
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, morphological, electrical, and conduction mechanism studies of a sodium cerium diphosphate compound
Fareh Hamrita,
Rabiaa Chtouroub,
Djedid Taloubc,
Imen Gharbi b and
Abderrazek Oueslati*b
b and
Abderrazek Oueslati*b
aLaboratoire de Caractérisation Spectroscopique et Optique des Matériaux, Faculté des Sciences, Université de Sfax, B. P. 1171, 3000 Sfax, Tunisia
bDepartment of Mechanic, Faculty of Technology, M. Boudiaf Univ. of M'sila, Algeria. E-mail: oueslatiabderrazek@yahoo.fr
cDepartment of Physics, Faculty of Sciences, M. Boudiaf Univ. of M'sila, Algeria
First published on 22nd May 2023
Abstract
A NaCeP2O7 compound was successfully synthesized by a high-temperature reaction with the solid-state method. Analyzing the XRD pattern, of the studied compound, confirms the orthorhombic phase with the Pnma space group. The examination of SEM images reveals that the majority of grains are around 500 to 900 nm with a uniform distribution. As for the EDXS analysis, all chemical elements were detected and found in the appropriate ratio. The curves of temperature-dependent imaginary modulus M′′ versus angular frequency reveal the presence of one peak at each temperature, proving that the dominant contribution is associated with the grains. The frequency dependence of the conductivity of alternating current is explained using Jonscher's law. The close values of the activation energies obtained from the jump frequency and extracted from the dielectric relaxation of the modulus spectra, as well as from the continuous conductivity imply that the transport takes place by the Na+ ions hopping mechanism. The charge carrier concentration in the title compound has been evaluated and shown to be independent of temperature. The exponent s increases with the increase in temperature; this behavior proves that the non-overlapping small polaron tunneling (NSPT) is the appropriate conduction mechanism model.
1. Introduction
In the last century, diphosphates have attracted a lot of attention due to their interesting optical and electrical properties, which are relevant to their structural diversity. Researchers have reported that the structure of this family of compounds is mainly presented in seven different crystallographic types.1However, in recent years, diphosphates with the general formula MILnP2O7 (where MI is a monovalent cation and Ln is a rare earth element or yttrium) have been well recognized as multifunctional materials.2 The remarkable optical properties were found to be related to the fact that the Ln–Ln distances are relatively large, leading to a very low fluorescence quenching concentration.3,4 Furthermore, thanks to their significant ionic conductivity, these materials were reported to be previously manufactured as ionic conductors, catalysts, sensors, and rechargeable alkaline batteries.5–10
The structure of the M1LnP2O7 diphosphates (where M1 = K, Cs, Na, and, Ln = Y, Ce, Yb) was found to change depending on the ionic radii of the alkali metal and the rare earth element.11,12 The single-crystal structure's resolution was firstly reported for the CsYbP2O7 (P21/c),13 KYP2O7 (Cmcm),14 and NaYP2O7 (P21).15 After a literature survey, Férid and coworkers have conducted several studies on sodium and Ln diphosphate's structure, and have synthesized new compounds belonging to the NaLnP2O7 family of diphosphates. They reported that the Ln diphosphate series is also defined by two different types of structure. They reported that the NaPrP2O7,16 NaEuP2O7 (ref. 17) NaHoP2O7,18 and NaYbP2O7 (ref. 19) crystallize in the P21/n space group of the monoclinic system. However, the NaCeP2O7 (ref. 2) was reported to be isostructural to the NaLaP2O7 (ref. 21), where it crystallizes in the Pnma space group of the orthorhombic system. This family of compounds shows structural transformation where the symmetry decreases with the decrease of Ln's ionic radius.16–23
Various phosphates containing trivalent cerium have been proposed as strong green emitters under UV excitation,24 as well as those containing sodium, have been found to have excellent ionic conductivity. For these reasons, we have selected the NaCeP2O7 compound for investigation.
NaCeP2O7 has been studied from the perspective of structure, but to our knowledge, there are no reports of their electrical response. Since electrical properties can be critical to understanding the behavior of the studied material, they also help define the nature and scope of application. The present paper is devoted to describing the process of synthesis using the solid-state method, as well as to reporting the X-ray diffraction (XRD), scanning electron microscopy (SEM), energy dispersive X-ray (EDXS), and infrared spectroscopy which were used to characterize the synthesized compound. In addition, the impedance spectroscopy measurement of the NaCeP2O7 compound was performed as a function of temperature. This study will expand the knowledge of electrical conductivity, shedding light on the transport mechanism within this material, and the correlation between structure and ionic conductivity provides insight into the path of the transport mechanism.
2. Experimental details
The NaCeP2O7 ceramic was synthesized using a solid-state reaction technique from high-purity oxides which are Na2CO3 (99%), CeO2 (99.9%), and NH4H2PO4 (99.9%). The raw materials were weighed according to the stoichiometric amounts and mixed using an agate mortar. The sample was dried at 373 K, then calcined at 573 K for 5 hours to eliminate NH3, CO2, and H2O. The sample was ground thoroughly to ensure that there was no agglomeration in the powder. The fine powder was pressed into cylindrical pellets of 8 mm in diameter and 1.3 mm in thickness. These pellets were sintered at a temperature of 1273 K for 6 hours. The structural analysis of the synthesized sample was performed at room temperature with the help of a Philips PW 1710 diffractometer with copper radiation λKα = 1.5418 Å over the 2θ range of 10°–60° with a step 0.02°. To analyze the microstructure and elemental composition of the samples, a scanning electron microscope (FEI Nova NanoSEM 200) coupled with an energy-dispersive X-ray (EDXS) analyzer was used. The IR spectra were performed for the NaCeP2O7 compound on powder at room temperature using a PerkinElmer spectrophotometer (FTIR-100). The flat polished surface of the sintered sample was electrode with air-drying silver paste. The study of the real and imaginary parts of the impedance was carried out at various temperatures from 473 to 573 K with a frequency range of 100 Hz–7 MHz using a Solartron 1260 impedance analyzer.3. Results and discussion
3.1. Crystalline parameters
The X-ray diffraction pattern of the NaCeP2O7 sample, recorded at room temperature, is illustrated in Fig. 1. The refinement leads to a rather good agreement between the experimental and calculated pattern. All the reflection peaks of the X-ray profile were indexed in the Pnma space group of the orthorhombic system with the refined lattice parameters: a = 8.620 (4) Å, b = 5.181(3) Å, c = 12.530 (6) Å, V = 577.218(5) Å3, Z = 4 and a reliability factor of χ2 = 2.73. The unit cell parameters are in good agreement with the literature values.17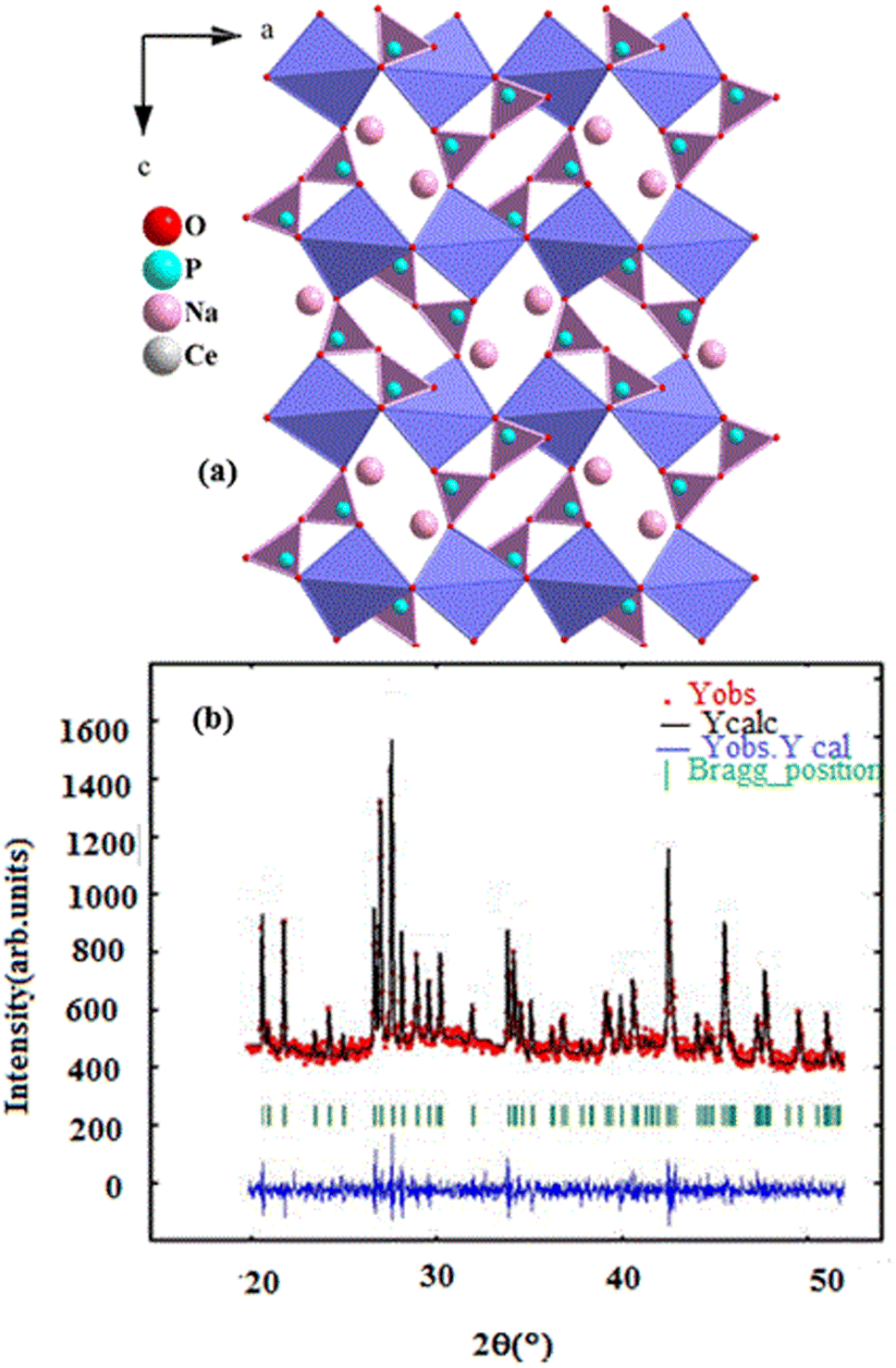 | ||
| Fig. 1 (a) Projection, along b direction, (b) X-ray diffraction pattern of the NaCeP2O7 diphosphate structure. | ||
The average crystallite size of the NaCeP2O7 was calculated using the Debye–Scherrer formula:25
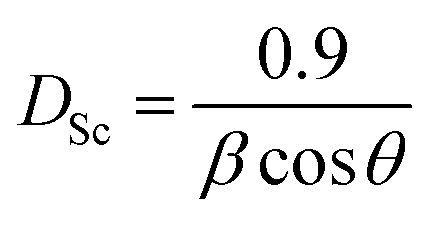 | (1) |
3.2. Structure description
The structure is characterized by a three-dimensional framework of PO4 tetrahedra (forming P2O7 groups via corner-sharing) and CeO9 polyhedra are deformed and form infinite ribbons parallel to the b-axis, which are occupied by Na atoms. This framework is composed of four crystallographic building blocks: four NaO9 polyhedral, four CeO9 polyhedral, and four P2O7 diphosphates in the unit cell where the cohesion of the structure is provided by bridges Ce–O–P and Na–O–P. The projection, along the b-axis of the structure, is shown in inset Fig. 1a.173.3. Scanning electron microscope analysis
SEM analysis is a technique to image surface morphology by providing information about the size, shape, and morphology of the studied sample is an analysis that scans a sample with an electron beam to produce a magnified image.26 Fig. 2a depicted a SEM image of grains at a 10 μm scale. The figure reveals a non-uniform distribution of small and large grains interconnected by pores in the observed morphology. Most of the grains are well connected with few pores between them. Fig. 2b presents the histogram particle size distribution of the NaCeP2O7 ceramic, where the average grain size was determined using ImageJ software. The size of the majority of grains is found to be around 500 to 900 nm. In addition, the histogram was fitted by a log-normal distribution, revealing a uniform distribution.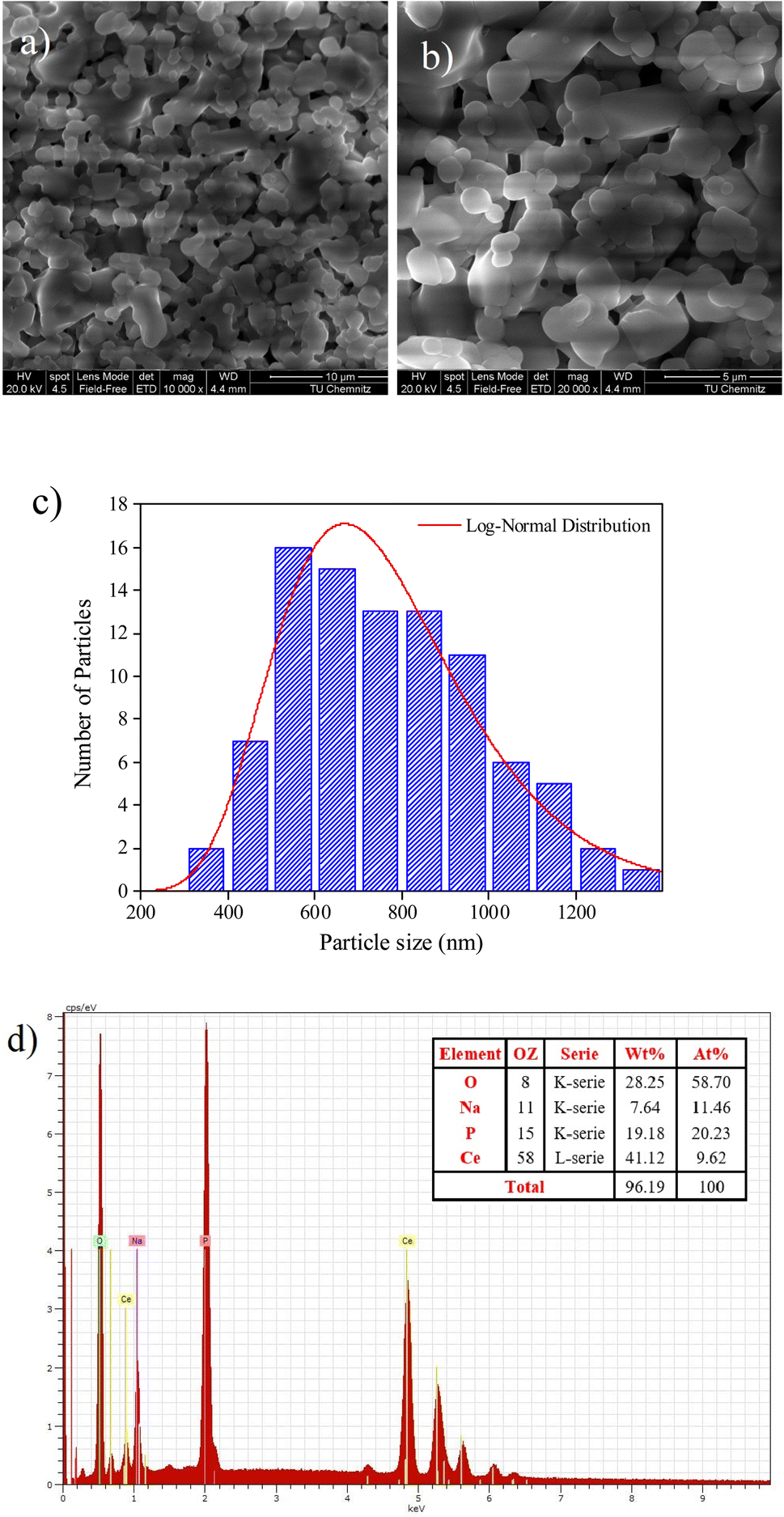 | ||
| Fig. 2 (a) and (b) SEM images (c) particle size distribution histogram with corresponding log-normal distribution fit, and (d) EDXS spectrum of the fractured surface of NaCeP2O7 diphosphate. | ||
The difference between the determined size by the XRD and SEM analysis can be ascribed to the fact that XRD gives the mean size of the crystallites, while SEM analysis takes into account the grains, which can be defined as the region of a polycrystalline material with the same crystallographic orientation and same structure.27 The size of the crystallites was found to be much smaller than the grain sizes of the NaCeP2O7. However, both the crystallites and the grains are on the nanometric scale. Nanocrystalline materials with particle or grain sizes in the nanometer range (typically less than 100 nm) have been attracting increasing attention in the last few years due to their remarkable physical and chemical properties that may significantly differ from those of the corresponding coarse-grained materials with the same composition.27
EDXS analysis is an elemental analysis of surfaces in SEM, which measures the energy and intensity distribution of X-ray signals generated by the electron beam striking the surface of the specimen. The elemental composition at a point, along a line, or in a defined area, can be easily determined to a high degree of precision.28
The EDXS spectrum showed in Fig. 2c the presence of Na, Ce, P, and O atoms, confirming that all constituent elements could be detected and there is no loss of any integrated element during the sintering. The weight and the atomic percentages of chemical elements of the studied compound are given in the table, which exists in the inset of Fig. 2c. The EDXS analysis indicates that the chemical composition of this compound is close to the nominal ones. The spectrum analysis affirms that the using reagents were in a suitable stoichiometry. Consequently, a perfect correlation between the XRD and EDXS analysis is assured.
3.4. Infrared spectroscopy analysis
Infrared spectroscopy (IR) analysis is based on irradiating the sample with a broad band of infrared frequencies, and the intensity of the reflected or transmitted infrared radiation is measured as a function of frequency. From the knowledge of incident intensity and reflected or transmitted intensity, as a function of infrared frequency, an infrared absorbance spectrum can be reconstructed. Absorption at specific frequencies is characteristic of certain bonds. Thus, the IR spectrum identifies the various bonds and functional groups within the molecule.29The IR spectrum of the studied compound is displayed in Fig. 3. The assignment of the bands was achieved by comparison with similar compounds.30–32 The frequencies of the [P2O7]4− anion are assigned based on the characteristic vibrations of the (P–O–P) bridge and (PO3) groups. As the (P–O) bond in the (PO3) group is stronger than that in the (P–O–P) bridge, the vibrational frequencies of (PO3) are expected to be higher than those of (P–O–P). The bands due to the symmetric and asymmetric stretching frequencies of (PO3) are usually seen in the region of 1143 cm−1 to 1087 cm−1.31 The bands observed at 950 cm−1 and from 750 cm−1 to 618 cm−1 (Fig. 3) are attributed to the asymmetric νa(POP) and symmetric νs(POP) stretching modes.31,32 The bands assigned to the asymmetric δa(PO3) and symmetric δs(POP) deformations are also identified at 550 cm−1 and from 485 cm−1 to 418 cm−1, respectively. Band assignments for the fundamental modes of this compound are given in Table 1, confirming the presence of the diphosphate group in the title compound.
 | ||
| Fig. 3 IR spectrum of NaCP2O7 diphosphate recorded at room temperature. | ||
| IR | Assignment |
|---|---|
| 1143 | νa(PO3) |
| 1087 | |
| 1050 | νs(PO3) + νa(PO3) |
| 1007 | |
| 950 | νa(POP) + νs(POP) |
| 750 | |
| 618 | |
| 550 | δa(PO3) |
| 528 | |
| 485 | δ(POP) |
| 468 | |
| 437 | |
| 418 |
3.5. Impedance spectroscopy analysis
Impedance spectroscopy is a very effective technique to study the electrical transport mechanism in materials. This technique allows the separation of resistances whether it is related to grains, grain boundaries, or electrode effects. This technique is versatile and gives information on the mobility of individual components within the system as well as on the occurrence of specific interactions.33,34This technique is a method to observe the way in which a system follows perturbations at a steady state. Impedance is evaluated from the response of the current to the varying frequency of the imposed alternating voltage over a wide range, and it is separated into real and imaginary components.35
Fig. 4 shows the variation of the imaginary part of the modulus (M′′), as a function of the angular frequency, in the measured temperature range. Each curve is characterized by an asymmetric relaxation peak 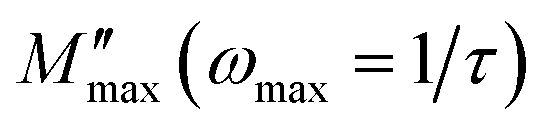 which shifts to higher frequencies when the temperature increases. This pattern provides wider information related to charge transport processes such as the mechanism of electrical transport, conductivity relaxation, and ion dynamics as a function of frequency and temperature.37,38 The frequency region below the peak maximum
which shifts to higher frequencies when the temperature increases. This pattern provides wider information related to charge transport processes such as the mechanism of electrical transport, conductivity relaxation, and ion dynamics as a function of frequency and temperature.37,38 The frequency region below the peak maximum  is where the charge carriers are mobile over long distances. As for the region above, it is where the charge carriers are spatially confined to potential wells and where they are mobile on short distances, making only the localized motion within the wells. The broadening of the peak points out the spread of relaxation with different meantime constants and a non-Debye type relaxation for the studied material. Further, the appearance of a peak in the modulus spectrum provides a clear indication of conductivity relaxation.39,40
is where the charge carriers are mobile over long distances. As for the region above, it is where the charge carriers are spatially confined to potential wells and where they are mobile on short distances, making only the localized motion within the wells. The broadening of the peak points out the spread of relaxation with different meantime constants and a non-Debye type relaxation for the studied material. Further, the appearance of a peak in the modulus spectrum provides a clear indication of conductivity relaxation.39,40
 | ||
| Fig. 4 Variation of imaginary part (M′′) of modulus with frequency at various temperatures. Inset: the ln(ωmax) vs. (1000/T) plot. | ||
Using the equation proposed by Bergman–Kohlrausch, Williams, and Watts (KWW), the curves of  have been well adjusted for this sample.41
have been well adjusted for this sample.41
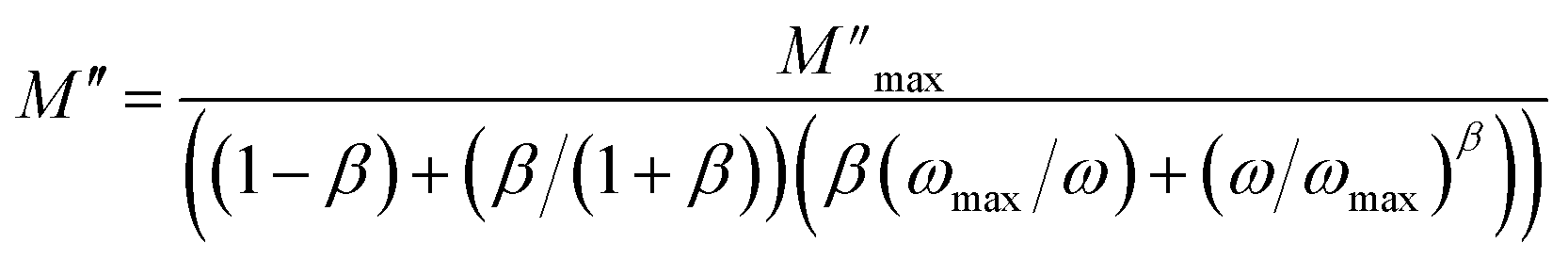 | (2) |
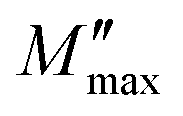 and ωmax are the peak maxima and the corresponding angular frequency, respectively. β (0 <β < 1) is the stretching factor that determines whether the relaxation in the dielectric is of a Debye or a non-Debye nature.41
and ωmax are the peak maxima and the corresponding angular frequency, respectively. β (0 <β < 1) is the stretching factor that determines whether the relaxation in the dielectric is of a Debye or a non-Debye nature.41
The fit values of β (β < 1) suggest a non-Debye-type dielectric relaxation and give evidence of dipole–dipole interactions in the sample.
On the other hand, the temperature dependence of the conductivity relaxation frequencies ωmax is plotted in inset Fig. 4, and the spectra obey the Arrhenius relation. The activation energy calculated from the slope of the plot is Em = (1.34 ± 0.05). This value is very close to the determined one from the dc-conductivity in literature,21 confirming that the transport is through the ion hopping mechanism in the NaCeP2O7 compound. This transport mechanism is thermally activated to overcome the potential barrier.
Fig. 5 shows the normalized plot of 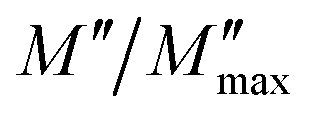 at different temperatures. The perfect overlap of the curves into a single master one, at different temperatures, indicates that the Kohlrausch parameter β is temperature independent. Moreover, dynamical processes occurring at different frequencies are independent of temperature or have the same thermal energy.18,22
at different temperatures. The perfect overlap of the curves into a single master one, at different temperatures, indicates that the Kohlrausch parameter β is temperature independent. Moreover, dynamical processes occurring at different frequencies are independent of temperature or have the same thermal energy.18,22
 | ||
Fig. 5 The variation of the normalized 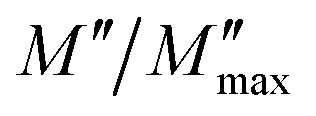 imaginary part versus ω/ωmax. imaginary part versus ω/ωmax. | ||
AC conductivity analysis helps to identify the type of charge carrier transport, which is responsible for the conduction process and its response as a function of frequency and temperature.42–44 The conductivity dispersion phenomenon is described by Jonscher's universal power.45
| σac(ω) = σdc + Aωs | (3) |
Fig. 6 illustrated the ac conductivity of the NaCeP2O7 compound with frequency at different temperatures. In the low-frequency regions, a nearly constant conductivity is observed, where charge transport can be explained by a hopping model.46 In this region, ions jump from one site to the adjacent vacancy successfully resulting in a long-range transitional motion of ions contributing to the dc conductivity. In contrast, in the high-frequency region, a dispersive region attributed to ac conductivity is noteworthy, where the conductivity relaxation is mainly caused by the forward–backward hopping mechanism of the ions. Different scaling models have been proposed.47,48 Among them, we indicated Ghosh's model:
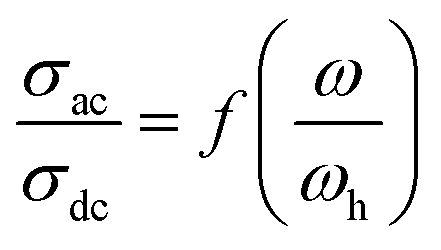 | (4) |
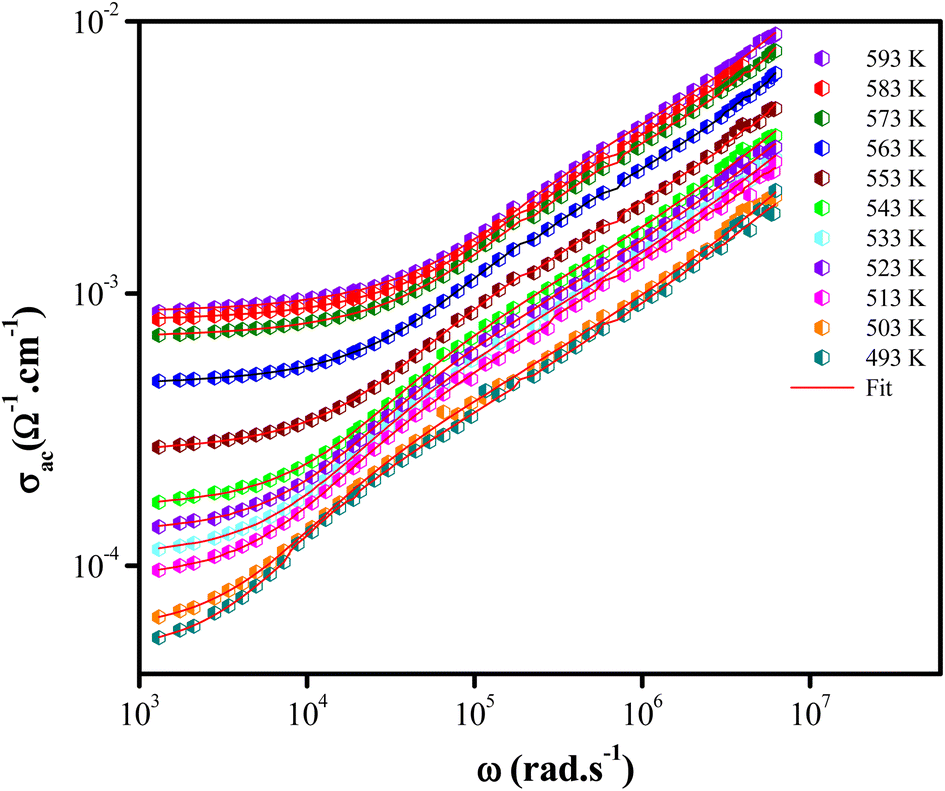 | ||
Fig. 6 Plots of conductivity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) σ) vs. frequency (logω) at different temperatures for NaCeP2O7. σ) vs. frequency (logω) at different temperatures for NaCeP2O7. | ||
The superposition of conductivity plots, of the NaCeP2O7 sample at different temperatures, is shown in Fig. 7. The conductivity spectra, presented in this way, merge on a single curve which implies that the relaxation dynamics of charge carriers are independent of temperature. The inset of Fig. 7 shows the temperature dependence of the hopping frequency, where an Arrhenius-type behavior is shown. The activation energy calculated from the slope of ln(ωh) versus 1000/T is found to be Ehop = 1.23 ± 0.05 eV. This value is different from that determined above from the relaxation mechanism and the dc-conductivity in the literature.
 | ||
| Fig. 7 Plot of (σac/σdc) versus (ω/ωh) at different temperatures. Inset: temperature dependence of the hopping frequency ωh. | ||
Four theoretical models have been developed to explain the frequency and temperature dependence of the AC conductivity and the exponent s(T). These models were based on relaxation and induced by the hopping or tunneling of electrons or ions between equilibrium sites.49 The behavior of the exponent “s” and the corresponding mechanisms have been discussed in several previous research papers.50 Accordingly, the correlated barrier hopping model “CBH” is predominant if “s” decreases with the rise of temperature. As for the quantum mechanical tunneling model “QMT”, “s” was reported to be very close to 0.8 and increases slightly with increasing temperature or can be independent of the temperature. In the overlapping large polaron tunneling model “OLPT ”, exponent “s” is frequency and temperature-dependent, where it decreases as the temperature increases to a minimum value at a given temperature and then increases with the increase of temperature. However, in the non-overlapping small polaron tunneling model “NSPT”, “s” is temperature-dependent and it increases with the increase of temperature.51
The temperature dependence of the s exponent is plotted in Fig. 8. The values are in the range of 0.72 to 0.86, where they increase with the increase in temperature. This result suggests that the non-overlapping small polaron tunneling (NSPT) model is a suitable model to characterize the electrical conduction mechanism in the NaCeP2O7 compound.52–54
 | ||
| Fig. 8 Variation of the frequency exponent s at different temperatures. | ||
To discover the temperature dependence of the electrical properties of the NaCeP2O7 compound, the ac conductivity is tacked out. It also outputs information about the conduct of the charge carriers.
The AC conductivity expression is given by:52–54
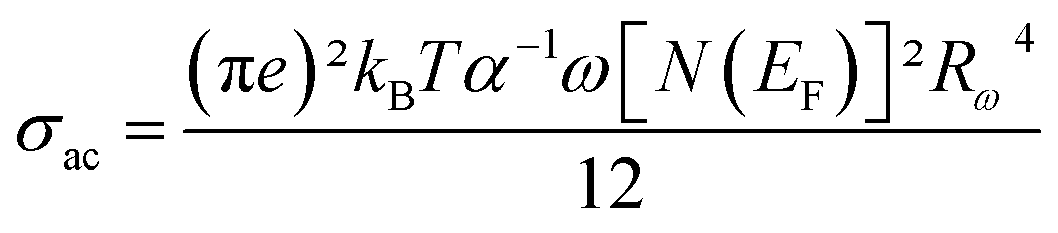 | (5) |
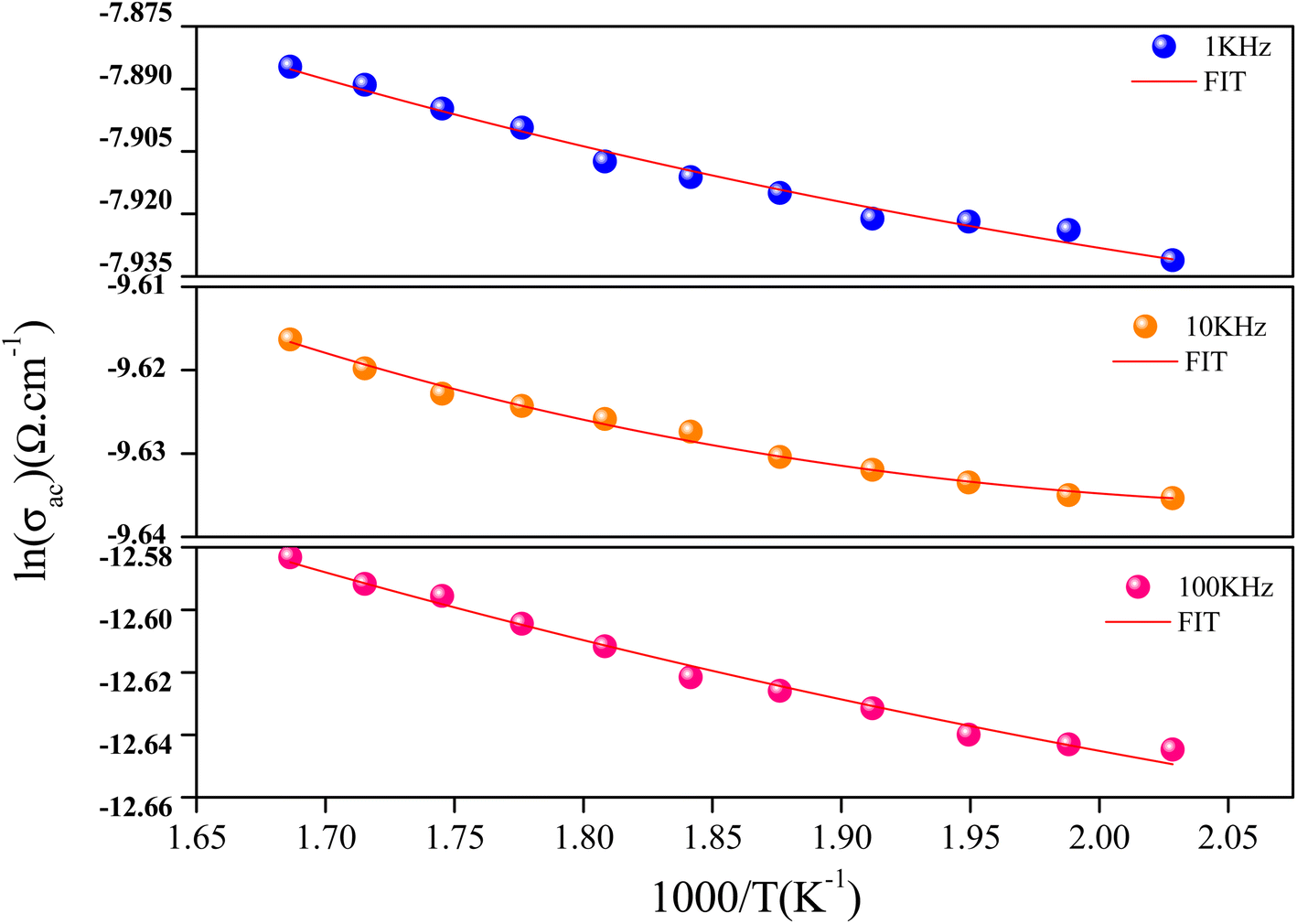 | ||
| Fig. 9 Temperature dependence of the ln(σac) at different frequencies. | ||
The values of the product of α−1 and N(EF) were adjusted to fit the calculated curves of ln(σac) versus T. The obtained values of α−1 and N(EF) are listed in Table 2.
| Frequency (kHz) | N (eV−1 m−1) (×1024) | α (A−1) |
|---|---|---|
| 1 | 0.268 | 0.920 |
| 10 | 0.623 | 1.202 |
| 100 | 0.771 | 1.370 |
| 1000 | 1.27 | 1.577 |
The values of the density of states N(EF) are reasonable for localized states. We observe the augmentation of both parameters α−1 and N(EF) with frequency, which is in good agreement with the literature.51,52
The variation of the tuning distance Rω, as a function of the frequency and temperature, is displayed in Fig. 10. The values of Rω are in the order of the inter-atomic spacing and these values vary in the range of (2.8 Å to 3.9 Å).
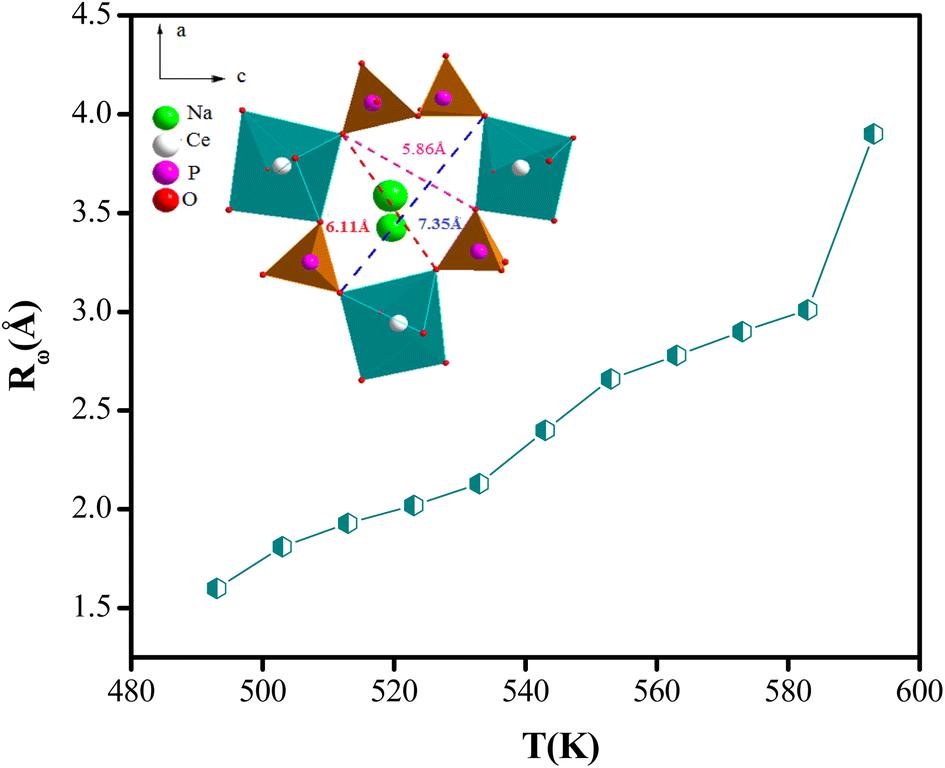 | ||
| Fig. 10 Variation of the Rω (Å) parameter according to the frequency (NSPT model). | ||
This fact can be explained by keeping in mind that at low and high temperatures, the structure of NaCeP2O7 was probably preserved and the conductivity was due to a single actuation of the conduction mechanism. Transport properties in this material appear to be due to the Na+ ions hopping mechanism.21 The Na+ mobility may occur through a one-dimensional channel along the b direction. Based on the atom positions, the Na+ within the chains is only 3.9 Å. The complex geometry of the (P2O7) group and CeO9 polyhedrons, makes the Na+ hopping difficult compared to intrachain mobility and therefore favors the transport of Na+ along a one-dimensional tunnel. Most likely, this feature wasn't favorable to good ionic conduction with the increase in temperature. Hence, we can deduce that the conduction transport properties, in this material, appear to be due to the Na+ ion's hopping mechanism along a one-dimensional tunnel.
In hopping transport, we can use the relation between the continuous conductivity σdc and the hopping frequency ωh:49
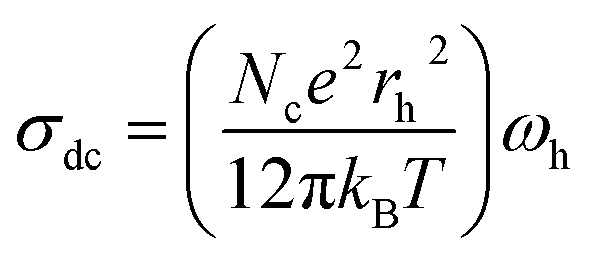 | (6) |
 | ||
| Fig. 11 Variation of the effective charge carrier Nc as a function of temperature. Inset: mobility μ for the NaCeP2O7 diphosphate. | ||
The charge carrier mobility μ can be obtained by involving eqn (7):
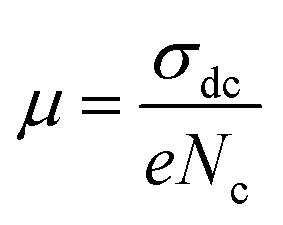 | (7) |
The temperature dependence of μ is exhibited in the inset of Fig. 11. An Arrhenius-type behavior is shown. The activation energy calculated from the slope of ln(μT) versus 1000/T (inset Fig. 11) is found to be Eμ = 1.29 ± 0.01 eV. This value is very close to that determined above from the hopping frequency (Ehopp = 1.23 ± 0.05 eV), implying that the mobility of the charge carrier is due to a hopping mechanism in the investigated material.
4. Conclusion
In the present work, we have carried out the synthesis of NaCeP2O7 using a solid-state reaction. X-ray analysis revealed that this material crystallizes in the Pnma space group of the orthorhombic symmetry. SEM images reveal the appearance of particles with a similar shape and an unequal size with a uniform distribution, where the majority of grains were found to be around 500 to 900 nm. All chemical elements Na, Ce, P, and O in the studied compound were mainly found in their appropriate ratios. As for the IR spectroscopy, the transmittance spectra recorded at room temperature have given information about the vibration modes of functional group P2O7.As for the impedance spectroscopy, the electrical study revealed that the activation energies, deduced from conductivity hopping frequency Ehop = 1.23 ± 0.05 eV and relaxation frequency Em= (1.34 ± 0.05), are close. Therefore, transport properties in this material appear to be due to Na+ ions' hopping mechanism along the b direction located between [P2O7]4− groups and CeO9 polyhedrons. In addition, the concentration of the charge carrier is independent of temperature. A comparison among the number of sites of Na+ ions and the number of effective charge carriers indicates that 56.34% of the Na+ is responsible for the conduction in this material.
The ac conductivity obeys the augmented Jonscher's power law at different temperatures. Scaling analysis of the conductivity provides the time–temperature superposition principle on the charge carrier dynamics in the material. The electrical conductivity of this sample has been theoretically supported by the NSPT model.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- H. Bih, I. Saadoune, L. Bih, M. Mansori, H. Toufik, H. Fuess and H. Ehrenberg, Synthesis, Rietveld refinements, infrared and Raman spectroscopy studies of the sodium diphosphate NaCryFe1-yP2O7 (0 ≤ y ≤ 1), J. Mol. Struct., 2016, 1103, 103–109, DOI:10.1016/j.molstruc.2015.09.014.
- M. Ajili, I. Gharbi and A. Oueslati, Synthesis, morphological, and ionic conduction studies of a copper potassium phosphate compound, Ionics, 2023, 29(1), 353–361, DOI:10.1007/s11581-022-04791-1.
- R. Ternane, M. Ferid, Y. Guyot, M. Trabelsi-Ayadi and G. Boulon, Spectroscopic properties of Yb3+ in NaYbP2O7 diphosphate single crystals, J. Alloys Compd., 2008, 464(1–2), 327–331, DOI:10.1016/j.jallcom.2007.09.104.
- S. M. V. Novais and Z. S. Macedo, Local atomic arrangement and scintillation properties of Eu- and Ce-doped NaYP2O7, J. Solid State Chem., 2016, 233(3), 103–107, DOI:10.1016/j.jssc.2015.10.017.
- R. Ternane, M. Ferid, Y. Guyot, M. Trabelsi-Ayadi and G. Boulon, Spectroscopic properties of Yb3+ in NaYbP2O7 diphosphate single crystals, J. Alloys Compd., 2008, 464(1–2), 327–331, DOI:10.1016/j.jallcom.2007.09.104.
- J. L. Yuan, J. Wang, Z. J. Zhang, J. T. Zhao and G. B. Zhang, Synthesis and luminescent properties of AGd1−xLnxP2O7 (A= Rb, Cs; Ln= Ce, Eu, Tb) under VUV and X-ray excitations, Opt. Mater., 2008, 30(8), 1327–1334, DOI:10.1016/j.optmat.2007.06.017.
- A. Akrim, D. Zambon and J. C. Cousseins, Optical properties of Tb3+ in the diphosphate CsYP2O7, J. Alloys Compd., 1994, 207, 99–101, DOI:10.1016/0925-8388(94)90186-4 –208.
- J. Zhu, W.-D. Cheng, D.-S. Wu, H. Zhang, Y.-J. Gong, H.-N. Tong and D. Zhao, Crystal and band structures, and optical characterizations of sodium rare earth phosphates NaLnP2O7 and NaLn(PO3)4 (Ln=Ce, Eu), J. Alloys Compd., 2008, 454(1–2), 419–426, DOI:10.1016/j.jallcom.2006.12.129.
- M. Daoud, D. Zambon, R. Mahiou, A. Ammar and B. Tanouti, Spectroscopic Properties of Trivalent Gadolinium in Diphosphate CsYP2O7, Mater. Res. Bull., 1998, 33(4), 597–603, DOI:10.1016/s0025-5408(98)00012-9.
- J. L. Yuan, X. J. Wang, D. B. Xiong, C. J. Duan, J. T. Zhao, Y. B. Fu, G. B. Zhang and C. S. Shi, VUV spectroscopic properties of Ce3+ and Pr3+-doped AREP2O7-type alkali rare earth diphosphates (A=Na, K, Rb, Cs; RE=Y, Lu), J. Lumin., 2007, 126(1), 130–134, DOI:10.1016/j.jlumin.2006.06.002.
- A. Boukhris, M. Hidouri, B. Glorieux and M. B. Amara, Na2BaMg(PO4)2: synthesis, crystal structure and europium photoluminescence properties, J. Rare Earths, 2013, 31(9), 849–856, DOI:10.1016/s1002-0721(12)60369-x.
- A. Boukhris, M. Hidouri, B. Glorieux and M. Ben Amara, Correlation between structure and photoluminescence of the europium doped glaserite-type phosphate Na2SrMg(PO4)2, Mater. Chem. Phys., 2012, 137(1), 26–33, DOI:10.1016/j.matchemphys.2012.07.002.
- A. Hamady, M. F. Zid and T. Jouini, Structure Cristalline de KYP2O7, J. Solid State Chem., 1994, 113(1), 120–124, DOI:10.1006/jssc.1994.1349.
- A. Hamady and T. Jouini, NaYP2O7, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52(12), 2949–2951, DOI:10.1107/s0108270196009626.
- A. Hamady, M. F. Zid and T. Jouini, Structure Cristalline de KYP2O7, J. Solid State Chem., 1994, 113(1), 120–124, DOI:10.1006/jssc.1994.1349.
- A. Jouini, J. C. Gâcon, M. Ferid and M. Trabelsi-Ayadi, Luminescence and scintillation properties of praseodymium poly and diphosphates, Opt. Mater., 2003, 24(1–2), 175–180, DOI:10.1016/s0925-3467(03)00122-8.
- M. Ferid, K. Horchani and J. Amami, Preparation, structure and infrared spectrum of NaEuP2O7, Mater. Res. Bull., 2004, 39(12), 1949–1955, DOI:10.1016/j.materresbull.2004.05.022.
- M. Férid, K. Horchani-Naifer and M. Trabelsi-Ayedi, Crystal structure of sodium ytterbium diphosphate, NaYbP2O7, Z. Kristallogr. – New Cryst. Struct., 2004, 219(1–4), 385–386, DOI:10.1524/ncrs.2004.219.14.385.
- A. Béjaoui, K. Horchani-Naifer and M. Férid, Ionic conduction, bond valence analysis of structure–property relationships of NaHoP2O7, J. Solid State Chem., 2013, 204, 224–232, DOI:10.1016/j.jssc.2013.05.032.
- M. Férid and K. Horchani-Naifer, Synthesis, crystal structure and vibrational spectra of a new form of diphosphate NaLaP2O7, Mater. Res. Bull., 2004, 39(14–15), 2209–2217, DOI:10.1016/j.materresbull.2004.08.007.
- K. Horchani-Naifer and M. FERID, Structure and ionic conductivity of NaCeP2O7, Solid State Ionics, 2005, 176(23–24), 1949–1953, DOI:10.1016/j.ssi.2005.05.015.
- J.-L. Yuan, H. Zhang, H.-H. Chen, X.-X. Yang, J.-T. Zhao and M. Gu, Synthesis, structure and X-ray excited luminescence of Ce3+-doped AREP2O7-type alkali rare earth diphosphates (A=Na, K, Rb, Cs; RE=Y, Lu), J. Solid State Chem., 2007, 180(12), 3381–3387, DOI:10.1016/j.jssc.2007.09.021.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32(5), 751–767, DOI:10.1107/S0567739476001551.
- I. Szczygieł, L. Macalik, E. Radomińska, T. Znamierowska, M. Maczka, P. Godlewska and J. Hanuza, Luminescence, electronic absorption and vibrational IR and Raman studies of binary and ternary cerium ortho-, pyro- and meta-phosphates doped with Pr3+ ions, Opt. Mater., 2007, 29(9), 1192–1205, DOI:10.1016/j.optmat.2006.04.015.
- S. Ben yahya and B. Louati, Vibrational analysis and AC electrical conduction behavior of lithium zinc orthogermanate, Ionics, 2021, 27(7), 3027–3034, DOI:10.1007/s11581-021-04061-6.
- P. Fischer and S. Roy, Characterizing magnetic skyrmions at their fundamental length and time scales, Magnetic Skyrmions and Their Applications, 2021, pp. 55–97, DOI:10.1016/b978-0-12-820815-1.00005-5.
- M. S. Khoshkhoo, S. Scudino, J. Thomas, K. B. Surreddi and J. Eckert, Grain and crystallite size evaluation of cryomilled pure copper, J. Alloys Compd., 2011, 509(suppl. 1), S343–S347, DOI:10.1016/j.jallcom.2011.02.066.
- L. T. Gibson, Archaeometry and Antique Analysis|Metallic and Ceramic Objects, Encyclopedia of Analytical Science, 2005, pp. 117–123, DOI:10.1016/b0-12-369397-7/00020-0.
- M. J. Kristo, Nuclear Forensics, Handbook of Radioactivity Analysis, 2012, pp. 1281–1304, DOI:10.1016/b978-0-12-384873-4.00021-9.
- Y.-W. Hu, I. D. Raistrick and R. A. Huggins, Ionic conductivity of lithium phosphate-doped lithium orthosilicate, Mater. Res. Bull., 1976, 11(10), 1227–1230, DOI:10.1016/0025-5408(76)90025-8.
- D. Morgan, A. Van der Ven and G. Ceder, Li Conductivity in LixMPO4 (M = Mn, Fe, Co, Ni) Olivine Materials, Electrochem. Solid-State Lett., 2004, 7(2), A30, DOI:10.1149/1.1633511.
- R. Ben Said, B. Louati and K. Guidara, Theoretical and experimental study of ac electrical conduction mechanisms of KZn1.5P2O7 compound, J. Alloys Compd., 2016, 672, 521–528, DOI:10.1016/j.jallcom.2016.02.162.
- I. Kosacki and H. U. Anderson, Grain Boundary Effects in Nanocrystalline Mixed Conducting Films, Encyclopedia of Materials: Science and Technology, 2001, pp. 3609–3617, DOI:10.1016/b0-08-043152-6/00642-2.
- R. A. M. Hikmet, Organic Electrolytes and Electrodes for Batteries, Encyclopedia of Materials: Science and Technology, 2001, pp. 6534–6545, DOI:10.1016/b0-08-043152-6/01155-4.
- C. Iwakura, H. Inoue and S. Nohara, Hydrogen–Metal Systems: Electrochemical Reactions (Fundamentals and Applications), Encyclopedia of Materials: Science and Technology, 2001, pp. 3923–3941, DOI:10.1016/b0-08-043152-6/00695-1.
- J. H. Ambrus, C. T. Moynihan and P. B. Macedo, Conductivity relaxation in a concentrated aqueous electrolyte solution, J. Phys. Chem., 1972, 76(22), 3287–3295, DOI:10.1021/j100666a030.
- K. Abdouli, W. Cherif, E. Kadri, K. Dhahri, P. R. Prezas, M. P. F. Graça, M. A. Valent and L. Ktari, Structural, electric, and dielectric characterizations of La0.5Pr0.2Sr0.3Mn1−xFexO3 perovskite prepared by self combustion method, J. Alloys Compd., 2018, 739, 1048–1058, DOI:10.1016/j.jallcom.2017.12.269.
- A. Dhahri, E. Dhahri and E. K. Hlil, Electrical conductivity and dielectric behaviour of nanocrystalline La0.6Gd0.1Sr0.3Mn0.75Si0.25O3, RSC Adv., 2018, 8(17), 9103–9111, 10.1039/c8ra00037a.
- J. Liu, C.-G. Duan, W.-G. Yin, W. N. Mei, R. W. Smith and J. R. Hardy, Dielectric permittivity and electric modulus in Bi2Ti4O11, J. Chem. Phys., 2003, 119(5), 2812–2819, DOI:10.1063/1.1587685.
- V. Thakur, A. Singh, A. M. Awasthi and L. Singh, Temperature dependent electrical transport characteristics of BaTiO3 modified lithium borate glasses, AIP Adv., 2015, 5(8), 087110, DOI:10.1063/1.4928339.
- R. Bergman, General susceptibility functions for relaxations in disordered systems, J. Appl. Phys., 2000, 88(3), 1356–1365, DOI:10.1063/1.373824.
- D. P. Almond and A. R. West, Anomalous conductivity prefactors in fast ion conductors, Nature, 1983, 306(5942), 456–457, DOI:10.1038/306456a0.
- P. Muralidharan, N. Nallamuthu, I. Prakash, N. Satyanarayana and M. Venkateswarlu, AC Conductivity and Electrical Modulus Studies on Lithium Vanadophosphate Glasses, J. Am. Ceram. Soc., 2007, 90(1), 125–131, DOI:10.1111/j.1551-2916.2006.01355.x.
- S. Selvasekarapandian and M. Vijayakumar, The ac impedance spectroscopy studies on LiDyO2, Mater. Chem. Phys., 2003, 80(1), 29–33, DOI:10.1016/s0254-0584(02)00510-2.
- A. K. Jonscher, Nature, 1977, 267, 673–679 CrossRef CAS.
- R. M. Hill and A. K. Jonscher, DC and AC conductivity in hopping electronic systems, J. Non-Cryst. Solids, 1979, 32(1–3), 53–69, DOI:10.1016/0022-3093(79)90064-4.
- B. Roling, A. Happe, K. Funke and M. D. Ingram, Carrier Concentrations and Relaxation Spectroscopy: New Information from Scaling Properties of Conductivity Spectra in Ionically Conducting Glasses, Phys. Rev. Lett., 1997, 78(11), 2160–2163, DOI:10.1103/physrevlett.78.2160.
- B. Louati, F. Hlel and K. Guidara, Ac electrical properties and dielectric relaxation of the new mixed crystal (Na0.8Ag0.2)2PbP2O7, J. Alloys Compd., 2009, 486(1–2), 299–303, DOI:10.1016/j.jallcom.2009.06.148.
- S. R. Elliott, A. c. conduction in amorphous chalcogenide and pnictide semiconductors, Adv. Phys., 1987, 36(2), 135–217, DOI:10.1080/00018738700101971.
- M. Ben Bechir, K. Karoui, M. Tabellout, K. Guidara and A. Ben Rhaiem, Alternative current conduction mechanisms of organic-inorganic compound [N(CH3)3H]2ZnCl4, J. Appl. Phys., 2014, 115, 203712, DOI:10.1063/1.4880735.
- I. Gharbi, M. H. Dhaou, A. Oueslati and M. Gargouri, Synthesis and investigation on the morphological, structural, and electrical proprieties of a cesium magnesium phosphate prepared using solid-state reaction, J. Mater. Sci.: Mater. Electron., 2023, 34, 872, DOI:10.1007/s10854-023-10288-y.
- J. T. Gudmundsson, H. G. Svavarsson, S. Gudjonsson and H. P. Gislason, Frequency-dependent conductivity in lithium-diffused and annealed GaAs, Phys. B, 2003, 340–342, 324–328, DOI:10.1016/j.physb.2003.09.082.
- A. Kahouli, A. Sylvestre, F. Jomni, B. Yangui and J. Legrand, Experimental and Theoretical Study of AC Electrical Conduction Mechanisms of Semicrystalline Parylene C Thin Films, J. Phys. Chem. A, 2012, 116(3), 1051–1058, DOI:10.1021/jp207114u.
- R. Ben Said, B. Louati and K. Guidara, Theoretical and experimental study of ac electrical conduction mechanisms of KZn1.5P2O7 compound, J. Alloys Compd., 2016, 672, 521–528, DOI:10.1016/j.jallcom.2016.02.162.
| This journal is © The Royal Society of Chemistry 2023 |