
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFull-dimensional neural network potential energy surface and dynamics of the CH2OO + H2O reaction
Hao Wu ab,
Yanlin Fua,
Wenrui Dongab,
Bina Fu*abc and
Dong H. Zhangabc
ab,
Yanlin Fua,
Wenrui Dongab,
Bina Fu*abc and
Dong H. Zhangabc
aState Key Laboratory of Molecular Reaction Dynamics and Center for Theoretical and Computational Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: bina@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cHefei National Laboratory, Hefei, 230088, China
First published on 2nd May 2023
Abstract
An accurate global full-dimensional machine learning-based potential energy surface (PES) of the simplest Criegee intermediate (CH2OO) reaction with water monomer was developed based on the high level of extensive CCSD(T)-F12a/aug-cc-pVTZ calculations. This analytical global PES not only covers the regions of reactants to hydroxymethyl hydroperoxide (HMHP) intermediates, but also different end product channels, which facilities both the reliable and efficient kinetics and dynamics calculations. The rate coefficients calculated by the transition state theory with the interface to the full-dimensional PES agree well with the experimental results, indicating the accuracy of the current PES. Extensive quasi-classical trajectory (QCT) calculations were performed both from the bimolecular reaction CH2OO + H2O and from HMHP intermediate on the new PES. The product branching ratios of hydroxymethoxy radical (HOCH2O, HMO) + OH radical, formaldehyde (CH2O) + H2O2 and formic acid (HCOOH) + H2O were calculated. The reaction yields dominantly HMO + OH, because of the barrierless pathway from HMHP to this channel. The computed dynamical results for this product channel show the total available energy was deposited into the internal rovibrational excitation of HMO, and the energy release in OH and translational energy is limited. The large amount of OH radical found in the current study implies that the CH2OO + H2O reaction can provide crucially OH yield in Earth's atmosphere.
1. Introduction
It is well-known that ozonolysis of alkenes proceeds via Criegee intermediates (CIs) in the troposphere.1 This ozonolysis process has two steps: (1) ozone and alkenes react via 1,3-cycloaddition to form a primary ozonide; (2) the primary ozonide rapidly decomposes to vibrationally excited CIs and a stable carbonyl compound. The vibrationally hot CIs can further undergo unimolecular dissociation or be collisionally deactivated to stabilized CIs,2,3 which have a long lifetime to undergo bimolecular reactions with other species, such as NOx,4–6 SO2,7–9 organic acids,10,11 inorganic acids12 and water vapor.13,14 The self-reaction of CIs have also been reported,15 indicating a cyclic dimeric intermediate with a terminal O atom of one CH2OO bonded to the C atom of the other CH2OO is formed with large exothermicity before further decomposition to formaldehyde and oxygen. Because of the high concentration of water vapor, the reaction with water vapor is considered to be dominant in the atmosphere,14 and considerable efforts have been devoted to the study of the reaction of CIs with water.16,17 In 2015, Lin and coworkers observed quadratic dependence of the decay rate of CH2OO on water concentration, implying a predominant reaction with water dimer.18 In subsequent experimental and theoretical studies,19,20 the effect of water monomer to the CH2OO reaction with water vapor was also investigated.Nonetheless, there is still much controversy over the end product of the reaction of the simplest CI (CH2OO) with single water molecule both experimentally and theoretically. Various products, such as hydroxymethyl hydroperoxide (HMHP), formic acid (HCOOH), ketones, H2O2, hydroxymethoxy radical (HOCH2O, HMO), and OH have been observed experimentally through yield measurements of the gas-phase ozonolysis of ethene under humid conditions.21–25 These indirect determinations of reaction kinetics of CIs provided derived rate coefficients with large uncertainties. Therefore, these early experiments without direct detection of CIs can not be sufficiently reliable to provide a definitive understanding of the CH2OO + H2O reaction.
In 2012, Welts et al. first reported direct photoionization mass spectrometric detection of stabilized CH2OO as a product of the reaction of CH2I with O2.26 That direct kinetic measurement provided an upper limit of 4 × 10−15 cm−3 s−1 for the rate coefficient of the CH2OO + H2O reaction. They found that the consumption of CH2OO in their experiment should produce formaldehyde, and also observed slight HMHP signal at the highest water concentration. Stone et al. obtained kinetics of CH2OO reactions with some molecules, including H2O at varying pressures at a temperature of 295 K, using photolysis of CH2I2–O2–N2 mixtures under pseudo-first-order conditions combined with monitoring of the formaldehyde reaction products by laser-induced fluorescence (LIF) spectroscopy.27 The resulting upper limit on the rate coefficient in that work for CH2OO + H2O is 9 × 10−17 cm−3 s−1, which is significantly lower than the previously reported value.26 These outcomes indicate whether the dominant product of the reaction is formaldehyde (CH2O) still remains a question. Nakajima and Endo observed HMHP, dioxirane and formic acid in a reaction system containing CH2OO and water vapor through pure rotational spectroscopy in 2015.28
Theoretical studies have different opinions on the end product of the reaction as well. Those theoretical work was focused on the geometry optimization and single-point energy calculations at various ab initio levels for the formation of HMHP from CH2OO + H2O and the unimolecular HMHP decomposition pathways.29–32 These pathways include HCOOH + H2O, CH2O + H2O2, and HMO + OH.30–32 A recent work indicated HMO + OH can be the most feasible channel for the CH2OO + H2O reaction, but in the absence of global PES and dynamics calculations.32 In 2016, Lin et al.19 performed electronic calculations at the level of QCISD(T)/CBS//B3LYP/6-311+G(2d,2p) for the stationary points and pathways from CH2OO + H2O to HMHP, but the further dissociation pathways associated with end products of this reaction were not considered. The rate coefficients for the CH2OO + H2O reaction were also calculated by the transition state theory, which agree well with the experimental results.
To better understand the true reaction mechanism of CH2OO + H2O associated with end products of this reaction, we report here the first dynamical study by developing the first full-dimensional potential energy surface (PES) of CH2OO + H2O based on fundamental invariant-neural network (FI-NN) fitting to a large number of UCCSD(T)/aug-cc-pVTZ data points.33–35 The full-dimensional PES not only covers the CH2OO + H2O reaction involving HMHP intermediates, but also the end product channels. Section 2 presents the detailed computational details and properties of the new PES for CH2OO + H2O. Section 3 gives the results and discussion of rate coefficients, product branching ratios and dynamics information. The conclusions are summarized in Section 4.
2. Potential energy surface
The early studies show that CH2OO behaves more like a zwitterion than a biradical, and the single-reference methodologies can describe the reaction quite well.36,37 Thus, the “gold standard” level of CCSD(T)-F12a/aug-cc-pVTZ was used in ab initio calculations for most data points in a large configuration space. The T1 diagnostic values were also computed in this work for CH2OO + H2O in different regions and those values are small, indicating the reliability of CCSD(T)-F12a calculations. Due to the two radical fragments in the OHCH2O + OH channel, the single-reference method has problems in describing this channel. We thus employed the approach of mixing as incorporated in Gaussian program,38 which mixes some amount of the HOMO with the LUMO of the same spin. This is the most straightforward approach to get the correct initial guess for Hartree–Fock calculation that requires no prior knowledge of the system. All ab initio calculations were carried out using the MOLPRO 2018 (ref. 39) and Gaussian 16 (ref. 38) in the current work.In this work, we used the FI-NN method33–35 to fit the full-dimensional PES of CH2OO + H2O. The FI-NN approach uses the fundamental invariants as the input layer of the neural network, which is proposed based on the permutationally invariant polynomial (PIP)40 and PIP-NN41 approaches. The PIP method proposed by Bowman and co-workers uses primary and secondary invariants as the fitting basis to generate permutation invariant polynomials while the coefficients are obtained by linear least squares fitting.40 Another mathematically equivalent PIP method based on the symmetrized monomials was also developed.42 The PIP-NN method uses a set of PIPs instead of pairwise distances as input vector in the neural network, including all the polynomials truncated by a given degree.41 The FI-NN approach minimizes the size of polynomial and efficiently reduces the evaluation time of the potential energy, in particular for larger molecular systems with more identical atoms.43–46 Moreover, FIs are polynomials based on the internuclear distances, which are invariant for translation, rotation, and inversion.
The feed-forward network has two hidden layers. The output of the hidden layer is
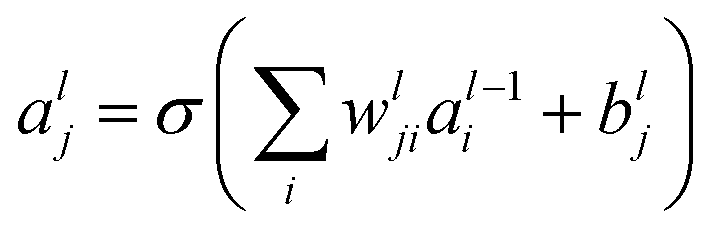 | (1) |

 | (2) |
We used Levenberg Marquardt algorithm47 as the optimization algorithm and root-mean-square error (RMSE) as the loss function to update the parameters of the neural network.
Since the configuration space of the investigated system is large, we split the dataset into the reactant asymptotic region (ASY) and the rest region (interaction region (INT), hereafter) with some overlaps (switch zone) between the two parts, to overcome the huge difficulties of fitting all the data points together. We used the distance (ROO) between the two oxygen atoms (one from H2O, the other from CH2OO attached to the carbon) as the condition:
| INT: ROO ≤ 7.0 Å |
| ASY: ROO ≥ 8.0 Å |
| Switch: 7.0 Å < ROO < 8.0 Å |
Finally, the two parts were connected by a smooth switch function. The final global PES is a weighted sum of the two parts:
| E = WINT × EINT + WASY × EASY | (3) |
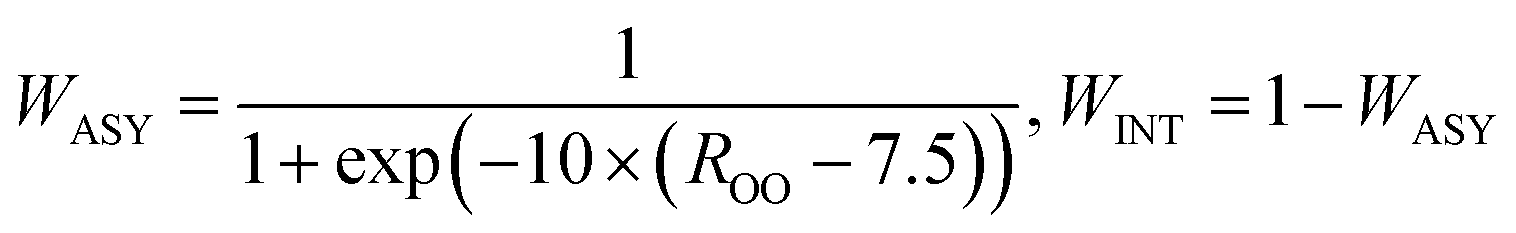 | (4) |
The Multi State Empirical Valance Bond (MSEVB) method is an efficient procedure to fit the interaction and asymptote regions,48 which can be considered and employed in future work. The architectures of the two NN PES are listed in Table 1. The FIs are calculated by King's algorithm49 using the homemade code of our group. Since, in the reactant asymptotic region, there is no need to consider atomic permutative symmetries between H2O and CH2OO. The total number of FIs in the asymptotic region is 143; however, the number is 505 for the interaction region, which is truncated to fifth-order terms.
| Structure | Fitting points | RMSE (meV) | |
|---|---|---|---|
| ASY | 143-10-10-1 (1561) | 8024 | 4.78 |
| INT | 505-30-30-1 (16141) | 106![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 848 848 |
15.82 |
| Global | 111076 |
15.52 |
The PES was constructed by the FI-NN method based on 111076 ab initio energy points. We initially obtained 10000 configurations by performing ab initio molecular dynamics (AIMD) simulations at the B3LYP/6-31g level of theory. For these calculations, we ran about 300 trajectories from the reagents CH2OO + H2O at the collision energy of 15.0 kcal mol−1. Additionally, roughly 300 trajectories were run initiated from the transition states leading to different product channels. Our first PES was fitted based on the initial dataset and more data points were added iteratively by doing QCT calculations on the preliminary PES. On every cycle, we collected data points with significant energy deviations and errors. To avoid adding too many similar data points, we define the following criterion to measure the “distance” between two geometries, a and b.
 | (5) |
076 energy points were computed and included in the fitting base, leading to the final RMSE of 15.5 meV.
Fig. 1 shows the schematics of energies and configurations of stationary points as well as different pathways and product channels for the CH2OO + H2O reaction. Comparisons between the CCSD(T)-F12a/aug-cc-pVTZ and PES energies for all stationary points of the different pathways are made, which shows good agreement between the two results. As depicted in Fig. 1, the collision between CH2OO and H2O can either proceed via the pre-reaction complex (IM1) and the transition state (TS1) and sequently form an intermediate (HMHP1), or proceed via IM2 and TS2, followed by the formation of HMHP2. The two HMHP intermediates (HMHP1 and HMHP2) can further dissociate into three end product channels, which is HMO + OH, HCOOH + H2O and CH2O + H2O2. There is a transition state and post-reaction well respectively for the highly exothermic HCOOH + H2O channel and the CH2O + H2O2 channel, but the HMO + OH channel is barrierless.
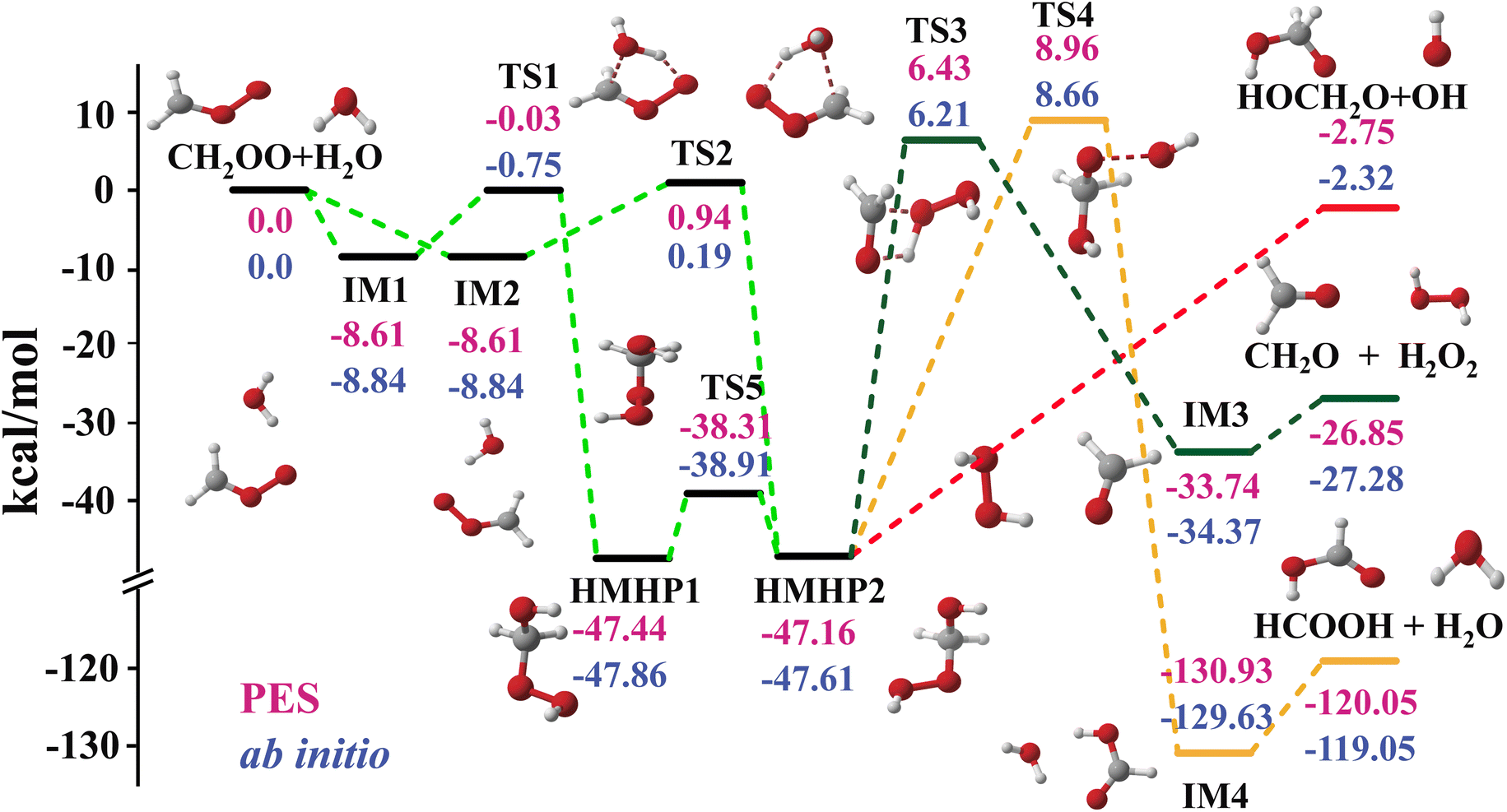 | ||
| Fig. 1 PES schematic of the CH2OO + H2O reaction. All energies are in kcal mol−1 and relative to the CH2OO + H2O asymptote at various levels: FI-NN PES (red fonts), CCSD(T)-F12a/AVTZ (blue fonts). | ||
The fitting error distribution as a function of potential energy is shown in Fig. 2, representing good performance of fitting results. Fig. 4 depicted four minimum energy paths corresponding to CH2OO + H2O → IM1 → TS1 → HMHP1, CH2OO + H2O → IM2 → TS2 → HMHP2, HMHP2 → IM3 → CH2O + H2O2, and CH2OO + H2O → IM2 → TS2 → HMHP2 → HMO + OH determined by the string method50 on the PES, together with those energies calculated directly by the CCSD(T)-F12a/AVTZ level of theory. The reaction coordinates for the transition states are all set to zero, and those for the rest configurations are the Euclidean distances from the corresponding transition state. The minimum energy paths are very smooth. The PES reproduces the energies of CCSD(T)-F12a/AVTZ quite well along the minimum energy paths, indicating the accuracy of full-dimensional PES.
 | ||
| Fig. 2 The fitting errors for all data points in the FI-NN PES, as a function of their corresponding ab initio energies relative to the energy of CH2OO + H2O. | ||
Fig. 3 shows the potential energy curve of the HMO + OH channel, including CCSD(T)-F12a/AVTZ energies, the CCSD(T)-AVTZ with Gaussian-mix Hartree–Fock, and fitted potential energies. One can see the direct CCSD(T)-F12a/AVTZ calculations have severe problems in the exit channel of HMO + OH, while the fitted PES accurately and smoothly describe the exit channel, by combining the CCSD(T)-F12a/AVTZ energies and CCSD(T)-AVTZ with Gaussian-mix energies.
 | ||
| Fig. 3 Comparisons of potential energies as a function of R (distance between two O atoms from OH and OHCH2O) obtained from the PES (solid lines) and from direct CCSD(T)-f12a/AVTZ (green circle), and mixing calculations (blue triangle). | ||
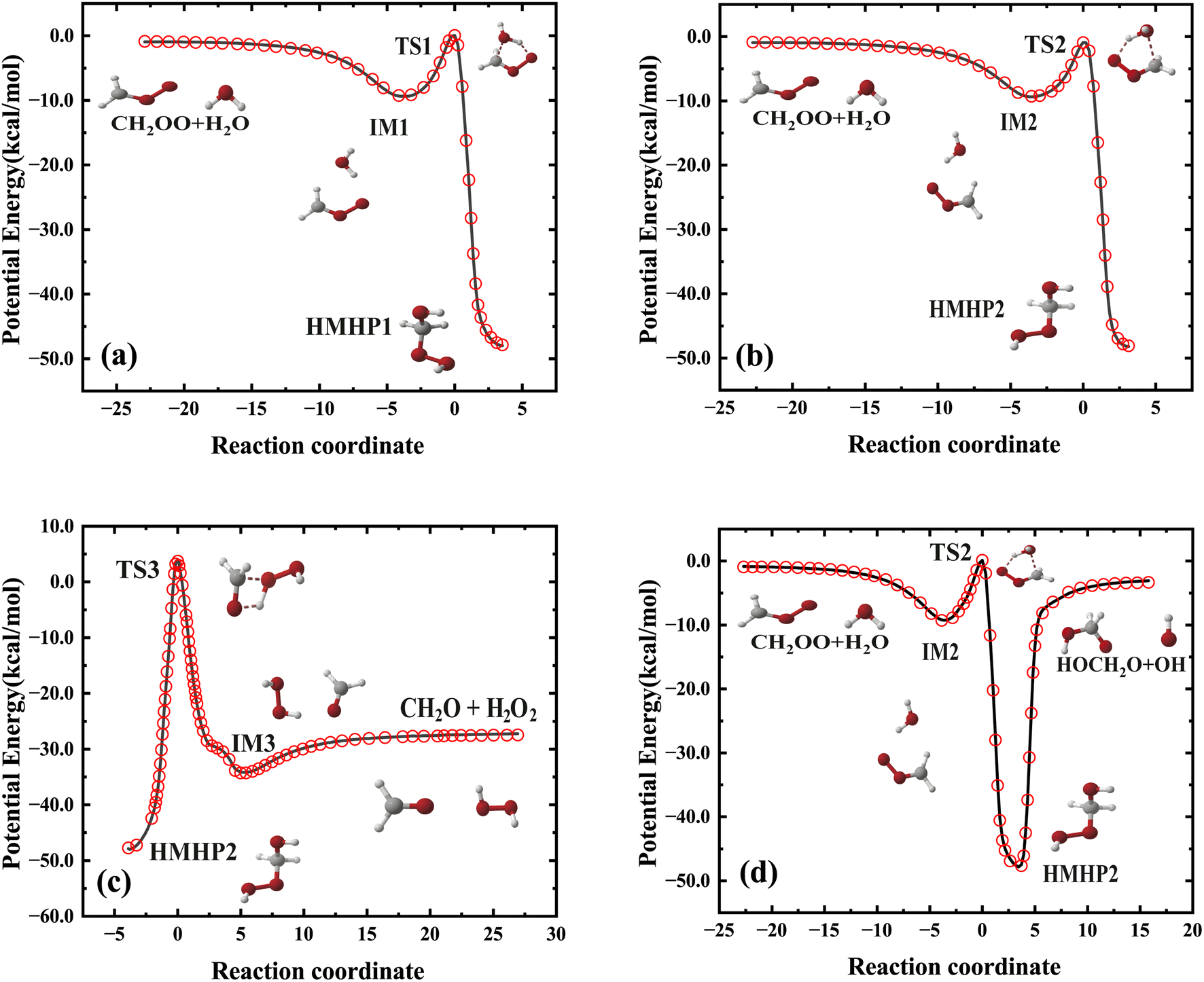 | ||
| Fig. 4 Comparisons of potential energies along minimum energy paths obtained from the PES (solid line) and calculated from CCSD(T)-f12a/AVTZ level of theory (red circle). The reaction coordinate is the signed distance along the reaction path from the saddle points. | ||
3. Kinetics and dynamics
The rate coefficients were calculated using canonical variational transition state theory (CVT) in conjunction with the small curvature tunneling (SCT) implemented in the POLYRATE-2017 software.51 The full-dimensional PES was used as the interface to rate constant calculations. The total rate coefficient is the sum of the two independent pathways, which correspond to IM1-TS1-HMHP1 and IM2-TS2-HMHP2. The computed rate coefficients of the CH2OO + H2O reaction are displayed in Fig. 5, together with those available experimental results for this reaction and theoretical results from Lin et al.19 The current results agree reasonably well with the experimental results in a wide temperature region. Our results based on CCSD(T) electron structure calculations are about 13% lower than the theoretical results based on QCISD(T), due to slightly higher barrier heights and lower pre-reaction wells of current theory. Our pre-reaction well with ZPE corrected (−6.8 kcal mol−1) is roughly 0.3 kcal mol−1 lower than that of Lin et al. (−6.53 kcal mol−1), and the two transition states with ZPE corrected (3.72 kcal mol−1 and 2.88 kcal mol−1) are slightly higher than that of Lin et al. (3.68 kcal mol−1 and 2.82 kcal mol−1). The Arrhenius form (Aexp(Ea/RT)) of fitted results in this work is 1.12 × 10−14 × exp(−2.15/RT) (A is in cm−3 s−1, Ea is in kcal mol−1).
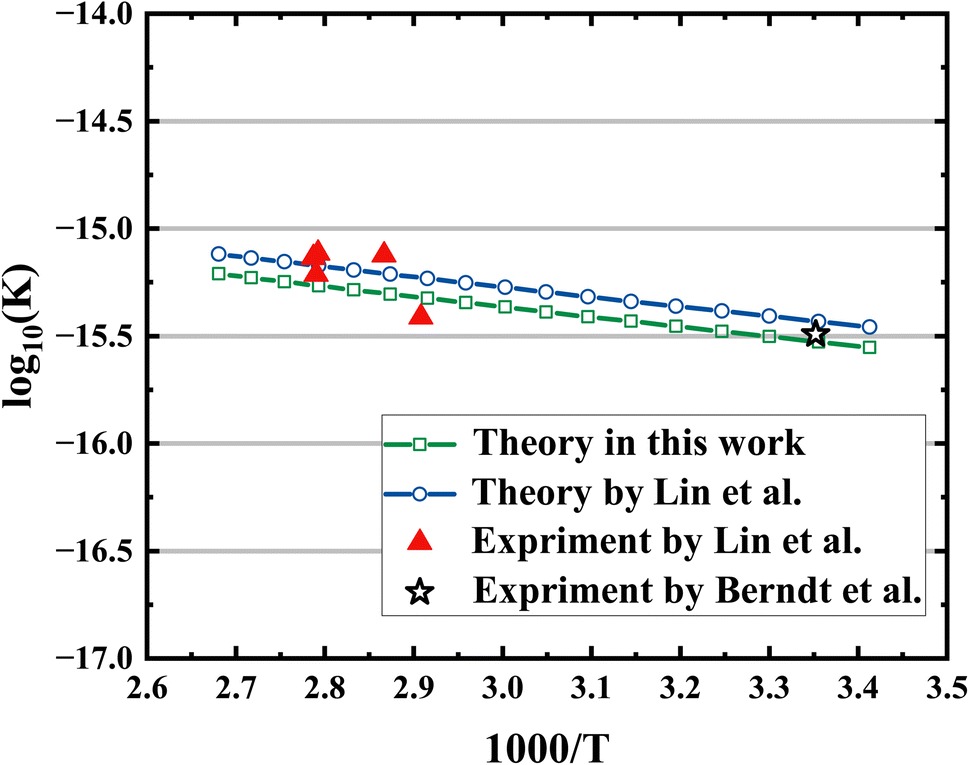 | ||
| Fig. 5 Comparisons of rate coefficients: this work (green line); theory by Lin et al.19 (blue line); experiment by Lin et al.19 (red triangle); experiment by Berndt et al.14 (black star). | ||
The standard QCT calculations were carried out on the full-dimensional PES for the bimolecular reaction CH2OO + H2O at the temperatures of 300 K and 358 K, respectively. The initial coordinates and momenta of the system were obtained by sampling a Boltzmann distribution at a given temperature. Due to the long-range interaction in the entrance channel, the initial distance between the center of mass of the two reactants was sufficiently large  here b is the impact parameter and x was set to 23 Bohr. The impact parameter was selected randomly from the distribution
here b is the impact parameter and x was set to 23 Bohr. The impact parameter was selected randomly from the distribution  where r is a random number uniformly distributed from 0 to 1. The maximum impact parameter (bmax) was determined by running a set of trajectories at a given temperature. The trajectories were propagated using the velocity-Verlet integration algorithm with a time step of 0.1 fs for a maximum time of 2 ns. The trajectories are terminated if the distance between any two atoms is greater than 20 Bohr. The trajectories either result in one of the product channels or return to the reactants.
where r is a random number uniformly distributed from 0 to 1. The maximum impact parameter (bmax) was determined by running a set of trajectories at a given temperature. The trajectories were propagated using the velocity-Verlet integration algorithm with a time step of 0.1 fs for a maximum time of 2 ns. The trajectories are terminated if the distance between any two atoms is greater than 20 Bohr. The trajectories either result in one of the product channels or return to the reactants.
A total of 10400000 trajectories were run from CH2OO and H2O at the temperature of 300 K and 358 K, respectively, up to the maximum time of 2 ns. Among those trajectories, we only obtained 138 reactive trajectories, with 134 trajectories leading to HMO + OH and 4 trajectories leading to HCOOH + H2O at 300 K. At 358 K, the reactive trajectories were 105, with 98 trajectories producing HMO + OH and 7 trajectories producing HCOOH + H2O. Although the number of total trajectories has been already huge, it was found that the product branching ratio is far from convergence, owing to the extremely small reaction probability for the bimolecular reaction.
Because the reaction probably at the temperature of atmosphere relevance is extremely small, it is computationally unaffordable to get the converged branching ratios of end products from the initial collision between CH2OO and H2O. To get rid of unnecessary computational time in running nonreactive trajectories, an alternative approach that launching trajectories from HMHP was employed. The bimolecular reaction should first proceed through the intermediate HMHP1 or HMHP2, which further decomposes to different end products, and thus we initiated trajectories at HMHP to run unimolecular reaction dynamics. The initial velocities was sampled by a Boltzmann distribution around the total available energy, which has the same initial energy distribution as the bimolecular collision.
A total of 180000 trajectories were run initiated respectively from HMHP (HMHP1 and HMHP2) with the initial condition relevance to the bimolecular collision, to obtain well converged product branching ratio. The von Neumann acceptance–rejection method52 was used to generate initial conditions of normal-mode coordinates and momenta, subject to the same total energy distribution as initialized from CH2OO + H2O. As shown in Fig. 6, the total energy distribution of the trajectories initialized from HMHP and CH2OO + H2O at 300 K and 358 K are consistent with each other, indicating that the total energy distribution of CH2OO + H2O has been successfully achieved on HMHP.
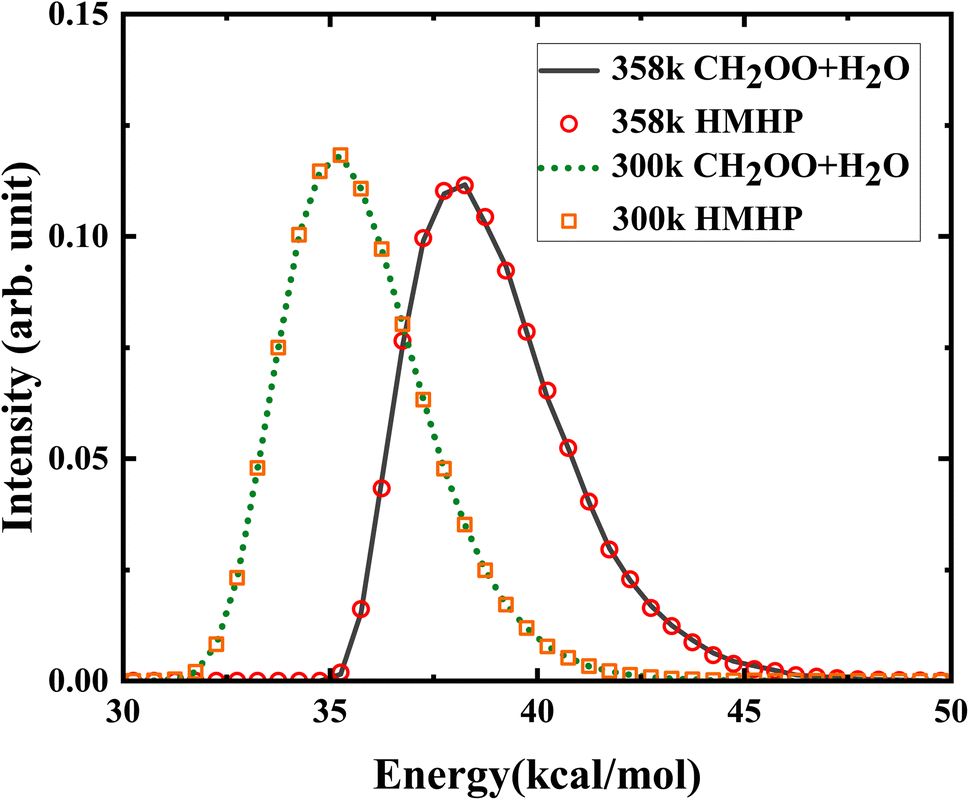 | ||
| Fig. 6 The initial total energy distributions initiated from CH2OO + H2O and from HMHP at 300 K and 358 K. | ||
Table 2 shows the product branching ratios at the temperature of 300 K and 358 K initiated from HMHP1 and HMHP2, respectively. One can see that the branching ratios obtained from HMHP1 and HMHP2 at the specified temperature are basically the same, indicating the isomerization between the two isomers is very efficient. This is because the potential barrier between the two isomers is only 9 kcal mol−1, which is significantly lower than the available energy that can be used for isomerization. Nearly all trajectories decompose to end product channels or return to reactants after the maximum propagation time of 2 ns, although at 300 K a faint, negligible fraction (0.01% or 0.04%) was trapped in the HMHP well. The HMO + OH product channel is dominant, which accounts for ∼94% of the overall reaction yield, because this dissociation channel is a direct dissociation channel without a barrier. Although the dissociation barrier of HCOOH + H2O is about 2.5 kcal mol−1 higher than that of HCOH + H2O2, the HCOOH + H2O channel accounts for about 6% of total yield, but a tiny fraction (<0.1%) results in HCOH + H2O2. This is presumably due to significantly larger exothermic energy of the HCOOH + H2O channel.
| Temperature | Initial geometry | HMHP (%) | HMO + OH (%) | HCOOH + H2O (%) | CH2O + H2O2 (%) |
|---|---|---|---|---|---|
| 300 K | HMHP1 | 0.01 | 94.0 | 5.93 | 0.06 |
| 300 K | HMHP2 | 0.04 | 93.92 | 5.97 | 0.07 |
| 358 K | HMHP1 | 0.0 | 93.99 | 5.92 | 0.09 |
| 358 K | HMHP2 | 0.0 | 94.01 | 5.90 | 0.09 |
As the HMO + OH channel has the predominant contribution to the total reaction yield, the detailed dynamical information for this channel was also calculated by the QCT calculations. The center of mass product translational energy distribution of HMO + OH at 358 K is displayed in Fig. 7. As seen, the translational energy distribution peaks at around 1.0 kcal mol−1, with a tail up to the energy of about 15 kal mol−1. This behavior of narrow translational energy distribution supports that this channel leading to HMO + OH is barrierless. The average translational energy release of HMO + OH is around 3.5 kcal mol−1, corresponding to a small fraction (0.09) of the total available energy (∼38 kcal mol−1). This implies that most of the available energy in this channel is deposited into the internal degrees of freedom.
 | ||
| Fig. 7 Center-of-mass translational energy distribution of HMO + OH obtained from the QCT calculations at 358 K. | ||
Further analysis reveals that the product OH is exclusively populated in the vibrational ground state. The rotational state distribution of OH presented in Fig. 9 peaks at j = 1, indicates that the rotation excitation of OH is insignificant too. It was found that most of the available energy goes into the internal energy of HMO. As verified in Fig. 8, the internal energy distribution of HMO reaches the maximum of about 45 kcal mol−1 and peaks at 33 kcal mol−1, which is very close to the average total available energy (∼38 kcal mol−1).
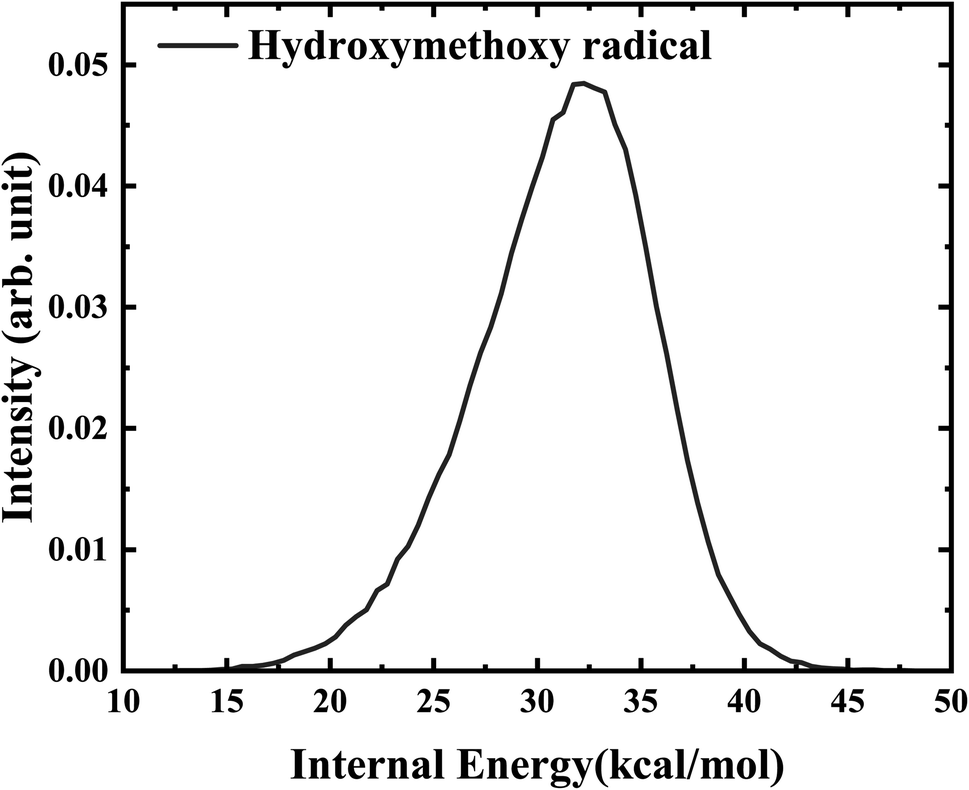 | ||
| Fig. 8 The internal energy distribution of HMO obtained from the QCT calculations at 358 K. | ||
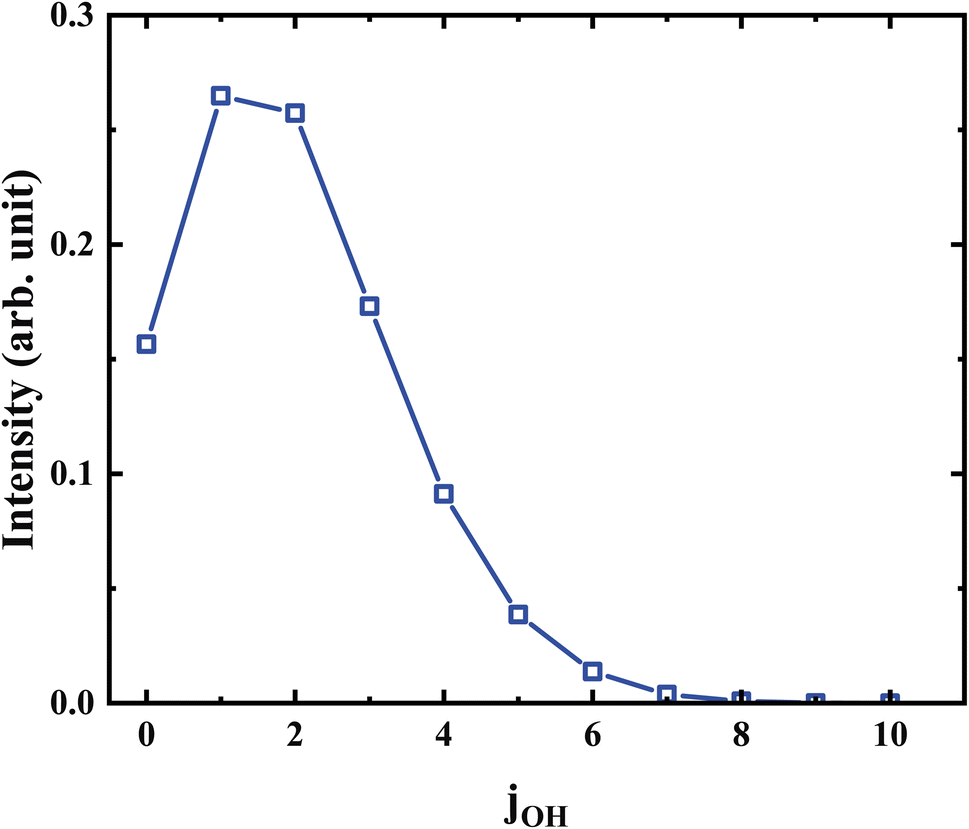 | ||
| Fig. 9 The distribution of the rotational state of OH at 358 K. | ||
4. Conclusions
To summarize, an accurate full-dimensional analytical PES for the eight-atom CH2OO + H2O reaction was developed by the FI-NN fitting approach at the level of CCSD(T)-F12a/aug-cc-pVTZ level of theory. The pathways from CH2OO + H2O to HMHP, as well as the multiple end product channels were considered in mapping the PES. With the aid of this full-dimensional analytical PES, the kinetics and dynamics information was successfully archived. The total rate coefficient of CH2OO + H2O → HMHP was computed by the CVT/SCT calculations with the interface of PES, which agree well with the experimental results and previous theoretical results based on stationary point calculations. Extensive QCT calculations were performed to get the converged product branching ratios. The barrierless hydroxymethoxy radical (HOCH2O, HMO) + OH channel plays the dominant role, which account for more than 90% of the total yield. The highly exothermic channel HCOOH + H2O account for about 6% of overall yield, while a tiny fraction (<1%) leads to CH2O + H2O2. The detailed dynamical results for HMO + OH indicate the majority of total available energy was channeled into the rovibrationally excitation of HMO but not the translation energy of products, consistent with the feature of a barrierless reaction. The OH product is exclusively in the ground state and the rotation of OH is very limited. The full-dimensional quantum dynamics calculations for this system are currently formidable, and thus the current QCT calculations are valuable and important. This is the first full-dimensional dynamics study based on a full-dimensional PES, which has provided valuable insight into the end product channels for the reaction between the simplest CI and water monomer as well as the kinetics and dynamics information. The OH radical is one of the main chemical species controlling the oxidizing capacity of the global Earth atmosphere. Thus, the current study for the CI reaction with water monomer producing the dominantly OH radical can have a major impact on the concentrations and distribution of greenhouse gases and pollutants in the Earth atmosphere. Our study opens the door for accurate dynamical characterization for many CI related reactions, which ultimately provides deep insight into the important CI associated atmospheric chemistry and environmental chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22173099, and 22288201), and the Innovation Program for Quantum Science and Technology (2021ZD0303305).References
- R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745 CrossRef.
- O. Horie and G. Moortgat, Atmos. Environ., Part A, 1991, 25, 1881 CrossRef.
- Z. Hassan, M. Stahlberger, N. Rosenbaum and S. Bräse, Angew. Chem., Int. Ed., 2021, 60, 15138 CrossRef CAS PubMed.
- L. Vereecken, H. Harder and A. Novelli, Phys. Chem. Chem. Phys., 2012, 14, 14682 RSC.
- B. Ouyang, M. W. McLeod, R. L. Jones and W. J. Bloss, Phys. Chem. Chem. Phys., 2013, 15, 17070 RSC.
- R. L. Caravan, M. A. H. Khan, B. Rotavera, E. Papajak, I. O. Antonov, M.-W. Chen, K. Au, W. Chao, D. L. Osborn, J. J.-M. Lin, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Faraday Discuss., 2017, 200, 313 RSC.
- R. L. M. III, T. Berndt, M. Sipilä, P. Paasonen, T. Petäjä, S. Kim, T. Kurtén, F. Stratmann, V.-M. Kerminen and M. Kulmala, Nature, 2012, 488, 193 CrossRef PubMed.
- M. Sipilä, T. Jokinen, T. Berndt, S. Richters, R. Makkonen, N. M. Donahue, R. L. M. III, T. Kurtén, P. Paasonen, N. Sarnela, M. Ehn, H. Junninen, M. P. Rissanen, J. Thornton, F. Stratmann, H. Herrmann, D. R. Worsnop, M. Kulmala, V.-M. Kerminen and T. Petäjä, Atmos. Chem. Phys., 2014, 14, 12143 CrossRef.
- K. T. Kuwata, E. J. Guinn, M. R. Hermes, J. A. Fernandez, J. M. Mathison and K. Huang, J. Phys. Chem. A, 2015, 119, 10316 CrossRef CAS PubMed.
- O. Welz, A. J. Eskola, L. Sheps, B. Rotavera, J. D. Savee, A. M. Scheer, D. L. Osborn, D. Lowe, A. Murray Booth, P. Xiao, M. A. H. Khan, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Angew. Chem., 2014, 126, 4635 CrossRef.
- R. Zhao, C. M. Kenseth, Y. Huang, N. F. Dalleska, X. M. Kuang, J. Chen, S. E. Paulson and J. H. Seinfeld, J. Phys. Chem. A, 2018, 122, 5190 CrossRef CAS PubMed.
- E. S. Foreman, K. M. Kapnas and C. Murray, Angew. Chem., 2016, 128, 10575 CrossRef.
- C. A. Taatjes, D. E. Shallcross and C. J. Percival, Phys. Chem. Chem. Phys., 2014, 16, 1704 RSC.
- T. Berndt, R. Kaethner, J. Voigtländer, F. Stratmann, M. Pfeifle, P. Reichle, M. Sipilä, M. Kulmala and M. Olzmann, Phys. Chem. Chem. Phys., 2015, 17, 19862 RSC.
- Y.-T. Su, H.-Y. Lin, R. Putikam, H. Matsui, M. C. Lin and Y.-P. Lee, Nat. Chem., 2014, 6, 477 CrossRef CAS PubMed.
- B. Long, J. L. Bao and D. G. Truhlar, J. Am. Chem. Soc., 2016, 138, 14409 CrossRef CAS PubMed.
- C. Yin and K. Takahashi, Phys. Chem. Chem. Phys., 2018, 20, 20217 RSC.
- W. Chao, J. T. Hsieh, C. H. Chang and J. J. M. Lin, Science, 2015, 347, 751 CrossRef CAS PubMed.
- L. C. Lin, H. T. Chang, C. H. C. H. Chang, W. Chao, M. C. Smith, C. H. C. H. Chang, J. Min Lin Jr and K. Takahashi, Phys. Chem. Chem. Phys., 2016, 18, 4557 RSC.
- L. Sheps, B. Rotavera, A. J. Eskola, D. L. Osborn, C. A. Taatjes, K. Au, D. E. Shallcross, M. A. H. Khan and C. J. Percival, Phys. Chem. Chem. Phys., 2017, 19, 21970 RSC.
- S. Hatakeyama, H. Bandow, M. Okuda and H. Akimoto, J. Phys. Chem., 1981, 85, 2249 CrossRef CAS.
- K. Becker, J. Bechara and K. Brockmann, Atmos. Environ., Part A, 1993, 27, 57 CrossRef.
- P. Neeb and G. K. Moortgat, J. Phys. Chem. A, 1999, 103, 9003 CrossRef CAS.
- A. S. Hasson, M. Y. Chung, K. T. Kuwata, A. D. Converse, D. Krohn and S. E. Paulson, J. Phys. Chem. A, 2003, 107, 6176 CrossRef CAS.
- K. E. Leather, M. R. McGillen, M. C. Cooke, S. R. Utembe, A. T. Archibald, M. E. Jenkin, R. G. Derwent, D. E. Shallcross and C. J. Percival, Atmos. Chem. Phys., 2012, 12, 469 CrossRef CAS.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Science, 2012, 335, 204 CrossRef CAS PubMed.
- D. Stone, M. Blitz, L. Daubney, N. U. Howes and P. Seakins, Phys. Chem. Chem. Phys., 2014, 16, 1139 RSC.
- M. Nakajima and Y. Endo, J. Chem. Phys., 2015, 143, 164307 CrossRef PubMed.
- P. Aplincourt and M. F. Ruiz-Lopez, J. Am. Chem. Soc., 2000, 122, 8990 CrossRef CAS.
- R. Crehuet, J. M. Anglada and J. M. Bofill, Chem.–Eur. J., 2001, 7, 2227 CrossRef CAS PubMed.
- L. Chen, W. Wang, W. Wang, Y. Liu, F. Liu, N. Liu and B. Wang, Theor. Chem. Acc., 2016, 135, 1 Search PubMed.
- M. W. Wen, S. Hong, W. Fang, R. Zheng and Y. Qin, Theor. Chem. Acc., 2019, 138, 1 Search PubMed.
- K. Shao, J. Chen, Z. Zhao and D. H. Zhang, J. Chem. Phys., 2016, 145, 071101 CrossRef PubMed.
- B. Fu and D. H. Zhang, J. Chem. Theory Comput., 2018, 14, 2289 CrossRef CAS PubMed.
- R. Chen, K. Shao, B. Fu and D. H. Zhang, J. Chem. Phys., 2020, 152, 204307 CrossRef CAS PubMed.
- L. Vereecken, D. R. Glowacki and M. J. Pilling, Chem. Rev., 2015, 115, 4063 CrossRef CAS PubMed.
- E. Miliordos and S. S. Xantheas, Angew. Chem., 2016, 128, 1027 CrossRef.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson, H. Nakatsuji and et al., Gaussian 16, 2016 Search PubMed.
- H. Werner, P. Knowles, G. Knizia, F. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh and et al., 2018, see https://www.molpro.net.
- B. J. Braams and J. M. Bowman, Int. Rev. Phys. Chem., 2009, 28, 577 Search PubMed.
- B. Jiang and H. Guo, J. Chem. Phys., 2013, 139, 054112 CrossRef PubMed.
- Z. Xie and J. M. Bowman, J. Chem. Theory Comput., 2010, 6, 26 CrossRef CAS PubMed.
- Y.-L. Fu, X. Lu, Y.-C. Han, B. Fu, D. H. Zhang and J. M. Bowman, Chem. Sci., 2020, 11, 2148 RSC.
- Y.-L. Fu, Y. Bai, Y.-C. Han, B. Fu and D. H. Zhang, J. Phys. Chem. Lett., 2021, 12, 4211 CrossRef CAS PubMed.
- X. Lu, L. Li, X. Zhang, B. Fu, X. Xu and D. H. Zhang, J. Phys. Chem. Lett., 2022, 13, 5253 CrossRef CAS PubMed.
- X. Lu, C. Shang, L. Li, B. Fu, X. Xu and D. H. Zhang, Nat. Commun., 2022, 13, 4427 CrossRef CAS PubMed.
- M. T. Hagan and M. B. Menhaj, IEEE transactions on Neural Networks, 1994, 5, 989 CrossRef CAS PubMed.
- I. S. Ufimtsev, A. G. Kalinichev, T. J. Martinez and R. James Kirkpatrick, Phys. Chem. Chem. Phys., 2009, 11, 9420 RSC.
- S. A. King, J. Symb. Comput., 2013, 48, 101 CrossRef.
- A. Samanta and W. E, Communications in Computational Physics, 2013, 14, 265 CrossRef.
- J. Zheng, J. L. Bao, R. Meana-Pañeda, S. Zhang, B. Lynch, J. Corchado, Y. Chuang, P. Fast, W. Hu, Y. Liu, et al., Polyrate-2017, University of Minnesota, Minneapolis, MN, 2017 Search PubMed.
- R. Y. Rubinstein and D. P. Kroese, Simulation and the Monte Carlo method, John Wiley & Sons, 2016 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |