
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural, magnetic and optical properties of disordered double perovskite Gd2CoCrO6 nanoparticles†
M. J. Hosenab,
M. A. Basith *b and
I. M. Syedacd
*b and
I. M. Syedacd
aDepartment of Physics, University of Dhaka, Dhaka-1000, Bangladesh
bNanotechnology Research Laboratory, Department of Physics, Bangladesh University of Engineering and Technology, Dhaka-1000, Bangladesh. E-mail: mabasith@phy.buet.ac.bd
cSemiconductor Technology Research Center, University of Dhaka, Dhaka-1000, Bangladesh
dCentre for Advanced Research in Sciences, University of Dhaka, Dhaka-1000, Bangladesh
First published on 12th June 2023
Abstract
We have synthesized disordered double perovskite Gd2CoCrO6 (GCCO) nanoparticles with an average particle size of 71 ± 3 nm by adopting a citrate sol–gel method to investigate their structural, magnetic, and optical properties. Rietveld refinement of the X-ray diffraction pattern showed that GCCO is crystallized in a monoclinic structure with space group P21/n, which is further confirmed by Raman spectroscopic analysis. The absence of perfect long-range ordering between Co and Cr ions is confirmed by the mixed valence states of Co and Cr. A Néel transition was observed at a higher temperature of TN = 105 K compared to that of an analogous double perovskite Gd2FeCrO6 due to a greater degree of magnetocrystalline anisotropy of Co than Fe. Magnetization reversal (MR) behavior with a compensation temperature of Tcomp = 30 K was also observed. The hysteresis loop obtained at 5 K exhibited the presence of both ferromagnetic (FM) and antiferromagnetic (AFM) domains. Super-exchange and Dzyaloshinskii–Moriya (DM) interactions between various cations via oxygen ligands are responsible for the observed FM or AFM ordering in the system. Furthermore, UV-visible and photoluminescence spectroscopy demonstrated the semiconducting nature of GCCO with a direct optical bandgap of 2.25 eV. The Mulliken electronegativity approach revealed the potential applicability of GCCO nanoparticles in photocatalytic H2 and O2 evolution from water. Due to a favorable bandgap and potentiality as a photocatalyst, GCCO can be a promising new member of double perovskite materials for photocatalytic and related solar energy applications.
1 Introduction
Perovskite oxides have attracted notable research interest in the last few decades because of their fascinating structure and physical properties like ferroelectric, ferromagnetic, multiferroic, photocatalytic, etc1–5. It is noticeable that among different types of perovskites, GdCoO3 and GdCrO3 possess interesting properties along with potential applications.6–11 For instance, GdCoO3 exhibits a giant magnetocaloric effect near the Néel transition TN = 3.1 K with a significant refrigerant capacity of 278 J kg−1 for a 0–70 kOe field change which belongs to the half-filled 4f orbital of Gd3+ ions.6 It also shows impressive optoelectronic properties like a suitable bandgap of ∼1.83 eV, excellent absorbance in the visible region of the solar spectrum, and noticeable photocatalytic efficiency in a decrease of the recombination rate of the electron–hole pairs.7,8 On the other hand, GdCrO3 shows temperature-dependent magnetization reversal (TMR), spin reorientation (SR), and spin flipping (SF) as a result of competitive magnetic interactions between Gd and Cr-sublattices that makes it a strong candidate for potential applications.9 Furthermore, GdCrO3 exhibits antiferromagnetic (AFM) ordering at a comparatively higher temperature of 167 K than that of GdCoO3 because of the Cr3+–Cr3+ interaction followed by weak ferromagnetic (FM) ordering of Cr3+–Gd3+ interaction and magnetization reversal below 95 K due to interaction between Gd3+ and Cr3+ ions.10 Notably, GdCrO3 also shows excellent absorbance in UV-visible regions with an optical bandgap of 3.15 eV.11 Such diversified magnetic and optical properties of parent single perovskites GdCoO3 and GdCrO3 motivated us to synthesize nanoparticles of their double perovskite Gd2CoCrO6.It is worth mentioning that a number of previous investigations demonstrated double perovskites of general formula A2BB'O6 (A = rare earth elements, B and B′ = transition metal elements) exhibit better flexibility and show a wide variety of properties compared to single perovskites due to two transition metal elements at B sites.2,12,13 These perovskite materials show superconductivity, magnetic ordering from ferromagnetic to antiferromagnetic, multiferroic, and catalytic properties.14–16 As a result, they can be exploited in different fields of applications such as spintronic devices, information storage devices, photocatalytic systems, photovoltaic devices, photochemical energy storage devices, etc17–22.
Notably, due to various cationic ordering and compositions at B and B′ sites along with octahedral distortion in B/B′O6 octahedral rare-earth-based double perovskites exhibit complex magnetic behavior like coexisting ferromagnetic and antiferromagnetic states, high curie temperature/Néel transition, magnetic reversibility, spin reorientation, etc16,22–24. For instance, Gd2CoMnO6 shows ferromagnetic transition at a higher temperature TC = 112 K, whereas its parent materials GdMnO3 and GdCoO3 exhibit ferromagnetic and antiferromagnetic behavior with very low transition temperatures 20 K and 3.2 K, respectively.6,23,25 Another investigation on Pr2FeCrO6 exhibits a magnetically ordered state below the transition temperature of 240 K due to 180° super-exchange interactions between Fe3+(t32ge2g)–O–Cr3+(t32ge0g).16 A separate investigation on Gd2FeCrO6 shows complex behavior of both FM and AFM states at 5 K due to octahedral distortion on Fe/CrO6 octahedral.22 Recent investigation on Y2FeCrO6 double perovskite shows magnetization reversal with higher compensation temperature Tcomp = 192 K, and spin reorientation behavior with transition temperature TSR = 30 K followed by DM interaction and single-ion anisotropy.24
Furthermore, double perovskites exhibit great catalytic efficiency due to favorable bandgap and strong absorbance in the visible regions of the solar spectrum.26,27 For instance, La2CuNiO6 shows much higher catalytic activity for methane combustion compared to its parent material LaNiO3 and the mixture of LaNiO3 and LaCuO3 due to different structures and surface morphologies.26 Another investigation on La2FeTiO6 and LaFeO3 reveals that La2FeTiO6 shows the higher photocatalytic activity of p-chlorophenol degradation compared to LaFeO3 under visible light irradiation, which is also responsible for different structures and surface morphologies.27 These fascinating magnetic and optical properties of double perovskites compared to single perovskites also motivated us to investigate the Gd2CoCrO6 double perovskite.
However, it is difficult to synthesize the ordered structure of Gd2CoCrO6 double perovskite owing to antisite disorder which arises due to the almost identical ionic radii of Co2+ and Cr3+ ions 0.65 Å and 0.62 Å, respectively.28,29 The comparable ionic radii of these two ions disrupt the ideal repeating order of Co and Cr ions.28,30 It is well known that the physical properties of A2BB'O6 double perovskites are closely connected to the fluctuation of antisite disorder.28 Because of difficulties to control the order–disorder effect during synthesis, the family of A2CoCrO6 has not been investigated yet.
Therefore, in the present investigation, Gd2CoCrO6 nanoparticles were synthesized via the citrate sol–gel method, and their structural, morphological, magnetic, and optical properties were investigated extensively. Finally, the potential of photocatalytic water splitting of GCCO was discussed in terms of bandgap and band edge potential.
2 Experimental details
2.1 Sample preparation
The Gd2CoCrO6 double perovskite was synthesized by adopting a citrate sol–gel method due to the simplicity, higher phase purity, and higher production efficiency of the sol–gel method.22,31 Fig. 1 depicts schematically the synthesis steps of Gd2CoCrO6 double perovskite. At first, stoichiometric amounts of analytical grade Gd(NO3)3·6H2O (Sigma-Aldrich, 99.9%), Cr(NO3)3·9H2O (Sigma-Aldrich, 99%), and Co(NO3)2·6H2O (Merck, 99%) were dissolved separately in 50 mL of deionized (DI) water. Then, the solutions were mixed in a beaker and vigorously stirred for 25 minutes at 50 °C for better mixing. To avoid the formation of multiple phases of binary oxide, citric acid (CA), a chelating agent that binds metal ions together, was used. Aqueous ammonia was used to maintain the pH value of the solution at approximately 7. Drop by drop, Ethylene glycol (EG) was added to the solution following the molar ratio of {Gd3+, Co2+ + Cr3+}![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :{CA}:{EG} = 1:1:4 to create a polymeric metal-cation network.22,32 The mixture was then heated at 80 °C for 4 hours to produce the gel precursor. The resulting gel was burned at a high temperature until auto-combustion was initiated. The resultant fluffy powder was then ground in an agate mortar and finally calcinated at 800 °C for 6 hours in air.22,33
:{CA}:{EG} = 1:1:4 to create a polymeric metal-cation network.22,32 The mixture was then heated at 80 °C for 4 hours to produce the gel precursor. The resulting gel was burned at a high temperature until auto-combustion was initiated. The resultant fluffy powder was then ground in an agate mortar and finally calcinated at 800 °C for 6 hours in air.22,33
 | ||
| Fig. 1 Schematically the synthesis steps of Gd2CoCrO6 double perovskite using the sol–gel method. | ||
2.2 Characterization details
In this investigation, the X-ray diffraction (XRD) was carried out by using a PW3040 X'pert PRO Philips XRD diffractometer ranging from 20° to 80° with a Cu X-ray source of wavelength, λ: Kα = 1.5405 Å. The FullProf software package was used to analyze the Rietveld profile using the powder XRD data. Raman spectroscopy was carried out by MonoVista CRS+ Raman microscope systems (S & I) to further verify the crystal structure of GCCO nanomaterials. Fourier transform infrared spectroscopy (FTIR) was conducted via a Shimadzu (IRSpirit) FTIR spectrometer to observe the presence of the functional group in the as-synthesized GCCO nanomaterials. Thermogravimetric analysis (TGA) was performed using a thermogravimetric analyzer (Hitachi TG/DTA7200) with a simultaneous thermal analysis (TGA + DTA) module and an Al2O3 pan in a nitrogen atmosphere at a heating rate of 10 °C min−1 ranging from 30 °C to 900 °C to investigate the thermal stability of GCCO nanoparticles. Both field emission electron microscope (FESEM) (JEOL JSM 7600F) and transmission electron microscope (TEM) (Talos F200X) were carried out to scrutinize the surface morphology of as-prepared GCCO nanomaterials. X-ray photoelectron spectroscopy (XPS) was performed by a K-alpha XPS (Thermo Fisher Scientific) with an Al Kα (1486.6 eV) X-ray source to see the valence state of all elements present in the GCCO. Magnetic properties were investigated using a quantum design physical properties measurement system (PPMS) DynaCool. Finally, to see the optical properties of GCCO nanomaterial, the optical absorption spectrum was investigated by using an ultraviolet-visible (UV-visible) spectrophotometer (UV-2600, Shimadzu) and steady-state photoluminescence (PL) spectrum was investigated using a Spectro Fluorophotometer (RF-6000, Shimadzu).3 Results and discussion
3.1 Crystallographic structure analysis
Prior to conducting experimental analysis, the tolerance factor and global instability index (GII) were calculated to predict the crystal structure of GCCO double perovskite by using the Structure Prediction Diagnostic Software (SPuDS) to get a notion about its crystal structure.34 The tolerance factor (t) for GCCO double perovskite can be written as,22,35where rGd, rCo, rCr, and rO represent the ionic radius of Gd, Co, Cr, and O ions, respectively. For double perovskites, generally, the value of the tolerance factor t ≈ 1 is related to perfect cubic structure. If t > 1.05, the crystal could have a hexagonal structure, and if it is less than 0.97, the structure will be either monoclinic or orthorhombic.22,35 Noticeably, at room temperature (RT), the tolerance factor t of GCCO double perovskite was computed to be 0.94, which suggests that the crystal structure of GCCO is either monoclinic or orthorhombic.

The overall structural stability of a particular phase of double perovskites can be predicted by calculating their global instability indices (GII), which are the difference between the formal valence of cations and anions and the calculated bond-valence sum (BVS). The global instability index (GII) is defined as the root mean square value of the bond discrepancy factor.34,36

Fig. 2 demonstrates the Rietveld refined powder XRD pattern of as-synthesized GCCO nanoparticles at room temperature (RT). The crystalline size of GCCO double perovskite was found to be 36 nm using the Scherrer formula.39 No undesired peak in the XRD pattern confirms the high phase purity of the as-synthesized GCCO nanoparticles. As was mentioned earlier, the tolerance factor was calculated to be 0.94, which means that the crystal structure would be either monoclinic or orthorhombic. Hence, Rietveld refinement was performed by considering both monoclinic and orthorhombic structures, and it was found that the monoclinic structure of the P21/n space group fitted more accurately compared to the orthorhombic structure of the Pbnm space group. Cation ordering can be verified by checking the superstructure peak around the diffraction angle 20°.40,41 The presence of a superstructure peak at an angle of 20.6° is almost negligible, which demonstrates the absence of proper long-range ordering between Co and Cr ions. Mixed valence states of Co and Cr elements confirmed by the XPS analysis, which will be discussed further also revealed the absence of perfect long-range ordering between Co and Cr cations. The lattice parameters of the GCCO double perovskite from Rietveld refinement were observed to be a = 5.266 Å, b = 5.455 Å, c = 7.525 Å, α = γ = 90° and β = 90.083° with unit cell volume 216.17 Å3, which were consistent with similar double perovskites.16,22,42 Exploiting these parameters, the GCCO unit cell with Co/CrO6 octahedra was demonstrated by the structure visualization software VESTA.43 Visualized unit cell of GCCO is shown in Fig. 3(a).
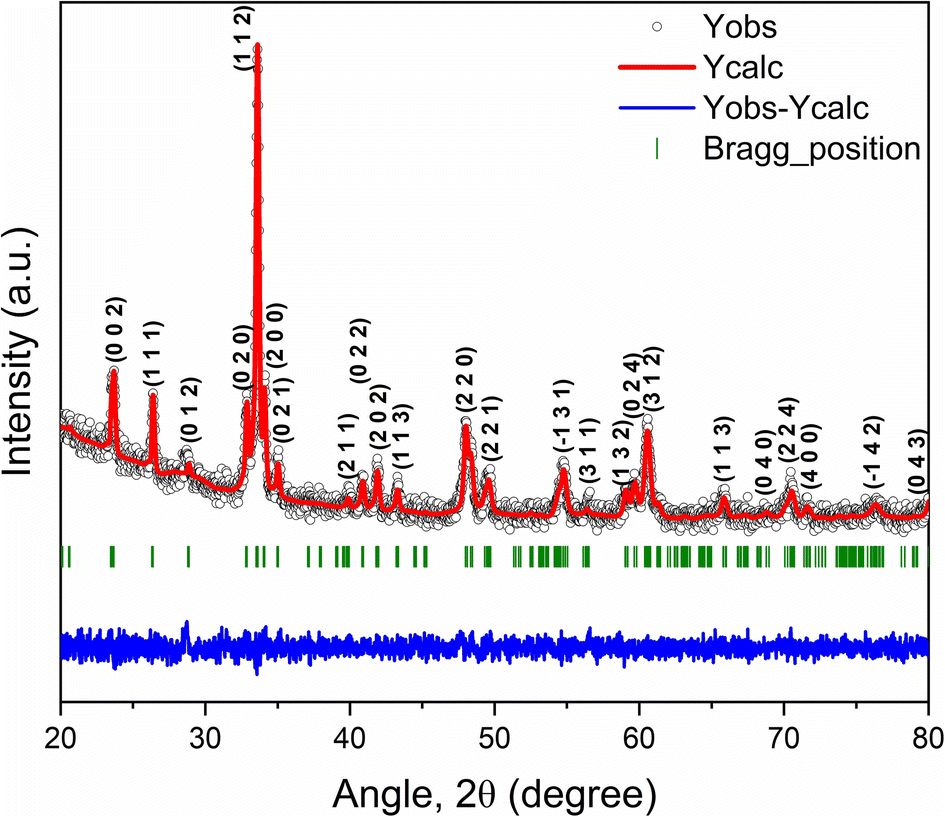 | ||
| Fig. 2 Rietveld refined powder XRD pattern of Gd2CoCrO6 nanoparticles at the room temperature. | ||
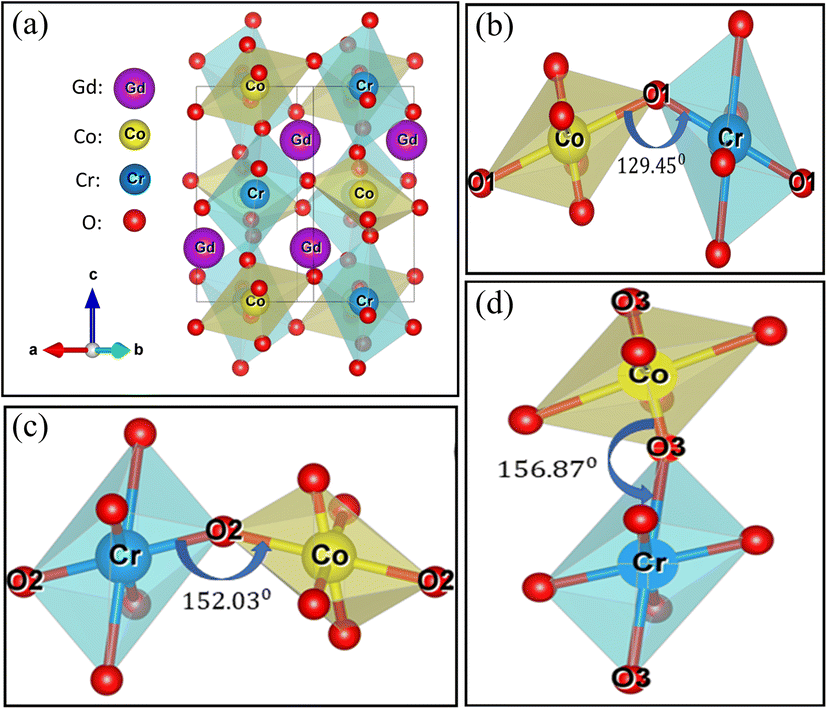 | ||
| Fig. 3 GCCO unit cell (a) and magnified view of Co/CrO6 octahedral (b, c, and d). | ||
In Fig. 3(a), the deviation of Co and Cr ions from the center of the octahedron indicates the octahedral distortion.44 For further insight, the magnified view of three Co/CrO6 octahedral is presented in Fig. 3(b–d). The distortion can be estimated by calculating tilt angle Φ, which is equal to Φ = (180 − θ)/2, where θ is Co/Cr–O–Cr/Co bond angle.16 Unequal bond distances further verify the octahedral distortion in the GCCO unit cell. The mean value of the tilt angle Φ was calculated to be 16.94°. Table 1 represents the Rietveld refined structural parameters of GCCO double perovskite.
| Atom | Wyckoff position | X | Y | Z | Bond distance (Å) | Bond angle (°) | Tilt angle (°) | Reliability factors |
|---|---|---|---|---|---|---|---|---|
| Gd | 4e | 0.511 | 0.554 | 0.251 | Co–O1 = 2.25 | Co–O1–Cr = 129.45 | 25.28 | Rexp = 5.73 |
| Co | 2c | 0 | 0.500 | 0 | Co–O2 = 2.01 | |||
| Cr | 2d | 0.500 | 0 | 0 | Co–O3 = 1.50 | Co–O2–Cr = 152.03 | 13.99 | Rwp = 6.00 |
| O1 | 4e | 0.213 | 0.223 | −0.088 | Cr–O1 = 1.94 | |||
| O2 | 4e | 0.258 | 0.755 | 0.053 | Cr–O2 = 1.89 | Co–O3–Cr = 156.87 | 11.56 | χ2 = 1.10 |
| O3 | 4e | 0.416 | −0.043 | 0.278 | Cr–O3 = 2.33 |
3.2 Raman spectrum analysis
Raman spectroscopy was performed at room temperature (RT) to further verify the as-synthesized GCCO crystal structure. Vibrational modes that arise in the Raman spectroscopy are mainly due to the vibration of Co/CrO6 octahedral and Co/Cr–O bonds. Fig. 4 shows the Raman spectrum of as-synthesized GCCO nanoparticles.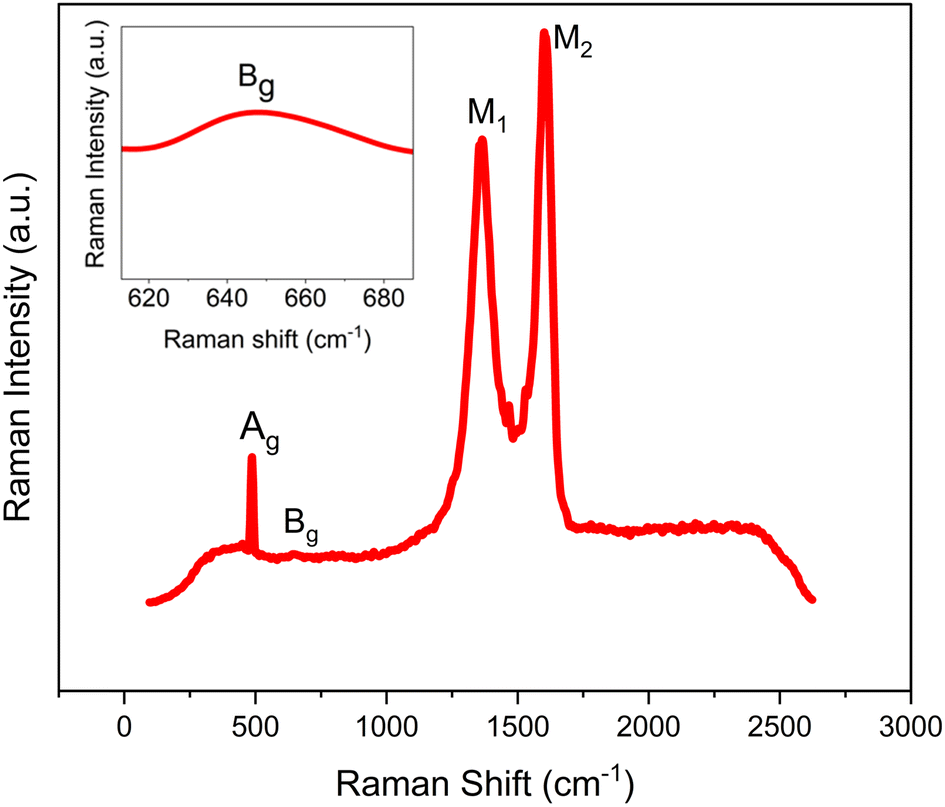 | ||
| Fig. 4 Raman spectrum of GCCO nanoparticles observed at RT. Inset of the figure shows magnified view of Bg mode. | ||
Since GCCO possesses a monoclinic structure with space group P21/n, hence it is expected to the presence of two vibrational modes Ag and Bg in the Raman spectrum of GCCO.45,46 The presence of Ag and Bg modes can be seen clearly in the Raman spectrum of GCCO at Raman shifts of 487 cm−1 and 653 cm−1 respectively. Here, Ag mode arises due to the anti-symmetric stretching vibration of Co/CrO6 octahedra, and Bg mode arises due to the symmetric stretching vibration of Co/CrO6 octahedra.45 The presence of these two modes further confirms the monoclinic structure with space group P21/n. There is also the presence of two intense peaks, M1 and M2, at Raman shifts of 1361 cm−1 and 1604 cm−1. The Raman peak at 1361 cm−1 arises due to the overtone of two Bg modes, and the Raman peak at 1604 cm−1 arises due to the linear combination of 2Ag and Bg modes.41 The average bond distance can also be calculated using the stretching mode through the following formula,47,48
| v (cm−1) = 21349exp(−1.9176R (Å)) |
3.3 FTIR spectrum analysis
FTIR spectroscopy was performed at room temperature to identify the infrared (IR) active modes and the presence of different functional groups in the as-synthesized GCCO nanoparticles, as demonstrated in the ESI Fig. S1.† It is expected that there will be six normal modes of vibration (V1–V6) of the Co/CrO6 octahedral complex in the GCCO unit cell. Among these V3 and V4 are IR active, V1, V2, and V5 are Raman active, and V6 is inactive.22,42,49The broad transmission mode around 516 cm−1 arises basically due to the bending vibration of Co/Cr–O bonds in Co/CrO6 octahedral in the GCCO unit cell and is denoted by V3.22 The transmission mode at 571 cm−1 is denoted by V4 and arises basically due to the stretching vibration of Co/Cr–O bonds.42 The transmission band at 918 cm−1 is assigned to the NO3− ions trapped in the GCCO nanoparticles, which arises due to the use of nitrate salt as precursor materials.22,49 C–O bonds arise at the range of 1384–1681 cm−1 and 2355 cm−1 due to the chemisorption of atmospheric CO2 by the GCCO nanomaterials.22,42 Finally, O–H bonds arise in the range of 3663–3868 cm−1 because of the absorption of the water at the surface of GCCO nanoparticles.42
3.4 TGA and DTA analysis
Thermogravimetric analysis (TGA) was performed to see the thermal stability of GCCO double perovskite. The TGA curve demonstrated in the ESI Fig. S2† shows that the weight loss of GCCO powders is less than ∼1% up to temperatures of 900 °C, which indicates the great thermal stability of GCCO nanoparticles over a wide range of temperatures. The weight loss at a lower temperature is most probably due to the removal of absorbed water and gases from the surface of GCCO nanoparticles. The weight loss at higher temperatures might be due to the loss of Co and Cr ions in the lattice because of the loss of lattice oxygen.22,50 Thermal stability up to 900 °C indicates that the calcination at a temperature of 800 °C was reasonable.In the Differential Thermal Analysis (DTA) curve illustrated in the ESI Fig. S2,† initially enthalpy increases due to the increase of operating temperature. No intense peak was observed corresponding to a significant phase transition. The weak endothermic peak at the temperature of 720 °C was observed, which was due to the dissociation of C–O bonds present in the GCCO nanoparticles.51 The presence of C–O bonds was also confirmed by the FTIR analysis in the previous section.
3.5 FESEM imaging
The surface morphology of as-synthesized GCCO double perovskite was investigated initially by conducting field emission scanning electron microscopy (FESEM). Fig. 5(a) shows the surface morphology of as-synthesized GCCO nanoparticles obtained from FESEM measurement. The surface of the GCCO nanomaterials calcined at 800 °C is not homogeneously distributed and the particles are agglomerated to some extent. This agglomeration occurs due to the relatively higher surface-to-volume ratio and higher surface energy of nanoparticles. After that, agglomeration occurs to lower its surface energy.52,53 Again, the precursors, chelating agent, pH of the solution, and solvent affect the particles to agglomerate to some extent.54,55 Finally, the agglomeration can take place due to the larger enthalpy changes due to bond formation than entropy, which is responsible for negative Gibbs free energy. Agglomeration tends to eliminate this negative free energy (ΔG) to be thermodynamically stable.56,57 Inset of Fig. 5(a) shows the particle size distribution of GCCO nanoparticles obtained from a FESEM image, which shows that the majority of the particles are in the range of 40 to 140 nm with an average particle size of 71 ± 3 nm, which is well in agreement with the similar material Gd2FeCrO6 double perovskite.22 The energy dispersive spectrum (EDS) illustrated in the Fig. 5(b) shows the presence of all required elements.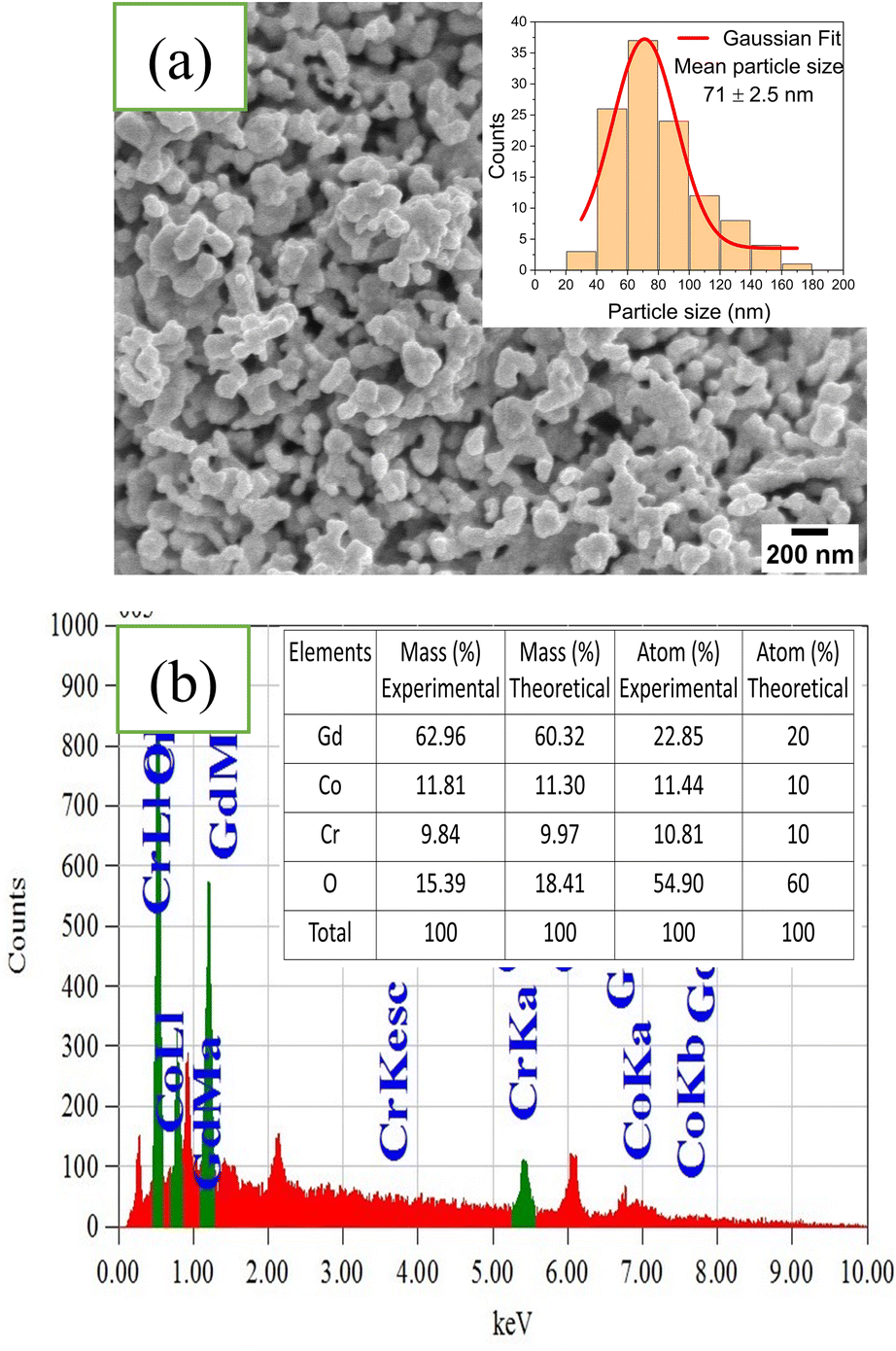 | ||
| Fig. 5 (a) FESEM image of Gd2CoCrO6 nanoparticles. Inset of figure (a) shows particle size distribution histogram. (b) Energy dispersive spectrum with inset showing the percentage of all required elements. | ||
The mass and atomic percentage of all required elements found from EDS spectroscopy and calculated theoretically are demonstrated in the inset of Fig. 5(b). It is seen that the experimentally obtained values are analogous to the theoretically calculated values, which further demonstrated the successful synthesis of GCCO nanomaterials.
3.6 TEM imaging
In the morphological investigation, transmission electron microscopy (TEM) shows a better insight into particle morphology. Fig. 6(a) exhibits the bright field TEM image of the GCCO nanoparticles. The particle sizes obtained from TEM are in the range of 40–120 nm, which is well in agreement with the particle sizes obtained from FESEM analysis. Fig. 6(b) demonstrates the high-resolution TEM image with an interplanar spacing of 0.31 nm. The Rietveld analysis demonstrated that the interplanar spacing of 0.31 nm is associated with the (−1 1 2) lattice plane. The ring shape pattern of selected area electron diffraction (SAED) in the inset of Fig. 6(b) demonstrates the polycrystallinity of GCCO double perovskite with the presence of (−1 2 4), (0 1 4), (1 1 3), (0 2 1), and (1 1 1) lattice planes.7 | ||
| Fig. 6 (a) Bright field TEM image of GCCO nanoparticles, (b) high resolution TEM image. Inset of figure (b) shows SAED pattern of as-synthesized GCCO nanomaterials. | ||
3.7 X-ray photoelectron spectroscopy (XPS) analysis
The oxidation states and chemical binding energies of individual atoms of as-prepared GCCO double perovskite were scrutinized by performing X-ray photoelectron spectroscopy (XPS). The high-resolution XPS spectra of Gd-4d, Co-2p, Cr-2p, and O-1s core levels of the as-prepared sample are shown in Fig. 7(a–d), respectively. The XPS spectrum of Gd (Fig. 7(a)) exhibits two characteristic doublet peaks due to spin–orbit splitting at 4d5/2 and 4d3/2, which are associated with the binding energies of 141.63 eV and 146.02 eV, respectively. The binding energies of Gd-4d5/2 and Gd-4d3/2 indicate the presence of the Gd3+ ion in the GCCO double perovskite.5,58 In the XPS spectrum of Co content (Fig. 7(b)), the spin–orbit splitting of the Co-2p orbital is composed of two major peaks at 780.28 eV and 795.78 eV, related to the Co-2p3/2 and Co-2p1/2 states, respectively, with a satellite peak at the binding energy of 803.49 eV due to interaction between the photo-ejected electron and the valence electron. The deconvolution peaks of Co-2p3/2 at binding energies of 780.22 eV and 784.49 eV represent the Co3+ and Co2+ states, respectively, and the deconvolution peak of Co-2p1/2 at 795.78 eV represents the Co3+ state.59,60 Similarly, the XPS spectrum of the Cr element also exhibits spin–orbit splitting at 575.88 eV and 585.85 eV, which correspond to the 2p3/2 and 2p1/2 states, respectively. Further deconvolution of state 2p3/2 at 575.54 eV and 576.98 eV reveals the presence of mixed states Cr2+ and Cr3+, respectively, and deconvolution of state 2p1/2 at 585.68 eV and 586.88 eV represents the mixed state of Cr3+ and Cr6+ states.61,62 The presence of Cr6+ is well consistent with the previous investigation.62 | ||
| Fig. 7 (a–d) High resolution XPS spectra of Gd-4d, Co-2p, Cr-2p and O-1s of Gd2CoCrO6 nanoparticles, respectively. | ||
Fig. 7(d) shows the core level XPS spectrum of O 1s1/2, the peak at 528.92 eV corresponds to the characteristic peak of O2− ions of the lattice oxygen, while the wide peak at 530.74 eV denotes the ionization of oxygen ions associated with the subsurface of GCCO nanomaterials. This suggests that the subsurface is formed of oxygen ions with a lower electron concentration than the O2− ions. These oxide ions can be ascribed as excess oxygen or O− species.16 The presences of mixed states of Co and Cr elements indicate the absence of perfect long-range ordering between Co and Cr elements in the GCCO double perovskite.
3.8 Magnetic characterization
The temperature-dependent direct-current (dc) magnetization measurements of Gd2CoCrO6 nanoparticles were performed under an applied magnetic field of 100 Oe using both ZFC and FC modes as shown in Fig. 8(a). The FC magnetization curve increases as temperature decreases without any sharp transition, indicating the absence of proper long-range ordering among Co3+ and Cr3+ ions which is consistent with XRD analysis.16,22 Notably, the ZFC curve displays a cusp-like feature at 105 K (shown by the red arrow in the inset of Fig. 8(a)), which suggests a Néel transition at TN = 105 K. In a similar type of double perovskite Gd2FeCrO6, we observed previously a Néel transition at a temperature around ∼15 K.22 Interestingly, the replacement of Fe by Co enhanced Néel transition temperature significantly probably due to the higher degree of magnetocrystalline anisotropy of Co owing to strong spin–orbit coupling and stronger exchange interactions among the 3d electrons compared to Fe.63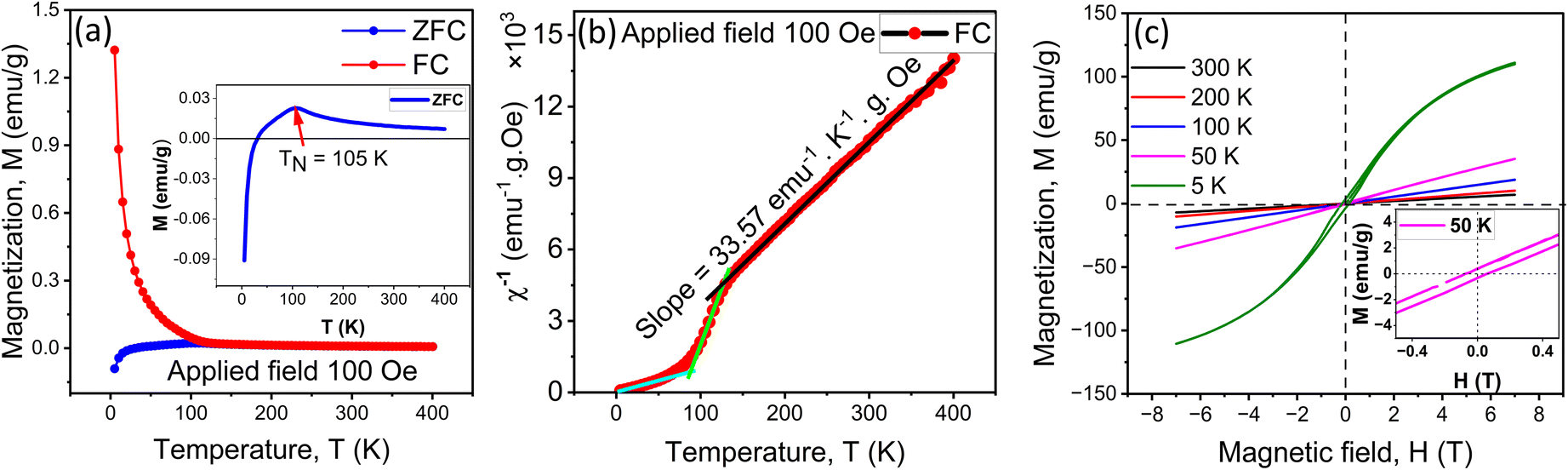 | ||
| Fig. 8 (a) ZFC and FC magnetization curves at an applied field of 100 Oe. Inset of (a) shows enlarged view of ZFC curve. (b) Curie–Weiss plot of FC magnetization curve. (c) M–H loops obtained at 5 K, 50 K, 100 K, 200 K and 300 K. Inset shows magnified view of M–H loop obtained at 50 K. | ||
Magnetization reversal (MR) behavior was also observed in the ZFC curve of Fig. 8(a) with a compensation temperature of Tcomp = 30 K. In a recent investigation,33 we observed such a magnetization reversal in an analogous double perovskite Y2FeCrO6. We also found that the temperature range/region of this negative magnetization is decreasing with increasing field and eventually vanishes for a higher applied magnetic field. Notably, a small negative field, trapped in the superconducting magnet can mimic a negative magnetization under the ZFC condition and that might be misleading. Therefore, in our investigation, the magnitude of the remanence in the magnet was reduced to 1–2 gauss by setting the field to zero from an initial field above 1 Tesla in the oscillator mode of the magnetometer. This also attenuated the flux creep effects. The remanence was further reduced to <0.05 gauss by using the ultra-low field option in the magnet to compensate for the trapped flux from the magnet. Therefore, we think that in our investigation, the observed negative magnetization is not an artifact due to the trapped field in the superconducting magnet.
For the existence of negative magnetization in the GCCO, we think that at around TN = 105 K, canted-antiferromagnetic ordering arises in the |Co + Cr| sublattice according to the antisymmetric Dzyaloshinskii–Moriya (DM) interaction, while Gd-sublattice remains its paramagnetic state.9 Below 105 K magnetization gradually decreases due to the polarization of paramagnetic Gd ions along anti-parallel to the canted antiferromagnetic state of |Co + Cr| sublattice and becomes zero at Tcomp = 30 K. At T < Tcomp, polarized Gd3+ moment dominant over the canted antiferromagnetic state of |Co + Cr| sublattice and magnetization become negative.33,64
Fig. 8(b) displayed the inverse susceptibility χ−1 vs. temperature curve obtained at 100 Oe using the FC technique. There are three regions in the inverse susceptibility curve. Beyond 130 K is region I, where antiferromagnetic-like inverse susceptibility shows a negative intercept, region II from 85 to 130 K indicates the presence of additional magnetic interaction between Co and Cr ions and shows ferromagnetic-like inverse susceptibility with a positive intercept, and region III from 0 to 85 K, which shows paramagnetic like inverse susceptibility with zero intercept.65 We have also calculated the paramagnetic moment of GCCO nanoparticles by Curie Weiss fitting in the linear region 130–400 K. According to Curie Weiss law,66

where C is the inverse of the slope in emu K per mole. The calculated effective paramagnetic moment μeff was found to be 11.15 μB. Theoretically, the magnetic moment of Co and Cr ions can be expressed as

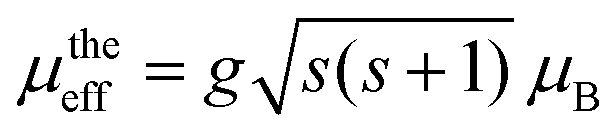 , considering their orbital angular momentum quenched by interatomic forces,66 where g is the gyromagnetic ratio. The calculated μeff values of Co2+, Co3+, Cr2+, Cr3+, and Cr6+ are 3.88, 4.90, 4.90, 3.88, and 0 μB, respectively. Moreover, μeff (Gd3+) is given by
, considering their orbital angular momentum quenched by interatomic forces,66 where g is the gyromagnetic ratio. The calculated μeff values of Co2+, Co3+, Cr2+, Cr3+, and Cr6+ are 3.88, 4.90, 4.90, 3.88, and 0 μB, respectively. Moreover, μeff (Gd3+) is given by  , where gyromagnetic ratio g = 2 and total angular momentum J = 7/2 for the Gd3+ ion. Then, the magnetic moment of Gd3+ is 7.9 μB. Thus, the theoretical paramagnetic moment of Gd2CoCrO6 is,
, where gyromagnetic ratio g = 2 and total angular momentum J = 7/2 for the Gd3+ ion. Then, the magnetic moment of Gd3+ is 7.9 μB. Thus, the theoretical paramagnetic moment of Gd2CoCrO6 is,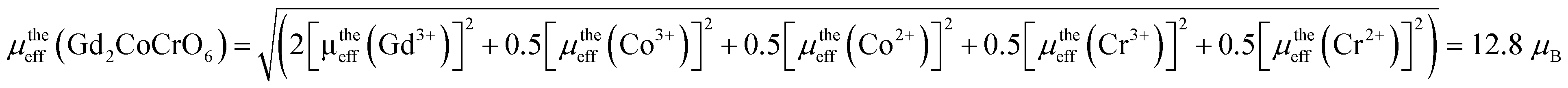
The experimentally obtained paramagnetic moment experienced a little deviation from the theoretical one, which is consistent with a similar investigation of Gd2NiMnO6 (ref. 41) that might be due to the lattice distortion present in the GCCO double perovskite. Further, theoretically obtained effective paramagnetic moment also suggest the presence of mixed states of Co2+, Co3+, Cr2+, and Cr3+ states in the GCCO double perovskite.
Additionally, the field-dependent magnetization of GCCO double perovskite was carried out at temperatures 300 K, 200 K, 100 K, 50 K, and 5 K under an external magnetic field of up to ±7 T. The magnetization vs. magnetic field (M–H) curves are displayed in Fig. 8(c). The linear M–H curves obtained at 300 K, 200 K, and 100 K indicate the paramagnetic nature of GCCO nanoparticles at these temperatures. An enlarged image of the magnetic hysteresis loop obtained at 50 K is shown in the inset of Fig. 8(c), which is also linear with applied field along with some remanent and coercivity and indicates the presence of both AFM and FM states. The hysteresis loop obtained at 5 K is nearly saturated with a maximum magnetization of 110 emu g−1 at an applied field of 7 T. From this hysteresis loop, the remanent magnetization (Mr) and coercive field (Hc) are determined to be ∼3.37 emu g−1 and ∼0.14 T, respectively. Noticeably, the narrow hysteresis loop was not saturated even at a higher applied field of 7 T, which also indicates the presence of both FM and AFM states at a lower temperature (<50 K). This phenomenon of multiple magnetic domain states may be explained in the following ways. From structural analysis, we have seen that Co3+ and Cr3+ are not perfectly ordered at B sites (B and B′). Further, the XPS studies reveal the presence of mixed states of Co and Cr elements. Thereby, in the absence of long-range ordering between Co3+ and Cr3+ sites, there may exist several configurations of B–B′ ions in GCCO: Co2+(e2g)–Co2+(e2g), Co2+(e2g)–Co3+(e1g), Co3+(e1g)–Co3+(e1g), Cr2+(e1g)–Cr2+(e1g), Cr2+(e1g)–Cr3+(e0g), Cr3+(e0g)–Cr3+(e0g), Co2+(e2g)–Cr2+(e1g), Co2+(e2g)–Cr3+(e0g), Co3+(e1g)–Cr2+(e1g), and Co3+(e1g)–Cr3+(e0g). Among them, only Co2+(e2g)–Cr3+(e0g) pair is responsible for the FM state according to the Goodenough–Kanamori rule,68 and others are responsible for the AFM state.16,22 This rule states that the FM state is produced by a 180° super-exchange interaction between an empty Cr3+-d(e0g) orbital and a half-filed Co2+-d(e2g) orbital. However, due to lattice distortion, this magnetic interaction changes into an AFM when the d(e2g)–O–d(e0g) angle is less than 180°.69 Particularly for the systems like GCCO, the super-exchange interaction differs from FM to AFM for a span of 125° to 150° bond angles.69 As previously shown in XRD analysis, octahedral distortion causes the Co–O–Cr bond in GCCO double perovskite to bend to 146°. This distortion is also attributed to the formation of the AFM interaction alongside the FM state.
3.9 Optical properties
To analyze the optical properties of as-synthesized GCCO nanomaterials, UV-visible absorbance spectroscopy was performed. Fig. 9(a) shows the absorbance spectrum with corresponding band fittings of as-synthesized GCCO nanoparticles. Band fittings were carried out using Gaussian functions by employing the fitting program integrated into Origin 10.0. In the absorbance spectrum three absorption bands A1, A2, and A3 at 300 nm, 430 nm, and 720 nm, respectively, suggest the multi-band electronic structure of GCCO double perovskite.22,42 The absorption bands (A1 and A2) around 300 nm and 430 nm, respectively, are due to the p–d charge-transfer (CT) transition [O(2p) → Co/Cr(3d)] in the centers of the Co/CrO6 octahedral structure of GCCO nanomaterials.42,70 The low energy absorption band (A3) close to 720 nm is ascribed by the d–d electronic transition in the Co3+ ions.71 Moreover, the three absorption bands in Fig. 9(a) show a wide solar harvesting potential of GCCO, which may increase the photocatalytic efficiency of GCCO nanomaterial.72,73 The direct optical bandgap was estimated by a Tauc plot using the Tauc relation,| αhν = A(hν − Eg)½ |
 | ||
| Fig. 9 (a) Absorbance (Abs.) spectrum and corresponding band fittings, (b) bandgap estimated by Tauc plot and (c) PL spectrum of GCCO nanomaterials. | ||
Finally, we have made a comparative analysis of structural, magnetic, and optical properties of as-synthesized Gd2CoCrO6 and previously investigated rare-earth and transition metal-based double perovskite materials. The summary of the comparative analysis is presented in Table 2. It is seen from Table 2 that GCCO possesses a higher Néel transition temperature compared to other rare-earth chromite except for Y2FeCrO6 double perovskite.24 The optical bandgap of this class of double perovskite materials is close to 2.0 eV, which might have potential optoelectronic applications.
| Materials | Lattice parameters (Å) | Space group | Structure | B site order | TN (K) | Bandgap (eV) | Ref |
|---|---|---|---|---|---|---|---|
| Gd2CoCrO6 | a = 5.27 | P21/n | Monoclinic | Disorder | 105 | 2.25 | Present work |
| b = 5.46 | |||||||
| c = 7.53 | |||||||
| Gd2FeCrO6 | a = 5.36 | P21/n | Monoclinic | Disorder | 15 | 2.0 | 22 |
| b = 5.59 | |||||||
| c = 7.68 | |||||||
| Er2FeCrO6 | a = 5.25 | Pbnm | Hexagonal | Disorder | 11.7/5.7 | — | 76 |
| b = 5.55 | |||||||
| c = 7.57 | |||||||
| Y2FeCrO6 | a = 5.55 | Pbnm | Orthorhombic | Disorder | 327 | 1.90 | 24,33 |
| b = 7.57 | |||||||
| c = 5.26 | |||||||
| La2FeCrO6 | — | — | — | Order | 45 | 1.6 | 77 |
3.10 Potentiality as a photocatalyst
The applicability of GCCO nanoparticles as a photocatalyst was assessed by calculating the valence band maximum (VBM) and conduction band minimum (CBM) by the Mulliken electronegativity approach.78,79 According to the approach,
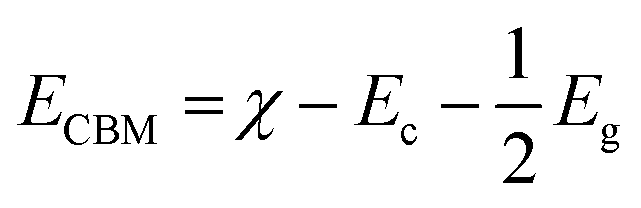 | (1) |
| EVBM = ECBM + Eg | (2) |
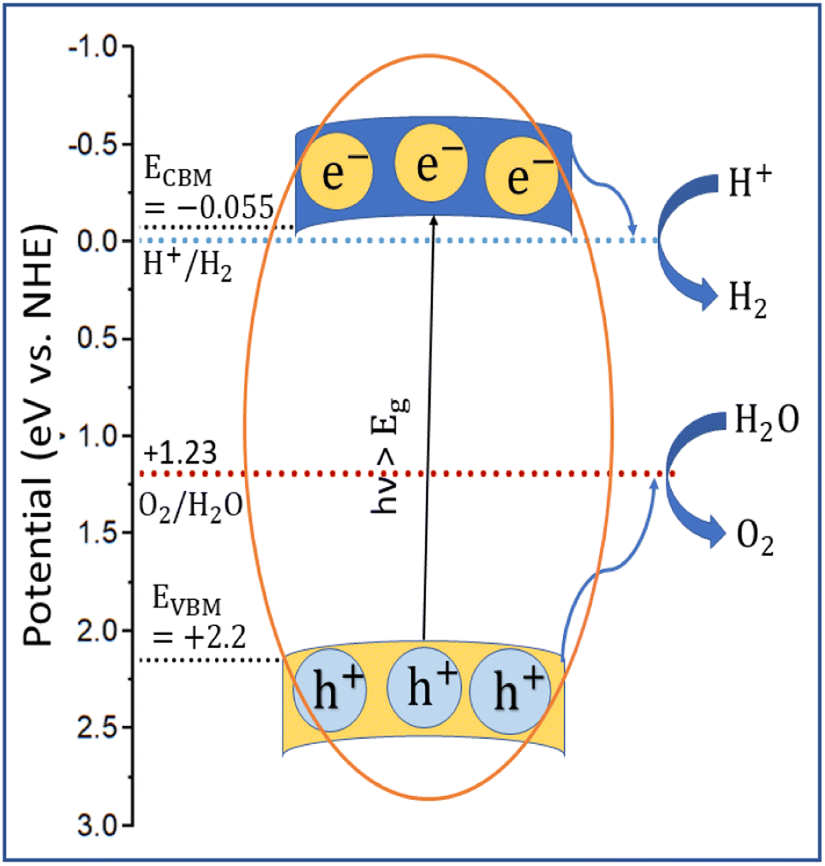 | ||
| Fig. 10 Schematic representation of estimated band edge position of GCCO nanoparticles. | ||
According to the literature, if the conduction band minimum of a semiconductor is lower than the reduction potential of hydrogen, then it can produce photocatalytic hydrogen by splitting water, and if the valence band maximum is higher than the oxidation potential of oxygen, then it can produce O2 from water.22,80,81 As illustrated in Fig. 10, the conduction band minimum of GCCO is lower than the reduction potential of H2, and the valence band maximum is higher than the oxidation energy of oxygen. Therefore, according to a theoretical perspective, GCCO nanoparticles are potential candidates for photocatalytic H2 and O2 evolution through the splitting of water.22,81
4 Conclusions
Powder X-ray diffraction, Raman spectroscopy, X-ray photoelectron spectroscopy, and electron microscopy imaging ensured the successful synthesis of double perovskite Gd2CoCrO6 nanoparticles with an average particle size of 71 ± 3 nm. A further decrease of temperature from Néel transition at TN = 105 K demonstrated a magnetization reversal behavior with a compensation temperature of Tcomp = 30 K. Low-temperature hysteresis loops demonstrated the presence of both FM and AFM domains in the GCCO double perovskite. Strong anisotropy along with different kinds of exchange interactions like super-exchange and Dzyaloshinskii–Moriya interactions between various cations via oxygen ions are responsible for the presence of multiple magnetic states in this perovskite system. Finally, the favorable optical bandgap (∼2.25 eV) unveiled the promising potential of GCCO nanoparticles in photocatalytic H2 and O2 evolution from water. The fascinating structural, magnetic, and optical properties revealed that GCCO double perovskite might be a potential new member of rare earth and transition metal-based perovskite material for applications in spintronics, optoelectronics, and photocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial assistance from the Semiconductor Technology Research Center (STRC), University of Dhaka and CASR, Bangladesh University of Engineering and Technology (BUET). We also acknowledge M. D. I. Bhuyan, a former PhD student of Nanotechnology Research Laboratory, Dept. of Physics, BUET for sample preparation.Notes and references
- A. S. Bhalla, R. Guo and R. Roy, Mater. Res. Innovations, 2000, 4, 3–26 CrossRef CAS.
- M. A. Basith, O. Kurni, M. S. Alam, B. L. Sinha and B. Ahmmad, J. Appl. Phys., 2014, 115, 024102 CrossRef.
- B. Ahmmad, M. Z. Islam, A. Billah and M. A. Basith, J. Phys. D: Appl. Phys., 2016, 49, 095001 CrossRef.
- M. Shang, C. Zhang, T. Zhang, L. Yuan, L. Ge, H. Yuan and S. Feng, Appl. Phys. Lett., 2013, 102, 062903 CrossRef.
- F. Sharmin and M. A. Basith, J. Alloys Compd., 2022, 901, 163604 CrossRef CAS.
- Q. Y. Dong, K. Y. Hou, X. Q. Zhang, L. Su, L. C. Wang, Y. J. Ke, H. T. Yan and Z. H. Cheng, J. Appl. Phys., 2020, 127, 033904 CrossRef CAS.
- L. Gildo-Ortiz, V. M. Rodríguez-Betancourtt, O. Blanco-Alonso, A. Guillén-Bonilla, J. T. Guillén-Bonilla, A. Guillén-Cervantes, J. Santoyo-Salazar and H. Guillén-Bonilla, Results Phys., 2019, 12, 475–483 CrossRef.
- P. Mahata, T. Aarthi, G. Madras and S. Natarajan, J. Phys. Chem. C, 2007, 111, 1665–1674 CrossRef CAS.
- S. Mahana, U. Manju and D. Topwal, J. Phys. D: Appl. Phys., 2018, 51, 305002 CrossRef.
- A. Jaiswal, R. Das, K. Vivekanand, T. Maity, P. M. Abraham, S. Adyanthaya and P. Poddar, J. Appl. Phys., 2010, 107, 013912 CrossRef.
- K. D. Singh, R. Pandit and R. Kumar, Solid State Sci., 2018, 85, 70–75 CrossRef CAS.
- T. Saha-Dasgupta, Mater. Res. Express, 2020, 7, 014003 CrossRef CAS.
- M. A. Basith, M. A. Islam, B. Ahmmad, M. D. Sarowar Hossain and K. Mølhave, Mater. Res. Express, 2017, 4, 075012 CrossRef.
- R. J. Cava, B. Batlogg, J. J. Krajewski, R. Farrow, L. W. Rupp, A. E. White, K. Short, W. F. Peck and T. Kometani, Nature, 1988, 332, 814–816 CrossRef CAS.
- H. Tanaka and M. Misono, Curr. Opin. Solid State Mater. Sci., 2001, 5, 381–387 CrossRef CAS.
- N. Das, S. Singh, A. G. Joshi, M. Thirumal, V. R. Reddy, L. C. Gupta and A. K. Ganguli, Inorg. Chem., 2017, 56, 12712–12718 CrossRef CAS PubMed.
- S. Halder, M. S. Sheikh, R. Maity, B. Ghosh and T. P. Sinha, Ceram. Int., 2019, 45, 15496–15504 CrossRef CAS.
- P. Shirazi, M. Rahbar, M. Behpour, M. Ashrafi and M. Behpour, New J. Chem., 2019, 44, 231–238 RSC.
- M. A. Basith, N. Yesmin and R. Hossain, RSC Adv., 2018, 8, 29613–29627 RSC.
- M. A. Basith, S. Manjura Hoque, M. Shahparan, M. A. Hakim and M. Huq, Phys. B, 2007, 395, 126–129 CrossRef CAS.
- S. Vasala and M. Karppinen, Prog. Solid State Chem., 2015, 43, 1–36 CrossRef CAS.
- M. D. I. Bhuyan, S. Das and M. A. Basith, J. Alloys Compd., 2021, 878, 160389 CrossRef CAS.
- J. K. Murthy and A. Venimadhav, J. Alloys Compd., 2017, 719, 341–346 CrossRef CAS.
- K. P. Patra and S. Ravi, J. Appl. Phys., 2022, 132, 213903 CrossRef CAS.
- X. L. Wang, D. li, T. Y. Cui, P. Kharel, W. Liu and Z. D. Zhang, J. Appl. Phys., 2010, 107, 09B510 CrossRef.
- R. Hu, R. Ding, J. Chen, J. Hu and Y. Zhang, Catal. Commun., 2012, 21, 38–41 CrossRef CAS.
- R. Hu, C. Li, X. Wang, Y. Sun, H. Jia, H. Su and Y. Zhang, Catal. Commun., 2012, 29, 35–39 CrossRef CAS.
- B. Gray, H. N. Lee, J. Liu, J. Chakhalian and J. W. Freeland, Appl. Phys. Lett., 2010, 97, 013105 CrossRef.
- M. F. Zaranyika and T. Chirinda, J. Environ. Chem. Ecotoxicol., 2011, 3, 103–115 CAS.
- K. Miura and K. Terakura, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 7 CrossRef.
- D. Bokov, A. Turki Jalil, S. Chupradit, W. Suksatan, M. Javed Ansari, I. H. Shewael, G. H. Valiev and E. Kianfar, Adv. Mater. Sci. Eng., 2021, 2021, 1–21 CrossRef.
- S. Chanda, S. Saha, A. Dutta and T. P. Sinha, Mater. Res. Bull., 2015, 62, 153–160 CrossRef CAS.
- M. A. Islam, T. Sato, F. Ara and M. A. Basith, J. Alloys Compd., 2023, 944, 169066 CrossRef CAS.
- M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 725–738 CrossRef CAS PubMed.
- M. T. Anderson, K. B. Greenwood, G. A. Taylor and K. R. Poeppelmeier, Prog. Solid State Chem., 1993, 22, 197–233 CrossRef CAS.
- I. Yamada, A. Takamatsu and H. Ikeno, Sci. Technol. Adv. Mater., 2018, 19, 101–107 CrossRef CAS PubMed.
- Z. Li, Y. Cho, X. Li, X. Li, A. Aimi, Y. Inaguma, J. A. Alonso, M. T. Fernandez-Diaz, J. Yan, M. C. Downer, G. Henkelman, J. B. Goodenough and J. Zhou, J. Am. Chem. Soc., 2018, 140, 2214–2220 CrossRef CAS PubMed.
- S. H. Byeon, M. W. Lufaso, J. B. Parise, P. M. Woodward and T. Hansen, Chem. Mater., 2003, 15, 3798–3804 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- C. Shi, Y. Hao and Z. Hu, J. Phys. D: Appl. Phys., 2011, 44, 245405 CrossRef.
- M. Nasir, S. Kumar, N. Patra, D. Bhattacharya, S. N. Jha, D. R. Basaula, S. Bhatt, M. Khan, S. W. Liu, S. Biring and S. Sen, ACS Appl. Electron. Mater., 2019, 1, 141–153 CrossRef CAS.
- V. M. Gaikwad, M. Brahma, R. Borah and S. Ravi, J. Solid State Chem., 2019, 278, 120903 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3384–3392 CrossRef CAS.
- N. Bajpai, M. Saleem and A. Mishra, J. Mater. Sci.: Mater. Electron., 2020, 32, 12890–12902 CrossRef.
- M. N. Iliev, M. v. Abrashev, A. P. Litvinchuk, V. G. Hadjiev, H. Guo and A. Gupta, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 104118 CrossRef.
- S. Obregón and G. Colón, Catal. Sci. Technol., 2014, 4, 2042–2050 RSC.
- F. D. Hardcastle and I. E. Wachs, J. Phys. Chem., 1991, 95, 5031–5041 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part A: Theory and Applications in Inorganic Chemistry, 6th edn, 2008, pp. 1–419 Search PubMed.
- H. Ding, Z. Tao, S. Liu and Y. Yang, J. Power Sources, 2016, 327, 573–579 CrossRef CAS.
- P. Düngen, R. Schlögl and S. Heumann, Carbon, 2018, 130, 614–622 CrossRef.
- S. J. Jung, T. Lutz, M. Boese, J. D. Holmes and J. J. Boland, Nano Lett., 2011, 11, 1294–1299 CrossRef CAS PubMed.
- A. Kocjan, M. Logar and Z. Shen, Sci. Rep., 2017, 7, 1–8 CrossRef CAS PubMed.
- F. Loosli and S. Stoll, Environ. Sci.: Nano, 2017, 4, 203–211 RSC.
- W. Y. Maeng and M. Yoo, J. Nanosci. Nanotechnol., 2015, 15, 8429–8433 CrossRef CAS PubMed.
- S. v. Sokolov, E. Kätelhön and R. G. Compton, J. Phys. Chem., 2015, 119, 25093–25099 CAS.
- E. Kätelhön, S. v. Sokolov, T. R. Bartlett and R. G. Compton, Chemphyschem, 2017, 18, 51–54 CrossRef PubMed.
- J. S. Punitha, A. Nataraj, V. Anbarasu, M. Dhilip and K. S. Kumar, J. Mater. Sci.: Mater. Electron., 2021, 32, 10266–10276 CrossRef CAS.
- G. Muscas, K. Prabahar, F. Congiu, G. Datt and T. Sarkar, J. Alloys Compd., 2022, 906, 164385 CrossRef CAS.
- M. N. Ha, G. Lu, Z. Liu, L. Wang and Z. Zhao, J. Mater. Chem. A, 2016, 4, 13155–13165 RSC.
- R. P. Maiti, S. Dutta, M. K. Mitra and D. Chakravorty, J. Phys. D: Appl. Phys., 2013, 46, 415303 CrossRef.
- P. Gupta, R. Bhargava, R. Das and P. Poddar, RSC Adv., 2013, 3, 26427–26432 RSC.
- J. Mohapatra, M. Xing, J. Elkins and J. P. Liu, J. Alloys Compd., 2020, 824, 153874 CrossRef CAS.
- M. P. Sharannia, S. De, R. Singh, A. Das, R. Nirmala and P. N. Santhosh, J. Magn. Magn. Mater., 2017, 430, 109–113 CrossRef CAS.
- Y. Qiao, Y. Zhou, S. Wang, L. Yuan, Y. Du, D. Lu, G. Che and H. Che, Dalton Trans., 2017, 46, 5930–5937 RSC.
- S. Das, B. Ahmmad and M. A. Basith, AIP Adv., 2020, 10, 095135 CrossRef CAS.
- D. Kumar and A. K. Singh, J. Magn. Magn. Mater., 2019, 469, 264–273 CrossRef CAS.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87–98 CrossRef CAS.
- H. L. Feng, M. Arai, Y. Matsushita, Y. Tsujimoto, Y. Guo, C. I. Sathish, X. Wang, Y. H. Yuan, M. Tanaka and K. Yamaura, J. Am. Chem. Soc., 2014, 136, 3326–3329 CrossRef CAS PubMed.
- T. Arima, Y. Tokura and J. B. Torrance, Phys. Rev. B, 1993, 48, 17006 CrossRef CAS PubMed.
- S. Sun, W. Wang, L. Zhang and M. Shang, J. Phys. Chem. C, 2009, 113, 12826–12831 CrossRef CAS.
- Q. L. Liu, Z. Y. Zhao, R. D. Zhao and J. H. Yi, J. Alloys Compd., 2020, 819, 153032 CrossRef CAS.
- K. P. Ong, S. Wu, T. H. Nguyen, D. J. Singh, Z. Fan, M. B. Sullivan and C. Dang, Sci. Rep., 2019, 9, 1–8 CrossRef CAS PubMed.
- R. Bairy, P. S. Patil, S. R. Maidur, H. Vijeth, M. S. Murari and U. K. Bhat, RSC Adv., 2019, 9, 22302–22312 RSC.
- A. Chanda, S. Gupta, M. Vasundhara, S. R. Joshi, G. R. Mutta and J. Singh, RSC Adv., 2017, 7, 50527–50536 RSC.
- Z. Dong, Z. Wang and S. Yin, Ceram. Int., 2020, 46, 26632–26636 CrossRef CAS.
- A. Ohtomo, S. Chakraverty, H. Mashiko, T. Oshima and M. Kawasaki, J. Mater. Res., 2013, 28, 689–695 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 2004, 3, 573 CrossRef.
- R. S. Mulliken, J. Chem. Phys., 2004, 2, 782 CrossRef.
- S. Lin, H. Huang, T. Ma and Y. Zhang, Adv. Sci., 2020, 8, 2002458 CrossRef PubMed.
- F. Sharmin, D. Chandra Roy and M. A. Basith, Int. J. Hydrogen Energy, 2021, 46, 38232–38246 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02233a |
| This journal is © The Royal Society of Chemistry 2023 |