
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium catalysed C2 site-selective methylation of indoles using a pivaloyl directing group through weak chelation-assistance†
Subban Kathiravan *a,
Tianshu Zhangb and
Ian A. Nichollsb
*a,
Tianshu Zhangb and
Ian A. Nichollsb
aAttana AB, Greta Arwidssons Väg 21, 11419 Stockholm, Sweden. E-mail: suppan.kathiravan@lnu.se
bBioorganic & Biophysical Chemistry Laboratory, Linnaeus University Centre for Biomaterials Chemistry, Department of Chemistry & Biomedical Sciences, Linnaeus University, Kalmar SE-39182, Sweden
First published on 11th April 2023
Abstract
Here we present an iridium catalysed C2-selective methylation of indoles using methyltrifluoroborate as a source of methyl group. The iridium catalyst selectively discriminates the indole C2 and C4 C–H bonds by coordination with a pivaloyl directing group.
Numerous directing groups have been utilized to develop transition metal-catalysed C–C bond formation through C–H activation.1 However, despite these advances, site-selective C–H bond activation, particularly for bonds of similar character, remains a challenge for transition metal catalysis.2 This challenge has prompted the exploration of new synthetic disconnections and transformations. Indoles and their derivatives play a significant role as structural components in both medicinal chemistry and materials science.3 Among them, C2 functionalized indoles are particularly noteworthy due to their prevalence in bioactive natural products and drugs, making new selective synthesis strategies extremely desirable to synthetic and medicinal chemists (Fig. 1).4
 | ||
| Fig. 1 Examples of bioactive C2-functionalized indole derivatives. | ||
Over the last decade, several techniques have been devised to enable C2 C–H functionalization of indoles.5 One well explored approach involves incorporating a directing group on the indole nitrogen to provide C2 selectivity.6 Furthermore, directing groups at the C3 position have been used as tools for targeting the C4 and C2 positions,7 where competition between the formation of five- and six-membered metallocycle is possible, though favoring functionalization at C4 over C2. Accordingly, methods for the regioselective functionalization of the C2 C–H bond in the presence of the C4 C–H bond is a challenge, and most attractive due their potential in synthetic applications. Previous efforts towards this goal have included an iridium-catalyzed C2/C4 regioselective heteroarylation, developed by You et al. that employed a pivaloyl directing group and carefully selected catalytic systems.8 In a separate study, Ravikumar et al. reported a cobalt catalyzed regioselective C4 C–H functionalization of 3-pivaloyl indole, where the directing group.9 Li et al. demonstrated the possibility of rhodium-catalyzed site-selective coupling of indoles with diazo esters to achieve C4 alkylation versus C2 annulation.10 Moreover, Shi et al. have established an effective strategy for site-selective C4 arylation, utilizing a pivaloyl directing group positioned at the C3 position.11 Additionally, Prabhu and colleagues have shown the crucial role of the electronic nature of the ketone directing groups in regulating the C2 and C4 functionalization of indoles.12
The introduction of the magic methyl group through transition metal catalyzed C–H methylation has been the focus of extensive research effort due to the importance of methylation for exploring the pharmacological properties of organic molecules.13 This fundamental research has had a profound impact on the design of drug candidates with tailored physical properties in medicinal chemistry. According to a 2018 survey conducted by Njardarson, at least 73% of the top 200 marketed drugs contain a methyl substituent.14 Various transition metals have been utilized in ortho-C–H methylation reactions using directing group strategies, with MeB(OH)2, MeBF3K, Me3BO3, DTBP, DCP, MeI, thiophenium salt serving as methyl sources.15 For instance, Ackermann et al. have reported a cationic ruthenium(II)-complex enabled C2-C–H methylation on indoles using 2-pyrdine as a strongly coordinating directing group.16 Li and co-workers developed rhodium(III) catalyzed C2-C–H methylation of arenes with alkylboron reagents.17 To the best of our knowledge transition metal catalyzed regioselective C2 methylation of indoles with a directing group at the C3 position has not previously been demonstrated. Herein we disclose an efficient protocol via bench stable [Cp*IrCl2]2 catalyzed C2 selective C–H methylation using commercially available potassium trifluoromethylborate as a methyl source under mild reaction conditions and with wide substrate scope.
Recently, we reported a tandem iridium-catalysed decabonylative C–H activation of indoles using a sacrificial electron rich ketone for bis-arylsulfenylation reactions.18 Using this efficient iridium(III) catalytic system as a starting point, we pondered whether the catalytic conditions could be applicable to the C2-selective methylation of indoles by switching the inherently preferred regioselectivity. We initiated our studies with a screening of potential directing groups (ESI, Table S1,†) using potassium trifluoromethyl borate (2) to investigate potential for the iridium catalysed C2-selective methylation reactions. The dominant 3-pivaloyl-N-methyl indole (1a) was used in subsequent studies (Table 1).
|
||
|---|---|---|
| Variation from standard conditions | ||
| a Reaction conditions: 1 (0.34 mmol), 2 (5 equiv.), [IrCp*Cl2]2 (5 mol%), AgNTf2 (20 mol%), AgOAc (2 equiv.), 1,2-DCE (1 mL), 115 °C, 18 h, N2 atmosphere; NR = no reaction; yields are isolated yields.b Isolated yields.c 50% dissolved in THF (5 equiv.). | ||
| 1 | None | 89%b |
| 2 | HFIP as solvent | 76% |
| 3 | TFE as solvent | 75% |
| 4 | t-AmylOH as solvent | 70% |
| 5 | Acetonitrile as solvent | NR |
| 6 | DME as solvent | 58% |
| 7 | No AgNTf2 | NR |
| 8 | Reaction at 80 °C | 75% |
| 9 | Reaction at RT | Trace |
| 10 | MeB(OH)2 instead of MeBF3K | NR |
| 11 | Me(BO)3 instead of MeBF3K | 49%c |
| 12 | Under air | 80% |
| 13 | [Ru(p-cymene)Cl2]2 as catalyst | NR |
| 14 | Pd(OAc)2 | NR |
| 15 | [Rh(Cp*Cl2)]2 as catalyst | 67% |
| 16 | [Co(Cp*CO)I2] as catalyst | NR |
| 17 | No AgOAc | NR |
| 18 | Ag2CO3 instead of AgOAc | 63% |
| 19 | Ag2O instead of AgOAc | NR |
| 20 | No [IrCp*Cl2]2 | NR |
| 21 | 3 equiv. of MeBF3K | 80% |

The best identified conditions for this reaction were 5 mol% of [Cp*IrCl2]2 as catalyst, 20 mol% of silver bistrifluoromethanesulfonate as additive, 2 equivalents of silver acetate as oxidant, in 1,2-dichloroethane as solvent at 115 °C for 23 h under nitrogen atmosphere. To our delight the desired product was obtained in 89% isolated yield (Table 1, entry 1). When we screened fluoro-alcohol-based solvents, which are known to promote C–H activation reactions19 the isolated yields decreased from 89% to 70% (Table 1, entries 2–3). Reactions performed using the solvents t-amylOH, acetonitrile and dimethoxyethane all produced lower yields (Table 1, entries 4–6). As expected, the absence of a silver additive resulted in no product formation, as it is crucial for the formation of the iridium complex with the indole substrate (Table 1, entry 7). Conducting the reaction at lower temperature (80 °C) did not improve the yield and at RT the reaction was highly compromised, producing only traces of the product (Table 1, entries 8–9). Next, we focused our attention on screening potential methyl donors. While we didn't observe product formation with methylboronic acid, due to poor solubility, the soluble boroxine delivered the product in 49% yield (Table 1, entry 10–11). When reactions were performed under air, the product was obtained in 80% yield, which is sufficient for use in up-scaled syntheses (Table 1, entry 12). On account of the cost of iridium, less expensive ruthenium and palladium catalysts were tested, though found to be impotent with respect to the reaction under these conditions used (Table 1, entries 13–14). In the case of rhodium(III) catalysis, the product was obtained in 67% yield (Table 1, entry 15). Unfortunately, the non-noble metal Co(III) catalyst did not support the reaction (Table 1, entry 16). Moreover, in the absence of AgOAc no product was obtained (Table 1, entry 17), and the use of silver carbonate in place of silver acetate resulted in lower yield. Even the use of silver oxide was not efficient (Table 1, entries 18–19). Control experiments verified that the iridium catalyst and an excess of potassium trifluoromethyl borate are both essential to obtain the very good, isolated yields (Table 1, entries 20–21).
Next, with optimized conditions in hand, we explored the substrate scope using potassium methyltrifluoroborate and its use for targeting a range of methylated indole derivatives was investigated with respect to substitution of the arene ring (Scheme 1). Initial studies showed that the unsubstituted indole could react under the standard reaction conditions, furnishing the C4-methylated indole (3a) in good yield. Subsequent studies with a range of substituents indicated a subtle interplay between steric and electronic effects on reaction yields. The presence of a strongly electron donating methoxy substituent at the 5-position of indole gave the desired product (3b) in 39% yield with a comparable yield in the case of reactions performed with the 6-methoxy derivative (3c, 31%), albeit lower than that of the unsubstituted indole. In contrast, electron-withdrawing substituents were observed to be better tolerated, with the 5- and 6-methyl carboxylate substituted indoles (1d–1e) affording the C4-methylated products (3d–3e) in 80% and 60% yield, and the NO2 group substituted indole yielding the product (3f) in 60% yield. However, the electron withdrawing cyano (CN) group at 5-position gave low yield (30%, 3g), this is suspected to arise from competition by the cyano group for coordination with the metal center. Some support for this hypothesis is provided by studies using halogens at the 5- and 6-position, where the fluoro (1h), chloro (1i–j), bromo (1k) and iodo (1l) derivatives, afforded good to very good yields (3h–l, 61–78%) of the corresponding methylated products. The ready accessibility of halogenated derivatives highlights the potential of this methodology for applications in late-stage transformations. A fused ring substrate (1m) delivered the corresponding expected product (3m) in moderate yield. C7 bromo- and methyl-substituted indoles afforded the products (3n–o) in good yield. Subsequently the effect of substituents on the nitrogen was studied (1p–1u). Interestingly, and to our surprise, the N-ethyl indole provided C4 methylated product (3p) in 96% isolated yield. When we used N-pentyl and N-hexyl substituted indoles, the products (3q–r) were isolated in 61% and 84% respectively. Finally, various benzyl and phenyl derivatives were also incorporated to provide the target products (3s–3w) in very good to moderate yields.
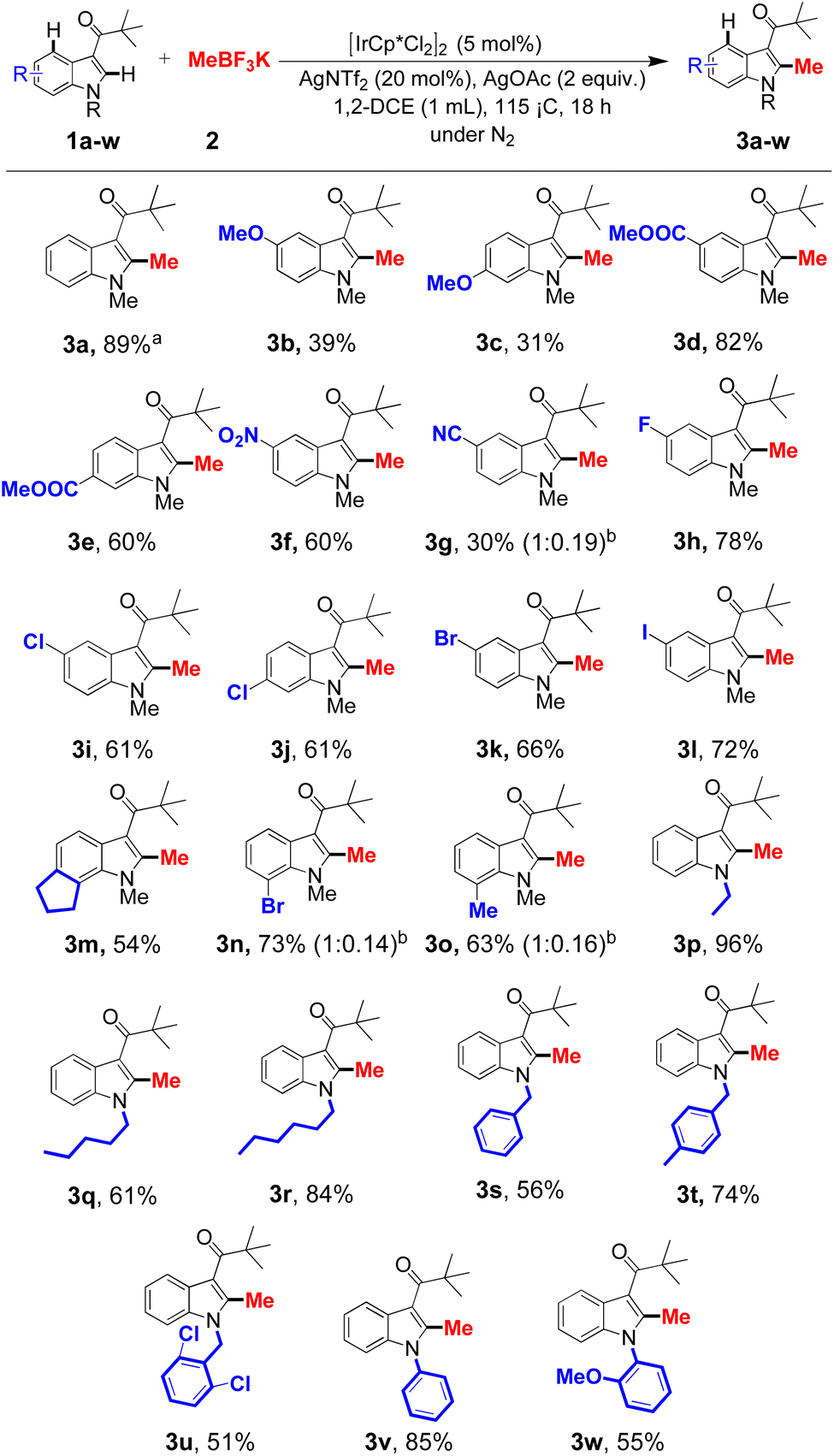 | ||
| Scheme 1 Reaction conditions: 1 (0.34 mmol), 2 (5 equiv.), [IrCp*Cl2]2 (5 mol%), AgNTf2 (20 mol%), AgOAc (2 equiv.), 1,2-DCE (1 mL), 115 °C, 18 h, N2 atmosphere. aIsolated yields. bRatio of C2 & C4 | ||
We then focussed on the mechanism of the iridium catalysed methylation reaction using a series of isotope experiments (Scheme SI-1†). Based upon our supporting experimental findings together with previous studies,18 a plausible catalytic cycle for the iridium(III) catalysed methylation reaction can be presented (Fig. 2). The first step being the coordination of the iridium catalyst by the pivaloyl carbonyl group through weak coordination, followed by the formation of either a five- or a six-membered iridiacycle via C–H metalation at C2 and C4 positions, respectively. However, the iridium catalyst preferably defaults to the energetically more favourable five-membered iridacycle for the subsequent transmetalation with potassium methyltrifluoroborate (2). Thereafter, the reductive elimination liberates the product (3) and finally the reduced iridium(I) was re-oxidized with silver to regenerate the catalytically active iridium(III) catalyst.
 | ||
| Fig. 2 Plausible reaction mechanism. | ||
In conclusion, we have developed the first iridium(III) catalyzed C2-selective methylation of an indole, with the reaction performed via a transmetalation reaction. The iridium catalyst, with the help of a simple carbonyl directing group, facilitates the preferential transformation of the five-membered iridacycle over its six-membered counterpart, thus providing discrimination between two indole C–H bonds. Mechanistic studies indicated that the C–H bond iridium metalation step is reversible. Collectively, this opens for the possibility to use this reaction as a tool for use in the efficient, selective methylation reaction in drug development and other areas. Further studies to apply this catalysis to various other substrates is undergoing in our laboratory.
Author contributions
SK and TZ performed the synthesis and analysis. IAN and SK conceived the project. All authors contributed to the writing of the manuscript and gave approval for submission.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Linnaeus University, the Swedish Research Council, (2014-4573), the Crafoord Foundation (2019-0925) (2020-0775) and the Helge Ax:son Johnsons Foundation (2019-0318 and 2022-0317) for generous funding.Notes and references
- R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef CAS PubMed; T. Gensch, M. N. Hopkinson, F. Glorius and J. W. Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC; D. Alberico, M. E. Scott and M. Lautans, Chem. Rev., 2007, 107, 174–238 CrossRef PubMed; O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef PubMed; Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef PubMed; C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef PubMed; Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef PubMed; C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef PubMed; K. Padala and M. Jeganmohan, Org. Lett., 2011, 13, 6144–6147 CrossRef PubMed; R. B. Bedford, S. J. Durrant and M. Montgomery, Angew. Chem., Int. Ed., 2015, 54, 8787–8790 CrossRef PubMed; Y.-J. Wang, C.-H. Yuan, D.-Z. Chu and L. Jiao, Chem. Sci., 2020, 11, 11042–11054 RSC; V. K. Tiwari and M. Kapur, Org. Biomol. Chem., 2019, 17, 1007–1026 RSC; L. Ping, D. S. Chung, J. Bouffard and S.-G. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC; L. Han, X. Ma, Y. Liu, Z. Yu and T. Liu, Org. Chem. Front., 2018, 5, 725–733 RSC.
- Y. Wan, Y. Li, C. Yan, M. Yan and Z. Tang, Eur. J. Med. Chem., 2019, 183, 111691 CrossRef CAS PubMed; R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef PubMed; C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef PubMed; M.-Z. Zhang, Q. Chen and F.-F. Yang, Eur. J. Med. Chem., 2015, 89, 421–441 CrossRef PubMed; A. J. Kochanowsks-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef PubMed; S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef PubMed; D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef PubMed; M. d'Ischia, A. Napolitano and A. Pezzella, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 3, pp. 353–388 CrossRef PubMed; G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef PubMed.
- Y. Wan, Y. Li, C. Yan, M. Yan and Z. Tang, Eur. J. Med. Chem., 2019, 183, 111691 CrossRef CAS PubMed; R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef PubMed; H. Lu, G. Zhu, T. Tang, Z. Ma, Q. Chen and Z. Chen, iScience, 2019, 22, 214–218 CrossRef PubMed.
- D. R. Stuart, E. Villemure and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 12072–12073 CrossRef CAS PubMed; Y. Xu, X. Zhou, G. Zheng and X. Li, Org. Lett., 2017, 19, 5256–5259 CrossRef PubMed; E. M. Back, N. P. Grimster, R. Hatley and M. J. Gaunt, J. Am. Chem. Soc., 2006, 128, 2528–2529 CrossRef PubMed; R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172–8174 CrossRef PubMed.
- L. Jiao, E. Herdtweck and T. Bach, J. Am. Chem. Soc., 2012, 134, 14563–14572 CrossRef CAS PubMed; C. Wang, B. Maity, L. Cavallo and M. Rueping, Org. Lett., 2018, 10, 3105–3108 CrossRef PubMed; G. S. Kumar and M. Kapur, Org. Lett., 2016, 18, 1112–1115 CrossRef PubMed; K. Ramachandran, and P. Anbarasan, Org. Lett. 2022, 24, 6745–6749 CrossRef PubMed; Z. Ding and N. Yoshikai, Angew. Chem., Int. Ed., 2012, 51, 4698–4701 CrossRef PubMed; T. A. Shah, P. B. De, S. Pradhan, S. Banerjee and T. Punniyamurthy, J. Org. Chem., 2019, 84, 16278–16285 CrossRef PubMed; L. Ackermann and A. V. Lygin, Org. Lett., 2011, 13, 3332–3335 CrossRef PubMed; V. Soni, R. A. Jagtap, R. G. Gonnade and B. Punji, ACS Catal., 2016, 6, 5666–5672 CrossRef; Z.-Z. Zhang, B. Liu, C.-Y. Wang and B.-F. Shi, Org. Lett., 2015, 17, 4094–4097 CrossRef PubMed; Z.-L. Yan, W.-L. Chen, Y.-R. Gao, S. Mao, Y.-L. Zhang and Y.-Q. Wang, Adv. Synth. Catal., 2014, 356, 1085–1092 CrossRef; A. G. Rubia, R. G. Arrayas and J. C. Carretero, Angew. Chem., Int. Ed., 2009, 48, 6511–6515 CrossRef PubMed; Z. Wang, F. Song, Y. Zhao, Y. Huang, L. Yang, D. Zhao, J. Lan and J. You, Chem.–Eur. J., 2012, 18, 16616–16620 CrossRef PubMed.
- M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed; H. A. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403–2435 CrossRef; M. Petrini, Chem.–Eur. J., 2017, 23, 16115–16151 CrossRef PubMed; J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618–5627 CrossRef; J. Kalepu, P. Gandeepan, L. Ackermann and L. T. Pilarski, Chem. Sci., 2018, 9, 4203–4216 RSC; J. Wen and Z. Shi, Acc. Chem. Res., 2021, 54, 1723–1736 CrossRef PubMed; S. Chen, B. Feng, X. Zheng, J. Yin, S. Yang and J. You, Org. Lett., 2017, 19, 2502–2505 CrossRef PubMed; C. Pan, G. Huang, Y. Shan, Y. Li and J.-T. Yu, Org. Biomol. Chem., 2018, 18, 3038–3042 RSC.
- S. Chen, M. Zhang, R. Su, X. Chen, B. Feng, Y. Yang and J. You, ACS Catal., 2019, 9, 6372–6379 CrossRef CAS.
- S. K. Banjare, T. Nanda, B. V. Pati, G. K. D. Adhikari, J. Dutta and P. C. Ravikumar, ACS Catal., 2021, 11, 11579–11587 CrossRef CAS.
- X. Chen, G. Zheng, Y. Li, G. Song and X. Li, Org. Lett., 2017, 19, 6184–6187 CrossRef CAS PubMed.
- Y. Yang, P. Gao, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 3966–3971 CrossRef CAS PubMed.
- V. Lanke, K. R. Bettadapur and K. R. Prabhu, Org. Lett., 2016, 18, 5496–5499 CrossRef CAS PubMed.
- E. J. Barreiro, A. E. Kummerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed; H. Schcnherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed; S. Sun and J. Fu, Bioorg. Med. Chem. Lett, 2018, 28, 3283–3289 CrossRef PubMed; L. F. T. Novaes, J. S. K. Ho, K. Mao, K. Liu, M. Tanwar, M. Neurock, E. Vilemure, J. A. Terrett and S. Lin, J. Am. Chem. Soc., 2022, 144, 1187–1197 CrossRef PubMed; S. D. Friiss, M. J. Johansson and L. Ackermann, Nat. Chem., 2020, 12, 511–519 CrossRef PubMed.
- E. Vitaku, E. A. Ilardi and J. T. Njardarson, Top 200 Small Molecule Pharmaceuticals by Retail Sales in 2021, 2021, https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/Top%20200%20Small%20Molecule%20Pharmaceuticals%202018_0.pdf, accessed on 2022-06-07.
- D. Aynetdinova, M. C. Callens, H. B. Hicks, C. Y. X. Poh, B. D. A. Shennan, A. M. Boyd, Z. H. Lim, J. A. Leitch and D. J. Dixon, Chem. Soc. Rev., 2021, 50, 5517–5563 RSC.
- M. D. L. Tonin, D. Zell, V. Muller and L. Ackermann, Synthesis, 2017, 49, 127–134 CAS.
- H. Wang, S. Yu, Z. Qi and X. Li, Org. Lett., 2015, 17, 2812–2815 CrossRef CAS PubMed.
- S. Kathiravan, P. Anaspure, Z. Tianshu and I. A. Nicholls, Org. Lett., 2021, 23, 3331–3336 CrossRef CAS PubMed.
- T. Bhattacharya, A. Ghosh and D. Maiti, Chem. Sci., 2021, 12, 3857–3870 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR data, and spectra. See DOI: https://doi.org/10.1039/d3ra02031b |
| This journal is © The Royal Society of Chemistry 2023 |