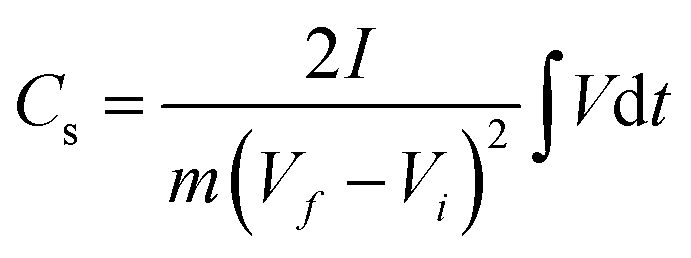DOI:
10.1039/D3RA02314A
(Paper)
RSC Adv., 2023,
13, 16136-16144
Introducing micropores into carbon nanoparticles synthesized via a solution plasma process by thermal treatment and their charge storage properties in supercapacitors†
Received
7th April 2023
, Accepted 15th May 2023
First published on 8th June 2023
Abstract
Carbon materials synthesized via a solution plasma process (SPP) have recently shown great potential for various applications. However, they mainly possess a meso–macroporous structure with a lack of micropores, which limits their applications for supercapacitors. Herein, carbon nanoparticles (CNPs) were synthesized from benzene via SPP and then subjected to thermal treatment at different temperatures (400, 600, 800, and 1000 °C) in an argon environment. The CNPs exhibited an amorphous phase and were more graphitized at high treatment temperatures. A small content of tungsten carbide particles was also observed, which were encapsulated in CNPs. An increase in treatment temperature led to an increase in the specific surface area of CNPs from 184 to 260 m2 g−1 through the development of micropores, while their meso–macropore structure remained unchanged. The oxygen content of CNPs decreased from 14.72 to 1.20 atom% as the treatment temperature increased due to the degradation of oxygen functionality. The charge storage properties of CNPs were evaluated for supercapacitor applications by electrochemical measurements using a three-electrode system in 1 M H2SO4 electrolyte. The CNPs treated at low temperatures exhibited an electric double layer and pseudocapacitive behavior due to the presence of quinone groups on the carbon surface. With increasing treatment temperature, the electric double layer behavior became more dominant, while pseudocapacitive behavior was suppressed due to the quinone degradation. Regarding cycling stability, the CNPs treated at high temperatures (with a lack of oxygen functionality) were more stable than those treated at low temperatures. This work highlights a way of introducing micropores into CNPs derived from SPP via thermal treatment, which could be helpful for controlling and adjusting their pore structure for supercapacitor applications.
Introduction
Supercapacitors have emerged as a promising class of electrochemical energy-storage devices that could bridge the gap between conventional capacitors and batteries.1 They offer several advantages over other energy storage devices, including high power density, fast charge and discharge, long cycle life, and environmental friendliness.2,3 They can be classified into three main types based on their charge storage mechanism: (i) electric double layer capacitors (EDLCs), which store charges through electrostatic interaction on the surface of the electrode (non-faradaic process),4–6 (ii) pseudocapacitors (PCs), which store charges through reversible redox reactions at the electrode–electrolyte interface (faradaic process),7,8 and (iii) hybrid supercapacitors, which combine both EDLC and PC behaviors to achieve high energy and power density.9–12 The electrode materials have been proven to play a vital role in determining the charge storage performance and stability of supercapacitors.1–3 Various families of materials have been investigated and used as electrode materials in both research and commercial products, including porous carbons,13–16 metal oxide,17 metal sulfide,18 metal nitride,19 MXenes,20 metal–organic frameworks,21 and mixtures thereof.22–25 Among them, carbon-based materials are the most popular and attractive materials for supercapacitor electrodes because of their unique properties, such as low cost, abundance, large specific surface area, microporosity, high conductivity, and high thermal and chemical stability.13–16
Carbon materials with a broad range of nanostructures, porosity, and functionality have successfully been synthesized from either natural or synthetic sources via various synthesis approaches.26–30 Most of the research efforts in this field have concentrated on synthesizing porous carbons from natural biomass via carbonization and activation processes.15,29,30 However, the use of synthetic sources has received relatively little attention so far. Over the past decade, the solution plasma process (SPP) has emerged as a powerful and interesting method for synthesizing carbon materials from various synthetic carbon precursors (e.g., benzene,31–35 toluene,36 xylene37,38) at room temperature and atmospheric pressure. Benzene was found to be the most suitable precursor for synthesizing carbon materials via SPP in terms of the highest yield since its unsaturated ring structure could induce a faster formation rate than the others.35
Typically, carbon materials synthesized via SPP are in a particulate form with an amorphous structure and possess a hierarchical meso–macroporous structure with a lack of micropores.31–34 Due to these properties, they have been applied for various energy and environmental applications, such as electrocatalysts in fuel cells,33,36,39 anode materials in lithium and sodium-ion batteries,37,38 catalysts for biomass conversion,40,41 and adsorbents for CO2 capture and dye removal.34,42 Although significant progress has been made in various applications, there has been no study on the applications of SPP-derived carbon materials in supercapacitors due to the limitations of low surface area and lack of micropores. Furthermore, their charge storage properties are still questionable and require more study. Therefore, it is necessary to address this limitation to broaden their potential application in supercapacitors.
Herein, carbon nanoparticles (CNPs) were synthesized from benzene via SPP. A thermal treatment process was then conducted at 400, 600, 800, and 1000 °C in an inert atmosphere to improve the surface area through the development of micropores. The effect of treatment temperature on the structural properties, morphology, and surface chemical functionality of CNPs was investigated and discussed. The electrochemical charge storage properties of CNPs were tested using a three-electrode system at room temperature in 1 M H2SO4 electrolyte, and their charge storage properties were correlated with their physicochemical properties.
Experimental
Materials
Benzene (C6H6, purity >99%) was purchased from Qrec Chemical, Co., Ltd, and tungsten wire (1 mm diameter, 99.99% purity) was purchased from Nilaco Corporation. Ethanol (C2H5OH, purity 99.9%), isopropanol (C3H8O, purity 99.8%), and sulfuric acid (H2SO4, purity 98%) were purchased from RCI Labscan Ltd. Nafion® DE 521 solution (5 wt% in a mixture of lower aliphatic alcohols and water) was purchased from Sigma-Aldrich. Ultrapure water (18.2 MΩ cm at 25 °C) was obtained from a Direct-Q™ 5 UV Millipore water purification system. All chemicals used in this work were of analytical grade and used without purification.
Synthesis of CNPs via SPP
A pair of symmetric tungsten wire electrodes (1 mm diameter) covered with an insulating ceramic tube was inserted in the center of a 100 mL glass vessel reactor. Benzene (80 mL) was poured into the glass reactor and used as a precursor for CNP synthesis. Plasma was generated at the electrode gap submerged in benzene by connecting them to a bipolar pulse power supply (MPP-HV02, Kurita Seisakusho Co., Ltd, Kyoto, Japan). The voltage amplitude was about 1 kV, and the frequency and pulse duration were 20 kHz and 1 μs, respectively. Once plasma occurred under vigorous stirring, black solid particles were immediately produced from the plasma region, and colorless benzene turned black in a few minutes. The formation mechanism of carbons from SPP has already been reported and described in the literature.31,32,35 After synthesis for 20 min, the solid carbon product was collected by vacuum filtration through a filter paper (Whatman No. 1) and repeatedly washed with ethanol until the washed solvent was colorless. The washed samples were subsequently dried at 80 °C for 24 h in an oven (Memmert, UN55). On average, the yield of carbon product was ∼500 mg (synthesis rate ∼25 mg min−1). The as-synthesized CNPs from SPP were denoted as CNP-RT. The CNP-RT was then loaded into a ceramic boat and subjected to thermal treatment in a tube furnace (Vecstar, VCTF4) at 400, 600, 800, and 1000 °C for 2 h with a heating rate of 10 °C min−1 under a flow of argon gas (0.2 L min−1, purity 99.99%), followed by natural cooling to room temperature. The thermally treated CNP-RT was denoted as CNP-T, where T represents the treatment temperature (i.e., 400, 600, 800, and 1000 °C).
Characterization
To investigate the properties and structure of CNPs, various analytical techniques were employed. The phase structure of CNPs was verified using a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) operated at 40 kV and 30 mA. Raman spectra were collected using a JASCO NRS-5100 spectrometer with a laser excitation wavelength of 532.1 nm. Morphological features of the CNPs were examined with a JEOL JSM-7600F field-emission scanning electron microscope at an acceleration voltage of 1 kV. Energy dispersive spectroscopy (EDS) was performed using an Oxford Instrument spectrometer equipped with a Hitachi SU3500 scanning electron microscope. The microstructural details of CNPs were evaluated using a JEOL JEM-3100F transmission electron microscope at an acceleration voltage of 300 kV. Thermogravimetric analysis of CNPs was performed on a Mettler Toledo TGA 2 analyzer under N2 and O2 flows from 50 to 800 °C with a heating rate of 10 °C min−1. N2 adsorption–desorption isotherms of CNPs were recorded using a Micromeritics 3Flex surface characterization analyzer at −196 °C to evaluate their specific surface area and pore structure. Prior to the measurement of N2 adsorption–desorption isotherms, the CNPs were degassed at 150 °C for 12 h under a vacuum using a Smart VacPrep instrument. The elemental composition and chemical bonding states were determined using a Kratos Axis Ultra DLD X-ray photoelectron spectrometer (XPS) equipped with an Mg Kα radiation as an X-ray source (1253.6 eV). The binding energy was calibrated using the C 1s peak at 284.5 eV.
Preparation of working electrode
The CNPs (5 mg) were dispersed in a mixture of 475 μL of ultrapure water, 475 μL of isopropanol, and 50 μL of Nafion® DE 521. The mixture was sonicated for 60 min to obtain a homogeneous suspension. The glassy carbon (GC) electrode (3 mm disk diameter, ALS Co., Ltd) was thoroughly polished on a polishing pad with 0.1 μm diamond and 0.05 μm alumina slurries. The GC electrode was finally polished on an alumina polishing pad with only water to remove the alumina particles remaining on the electrode surface. The polished and cleaned GC electrodes were rinsed with ultrapure water and air-dried. After that, 3 μL of the prepared suspension was drop-cast onto the GC electrode, which was then air-dried, yielding a mass loading of 0.015 mg, before electrochemical measurements.
Electrochemical measurements
Electrochemical measurements were performed using a three-electrode system in 1 M H2SO4 electrolyte at room temperature. Platinum wire (ALS Co., Ltd) and Ag/AgCl in saturated KCl solution (ALS Co., Ltd) were used as the counter and reference electrodes, respectively. The CNP-modified GC electrodes were used as the working electrodes. Three electrodes were connected to a Biologic VSP potentiostat/galvanostat station controlled by EC-Lab software. Cyclic voltammetry (CV) measurement was performed in the potential range of 0–1 V vs. Ag/AgCl in 1 M H2SO4 solution at different scan rates from 10 to 100 mV s−1. Additionally, galvanostatic charge–discharge (GCD) test was conducted in the same potential range corresponding to the CV measurements at different current densities ranging from 1 to 20 A g−1. A stability test was done by charge–discharge cycling for 5000 cycles at a current density of 10 A g−1.
Results and discussion
Structural properties
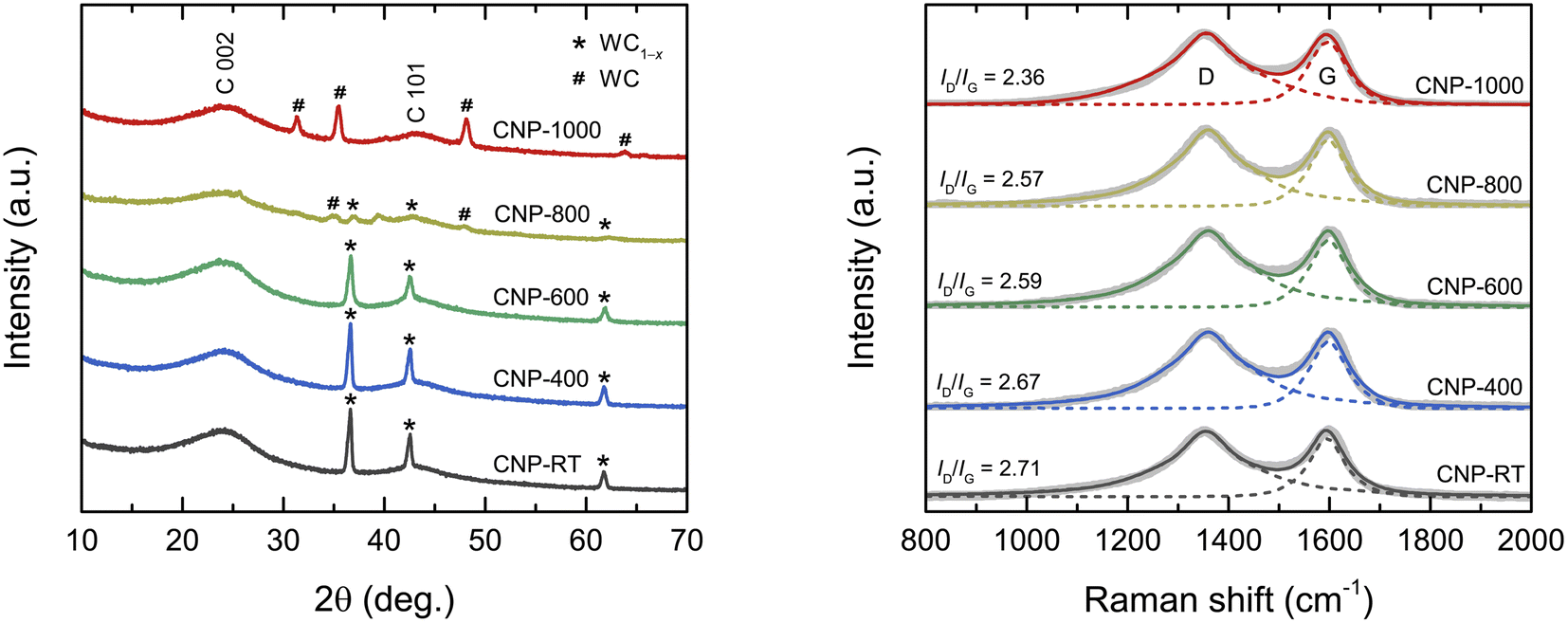
The XRD patterns of all CNPs are shown in Fig. 1a. The major broad peak at 24° and a small hump at 43° corresponded to the (002) and (101) planes of the amorphous carbon, respectively. The d002 lattice spacing was calculated from the C 002 peak using Bragg's law as about 0.370 nm, which was larger than that of ideal graphite (0.335 nm). A broad diffraction peak and large d002 values indicate an amorphous structure of CNPs. Additionally, for CNP-RT, CNP-400, and CNP-600, a set of sharp diffraction peaks detected at 36.7°, 42.6°, and 61.8° were identified as the (111), (200), and (220) planes of the WC1−x phase (JCPDS no. 20-1316), respectively. Although the WC1−x phase showed a more intense peak than the carbon phase, it cannot be used to indicate a higher amount of WC1−x in CNPs. According to the TGA analysis under O2 flow, the amount of tungsten was only 2.25 wt%, as indicated by the remaining weight at 800 °C (Fig. S1†). For CNP-800, the peak intensity of WC1−x significantly decreased, while new peaks appeared at 31.4°, 35.4°, 48.1°, and 63.8°, which correspond to the (001), (100), (101), and (110) planes of WC (JCPDS no. 65-8828). The peaks of the WC phase became more intense for CNP-1000, whereas those of the WC1−x phase disappeared completely. This result indicates that the WC1−x began to transform into the WC phase at 800 °C, which was completed at 1000 °C. The WC1−x phase is typically found in CNPs synthesized from SPP due to the interaction between tungsten eroded from the electrodes and carbon radicals generated from carbon precursors during synthesis.43–45
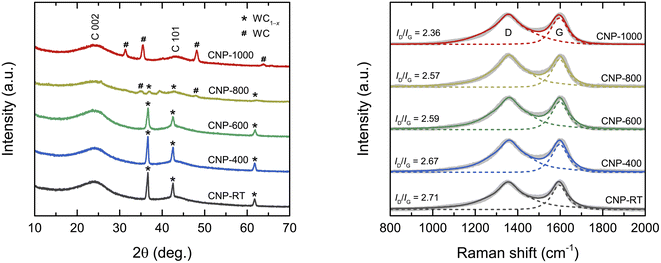 |
| | Fig. 1 (a) XRD patterns and (b) Raman spectra of CNPs. | |
Raman spectroscopic measurement was carried out to obtain more structural information on the degree of graphitization of CNPs. The Raman spectra of all CNPs displayed two distinct peaks at 1355 cm−1 and 1600 cm−1, corresponding to the D and G bands, respectively (Fig. 1b). The G band corresponds to the vibration mode of sp2 carbon bonds in the hexagonal lattice of graphitic structure (ordered structure), while the D band is associated with the disordered structure in the carbon lattice.46,47 The ratio of the integrated intensity of the D band to the G band (ID/IG) is typically used to determine the degree of graphitization of carbon materials.46–49 The ID/IG values decreased from 2.71 for CNP to 2.36 for CNP-1000, indicating that the CNPs were more crystallized or had a higher degree of graphitization as the treatment temperature increased.
Morphology and microstructure
The morphology and particle size of CNPs were observed using FESEM and TEM, as displayed in Fig. 2. FESEM images show that all CNPs were composed of aggregated nanosized particles that formed a meso–macroporous structure.32 In the EDS area analysis (Fig. S2†), a small tungsten content was detected at about 0.48 ± 0.14 atom%, which is consistent with the TGA result. TEM images revealed that all CNPs had a round shape with a primary particle size of 20–40 nm, which was consistent with the FESEM images. There was no significant difference in morphology and particle size observed among CNPs treated at different temperatures, indicating that the thermal treatment process had no influence in altering the morphology and particle size of CNPs. High-resolution TEM (HRTEM) images were acquired to gain more microstructural information. A short-range ordered structure of nanocrystalline graphite surrounded by a disordered structure of amorphous carbon was clearly observed. Additionally, small particles of tungsten carbide (dark contrast) were occasionally observed in CNPs (Fig. S3†). The selected-area electron diffraction (SAED) patterns in the inset of the corresponding TEM images show a diffused ring pattern, which confirms the amorphous structure of CNPs. The results obtained from HRTEM and SAED patterns of CNPs are consistent with the XRD results.
 |
| | Fig. 2 FESEM, TEM, and HRTEM images: (a) CNP-RT, (b) CNP-400, (c) CNP-600, (d) CNP-800, and (e) CNP-1000. The corresponding SAED patterns are shown in the inset of TEM images (a-2)–(e-2). | |
Thermal properties
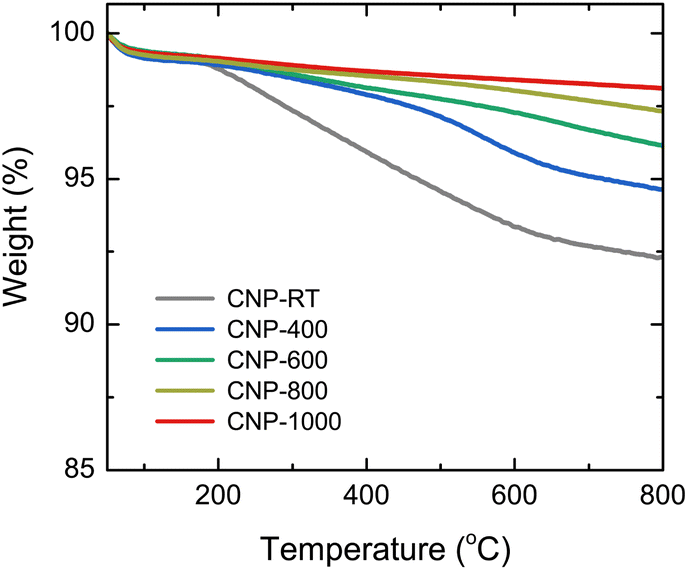
The mass change during the heating of CNPs was examined using TGA analysis at the temperature range of 50–800 °C in an N2 flow to avoid oxidation and burning out of CNPs, as shown in Fig. 3. Two steps of weight loss were observed for all CNPs. The first step occurred between 50 and 150 °C, which was caused by the evaporation of adsorbed water molecules from the CNP surface. The second step ranging from 150 to 600 °C was possibly due to the degradation of residual organic fractions produced from SPP and oxygen functional groups on the CNP surface.50–52 While there was no significant difference in mass change at the first step (∼1 wt%) between CNPs before and after thermal treatment, weight loss at the second step decreased as the treatment temperatures increased. For instance, CNP-400, CNP-600, CNP-800, and CNP-1000 had weight losses of 5.5, 3.9, 2.7, and 1.9 wt% at 800 °C, respectively, in comparison to 7.8 wt% loss for CNP-RT. This decrease suggests that the amount of residual organic fraction and oxygen functional groups in CNPs decreased with increasing treatment temperature. Degradation of organic fractions and oxygen functional groups during thermal treatment could result in the formation of micropores in CNPs.
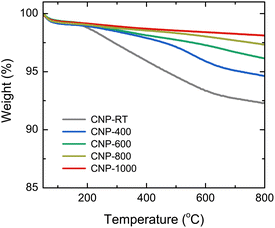 |
| | Fig. 3 TGA curves of CNPs in the temperature range of 50 to 800 °C under an N2 flow. | |
Surface area and porosity
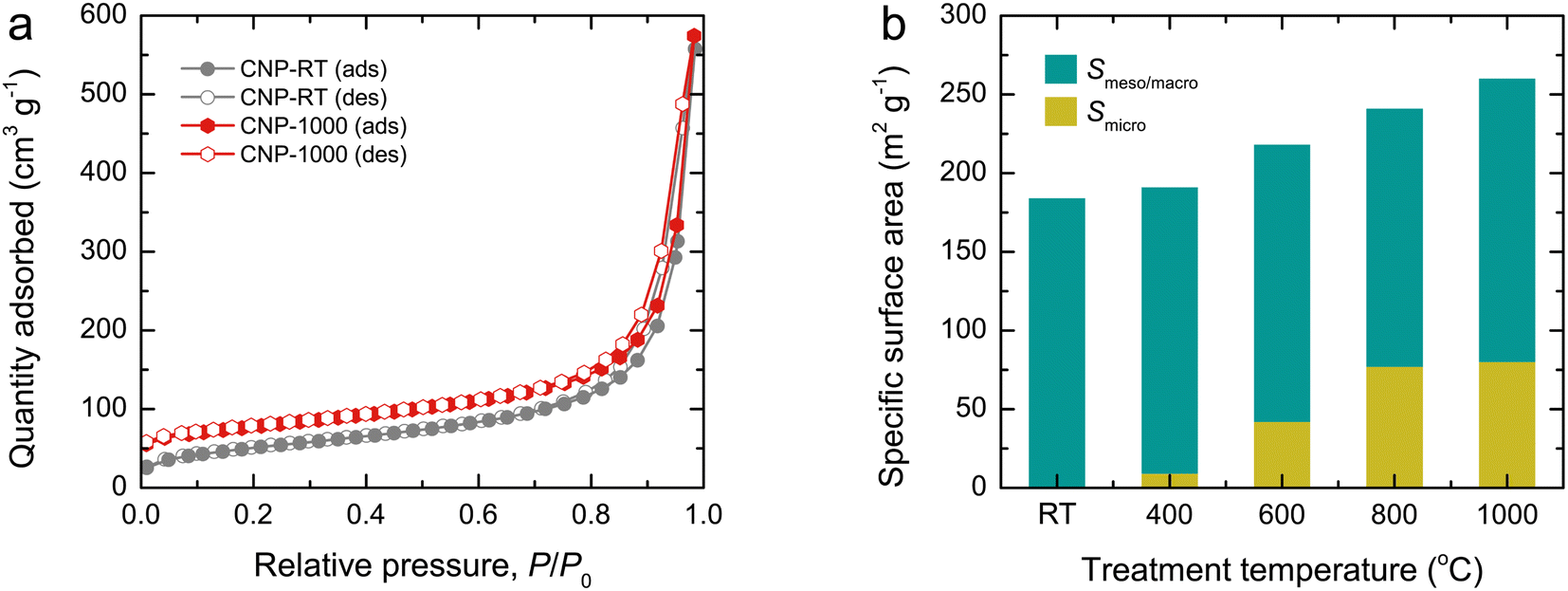
N2 adsorption–desorption isotherms were used to examine the surface area and porous structure of CNPs, as shown in Fig. 4a and S4.† All isotherms exhibited mixed type II and IV features with a narrow hysteresis loop, suggesting the predominance of mesopores and macropores formed by interparticle spacing. Although the isotherm feature remained unchanged as the treatment temperature increased, the quantity adsorbed at an initial relative pressure gradually increased due to a greater quantity being adsorbed in monolayers. This result indicates the development of micropores in CNPs after thermal treatment. Using the Brunauer–Emmett–Teller (BET) method, the specific surface area (SBET) of CNPs was determined from the adsorption data at relative pressures of 0.05–0.30. The SBET values increased from 184 m2 g−1 for CNP-RT to 260 m2 g−1 for CNP-1000 as the treatment temperature increased (Fig. 4b). The t-plot analysis indicated that CNP-RT had only meso–macropores with no micropores, whereas the micropore surface area increased by 5–32% as the treatment temperature increased. The surface areas contributed by meso–macropores of CNPs remained at almost the same value without significant change. This result suggests that the increased SBET of CNPs was due to the development of micropores created by the thermal treatment process. The decomposition of less stable organic compounds remaining in CNPs after synthesis and oxygen functional groups could lead to the development of micropores on the CNP surface. The range of total pore volumes was 0.445–0.472 cm3 g−1. The trend of micropore volume was consistent with the specific micropore surface area. The pore size distribution of CNPs was also determined by the Barrett–Joyner–Halenda (BJH) method using the desorption branch isotherm (Fig. S5†). Evidently, all CNPs revealed a broad distribution covering the mesopore and macropore regions. The average pore diameter was estimated to be about 20 nm for all CNPs. A summary of all textural parameters discussed is presented in Table 1.
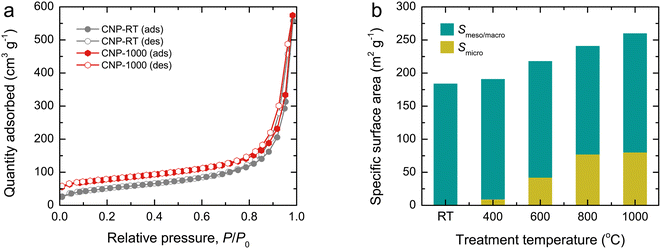 |
| | Fig. 4 (a) N2 adsorption–desorption isotherms and (b) bar plots showing the contribution of specific surface area (SBET) of CNPs by micropores (Smicro) and meso–macropores (Smeso/macro). | |
Table 1 Textural parameters obtained from analysis of the N2 adsorption–desorption isotherms for CNPs
| Samples |
SBETa (m2 g−1) |
Smicrob (m2 g−1) |
Smeso/macroc (m2 g−1) |
Vtotald (cm3 g−1) |
Vmicroe (cm3 g−1) |
Vmeso/macrof (cm3 g−1) |
Dg (nm) |
| SBET is the specific surface area determined by the BET method. Smicro is the micropore-specific surface area determined by the t-plot method. Smeso/macro is the specific surface area contributed by mesopores and macropores (Smeso/macro = SBET − Smicro). Vtotal is the total pore volume determined at a relative pressure (P/P0) of 0.95. Vmicro is the micropore volume determined by the t-plot method. Vmeso/macro is the pore volume contributed by mesopores and macropores (Vmeso/macro = Vtotal − Vmicro). D is the average pore diameter determined by BJH using the desorption isotherm. |
| CNP-RT |
184 |
0 |
184 |
0.455 |
0 |
0.455 |
19.3 |
| CNP-400 |
191 |
9 |
182 |
0.450 |
0.003 |
0.447 |
20.3 |
| CNP-600 |
218 |
42 |
176 |
0.471 |
0.021 |
0.450 |
22.5 |
| CNP-800 |
241 |
67 |
174 |
0.447 |
0.039 |
0.408 |
21.2 |
| CNP-1000 |
260 |
80 |
180 |
0.485 |
0.041 |
0.444 |
19.6 |
Surface composition and chemical bonding states
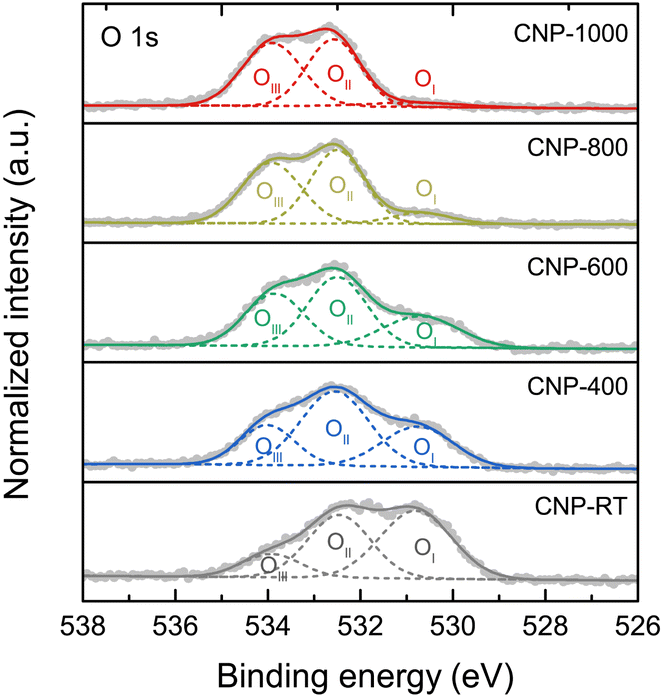
XPS measurement was employed to quantitatively extract the surface chemical composition and bonding states of CNPs. The XPS survey scans were composed of the C 1s and O 1s peaks with no W 4f peaks (Fig. S6†). Although the CNPs contained WC1−x and WC particles, the W 4f peak was not detected because they were encapsulated in the carbon matrix (Fig. S3†). The chemical compositions (C and O contents) for all CNPs were determined through quantitative XPS analysis and are listed in Table 2. The O content decreased from 14.72 atom% for CNP-RT to 1.20 atom% for CNP-1000 as the treatment temperature increased, which was due to the removal of oxygen functional groups at high temperatures. High-resolution XPS O 1s spectra were used to distinguish the type of oxygen functional groups. The XPS O 1s spectra (Fig. 5) were deconvoluted into three sub-peaks, including OI at 530.7 ± 0.1 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O: carbonyl and quinone), OII at 532.3 ± 0.1 eV (C–O: phenol, ether, and epoxide groups), and OIII at 534.5 ± 0.1 eV (carboxylic groups and water adsorbed molecules).53–57 With increasing treatment temperature, the OI peak decreased and nearly disappeared at 1000 °C, while the other two peaks (OII and OIII) were still observed. This finding can be explained that the C–O bonds are more thermally stable than the CO bonds.58,59 The formation of the oxygen functional groups on the CNP surface during SPP could be attributed to two possible causes: (i) the interaction between reactive carbon species and oxygen dissolved in benzene during synthesis60,61 and (ii) surface exposure to ethanol and air during washing and drying processes.
O: carbonyl and quinone), OII at 532.3 ± 0.1 eV (C–O: phenol, ether, and epoxide groups), and OIII at 534.5 ± 0.1 eV (carboxylic groups and water adsorbed molecules).53–57 With increasing treatment temperature, the OI peak decreased and nearly disappeared at 1000 °C, while the other two peaks (OII and OIII) were still observed. This finding can be explained that the C–O bonds are more thermally stable than the CO bonds.58,59 The formation of the oxygen functional groups on the CNP surface during SPP could be attributed to two possible causes: (i) the interaction between reactive carbon species and oxygen dissolved in benzene during synthesis60,61 and (ii) surface exposure to ethanol and air during washing and drying processes.
Table 2 Surface composition, O/C ratio, and relative percentage of oxygen bonding states from deconvolution of XPS O 1s spectra for CNPs
| Sample |
Composition (atom%) |
O/C |
Deconvoluted peak composition (%) |
| C |
O |
OI |
OII |
OIII |
| CNP-RT |
85.28 |
14.72 |
0.17 |
47.88 |
38.71 |
13.41 |
| CNP-400 |
89.92 |
10.08 |
0.11 |
28.32 |
49.91 |
21.77 |
| CNP-600 |
92.77 |
7.23 |
0.08 |
24.64 |
43.01 |
32.35 |
| CNP-800 |
96.96 |
3.04 |
0.03 |
16.12 |
44.12 |
39.76 |
| CNP-1000 |
98.80 |
1.20 |
0.01 |
8.04 |
48.05 |
43.91 |
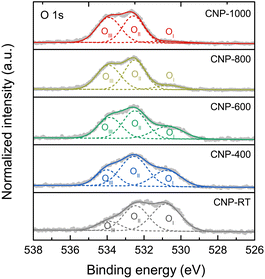 |
| | Fig. 5 High-resolution XPS O 1s spectra of CNPs with deconvolution. | |
Electrochemical charge storage properties
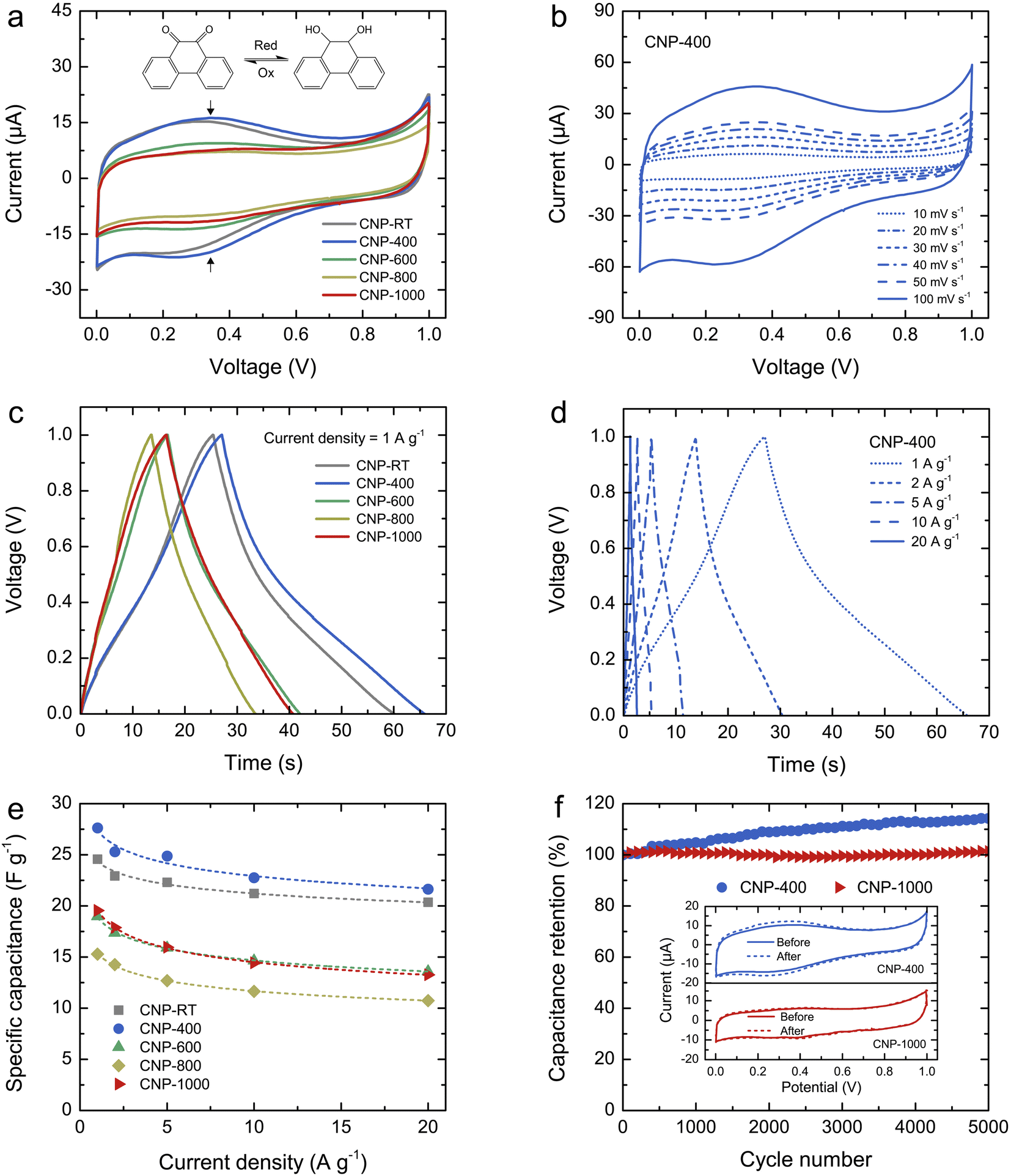
The charge storage properties of CNPs for supercapacitor applications were electrochemically tested in 1 M H2SO4 solution using CV and GCD measurements. Fig. 6a shows the comparative CV curves of CNPs within the potential window of 0–1 V at a scan rate of 20 mV s−1. The CV curves of CNP-RT and CNP-400 displayed a quasi-rectangular feature along with reversible redox peaks (oxidation/reduction) in the potential range of 0.1–0.6 V. This indicates the combined EDLC and PC behaviors. The appearance of redox peaks in CNP-RT and CNP-400 is attributed to the presence of the quinone groups on the carbon surface, which can be reduced to hydroquinone through reaction with hydronium ions (H3O+) in the electrolyte (inset of Fig. 6a).62–66 However, other types of oxygen functionalities seem to have a negligible effect on electrochemical PC behavior in acid electrolytes.62–64 The redox peak was substantially suppressed with increasing treatment temperature to 600–1000 °C, indicating that the PC contribution became less prominent due to the substantial loss of quinone groups, which is consistent with the XPS result. As the scan rate was increased from 10 to 100 mV s−1, the CV curves of CNPs showed an increase in current without any distortion in shape (Fig. 6b and S7†), indicating excellent rate capability.
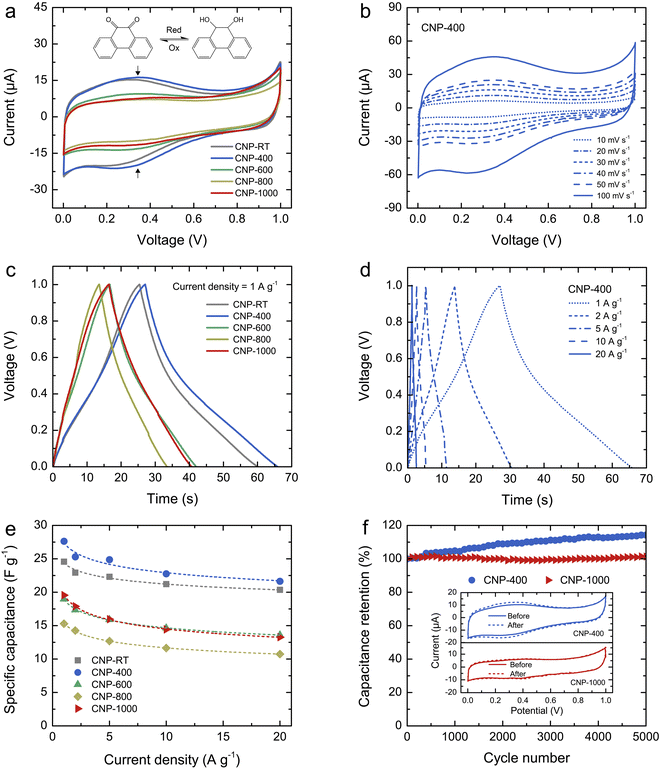 |
| | Fig. 6 (a) Comparative CV curves of CNPs within the potential window of 0–1 V at a scan rate of 20 mV s−1: the vertical arrows indicate the faradaic redox peak, and the inset shows the quinone redox reaction in the potential from 0.1 to 0.6 V. (b) CV curves of CNP-400 at different scan rates from 10 to 100 mV s−1, (c) comparative GCD curves of CNPs at a current density of 1 A g−1, (d) GCD curves of CNP-400 at different current densities from 1 to 20 A g−1, (e) specific capacitance (Cs) of CNPs as a function of current density, and (f) capacitance retention of CNP-400 and CNP-1000 measured at a current density of 10 A g−1 over 5000 cycles. The inset shows the CV curves of CNP-400 and CNP-1000 before and after 5000 cycles. | |
The GCD curves of CNPs were compared at a current density of 1 A g−1 (Fig. 6c). CNP-RT and CNP-400 displayed nonlinear behavior in their charge and discharge curves, and the slope (dV/dt) changed with the voltage due to the PC effect. CNP-600, CNP-800, and CNP-1000 had a more linear behavior due to a decrease in the PC effect. The GCD curves of all CNPs remained similar in shape with faster charge and discharge times as the current density increased from 1 to 20 A g−1 (Fig. 6d and S8†). Typically, the specific capacitance (Cs) can be determined using the slope of the discharge curve when the charge and discharge curves are linear within the potential window investigated. However, due to the nonlinear GCD curve, the Cs values were calculated by integrating the area under the discharge curve using the following equation:
| |
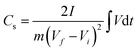 | (1) |
where ∫
Vd
t is the integrated area under the discharge curve,
I is the applied constant current (A),
m is the mass of active materials (g), and
Vf ¬
Vi is the potential window.
67
The calculated Cs values of CNPs were plotted as a function of current density and are displayed in Fig. 6e. The Cs values decreased as the current density increased. The decreasing tendency can be explained by the diffusion limit of the electrolyte ions. At low current density, the ions have sufficient time to diffuse and penetrate into the deeper pores of CNPs, resulting in more interaction with a large surface area. In contrast, due to fast charging and discharging at high current density, the ions had limited access to the pores, leading to a lower number of charge storage. The Cs values were found to be 24.6, 27.6, 19.0, 15.3, and 19.5 F g−1 for CNP-RT, CNP-400, CNP-600, CNP-800, and CNP-1000 at a current density of 1 A g−1, respectively. CNP-400 showed the highest Cs value among CNPs, which was mainly due to PC behavior through the quinone groups on its surface. Although CNPs treated at high temperatures had larger surface areas with more contribution of micropores, their Cs values were not improved as expected. This finding suggests that the oxygen functional groups had a stronger influence on enhancing the Cs value of CNPs than the surface area and microporous structure. It should be noted here that the effect of WC1−x and WC phases on charge storage could be ignored due to their small contents in CNPs.
To assess the cycling stability of CNPs, a cyclic charge–discharge test was carried out for 5000 cycles at a current density of 10 A g−1. CNP-400 and CNP-1000 were selected as representative PC and EDLC electrodes, respectively. After 5000 cycles, the capacitance retention of CNP-400 gradually increased up to 114% of its initial value. This increase in capacitance retention over 100% could be attributed to gradual surface activation that occurred during the prolonged charge–discharge process, which is indicated by the stronger redox peak in its CV curve after 5000 cycles (inset of Fig. 6f). This could result in additional PC with increasing charge–discharge cycles. Moreover, the higher surface wettability caused by surface activation led to more accessibility of electrolyte ions into the micro–mesopores of the carbon electrodes.68–70 In contrast, the capacitance retention of CNP-1000 exhibited excellent stability, with almost 100% retention after 5000 cycles. There was no change in the CV curves for CNP-1000 before and after the cycling test (inset of Fig. 6f). This result confirms that CNP-1000 exhibited better cyclic performance and stability than CNP-400 because the surface of CNP-1000 was more stable in acid electrolyte than CNP-400.
Conclusions
CNPs were synthesized from benzene via SPP and then subjected to thermal treatment under an inert atmosphere at varying temperatures. The CNPs exhibited an amorphous phase and became more graphitized at high treatment temperatures. Although the morphology and particle size of CNPs remained unchanged after thermal treatment, it could lead to a moderate increase in the specific surface area through the development of micropores in primary particles. The oxygen content of CNPs decreased significantly (from 14.72 to 1.20 atom%) due to the removal of oxygen functionality. Electrochemical measurements revealed that CNP-RT and CNP-400 exhibited EDLC along with PC behavior (reversible redox peak). The redox reactions occurred through the oxidation/reduction of the quinone groups on the CNP surface. With increasing treatment temperature, EDLC became the main contribution to charge storage due to the PC suppression, resulting from quinone degradation. After the stability test for 5000 cycles, CNP-1000 showed better cycling performance than CNP-400, which was likely due to its higher stable surface or lack of oxygen functional groups of CNP-1000. These findings suggest that thermal treatment under an inert atmosphere can create micropores on SPP-derived CNPs to enhance their suitability for supercapacitor applications. Although the number of introduced micropores in CNPs via thermal treatment was not as high as expected, there is ample opportunity to enhance and regulate the porosity of CNPs to achieve further progress for supercapacitor applications.
Author contributions
Myo Myo Thu: investigation, formal analysis, methodology, data curation, writing – original draft. Nattapat Chaiammart: investigation, formal analysis. Oratai Jongprateep: validation, funding acquisition. Ratchatee Techapiesancharoenkij: validation, funding acquisition. Aye Aye Thant: validation, Nagahiro Saito: resource. Gasidit Panomsuwan: conceptualization, methodology, formal analysis, data curation, validation, visualization, funding acquisition, project administration, supervision, writing – review & editing.
Conflicts of interest
The authors declare no conflict of interest in this work.
Acknowledgements
This work was financially supported by the ICE-Matter Consortium under the AUN/SEED-Net Collaborative Education Program (CEP) Scholarship with support from JICA and the Kasetsart University Research and Development Institute (KURDI, grant no. FF(KU) 25.64). The authors are thankful to the Department of Materials Engineering and Materials Innovative Center, Kasetsart University, for the facilities and instrument used in this work.
Notes and references
- A. Gonzalez, E. Goikolea, J. A. Barrena and R. Mysyk, Renewable Sustainable Energy Rev., 2016, 58, 1189–1206 CrossRef CAS.
- J. Zhao and A. F. Burke, J. Energy Chem., 2021, 59, 276–291 CrossRef CAS.
- Q. Wu, T. He, Y. Zhang, Z. Wang, Y. Liu, L. Zhao, Y. Wu and F. Ran, J. Mater. Chem. A, 2021, 9, 24094–24147 RSC.
- P. Sharma and T. S. Bhatti, Energy Convers. Manage., 2010, 51, 2901–2912 CrossRef CAS.
- S. Najib and E. Erdem, Nanoscale Adv., 2019, 1, 2817–2827 RSC.
- H. Zhang, L. Zhang, H. Chen, H. Su, F. Liu and W. Yang, J. Power Sources, 2016, 315, 120–126 CrossRef CAS.
- S. Fleischmann, J. B. Mitchell, R. Wang, C. Zhan, D. Jiang, V. Presser and V. Augustyn, Chem. Rev., 2020, 120, 6738–6782 CrossRef CAS PubMed.
- P. Bhojane, J. Energy Storage, 2022, 45, 103654 CrossRef.
- J. Dang, W. Hu, E. Paek and D. Mitlin, Chem. Rev., 2018, 118, 6457–6498 CrossRef PubMed.
- X. Zheng, L. Miao, Z. Song, W. Du, D. Zhu, Y. Lv, L. Gan and M. Liu, J. Mater. Chem. A, 2022, 10, 611–621 RSC.
- Z. Song, L. Miao, L. Ruhlmann, Y. Lv, D. Zhu, L. Li, L. Gan and M. Liu, Adv. Funct. Mater., 2022, 32, 2208049 CrossRef CAS.
- Z. Huang, R. Zhang, S. Zhang, P. Li, C. Li and C. Zhi, Mater. Future, 2022, 1, 022101 CrossRef.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- X. Zhang, H. Zhang, C. Li, K. Wang, X. Sun and Y. Ma, RSC Adv., 2014, 4, 45862–45884 RSC.
- S. Saini, P. Chand and A. Joshi, J. Energy Storage, 2021, 39, 102646 CrossRef.
- Y.-R. Zhao, C.-C. Liu, Q.-Q. Lu, O. Ahmad, Z.-J. Pan and M. Daria, New Carbon Mater., 2022, 37, 875–897 CrossRef.
- M. Kandasamy, S. Sahoo, S. K. Nayak, B. Chakraborty and C. S. Rout, J. Mater. Chem. A, 2021, 9, 17643–17700 RSC.
- R. Barik and P. P. Ingole, Curr. Opin. Electrochem., 2020, 21, 327–334 CrossRef CAS.
- S. Mahadik, S. Surendran, J. Y. Kim, G. Janani, D.-K. Lee, T.-H. Kim, J. K. Kim and U. Sim, J. Mater. Chem. A, 2022, 10, 14655–14673 RSC.
- G. Murali, J. Rawal, J. K. R. Modigunta, Y. H. Park, J.-H. Lee, S.-Y. Lee, S.-J. Park and I. In, Sustainable Energy Fuels, 2021, 5, 5672–5693 RSC.
- W. Li, X. Zhao, Q. Bi, Q. Ma, L. Han and K. Tao, Dalton Tran., 2021, 50, 11701–11710 RSC.
- J. Shi, B. Jiang, C. Li, F. Yan, D. Wang, C. Yang and J. Wan, Mater. Chem. Phys., 2020, 245, 122533 CrossRef CAS.
- J. G. Ruiz-Montoya, L. V. Quispe-Garrido, J. C. Calderón Gómez, A. M. Baena-Moncada and J. M. Gonçalves, Sustainable Energy Fuels, 2021, 5, 5332–5365 RSC.
- P. Veerakumar, A. Sangili, S. Manavalan, P. Thanasekaran and K.-C. Lin, Ind. Eng. Chem. Res., 2020, 59, 6347–6374 CrossRef CAS.
- M. F. Iqbal, M. N. Ashiq and M. Zhang, Energy Technol., 2021, 9, 2000987 CrossRef CAS.
- M. Xu, Q. Yu, Z. Liu, J. Lv, S. Lian, B. Hu, L. Mai and L. Zhou, Nanoscale, 2018, 10, 21604–21616 RSC.
- C. Young, T. Park, J. Woo, J. Kim, M. S. A. Hossain, Y. V. Kaneti and Y. Yamauchi, ChemSusChem, 2018, 11, 3546–3558 CrossRef CAS PubMed.
- J. Yin, W. Zhang, N. A. Alhebshi, N. Salah and H. Alshareef, Small, 2020, 4, 1900853 CrossRef CAS.
- Z. Bi, Q. Kong, Y. Cao, G. Sun, F. Su, X. Wei, X. Li, A. Ahmad and C.-M. Chen, J. Mater. Chem. A, 2019, 7, 16028–16045 RSC.
- L. Sun, Y. Gong, D. Li and C. Pan, Green Chem., 2022, 24, 3864–3894 RSC.
- J. Kang, O. L. Li and N. Saito, Carbon, 2013, 60, 292–298 CrossRef CAS.
- J. Kang, O. L. Li and N. Saito, J. Power Sources, 2014, 261, 156–161 CrossRef CAS.
- G. Panomsuwan, S. Chiba, Y. Kaneko, N. Saito and T. Ishizaki, J. Mater. Chem. A, 2014, 2, 18677–18686 RSC.
- H. Kim and N. Saito, Sci. Rep., 2018, 8, 4342 CrossRef PubMed.
- T. Morishita, T. Ueno, G. Panomsuwan, J. Hieda, A. Yoshida, M. A. Bratescu and N. Saito, Sci. Rep., 2016, 6, 36880 CrossRef CAS PubMed.
- G. Panomsuwan, N. Saito and T. Ishizaki, J. Mater. Chem. A, 2015, 3, 9972–9981 RSC.
- J. Kang, H. V. Kim, S. A. Chae and K. H. Kim, Small, 2018, 14, 1704394 CrossRef PubMed.
- J. Kang, D.-Y. Kim, S.-A. Chae, N. Saito, S.-Y. Choi and K.-H. Kim, J. Mater. Chem. A, 2019, 7, 16149–16160 RSC.
- G. Panomsuwan, N. Saito and T. Ishizaki, Carbon, 2016, 98, 411–420 CrossRef CAS.
- O. L. Li, R. Ikura and T. Ishizaki, Green Chem., 2017, 19, 4774–4777 RSC.
- L. Qin, S. Lee, J. Ha and O. L. Li, Selective production of value-added chemicals from cellulosic biomass waste via plasma-synthesized catalysts, Catal. Today, 2023, 114030 CrossRef CAS.
- P. Pornarootham, G. Panomsuwan, S. Chae, N. saito, N. Thouchprasitchai, Y. Phongboonchoo and S. Pongstabodee, Nanomaterials, 2019, 9, 1776 CrossRef PubMed.
- D.-W. Kim, O. L. Li, P. Pootawang and N. Saito, RSC Adv., 2014, 4, 16813–16819 RSC.
- O. L. Li, S. Chiba, Y. Wada, G. Panomsuwan and T. Ishizaki, J. Mater. Chem. A, 2017, 5, 2073–2082 RSC.
- H. Lee, Y. Wada, A. Kaneko, O. L. Li and T. Ishizaki, Jpn. J. Appl. Phys., 2018, 57, 0102BD CrossRef.
- A. Jorio and A. G. Souza Filho, Annu. Rev. Mater. Res., 2016, 46, 357–382 CrossRef CAS.
- V. Thapliyal, M. E. Alabdulkarim, D. R. Whelan, B. Mainali and J. L. Maxwell, Diamond Relat. Mater., 2022, 127, 109180 CrossRef CAS.
- J. Yan, L. Miao, H. Duan, D. Zhu, Y. Lv, L. Li, L. Gan and M. Liu, Chin. Chem. Lett., 2022, 33, 2681–2686 CrossRef CAS.
- Y. Qin, L. Miao, M. Mansuer, C. Hu, Y. Lv, L. Gan and M. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 33328–33339 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Carbon, 2012, 50, 914–921 CrossRef CAS.
- Y. J. Oh, J. J. Yoo, Y. I. Kim, J. K. Yoon, H. N. Yoon, J.-H. Kim and S. B. Park, Electrochim. Acta, 2014, 116, 118–128 CrossRef CAS.
- S. Kundu, Y. Wang, X. Xia and M. Muhler, J. Phys. Chem. C, 2008, 112, 16869–16878 CrossRef CAS.
- M. Demir, B. Ashourirad, J. H. Mugumya, S. K. Saraswat, H. M. El-Kaderi and R. B. Gupta, Int. J. Hydrogen Energy, 2008, 43, 18549–18558 CrossRef.
- Y.-H. Hwang, S. M. ee, Y. J. Kim, Y. H. Kahng and K. Lee, Carbon, 2016, 100, 7–15 CrossRef CAS.
- G. Xia, C. Wang, P. Jiang, J. Lu, J. Diao and Q. Chen, J. Mater. Chem. A, 2019, 7, 12317–12324 RSC.
- H. Zhao, J. Ye, W. Song, D. Zhao, M. Kang, H. Shen and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 6991–7000 CrossRef CAS PubMed.
- G. de la Puente, J. J. Pis, J. A. Menendez and P. Grange, J. Anal. Appl. Pyrolysis, 1997, 43, 125–138 CrossRef CAS.
- L. Li, X. Yao, H. Li, Z. Liu, W. Ma and X. Liang, J. Chem. Eng. Jpn., 2014, 47, 21–27 CrossRef CAS.
- T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Ind. Eng. Chem. Res., 2014, 53, 19331–19337 CrossRef CAS.
- T. Tsuji, K. Ohya, A. J. X. Lai, N. B. A. Manaf, T. Hoshina and S. Oba, Fluid Ph. Equilib., 2019, 492, 34–40 CrossRef CAS.
- H. A. Andreas and B. E. Conway, Electrochim. Acta, 2006, 51, 6510–6520 CrossRef CAS.
- Y. J. Oh, J. J. Yoo, Y. I. Kim, J. K. Yoon, H. N. Yoon, J.-H. S. Kim and B. Park, Electrochim. Acta, 2014, 116, 118–128 CrossRef CAS.
- Y. He, Y. Zhang, X. Li, X. Wang, Z. Liu and X. Huang, Electrochim. Acta, 2018, 282, 618–625 CrossRef CAS.
- G.-F. Han, W. Zou, M. Karamad, J.-P. Jeon, S.-W. Kim, S.-J. Kim, Z. Fu, Y. Lu, S. Siahrostami and J.-B. Baek, Nat. Commun., 2020, 11, 2209 CrossRef CAS PubMed.
- S. P. Ega and P. Srinivasan, J. Energy Storage, 2022, 47, 103700 CrossRef.
- B. Pandit, D. P. Dubal and B. R. Sankapal, Large scale flexible solid state symmetric supercapacitor through inexpensive solution processed V2O5 complex surface architecture, Electrochim. Acta, 2017, 242, 382–389 CrossRef CAS.
- J. Zhou, J. Lian, L. Hou, J. Zhang, H. Gou, M. Xia, Y. Zhao, T. A. Strobel, L. Tao and F. Gao, Nat. Commun., 2015, 6, 8503 CrossRef CAS PubMed.
- C. Zequine, C. K. Ranaweera, Z. Wang, S. Singh, P. Tripathi, O. N. Srivastava, B. K. Gupta, K. Ramasamy, P. K. Kahol, P. R. Dvornic and R. K. Gupta, Sci. Rep., 2016, 6, 2016 Search PubMed.
- D. Gandla, X. Wu, F. Zhang, C. Wu and D. Q. Tan, ACS Omega, 2021, 6, 7615–7625 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA curve of CNT-RT under O2 flow, EDS area analysis of CNP-RT, TEM images showing the presence of WC1−x particles embedded in carbon particles, N2 adsorption–desorption isotherm of CNPs, BJH pore size distribution obtained from desorption isotherm of CNPs, XPS survey spectra of CNPs, CV curves of CNPs at different scan rates from 10 to 100 mV s−1, and GCD curves of CNPs at different current densities from 1 to 20 A g−1. See DOI: https://doi.org/10.1039/d3ra02314a |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Ratchatee Techapiesancharoenkijab,
Aye Aye Thantc,
Nagahiro Saito
ab,
Ratchatee Techapiesancharoenkijab,
Aye Aye Thantc,
Nagahiro Saito