
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional activity-based chemical probes for sirtuins†
Chiranjeev Sharmaa,
Dickson Donua,
Alyson M. Currya,
Elizabeth Bartona and
Yana Cen *ab
*ab
aDepartment of Medicinal Chemistry, Virginia Commonwealth University, Richmond, VA 23219, USA. E-mail: ceny2@vcu.edu; Tel: +1-804-828-7405
bInstitute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA 23219, USA
First published on 14th April 2023
Abstract
The sirtuin family of NAD+-dependent protein deacylases has gained significant attention during the last two decades, owing to their unique enzymatic activities as well as their critical roles in a broad array of cellular events. Innovative chemical probes are heavily pursued for the functional annotation and pharmacological perturbation of this group of “eraser” enzymes. We have developed several series of activity-based chemical probes (ABPs) to interrogate the functional state of active sirtuins in complex biological samples. They feature a simple Ala–Ala–Lys tripeptide backbone with a thioacyl “warhead”, a photoaffinity group (benzophenone or diazirine), and a bioorthogonal group (terminal alkyne or azido) for conjugation to reporters. When applied in a comparative fashion, these probes reveal the changes of active sirtuin contents under different physiological conditions. Additionally, they can also be utilized in a competitive manner for inhibitor discovery. The Nobel-winning “click” conjugation to a fluorophore allows the visualization of the active enzymes, while the covalent adduct to a biotin leads to the affinity capture of the protein of interest. Furthermore, the “clickable” tag enables the easy access to proteolysis targeting chimeras (PROTACs) that effectively degrade human SIRT2 in HEK293 cells, albeit at micromolar concentrations. These small molecule probes offer unprecedented opportunities to investigate the biological functions and physiological relevance of the sirtuin family.
Introduction
The NAD+-dependent protein deacylases, known as sirtuins, were initially identified as histone deacetylases (HDACs).1 The removal of acetyl group from lysine residues causes chromatin condensation, leading to transcriptional silencing. In addition to histone proteins, other cellular enzymes and proteins can also be targeted by sirtuins as endogenous substrates,2,3 many of which are involved in carbon fuel metabolism,4 cell cycle regulation,5 mitochondrial biogenesis,6 and genome integrity.7 Recent studies reveal the pleiotropic enzymatic activities of sirtuins: they remove propionyl,8 crotonyl,9 butyryl,8,9 succinyl,10 malonyl,10,11 glutaryl,12 and long chain fatty acyl groups13,14 from lysines of diverse cellular targets.From a mechanistic perspective, sirtuins exert their biological functions through a unique chemical process (Scheme 1). It starts with the cleavage of the N-glycosidic bond of NAD+ to form an imidate intermediate.15,16 Subsequently, the nucleophilic attack of the imidate by the 2′-OH group leads to the formation of a bicyclic intermediate. Ultimately, the collapse of the bicyclic intermediate results in the deacylated lysine product. In the meantime, the acyl group is transferred to the ribose moiety to generate a novel metabolite called acyl-ADP-ribose (AADPR). The imidate can either proceed forward for deacylation, or combine with nicotinamide (NAM) to regenerate NAD+, the latter is often referred as a “base exchange” reaction.17 Interestingly, when a thioacyllysine is introduced (replacing the carbonyl oxygen with a sulfur), the formation of the thioimidate becomes essentially irreversible, and it undergoes extremely slow forward deacylation, causing mechanism-based inhibition.18–20
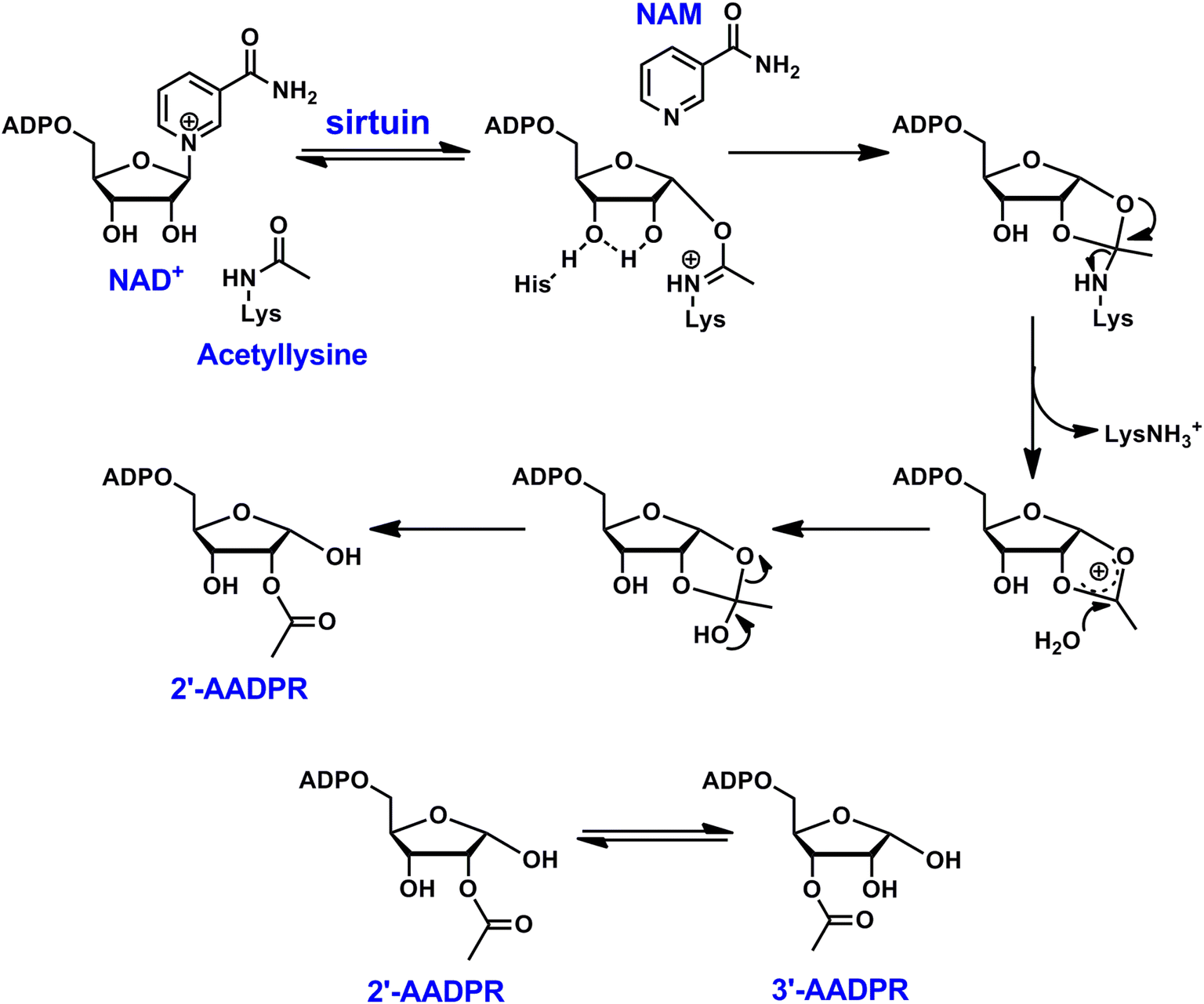 | ||
| Scheme 1 Reaction scheme of sirtuin-catalyzed NAD+-dependent deacetylation. In aqueous solutions, 2′-AADPR and 3′-AADPR equilibrate rapidly. | ||
We took advantage of the distinct reactivity of thioacyllysines to develop several series of activity-based chemical probes (ABPs) for human sirtuins.21–23 These probes are Ala–Ala–Lys tripeptide featuring a thioacyl “warhead”. A benzophenone or diazirine photoaffinity group is included for the covalent tethering of the ABP with the enzyme target upon UV irradiation. Additionally, a terminal alkyne or an azido group is incorporated for the subsequent bioorthogonal conjugation of the enzyme–probe complex to fluorescent or affinity reporters. These ABPs allow the visualization or affinity capture of active sirtuins in their native environment. When used in a comparative manner, they report the dynamic change of sirtuin activity in response to various cellular stimuli. When applied in a competitive fashion, they provide access to selective sirtuin inhibitors. These ABPs can be further elaborated to PROTACs via “click” conjugation to an E3 ligase ligand. The treatment of HEK293 cells with a representative PROTAC causes effective degradation of SIRT2. These multifaceted small molecules should empower the functional annotation and pharmacological perturbation of these “eraser” enzymes.
Methods and materials
Reagents and instruments
All reagents were purchased from Aldrich or Fisher Scientific and were of the highest purity commercially available. UV spectra were obtained with a Varian Cary 300 Bio UV-visible spectrophotometer. HPLC was performed on a Dionex Ultimate 3000 HPLC system equipped with a diode array detector using Macherey-Nagel C18 reverse-phase column. NMR spectra were acquired on a Bruker AVANCE III 400 MHz high-field NMR spectrometer and the data were processed using Topspin software. HRMS spectra were acquired with either a Waters Micromass Q-tof Ultima or a Thermo Scientific Q-Exactive hybrid Quadrupole Orbitrap. Fluorescence scanning was performed on a Biorad Versa Doc 4000 MP Imaging System or a Biorad ChemiDoc MP imaging system.Synthetic peptides
Synthetic peptides H3K9Ac: ARTKQTAR(K-Ac)STGGKAPRKQLAS, p53K382Ac: KKGQSTSRHK(K-Ac)LMFKTEG, H3K9Suc: ARTKQTAR(K-Suc)STGGKAPRKQLA were synthesized and purified by Genscript. The peptides were purified by HPLC to a purity >95%.Protein expression and purification
Plasmids of SIRT1 (full length), SIRT2 (38–356), SIRT3 (102–399), SIRT5 (34–302) and SIRT6 (1–314) were generous gifts from Dr Hening Lin (Cornell University). The proteins were expressed and purified according to previously published protocols.24 PET11a-Af2Sir2 (Addgene plasmid # 8383) was a gift from Dr Cynthia Wolberger (Johns Hopkins University). The protein was expressed and purified as described before.25 The identity of the protein was confirmed by tryptic digestion followed by LC-MS/MS analysis performed at the Vermont Biomedical Research Network (VBRN) Proteomics Facility. Protein concentrations were determined by Bradford assay.Sirtuin inhibition assay
A typical reaction contained 500 μM NAD+, 500 μM peptide substrate (H3K9Ac for SIRT2, SIRT3 and SIRT6, p53K382Ac for SIRT1 and SIRT5), varying concentrations of small molecule probe in 100 mM phosphate buffer pH 7.5. The reactions were initiated by the addition of 10 μM of sirtuin and were incubated at 37 °C before being quenched by 8 μL of 10% TFA. The incubation time was controlled to achieve less than 15% substrate conversion. The samples were then injected on an HPLC fitted to a Macherey-Nagel Nucleosil C18 column. NAD+, NAM, and AADPR peaks were resolved using a gradient of 0 to 20% methanol in 20 mM ammonium acetate. Chromatograms were analyzed at 260 nm. Reactions were quantified by integrating areas of peaks corresponding to NAD+ and AADPR. Rates were plotted as a function of small molecule probe concentration, and points were fitted to the following equation:| ν(%) = ν0(%) − [ν0(%)(10x)/(10x + IC50)] |
Labeling of recombinant sirtuin
A typical labeling experiment was performed as follows: in a 0.7 mL Eppendorf tube, purified recombinant human sirtuin (10 μM) was incubated with NAD+ (500 μM) and activity-based probe at 37 °C for 10 min. The sample was transferred to a clear-bottom 96-well plate, placed on ice, and irradiated at 365 nm with a UV-lamp in a cold room. Subsequently, ingredients of the “click-chemistry” including TAMRA azide, CuSO4, reducing agent (tris(2-carboxyethyl)phosphine, TCEP) and stabilizing agent (tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, TBTA) were added, and the sample was gently agitated at 250 rpm on a microshaker at room temperature for 30 min. Then, the sample was resolved by SDS-PAGE. To reduce the signal to noise ratio, the gel was destained to eliminate non-specific binding of free dyes. This was done in a mixture of methanol/distilled water/acetic acid (v/v/v = 4/5/1) at ambient temperature for 4 h. The destained gel was analyzed with in-gel fluorescence scanning using a Biorad ChemiDoc MP imager (excitation 532 nm, 580 nm cut-off filter and 30 nm band-pass). Finally, Coomassie blue staining was applied to provide loading control.Cell culture
HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin and 100 mg mL−1 streptomycin. Cells were maintained in a humidified 37 °C incubator with 5% CO2.Metabolic labeling
Cells were treated with 20 μM azido myristic acid for 18 h. Cells treated with DMSO were used as controls. Cells were harvested, re-suspended in PBS, and pelleted at 1000 g for 5 min at 4 °C. The cell pellet was dissolved in lysis buffer (RIPA buffer, Thermo Scientific) with halt protease phosphatase inhibitor cocktail (Thermo Scientific). The lysate was centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 5 min at 4 °C. The supernatant was used for the de-fatty acylation analysis.
000 g for 5 min at 4 °C. The supernatant was used for the de-fatty acylation analysis.
De-fatty acylation assay
Cell lysate was incubated with 500 μM NAD+, 10 μM SIRT6 with or without 20 μM probe 2B at 37 °C for 1 h. The samples were then incubated with 15 mM iodoacetamide at room temperature for 30 min before TAMRA-DBCO was introduced. The samples were incubated at room temperature for another 30 min. Finally, the samples were heated with NH2OH (60 mM, pH 7.2) at 95 °C for 7 min. Subsequently, the samples were resolved by SDS-PAGE. The destained gel was analyzed with in-gel fluorescence scanning using a Biorad ChemiDoc MP imager (excitation 532 nm, 580 nm cut-off filter and 30 nm band-pass).Competitive assay for inhibitor discovery
A typical reaction contained 500 μM NAD+, 10 μM SIRT6, and the candidate compound at the indicated concentration in 100 mM phosphate buffer pH 7.5. The samples were incubated at 37 °C for 30 min, followed by the addition of probe 1B (25 μM) and labeling as described above. The samples were resolved by SDS-PAGE and analyzed with in-gel fluorescence scanning using a Biorad ChemiDoc MP imager (excitation 532 nm, 580 nm cut-off filter and 30 nm band-pass).Detection of SIRT2 degradation
Cells were incubated with the vehicle (DMSO) or increasing concentrations of PRO-SIRT2 for 12 h. For the rescue experiment with MG132, cells were treated with 10 μM MG132 for 2 h, followed by the co-treatment of MG132 and PRO-SIRT2 for 12 h. Cells were harvested and lysed with lysis buffer (50 mM Tris–HCl, pH 7.5, 300 mM NaCl, 1% Triton X-100) supplemented with protease inhibitor cocktail (Thermo Scientific). Protein concentration was determined by Bradford assay. SIRT2 levels were detected by western blot using an anti-SIRT2 antibody (Cell Signaling Technology). α-Tubulin was used as a loading control.Overexpression of SIRT5 in HEK293 cells
SIRT5 Flag was a gift from Eric Verdin (Addgene plasmid #13816).26 The vector was transfected into HEK293 cells with lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer's protocol. Overexpression efficiency was determined via western blot.Mitochondria fractionation
Mitochondria from HEK293 cells overexpressing human SIRT5 were isolated using the mitochondrial isolation kit for cultured cells (Thermo Scientific) following the manufacturer's protocol. Protein concentration was determined by Bradford assay.Sirtuin enrichment in cell lysate
Mitochondrial fraction (20 μg) was incubated with or without 10 μM probe 2C. After the UV irradiation, the sample was conjugated to cleavable diazo biotin azide (Click Chemistry Tools). The biotinylated proteins were captured by high capacity streptavidin beads (Thermo Fisher Scientific). The captured protein can be eluted by incubation with 25 mM of Na2S2O4, 250 mM of NH4HCO3 and 0.05% SDS for 1 h. The eluent was concentrated by lyophilization and analyzed by western blot using an anti-SIRT5 antibody (Cell Signaling Technology).Western blot
The samples were resolved on a 10% SDS-PAGE gel and transferred to the immun-blot PVDF membrane (Biorad). The blot was blocked with 5% nonfat milk in TBST, probed with the primary antibody, washed with TBST, followed by incubation with the anti-rabbit HRP conjugated secondary antibody. The signal was then detected by Clarity™ western ECL substrate (Biorad).Results and discussion
Design of three generations of ABPs
Several groups have developed thioacyllysine peptides as sirtuin inhibitors.18,20,27,28 These peptides normally contain 5 to 20 amino acids (AAs). The sequences are based on the known sirtuin substrates. We took a “less is more” strategy to engineer a simple Ala–Ala–Lys tripeptide as the backbone.21–23 The epsilon-amino of the lysine residue was functionalized with a thioacyl group. The first generation ABPs harbor a benzophenone photoaffinity group for its synthetic easiness (Fig. 1).21 To improve the labeling efficiency and sensitivity, the benzophenone group was replaced by a diazirine photocrosslinkable group in the second generation ABPs (Fig. 1).22 These two series also carry the terminal alkyne to enable Cu(I)-catalyzed “click” conjugation to a reporter. Most recently, we reported the synthesis and characterization of the third generation ABPs featuring an azido group (Fig. 1).23 These biocompatible probes not only demonstrate isoform-selective labeling of an individual sirtuin isoform, but also allow the Cu-free conjugation to a fluorophore or a biotin. In the following sections, the utility of these ABPs will be further illustrated.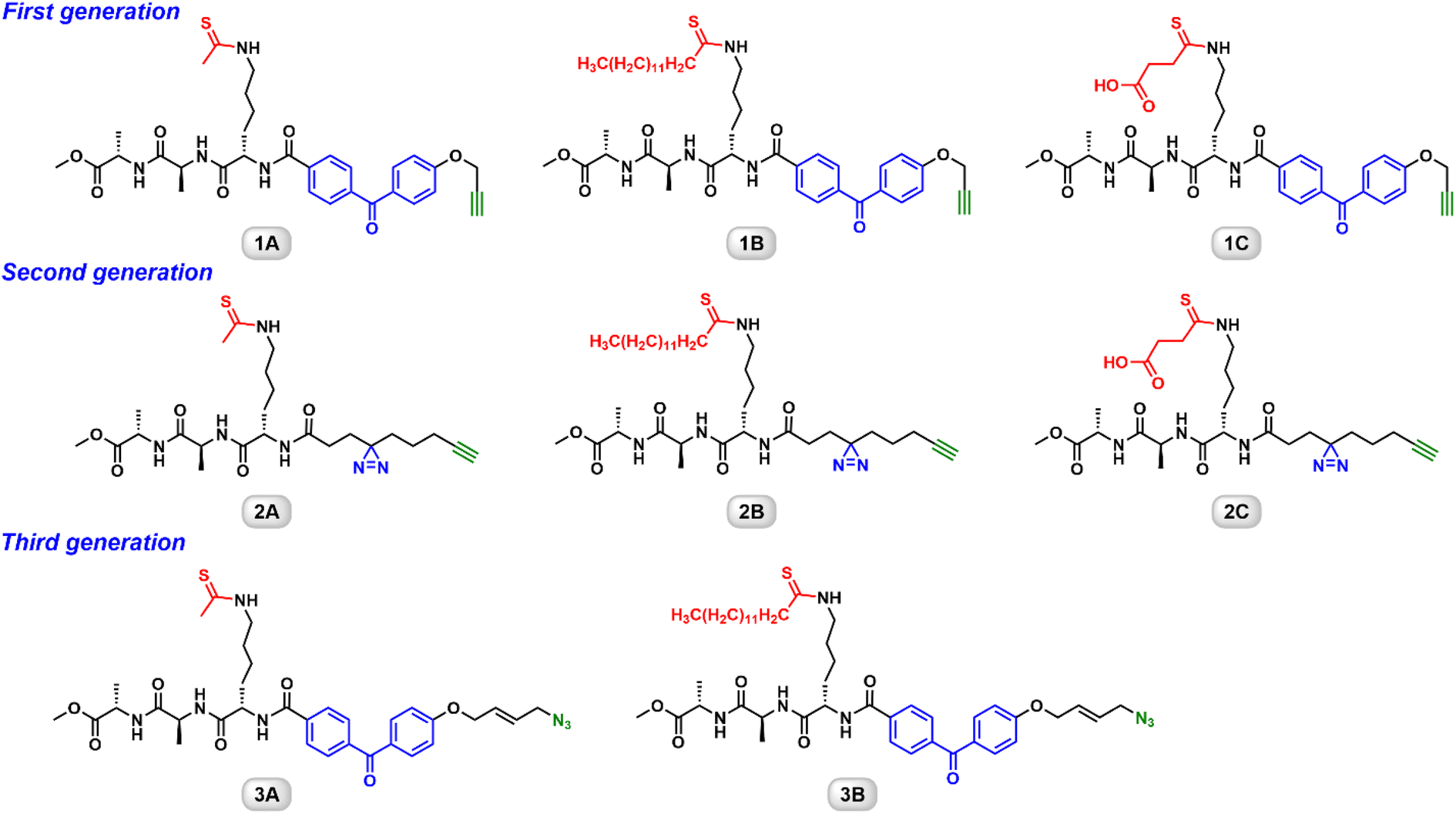 | ||
| Fig. 1 Chemical structures of the three generation ABPs. | ||
Thioacyllysine-based ABPs are selective and cell permeable inhibitors of human sirtuins
In our initial studies, the ABPs were evaluated using an HPLC-based biochemical assay for the respective recombinant sirtuin, where some of the probes exhibited on-target potency with IC50 values in the low micromolar range (Table 1). The probes were further assessed in cellular assays to demonstrate their ability to increase the acetylation level of endogenous sirtuin substrates. For example, probe 1A, when combined with the class I and class II HDAC inhibitor trichostatin A (TSA), was able to increase the acetylation level of p53K382, a physiological substrate of SIRT1.21| Sirtuin | Substrate | IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1A | 1B | 1C | 2A | 2B | 2C | 3A | 3B | ||
| a IC50 values were determined by HPLC assay as described in “Materials and methods”.b Not inhibited. | |||||||||
| SIRT1 | p53K382Ac | 39 ± 2.8 | >1000 | >5000 | 11.4 ± 2.7 | 79.9 ± 7.6 | >2000 | NI | NI |
| SIRT2 | H3K9Ac | 7 ± 0.5 | 21.8 ± 4.0 | >5000 | 17.2 ± 3.3 | 7.4 ± 1.3 | >5000 | 2.98 ± 0.04 | 9.14 ± 0.98 |
| SIRT3 | H3K9Ac | 166 ± 23 | >5000 | >5000 | 77.9 ± 5.4 | >2000 | >5000 | 35.9 ± 5.82 | 303.1 ± 52.5 |
| SIRT5 | p53K382Ac | NIb | >5000 | 3.2 ± 0.4 | NI | >5000 | 1.9 ± 0.6 | NI | NI |
| SIRT6 | H3K9Ac | NI | 7.8 ± 1.1 | >5000 | 151.2 ± 21.9 | 13.7 ± 3.2 | >5000 | NI | 618.7 ± 49 |
SIRT6 is known to be a robust de-fatty acylase.29 This novel enzymatic activity gives SIRT6 its far-reaching functions in genomic stability through the regulation of DNA repair and telomere integrity, and in metabolism through the control of glucose and lipid homeostasis.30 It is of great interest to explore whether the ABPs can influence lysine fatty-acylation via SIRT6 inhibition, especially in a physiologically relevant setting. Pan and specific lysine fatty acylation antibodies are not readily available. Instead, a metabolic labeling strategy using azido myristic acid was employed.31 HEK293 cells were cultured in the presence of azido myristic acid, leading to fatty-acylation of cellular proteins (Fig. 2A). The cell lysate was treated with recombinant SIRT6 with or without probe 2B. Subsequently, the labeled proteins were conjugated to TAMRA-DBCO via strain-promoted “click” reaction. NH2OH was then used to remove Cys acylation. The Lys acylated proteins were resolved by SDS-PAGE and visualized by in-gel fluorescence scanning (Fig. 2B). The lysate from azido myristic acid-treated cells had significantly increased labeling compared to the control cells. Incubation of the lysate with SIRT6 caused the reduction of fluorescence signals of several protein bands, consistent with the previous report that SIRT6 is a lysine defatty-acylase.29 Additionally, the incubation of lysate with SIRT6 in the presence of probe 2B increased the labeling intensity of numerous proteins, suggesting that SIRT6 regulates the fatty-acylation level of cellular target and this function can be inhibited by probe 2B.
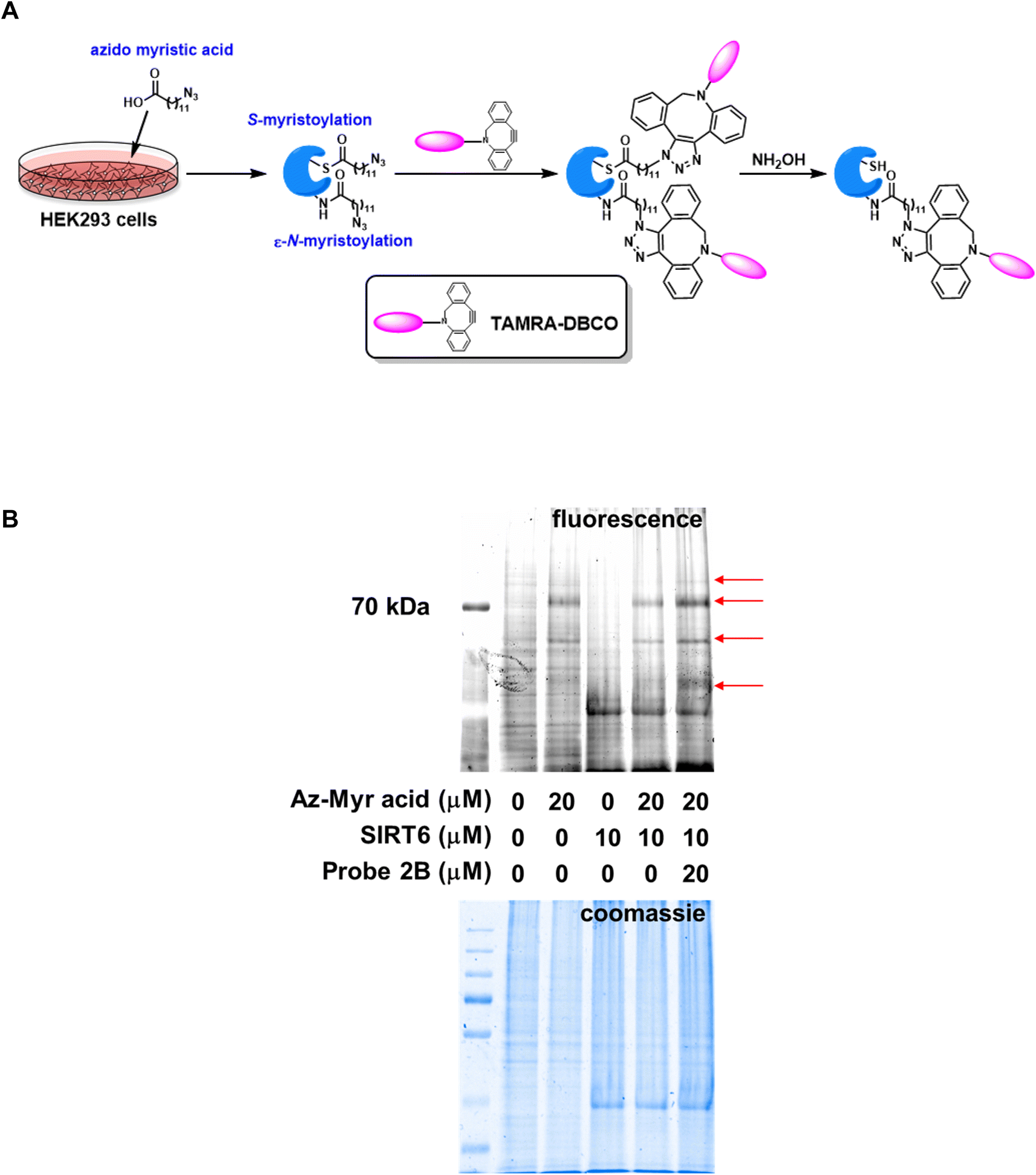 | ||
| Fig. 2 SIRT6 regulates the fatty acylation of cellular proteins. (A). Schematic representation of the fatty acylation labeling protocol using azido myrsitic acid; (B). Profiling of fatty acylated proteins in HEK293 cell lysates. The lysate from azido myristic acid-treated cells had significantly increased labeling compared to the control cells. Incubation of the lysate with SIRT6 in the presence of probe 2B increased the labeling intensity of cellular proteins. Arrows highlight proteins with elevated fluorescence due to probe 2B exposure. | ||
ABPs can be used in a competitive manner for inhibitor discovery
When the ABPs are used in a comparative fashion, the functional states of enzymes are closely associated with distinct physiological or pathophysiological conditions.32,33 Furthermore, they can be adapted for inhibitor discovery in a competitive manner.34,35 This type of study is normally performed in a parallel fashion in multi-well plates (Fig. 3A). To each of the wells, same concentrations of target protein and identical buffer condition are introduced. Subsequently, a different inhibitor candidate is added to each well, followed by the addition of an ABP. There are two possible scenarios: if the candidate compound is indeed an inhibitor, it will block the active site of the enzyme to prevent the ABP from interacting with the target, leading to reduced or complete loss of labeling (Fig. 3A, top). Alternatively, the candidate compound may not be an inhibitor for the protein target. If so, the active site will remain available for the normal labeling by the ABP (Fig. 3A, bottom). This method can be applied to cell lysates, so there is no need to recombinantly express and purify the protein target. It does not require a substrate, which renders this approach suitable for enzymes without known substrates. | ||
| Fig. 3 Competitive application of ABPs for inhibitor discovery. (A). Schematic representation of the competitive screening assay; (B). Representative gel images of SIRT6 inhibitor screening. SIRT6 was incubated with 100 μM candidate compound or DMSO (vehicle) for 30 min before the addition of 25 μM probe 1B. The samples were then labeled as described in “Methods and materials” and analyzed on gel; (C). Representative gel images of SIRT2 inhibitor screening. Partially purified SIRT1, SIRT2, SIRT3, SIRT5, and SIRT6 were mixed together and incubated with 100 μM candidate compound before the addition of probe 3B. The samples were labeled as described in “Methods and materials”. | ||
In an effort to search for small molecule SIRT6 inhibitors, a focused library of epigenetic inhibitors were screened using probe 1B competitively (Table S1, Fig. S1†). As shown in Fig. 3B, the addition of candidate compound 1 markedly reduced the labeling intensity of SIRT6 by probe 1B, suggesting that compound 1 may be a SIRT6 inhibitor. Compound 2, on the contrary, failed to alter the labeling intensity to any appreciable levels. The compounds scored favorably in the initial screening were further characterized using HPLC-based assay. Compound 1 turned out to be a selective SIRT6 inhibitor with an IC50 value of 2.6 ± 0.3 μM (Table 2, Fig. S2†). It showed no apparent inhibition towards other human sirtuins or yeast Sir2 at up to 50 μM concentration. This compound also inhibited Archaeoglobus fulgidus Sir2 (Af2Sir2), an archeal sirtuin, with an IC50 value of 55.6 ± 3.8 μM (Fig. S2†), 21-fold higher than that of SIRT6. Compound 1 is trichostatin A (TSA), a molecule that has been shown to selectively inhibit SIRT6 in the mammalian sirtuin family.36,37 Our previous study has indicated that SIRT6 deacetylase activity can be inhibited by TSA for both synthetic peptides and endogenous physiological substrates.36
| Sirtuin | Substrate | IC50 (μM) |
|---|---|---|
| a IC50 values were determined by HPLC assay as described in “Materials and methods”. | ||
| SIRT6 | H3K9Ac | 2.6 ± 0.3 |
| SIRT1 | p53K382Ac | >50 |
| SIRT2 | H3K9Ac | >50 |
| SIRT3 | H3K9Ac | >50 |
| SIRT5 | p53K382Ac | >50 |
| Yeast Sir2 | H3K9Ac | >50 |
| Af2Sir2 | H3K9Ac | 55.6 ± 3.8 |
The power of this competitive approach was further demonstrated in the screen of SIRT2 inhibitors. Probe 3B is a selective SIRT2 inhibitor with an IC50 value of 9.14 ± 0.98 μM.23 This ABP was applied to a mixture of partially purified recombinant sirtuins including SIRT1, SIRT2, SIRT3, SIRT5, and SIRT6. SIRT2 was the only isoform that can be labeled (Fig. 3C), and this labeling was outcompeted by thiomyristoyl (candidate compound 8), a known SIRT2 inhibitor.38 Collectively, these results suggest that competitive ABP application offers a versatile and substrate-free strategy for identifying inhibitors of individual enzyme isoform.
Thioacyllysine-based ABPs offer easy access to sirtuin-targeting PROTACs
The last few decades have witnessed remarkable advances in discovering disease-relevant protein targets and translating these discoveries into therapeutic benefits. Unfortunately, as much as 85% of the human genome by some estimate is still considered “undruggable” due to lack of functional and ligandable “hotspots” that small molecules can bind to manipulate protein function and activity.39 Beyond the classical host-substrate paradigm driven by drug occupancy parameters, new modes of action were also developed to modulate protein homeostasis such as targeted protein degradation through proteolysis targeting chimeras (PROTACs).40 PROTACs are heterobifunctional small molecules consisting of a binding ligand for a protein of interest (POI) and an E3 ligase-recruiting ligand that are connected through a chemical linker (Fig. 4A). Binding of a PROTAC to its target protein brings an E3 ligase in close proximity to initiate polyubiquitination of the POI ensuring its proteasome-mediated degradation.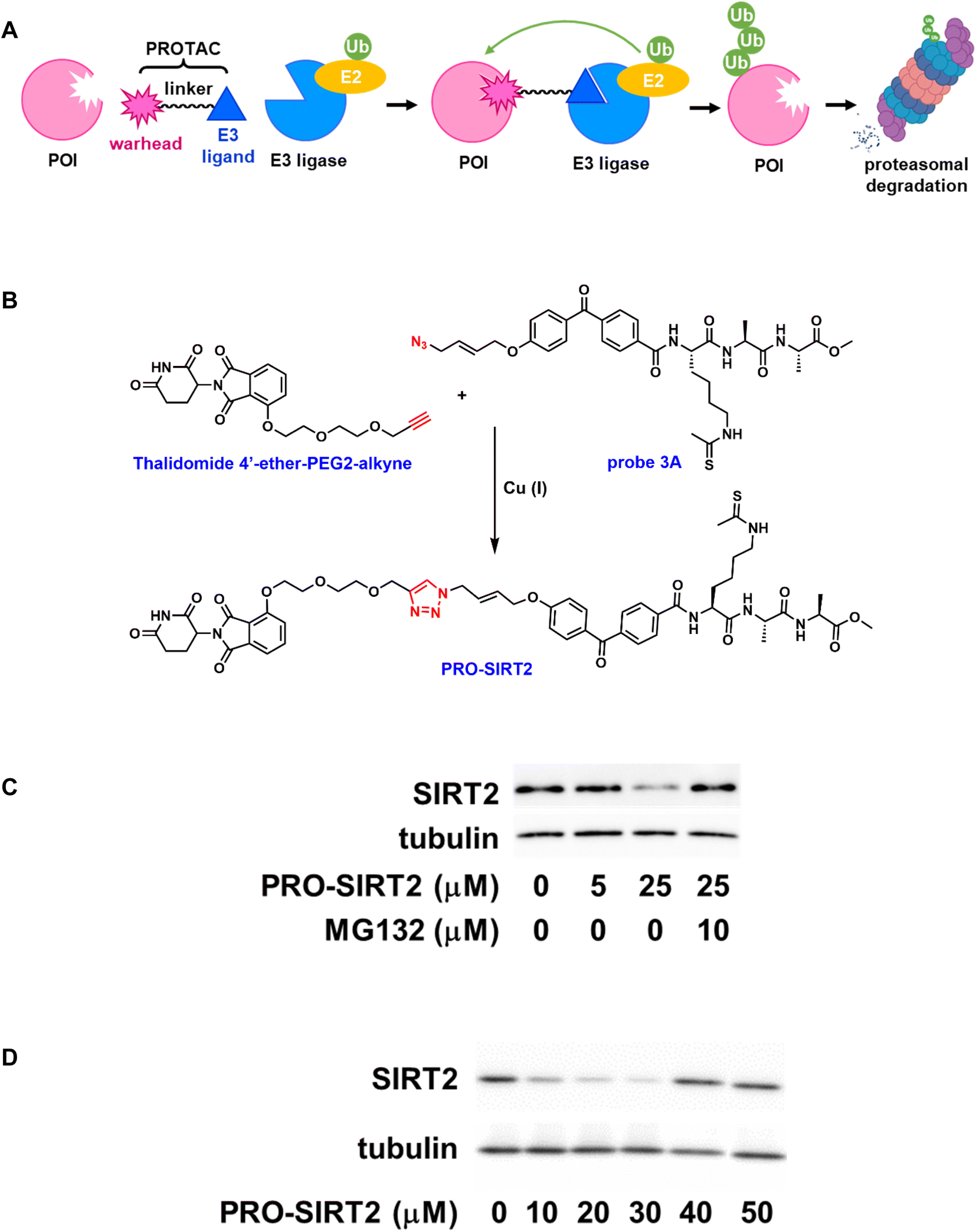 | ||
| Fig. 4 ABP-based PROTAC. (A). Schematic representation of PROTAC; (B). Synthesis of PRO-SIRT2; (C). PRO-SIRT2-induced SIRT2 degradation. Representative western blot for SIRT2 after incubation with indicated concentrations of PRO-SIRT2 for 12 hours in HEK293 cells. SIRT2 degradation can be rescued by the co-treatment of PRO-SIRT2 and MG132; (D). Concentration-dependent degradation of SIRT2 by PRO-SIRT2. | ||
To design ligands suitable for conversion to effective PROTACs, high permeability is considered a primary requirement in addition to cellular potency and selectivity. Cell permeable APBs such as probe 3A served as the basis for further development of PROTACs. Moreover, the azido group in probe 3A allowed convenient elaboration through Cu(I)-catalyzed “click” reactions. Thalidomide 4′-ether-PEG2-alkyne (Fig. 4B), a cereblon-recruiting ligand with a PEG linker and terminal alkyne group, was readily available through commercial vendors. The “click” conjugation between probe 3A and thalidomide 4′-ether-PEG2-alkyne with the addition of Cu(I) catalyst, followed by purification afforded the desired PROTAC (PRO-SIRT2) in good yield.
The inhibitory effect of PRO-SIRT2 on human sirtuin isoforms was then evaluated. Similar to its parent compound, probe 3A, the PROTAC inhibited human SIRT2 and SIRT3 with IC50 values of 3.5 ± 0.6 μM and 38.2 ± 2.8 μM, respectively, a 10-fold preference for SIRT2. Meanwhile, it did not exhibit any significant inhibition on SIRT1, SIRT5, or SIRT6, suggesting that PRO-SIRT2 retained SIRT2 selectivity.
The ability of PRO-SIRT2 to induce SIRT2 degradation was assessed in HEK293 cells. The cells were treated with increasing concentrations of PRO-SIRT2 for 12 hours. As shown in Fig. 4C, the endogenous level of SIRT2 was markedly reduced by PRO-SIRT2 at 25 μM. This depletion can be rescued with the proteasome inhibitor MG132. Fig. 4D illustrates the concentration-dependent degradation of SIRT2 by PRO-SIRT2. There is more degradation of SIRT2 at 30 μM than 40 μM, likely due to the hook effect.41 Overall, our results indicate that PRO-SIRT2 led to efficient SIRT2 degradation in a cereblon-mediated, proteasome-dependent manner.
ABPs enable affinity capture of sirtuins
In the previous studies, the ABPs demonstrated isoform-selective labeling of individual sirtuin isoform in purified proteins as well as cell lysates.21–23 The labeled proteins were visualized on a SDS-PAGE gel or by cellular imaging upon conjugation to a fluorophore. The versatility of the probes can be further illustrated in affinity enrichment of a particular sirtuin isotype. HEK293 cells were transfected to overexpress human SIRT5. The cells were harvested and the mitochondrial fraction was isolated. The mitochondrial fraction was then treated in the presence or absence of probe 2C (Fig. 5), a selective SIRT5 inhibitor. After photocrosslinking, the samples were incubated with the cleavable diazo biotin azide with the addition of Cu(I) catalyst. The biotinylated proteins can then be captured by streptavidin beads. Subsequently, the enriched proteins were released with Na2S2O4, resolved by SDS-PAGE, and analyzed with an anti-SIRT5 antibody. SIRT5 was selectively enriched by probe 2C, and this affinity capture was competed off with the addition of excess amount of a synthetic peptide H3K9Suc (Fig. 5), a known SIRT5 substrate. Taken together, we envision that the ABPs with good on-target effect will empower the enrichment-MS analysis for activity profiling in complex native proteomes.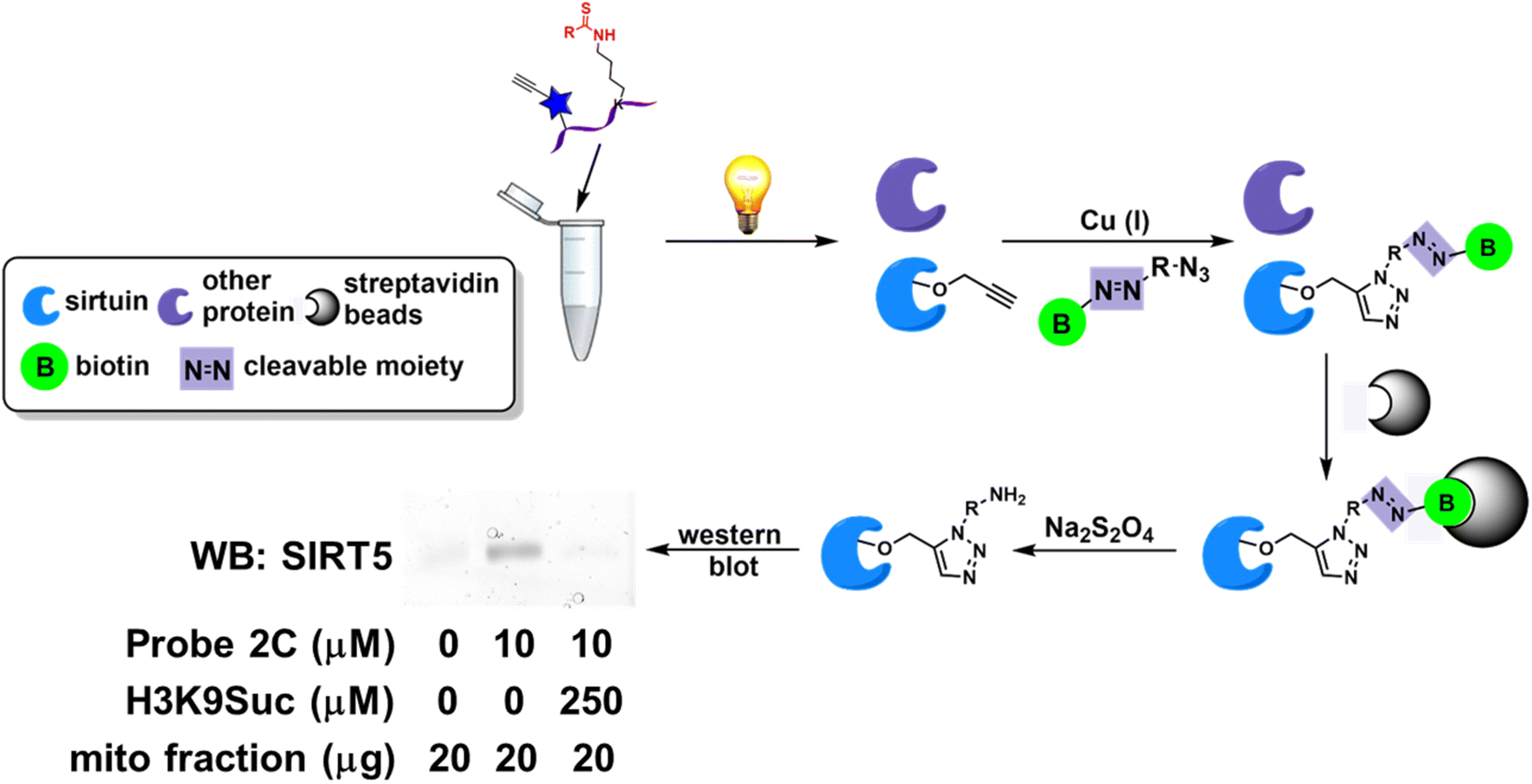 | ||
| Fig. 5 ABPs enable affinity enrichment of sirtuins. Affinity capture of SIRT5 by probe 2C in mitochondrial fraction of HEK293 cells. This enrichment can be disrupted with the co-treatment of H3K9Suc. | ||
Conclusions and perspectives
The human genome encodes 20000–25000 genes.42 Based on alternative promoters or alternative splicing as well as mRNA editing, it is estimated that the transcriptome harbors ∼100000 transcripts.43 The complexity increases exponentially via posttranslational modifications. The proteome may contain more than 1000000 proteins.44 The functional assignment of these proteins in the so-called “post-genomic era” can be challenging.45
Recent studies suggest that the functional interrogation of enzymes in complex native matrix can be accomplished by ABPs, pioneered by Ben Cravatt's group at the Scripps Research Institute. This approach utilizes active site-directed small molecules to directly confer the functional state of a particular enzyme in a complex biological sample.35,46 The small molecule probe normally has three major components: (1) a reactive group (or “warhead”) that targets the active site of an enzyme; (2) a tag that allows the visualization or affinity capture of the active enzyme; (3) a linker that connects the “warhead” with the tag. The ABPs label the active enzyme, but not the inactive or inhibitor-bound ones. Comparison of the labeling intensity and labeling pattern in normal cells/tissues vs. disease cells/tissues will reveal the role a specific protein plays in certain physiological or pathophysiological process, and may also suggest potential therapeutic targets for drug discovery. The past two decades have witnessed an ever-growing interest in developing ABPs for functional proteomics analysis.35,46–48 These probes greatly expanded our scope of the “druggable” proteomes and led to the discovery of some potent and selective enzyme inhibitors.35,49
We have constructed a small library of ABPs for human sirtuins.21–23 These powerful small molecules are based on a simple tripeptide backbone with a thioacyl group on the ε-amino group of the Lys residue as the “warhead”, a photoactivatable group for target crosslinking, and a bioorthogonal group for conjugation to reporters. Some of them have been proven to be isoform-selective and cell permeable sirtuin inhibitors. They demonstrate robust labeling of individual sirtuin isotypes in protein mixtures and cell lysates. When used comparatively, they report how sirtuin activity responds to various cellular stimuli. They can also be applied competitively for the discovery of sirtuin inhibitors. The ABPs can be readily elaborated to PROTACs, bifunctional small molecules capable of inducing targeted sirtuin degradation. Labeling of the target protein can be visualized when the probe-protein complex is “click” conjugated to a fluorophore. Alternatively, deployment of biotin azide allows the enrichment of tagged protein using affinity resins. These multifunctional ABPs offer a reliable platform to better analyze the human sirtuin family, an intriguing group of posttranslational modification enzymes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by 1R15GM1233931 and R01GM143176-01A1 from NIH/NIGMS (to Y. C.), and VCU CCTR Endowment Fund (sub-award of UL1TR002649 from National Center for Advancing Translational Sciences to VCU) (to Y. C.). MS analysis reported in this manuscript was performed at the VBRN Proteomics Facility supported by P20GM103449 (NIGMS/NIH).References
- S. Imai, C. M. Armstrong, M. Kaeberlein and L. Guarente, Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase, Nature, 2000, 403(6771), 795–800 CrossRef CAS.
- P. Martinez-Redondo and A. Vaquero, The diversity of histone versus nonhistone sirtuin substrates, Genes Cancer, 2013, 4(3–4), 148–163 CrossRef CAS PubMed.
- M. Wang and H. Lin, Understanding the Function of Mammalian Sirtuins and Protein Lysine Acylation, Annu. Rev. Biochem., 2021, 90, 245–285 CrossRef CAS PubMed.
- S. Shahgaldi and F. R. Kahmini, A comprehensive review of Sirtuins: With a major focus on redox homeostasis and metabolism, Life Sci., 2021, 282, 119803 CrossRef CAS.
- P. Maissan, E. J. Mooij and M. Barberis, Sirtuins-Mediated System-Level Regulation of Mammalian Tissues at the Interface between Metabolism and Cell Cycle: A Systematic Review, Biology, 2021, 10(3), 194 CrossRef CAS PubMed.
- C. Zeng and M. Chen, Progress in Nonalcoholic Fatty Liver Disease: SIRT Family Regulates Mitochondrial Biogenesis, Biomolecules, 2022, 12(8), 1079 CrossRef CAS PubMed.
- L. Bosch-Presegue and A. Vaquero, Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity, FEBS J., 2015, 282(9), 1745–1767 CrossRef CAS PubMed.
- B. C. Smith and J. M. Denu, Acetyl-lysine analog peptides as mechanistic probes of protein deacetylases, J. Biol. Chem., 2007, 282(51), 37256–37265 CrossRef CAS PubMed.
- E. M. Avrahami, S. Levi, E. Zajfman, C. Regev, O. Ben-David and E. Arbely, Reconstitution of Mammalian Enzymatic Deacylation Reactions in Live Bacteria Using Native Acylated Substrates, ACS Synth. Biol., 2018, 7(10), 2348–2354 CrossRef CAS PubMed.
- J. Du, Y. Zhou, X. Su, J. J. Yu, S. Khan, H. Jiang, J. Kim, J. Woo, J. H. Kim, B. H. Choi, B. He, W. Chen, S. Zhang, R. A. Cerione, J. Auwerx, Q. Hao and H. Lin, Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase, Science, 2011, 334(6057), 806–809 CrossRef CAS.
- C. Peng, Z. Lu, Z. Xie, Z. Cheng, Y. Chen, M. Tan, H. Luo, Y. Zhang, W. He, K. Yang, B. M. Zwaans, D. Tishkoff, L. Ho, D. Lombard, T. C. He, J. Dai, E. Verdin, Y. Ye and Y. Zhao, The first identification of lysine malonylation substrates and its regulatory enzyme, Mol. Cell. Proteomics, 2011, 10(12), M111012658 CrossRef PubMed.
- M. Tan, C. Peng, K. A. Anderson, P. Chhoy, Z. Xie, L. Dai, J. Park, Y. Chen, H. Huang, Y. Zhang, J. Ro, G. R. Wagner, M. F. Green, A. S. Madsen, J. Schmiesing, B. S. Peterson, G. Xu, O. R. Ilkayeva, M. J. Muehlbauer, T. Braulke, C. Muhlhausen, D. S. Backos, C. A. Olsen, P. J. McGuire, S. D. Pletcher, D. B. Lombard, M. D. Hirschey and Y. Zhao, Lysine glutarylation is a protein posttranslational modification regulated by SIRT5, Cell Metab., 2014, 19(4), 605–617 CrossRef CAS.
- H. Jiang, S. Khan, Y. Wang, G. Charron, B. He, C. Sebastian, J. Du, R. Kim, E. Ge, R. Mostoslavsky, H. C. Hang, Q. Hao and H. Lin, SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine, Nature, 2013, 496(7443), 110–113 CrossRef CAS.
- Y. B. Teng, H. Jing, P. Aramsangtienchai, B. He, S. Khan, J. Hu, H. Lin and Q. Hao, Efficient demyristoylase activity of SIRT2 revealed by kinetic and structural studies, Sci. Rep., 2015, 5, 8529 CrossRef CAS.
- A. A. Sauve, I. Celic, J. Avalos, H. Deng, J. D. Boeke and V. L. Schramm, Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions, Biochemistry, 2001, 40(51), 15456–15463 CrossRef CAS PubMed.
- A. A. Sauve and V. L. Schramm, SIR2: the biochemical mechanism of NAD(+)-dependent protein deacetylation and ADP-ribosyl enzyme intermediates, Curr. Med. Chem., 2004, 11(7), 807–826 CrossRef CAS PubMed.
- A. A. Sauve and V. L. Schramm, Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry, Biochemistry, 2003, 42(31), 9249–9256 CrossRef CAS PubMed.
- B. C. Smith and J. M. Denu, Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide, Biochemistry, 2007, 46(50), 14478–14486 CrossRef CAS PubMed.
- Y. Cen, J. N. Falco, P. Xu, D. Y. Youn and A. A. Sauve, Mechanism-based affinity capture of sirtuins, Org. Biomol. Chem., 2011, 9(4), 987–993 RSC.
- D. G. Fatkins, A. D. Monnot and W. Zheng, Nepsilon-thioacetyl-lysine: a multi-facet functional probe for enzymatic protein lysine Nepsilon-deacetylation, Bioorg. Med. Chem. Lett., 2006, 16(14), 3651–3656 CrossRef CAS PubMed.
- E. Graham, S. Rymarchyk, M. Wood and Y. Cen, Development of Activity-Based Chemical Probes for Human Sirtuins, ACS Chem. Biol., 2018, 13(3), 782–792 CrossRef CAS.
- A. M. Curry, E. Barton, W. Kang, D. V. Mongeluzi and Y. Cen, Development of Second Generation Activity-Based Chemical Probes for Sirtuins, Molecules, 2020, 26(1), 11 CrossRef.
- A. M. Curry, I. Cohen, S. Zheng, J. Wohlfahrt, D. S. White, D. Donu and Y. Cen, Profiling sirtuin activity using Copper-free click chemistry, Bioorg. Chem., 2021, 117, 105413 CrossRef CAS PubMed.
- J. Du, H. Jiang and H. Lin, Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD, Biochemistry, 2009, 48(13), 2878–2890 CrossRef CAS PubMed.
- J. S. Smith, J. Avalos, I. Celic, S. Muhammad, C. Wolberger and J. D. Boeke, SIR2 family of NAD(+)-dependent protein deacetylases, Methods Enzymol., 2002, 353, 282–300 CAS.
- B. J. North, B. L. Marshall, M. T. Borra, J. M. Denu and E. Verdin, The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase, Mol. Cell, 2003, 11(2), 437–444 CrossRef CAS PubMed.
- B. He, J. Du and H. Lin, Thiosuccinyl peptides as Sirt5-specific inhibitors, J. Am. Chem. Soc., 2012, 134(4), 1922–1925 CrossRef CAS PubMed.
- C. J. Goetz, D. J. Sprague and B. C. Smith, Development of activity-based probes for the protein deacylase Sirt1, Bioorg. Chem., 2020, 104, 104232 CrossRef CAS PubMed.
- X. Zhang, S. Khan, H. Jiang, M. A. Antonyak, X. Chen, N. A. Spiegelman, J. H. Shrimp, R. A. Cerione and H. Lin, Identifying the functional contribution of the defatty-acylase activity of SIRT6, Nat. Chem. Biol., 2016, 12(8), 614–620 CrossRef CAS.
- A. Korotkov, A. Seluanov and V. Gorbunova, Sirtuin 6: linking longevity with genome and epigenome stability, Trends Cell Biol., 2021, 31(12), 994–1006 CrossRef CAS PubMed.
- G. Charron, M. M. Zhang, J. S. Yount, J. Wilson, A. S. Raghavan, E. Shamir and H. C. Hang, Robust fluorescent detection of protein fatty-acylation with chemical reporters, J. Am. Chem. Soc., 2009, 131(13), 4967–4975 CrossRef CAS PubMed.
- N. Jessani and B. F. Cravatt, The development and application of methods for activity-based protein profiling, Curr. Opin. Chem. Biol., 2004, 8(1), 54–59 CrossRef CAS PubMed.
- K. Wang, T. Yang, Q. Wu, X. Zhao, E. C. Nice and C. Huang, Chemistry-based functional proteomics for drug target deconvolution, Expert Rev. Proteomics, 2012, 9(3), 293–310 CrossRef CAS PubMed.
- D. Leung, C. Hardouin, D. L. Boger and B. F. Cravatt, Discovering potent and selective reversible inhibitors of enzymes in complex proteomes, Nat. Biotechnol., 2003, 21(6), 687–691 CrossRef CAS PubMed.
- A. M. Roberts, C. C. Ward and D. K. Nomura, Activity-based protein profiling for mapping and pharmacologically interrogating proteome-wide ligandable hotspots, Curr. Opin. Biotechnol., 2017, 43, 25–33 CrossRef CAS PubMed.
- M. Wood, S. Rymarchyk, S. Zheng and Y. Cen, Trichostatin A inhibits deacetylation of histone H3 and p53 by SIRT6, Arch. Biochem. Biophys., 2018, 638, 8–17 CrossRef CAS.
- W. You and C. Steegborn, Structural Basis of Sirtuin 6 Inhibition by the Hydroxamate Trichostatin A: Implications for Protein Deacylase Drug Development, J. Med. Chem., 2018, 61(23), 10922–10928 CrossRef CAS.
- H. Jing, J. Hu, B. He, Y. L. Negron Abril, J. Stupinski, K. Weiser, M. Carbonaro, Y. L. Chiang, T. Southard, P. Giannakakou, R. S. Weiss and H. Lin, A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity, Cancer Cell, 2016, 29(5), 767–768 CrossRef CAS.
- C. M. Crews, Targeting the undruggable proteome: the small molecules of my dreams, Chem. Biol., 2010, 17(6), 551–555 CrossRef CAS PubMed.
- Y. Prozzillo, G. Fattorini, M. V. Santopietro, L. Suglia, A. Ruggiero, D. Ferreri and G. Messina, Targeted Protein Degradation Tools: Overview and Future Perspectives, Biology, 2020, 9(12), 421 CrossRef CAS PubMed.
- M. Pettersson and C. M. Crews, PROteolysis TArgeting Chimeras (PROTACs) - Past, present and future, Drug Discovery Today: Technol., 2019, 31, 15–27 CrossRef PubMed.
- C. International Human Genome Sequencing, Finishing the euchromatic sequence of the human genome, Nature, 2004, 431(7011), 931–945 CrossRef PubMed.
- G. K. Marinov, B. A. Williams, K. McCue, G. P. Schroth, J. Gertz, R. M. Myers and B. J. Wold, From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing, Genome Res., 2014, 24(3), 496–510 CrossRef CAS PubMed.
- E. A. Ponomarenko, E. V. Poverennaya, E. V. Ilgisonis, M. A. Pyatnitskiy, A. T. Kopylov, V. G. Zgoda, A. V. Lisitsa and A. I. Archakov, The Size of the Human Proteome: The Width and Depth, Int. J. Anal. Chem., 2016, 2016, 7436849 Search PubMed.
- D. Eisenberg, E. M. Marcotte, I. Xenarios and T. O. Yeates, Protein function in the post-genomic era, Nature, 2000, 405(6788), 823–826 CrossRef CAS PubMed.
- A. E. Speers and B. F. Cravatt, Chemical strategies for activity-based proteomics, Chembiochem, 2004, 5(1), 41–47 CrossRef CAS PubMed.
- H. Deng, Q. Lei, Y. Wu, Y. He and W. Li, Activity-based protein profiling: Recent advances in medicinal chemistry, Eur. J. Med. Chem., 2020, 191, 112151 CrossRef CAS PubMed.
- H. J. Benns, C. J. Wincott, E. W. Tate and M. A. Child, Activity- and reactivity-based proteomics: Recent technological advances and applications in drug discovery, Curr. Opin. Chem. Biol., 2021, 60, 20–29 CrossRef CAS PubMed.
- S. Wang, Y. Tian, M. Wang, M. Wang, G. B. Sun and X. B. Sun, Advanced Activity-Based Protein Profiling Application Strategies for Drug Development, Front. Pharmacol., 2018, 9, 353 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02133e |
| This journal is © The Royal Society of Chemistry 2023 |