13H); 13C NMR (CDCl3, 100.6 MHz): δ 159.9 (CO), 154.2, 150.6, 146.0, 137.9, 137.6, 137.5, 131.3, 129.7, 128.8, 128.6, 128.5, 128.3, 128.1, 128.0, 127.9, 121.8, 118.1, 116.0 and 104.0 (ArC), 97.7 (C-7), 80.5 (C-10), 78.1 (C-9), 75.0 (OCH2Ph), 73.8 (OCH2Ph), 73.1 (C-11), 72.6 (OCH2Ph), 68.0 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H32ClO7 [M + H]+: 623.1831; found: 623.1841.(9R,10S,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-3-methoxy-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (4f)
It was obtained as light-yellow viscous in 95% yield. Rf = 0.40 (15% ethyl acetate in petroleum ether); [α]24D = +38.10 (c 0.1, MeOH); IR (KBr, cm−1): 3090, 2932, 2862, 1713, 1424, 1357, 1082, 849, 760, 645, 560; 1H NMR (CDCl3, 400 MHz): δ 7.53 (1H, d, J = 8.8 Hz, ArH), 7.47 (2H, d, J = 7.2 Hz, ArH), 7.28–7.42 (13H, m, ArH), 6.82 (1H, dd, J = 8.8, 2.4 Hz, ArH), 6.78 (1H, d, J = 2.3 Hz, ArH), 6.03 (1H, d, J = 2.5 Hz, C-7H), 5.50 (1H, dd, J = 9.4, 2.5 Hz, C-11aH), 4.96 (1H, d, J = 11.3 Hz, OCH2Ph), 4.88 (2H, q, J = 12.1 Hz, OCH2Ph), 4.64 (1H, d, J = 11.3 Hz, OCH2Ph), 4.49 (2H, q, J = 11.7 Hz, OCH2Ph), 4.15 (1H, d, J = 1.8 Hz, C-10H), 4.05 (1H, dd, J = 9.4, 2.6 Hz, C-11H), 3.91 (1H, t, J = 6.5 Hz, C-9H), 3.85 (3H, s, OCH3), 3.68 (2H, dd, J = 6.5, 3.9 Hz, C-13H); 13C NMR (CDCl3, 100.6 MHz): δ 162.7 (C-3), 160.8 (CO), 156.3, 154.2, 144.3, 138.0, 137.9, 137.6, 131.4, 128.7, 128.6, 128.4, 128.3, 128.1, 128.0, 127.7, 127.0, 123.4, 112.6, 108.2, 100.6 and 100.1 (ArC), 98.2 (C-7), 80.8 (C-10), 78.0 (C-9), 77.3 (C-11a), 75.0 (OCH2Ph), 73.7 (OCH2Ph), 73.6 (C-11), 72.9 (OCH2Ph), 68.1 (C-13), 55.8 (OCH3); HR-ESI-TOF-MS: m/z cal. for C38H35O8 [M + H]+: 619.2326; found: 619.2335.(9R,10S,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-3-fluoro-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (4g)
It was obtained as light-yellow viscous in 91% yield. Rf = 0.49 (15% ethyl acetate in petroleum ether); [α]24D = +127.67 (c 0.1, MeOH); IR (KBr, cm−1): 3162, 2987, 2864, 1730, 1432, 1362, 1065, 867, 730, 743, 511; 1H NMR (CDCl3, 400 MHz): δ 7.58 (1H, dd, J = 8.7, 6.0 Hz, ArH), 7.46 (2H, d, J = 6.9 Hz, ArH), 7.30–7.42 (13H, m, ArH), 6.96–7.02 (2H, m, ArH), 6.02 (1H, d, J = 2.5 Hz, C-7H), 5.51 (1H, dd, J = 9.4, 2.5 Hz, C-11aH), 4.96 (1H, d, J = 11.3 Hz, OCH2Ph), 4.86 (2H, s, OCH2Ph), 4.65 (1H, d, J = 11.3 Hz, OCH2Ph), 4.50 (2H, q J = 11.7 Hz, OCH2Ph), 4.17 (1H, d, J = 1.7 Hz, C-10H), 4.06 (1H, dd, J = 9.4, 2.6 Hz, C-11H), 3.93 (1H, t, J = 6.6 Hz, C-9H), 3.68 (2H, dd, J = 6.6, 4.0 Hz, C-13H); 13C NMR (CDCl3, 100.6 MHz): δ 165.6 (C-3), 163.1 (CO), 160.2, 155.2, 153.4, 153.3, 145.2, 137.9, 137.8, 137.5, 128.7, 128.6, 128.4, 128.3, 128.2, 128.1, 128.0, 127.7, 127.0, 124.0, 123.9, 112.5, 112.2, 111.6, 104.4, 104.2, 101.9, 97.8 (C-7), 80.7 (C-10), 78.1 (C-9), 75.0 (OCH2Ph), 73.8 (OCH2Ph), 73.4 (C-11), 72.8 (OCH2Ph), 68.0 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H32FO7 [M + H]+: 607.2127; found: 607.2137.(9R,10S,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-3-chloro-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (4h)
It was obtained as light-yellow viscous in 94% yield. Rf = 0.50 (15% ethyl acetate in petroleum ether); [α]24D = +156.12 (c 0.1, MeOH); IR (KBr, cm−1): 3169, 3078, 2985, 2849, 1743, 1439, 1324, 1037, 840, 737, 687, 524; 1H NMR (CDCl3, 400 MHz): δ 7.51 (1H, d, J = 8.5 Hz, ArH), 7.46 (2H, d, J = 6.9 Hz, ArH), 7.29–7.40 (14H, m, ArH), 7.21 (1H, dd, J = 8.5, 1.9 Hz, ArH), 6.01 (1H, d, J = 2.5 Hz, C-7H), 5.51 (1H, dd, J = 9.4, 2.5 Hz, C-11aH), 4.95 (1H, d, J = 11.3 Hz, OCH2Ph), 4.86 (2H, s, OCH2Ph), 4.64 (1H, d, J = 11.3 Hz, OCH2Ph), 4.50 (2H, q, J = 11.7 Hz, OCH2Ph), 4.17 (1H, d, J = 1.8 Hz, C-10H), 4.07 (1H, dd, J = 9.4, 2.6 Hz, C-11H), 3.94 (1H, t, J = 6.7 Hz, C-9H), 3.68 (2H, dd, J = 6.6, 3.6 Hz, C-13H); 13C NMR (CDCl3, 100.6 MHz): δ 159.9 (CO), 154.9, 152.5, 145.6, 137.9, 137.8, 137.6, 137.2, 128.7, 128.6, 128.5, 128.3, 128.2, 128.1, 128.0, 127.7, 124.8, 123.2, 117.0, 113.5 and 103.1 (ArC), 97.8 (C-7), 80.6 (C-10), 78.1 (C-9), 75.0 (OCH2Ph), 73.8 (OCH2Ph), 73.4 (C-11), 72.8 (OCH2Ph), 68.1 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H32ClO7 [M + H]+: 623.1831; found: 623.1840.(9R,10S,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-3-hydroxy-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (4i)
It was obtained as light-yellow viscous in 91% yield. Rf = 0.22 (30% ethyl acetate in petroleum ether); [α]24D = +198.22 (c 0.1, MeOH); IR (KBr, cm−1): 3321, 3212, 3193, 2988, 2857, 1738, 1459, 1357, 1034, 859, 739, 738, 516; 1H NMR (CDCl3, 400 MHz): δ 7.45 (3H, dd, J = 7.8, 5.4 Hz, ArH), 7.27–7.38 (13H, m, ArH), 6.98 (1H, d, J = 2.1 Hz, ArH), 6.78 (1H, dd, J = 8.7, 2.2 Hz, ArH), 5.99 (1H, d, J = 2.5 Hz, C-7H), 5.47 (1H, dd, J = 9.4, 2.5 Hz, C-11aH), 4.95 (1H, d, J = 11.3 Hz, OCH2Ph), 4.86 (2H, q, J = 12.1 Hz, OCH2Ph), 4.63 (1H, d, J = 11.3 Hz, OCH2Ph), 4.48 (2H, dd, J = 26.7, 11.7 Hz, OCH2Ph), 4.13 (1H, d, J = 2.8 Hz, C-10H), 4.06 (1H, dd, J = 9.4, 2.6 Hz, C-11H), 3.92 (1H, t, J = 6.7 Hz, C-9H), 3.65 (2H, dd, J = 6.4, 4.4 Hz, C-13H), 2.29 (1H, s, OH); 13C NMR (CDCl3, 100.6 MHz): δ 162.0 (CO), 160.7, 157.4, 153.9, 144.1, 138.0, 137.9, 137.6, 128.7, 128.6, 128.4, 128.3, 128.1, 127.9, 127.7, 127.1, 123.7, 113.9, 107.4, 103.1 and 99.2 (ArC), 97.9 (C-7), 80.7 (C-10), 78.0 (C-9), 75.0 (OCH2Ph), 73.7 (OCH2Ph), 73.7 (C-11), 73.0 (OCH2Ph), 68.2 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H33O8 [M + H]+: 605.2170; found: 605.2184.(9R,10R,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (5a)
It was obtained as light-yellow viscous in 91% yield. Rf = 0.49 (15% ethyl acetate in petroleum ether); [α]24D = +96.01 (c 0.1, MeOH); IR (KBr, cm−1): 3021, 2998, 2845, 1704, 1540, 1302, 742, 710, 601, 548; 1H NMR (CDCl3, 400 MHz): δ 7.53 (1H, d, J = 7.9 Hz, ArH), 7.45–7.48 (3H, m, ArH), 7.29–7.39 (12H, m, ArH), 7.17–7.22 (3H, m, ArH), 6.16 (1H, d, J = 2.3 Hz, C-7H), 5.19 (1H, dd, J = 8.7, 2.3 Hz, C-11aH), 5.11 (1H, d, J = 11.3 Hz, –OCH2Ph), 4.98 (1H, d, J = 11.3 Hz, –OCH2Ph), 4.88 (1H, d, J = 10.7 Hz, –OCH2Ph), 4.63 (2H, dd, J = 15.7, 11.4 Hz, –OCH2Ph), 4.55 (1H, d, J = 12.0 Hz, –OCH2Ph), 4.11 (1H, t, J = 9.0 Hz, C-11H), 3.90 (1H, t, J = 9.4 Hz, C-10H), 3.82 (2H, d, J = 1.8 Hz, C-13H), 3.70 (1H, d, J = 9.7 Hz, C-9H); 13C NMR (CDCl3, 100.6 MHz): δ 160.4 (CO), 155.4, 152.4, 144.9, 138.2, 137.8, 137.7, 131.6, 128.7, 128.5, 128.1, 128.0, 127.9, 124.2, 122.3, 116.8, 114.7 and 102.3 (ArC), 99.3 (C-7), 84.2 (C11), 79.7 (C-9), 79.1 (C11a), 75.8 (OCH2Ph), 75.7 (C-10), 75.5 (OCH2Ph), 73.7 (OCH2Ph), 68.0 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H33O7 [M + H]+: 589.2221; found: 589.2242.(9R,10R,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-2-methyl-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (5b)
It was obtained as light-yellow viscous in 94% yield. Rf = 0.50 (15% ethyl acetate in petroleum ether); [α]24D = +88.17 (c 0.1, MeOH); IR (KBr, cm−1): 3088, 2976, 2890, 1710, 1601, 1543, 1452, 1271, 921, 880, 632, 609, 549; 1H NMR (CDCl3, 400 MHz): δ 7.47 (2H, dd, J = 4.1, 3.7 Hz, ArH), 7.38 (2H, d, J = 7.1 Hz, ArH), 7.27–7.36 (11H, m, ArH), 7.17–7.21 (3H, m, ArH), 6.15 (1H, d, J = 2.4 Hz, C-7H), 5.17 (1H, dd, J = 8.8, 2.5 Hz, C-11aH), 5.10 (1H, d, J = 11.3 Hz, OCH2Ph), 5.00 (1H, d, J = 11.3 Hz, OCH2Ph), 4.90 (1H, d, J = 10.8 Hz, OCH2Ph), 4.64 (2H, dd, J = 11.3, 9.8 Hz, OCH2Ph), 4.55 (1H, d, J = 12.0 Hz, OCH2Ph), 4.11 (1H, t, J = 9.0 Hz, C-11H), 3.91 (1H, t, J = 9.4 Hz, C-10H), 3.82 (2H, m, C-13H), 3.71 (1H, dt, J = 9.7, 2.6 Hz, C-9H), 2.35 (3H, s, CH3); 13C NMR (CDCl3, 100.6 MHz): δ 160.6 (CO), 155.3, 150.6, 144.8, 138.2, 137.8, 137.7, 133.9, 132.7, 128.7, 128.5, 128.1, 128.0, 127.9, 127.8, 122.0, 116.5, 114.4 and 102.1 (ArC), 99.3 (C-7), 84.2 (C-11), 79.7 (C-9), 78.9 (C-11a), 75.8 (OCH2Ph), 75.7 (C-10), 75.5 (OCH2Ph), 73.7 (OCH2Ph), 68.0 (C-13), 21.1 (CH3); HR-ESI-TOF-MS: m/z cal. for C38H35O7 [M + H]+: 603.2377; found: 603.2391.(9R,10R,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-2-methoxy-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (5c)
It was obtained as light-yellow viscous in 92% yield. Rf = 0.45 (15% ethyl acetate in petroleum ether); [α]24D = +27.67 (c 0.1, MeOH); IR (KBr, cm−1): 3095, 2967, 2761, 1706, 1459, 1387, 1078, 849, 739, 710, 554; 1H NMR (CDCl3, 400 MHz): δ 7.45 (2H, d, J = 6.7 Hz, ArH), 7.30–7.36 (11H, m, ArH), 7.18–7.22 (3H, m, ArH), 7.04 (2H, dd, J = 8.3, 2.3 Hz, ArH), 6.16 (1H, d, J = 2.4 Hz, C-7H), 5.20 (1H, dd, J = 8.8, 2.4 Hz, C-11aH), 5.13 (1H, d, J = 11.1 Hz, OCH2Ph), 4.99 (1H, d, J = 11.1 Hz, OCH2Ph), 4.88 (1H, d, J = 10.8 Hz, OCH2Ph), 4.67 (1H, d, J = 6.5 Hz, OCH2Ph), 4.62 (1H, d, J = 11.0 Hz, OCH2Ph), 4.54 (1H, d, J = 12.1 Hz, OCH2Ph), 4.11 (1H, t, J = 9.0 Hz, C-11H), 3.91 (1H, t, J = 9.4 Hz, C-10H), 3.82 (2H, d, J = 2.3 Hz, C-13H), 3.71 (1H, dt, J = 9.7, 2.4 Hz, C-9H), 3.65 (3H, s, OCH3); 13C NMR (CDCl3, 100.6 MHz): δ 160.5 (CO), 156.0, 155.0, 146.9, 145.0, 138.1, 137.7, 137.6, 128.6, 128.5, 128.0, 127.9, 127.6, 127.0, 120.0, 117.9, 114.9, 103.7 and 102.3 (ArC), 99.3 (C-7), 84.0 (C-11), 79.7 (C-9), 79.0 (C-11a), 75.6 (OCH2Ph), 75.6 (C-10), 75.5 (OCH2Ph), 73.7 (OCH2Ph), 67.9 (C-13), 55.6 (OCH3); HR-ESI-TOF-MS: m/z cal. for C38H35O8 [M + H]+: 619.2326; found: 619.2341.(9R,10R,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-2-bromo-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (5d)
It was obtained as light-yellow solid in 90% yield. Rf = 0.55 (15% ethyl acetate in petroleum ether); m.p.: 120–123 °C; [α]24D = +56.17 (c 0.1, MeOH); IR (KBr, cm−1): 3088, 2970, 2862, 1715, 1563, 1465, 1379, 1078, 741, 631, 605, 550; 1H NMR (CDCl3, 400 MHz): δ 7.72 (1H, d, J = 2.3 Hz, ArH), 7.55 (1H, dd, J = 8.8, 2.4 Hz, ArH), 7.40–7.47 (4H, m, ArH), 7.29–7.36 (9H, m, ArH), 7.22 (2H, dd, J = 7.3, 2.1 Hz, ArH), 7.16 (1H, d, J = 8.8 Hz, ArH), 6.12 (1H, d, J = 2.5 Hz, C-7H), 5.15 (1H, dd, J = 8.9, 2.5 Hz, C-11aH), 5.02 (2H, q, J = 11.2 Hz, OCH2Ph), 4.91 (1H, d, J = 10.8 Hz, OCH2Ph), 4.66 (1H, d, J = 2.0 Hz, OCH2Ph), 4.63 (1H, s, OCH2Ph), 4.55 (1H, d, J = 12.0 Hz, OCH2Ph), 4.10 (1H, t, J = 9.0 Hz, C-11H), 3.91 (1H, t, J = 9.4 Hz, C-10H), 3.82 (2H, d, J = 2.9 Hz, C-13H), 3.73 (1H, dt, J = 9.6, 2.4 Hz, C-9H); 13C NMR (CDCl3, 100.6 MHz): δ 159.7 (CO), 153.9, 151.0, 145.6, 137.8, 137.7, 137.6, 134.3, 128.8, 128.6, 128.5, 128.2, 128.1, 128.0, 127.9, 124.8, 118.5, 117.0, 116.3 and 103.3 (ArC), 98.9 (C-7), 83.6 (C-11), 79.7 (C-9), 78.8 (C-11a), 75.9 (OCH2Ph), 75.7 (C-10), 75.4 (OCH2Ph), 73.7 (OCH2Ph), 67.9 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H32BrO7 [M + H]+: 667.1326; found: 667.1354.(9R,10R,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-2-chloro-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (5e)
It was obtained as light-yellow solid in 91% yield. Rf = 0.56 (15% ethyl acetate in petroleum ether); m.p.: 108–110 °C; [α]24D = +22.50 (c 0.1, MeOH); IR (KBr, cm−1): 3091, 2980, 2822, 1710, 1434, 1365, 1054, 893, 731, 721, 551; 1H NMR (CDCl3, 400 MHz): δ 7.54 (1H, d, J = 2.4 Hz, ArH), 7.40–7.47 (5H, m, ArH), 7.29–7.36 (9H, m, ArH), 7.20–7.23 (3H, m, ArH), 6.13 (1H, d, J = 2.4 Hz, C-7H), 5.15 (1H, dd, J = 8.8, 2.4 Hz, C-11aH), 5.02 (2H, q, J = 11.3 Hz, OCH2Ph), 4.90 (1H, d, J = 10.8 Hz, OCH2Ph), 4.66 (1H, d, J = 4.7 Hz, OCH2Ph), 4.63 (1H, d, J = 3.4 Hz, OCH2Ph), 4.55 (1H, d, J = 12.0 Hz, OCH2Ph), 4.10 (1H, t, J = 9.0 Hz, C-11H), 3.91 (1H, t, J = 9.4 Hz, C-10H), 3.81–3.82 (2H, m, C-13H), 3.70–3.74 (1H, m, C-9H); 13C NMR (CDCl3, 100.6 MHz): δ 159.8 (CO), 154.0, 150.6, 145.6, 137.9, 137.6, 137.6, 131.5, 129.7, 129.0, 128.6, 128.1, 128.1, 128.0, 121.8, 118.2, 115.8, 103.3, 98.9 (C-7), 83.7 (C-11), 79.7 (C-9), 78.8 (C-11a), 75.9 (OCH2Ph), 75.7 (C-10), 75.5 (OCH2Ph), 73.7 (OCH2Ph), 67.9 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H32ClO7 [M + H]+: 623.1831; found: 623.1850.(9R,10R,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-3-methoxy-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (5f)
It was obtained as light-yellow viscous in 93% yield. Rf = 0.45 (15% ethyl acetate in petroleum ether); [α]24D = +90.43 (c 0.1, MeOH); IR (KBr, cm−1): 3107, 2990, 2797, 1708, 1545, 1405, 1310, 1020, 861, 762, 602, 540; 1H NMR (CDCl3, 400 MHz): δ 7.38–7.45 (4H, m, ArH), 7.29–7.36 (10H, m, ArH), 7.17–7.19 (2H, m, ArH), 6.78–6.80 (2H, m, ArH), 6.14 (1H, d, J = 2.3 Hz, C-7H), 5.15 (1H, dd, J = 8.7, 2.3 Hz, C-11aH), 5.09 (1H, d, J = 11.3 Hz, OCH2Ph), 4.97 (1H, d, J = 11.3 Hz, OCH2Ph), 4.87 (1H, d, J = 10.7 Hz, OCH2Ph), 4.63 (2H, dd, J = 17.3, 11.4 Hz, OCH2Ph), 4.55 (1H, d, J = 12.1 Hz, OCH2Ph), 4.08 (1H, t, J = 8.9 Hz, C-11H), 3.89 (1H, t, J = 9.5 Hz, C-10H), 3.85 (3H, s, OCH3), 3.81 (2H, d, J = 2.0 Hz, C-13H), 3.67 (1H, d, J = 9.7 Hz, C-9H); 13C NMR (CDCl3, 100.6 MHz): δ 162.8 (CO), 160.8, 156.1, 154.3, 143.8, 138.2, 137.8, 137.7, 128.6, 128.5, 128.1, 128.0, 127.9, 123.4, 112.7, 108.0, 100.6 and 99.5 (ArC), 99.4 (C-7), 84.3 (C-11), 79.6 (C-9), 79.0 (C-11a), 75.9 (OCH2Ph), 75.6 (C-10), 75.5 (OCH2Ph), 73.7 (OCH2Ph), 68.0 (C-13), 55.8 (OCH3); HR-ESI-TOF-MS: m/z cal. for C38H35O8 [M + H]+: 619.2326; found: 619.2345.(9R,10R,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-3-fluoro-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (5g)
It was obtained as light-yellow viscous in 89% yield. Rf = 0.54 (15% ethyl acetate in petroleum ether); [α]24D = +34.12 (c 0.1, MeOH); IR (KBr, cm−1): 3101, 2987, 2861, 1701, 1451, 1295, 991, 854, 730, 700, 543; 1H NMR (CDCl3, 400 MHz): δ 7.29–7.45 (14H, m, ArH), 7.18 (2H, dd, J = 6.8, 2.6 Hz, ArH), 7.01 (1H, dd, J = 9.0, 2.4 Hz, ArH), 6.94 (1H, td, J = 8.5, 2.4 Hz, ArH), 6.12 (1H, d, J = 2.4 Hz, C-7H), 5.17 (1H, dd, J = 8.8, 2.4 Hz, C-11aH), 5.06 (1H, d, J = 11.4 Hz, OCH2Ph), 4.99 (1H, d, J = 11.4 Hz, OCH2Ph), 4.87 (1H, d, J = 10.7 Hz, OCH2Ph), 4.63 (2H, dd, J = 15.6, 11.4 Hz, OCH2Ph), 4.55 (1H, d, J = 12.0 Hz, OCH2Ph), 4.09 (1H, t, J = 9.0 Hz, C-11H), 3.91 (1H, t, J = 9.4 Hz, C-10H), 3.82 (2H, s, C-13H), 3.70 (1H, dt, J = 9.6, 2.3 Hz, C-9H); 13C NMR (CDCl3, 100.6 MHz): δ 165.7 (C-3), 160.2 (CO), 155.0, 153.4, 144.7, 138.2, 137.7, 137.6, 128.7, 128.5, 128.1, 128.0, 127.9, 124.1, 124.0, 112.5, 112.3, 111.4, 104.5, 104.2 and 101.2 (ArC), 99.1 (C-7), 84.2 (C-11), 79.6 (C-9), 78.9 (C-11a), 75.8 (OCH2Ph), 75.7 (C-10), 75.5 (OCH2Ph), 73.7 (OCH2Ph), 67.9 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H32FO7 [M + H]+: 607.2127; found: 607.2139.(9R,10R,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-3-chloro-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (5h)
It was obtained as light-yellow viscous in 91% yield. Rf = 0.56 (15% ethyl acetate in petroleum ether); [α]24D = +7.51 (c 0.1, MeOH); IR (KBr, cm−1): 3045, 2923, 2829, 1712, 1451, 1350, 1071, 861, 730, 776, 550; 1H NMR (CDCl3, 400 MHz): δ 7.44 (2H, dd, J = 7.8, 1.6 Hz, ArH), 7.33–7.39 (8H, m, ArH), 7.29–7.32 (5H, m, ArH), 7.16–7.19 (3H, m, ArH), 6.12 (1H, d, J = 2.5 Hz, C-7H), 5.16 (1H, dd, J = 8.8, 2.5 Hz, C-11aH), 5.02 (2H, q, J = 11.5 Hz, OCH2Ph), 4.87 (1H, d, J = 10.7 Hz, OCH2Ph), 4.65 (1H, d, J = 12.1 Hz, OCH2Ph), 4.62 (1H, d, J = 10.8 Hz, OCH2Ph), 4.55 (1H, d, J = 12.0 Hz, OCH2Ph), 4.09 (1H, t, J = 9.0 Hz, C-11H), 3.90 (1H, t, J = 9.4 Hz, C-10H), 3.81–3.82 (2H, m, C-13H), 3.70 (1H, dt, J = 9.9, 2.6 Hz, C-9H); 13C NMR (CDCl3, 100.6 MHz): δ 159.8 (CO), 154.7, 152.5, 145.1, 138.2, 137.7, 137.6, 137.4, 128.7, 128.6, 128.1, 128.0, 127.9, 124.8, 123.2, 117.0, 113.3 and 102.3 (ArC), 99.1 (C-7), 84.1 (C-11), 79.7 (C-9), 78.9 (C-11a), 75.8 (OCH2Ph), 75.7 (C-10), 75.5 (OCH2Ph), 73.7 (OCH2Ph), 67.9 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H32ClO7 [M + H]+: 623.1831; found: 623.1845.(9R,10R,11S,11aS)-10,11-Bis(benzyloxy)-9-((benzyloxy)methyl)-3-hydroxy-9,10,11,11a-tetrahydro-6H-pyrano[2′,3′:5,6]pyrano[3,2-c]benzopyran-6-one (5i)
It was obtained as light-yellow viscous in 91% yield. Rf = 0.21 (30% ethyl acetate in petroleum ether); [α]24D = +102.17 (c 0.1, MeOH); IR (KBr, cm−1): 3345, 3032, 2946, 2837, 1712, 1534, 1470, 1120, 840, 721, 988, 534; 1H NMR (CDCl3, 400 MHz): δ 7.43 (2H, d, J = 6.6 Hz, ArH), 7.27–7.36 (12H, m, ArH), 7.16 (2H, dd, J = 6.9, 2.6 Hz, ArH), 6.98 (1H, dd, J = 2.1 Hz, ArH), 6.76 (1H, dd, J = 8.7, 2.2 Hz, ArH), 6.08 (1H, d, J = 2.4 Hz, C-7H), 5.12 (1H, dd, J = 8.7, 2.4 Hz, C-11aH), 5.06 (1H, d, J = 11.3 Hz, OCH2Ph), 4.95 (1H, d, J = 11.3 Hz, OCH2Ph), 4.86 (1H, d, J = 10.8 Hz, OCH2Ph), 4.63 (1H, d, J = 12.0 Hz, OCH2Ph), 4.58 (1H, d, J = 10.8 Hz, OCH2Ph), 4.53 (1H, d, J = 12.1 Hz, OCH2Ph), 4.08 (1H, t, J = 9.0 Hz, C-11H), 3.86 (1H, t, J = 9.4 Hz, C-10H), 3.79 (2H, d, J = 2.3 Hz, C-13H), 3.68 (1H, dt, J = 9.8, 2.4 Hz, C-9H), 2.59 (1H, s, OH); 13C NMR (CDCl3, 100.6 MHz): δ 162.1 (CO), 161.0, 157.2, 153.9, 143.6, 138.1, 137.7, 137.6, 128.6, 128.5, 128.1, 128.0, 127.9, 123.7, 114.1, 107.1, 103.0 and 99.2 (ArC), 98.5 (C-7), 84.2 (C-11), 79.4 (C-9), 79.0 (C-11a), 75.8 (OCH2Ph), 75.7 (C-10), 75.5 (OCH2Ph), 73.7 (OCH2Ph), 68.0 (C-13); HR-ESI-TOF-MS: m/z cal. for C37H33O8 [M + H]+: 605.2170; found: 605.2198.MTT assay (cells viability assay)
Efficacies of 18 compounds are evaluated by using MTT-assay for their anticancer role in breast cancer cell lines in MCF-7 & MDA-MB-231. DMSO used to make a 100 mM stock of compounds. MCF-7 and MDA-MB-231 cells (5 × 103 cells per well) were seeded into 96 well plates and incubate for 24 hours. A gradient of drug concentrations (20 μM, 40 μM, 60 μM, 80 μM, 100 μM, 120 μM) were added for 24 hours of treatment. Treated cells and controls were stained with MTT dye (5 mg mL−1) for 4 hours then cell lysis done by using cell lysis solution (SDS and HCL solution). Optical density (OD) value was measured at 570 nm wavelength in micro-plate reader. Experiments were performed in triplicate. The percent cell viability was calculated by using the following formula:46| Percentage cell viability = OD of experiment sample × 100/OD of the control. |
Cell morphology analysis (cytotoxic assay)
In the continuation cytotoxicity of all these compounds, we have evaluated cell morphology. MCF-7 & MDA-MB-231 breast cancer cells were cultured and 1 × 103 cells per well were seeded into six well plates in DMEM media with 10% FBS and 1% antibiotics, incubated for 24 hours. Further cells were treated with these compounds with various concentrations (0 μM, 20 μM, 40 μM, 60 μM, 80 μM, 100 μM, 120 μM) for 24 hours and images were captured by inverted phase contrast microscope at 10× resolution. Cytotoxicity of these compounds was evaluated by morphology of these cells. Adhere, irregular and triangle shape structures are live cells whereas round spherical shape structures denote stressed and death induced cells. Whereas, round and floating cells in the medium are dead cells.47Apoptosis assay by (Etbr/acridin orange staining)
MCF-7 & MD-MB-231 cells (5 × 103 cells per well) were seeded into 96-well plate and incubated for 24 hours. A gradient of active compound concentrations (0 μM, 20 μM, 40 μM, 60 μM, 80 μM, 100 μM, 120 μM) were used for treatment of seeded cells for 24 hours at 37 °C at 5% CO2 conditions. Cells were fixed using 4% paraformaldehyde (PFA) for 30 min, cells were washed with 1× PBS followed by staining with 1 mg mL−1 AO/EtBr for 10 minutes and washed again with 1× PBS. Images of the cells were captured by an inverted fluorescence microscope at 485 nm wavelength and at 10× magnification. Images were analysed by ImageJ software and apoptosis cells no. were counted and graphs were plotted by using GraphPad Prism 5.48Migration assay (wound healing assay)
MCF-7 & MDA-MB-231 breast cancer cells (5 × 105 cells per well) are seeded into 6-well plate, maintained till 100% confluence. In a sterile environment, in each well make a vertical wound down through the cell monolayer by using sterile 10 μl tips. Carefully aspirate the media and cell debris and washed the cells gently with 1× PBS to wash away the detached cells, slowly added enough culture media with different concentrations (0 μM, 20 μM, 60 μM) of active compounds. Image of the scratch was captured after 24 h of treatment by inverted phase contrast microscopy at 10× magnification. The distance of migration was calculated using ImageJ software and graph was plotted to see migration inhibition of the treated cells.44Conclusion
A series of novel sugar fused pyrano[3,2-c]pyranone derivatives were achieved using an efficient and facile one pot condensation reaction between 1-C-formyl glycals and substituted 4-hydroxy coumarins. Reactions were carried out at room temperature (25 °C) in ethyl acetate by stirring for ∼1–2 h in presence of L-proline to obtain exclusively single diastereomer of pyrano[3,2-c]pyranone derivatives. The reactions went smoothly in a stereoselective manner and produced only one diastereomer exclusively for each of these derivatives. No requirement of high temperature, shortened reaction time and high yields made the protocol attractive. A total of eighteen compounds were synthesized following the methodology. We have screened all eighteen compounds for the anticancer activity. In this study we have selected MCF-7 cell line as an Estrogen Receptor (ER) and Progesterone Receptor (PR) +ve cells with Her-2 gene −ve breast cancer subtype against that these active compounds (5c, 5g, 5h, and 5i) showed significant antitumor effect. However, MDA-MB-231 is an ER, PR, and Her-2 gene −ve cell lines represent triple −ve subtype of breast cancer is known as highly aggressive subtype where these compounds are not effective.Author contributions
S. Kumar, A. K. Prasad and B. K. Singh designed the project. S. Kumar carried out synthesis of all the compounds. R. K. Sahu and B. Kumar performed anticancer activity. S. Kumar, P. Kumari, J. Maity, R. J. Chhatwal and B. K. Singh wrote and compiled the paper.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful to Institute of Eminence, University of Delhi for providing financial support to strengthen research and development. We are thankful to DST-FIST program, CIF–USIC and Department of Chemistry, University of Delhi for providing the NMR spectral and HRMS recording facilities. Sandeep Kumar thanks CSIR, New Delhi for award of SPM Research Fellowship [File No. 09/045(0269)/2018-EMR-1]. Priti Kamari thankful to UGC for providing DS Kothari postdoctoral fellowship (F.4-2/2006(BSR)/CH/18-19/0314). This article is dedicated to Late Professor Ashok Kumar Prasad (1961–2023) who fought with cancer till his last breath. He was a person of high academic achievements, designated as Head of Department and Dean of Science faculty in University of Delhi. We are solely dedicating this work to him with a little hope towards possible contribution to cancer treatment.Notes and references
- D. Kumar, P. Sharma, H. Singh, K. Nepali, G. K. Gupta, S. K. Jain and F. N. Kang, RSC Adv., 2017, 7, 36977–36999, 10.1039/C7RA05441F.
- J. Ferlay, H. R. Shin, F. Bray, D. Forman, C. Mathers and D. M. Parkin, GLOBOCAN 2008 v1.2. Cancer Incidence and Mortality Worldwide: IARC Cancer Base No. 10, International Agency for Research on Cancer, Lyon, France, 2010 Search PubMed.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61, 69–90, DOI:10.3322/caac.20107.
- S. K. Kim, Handbook of Anticancer Drugs from Marine Origin, Springer International Publishing, Switzerland, 2015 Search PubMed.
- S. E. S. Dadashpour, Eur. J. Med. Chem., 2015, 102, 611–630, DOI:10.1016/j.ejmech.2015.08.033.
- G. M. Cragg and J. Newman, Phytochem. Rev., 2009, 8, 313–331, DOI:10.1007/s11101-009-9123-y.
- A. C. W. Wilson, Nat. Prod. Rep., 1997, 14, 99–110, 10.1039/NP9971400099.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661–677, DOI:10.1038/nrd2852.
- H. Miyabe, Molecules, 2015, 20, 12558–12575, DOI:10.3390/molecules200712558.
- S. Hoon, A. M. Smith, I. M. Wallace, S. Suresh, M. Miranda, E. Fung, M. Proctor, K. M. Shokat, C. Zhang, R. W. Davis, G. Giaever, R. P. S. Onge and C. Nislow, Nat. Chem. Biol., 2008, 4, 498–506, DOI:10.1038/nchembio.100.
- D. N. Nicolaides, D. R. Gautam, K. E. Litinas, D. J. Hadjipavlou-Litina and K. C. Fylaktakidou, Eur. J. Med. Chem., 2004, 39, 323–332, DOI:10.1016/j.ejmech.2004.01.003.
- R. S. Mali, P. P. Joshi, P. K. Sandhu and A. Manekar-Tilve, J. Chem. Soc., Perkin Trans. 1, 2002, 371–376, 10.1039/B109597H.
- L. Xie, Y. Takeuchi, L. M. Cosentino, A. T. McPhail and K. H. Lee, J. Med. Chem., 2001, 44, 664–671, DOI:10.1021/jm000070g.
- D. P. Chakraborty, A. Das Gupta and P. K. Bose, Ann. Biochem. Exp. Med., 1957, 17, 59–62 CAS.
- D. L. Galinis, R. W. Fuller, T. C. McKee, J. H. Cardellina, R. J. Gulakowski, J. B. McMahon and M. R. Boyd, J. Med. Chem., 1996, 39, 4507–4510, DOI:10.1021/jm9602827.
- R. S. Mali, N. A. Pandhare and M. D. Sindkhedkar, Tetrahedron Lett., 1995, 36, 7109–7110, DOI:10.1016/0040-4039(95)01410-J.
- L. Pokhrel, I. Maezawa, T. D. T. Nguyen, K. O. Chang, L. W. Jin and D. H. Hua, J. Med. Chem., 2012, 55, 8969–8973, DOI:10.1021/jm3012189.
- A. Basile, S. Sorbo, V. Spadaro, M. Bruno, A. Maggio, N. Faraone and S. Rosselli, Molecules, 2009, 14, 939–952, DOI:10.3390/molecules14030939.
- I. Kostova, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 29–46, DOI:10.2174/1568011053352550.
- E. Melliou, P. Magiatis, S. Mitaku, A. L. Skaltsounis, E. Chinou and I. Chinou, J. Nat. Prod., 2005, 68, 78–82, DOI:10.1021/np0497447.
- V. A. Osyanin, D. V. Osipov, I. A. Semenova, K. S. Korzhenko, A. V. Lukashenko, O. P. Demidov, N. Yuri and Y. N. Klimochkin, RSC Adv., 2020, 10, 34344–34354, 10.1039/D0RA06450E.
- M. Bittner, J. Jakupovic, F. Bohlmann, M. Grenz and M. Silva, Phytochemistry, 1988, 27, 3263–3266, DOI:10.1016/0031-9422(88)80039-6.
- E. K. Seo, M. C. Wani, M. E. Wall, H. Navarro, R. Mukherjee, N. R. Farnsworth and A. D. Kinghorn, Phytochemistry, 2000, 55, 35–42, DOI:10.1016/S0031-9422(00)00208-9.
- C. Prompanya, C. Fernandes, S. Cravo, M. M. M. Pinto, T. Dethoup, A. M. S. Silva and A. A. Kijjoa, Mar. Drugs, 2015, 13, 1432–1450, DOI:10.3390/md13031432.
- H. Leutbecher, L. A. D. Williams, H. Rosner and U. Beifuss, Bioorg. Med. Chem. Lett., 2007, 17, 978–982, DOI:10.1016/j.bmcl.2006.11.045.
- S. Rana, H. S. Hong, L. Barrigan, L. W. Jin and D. H. Hua, Bioorg. Med. Chem. Lett., 2009, 19, 670–674, DOI:10.1016/j.bmcl.2008.12.060.
- D. H. Hua, Y. Chen, H. S. Sin, M. J. Maroto, P. D. Robinson, S. W. Newell, E. M. Perchellet, J. B. Ladesich, J. A. Freeman, J. P. Perchellet and P. K. Chiang, J. Org. Chem., 1997, 62, 6888–6896, DOI:10.1021/jo970642d.
- R. Sagar, J. Park, M. Koh and S. B. Park, J. Org. Chem., 2009, 74, 2171–2174, DOI:10.1021/jo8023889.
- P. Kumari, S. Gupta, C. Narayana, S. Ahmad, S. Singh and R. Sagar, New J. Chem., 2018, 42, 13985–13997, 10.1039/C8NJ01395K.
- M. Brito-Arias, Synthesis and Characterization of Glycosides, Springer, Boston, MA, 2007, DOI:10.1007/978-0-387-70792-1_1.
- M. Ikawa, M. A. Stahmann and K. P. Link, J. Am. Chem. Soc., 1944, 66, 902–906, DOI:10.1021/ja01234a019.
- M. Seidman, D. N. Robertson and K. P. Link, J. Am. Chem. Soc., 1950, 72, 5193–5195, DOI:10.1021/ja01167a113.
- T. Sunazuka, M. Handa, K. Nagai, T. Shirahata, Y. Harigaya, K. Otoguro, I. Kuwajima and S. Omura, Tetrahedron, 2004, 60, 7845–7859, DOI:10.1016/j.tet.2004.06.059.
- R. Sagar, P. Singh, R. Kumar, P. R. Maulik and A. K. Shaw, Carbohydr. Res., 2005, 340, 1287–1300, DOI:10.1016/j.carres.2005.02.018.
- R. P. Hsung, A. V. Kurdyumor and N. Sydorenko, Eur. J. Org. Chem., 2005, 2005, 23–44, DOI:10.1002/ejoc.200400567.
- A. Mukherjee, S. Mahato, G. V. Zyryanov, A. Majee and S. Santra, New J. Chem., 2020, 44, 18980–18993, 10.1039/D0NJ03846F.
- V. Khatri, A. Kumar, B. Singh, S. Malhotra and A. K. Prasad, J. Org. Chem., 2015, 80, 11169–11174, DOI:10.1021/acs.joc.5b01933.
- J. C. Jung, Y. J. Jung and O. S. Park, Synth. Commun., 2001, 31, 1195–1200, DOI:10.1081/SCC-100104003.
- S. Sipos and I. Jablonkai, Carbohydr. Res., 2011, 346, 1503–1510, DOI:10.1016/j.carres.2011.04.019.
- X. Y. Lei, S.-q. Yao, X.-Y. Zu, Z. X. Huang, L. J. Liu, M. Zhong, B. Y. Zhu, S. Tang and D. F. Liao, Acta Pharmacol. Sin., 2008, 29, 1233–1239, DOI:10.1111/j.1745-7254.2008.00851.x.
- R. C. Osthus, H. Shim, S. Kim, Q. Li, R. Reddy, M. Mukherjee, Y. Xu, D. Wonsey, L. A. Lee and C. V. Dang, J. Biol. Chem., 2000, 275, 21797–21800, DOI:10.1074/jbc.C000023200.
- D. Zheng, J. H. Sussman, M. P. Jeon, S. T. Parrish, M. A. MacMullan, A. Delfarah and N. A. Graham, J. Cell Sci., 2020, 133, jcs239277, DOI:10.1242/jcs.239277.
- M. L. Sheu, F. M. Ho, R. S. Yang, K. F. Chao, W. W. Lin, S. Y. Lin-Shiau and S.-H. Liu, Arterioscler., Thromb., Vasc. Biol., 2005, 25, 539–545, DOI:10.1161/01.ATV.0000155462.24263.e4.
- J. E. N. Jonkman, J. A. Cathcart, F. Xu, M. E. Bartolini, J. E. Amon, K. M. Stevens and P. Colarusso, Cell Adhes. Migr., 2014, 5, 440–451, DOI:10.4161/cam.36224.
- A. Wolfer and S. Ramaswamy, Cell Cycle, 2010, 9, 3639, DOI:10.4161/cc.9.18.13220.
- X. Yang, Y. Xu and T. Wang, Redox Biol., 2017, 11, 653–662, DOI:10.1016/j.redox.2017.01.016.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131, DOI:10.1016/S0142-9612(02)00445-3.
- D. Ribble, N. B. Goldstein, D. A. Norris and Y. G. Shellman, BMC Biotechnol., 2005, 10, 5–12, DOI:10.1186/1472-6750-5-12.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02371k |
| ‡ Ashok Kumar Prasad “author passed away, please see acknowledgements”. |
| This journal is © The Royal Society of Chemistry 2023 |