
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting the capacity and stability of a MoO3 cathode via valence regulation and polypyrrole coating for a rechargeable Zn ion battery†
Yachen Hea,
Weiwei Xuea,
Yifeng Huanga,
Hongwei Tanga,
Guangxia Wang a,
Dezhou Zheng*a,
Wei Xua,
Fuxin Wang*a and
Xihong Luab
a,
Dezhou Zheng*a,
Wei Xua,
Fuxin Wang*a and
Xihong Luab
aSchool of Applied Physics and Materials, Wuyi University, Jiangmen 529020, PR China. E-mail: wangfux91@126.com
bMOE of the Key Laboratory of Bioinorganic and Synthetic Chemistry, The Key Lab of Low-carbon Chem & Energy Conservation of Guangdong Province, School of Chemistry, Sun Yat-Sen University, Guangzhou 510275, PR China
First published on 19th May 2023
Abstract
Molybdenum trioxide (MoO3) is emerging as a hugely competitive cathode material for aqueous zinc ion batteries (ZIBs) for its high theoretical capacity and electrochemical activity. Nevertheless, owing to its undesirable electronic transport capability and poor structural stability, the practical capacity and cycling performance of MoO3 are yet unsatisfactory, which greatly blocks its commercial use. In this work, we report an effective approach to first synthesise nanosized MoO3−x materials to provide more active specific surface areas, while improving the capacity and cycle life of MoO3 by introducing low valence Mo and coated polypyrrole (PPy). MoO3 nanoparticles with low-valence-state Mo and PPy coating (denoted as MoO3−x@PPy) are synthesized via a solvothermal method and subsequent electrodeposition process. The as-prepared MoO3−x@PPy cathode delivers a high reversible capacity of 212.4 mA h g−1 at 1 A g−1 with good cycling life (more than 75% capacity retention after 500 cycles). In contrast, the original commercial MoO3 sample only obtains a capacity of 99.3 mA h g−1 at 1 A g−1, and a cycling stability of 10% capacity retention over 500 cycles. Additionally, the fabricated Zn//MoO3−x@PPy battery obtains a maximum energy density of 233.6 W h kg−1 and a power density of 11.2 kW kg−1. Our results provide an efficient and practical approach to enhance commercial MoO3 materials as high-performance cathodes for AZIBs.
Introduction
With the rapid development of wind, water, and solar energy, along with other forms of clean energy, the need for corresponding electrochemical energy storage technology has become extremely urgent.1,2 Additionally, increased demands for the development of this technology have been raised due to the electrification of transportation tools and the growing intelligence of electronic devices.3 Rechargeable lithium-ion batteries are already extensively utilized in energy storage devices on account of the advantages of their high energy density.4 Nevertheless, the limited resources of lithium elements, as well as safety concerns associated with the use of high-cost organic electrolytes, have restricted the further development of lithium-ion batteries.5 Compared with organic batteries, aqueous batteries use an aqueous solution as the electrolyte,6 which have remarkable merits of high safety, inexpensive, eco-friendly, and simple packaging.7 The superior ionic conductivity of aqueous solutions gives them excellent rate performance.8 In recent years, aqueous secondary batteries, including monovalent ion (Li+, Na+, K+) and multivalent ion (Ca2+, Zn2+, Mg2+, Al3+) batteries, have attracted a lot of attention.9,10 Among them, aqueous zinc ion batteries (AZIBs) are growing rapidly for the advantages of inexpensive,11 low potential (−0.76 V vs. SHE),12,13 high capacity (820 mA h g−1),14 and good stability in aqueous solutions of zinc anode,15 making them suitable for the broad scope of use in large-scale grid energy storage as well as flexible and wearable electronic devices.16As an innovative energy storage device, AZIBs currently have limited range of materials suitable as cathode materials for zinc ion storage.17 They are mainly consist of manganese-based oxides,18 vanadium-based oxides,19 molybdenum-based oxides,1 Prussian blue and its analogues,20 and organic oxides.21 Among them, molybdenum trioxide (MoO3) has the advantages of high abundance,22 low toxicity and easy recovery,23 as well as its multivalent states and two-dimensional layered structure.24 Nevertheless, MoO3 suffers from a serious drawback that it tends to dissolve in neutral or weakly acidic electrolytes to destroy its structure,25 resulting in low actual capacity and poor cycling stability.26 In previous studies, the electrochemical performance of electrode materials have been generally boosted by means of modifications such as morphology modulation,27 interfacial engineering,28 valence modulation,29 doping and coating,30 and composites.31 As an instance, Liu et al. reported that MoO3 can exhibit an initial discharge capacity of 120 mA h g−1, but its capacity decreases sharply over 5 cycles of charging/discharging, mainly due to the structure damage.26 Liu et al. used a surface engineering to avoid the structure damage of electrode material through coating a layer of Al2O3 on the surface of MoO3. Through the DFT calculations, MoO3−x obtain a lower Gibbs free energy of Zn2+ adsorption on the surface of MoO3−x, indicating that the desorption of Zn2+ on MoO3−x is thermodynamically more beneficial and its adsorption/desorption of Zn2+ is more reversibility. In addition, it leads to a narrower of the band gap between the valence band and the conduction band, which facilitates the excitation of charge carriers to the conduction band and thus improves the electrical conductivity of MoO3−x.32 The assembled Zn//P-MoO3−x@Al2O3 battery shows improved cycling stability. The capacity retention of Zn//P-MoO3−x@Al2O3 battery was 69.2% over 100 cycles, which is better than that of that of Zn//P-MoO3−x battery (only 40.8%).32 He et al. developed an effective strategy for quasi-solid PVA/ZnCl2 gel electrolyte to stabilize MoO3 nanowires instead of aqueous electrolyte, and the capacity retention of the assembled MoO3//Zn battery reach to 70.4% in gel electrolyte over 400 cycles, but only 27.1% in aqueous electrolyte.33 In addition, Zhang et al. developed WP-MoO3 electrodes by a water-proton co-pre-insertion strategy. WP-MoO3 electrode achieve selective hydrated proton insertion by using water molecules between the layers to increase the insertion energy barrier of zinc ions by blocking the transport path of zinc ions. Based on the above merits, the capacity retention of the prepared battery is 83% after 1000 cycles.34 Despite the impressive progress of these works, there is still room to enhance the capacity and cycle stability of MoO3. Moreover, exploring simpler and practical ways to boost the electrochemical performance of MoO3 electrodes remains a meaningful challenge.35
In this work, we focus on improving the capacity and cycling stability of commercial MoO3 through a simple and effective approach. Low-valent state enriched and polymer coated MoO3 electrode materials (MoO3−x@PPy) are synthesized by a simple solvothermal method and subsequent electrodeposition process. The reactivity and structural stability of MoO3−x@PPy electrodes are boosted with the help of low-valence-state Mo introduction and coating strategies. Firstly, the solvothermal reaction allows the MoO3 particles to be nanosized to obtain more active sites, while the introduction of low-valent molybdenum improves the reactivity of the electrode material. Secondly, the coated PPy layer improves the stability of the electrode material in the electrolyte. Combing the above advantages, the assembled Zn//MoO3−x@PPy battery obtain a high reversible capacity of 212.4 mA h g−1 at 1 A g−1 and favorable cycle life of 75% capacity retention over 500 cycles, while that of Zn//MoO3 battery is only 99.3 mA h g−1 at 1 A g−1 and 10% capacity retention over 500 cycles. Moreover, the fabricated Zn//MoO3−x@PPy battery exhibits a maximum energy density of 233.6 W h kg−1 and power density of 11.2 kW kg−1.
Experimental section
Preparation of MoO3−x electrode
All chemicals in this work were utilized without any purification (the chemicals were 99% pure). 0.3 g of commercial MoO3 powder was added to a mixed solution of 10 ml of water and 10 ml of ethylene glycol. After stirring magnetically for 30 min at room temperature, the homogeneous mixed solution was poured into an autoclave lined with polytetrafluoroethylene and then placed in a blast drying oven at 180 °C for 12 h. After it cooled down, the powder was washed repeatedly with deionized water and anhydrous ethanol after centrifugation, and then set it in an oven at 60 °C for 6 h to obtain black MoO3−x powder. After that, the obtained MoO3−x powder, acetylene black and polyvinylidene fluoride (PVDF) were mixed and ground in the ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 by mass. Then, the above mixture powder was added to N-methylpyrrolidone (NMP) solution to form a uniform slurry. Then the obtained slurry was homogeneously coated on carbon paper and dried in a vacuum oven for 6 h to obtain MoO3−x electrode.
:1:1 by mass. Then, the above mixture powder was added to N-methylpyrrolidone (NMP) solution to form a uniform slurry. Then the obtained slurry was homogeneously coated on carbon paper and dried in a vacuum oven for 6 h to obtain MoO3−x electrode.
Preparation of MoO3−x@PPy electrode
The above prepared MoO3−x electrode (1 × 2 cm) was electrodeposited in a homogeneous electrolyte formed by mixing and stirring 0.1 M sodium sulfate and 0.145 M pyrrole. The deposition process was performed using cyclic voltammetry with a voltage window of −0.5 to 0.8 V for 4 segments at 50 mV s−1, resulting in the electrodeposition of PPy coating on the surface of the MoO3−x electrode material (MoO3−x@PPy).Characterization of the material
The surface morphology, microstructure, and elemental distribution of electrode materials are characterized by Scanning electron microscopy (SEM, Sigma500, ZEISS) equipped with X-ray energy spectrometry (EDS, INCA 300, Oxford Instruments) and transmission electron microscope (TEM, JEM-F200, JEOL) analysis. The crystal structure of the prepared electrode materials is measured by X-ray diffraction analysers (XRD, Bruker Germany) equipped with a Cu-Kα radiation source. X-ray photoelectron spectroscopy (XPS, NEXSA, Thermo VG) and Fourier Transform infrared spectroscopy (FTIR, NICOLET 6700) were utilized to investigate the elemental composition and functional groups of prepared electrode materials.Electrochemical tests
The cathode material has an active substance loading of about 2 mg cm−2. Electrochemical characterization techniques such as cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), and galvanostatic charge/discharge (GCD) tests were conducted on an electrochemical workstation (CHI660E) with a potential window of 0.2–1.3 V using AZIBs packed in air with pure MoO3, fabricated MoO3−x, and fabricated MoO3−x@PPy as cathodes, zinc plates as anodes, and 2 M ZnSO4 as the electrolyte.Results and discussion
The synthesis of MoO3−x@PPy electrode consists of a two-step synthesis process as shown in Fig. 1a. Firstly, the MoO3−x electrode material enriched in the low-valence-state Mo was synthesized by solvothermal reaction of commercial MoO3 with ethylene glycol. The free molybdenum produced by the dissolving of commercial MoO3 is partially reduced by ethylene glycol in solution, which resulted in the synthesis of MoO3−x electrode material. Subsequently, it was coated with acetylene black and PVDF in the ratio of 8:1:1 on carbon paper to produce MoO3−x electrode. A comparison of SEM image of commercial MoO3 electrode (Fig. S1†) and MoO3−x electrode (Fig. 1b and d) shows that the particles of MoO3−x electrode materials become smaller after the solvent heat treatment. Secondly, the conducting polymer PPy was deposited on the surface of the MoO3−x electrode with an electrodeposition process. As shown in Fig. 1c and e, the morphology of the electrode material is almost unchanged after PPy deposition, implying that the polymer coating has not changes the microscopic morphology of the MoO3−x electrode material.
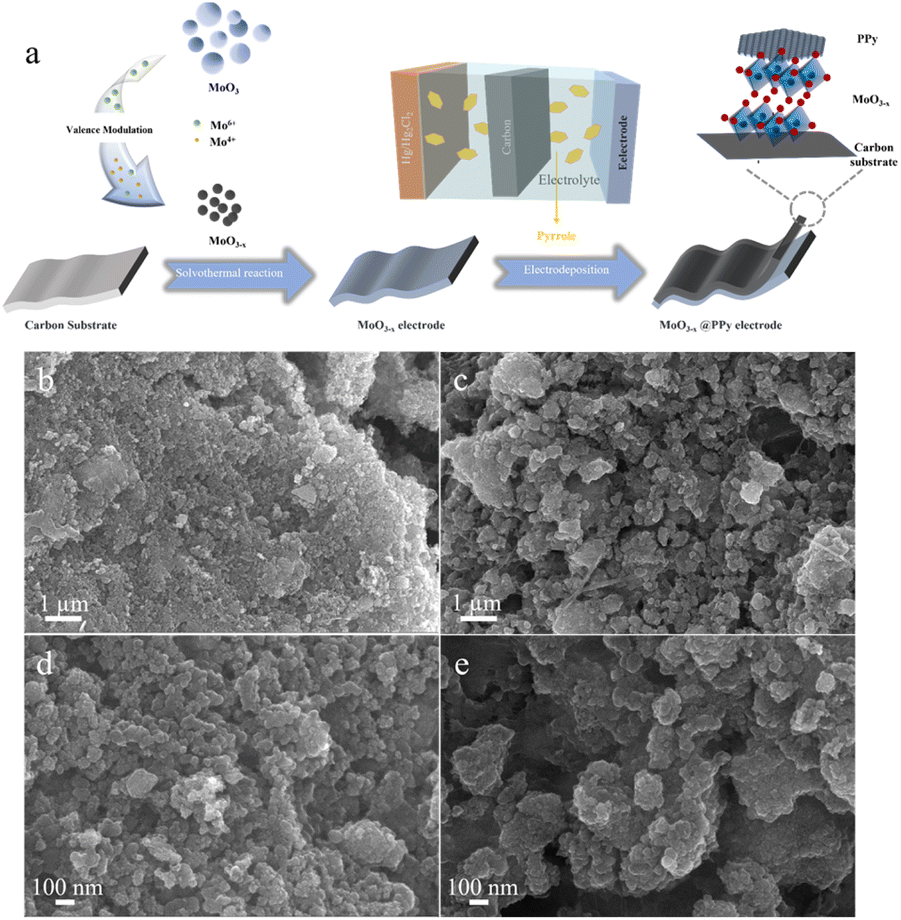 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of MoO3−x@PPy electrode material. (b and d) SEM images of MoO3−x electrode material. (c and e) SEM images of MoO3−x@PPy electrode material. | ||
To further investigate the microstructure and crystal type of the material, the synthesized material was characterized by TEM and XRD. Fig. S2† shows a TEM image of commercial MoO3 showing a particle size of approximately 150 nm. In addition, a lattice stripe of 0.133 nm is visible in the HRTEM image of MoO3, which corresponds to the (202) crystal plane of MoO3 (JCPDF # 05-0508), while the calibrated diffraction points in selected electron diffraction (SAED) pattern of the MoO3 material correspond to the to the (150), (210) and (110) crystal planes of MoO3 (JCPDF # 05-0508). Fig. 2a and b shows a TEM image of MoO3−x, showing a particle size of approximately 50 nm, which is much smaller than the commercial MoO3 material and in line with the SEM results. However, SAED pattern of the MoO3−x material shows two distinct diffraction rings corresponding to the (−321) crystal plane of MoO2 (JCPDS # 32-0671) and the (041) crystal plane of MoO3 (JCPDF # 05-0508), indicating the introduction of low-valent-state Mo after solvothermal reaction. Fig. 2c shows a TEM image of MoO3−x@PPy showing a thin film outside the MoO3−x material, demonstrating that PPy is encapsulated on the surface of the MoO3−x electrode material. Subsequently, the elemental distribution in the MoO3−x@PPy material was analyzed by energy density spectroscopy (EDS). The results display that the O, Mo and N elements are evenly dispersed on the electrode material, again proving that PPy is evenly coated on the MoO3−x electrode material. The crystallization pattern of the prepared materials was investigated more thoroughly by XRD test. Fig. 3a shows the XRD patterns of commercial MoO3, MoO3−x, and MoO3−x@PPy materials. It is noteworthy that the MoO3−x sample synthesized after the solvothermal reaction contains a large number of characteristic peaks (−111), (200), and (211) of MoO2 (JCPDS # 32-0671) peaks and a small number of characteristic peaks (110), (040), and (112) of MoO3, while only characteristic peaks of MoO3 (JCPDF # 05-0508) are present in the commercial MoO3, which implies the successful introduction of low-valent-state Mo in the commercial MoO3 by the solvothermal reaction. In addition, the XRD pattern of MoO3−x@PPy material after electrodeposition of PPy shows a decrease in peak intensity, but characteristic peaks of MoO2 (−111), (200), (211) and MoO3 (112) still can be found in the sample, which indicates that the coated amorphous PPy do not change the crystal structure, which is in line with the results of TEM test. Furthermore, XPS was utilized to analyses the valence and elemental composition of the material. As shown in Fig. 3b and S3b,† the Mo 3d spectrum of commercial MoO3 matches the two characteristic peaks of 3d5/2 and 3d3/2 of Mo6+ at 232.75 and 235.93 eV respectively,36 indicating that molybdenum is predominantly present in commercial MoO3 at +6 valence. However, the MoO3−x sample produced by solvothermal reduction with ethylene glycol showed two additional peaks in its Mo 3d spectrum. Of these, 230.43 and 233.13 eV correspond to the 3d3/2 and 3d5/2 characteristic peaks of Mo4+.37 The presence of Mo4+ indicates the introduction of low-valence-state Mo into the material. Furthermore, according to the analysis of the XPS test, the two characteristic peaks of Mo6+ are still found at 232.64 and 235.41 eV, which implies that the commercial MoO3 has not been completely reduced to MoO2. Fig. S3a† displays the XPS survey spectra of MoO3 and MoO3−x@PPy electrode material, which shows that only the material of MoO3@PPy contains N elements, indicating the successful coated of PPy.38 Also Fig. S3c† shows the Mo 3p spectra of MoO3 and MoO3−x@PPy. The Mo 3p peak is sharper for MoO3, while the distinct hump at 397.5 eV in the Mo 3p XPS spectrum of MoO3−x@PPy further confirms the presence of N on the MoO3−x@PPy surface.39 The O 1s spectra of the MoO3 and MoO3−x samples in Fig. 3c show that for 530.8 ± 0.1 eV (green area fraction) the binding energy component is derived from the lattice oxygen (Oa) of MoO3, for the MoO3−x sample the peak at 532.2 ± 0.1 eV (yellow area fraction) is associated with a chemically absorbed oxygen site (Oc).38 Fig. 3d shows the Fourier transform infrared spectra (FT-IR) of MoO3−x and MoO3−x@PPy electrode materials. The FT-IR spectra of both electrode materials show the core characteristic peaks of molybdenum oxide (Mo–O–Mo), while the main characteristic peaks of PPy (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–N) are also observed in the MoO3−x@PPy electrode,40 again demonstrating that PPy was successfully coated on the surface of the MoO3−x material.
C and C–N) are also observed in the MoO3−x@PPy electrode,40 again demonstrating that PPy was successfully coated on the surface of the MoO3−x material.
 | ||
| Fig. 2 (a) TEM and (b) HRTEM images of MoO3−x material (inset the SAED pattern). (c) TEM image of MoO3−x@PPy material (inset the SAED pattern). (d) The corresponding area EDS mapping. | ||
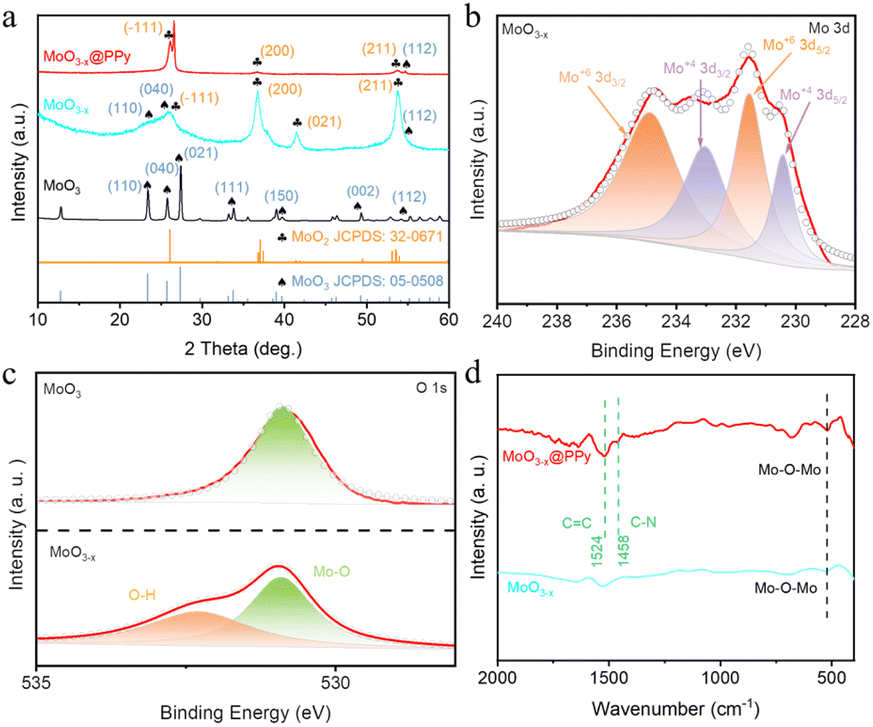 | ||
| Fig. 3 (a) XRD patterns corresponding to MoO3, MoO3−x and MoO3−x@PPy samples. (b) Mo 3d spectra of the synthesized MoO3−x sample. (c) O 1s spectra of MoO3 and MoO3−x samples. (d) FI-IR of MoO3−x and MoO3−x@PPy samples. | ||
To further investigate the effects of low-valence-state Mo introduction and PPy coating strategies on the zinc storage property of commercial MoO3 materials, a series of AZIBs based on the synthesized MoO3, MoO3−x, and MoO3−x@PPy materials MoO3 as cathode, zinc plate as anode and 2 M ZnSO4 as electrolyte were assembled. First, we optimised the thickness of the electrodeposition PPy layers. As shown in Fig. S4,† its electrochemical property decreases as the number of deposition cycles increases, indicating that too thick PPy will hinder MoO3−x from participating in the electrochemical reaction. However, PPy layer with appropriate thickness (2 cycles of electrodeposition) can not only stabilize the structural stability of MoO3−x in electrolyte, but also provide additional zinc storage performance. Fig. 4a shows the CV curves of assembled Zn//MoO3, Zn//MoO3−x, and Zn//MoO3−x@PPy batteries at 20 mV s−1 within the operating window of 0.2–1.3 V.
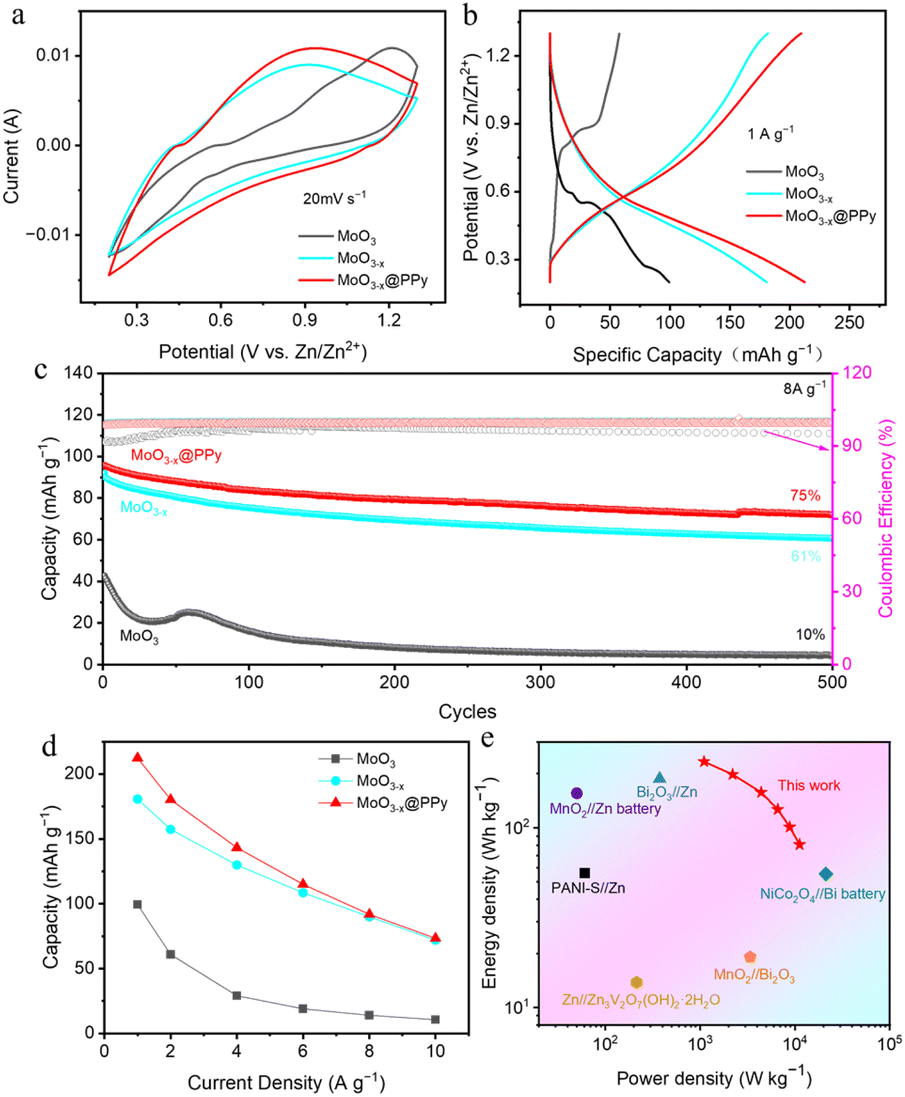 | ||
| Fig. 4 (a) CV curves of Zn//MoO3, Zn//MoO3−x and Zn//MoO3−x@PPy batteries at 20 mV s−1. (b) GCD curves of Zn//MoO3, Zn//MoO3−x and Zn//MoO3−x@PPy batteries at 1A g−1. (c) Cycle performance of Zn//MoO3, Zn//MoO3−x and Zn//MoO3− x@PPy batteries at 8 A g−1. (d) Rate performance of Zn//MoO3, Zn//MoO3−x and Zn//MoO3−x@PPy batteries. (e) Ragone plots of the fabricated Zn//MoO3−x@PPy battery in this work compared to other reported batteries. | ||
The graph clearly shows that the CV curves of the three batteries are almost identical in shape, but the current response signal gradually enlarges as the introduction of the low-valence-state Mo and the PPy coating, indicating that their zinc storage performance gradually enhanced. Then, the GCD curves in Fig. 4b shows more clearly that the Zn//MoO3−x@PPy battery achieves a highest discharge capacity of 212.4 mA h g−1 (based on the mass of the cathode active material) at 1 A g−1, while that of Zn//MoO3−x and Zn//MoO3 batteries are only 181.7 mA h g−1 and 99.3 mA h g−1, respectively. These results demonstrate that the introduce of low-valence-state Mo and PPy coating strategies can boost the Zn storage capacity of commercial MoO3. Furthermore, the rate performance of the fabricated three kinds of batteries was characterized at different current densities, illustrated in Fig. 4d, where the Zn//MoO3−x@PPy shown highest capacities at all currents. The reversible specific capacities of Zn//MoO3−x@PPy battery can reach 212.4 mA h g−1 to 73.3 mA h g−1 at the current density of 1 to 10 A g−1, respectively. In particular, Zn//MoO3−x@PPy battery exhibited 35% capacitance retention even at a high current density of 10 A g−1. By contrast, Zn//MoO3 battery achieved specific capacities of 99.3 mA h g−1 to 10.5 mA h g−1 at 1 to 10 A g−1, respectively. Its rate property is only 10.6%, again indicating that the low-valence-state Mo introduction and PPy coating strategies can boost the capacity and rate property of commercial MoO3 for zinc storage. Much encouragingly, the Zn//MoO3−x@PPy battery maintained 75% capacity retention over 500 cycles, while that of Zn//MoO3 and Zn//MoO3−x batteries were not high, 61% and 10%, respectively, indicating that coating with a corrosion resistant polymer coating is a very simple and practical way to enhance the stability of electrode materials (Fig. 4c). It is worth noting that the specific capacity of MoO3 is improved over the first 100 cycles, which might attribute to the continuous activation of the electrode due to the surface chemical reactions between MoO3 and the ZnSO4 electrolyte.41 Energy density and power density are the two vital parameters for evaluating the energy storage capacity of batteries. As shown in Fig. 4e, the fabricated Zn//MoO3−x@PPy battery obtained an energy density of up to 233.6 W h kg−1 (power density of 1.1 kW kg−1) and the maximum power density of 11.2 kW kg−1 (energy density of 80.6 W h kg−1), exceed those of some newly published aqueous batteries, as shown in Table 1. This satisfactory energy density and power density are expected to enable our designed Zn//MoO3−x@PPy battery to become a more practical alternative power source.
The electrochemical kinetics of the fabricated electrode were further investigated by conducting CV measurements at various scan rates ranging from 1 to 10 mV s−1 (displayed in Fig. 5a and S5†). As can clearly be seen the figure of the CV curve at any scan rates is essentially the same, but the peaks are slightly shifted. The diffusion and pseudocapacitance behaviour of the battery during the charge/discharge cycle was distinguished by adjusting the scan rate to correspond to the response of the peak current. In the case of a dominant battery behaviour, the relationship between peak current i and scan voltage v follows a power function of 0.5, indicating that diffusion plays an important role throughout the process. When analyzing battery behavior during charge/discharge cycles, it is possible to distinguish between diffusive and pseudo-capacitive behavior by adjusting the scan rate to match peak current response. In cases where the process is dominated by capacitance, peak current i varies linearly with scanning voltage v. To determine the presence of pseudo-capacitive behavior, researchers can calculate the b value using the equation i = avb. This allows for a more accurate understanding of electrode material behavior during charging and discharging. When the value of b equals 0.5, the electrode material displays battery characteristics. If the value of b falls among 0.5 to 1, both battery and pseudocapacitive properties can be observed in the electrode material. When b is greater than or equal to 1, the electrode material mainly displays pseudocapacitive behavior. Fig. 5b displays the fitted CV curves for the MoO3−x@PPy samples at different scan rates. It is noteworthy that the calculated b value of the reduction peak is 0.79, suggesting that a blend of both diffusion-controlled and capacity-based kinetic phenomena occurred. To determine the pseudo-capacitance contribution of MoO3−x@PPy at various scan rates, the pseudo-capacitance equation was utilized: i = k1v + k2v1/2. This equation takes into account k1 and k2 as constants, with k1v representing the theoretical value of the pseudo-capacitance contribution. Fig. 5c and S5† showcase the outcomes of this calculation for Zn//MoO3, Zn//MoO3−x, and Zn//MoO3−x@PPy batteries with varying scan rates ranging from 1 to 10 mV s−1. Upon comparison of the pseudocapacitance contribution at a scan rate of 10 mV s−1, it is evident that the Zn//MoO3−x@PPy battery exhibits the highest pseudocapacitance contribution, indicating superior rate performance. This finding corroborates with the data presented in Fig. 4d. Additionally, it is worth noting that the Zn//MoO3−x@PPy battery achieves a significant capacitance contribution of 86% at 10 mV s−1, further highlighting the dominance of pseudocapacitance in determining its performance (Fig. S6†). Notably, this observation is consistent with the outcomes obtained from our experiments and analysis. The MoO3−x@PPy active material possesses a pseudocapacitive nature that facilitates selective storage of Zn2+ ions at the electrode surface. This results in a shortened ion transport distance and an enhanced electron transport rate, ultimately leading to improved rate capabilities and longer cycle life of the electrode material. Zn2+ diffusion coefficients DZn2+ in the MoO3, MoO3−x and MoO3−x@PPy cathodes were also investigated through the galvanostatic intermittent titration technique (GITT). From Fig. 5d and S7,† all three electrodes exhibit a curve shape similar to the GCD curve (Fig. 4b). The ion diffusion coefficient of the MoO3−x@PPy electrode consistently surpasses that of the MoO3 and MoO3−x electrode, indicating that the ion diffusion rate is enhanced by coating it with PPy and low-valence-state Mo introduction (Fig. 5e and S7†). The electrochemical impedance spectroscopy (EIS) data for all electrode materials is presented in Fig. 5f. This figure clearly illustrates that the MoO3−x@PPy electrode exhibits a smaller semicircle in the high frequency region, which indicates a lower charge transfer resistance (Rct). The Rct of MoO3−x@PPy is approximately 278.3 Ω, lower than that of MoO3−x (357.5 Ω) and commercial MoO3 (769.9 Ω), demonstrating that the Rct of the MoO3−x@PPy electrode material can be greatly boosted by the low-valence-state Mo introduction and PPy coating strategies. Furthermore, in the low frequency region, the MoO3−x@PPy electrode demonstrates a steep linear slope, implying efficient electron diffusion capability. This is in contrast to the other electrode materials tested, which exhibit a lower and less steep slope in this region.
 | ||
| Fig. 5 (a) CV curves for MoO3−x@PPy at a range of scan rates of 1, 2, 4, 6, 8 and 10 mV s−1. (b) log(i) vs. log(v) plots of the two peaks in the CV curve for MoO3−x@PPy. (c) Ratio of diffusion contribution to capacitance contribution for Zn//MoO3−x and Zn//MoO3−x@PPy. (d) Discharge GITT curves for Zn//MoO3−x and Zn//MoO3−x@PPy at a current density of 2 A g−1 and (e) corresponding Zn2+ coefficients DZn2+. (f) Nyquist plots of MoO3, MoO3−x and MoO3−x@PPy. | ||
Conclusions
In this work, we synthesized high capacity, long cycle life PPy coated low-valent-state Mo enriched composites MoO3−x@PPy as cathode materials for AZIBs by a simple and effective solvothermal and electrodeposition method in two steps. With the help of low-valence-state Mo introduction and PPy coating to synergistically enhanced the capacity and cycling life of commercial MoO3. The Zn//MoO3−x@PPy battery assembled based on MoO3−x@PPy as the cathode material exhibited a high capacity of 212.4 mA h g−1 at 1 A g−1 and outstanding rate performance (73.3 mA h g−1 capacity even at 10 A g−1). Encouragingly, the Zn//MoO3−x@PPy battery exhibits excellent cycling life with a capacity retention rate of 75% after 500 cycles. Meanwhile, the Zn//MoO3 battery assembled based on commercial MoO3 has only 99.3 mA h g−1 capacity at 1 A g−1 and 10% capacitance retention (after 500 cycles). Moreover, the prepared battery exhibited favorable energy density of 233.6 W h kg−1. This work shows practical strategies to boost the zinc storage property of commercial MoO3, providing new ideas and approaches for its commercialization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (22005222), Guangdong Basic and Applied Basic Research Foundation (2022A1515110136, 2020A1515110120), Innovation Foundation of Educational Commission Guangdong Province (2022KQNCX090), Guangdong province innovation and strong school project (2020ZDZX2004, and 2020KQNCX087), Joint Science Foundation of Wuyi University and HK and Macao (2019WGALH14), Basic and Applied Basic Research Foundation of Jiangmen (JZ202212, 2020030102940008548), and Science Foundation for High-Level Talents of Wuyi University (2019AL022, 2019AL029, and 5041700133).Notes and references
- Z. Fang, C. Liu, X. Li, L. Peng, W. Ding, X. Guo and W. Hou, Adv. Funct. Mater., 2022, 33, 2210010 CrossRef.
- X. Wang, Z. Zhang, B. Xi, W. Chen, Y. Jia, J. Feng and S. Xiong, ACS Nano, 2021, 15, 9244–9272 CrossRef CAS PubMed.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- X. Li, Z. Huang, C. E. Shuck, G. Liang, Y. Gogotsi and C. Zhi, Nat. Rev. Chem., 2022, 6, 389–404 CrossRef PubMed.
- M. Zhou, S. Guo, J. Li, X. Luo, Z. Liu, T. Zhang, X. Cao, M. Long, B. Lu and A. Pan, Adv. Mater., 2021, 33, 2100187 CrossRef CAS PubMed.
- C. Liu, Z. Neale, J. Zheng, X. Jia, J. Huang, M. Yan, M. Tian, M. Wang, J. Yang and G. Cao, Energy Environ. Sci., 2019, 12, 2273–2285 RSC.
- K. Ouyang, D. Ma, N. Zhao, Y. Wang, M. Yang, H. Mi, L. Sun, C. He and P. Zhang, Adv. Funct. Mater., 2022, 32, 2109749 CrossRef CAS.
- J. Cao, D. Zhang, X. Zhang, Z. Zeng, J. Qin and Y. Huang, Energy Environ. Sci., 2022, 15, 499–528 RSC.
- H. Chen, C. Dai, F. Xiao, Q. Yang, S. Cai, M. Xu, H. J. Fan and S. J. Bao, Adv. Mater., 2022, 34, 2109092 CrossRef CAS PubMed.
- X. Zhang, T. Xiong, B. He, S. Feng, X. Wang, L. Wei and L. Mai, Energy Environ. Sci., 2022, 15, 3750–3774 RSC.
- Y. Tian, Y. An, C. Wei, B. Xi, S. Xiong, J. Feng and Y. Qian, Adv. Energy Mater., 2021, 11, 2002529 CrossRef CAS.
- S. Zhou, Y. Wang, H. Lu, Y. Zhang, C. Fu, I. Usman, Z. Liu, M. Feng, G. Fang and X. Cao, Adv. Funct. Mater., 2021, 31, 2104361 CrossRef CAS.
- M. Chen, J. Chen, W. Zhou, X. Han, Y. Yao and C. P. Wong, Adv. Mater., 2021, 33, 2007559 CrossRef CAS PubMed.
- J. Huang, Z. Guo, Y. Ma, D. Bin, Y. Wang and Y. Xia, Small Methods, 2019, 3, 1800272 CrossRef.
- X. Yang, C. Li, Z. Sun, S. Yang, Z. Shi, R. Huang, B. Liu, S. Li, Y. Wu and M. Wang, Adv. Mater., 2021, 33, 2105951 CrossRef CAS PubMed.
- P. Wang, S. Liang, C. Chen, X. Xie, J. Chen, Z. Liu, Y. Tang, B. Lu and J. Zhou, Adv. Mater., 2022, 34, 2202733 CrossRef CAS PubMed.
- R. Guo, X. Liu, F. Xia, Y. Jiang, H. Zhang, M. Huang, C. Niu, J. Wu, Y. Zhao and X. Wang, Adv. Mater., 2022, 34, 2202188 CrossRef CAS PubMed.
- F. Gao, B. Mei, X. Xu, J. Ren, D. Zhao, Z. Zhang, Z. Wang, Y. Wu, X. Liu and Y. Zhang, Chem. Eng. J., 2022, 448, 137742 CrossRef CAS.
- W. Liang, D. Rao, T. Chen, R. Tang, J. Li and H. Jin, Angew. Chem., Int. Ed., 2022, 61, e202207779 CAS.
- M. Wu, G. Zhang, H. Yang, X. Liu, M. Dubois, M. A. Gauthier and S. Sun, InfoMat, 2022, 4, e12265 CAS.
- Y. Zhao, Y. Huang, F. Wu, R. Chen and L. Li, Adv. Mater., 2021, 33, 2106469 CrossRef CAS PubMed.
- X. Chen, J. Zhu, J. Cai, Y. Zhang and X. Wang, J. Energy Storage, 2021, 40, 102721 CrossRef.
- M. R. Dunkin, J. Kuang, S. Yan, S. T. King, L. M. Housel, L. Ma, S. N. Ehrlich, J. S. Okasinski, K. J. Takeuchi and E. S. Takeuchi, Adv. Mater. Interfaces, 2022, 9, 2201125 CrossRef CAS.
- W. Xu, K. Zhao and Y. Wang, Energy Storage Mater., 2018, 15, 374–379 CrossRef.
- Y. Liu, G. He, H. Jiang, I. P. Parkin, P. R. Shearing and D. J. Brett, Adv. Funct. Mater., 2021, 31, 2010445 CrossRef CAS.
- W. Liu, J. Hao, C. Xu, J. Mou, L. Dong, F. Jiang, Z. Kang, J. Wu, B. Jiang and F. Kang, Chem. Commun., 2017, 53, 6872–6874 RSC.
- D. Chen, M. Lu, D. Cai, H. Yang and W. Han, J. Energy Chem., 2021, 54, 712–726 CrossRef CAS.
- Z. Kang, C. Wu, L. Dong, W. Liu, J. Mou, J. Zhang, Z. Chang, B. Jiang, G. Wang and F. Kang, ACS Sustainable Chem. Eng., 2019, 7, 3364–3371 CrossRef CAS.
- P. He, Q. Chen, M. Yan, X. Xu, L. Zhou, L. Mai and C.-W. Nan, EnergyChem, 2019, 1, 100022 CrossRef.
- C. Li, X. Zhang, W. He, G. Xu and R. Sun, J. Power Sources, 2020, 449, 227596 CrossRef CAS.
- Y. Li, J. Zhang, Q. Chen, X. Xia and M. Chen, Adv. Mater., 2021, 33, 2100855 CrossRef CAS PubMed.
- Y. Liu, J. Wang, Y. Zeng, J. Liu, X. Liu and X. Lu, Small, 2020, 16, 1907458 CrossRef CAS PubMed.
- X. He, H. Zhang, X. Zhao, P. Zhang, M. Chen, Z. Zheng, Z. Han, T. Zhu, Y. Tong and X. Lu, Adv. Sci., 2019, 6, 1900151 CrossRef PubMed.
- H. Zhang, W. Wu, Q. Liu, F. Yang, X. Shi, X. Liu, M. Yu and X. Lu, Angew. Chem., Int. Ed., 2021, 60, 896–903 CrossRef CAS PubMed.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- W. Liu, Z. Zhang, J. Shi, Y. Zheng, Y. Wu, X. Fu, N. Liu, J. Su and Y. Gao, J. Mater. Chem. A, 2022, 10, 4043–4052 RSC.
- C. Xia, Y. Zhou, D. B. Velusamy, A. A. Farah, P. Li, Q. Jiang, I. N. Odeh, Z. Wang, X. Zhang and H. N. Alshareef, Nano Lett., 2018, 18, 1506–1515 CrossRef CAS PubMed.
- Y. Liu, Y. Wang, Y. Meng, R. Plamthottam, W. W. Tjiu, C. Zhang and T. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 4490–4499 CrossRef CAS PubMed.
- C. Tang, L. Zhong, B. Zhang, H. F. Wang and Q. Zhang, Adv. Mater., 2018, 30, 1705110 CrossRef PubMed.
- X. Mu, Y. Song, Z. Qin, J. Meng, Z. Wang and X.-X. Liu, Chem. Eng. J., 2023, 453, 139575 CrossRef CAS.
- Z. Yao, W. Zhang, X. Ren, Y. Yin, Y. Zhao, Z. Ren, Y. Sun, Q. Lei, J. Wang, L. Wang, T. Ji, P. Huai, W. Wen, X. Li, D. Zhu and R. Tai, ACS Nano, 2022, 16, 12095–12106 CrossRef CAS PubMed.
- L. Xu, N. Xu, C. Yan, W. He, X. Wu, G. Diao and M. Chen, J. Electroanal. Chem., 2021, 888, 115196 CrossRef CAS.
- D. Zhou, Q. Lu, Z. Li, M. Zhao, S. Li, K. Yin and C. Teng, Chem.–Eur. J., 2023, 29, e202203500 CrossRef CAS PubMed.
- H. Y. Shi, Y. J. Ye, K. Liu, Y. Song and X. Sun, Angew. Chem., Int. Ed., 2018, 130, 16597–16601 CrossRef.
- X. Wu, H. B. Wu, W. Xiong, Z. Le, F. Sun, F. Liu, J. Chen, Z. Zhu and Y. Lu, Nano Energy, 2016, 30, 217–224 CrossRef CAS.
- Y. Zeng, Z. Lin, Y. Meng, Y. Wang, M. Yu, X. Lu and Y. Tong, Adv. Mater., 2016, 28, 9188–9195 CrossRef CAS PubMed.
- B. Sambandam, V. Soundharrajan, S. Kim, M. H. Alfaruqi, J. Jo, S. Kim, V. Mathew, Y.-k. Sun and J. Kim, J. Mater. Chem. A, 2018, 6, 3850–3856 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02350h |
| This journal is © The Royal Society of Chemistry 2023 |